

Structural snapshots characterize six hundred million years of evolution of intrinsically disordered proteins.
Abstract
In every established species, protein-protein interactions have evolved such that they are fit for purpose. However, the molecular details of the evolution of new protein-protein interactions are poorly understood. We have used nuclear magnetic resonance spectroscopy to investigate the changes in structure and dynamics during the evolution of a protein-protein interaction involving the intrinsically disordered CREBBP (CREB-binding protein) interaction domain (CID) and nuclear coactivator binding domain (NCBD) from the transcriptional coregulators NCOA (nuclear receptor coactivator) and CREBBP/p300, respectively. The most ancient low-affinity “Cambrian-like” [540 to 600 million years (Ma) ago] CID/NCBD complex contained less secondary structure and was more dynamic than the complexes from an evolutionarily younger “Ordovician-Silurian” fish ancestor (ca. 440 Ma ago) and extant human. The most ancient Cambrian-like CID/NCBD complex lacked one helix and several interdomain interactions, resulting in a larger solvent-accessible surface area. Furthermore, the most ancient complex had a high degree of millisecond-to-microsecond dynamics distributed along the entire sequences of both CID and NCBD. These motions were reduced in the Ordovician-Silurian CID/NCBD complex and further redistributed in the extant human CID/NCBD complex. Isothermal calorimetry experiments show that complex formation is enthalpically favorable and that affinity is modulated by a largely unfavorable entropic contribution to binding. Our data demonstrate how changes in structure and motion conspire to shape affinity during the evolution of a protein-protein complex and provide direct evidence for the role of structural, dynamic, and frustrational plasticity in the evolution of interactions between intrinsically disordered proteins.
INTRODUCTION
The web of protein-protein interactions that governs cell function is very complex, often involving competing interactions as well as spatial and temporal regulation of the proteins. Since most of this regulation has evolved over tens or hundreds of millions of years, it is likely that any new protein-protein interaction resulting from, for example, gene duplication, point mutation, horizontal gene transfer, or up-regulation of expression will interfere with the biochemistry of the cell in a nonbeneficial way and disappear through purifying selection. However, sometimes, new interactions are sufficiently beneficial to be retained in the population by natural selection. This is what happened sometime between 600 and 540 million years (Ma) ago for two intrinsically disordered protein (IDP) domains: the nuclear coactivator binding domain (NCBD) from CREB-binding protein [CREBBP, also called CBP, and its paralog p300 (CREBBP/p300)] and the CREBBP interaction domain (CID) from the nuclear coactivator protein [nuclear receptor coactivator 1 (NCOA1), NCOA2, and NCOA3, also called Src1, Tif2, and ACTR, respectively]. The CREBBP/p300 and NCOA proteins contain histone acetyl transferase domains and are involved in transcriptional regulation, for example, by activating steroid receptors (1). The tighter association between NCOA and CREBBP/p300 likely resulted in a more efficient transcription, which was sufficiently beneficial to be retained by natural selection in an ancestral population leading to present-day deuterostome animals.
Little is known about the molecular details of protein evolution as it happened in nature. Today, the large number of genome and transcriptome sequencing efforts of extant species allows reconstruction of ancient proteins from evolutionary ancestors, which provides the possibility of studying the putative historical sequence of events leading up to present-day proteins (2–5). We recently published the reconstruction, biophysical characterization, and molecular dynamics simulation of CID and NCBD domains from animal species living approximately 390 to 600 Ma ago (6) and found that the affinity between CID and NCBD was relatively low in an early deuterostome ancestor, which likely existed at some point 540 to 600 Ma ago (i.e., before or at the beginning of the Cambrian period). However, the affinity of the CID/NCBD protein-protein complex had probably increased, along with decreased conformational heterogeneity, to that of present-day CID/NCBD complexes already in an early ancestor of present-day fish living around or before 500 Ma ago during Cambrian and at the end of Ordovician/beginning of Silurian at the time of the two whole-genome duplications in the vertebrate lineage around 440 Ma ago. This affinity was maintained in the bony fish/tetrapod ancestor 390 Ma ago and is retained in present-day species (6).
To gain a deeper insight into the molecular details shaping the evolution of this protein-protein interaction, we have used here nuclear magnetic resonance (NMR) to obtain structures of three historical CID/NCBD complexes and analyzed the dynamics of the respective interaction by Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion NMR spectroscopy. We find that the most ancient Cambrian-like CID/NCBD complex lacks some of the structural features of the younger complexes and that there are a reduction and reshuffling of the backbone dynamics as well as local frustration going from the low-affinity ancestral to the younger Ordovician-Silurian/extant human complexes. Furthermore, isothermal titration calorimetry (ITC) experiments show opposing enthalpic and entropic contributions to the total affinity. The structural heterogeneity and plasticity observed in our evolutionary snapshots are consistent with an affinity sensitive to changes in local frustration distribution and thermodynamics and shed light on fundamental questions regarding the evolution of protein-protein interactions.
RESULTS
Structures of CID/NCBD complexes through evolution
To investigate how structure and conformational dynamics modulate protein function in natural evolution, we solved three protein complexes of CID/NCBD domains spanning roughly 600 Ma of evolution (Protein Data Bank codes 6ES5, 6ES6, and 6ES7). The ancient NCBD and CID domain sequences were previously reconstructed, and the resurrected proteins were subjected to a biophysical analysis (6). Using our previous nomenclature, we denote these domains D/P NCBD, which was present in the common ancestor of present-day Eubilateria [i.e., deuterostome (D) and protostome (P) animals], a creature that likely lived at some point 540 to 600 Ma ago; 1R/2R NCBD and 1R CID, which were present in fish that lived close to the transition between Ordovician and Silurian ~440 Ma ago (1R and 2R are the first and second round of the whole-genome duplications that took place in the vertebrate lineage that later diversified into present-day vertebrates such as jawed fish, reptiles, and mammals) (7); and CREBBP NCBD and NCOA3 CID, which are two extant human domains (Fig. 1A). Since the CID domain could not be identified in species from protostome phyla (e.g., Arthropoda and Mollusca) and sequences were too few and different in the deuterostome phyla Echinodermata (e.g., sea stars and sea urchins) and Hemichordata (e.g., acorn worms), it was not possible to reconstruct a CID domain older than 1R and as ancient as the D/P NCBD. It is also important to point out that any reconstructed sequence is an approximation based on gene variants from selected present-day species for which sequence data are available. Nevertheless, on the basis of the data on alternative ancient variants, we deem our overall conclusion robust, namely, that the Cambrian-like complex has a significantly lower affinity than the two younger complexes around neutral pH (KD, ~1 μM versus 100 nM; table S1). Thus, the three complexes analyzed in the present work consisted of the most likely CID and NCBD variants: (i) 1R CID with D/P NCBD (most ancient Cambrian-like complex), (ii) 1R CID with 1R/2R NCBD (the Ordovician-Silurian 1R/2R complex), and (iii) extant human CID/NCBD complex (from NCOA3 and CREBBP). Note that the two whole-genome duplications 1R and 2R occurred in a relatively short but undefined time span. Thus, while it is theoretically possible to reconstruct two CID variants (since three of four NCOA genes are present today), in practice, the 1R/2R should be treated as one evolutionary node. In addition, ITC binding data show that the affinities of 1R and 2R CID for 1R/2R NCBD are similar (6).
Fig. 1. Reconstructed ancestral sequences, phylogenetic tree ancestral, and extant CID/NCBD complexes.
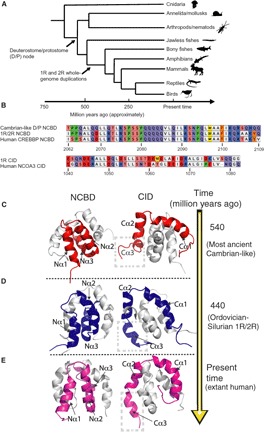
(A) The sequences were previously reconstructed (6) using 181 (NCBD) and 184 (CID) protein sequences from present-day protostome and deuterostome species of the animal phyla, superclasses, and classes depicted in the simplified phylogenetic tree. (B) The complexes used in this study are (i) 1R CID with D/P NCBD (most ancient Cambrian-like complex), (ii) 1R CID with 1R/2R NCBD (the Ordovician-Silurian 1R/2R complex), and (iii) extant human CID/NCBD complex (from NCOA3 and CREBBP, respectively). Note that the timing of the whole-genome duplications 1R and 2R is not resolved and might predate the divergence of jawed and jawless vertebrates (25). Animals were downloaded from phylopic.org. (C) The most ancient Cambrian-like complex (red) contains D/P NCBD and 1R CID. (D) The Ordovician-Silurian 1R/2R complex (blue) contains 1R/2R NCBD and 1R CID. (E) The extant human complex (magenta) contains human CREBBP NCBD and NCOA3 CID. In the three left panels, the solved structures of NCBD domains are displayed in cartoon representation and colored, while the CID domains are cartoon and gray. In the three right panels, the solved structures of CID domains are displayed as colored cartoons, and the NCBD domains are gray cartoons. The complexes in the left and right panels are rotated 180° with respect to each other.
First, we determined the structure of the extant human CID/NCBD complex by NMR using nuclear Overhauser effect (NOE) distance restraints, measured φ dihedral angles restraints from scalar couplings, chemical shift–derived φ/ψ dihedral angles, and 1D H-N residual dipolar couplings (RDCs) (8). The use of RDCs in structure calculations improves the overall quality by restraining the relative orientation of the structural elements (9). Comparison to a previously published NCOA3 CID/CREBBP NCBD structure (10) shows that the domains fold into similar overall structures with comparable compactness and total solvent-accessible surface area (SASA) [5730 Å2 (previous structure) versus 5940 Å2 (our structure)]. A few minor differences between the structures likely result from the different lengths of the CID and NCBD constructs used in our structure compared to the previous one.
We then solved the structures of the most ancient Cambrian-like complex and the Ordovician-Silurian 1R/2R complex to understand how the CID/NCBD interaction has evolved over time. The overall topology of the resurrected ancient complexes was similar to that of the extant human one (Fig. 1, C to E). Furthermore, all three complexes had a rotational correlation time (τc) of approximately 5 ns, as expected for a complex of this size and indicating that the domains gain structure upon binding. This is markedly different from the free CID domains, which had rotational correlation times of less than 1 ns, consistent with a disordered structure (11). We also observe that the SASA decreased from the Cambrian-like (7280 Å2) to the Ordovician-Silurian complex (6210 Å2) and the extant human complex (5940 Å2) (Fig. 2). This change in compactness is associated with changes in the orientation of the first and third α helices of NCBD (Nα1 and Nα3, respectively) and the third α helix of CID (Cα3) (Figs. 1 and 2 and fig. S1). 1R CID interacts with the Cambrian-like D/P NCBD using only Cα1 and Cα2, while, upon binding to Ordovician-Silurian 1R/2R NCBD, the third helix Cα3 makes more contacts with Nα2 of NCBD, permitting it to adopt a more helical structure (Figs. 1 to 3 and figs. S1 to S3). On more detailed inspection, we find that the additional contacts in this region in the extant human CID/NCBD complex result in an increase in helicity of both Nα3 and Cα3 (Figs. 2 and 3 and figs. S1 to S3). These additional structural elements increase the hydrophobic interaction area between CID and NCBD and provide new networks of interactions within the younger higher-affinity CID/NCBD complexes. For example, in the most ancient complex, Leu2074, Lys2075, Ser2076, Leu2087, and Ile2101 of Cambrian-like NCBD make direct contacts (as measured by NOEs) with Leu1049, Ile1050, and Ile1073 of 1R CID. In the Ordovician-Silurian 1R/2R complex, the number of detected direct NOEs tripled to 18, notably interactions between Ile2062, Leu2067, Gln2068, Lys2072, Gln2095, Gln2103, and Ala2105 of Ordovician-Silurian 1R/2R NCBD and Asp1044, Glu1045, Leu1048, Leu1056, Leu1067, and Gln1080 of 1R CID. These interactions are maintained in the extant human complex except Asn1079 of NCOA3 CID taking the place for Gln1080 of 1R CID. These results are further supported by the change in the amide chemical shift (ΔδHN), which shows increased bending of the Nα3 and Cα3 helices as they displayed alteration of the HN shift in a wave-like manner (fig. S4). Because bending will result in shorter hydrogen bonds between the carbonyl and amide groups of residues i, i + 4 at the concave side and longer hydrogen bonds at the convex side, amides at the concave side will show positive ΔδHN while those at the convex side will show more negative values (12). This behavior results in repeats of the amide proton secondary chemical shifts with periodicity of three to four along the sequence.
Fig. 2. Evolutionary snapshots highlighting structural heterogeneity.
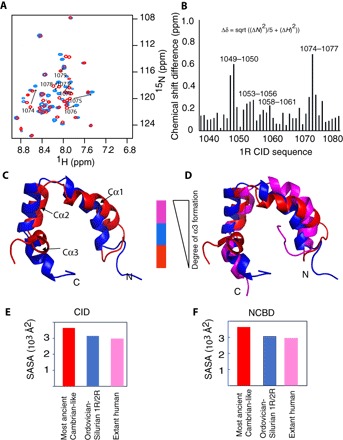
(A) Overlay of the heteronuclear single-quantum correlation spectra for 1R CID bound to Cambrian-like NCBD (red) and Ordovician-Silurian NCBD (blue). Arrows indicate the peak shifts of the amino acid residues 1073 to 1079 corresponding to the CID Cα3 helix (for clarity, only the assignments of residues 1073 to 1079 are shown; complete assignments can be found in fig. S7). In the most ancient low-affinity complex with Cambrian-like NCBD, these residues remain unstructured. However, in the younger high-affinity Ordovician-Silurian 1R/2R complex, the increased number of interactions in the region leads to increased α-helical content of Cα3 (see Fig. 4). The increase in dispersion for residues 1074 to 1077 is accompanied by an increase in the number of inter- and intramolecular NOEs (i, i + 3, indicative of helices; figs. S3 and S4), indicating that this region becomes more structured. ppm, parts per million. (B) Root mean square deviations (RMSDs) between the combined HN/N chemical shifts for the complexes of 1R CID with Cambrian-like D/P NCBD and Ordovician-Silurian 1R/2R NCBD, respectively. Large structural changes are seen for residues Leu1049 and Asp1050 located in Cα1 and for residues Asp1074, Lys1075, Leu1076, and Val1077 in Cα3. sqrt, square root. (C) Superposition of structures of 1R CID bound to Cambrian-like NCBD (red) and Ordovician-Silurian NCBD (blue), respectively. (D) 1R CID bound to Cambrian-like NCBD (red) and Ordovician-Silurian NCBD (blue), and human NCOA3 CID bound to CREBBP NCBD (magenta). For clarity, only the CID domains are displayed in (C) and (D). (E and F) Plots of total SASA calculated from the respective complex and plotted separately for each domain, CID (E) and NCBD (F).
Fig. 3. Evolution of Cα3 and Nα3 helical content and conformational rearrangements within the Cα3 helix of CID domains during evolution.
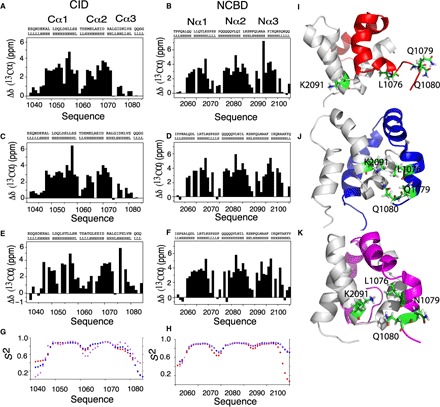
Plots of the difference of experimental Cα shifts from those of random coil values as a function of the amino acid sequences and TALOS prediction for ancestral and extant CID domains (A, C, and E) and NCBD domains (B, D, and F). The height of the bars indicates the degree of α helix formed with zero, indicating no α-helical content. There is a general increase in helical content for Cα3 and Nα3 as we evolve from Cambrian-like and Ordovician-Silurian (1R/2R) complexes to the present-day human CID/NCBD complex. (G and H) Chemical shift–based S2 values show that the flexibility in the Cα3 and Nα3 helices is higher in the most ancient low-affinity Cambrian-like complex (red) than in the younger high-affinity Ordovician-Silurian 1R/2R (blue) and present-day human (magenta) complexes. Specific interactions and rearrangements of K2091 of Nα2 and L1076, Q1079, and Q1080 of Cα3 (side chains are numbered and colored green) for (I) Cambrian-like D/P NCBD (gray) with 1R CID (red), (J) Ordovician-Silurian 1R/2R NCBD bound to 1R CID (blue), and (K) the extant human complex between CREBBP NCBD and NCOA3 CID (magenta). The three highlighted Cα3 residues are mainly solvent exposed in the most ancient Cambrian-like complex, while specific interactions are seen in both the Ordovician-Silurian 1R/2R and the extant human complex. For clarity, the structures have been slightly reoriented in relation to each other to show the specific interactions.
Further interesting details are provided by the side chains of residues 1070 to 1080, which are partly solvent-exposed and remain partially unstructured in the most ancient Cambrian-like complex (Figs. 2A and 3 and fig. S1) but form part of the interaction surface in the Ordovician-Silurian 1R/2R complex (Fig. 3H). The interaction surface is further increased by interactions involving helix Nα3 of the two younger NCBD domains. Specifically, Ala2105 and Lys2107 of 1R/2R NCBD interact with Ile1073 and Asp1068 of 1R CID, respectively, and Thr2105 of human CREBBP NCBD forms an interaction with His1053 of NCOA3 CID. In the most ancient complex, residues in Nα3 make interactions with only part of Cα3 of 1R CID, while most of the Cα3 remains exposed to the solvent. Thus, the three historical complexes demonstrate how an extensive reorganization of structure and interactions, rather than specific side-chain substitutions in an intact structure, can lead to a higher affinity in a protein-protein complex.
Redistribution of motions accompanies the evolution of higher affinity in the CID/NCBD complex
Since dynamics on different time scales is an inherent property of proteins and a modulator of protein function, we next examined the evolution of dynamics within the three complexes using NMR relaxation dispersion CPMG experiments. These experiments can probe slow motions (i.e., in the microsecond-to-millisecond time scale) that are often relevant for function (13). Equilibrium interconversion between different protein conformations in this time window will frequently result in an exchange contribution to the transverse relaxation rate constant (Rex), causing additional line broadening of the respective NMR spectral resonances with respect to the fast motion–driven R0. The exchange contributions, Rex, depend on the relative populations of the exchanging states, the chemical shift difference (Δω) between them, and the rate constant of exchange (kex) (Eq. 1). We observed that the Cambrian-like complex exhibited a varying degree of slow motions over the entire protein sequences (Fig. 4). In the Ordovician-Silurian 1R/2R complex, slow motions were markedly reduced in CID with a few exceptions. In particular, helices Cα1, Cα2, and Cα3 contribute little or no slow motions. In the extant human complex, slow motions appear in other locations, resulting in conformational dynamics mostly in the loop region between Cα1 and Cα2 and in the entire Cα3 region, which experiences increased Rex values. The chemical shift–derived order parameters, which are sensitive to motions faster than the inverse chemical shift difference in Hertz, (ca. 10 ms), also showed increased millisecond motions in this region (Fig. 3G). It is reasonable that such reshuffling of slow motions partly compensates for the loss of motion associated with the polypeptide going from disorder to order upon binding. In the two ancient complexes, the helix is not or only partially formed such that the conformational interconversion takes place in the fast-motion regime (faster than nanoseconds; Fig. 3G and fig. S4) (14). This process has been slowed down in the extant human complex because of the relatively stable Cα3 resulting in larger Rex values (Fig. 4). The trends in dynamics are less clear for NCBD, but Nα1 and Nα2 display a loss in dynamics slower than nanoseconds when comparing the most ancient with the two younger complexes. Rex in Nα3 changes from the N- to the C-terminal half of the helix. Thus, along with formation of more intermolecular CID/NCBD interactions as measured by NOEs, there is a general trend toward less and more localized conformational heterogeneity in the CID/NCBD complex upon going from the low-affinity Cambrian-like complex to the higher-affinity younger complexes (Figs. 3 and 4). We like to point out that localization and redistribution of motions could be highly dependent on particular side chains. Therefore, any erroneously predicted amino acid residue in the ancestral reconstruction could significantly influence the dynamics. Nevertheless, by comparing the Ordovician-Silurian 1R/2R complex with the extant human one, it is clear that high affinity is compatible with different dynamics profiles. Our data suggest that redistribution of motions may play a role in fine-tuning affinity upon mutations in diverging sequences. It is important to note that faster motions (nanoseconds to picoseconds) have also been implicated in function (15, 16). However, it was not possible to perform model-free dynamic analysis of the free domains, which would be needed for a conversion of NMR relaxation data into thermodynamic parameters as the local motions cannot be easily separated from global tumbling. Nevertheless, we provide information on the backbone subnanosecond motions in terms of R2(R1ρ)/R1 (fig. S5). The results indicate that, mostly, the N-terminal, C-terminal, and loop regions with low R2(R1ρ)/R1 values exhibit motions in the nanosecond-to-picosecond time scale.
Fig. 4. Redistribution of slow motions and flexibility during the evolution of the CID/NCBD complex.
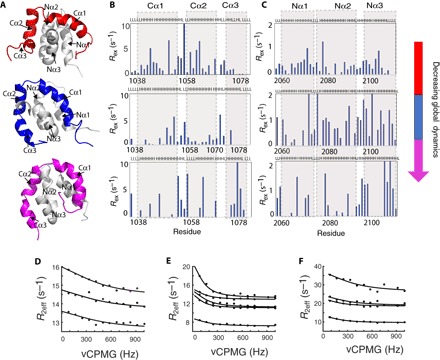
Plots of Rex from relaxation dispersion experiments as a function of position along the amino acid sequences for ancestral and extant CID domains (B) and NCBD domains (C). The CPMG probe (83 to 1000 Hz) used for these experiments detects conformational exchange occurring on the microsecond-to-millisecond time scale, and nonzero Rex values therefore indicate the presence of motions in this time regime. Notably, all three complexes show a distinct distribution. (A) Cartoon representation of the respective complex of CID/NCBD domains displayed in a similar orientation. R2eff determined as a function of different CPMG frequencies were fitted globally to Eq. 1 (see Methods), describing a two-site exchange process for selected residues. kex values of 3260 ± 1850 s−1, 1800 ± 245 s−1, and 2300 ± 1000 s−1 were obtained for (D) the most ancient Cambrian-like complex, (E) Ordovician-Silurian 1R/2R complex, and (F) the extant human CID/NCBD complex, respectively.
Thermodynamic basis for the different affinities of the CID/NCBD complexes
To investigate how the observed changes in structure, dynamics, and affinity are reflected in the binding thermodynamics, we performed ITC experiments for the three complexes at different pH and temperature (Fig. 5, fig. S5, and table S1). ITC experiments directly measure the change in enthalpy (ΔH) for the binding reaction and, from the shape of the titration profile, the equilibrium association constant (KA). From these parameters, KD (=1/KA), ΔS, and ΔG can be calculated.
Fig. 5. The pH and temperature dependence of interactions for ancestral and extant NCBD/CID.
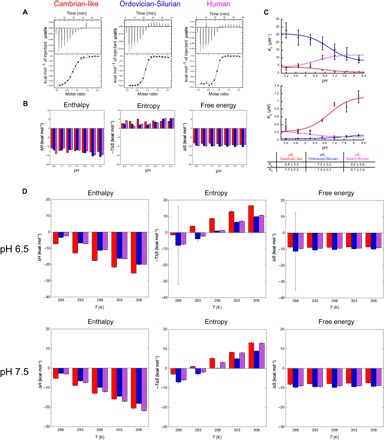
(A) Examples of ITC experiments conducted at pH 7.7 and 25°C for the three different complexes. (B) Contribution from enthalpy (ΔH, left) and entropy (−TΔS, middle) to the total free energy (ΔG, right) at different pH values for formation of Cambrian-like (red), Ordovician-Silurian (blue), and extant human (magenta) CID/NCBD complexes, respectively. (C) The association (KA, top) and dissociation (KD, bottom) equilibrium constants plotted against pH and fitted to a two-state equation (Eq. 4 in Methods), which yielded the apparent pKa values listed in the table. Because of the large noise in the original data for the extant human complex at pH 5.2, these points were not included in the curve fitting but are shown in the figure. (D) Bar diagrams showing the contribution of enthalpy (ΔH, left) and entropy (−TΔS, middle) to the free energy (ΔG, right) for formation of the respective CID/NCBD complex at different temperatures and at two different pH values (6.5 and 7.5, respectively). To determine ΔCp, the ΔH values were plotted against temperature and analyzed according to Eq. 2 (fig. S5).
First, we investigated the pH dependence of the interaction at a constant ionic strength and temperature (25°C). Thus, ITC experiments were conducted at pH values ranging from 5.2 to 8.2 (Fig. 5 and table S1). The results show that there is a favorable enthalpic and unfavorable entropic contribution to the affinity at all pH values and for all variants (Fig. 5). Furthermore, the affinities of all complexes have a transition around pH 6.5 and reach a plateau at higher pH values (Fig. 5). The Cambrian-like complex and the Ordovician-Silurian complex displayed similar pH dependences, where the affinities of both protein complexes increased toward lower pH values, maintaining a 10-fold difference in KA. In contrast, the extant human complex displayed an opposite pH dependence in which the affinity decreased toward lower pH values to equal that of the Cambrian-like complex around pH 5.2. KD and KA values were fitted to a two-state equation to estimate pKa values for the respective parameters in the different complexes. The pH dependence of KD reflects titrations of ionizable groups in the CID/NCBD complex, whereas the pH dependence of KA reflects titrations in free NCBD and CID (17). The data fitted reasonably well to the model, and the fitted pKa values for KA and KD were between 6.1 and 7.6 (Fig. 5). Except for one His in D/P NCBD, there are no obvious side chains with a pKa value in this range, and the most likely basis for the pH dependences of all three complexes is a general charge effect, corroborated by the relatively broad noncooperative transitions. These effects could lead to small structural perturbations of the molten globule-like NCBD domain, which, in turn, could modulate the affinity.
Next, we investigated the thermodynamic basis for the CID/NCBD affinity by performing ITC at different temperatures and at two different pH values, 7.5 (close to the affinity plateau) and 6.5 (in the pH transition region). We found that ΔH was negative (i.e., favorable) at temperatures above 285 K, whereas the entropy contribution −TΔS switched from being favorable (at lower temperatures) to nonfavorable between 289 and 297 K for the three complexes, both at pH 6.5 and 7.5 (Fig. 5). The calorimetric experiments show that the observed change in heat capacity upon binding ΔCp, (i.e., the temperature dependence of ΔH) for all three complexes was identical within error at pH 6.5 (−850 to −900 cal K−1 mol−1) and in the same range at pH 7.5 (−720 to −950 cal K−1 mol−1; fig. S5). These numbers suggest that the contribution from the change in solvent entropy to the binding energetics (i.e., the hydrophobic effect) is substantial and similar for the three complexes (18). However, there is an offset in the values of ΔH for the Ordovician-Silurian and human complexes as compared to the Cambrian-like complex at pH 6.5 such that the enthalpic contribution, which mirrors bond strength, is weakened in the higher-affinity complexes. Instead, the thermodynamic basis for the higher affinity is a less unfavorable or even favorable entropic contribution to the overall affinity, a result that is qualitatively very similar to another IDP interaction—that between FCP1 and Rap74 (19). At pH 7.5, this offset is smaller, and all three complexes display a similar contribution from ΔH. In addition, the enthalpic contribution in the human complex is accentuated at the higher temperatures at pH 7.5, which might reflect the more well-formed Cα3. From the calorimetric experiments, we conclude that (i) the net effect of bond breaking and formation (ΔH) is favorable at supposedly physiological temperatures (>12°C); (ii) the hydrophobic effect makes a major and favorable contribution to the free energy of binding; (iii) the higher affinity observed for the Ordovician-Silurian and extant human complexes as compared to the Cambrian-like complex at pH 6.5 is due to entropic effects, which are likely not related to the hydrophobic effect since ΔCp values are similar; and (iv) there is a delicate balance between factors contributing to affinity as shown by, for example, the large change in ΔH for the Cambrian-like complex at pH 6.5 compared to pH 7.5 and from the pH dependences of all three complexes. Although we cannot further dissect the entropic contribution in point (iii) into changes in rotational, translational, or conformational entropy (as explained above), the thermodynamic data underscore the impact of dynamics rather than noncovalent bonds for increasing the affinity of this interaction. In addition, we confirmed by urea and thermal denaturation experiments that the ancestral Cambrian-like complex was less stable as compared to the modern Ordovician-Silurian and extant human complexes, corroborating the affinity measurements by ITC (fig. S9).
Change in frustration parallels the experimental data
The concept of minimal frustration has been successfully applied to explain protein folding, and we were therefore interested in analyzing the evolution of frustration in the coupled binding-and-folding reaction of CID/NCBD. Frustration occurs when a physical system is not able to simultaneously achieve minimum energy for every part of it (20). It has been shown that, in protein-protein interactions, frustration of regions close to the binding site often changes upon association such that it is less frustrated in the complex, thus guiding specific association (20). It has been suggested that promiscuous IDP interactions display more frustration than those of well-folded proteins (21), but little is known, especially regarding evolution of frustration in IDPs. For our three CID/NCBD complexes, we therefore calculated and analyzed the mutational frustration index, for which the Frustratometer algorithm evaluates the energy of each contact of the native structure with respect to the distribution of the energies of decoys made by substituting residues at each position (Fig. 6) (22). It is apparent that there is a significant reduction and redistribution of highly frustrated interactions in the CID domain when comparing the most ancient Cambrian-like complex with the higher-affinity younger complexes (Fig. 6). In the most ancient Cambrian-like complex, the regions around residues 1056 to 1062 of the CID domain show a distinct frustrated patch, consistent with the location of a high-mobility region. It has been previously observed that regions of high local frustration correlate with extensive subnanosecond (23) and also slower time scale dynamics (24). In the extant human complex, the amount of frustration in this region is reduced and redistributed with respect to the ancient ones. Consistent with the pH dependence of the affinity, most of the frustration changes are related to electrostatic interactions, as the changes of frustration patterns are less evident when electrostatics is not considered in the calculation (figs. S6 and S9).
Fig. 6. Evolution of frustration in the CID/NCBD complex.
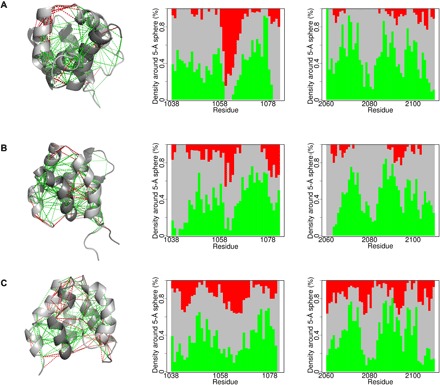
The local frustration patterns were calculated for each complex using the electrostatics mode of the Frustratometer (22). Left: CID/NCBD backbones are displayed as gray ribbons (darkest gray is NCBD), direct contacts with solid lines, and water-mediated interactions with dashed lines. Minimally frustrated interactions (green) and highly frustrated contacts (red) are shown. Middle and right: The proportion of contacts within 5 Å of the Cα atom of each residue is plotted and classified according to their frustration index. (A) The Cambrian-like complex contains 1R CID (middle) and D/P NCBD (right). (B) The Ordovician-Silurian complex contains 1R CID (middle) and 1R/2R NCBD (right). (C) The extant human complex contains NCOA3 CID (middle) and CREBBP NCBD (right).
DISCUSSION
Vertebrates originated during Cambrian (485 to 540 Ma), and one lineage gave rise to fishes during the subsequent Ordovician period (440 to 485 Ma). Sometime around the end of Ordovician or beginning of Silurian, a dramatic event occurred that shaped much of the following vertebrate evolution, namely, the two whole-genome duplications 1R and 2R (7, 25). This resulted in four copies of each gene and possibilities of evolutionary innovation of new functions for the encoded proteins. As the ancestral vertebrate population diversified into cartilaginous fish, bony fish, and later tetrapods including present-day amphibians, reptiles, birds, and mammals, many of the duplicated genes were subject to neo- or subfunctionalization. The genome duplications paved the way for extensive evolution of protein-protein interactions, which play a major role in cell signaling and regulation and often involve intrinsically disordered regions. Two examples are the protein domains involved in transcriptional regulation examined here—NCBD, which is present in extant CREBBP/p300 (two sister genes were lost during evolution) and CID, which is present in NCOA1 (Src1), NCOA2 (Tif2) and NCOA3 (ACTR) (one gene was lost). The analysis of this protein-protein interaction shed light on how a functional interaction can emerge from initially weak binding between IDPs (6) and the molecular complexity of increasing affinity.
One of the fundamental differences of IDPs as compared to ordered proteins is their ability to adopt alternative bound conformations, which is advantageous when recognizing different protein ligands and which would facilitate neofunctionalization as for CID recruiting NCBD as binding partner (6). Not only the disordered C-terminal domain of p53 is a prime example of such conformational plasticity (26) but also the NCBD domain, which adopts a distinct structure upon binding to a protein domain from the interferon regulatory factor (27), as compared to binding the CID domain. However, little is known regarding the evolution of conformational plasticity and how it modulates affinity in a protein-protein interaction. Our three historical structural and dynamic snapshots of the CID/NCBD complex provide a unique example of how increased affinity can be attained.
Overall, our findings support the notion that the high conformational plasticity of IDPs contributes to their protein-protein interactions (28). In the present case, the Cα3 of 1R CID adopts a helical conformation in the high-affinity complex with 1R/2R NCBD but not in the low-affinity complex with D/P NCBD. We speculate that the initial ancestral interaction between CID and NCBD was of lower affinity than our most ancient Cambrian-like complex yet achievable through the N-terminal leucine-rich binding motifs. Our NMR structures, NMR dynamics and thermodynamic data, and previous molecular dynamics simulations (6) illustrate how higher affinity in IDP interactions can be achieved by lowering the entropic penalty and increasing the enthalpic contribution of complex formation through a combination of specific interactions, conformational rearrangements, and redistribution of dynamics and frustration patterns. The most ancient Cambrian-like complex is more dynamic, which is probably a reflection of the fewer specific interactions and a very high local frustration not present in the higher-affinity variants. The high-affinity Ordovician-Silurian 1R/2R complex displays rearrangements of helices along with an increased hydrophobic interface. Thus, the conformational plasticity of the interacting IDPs permits formation of new favorable interactions between Cα3 and Nα2, leading to higher affinity in the Ordovician-Silurian and the extant human complex. Why are these features not clearly reflected in a larger enthalpic contribution to binding for the high-affinity complexes? For example, the affinity of the Ordovician-Silurian complex is always 10-fold higher than the Cambrian-like complex, but the enthalpy (supposedly reflecting structure) is less favorable and the entropy (supposedly reflecting dynamics) is less unfavorable except at low pH. The answer must lie in the large and opposing overall contributions from enthalpy and entropy and the hard-to-predict effect of water. Thus, while the overall thermodynamics of this coupled binding-and-folding reaction, which includes formation of a well-defined hydrophobic core, follows the same principles as observed in protein folding studies (29), they cannot easily be correlated to structural details. This observation is likely a reflection of a general property of IDP interactions, namely, they are plastic and thereby able to adapt to its binding partner.
The evolution of new protein-protein interactions involving intrinsically disordered regions of proteins is very common in nature as evident from virus biology. Viruses often hijack host protein-protein interactions by simply mimicking the host’s disordered interaction motifs (30). Because of the high mutation rate and short generation time of viral genomes, such evolution happens on a time scale much shorter than that for animals. However, the general molecular principles governing emergence of new protein-protein interactions are expected to be general. Our results demonstrate how protein-protein complexes involving intrinsically disordered domains can achieve stronger binding via an intricate interplay between structural and dynamical rearrangements that cannot easily be predicted.
METHODS
Protein expression
The constructs for the CID domains contained the complementary DNA corresponding to residues 1040 to 1080 of human NCOA3, and the constructs for the NCBD domains contained residues 2062 to 2109 of human CREBBP NCBD, as described (6). All constructs contained an N-terminal expression tag (6× His and lipoyl domain), followed by a thrombin protease site and were expressed from a pRSET vector. The CID and the NCBD domains were expressed in Escherichia coli BL21 (DE3) pLysS cells either unlabeled (in terrific broth medium) or labeled for NMR experiments (in M9 minimal medium enriched with 15N ammonium chloride and 13C d-glucose) and purified, as previously described (6). Concisely, on day 1, BL21 plyS cells containing transformed plasmid were grown overnight at 37°C on a plate containing ampicillin (100 μg/ml) and chloramphenicol (35 μg/ml). On day 2, transformed cells were grown in growth media containing ampicillin (50 μg/ml) at 37°C to an optical density of 0.6. Protein expression was induced by addition of 1 mM isopropyl-β-d-thiogalactopyranoside, and cells were allowed to grow overnight at 18°C. On day 3, these cells were harvested by centrifugation and resuspended in purification buffer [20 mM tris/Cl (pH 8.0) and 200 mM NaCl). The resuspended cells were then lysed by sonication and purified on a Ni Sepharose column (GE Healthcare). After desalting the partially pure fusion protein, ca. 0.5 units of thrombin protease per milligram of fusion protein was added to the sample and incubated at 37°C for 16 to 18 hours. The cleaved proteins were then passed through a second Ni Sepharose column, and unbound cleaved CID or NCBD was loaded on a reversed-phase C18 column equilibrated with 0.1 % trifluoroacetic acid. Bound proteins were eluted with a gradient of 0 to 70% acetonitrile in 0.1% trifluoroacetic acid. The purity and identity of the proteins were confirmed by SDS–polyacrylamide gel electrophoresis and mass spectrometry. Pure proteins were lyophilized and stored at −20°C until use. Before NMR experiments, the lyophilized samples were dissolved in a buffer containing 10 mM NaPO4 (pH 6.8) and 150 mM NaCl and were dialyzed against the same buffer overnight at 25°C.
NMR spectroscopy
Protein concentrations were estimated by absorbance measurement at 205 nm for all domains and double checked at 280 nm for 1R/2R NCBD and human CREBBP NCBD, which contain one tyrosine. The protein complexes were formed by adding saturating amounts of either unlabeled CID domain (14N/12C) with labeled NCBD domain (15N/13C) or vice versa. The final concentrations of the CID/NCBD complexes in the NMR tube ranged from 300 to 500 μM of the labeled protein and approximately a twofold excess of unlabeled protein. To these samples, 0.01% NaN3 and 5% D2O were added. All NMR experiments were recorded on Bruker spectrometers (600, 700, and 900 MHz, 1H frequencies), equipped with triple-resonance cryogenic temperature probes at 298 K. Assignment of the protein backbone was achieved by measuring two-dimensional (2D) [1H,15N]-HSQC (heteronuclear single-quantum coherence), 3D HNCACB, and [1H,1H]-NOESY-[1H,15N]-HSQC experiments (31). Side chains were assigned from the following 3D experiments: [1H,15N]-HSQC-[1H13C-13C1H]-TOCSY (total correlation spectroscopy; 60-ms mixing time), [1H,1H]-NOESY-[1H,13C]-HSQC, [1H,13C]-HSQC-[1H13C-13C1H]-TOCSY (60-ms mixing time) in combination with 2D [1H,15N]-HSQC, and 2D [1H,13C]-HSQC-CT (constant time; fig. S7) (31). The φ angle–restraining 3JHNHA couplings were determined from a 3D HNHA-type experiment using quantitative J coupling intensity evolution (32). 2D and 3D in-phase and anti-phase [1H-15N]-HSQC–based and [1H-15N/13C] HNCO–based experiments were used for the determination of 1DHN RDCs in a sample, with proteins aligned by pf1 phage. The differences between the apparent scalar couplings in the absence and presence of alignment media yielded the RDCs (fig. S8). For the collection of distance restraints, the following NOESY (NOE spectroscopy) experiments and parameters were used: 3D NOESY-[1H,15N]-HMQC (heteronuclear multiple-quantum coherence) and NOESY-[1H,13C]-HMQC with 60-ms mixing time and 128 (15N or 13C) × 200 (1H) × 2048 (1H, direct) number of points for intramolecular NOEs and a [14N-1H]-[12C-1H] double-filtered NOESY-[1H,15N/13C]-HSQC for intermolecular NOEs (33). R1, and R1ρ relaxation rates were measured with different randomized relaxation delays in an interleaved manner, with a recovery delay of 3 s using TROSY (transverse relaxation optimized spectroscopy)–based pulse schemes (34). For R1ρ, a spin lock of 2000 Hz was used. R2 values were then determined from R1ρ by correcting for the contribution of R1. The overall tumbling time τc was then estimated from the ratio of R2 (R1ρ)/R1. [1H,15N]-heteronuclear NOE experiments were recorded, with a 1H saturation time of 8 s with an equal recovery time for the reference experiment (34). CPMG dispersion profiles were determined from relaxation rates measured with a fixed CPMG transverse rotating-frame relaxation delay tCPMG (48 ms) with νCPMG frequencies of 83, 167, 250, 333, 417, 500, 583, 667, 750, 883, 917, and 1000 Hz applied in a randomized manner (35). The relaxation delay was set to 2 s. A reference experiment omitting the CPMG element was used to obtain the effective relaxation rate Reff from the expression −ln (I/I0)/tCPMG, where I and I0 are the intensities at different CPMG times and reference intensities, respectively. All experiments were processed with NMRPipe (36) and analyzed with CCPNmr (37). Curve fitting was done in MATLAB, NMRPipe, and KaleidaGraph. Errors were determined from Monte Carlo simulations. In the fast exchange limit, the effective relaxation rate as a function of νCPMG can be described by the following equation (13)
| (1) |
where, R20 is the transverse relaxation rate in the absence of exchange, pA and pB are the populations of the exchanging states, Δω is the chemical shift difference between the exchanging states, kex is the sum of the forward and reverse exchange rate constants, and τcp are the variable CPMG times. Chemical shift order parameters (S2) were predicted by submitting experimentally derived shifts to a web-based program from the Wishart laboratory (38). Rex was calculated by taking the difference between R2 (R2eff) without and with a CPMG radio pulse applied at a frequency of 1000 Hz (fig. S9).
Structure calculations
Structure calculations were done using the CYANA 3.97 package in two steps. First, the NOESY cross peaks were converted into upper distance restraints (fig. S7) following an automated process in CYANA. These distance restraints, together with φ/ψ dihedral angles determined from Cα chemical shifts and 3JHNHA (fig. S8), were used as input for the initial structure calculations. The structures were calculated with 200,000 torsion angle dynamics steps for 100 conformers, starting from random torsion angles by simulated annealing. Initially, the structures of the CID and NCBD domains were determined separately without the intermolecular NOEs. In a second step, the calculations were repeated using the intermolecular NOE-derived distance restraints (table S1) and a linker with a weak harmonic well potential with a bottom width of 1.2 Å that kept the two domains together (39). The separate structures calculated in step 1 were used as the starting structures. For refinement and cross-validation, calculations were repeated with 1DHN RDCs (fig. S8, F to K). The axially symmetric and rhombic components of the alignment tensor were optimized by an iterative process in CYANA in three steps. For representation and analysis, the 20 conformers with the lowest target function values were selected. The structural statistics, together with all input data for the structure calculations, are presented in Table 1 and figs. S8 and S9. The total SASA was determined using the program PyMol from the domains in the complex.
Table 1. Structural determination statistics.
| Most ancient | 1R/2R | Extant human | |
| NMR distance and dihedral restraints | |||
| Distance restraints | |||
| Total NOEs | 620 | 700 | 550 |
| Intraresidue | 222 | 265 | 213 |
| Sequential (|i − j| = 1) | 197 | 206 | 168 |
| Medium-range (1 < |i − j| < 5) | 140 | 164 | 97 |
| Long-range (|i − j | >5) | 45 | 55 | 38 |
| Total RDC restraints (15N-1HN) | 91 | 85 | 79 |
| Total dihedral angle restraints | |||
| 3JHNHA scalar couplings | 70 | 63 | 67 |
| 13Cα chemical shifts | 138 | 142 | 140 |
| Structure statistics | |||
| Average CYANA target function value (Å2) | 7.9 ± 0.2 | 4.2 ± 0.30 | 2.2 ± 0.2 |
| Violations | |||
| Distance constraints (>0.5 Å) | 0 | 3 | 0 |
| Dihedral angle constraints (>5°) | 0 | 0 | 0 |
| Deviations from idealized geometry | |||
| Bond lengths (Å) | 0 | 0 | 0 |
| Bond angles (o) | 0 | 0 | 0 |
| Impropers (o) | 0 | 0 | 0 |
| Average pairwise RMSD (Å)* | |||
| Backbone | 0.8 ± 0.2 | 0.6 ± 0.1 | 1.7 ± 0.6 |
| Heavy atoms | 1.3 ± 0.2 | 1.1 ± 0.1 | 2.2 ± 0.5 |
| RMSD of RDC-refined structure (Å)* | |||
| Backbone | 1.3 ± 0.3 | 0.8 ± 0.2 | 1.4 ± 0.7 |
| Heavy atoms | 1.8 ± 0.2 | 1.3 ± 0.2 | 1.9 ± 0.6 |
*RMSD for residues 1042 to 1079 and 2065 to 2106.
Isothermal titration calorimetry
ITC binding experiments were performed on an iTC200 (Malvern Instruments) and fitted using in-built software. Before performing the ITC experiments, all protein samples were dialyzed overnight in a buffer containing 20 mM sodium phosphate at pH 6.5 or 7.5. The protein samples were then filtered, and their concentrations were determined using absorbance at 205 nm. The ITC experiments were done at different temperatures (288, 293, 298, 303, and 308 K). Typically, 20 injections of 2 μl each of 300 μM CID domain solution were titrated into 280 μl of 30 μM NCBD domains. All samples were allowed to equilibrate for about 10 min at the experimental temperature before the actual measurements were initiated. For the determination of ΔCp, both ΔH and ΔS were plotted as a function of temperature following Eqs. 2 and 3.
| (2) |
| (3) |
ΔCp is the specific heat capacity at constant pressure. ΔxH and ΔxS are the temperature-independent contributions of enthalpy and entropy, respectively. The ITC measurements for pH dependence of affinity were carried out essentially as above, with the following differences: The protein samples were dialyzed against 20 mM sodium phosphate buffer (pH 8.2, 7.7, 7.2, 6.7, and 6.2) or, alternatively, 20 mM sodium acetate buffer (pH 5.7 and 5.2), and the ionic strength was adjusted with NaCl to 58 mM for all buffers. All measurements were conducted at 298 K. The measurement for the extant human complex at pH 5.2 was repeated four times because of problems with noise in the isotherms, and these data points were excluded from the fitting of pKa values. The data were fitted to a two-state equation (Eq. 4) to extract apparent pKa values for the association (KA) and dissociation (KD) constants for the respective protein complex.
| (4) |
Stability measurements
Urea and thermal denaturation experiments were performed on a JASCO J-1500 circular dichroism (CD) spectrophotometer, and the data were fitted using the program KaleidaGraph. All lyophilized protein samples were dissolved in a buffer containing 10 mM sodium phosphate (pH 6.8). For each complex, 40 μM of the CID domain was mixed with 15 μM NCBD domain. The temperature was set to 298 K for the urea denaturation experiments. The temperature was varied between 278 and 363 K for the thermal denaturation. The ellipticity at 222 nm was monitored for both measurements. The data were then fitted to the standard equations describing two-state unfolding by chemical denaturant and temperature, respectively (17).
Frustration index calculations
The frustration index was calculated for each contact in the complex using the Frustratometer, as described (22). A contact with an index of ≥0.78 is classified as minimally frustrated, contributing favorably to protein stability and affinity. On the other hand, interactions with a frustration index of ≤−1 are labeled as highly frustrated, and most substitutions at that location would be more favorable. In between these extremes, the interactions are labeled as neutral. Interactions are divided into classes according to this index and color coded in the figures. Considering that the CID/NCBD complexes are charged under the pH conditions used for the experiments, we computed the local frustration patterns using the electrostatics module of the Frustratometers, with an electrostatics k = 4.15 (22), and compared the results with calculations not considering the electrostatics.
Supplementary Material
Acknowledgments
We would like to thank D. Ferreiro for helpful contribution in performing the frustrational analysis. Funding: This work was supported by the Wenner-Gren Foundation WG-17 returning grants (to C.N.C.), the Swedish Research Council (to P.J.), and a start-up package from the University of Colorado at Denver (to B.V.). Author contributions: P.J. and C.N.C. conceived the study, designed the experiments, and wrote the paper with input from the other authors; C.N.C. conducted all the NMR experiments; C.N.C. and E.K. conducted the ITC and CD experiments; B.G. performed the frustration index analysis; E.A. expressed and purified all the protein samples; P.J., E.K., B.V., B.G., J.D., G.H., P.G., R.R., and C.N.C. analyzed and interpreted the data; all authors read and approved the final manuscript. Competing interests: All authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaau4130/DC1
Fig. S1. Structural changes taking place and chemical shift restraints.
Fig. S2. Strips from [1H-1H]-NOESY-[1H-15N]-HSQC spectra showing resonances emanating from residues 1074 and 1075 of the respective CID domains.
Fig. S3. Strips from [1H-1H]-NOESY [1H-15N]-HSQC spectra showing resonances emanating from residues 1076 and 1077 of the CID domains.
Fig. S4. 15N relaxation data for the bound CID and NCBD domains and amide secondary chemical shifts of the complexes.
Fig. S5. Backbone subnanosecond motions and ΔCp for the three CID/NCBD complexes.
Fig. S6. Frustration in the CID/NCBD complexes without electrostatics.
Fig. S7. [1H-15N]-HSQC correlation spectra for bound and free CID and NCBD domains and the number of restraints in the structure determination as a function of amino acid residue.
Fig. S8. Restraints used for structure calculations and refinement.
Fig. S9. Urea and temperature denaturation experiments and Coulomb surfaces of the ancestral and extant CID/NCBD complexes.
Table S1. ITC parameters and intermolecular NOEs.
REFERENCES AND NOTES
- 1.Leo C., Chen J. D., The SRC family of nuclear receptor coactivators. Gene 245, 1–11 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Harms M. J., Thornton J. W., Evolutionary biochemistry: Revealing the historical and physical causes of protein properties. Nat. Rev. Genet. 14, 559–571 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKeown A. N., Bridgham J. T., Anderson D. W., Murphy M. N., Ortlund E. A., Thornton J. W., Evolution of DNA specificity in a transcription factor family produced a new gene regulatory module. Cell 159, 58–68 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim S. A., Hart K. M., Harms M. J., Marqusee S., Evolutionary trend toward kinetic stability in the folding trajectory of RNases H. Proc. Natl. Acad. Sci. U.S.A. 113, 13045–13050 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nguyen V., Wilson C., Hoemberger M., Stiller J. B., Agafonov R. V., Kutter S., English J., Theobald D. L., Kern D., Evolutionary drivers of thermoadaptation in enzyme catalysis. Science 355, 289–294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hultqvist G., Åberg E., Camilloni C., Sundell G. N., Andersson E., Dogan J., Chi C. N., Vendruscolo M., Jemth P., Emergence and evolution of an interaction between intrinsically disordered proteins. eLife 6, e16059 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes T., Liberles D. A., Whole-genome duplications in the ancestral vertebrate are detectable in the distribution of gene family sizes of tetrapod species. J. Mol. Evol. 67, 343–357 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Tjandra N., Bax A., Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 278, 1111–1114 (1997). [DOI] [PubMed] [Google Scholar]
- 9.Mascioni A., Veglia G., Theoretical analysis of residual dipolar coupling patterns in regular secondary structures of proteins. J. Am. Chem. Soc. 125, 12520–12526 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Demarest S. J., Martinez-Yamout M., Chung J., Chen H., Xu W., Dyson H. J., Evans R. M., Wright P. E., Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature 415, 549–553 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Salvi N., Abyzov A., Blackledge M., Multi-timescale dynamics in intrinsically disordered proteins from NMR relaxation and molecular simulation. J. Phys. Chem. Lett. 7, 2483–2489 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Zhou N. E., Zhu B. Y., Sykes B. D., Hodges R. S., Relationship between amide proton chemical shifts and hydrogen bonding in amphipathic .alpha.-helical peptides. J. Am. Chem. Soc. 114, 4320–4326 (1992). [Google Scholar]
- 13.Palmer A. G., III, NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 104, 3623–3640 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Abyzov A., Salvi N., Schneider R., Maurin D., Ruigrok R. W. H., Jensen M. R., Blackledge M., Identification of dynamic modes in an intrinsically disordered protein using temperature-dependent NMR relaxation. J. Am. Chem. Soc. 138, 6240–6251 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Marlow M. S., Dogan J., Frederick K. K., Valentine K. G., Wand A. J., The role of conformational entropy in molecular recognition by calmodulin. Nat. Chem. Biol. 6, 352–358 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tzeng S.-R., Kalodimos C. G., Protein activity regulation by conformational entropy. Nature 488, 236–240 (2012). [DOI] [PubMed] [Google Scholar]
- 17.A. Fersht, Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding (Macmillan, 1999). [Google Scholar]
- 18.Baldwin R. L., Temperature dependence of the hydrophobic interaction in protein folding. Proc. Natl. Acad. Sci. U.S.A. 83, 8069–8072 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawrence C. W., Kumar S., Noid W. G., Showalter S. A., Role of ordered proteins in the folding-upon-binding of intrinsically disordered proteins. J. Phys. Chem. Lett. 5, 833–838 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Ferreiro D. U., Komives E. A., Wolynes P. G., Frustration, function and folding. Curr. Opin. Struct. Biol. 48, 68–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jemth P., Mu X., Engström Å., Dogan J. A., A frustrated binding interface for intrinsically disordered proteins. J. Biol. Chem. 289, 5528–5533 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parra R. G., Schafer N. P., Radusky L. G., Tsai M.-Y., Guzovsky A. B., Wolynes P. G., Ferreiro D. U., Protein Frustratometer 2: A tool to localize energetic frustration in protein molecules, now with electrostatics. Nucleic Acids Res. 44, W356–W360 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindström I., Dogan J., Dynamics, conformational entropy, and frustration in protein-protein interactions involving an intrinsically disordered protein domain. ACS Chem. Biol. 13, 1218–1227 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Fuglestad B., Gasper P. M., McCammon J. A., Markwick P. R. L., Komives E. A., Correlated motions and residual frustration in thrombin. J. Phys. Chem. B 117, 12857–12863 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuraku S., Meyer A., Kuratani S., Timing of genome duplications relative to the origin of the vertebrates: Did cyclostomes diverge before or after? Mol. Biol. Evol. 26, 47–59 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Oldfield C. J., Meng J., Yang J. Y., Yang M. Q., Uversky V. N., Dunker A. K., Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 9 (suppl. 1), S1 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qin B. Y., Liu C., Srinath H., Lam S. S., Correia J. J., Derynck R., Lin K., Crystal structure of IRF-3 in complex with CBP. Structure 13, 1269–1277 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Wright P. E., Dyson H. J., Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson C. M., Fersht A. R., Protein stability as a function of denaturant concentration: The thermal stability of barnase in the presence of urea. Biochemistry 34, 6795–6804 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Davey N. E., Travé G., Gibson T. J., How viruses hijack cell regulation. Trends Biochem. Sci. 36, 159–169 (2011). [DOI] [PubMed] [Google Scholar]
- 31.J. Cavanagh, W. J. Fairbrother, A. G. Palmer III, N. J. Skelton, Protein NMR Spectroscopy: Principles and Practice (Academic Press, ed. 2, 2007). [Google Scholar]
- 32.Vuister G. W., Bax A., Measurement of four-bond HN-Hα J-couplings in staphylococcal nuclease. J. Biomol. NMR 4, 193–200 (1994). [DOI] [PubMed] [Google Scholar]
- 33.Eichmüller C., Schüler W., Konrat R., Kräutler B., Simultaneous measurement of intra- and intermolecular NOEs in differentially labeled protein-ligand complexes. J. Biomol. NMR 21, 107–116 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Lakomek N.-A., Ying J., Bax A., Measurement of 15N relaxation rates in perdeuterated proteins by TROSY-based methods. J. Biomol. NMR 53, 209–221 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ban D., Mazur A., Carneiro M. G., Sabo T. M., Giller K., Koharudin L. M. I., Becker S., Gronenborn A. M., Griesinger C., Lee D., Enhanced accuracy of kinetic information from CT-CPMG experiments by transverse rotating-frame spectroscopy. J. Biomol. NMR 57, 73–82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A., NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 37.Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D., The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 59, 687–696 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Berjanskii M. V., Wishart D. S., A simple method to predict protein flexibility using secondary chemical shifts. J. Am. Chem. Soc. 127, 14970–14971 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Chi C. N., Vögeli B., Bibow S., Strotz D., Orts J., Güntert P., Riek R., A structural ensemble for the enzyme cyclophilin reveals an orchestrated mode of action at atomic resolution. Angew. Chem. Int. Ed. 54, 11657–11661 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaau4130/DC1
Fig. S1. Structural changes taking place and chemical shift restraints.
Fig. S2. Strips from [1H-1H]-NOESY-[1H-15N]-HSQC spectra showing resonances emanating from residues 1074 and 1075 of the respective CID domains.
Fig. S3. Strips from [1H-1H]-NOESY [1H-15N]-HSQC spectra showing resonances emanating from residues 1076 and 1077 of the CID domains.
Fig. S4. 15N relaxation data for the bound CID and NCBD domains and amide secondary chemical shifts of the complexes.
Fig. S5. Backbone subnanosecond motions and ΔCp for the three CID/NCBD complexes.
Fig. S6. Frustration in the CID/NCBD complexes without electrostatics.
Fig. S7. [1H-15N]-HSQC correlation spectra for bound and free CID and NCBD domains and the number of restraints in the structure determination as a function of amino acid residue.
Fig. S8. Restraints used for structure calculations and refinement.
Fig. S9. Urea and temperature denaturation experiments and Coulomb surfaces of the ancestral and extant CID/NCBD complexes.
Table S1. ITC parameters and intermolecular NOEs.