

Abstract
Clearance of dying cells is essential for development and homeostasis. Conserved genes mediate apoptotic cell removal, but whether these genes control non-apoptotic cell removal is a major open question. Linker cell-type death (LCD) is a prevalent non-apoptotic developmental cell death process with features conserved from C. elegans to vertebrates. Using microfluidics-based long-term in vivo imaging, we show that unlike apoptotic cells, the C. elegans linker cell, which dies by LCD, is competitively phagocytosed by two neighboring cells, resulting in cell splitting. Subsequent cell elimination does not require apoptotic engulfment genes. Rather, we find that RAB-35 GTPase is a key coordinator of competitive phagocytosis onset and cell degradation. RAB-35 binds CNT-1, an ARF-6 GTPase activating protein, and removes ARF-6, a degradation inhibitor, from phagosome membranes. This facilitates phosphatidylinositol-4,5-bisphosphate removal from phagosome membranes, promoting phagolysosome maturation. Our studies suggest that RAB-35 and ARF-6 drive a conserved program eliminating cells dying by LCD.
eTOC
Kutscher et al. discover a protein network for the engulfment and degradation of a C. elegans cell that dies by linker cell-type death (LCD). Two cells compete to engulf the cell corpse, relying on two small GTPases, RAB-35 and ARF-6 and their regulators to ensure timely corpse phagocytosis and clearance.
Introduction
Clearance of cells undergoing programmed cell death is important during development of multicellular organisms, and failure to remove dying cells is implicated in developmental abnormalities (Garlena et al., 2015) and autoimmune disease (Poon et al., 2014). Studies of the nematode C. elegans uncovered genes required for engulfment and degradation of cells dying by apoptosis (Ellis et al., 1991; Guo et al., 2010; Kinchen and Ravichandran, 2010; Kinchen et al., 2008; Mangahas et al., 2008; Yu et al., 2008). Homologous genes regulate apoptotic cell clearance in Drosophila and vertebrates, as do a number of species-specific genes (Franc et al., 1999; Park and Kim, 2017; Tosello-Trampont et al., 2001).
While caspase-dependent apoptosis is a common cell death mode, recent studies demonstrate that caspase-independent non-apoptotic cell death processes are equally relevant in development (Kutscher and Shaham, 2017) and in disease (Fuchs and Steller, 2015). While the molecular and genetic characterization of some non-apoptotic cell death processes has advanced considerably, whether common clearance mechanisms are used for apoptotic and non-apoptotic dying cells remains a major open question.
LCD (linker cell-type death) is a non-apoptotic developmental cell-death process that is morphologically conserved from C. elegans to mammals (Kutscher and Shaham, 2017). This cell death process is characterized by nuclear envelope crenellation, lack of chromatin condensation, and swelling of cytoplasmic organelles. These hallmarks of LCD are prevalent in vertebrate developmental settings, including degeneration of the Wolffian and Müllerian ducts during female and male gonadal development, respectively (Djehiche et al., 1994; Dyche, 1979; Price et al., 1977), and motor neuron elimination during spinal cord formation (Chu-Wang and Oppenheim, 1978). LCD morphology is also characteristic of dying cells in several disease states, including striatal neuron death in Huntington’s disease (Maat-Schieman et al., 1999).
During C. elegans development, the male-specific linker cell leads the elongation of the developing gonad, and eventually dies by LCD (Abraham et al., 2007). Linker cell death is independent of caspases and all known apoptosis, necrosis, and autophagy genes. A network of proteins governing linker cell death has been uncovered, converging on the stress-induced transcription factor HSF-1, acting in a stress-independent manner to promote expression of LET-70/UBE2D2, an E2 ubiquitin ligase. LET-70/UBE2D2, together with other components of the ubiquitin proteasome system, then drive LCD and linker cell clearance (Blum et al., 2012; Kinet et al., 2016; Malin et al., 2016). Following LCD initiation, the linker cell is engulfed (Sulston et al., 1980). The linker cell, therefore, is a particularly attractive in vivo model for investigating the clearance of a cell that normally dies non-apoptotically: the time of linker cell death onset is predictable, the process can be followed in live animals, and engulfment events can be dissected genetically.
Here we show that engulfment and degradation of the linker cell differs in mechanics and genetics from apoptotic cell clearance. We demonstrate that, unlike apoptotic cell corpses, the linker cell is simultaneously engulfed by two U cell-descendent phagocytes, resulting in cell splitting. Apoptotic engulfment genes are not required for this form of engulfment. Rather, we find that the GTPase RAB-35, not previously implicated in apoptotic corpse removal, is a key coordinator of at least twosteps in linker cell degradation. Early on, RAB-35 localizes to extending phagocyte pseudopods, and prevents premature onset of phagocytosis. Then, RAB-35 drives degradation of the linker cell by promoting the removal of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) from the nascent phagosome, thereby facilitating recruitment of RAB-5 and RAB-7 GTPases onto phagosome membranes. We demonstrate that both activities of RAB-35 require inactivation of another conserved small GTPase, ARF-6, which we show functions as a clearance inhibitor. RAB-35 physically interacts with the ARF-6 GTPase activating protein CNT-1, providing a plausible mechanism for ARF-6 inhibition. Furthermore, while RAB-35 localizes to engulfing-cell and phagosome membranes during most of linker cell clearance, ARF-6 only transiently persists at the membrane, and its removal coincides with the removal of PI(4,5)P2 from the phagosome membrane. Both ARF-6 and PI(4,5)P2 removal depend on RAB-35.
RAB-35 and ARF-6 have been previously suggested to promote engulfment of bacteria by cultured cells (Egami et al., 2011); however, neither protein has been studied for roles in removal of dying cells. Furthermore, the in vivo mechanisms by which these proteins promote engulfment have not been dissected. Our work establishes a powerful in vivo model to study phagocytosis of cells that die non-apoptotically in development, and suggests that different phagocytic pathways can target cells that die by different means.
Results
The dying linker cell is simultaneously engulfed by two neighboring cells
Linker cell death and clearance occurs during the transition from the fourth larval stage to the adult, and can take up to 8 hours to complete (Abraham et al., 2007; Keil et al., 2017). To examine this process at high spatiotemporal resolution, we used a long-term imaging microfluidic device we previously developed (Keil et al., 2017) to simultaneously image the linker cell (mig-24p::Venus) and the engulfing cells (lin-48p::mKate2) in live animals (Figure 1A,B). Animals were loaded into the device, and z-stacks were acquired every 8 min for > 20h (Figure 1A). Animal development and linker cell death kinetics were not affected by either imaging or fluorescent reporter identity (Figure 1B, Table S1, top, see also (Keil et al., 2017)). There was no significant photobleaching of the U.l/rp engulfing cells or other cells labeled by the lin-48p reporter over the duration of imaging (Figure 1A,B), but the intensity of YFP in the dying linker cell decreased as the cell was engulfed, degraded, and eventually removed, as expected (Figure 1C-K). The timings of characteristic events accompanying linker cell degradation were noted, with time zero corresponding to the time of first contact between the linker cell and the U.lp or U.rp engulfing cells (Figure 1C and Movie S1), and are consistent with previous reports (Abraham et al., 2007; Keil et al., 2017).
Figure 1. Linker cell death and degradation.
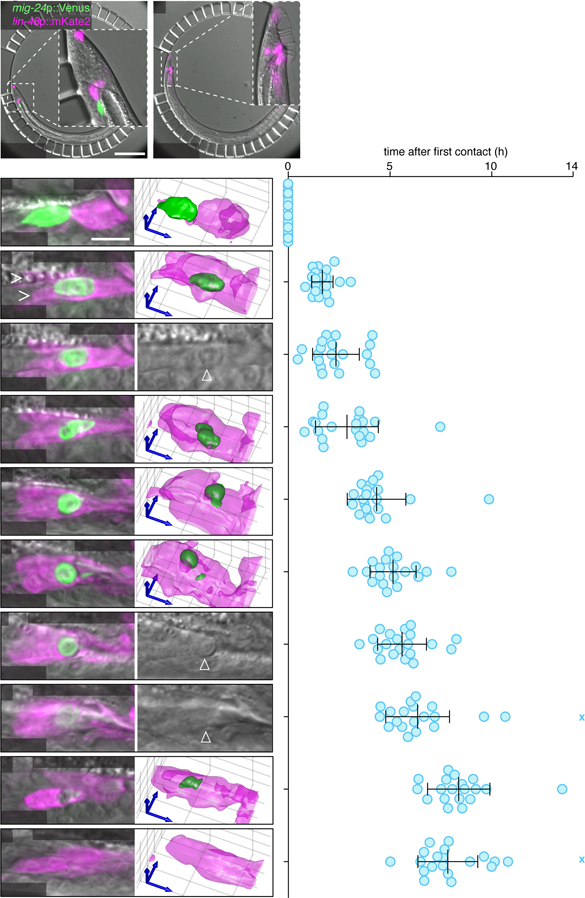
(A,B) C. elegans male immobilized in a microfluidic chamber (see (Keil et al., 2017)) at (A) first contact between linker cell (green) and engulfing U.l/rp cell (magenta, asterisk), and (B) after 10 hours. All males examined in all figures carry the male-producing him-5(e1490) or him-8(e1489) mutation. Scale bar, 100 µm. (C) Left: Maximum intensity projection showing linker cell at first contact with U.l/rp cells. Right: 3D rendering. Scale bar, 10 µm. (D-L) Stereotypical events during linker cell dismantling. Images as in (C), except that in (E,I,J) a single DIC slice is shown instead of 3D rendering. Right, time of events in individual animals (blue circles) after first contact. X, event did not occur during imaging. Bars, mean ± sd. Arrowhead, linker cell. (D) Linker cell between U.l/rp cells (caret) (E) Nuclear crenellation onset. (F) Competitive phagocytosis begins. (G) Linker cell rounding. (H) Linker cell corpse splits. (I) Onset of refractility. (J) End of refractility. (K) Small fragment disappears. (L) Large fragment disappears. See also Movies S1-S2, Figure S1, and Table S1.
We found that after first contact, the linker cell migrates so that it becomes sandwiched between the U.lp and U.rp cells (carets, Figure 1D, Movie S2A), whose cell nuclei are displaced anterior to the linker cell. Linker cell nuclear crenellation also becomes apparent at this time (Figure 1E). Remarkably, following linker cell nuclear changes, both U.l/rp cells simultaneously attempt to engulf the linker cell (Figure 1F, Figure S1A-D, Movie S2B). During this process, which we term competitive phagocytosis, linker cell rounding (Figure 1G) is followed by splitting of the linker cell into two fragments (Figure 1H, Figure S1E,F, Movie S2C). The larger linker cell remnant contains the nucleus, and is equally likely to be found within U.lp or U.rp (8/19 and 11/19, respectively; p=0.5, χ2-test). The cell engulfing the nucleus-containing fragment is more likely to be binucleate (17/19; p=0.0006, χ2-test), a result of an earlier unrelated U cell descendent fusion event (Sulston et al., 1980), suggesting that the larger U.l/rp cell engulfs a larger portion of the linker cell. The larger linker cell remnant, but not the smaller, then becomes refractile by DIC microscopy (Figure 1I,J). The two linker cell fragments are degraded with similar kinetics (Figure 1K,L). Competitive phagocytosis does not result in extensive leakage of linker cell cytoplasm, as we never observed reporter protein outside the cell (N=35).
To identify the precise time point at which the linker cell fragments become internalized, we followed linker cell death in animals expressing the mKate2-PH reporter, derived from PLC-δ1, which marks the plasma membrane and membranes of open phagosomes by binding phosphatidylinositol (4,5)-bisphosphate (PI(4,5)P2) (Botelho et al., 2000; Cheng et al., 2015). Localization of this reporter around the linker cell ceases 14 min ± 12.6 min prior to cell splitting (N = 8, Figure S1G-J), suggesting that cell splitting coincides with the onset of phagosome maturation.
The unique mechanics governing linker cell dismantling raise the question of whether U cell descendants are specifically equipped for linker cell clearance. To test this, we examined animals carrying a mutation in the gene him-4, in which linker cell migration is defective, and the cell ends up near the head (Abraham et al., 2007). In these animals, linker cell death occurs fairly reliably, as determined by onset of nuclear crenellation (Abraham et al., 2007). However, while engulfment and degradation by neighboring cells can occur, it is inefficient (62% ± 6.9% remaining corpses in 24h adult males, N=50, p<0.0001 compared to wild type, Fisher’s exact test). Thus, U.l/rp cells exhibit specialization for linker cell clearance.
Apoptotic engulfment genes are not required for linker cell clearance
It was previously reported that linker cell engulfment can occur in animals lacking apoptosis engulfment genes (Abraham et al., 2007); however, whether the cell is eventually degraded in these settings was not examined. To look at this in detail, we followed linker cell engulfment in animals lacking components of each or both parallel pathways that together drive apoptotic cell engulfment. Animals were scored 24h after the L4-to-adult transition, when the majority of animals have cleared the linker cell corpse (Keil et al., 2017). We found that linker cell removal is only weakly perturbed by mutations in some known engulfment genes (Table S2). Furthermore, double mutants between engulfment genes in different pathways do not strongly affect linker cell clearance. Consistent with this observation, when we expressed MFG-E8-GFP, a phosphatidylserine binding protein (Andersen et al., 2000; Venegas and Zhou, 2007), in U cell descendants, we did not observe fluorescence accumulation around the linker cell (5 lines, >100 animals scored in total). Nonetheless, we did find that mutations in the phagosome maturation gene sand-1, which is involved in the RAB-5 to RAB-7 conversion in phagosome maturation (Kinchen and Ravichandran, 2010), and in rab-7, a RAB protein responsible for the recruitment of late endosomes and lysosomes to the phagosome (Kinchen et al., 2008), significantly inhibited clearance of linker cell debris (Table S2).
Thus, while some genes promoting cell corpse degradation following engulfment are shared between LCD and apoptosis, upstream genes required for phagocytosis and the initiation of degradation are different. LCD-specific engulfment and degradation genes remain, therefore, to be discovered.
RME-4/DENND1 promotes linker cell engulfment and degradation
To identify components of the linker cell corpse removal machinery, we sought mutants in which linker cell engulfment and/or degradation is defective. To do so, we mutagenized hermaphrodites carrying a lag-2p::GFP linker cell reporter and a him-5 mutation, which increases the proportion of male progeny (Figure S1K). After two generations, males were screened for linker cell persistence. We previously showed that animals defective in linker cell death exhibit gonadal blockage, and can be sterile (Abraham et al., 2007). We therefore directly collected sperm from mutant males by impaling a needle into the gonad, and used this sperm to artificially inseminate wild-type hermaphrodites (LaMunyon and Ward, 1994) (Figure S1L-N, see Methods for details). Cross progeny were then used to establish mutant lines.
From this screen, we identified a mutant, ns410, which exhibits persistent refractile linker cell corpses (Figure 2A,B). Using standard genetic mapping, coupled with whole genome sequencing, we identified a mutation predicted to generate a G100E alteration in the DENN domain protein RME-4, homologous to vertebrate DENND1 (Sato et al., 2008) (Figure S2A). Linker cell degradation is restored to ns410 mutants by expressing a fosmid spanning the genomic region of rme-4 (Figure 2B). Furthermore, two other rme-4 alleles, ns412 (S217N; isolated in our screen) and b1001 (G257D; a previously characterized loss-of-function lesion (Sato et al., 2008)) also cause linker cell degradation defects, as does the rme-4(tm1865) deletion allele (Figure S2B). rme-4(tm1865) mutants exhibit a weaker linker cell survival defect, perhaps because the truncated RME-4 protein produced in this mutant is present at half wild-type levels (Sato et al., 2008). RNAi against rme-4 exacerbates the defect of animals carrying this allele (Figure S2B). Taken together, these results demonstrate that the rme-4 gene is required for linker cell corpse degradation.
Figure 2. RAB-35, RME-4 (DENND1), and TBC-10 (TBC10D1A) regulate linker cell clearance.
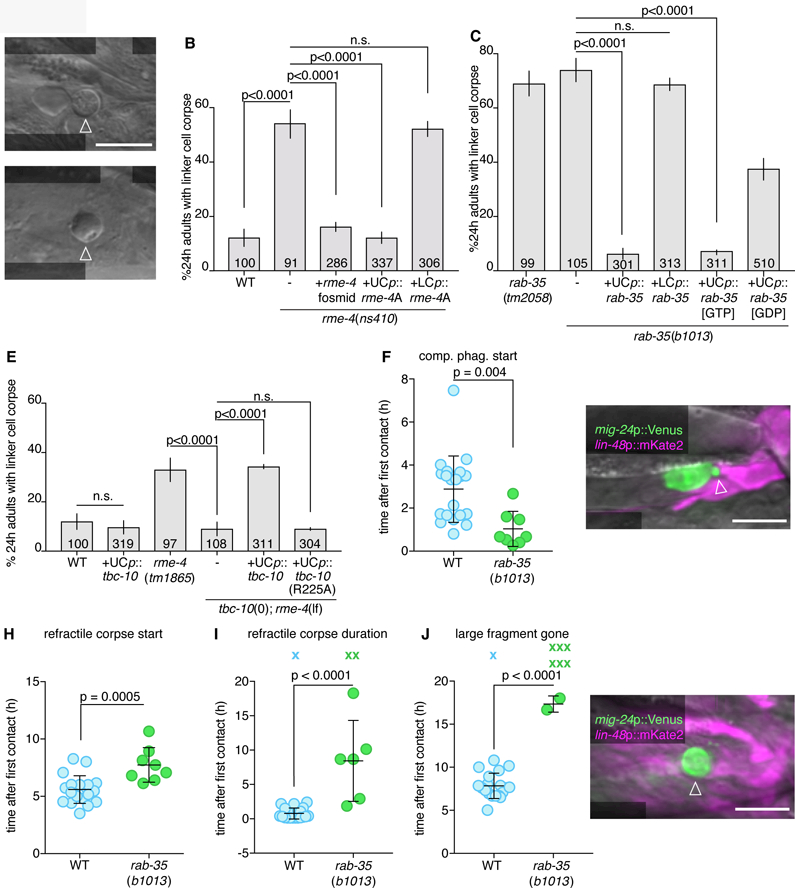
(A) DIC image of persistent linker cell corpse (arrowhead) in rme-4(ns410). Scale bar, 10 µm. (B,C) Linker cell degradation in indicated genotypes. Strains contain lag-2p::GFP linker cell reporter and him-5(e1490). UCp, lin-48p. LCp, mig-24p. Number of animals scoredinside bars. Average of at least three independent lines. Error bars, standard error of the proportion or standard error of the mean. n.s., p>0.05, Fisher’s exact test. (D) DIC image of persistent linker cell corpse (arrowhead) in rab-35(b1013). (E) Histogram details as in (B). (F) Competitive phagocytosis onset in individual animals (circles). Bars, mean ± sd. Student’s t-test. WT, wild-type. (G) Arrowhead, early competitive phagocytosis in rab-35(b1013). Details as in Figure 1. (H-J) Quantification of linker cell events as in (F). X, Event persisting or not observed by end of imaging; not included in statistical analysis. (K) Arrowhead, persistent large cell fragment in rab-35(b1013). Strain and image details as in (G). See also Movie S3, Figures S2, S3, and Table S1, S4.
RME-4 is widely expressed and found in the linker cell and in its engulfing cells (Figure S2C). To determine where RME-4 functions, we expressed cDNAs for either the rme-4A or rme-4B transcript (Figure S2A) in either the linker cell or the engulfing cells. We found that the longer A isoform restores linker cell degradation when expressed in U cell descendants, but not when expressed in the linker cell (Figure 2B). B isoform expression in the engulfing cells has no effect (Figure S2B). Thus, RME-4A is the active isoform driving linker cell engulfment, and it does so by acting in the engulfing cells.
RAB-35 GTPase is a key linker cell degradation regulator controlled by RME-4/DENND1 and TBC-10/TBC1D10A
The RME-4 DENN domain was previously proposed to drive the guanine nucleotide exchange reaction (GEF function) of the small GTPase RAB-35 (Allaire et al., 2010; Sato et al., 2008). The ns410, ns412, and b1001 rme-4 alleles are predicted to cause single amino-acid changes within this domain, suggesting that RME-4 could function as a GEF during linker cell degradation. To test this directly, we examined two rab-35 mutants, tm2058 and b1013, that carry a small deletion surrounding the start codon, and an early Q69Ochre stop mutation, respectively (Figure S2D). We found that both lesions inhibit linker cell corpse degradation, and persistent corpses resemble those seen in rme-4 mutants (Figure 2C,D). While rab-35 is broadly expressed (Figure S2E), we found that linker cell degradation is restored to rab-35 mutants by expression of rab-35 cDNA in engulfing cells, but not in the linker cell (Figure 2C). Thus, RAB-35 is required in engulfing cells for linker cell degradation.
RAB-35 functions were previously studied in in vitro models of macrophage phagocytosis of IgG-opsonized erythrocytes and of zymosan (Egami et al., 2011; 2015). Activation-inactivation cycles of RAB-35 were found to be important (Egami et al., 2015), but neither the guanine nucleotide exchange factor (GEF) nor the GTPase activating protein (GAP) driving these cycles were identified. Furthermore, downstream consequences of RAB-35 loss were not explored. We, therefore, sought to determine which regulatory proteins control RAB-35 activity, and how RAB-35 functions for phagosome maturation.
If RME-4 functions as a RAB-35 GEF, we expect RAB-35[GTP] to be the active form of the protein in linker cell degradation. Indeed, we found that expression of RAB-35(Q69L), predicted to lock the protein in the GTP-bound configuration, fully restores linker cell degradation to rab-35(b1013) mutants (Figure 2C). A GDP-bound mimetic, RAB-35(S24N), however, only partially rescues rab-35(b1013) linker cell defects. Furthermore, we found that RAB-35(S24N) protein selectively binds RME-4A (but not RME-4B) in a yeast two-hybrid assay (Sato et al., 2008) (Figure S2F). Thus, RAB-35 functions with RME-4 in engulfing cells, and RAB-35[GTP] is likely the relevant active form driving linker cell clearance.
We next sought to identify the GTPase activating protein (GAP) completing the RAB-35 GTPase cycle. We reasoned as follows: weak RME-4 (GEF) mutations should result in some RAB-35[GTP] production, but perhaps not enough for efficient linker cell degradation. Combining an rme-4(weak) mutation with a RAB-35 GAP mutation, which would block RAB-35[GTP] hydrolysis, could, however, allow sufficient accumulation of RAB-35[GTP] for efficient linker cell degradation. We therefore screened for the effects of mutations in C. elegans homologs of the known mammalian Rab35 GAP genes identified in the context of endocytosis, tbc-7/Tbc1D24, tbc-10/Tbc1D10A, and tbc-13/Tbc1D13 (Chaineau et al., 2013). Mutations or knockdown of these genes alone have no effect on linker cell corpse degradation, nor do they rescue linker cell degradation defects of animals carrying the strong rme-4(ns410) mutation, in which no RAB-35[GTP] is predicted to accumulate (Figure S2G). However, combining the tbc-10(gk388086) allele with the weak rme-4(tm1865) allele restores normal linker cell degradation (Figure 2E). Thus, TBC-10 is likely the RAB-35 GAP for linker cell engulfment and degradation.
Supporting this notion, while TBC-10 protein is widely expressed (Figure S2H), specific expression of TBC-10 in U cell descendants of rme-4(tm1865); tbc-10(gk388086) double mutants reintroduces the linker cell degradation defect. Similar expression of a TBC-10(R225A) protein, containing a lesion in the putative catalytic arginine finger, has no effect (Figure 2E).
Taken together, our results suggest that RAB-35[GTP] is a key regulator of linker cell degradation, and that its activity is controlled by RME-4/GEF and TBC-10/GAP.
RAB-35 promotes timely onset of linker cell engulfment and is required for subsequent phagosome maturation
To define more specifically the defects associated with loss of RAB-35, we imaged rab-35(b1013) mutants using the microfluidic setup, and quantified hallmark linker cell death and degradation events over > 24h (Movie S3 and Table S1). We found no significant differences compared to wild-type animals in the onset or duration of developmental milestones (e.g. tail-tip retraction or appearance of rays; Figure S3A-C), or in appearance of linker cell death hallmarks (e.g. nuclear crenellation; Figure S3D-I). However, competitive phagocytosis of the linker cell begins prematurely in rab-35 mutants (Figure 2F), initiating before the linker cell has the opportunity to intercalate between the U.l/rp cells (Figure 2G). While subsequent linker cell splitting occurs at the same time as in wild-type animals (Figure S3G), formation of the larger refractile corpse is delayed (Figure 2H), and refractility can last much longer (Figure 2I). Mutations in rab-35 delay degradation of the large fragment, if it occurs at all (Figure 2J,K). The smaller linker cell fragment is degraded as in the wild type (Figure S3H,I). Thus, RAB-35 plays roles in both engulfment initiation and phagosome maturation, but not phagosome closure.
Consistent with a role in engulfment initiation, we found that RAB-35, tagged with the fluorescent reporter YFP, localizes to extending pseudopods early on, and remains enriched around the larger linker cell-containing phagosome for an extended period of time, until late stages of linker cell degradation (Figure 3A-C). To track this quantitatively, we measured YFP fluorescence on the phagosomal membrane and in the cytoplasm of the U.l/rp cell containing the large fragment, and calculated their ratios over time (Figure 3D).
Figure 3. RAB-35 is required for PI(4,5)P2 removal and is enriched with RAB-5.
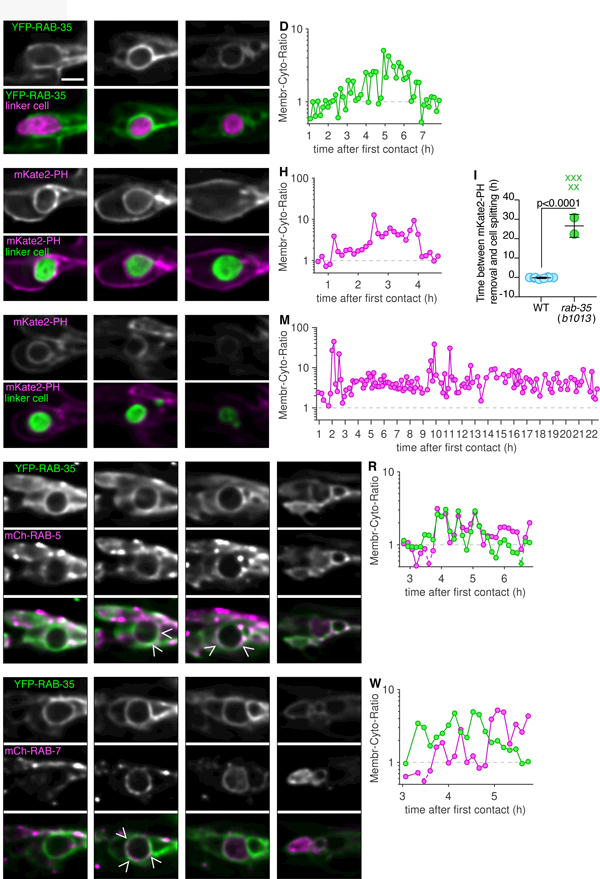
(A-C) Top: Localization of YFP-RAB-35 within U.l/rp cells. Bottom: YFP-RAB-35 (green), and linker cell (mig-24p::mKate2; magenta). Scale bar, 5 µm. (D) Ratio of average fluorescence intensity on phagosome membrane over the cytoplasm in hours after the first contact of animal shown in adjacent images. Logarithmic scale, where 1 indicates equal amount of cytoplasmic and phagosome membrane bound fluorescence. Any data point with a ratio below 0.5 indicated with a diamond and dotted line. (E-G) Same format as (A), except mKate2-PH imaged, and linker cell fluorescently labeled with YFP. (H) Same format as (D). (I) Timing of mKate2-PH removal in relation to linker cell splitting in individual animals (circles). Bars, mean ± sd. Student’s t-test. WT, wild-type. X, cell splitting occurred but mKate2-PH not removed and not included in statistical analysis. (J-L) Same format as (E) except in rab-35(b1013) animal. (M) Same format as in (D). (N-Q) Same format as (A), except mCherry-RAB-5 imaged, and linker cell not fluorescently labeled. Caret, site of colocalization. LC, linker cell. Asterisk, accumulation within phagosome. (R) Same format as in (D). (S-V) Same format as (A), except mCherry-RAB-7 imaged, and linker cell not fluorescently labeled. Caret, site of colocalization. LC, linker cell. Asterisk, accumulation within phagosome. (W) Same format as in (D). See also Figure S4 and Table S1.
Some aspects of phagosome maturation have been previously characterized. We therefore sought to address where in this process RAB-35 functions. The conversion of PI(4,5)P2 to PI(3)P at the phagosome membrane is necessary for phagosome maturation to proceed (Lu and Zhou, 2012). We therefore imaged an mKate2-PH reporter, marking PI(4,5)P2. While in wild-type animals, PI(4,5)P2 removal occurs just prior to cell splitting (Figure 3E-H), in the absence of rab-35, PI(4,5)P2, removal occurs much later, if it occurs at all (23.7 h ± 7.1 h, Figure 3i-m, note time stamps on image frames).
Because linker cell degradation in rab-35 mutants is blocked at a very early stage of phagosome maturation, we sought to determine whether defects in the recruitment of other maturation factors is also evident. To do so, we first characterized wild-type animals expressing both YFP-RAB-35 and either mCherry-RAB-5, marking early phagosomes, or mCherry-RAB-7, marking late phagosomes and phagolysosomes (Figure 3N-W, Table S1). We found that RAB-35 and RAB-5 were co-enriched on phagosome membranes in 5/6 animals examined (Figure 3O,P,R). mCherry-RAB-5 accumulates within the phagosome after co-enrichment (Figure 3Q, 6/6 animals). While RAB-35 and RAB-7 are also initially enriched on the membrane at a similar time, RAB-7 enrichment increases after RAB-35 is removed (Figure 3T,W, 7/7 animals). Once RAB-35 disappears from the phagosome membrane, mCherry-RAB-7 fluorescence accumulates within the phagosome (Figure 3U,V; 7/7 animals). Internalization initiates while the linker cell corpse is still round (Figure 3U), and is still evident as the linker cell Kutscher et al. phagosome changes shape and is degraded (Figure 3V; shape changes observed also by DIC microscopy). Such internalization of RAB-5 and RAB-7 had not been previously reported in other settings, and we believe it cannot be attributed to general differences in fluorescence levels during imaging, as our ratiometric quantification (e.g. Figure 3R and3W) is independent of overall fluorescence levels. Furthermore, RAB-35, RAB-5, and RAB-7 reporter fusion fluorescence is stable in other cells labeled in our strains.
Figure 6. EFA-6 (EFA6) and CNT-1 (ACAP2) regulate ARF-6.
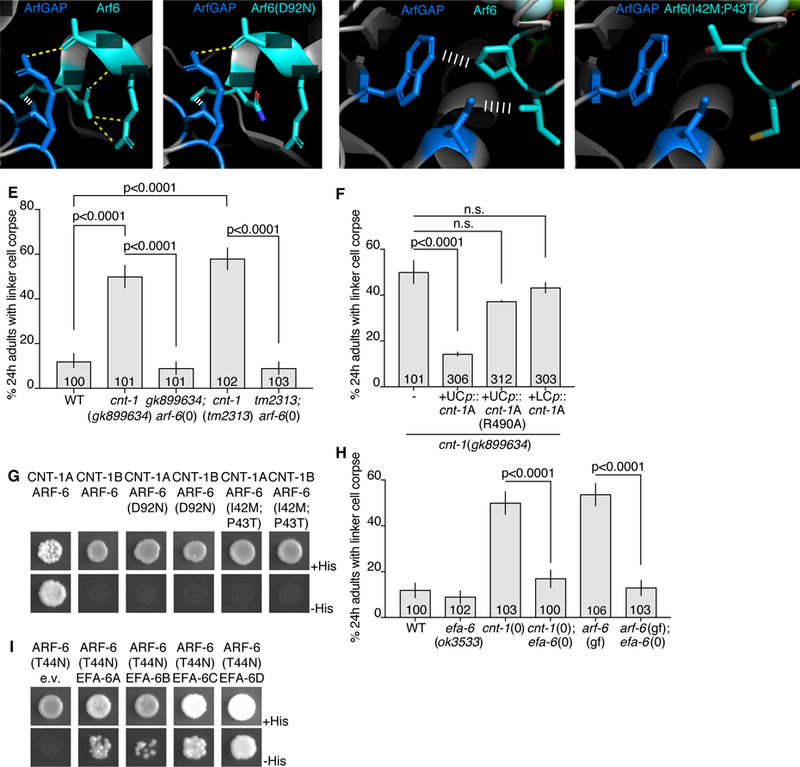
(A) Protein structures of human Arf6 (teal) and ArfGAP (blue) from (Ismail et al., 2010). PDB: 3LVQ. Relevant amino acids indicated in white. Yellow dotted line, hydrogen bonds. White ladder, hydrophobic interactions. Amino acids shown are conserved in C. elegans ARF-6 and CNT-1. (B) D92N mutation mapped onto Arf6. Protein structure information as in (A). (C) Different region of Arf6 and ArfGAP. Protein structure information as in (A). (D) I42M, P43T mutations mapped onto Arf6. (E,F) Histogram details as in Figure 2B. (G) Yeast two-hybrid assay with LexA-CNT-1A or LexA-CNT-1B as bait, GAD-ARF-6, GAD-ARF-6(D92N) or GAD-ARF-6(I42M,P43T) as prey. Top: histidine present. Bottom: histidine absence. Growth on -His plates indicates physical interaction. (H) Histogram details as in (E). (I) As in (G), with LexA-ARF-6[T44N] (GDP) as bait, GAD-EFA-6A-D as prey. See also Figure S5 and Table S3.
We next examined reporter localization in rab-35(b1013) mutants. We found that when the linker cell is not properly cleared, RAB-5 rarely accumulates on the phagosome surface, its enrichment there is not sustained, and it is not internalized into the phagosome (Figure S4A-D, 6/8 animals). RAB-7 does not accumulate on the phagosome surface at all, and is also not taken up into the phagosome (Figure S4E-H, 5/5 animals). These results suggest that RAB-35 is required for RAB-5 and RAB-7 recruitment to the phagosome membrane, consistent with an early block in phagosome maturation. Supporting this notion, rab-35(b1013); sand-1(ok1963) double mutants almost completely abrogate linker cell degradation, further implicating RAB-35 in phagosome maturation (sand-1(ok1963); rab-35(b1013): 95% ± 2.7%, N=63, p<0.0001 compared to single mutants, Fisher’s exact test).
Our studies, therefore, support the idea that RAB-35 acts early in phagosome maturation to remove PI(4,5)P2 and recruit RAB-5 and RAB-7 to the phagosome membrane for proper degradation to ensue.
The small GTPase ARF-6 blocks linker cell clearance
To delineate the molecular mechanism by which RAB-35 controls linker cell engulfment onset and degradation, we pursued studies of another mutant, ns388, isolated in our genetic screen. Animals carrying this lesion have similar linker cell defects to rab-35 mutants (Figure 4A,B), but do not harbor mutations in rab-35, tbc-10, or rme-4.
Figure 4. ARF-6 inhibits linker cell degradation.
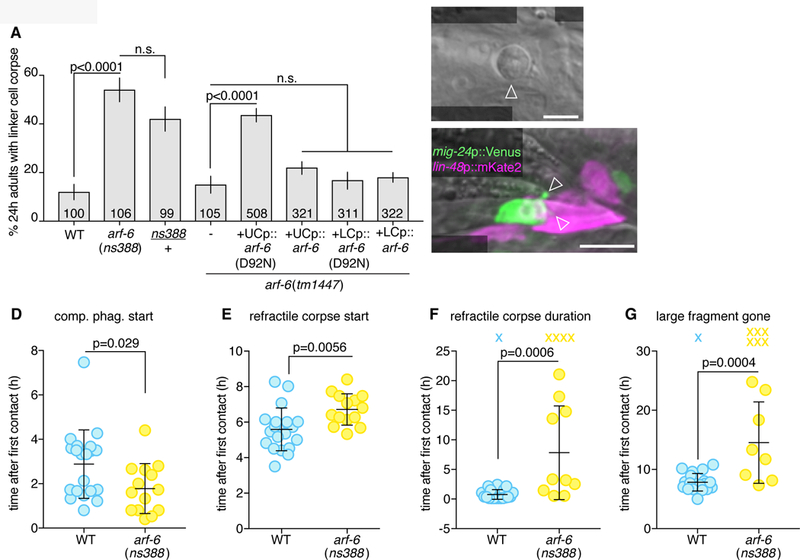
(A) Linker cell degradation in indicated genotypes. Details as in Figure 2B. (B) DIC image of persistent linker cell corpse (arrowhead) in arf-6(ns388) male. Scale bar, 5 µm. (C) Arrowheads, premature competitive phagocytosis in arf-6(ns388). Scale bar, 10 µm. (D-G) Timing of linker cell clearance events in arf-6(ns388). Quantification as in Figure 2F,H-J. See also Movie S4, Figure S3, S5 and Table S1.
Using whole genome sequencing and standard genetic mapping, we found that ns388 animals contain a point mutation predicted to cause a D92N mutation in the small GTPase ARF-6 (Figure S5A). Importantly, and unlike rab-35 mutations, a single arf-6(ns388) allele is sufficient to block linker cell degradation. Thus, arf-6(ns388) is a dominant allele (Figure 4A). The arf-6(tm1447) putative null allele, which lacks most of the coding region, and has no ARF-6 expression by Western blot (Shi et al., 2012), exhibits normal linker cell clearance (Figure 4A), suggesting that arf-6(ns388) is a gain-of-function and not a dominant-negative allele. To confirm that arf-6 is indeed the relevant gene, we generated two independent CRISPR alleles, ns751 and ns752, recreating the arf-6(ns388) lesion. Both promote linker cell defects similar to those of arf-6(ns388) mutants (Figure S5B). Furthermore, while arf-6 is widely expressed (Figure S5C,D), expression of an arf-6(ns388) cDNA in engulfing cells blocks linker cell clearance (Figure 4A). Wild-type arf-6 expression in engulfing cells does not affect linker cell death, nor does expression of either arf-6(ns388) or arf-6(+) cDNAs in the linker cell (Figure 4A).
Our results, therefore demonstrate that gain of ARF-6 function blocks linker cell clearance, and suggest that ARF-6 normally functions as a linker cell clearance inhibitor.
ARF-6(gf) promotes premature competitive phagocytosis onset and delays linker cell degradation
ARF-6 has also been previously implicated in macrophage phagocytosis of foreign particles, but the exact mechanism by which it does this was unexplored (Egami et al., 2011; 2015). To understand how ARF-6(gf) interferes with linker cell clearance, we imaged arf-6(ns388) animals in our microfluidic device and followed hallmark linker cell death and degradation events (Movie S4 and Table S1). Except for a slight advance in cuticle shedding, no defects in animal development or linker cell killing are seen in arf-6(ns388) mutants (Figure S3). However, as with rab-35 mutants, premature onset of competitive phagocytosis is observed (Figure 4C,D), formation of a refractile linker cell corpse is delayed (Figure 4E), and the period of linker cell refractility is prolonged (Figure 4F). Degradation of the large linker cell fragment is greatly delayed, if it occurs at all (Figure 4G).
The similar defects exhibited by arf-6(ns388) and rab-35 mutants suggests that the proteins encoded by these genes may localize in a similar manner. To test this idea, we expressed ARF-6-YFP in the U.l/rp cells and followed its localization during linker cell death and degradation (Figure 5A-D). ARF-6-YFP localizes to U.l/rp pseudopods (Figure 5A and Table S1), remains enriched on the phagosome membrane until immediately prior to cell splitting (removal occurs 18.7 min ± 14.9 min prior to cell splitting, Figure 4B), and then rapidly translocates to intracellular puncta (Figure 5C). Removal of ARF-6-YFP occurs at a similar time as PI(4,5)P2 removal, and coincides with enrichment of mCherry-RAB-5 around the phagosome (Figure 5J-N, 5/6 animals). RAB-5 also accumulates in the phagosome interior (Figure 5M, 6/6 animals). Similar torab-35 mutants, in arf-6(gf) mutants, removal of PI(4,5)P2 occurs slowly, if at all (11.5 h4.4 h, Figure 5E-L; note time stamps on images); and RAB-5 recruitment does not occur (Figure 5O-R, 4/5 animals), suggesting an early block in phagosome maturation. Thus, both ARF-6 and RAB-35 localize to the plasma membrane early and surround the dying linker cell. However, while RAB-35 normally promotes linker cell degradation, ARF-6 blocks this process.
Figure 5. ARF-6(d) affects removal of PI(4,5)P2 from the phagosome membrane and affects RAB-5 recruitment.
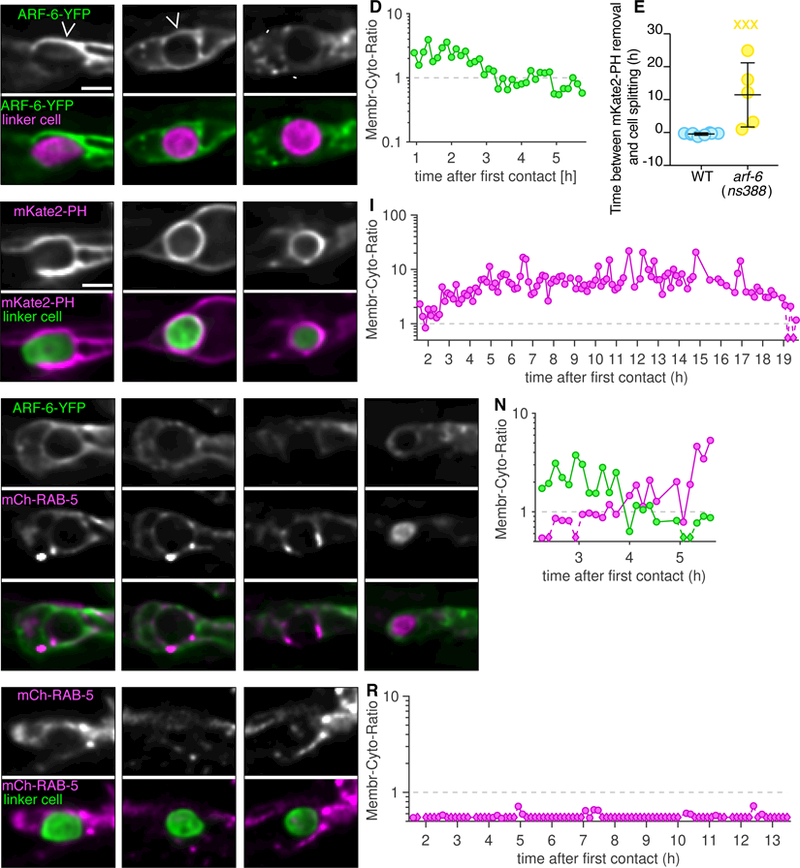
(A-C) Top: Localization of ARF-6-YFP within the U.l/rp cells. Caret, ARF-6 on phagosome membrane. Arrowhead, ARF-6 in intracellular puncta. Bottom: ARF-6-YFP (green), and linker cell (mig-24p::mKate2; magenta). Scale bar, 5 µm. (D) Ratio of average fluorescence intensity on phagosome membrane over the cytoplasm in hours after the first contact of animal shown in adjacent images. Logarithmic scale, where 1 indicates equal amount of cytoplasmic and phagosome membrane bound fluorescence. Any data point with a ratio below 0.5 indicated with a diamond and dotted line. (E) Timing of mKate2-PH removal in relation to linker cell splitting in individual animals (circles). Bars, mean ± sd. Student’s t-test. WT, wild-type, same data as in Figure 3i. X, cell splitting occurred but mKate2-PH not removed and not included in statistical analysis. (F-H) Same format as (A), except mKate2-PH imaged, and linker cell fluorescently labeled with YFP. (I) Same format as (D). (J-M) Localization of ARF-6-YFP (top) and mCherry-RAB-5 (middle). LC, linker cell. Asterisk, accumulation of RAB-5 within phagosome. (N) Same format as (D). (O-Q) Same format as (J), except in arf-6(ns388) animal (ARF-6-YFP not imaged). (R) Same format as (D). See also Table S1.
ARF-6 function is regulated by CNT-1/ACAP2 and EFA-6/EFA6
To understand the dominant nature of the ARF-6(D92N) lesion, we attempted to assess the consequences of locking ARF-6 protein in the GTP or GDP bound states (Macia et al., 2004). We first used CRISPR to generate arf-6 mutants encoding T44N and Q67L mutations (Figure S5A), predicted to accumulate ARF-6[GDP] or ARF-6[GTP] proteins, respectively. However, neither mutant exhibits linker cell defects, and the arf-6(Q67L) allele unexpectedly behaves as a null in genetic assays (see below). Nonetheless, we did serendipitously isolate from our CRISPR mutagenesis an arf-6(I42M,P43T) double mutant with linker cell clearance defects. Based on an Arf6:Arf6GAP co-crystal protein structure (Ismail et al., 2010), these lesions may interfere with GAP binding to ARF-6, and subsequent GTP hydrolysis. D92 of Arf6 forms intramolecular hydrogen bonds with R95 (Figure 6A). In the D92N mutant, these hydrogen bonds are lost (Figure 6B), which may result in loss of stability of adjacent interactions. Both P43 and I42 of Arf6 form hydrophobic interactions with W451 and I462 on the ArfGAP (Figure 6C). These interactions are lost in the P43T;I42M mutations (Figure 6D). Therefore, ARF-6[GTP] appears to inhibit phagosome maturation during linker cell clearance, and the mutants we isolated may hinder GAP binding.
If this idea is correct, then mutations in a relevant ARF-6 GAP should also exhibit linker cell clearance defects, as they would lock ARF-6 in the GTP-bound state. From a screen of candidate mutations (Figure 6E and Table S3), we found that two independent putative null alleles of cnt-1, encoding a ubiquitously-expressed protein similar to vertebrate Acap2, block linker cell clearance (Figure 6E and Figure S5E,F). Expression of the CNT-1A isoform in U.l/rp cells, but not in the linker cell, fully rescues linker cell defects of cnt-1 mutants (Figure 6F); whereas CNT-1B expression gives partial rescue (Figure S5G). Rescue is abolished by mutating the catalytic arginine of CNT-1A (Figure 6F). Importantly, ARF-6 is required for the linker cell defects of cnt-1 mutants, as an arf-6(tm1447) loss-of-function mutation restores linker cell clearance to cnt-1 mutants (Figure 6E). Consistent with the idea that the D92N and I42M;P43T mutations disrupt GAP binding, we found that while wild-type ARF-6 interacts with CNT-1A (but not CNT-1B) in a yeast two-hybrid assay; both ARF-6(D92N) and ARF-6(I42M;P43T) fail to interact with CNT-1A (Figure 6G). Thus, ARF-6[GTP] is likely the active form of the protein blocking linker cell removal, and CNT-1 is the ARF-6 GAP for linker cell clearance.
Similar reasoning used to identify TBC-10 as a RAB-35 GAP allowed us to identify EFA-6, homologous to the mammalian Arf6 GEF Efa6 (Macia et al., 2001), as the putative ARF-6 GEF for linker cell clearance. While the efa-6(ok3533) deletion allele has no linker cell degradation defect, this allele restores linker cell clearance to mutants carrying null alleles of cnt-1 (Figure 6H; cnt-1(tm2313); efa-6(ok3533): 14 ± 3.4%, N =102, p<0.0001 compared to cnt-1(tm2313), Fisher’s exact test). Furthermore, efa-6(ok3533) also blocks the dominant defects of arf-6(ns388) and arf-6(ns763[I42M;P43T]) mutants, strengthening our assessment that ARF-6 GTP loading is required for the function of these ARF-6 gain-of-function proteins (Figure 6H and Figure S5H). While EFA-6 is expressed in the linker cell and U.l/rp cells (Figure S5I), expression of EFA-6 isoforms in the U.l/rp cells partially restores linker cell clearance defects to efa-6(ok3533); arf-6(ns388) double mutants (Figure S5H). Furthermore, EFA-6 isoforms interact with ARF-6[GDP] in a yeast two-hybrid assay (Figure 6I).
Taken together, our results suggest that ARF-6 inhibits linker cell phagocytosis onset, and blocks linker cell degradation together with its regulators EFA-6/GEF and CNT-1A/GAP.
RAB-35 binds CNT-1A and drives ARF-6 removal from phagosome membranes
rab-35(0) and arf-6(gf) mutants exhibit similar linker cell clearance defects. We wondered, therefore, whether RAB-35 and ARF-6 proteins act in the same pathway, especially since these proteins are implicated together in vitro in phagocytosis of foreign particles by macrophages (Egami et al., 2011; 2015). We found that putative null mutations in arf-6 or efa-6 almost fully restore linker cell clearance to rab-35 mutants (Figure 7A and Figure S6A). The arf-6[Q67L] CRISPR mutant also suppresses rab-35(0) defects (Figure S6B), confirming this allele inactivates ARF-6 and is not an ARF-6[GTP] mimetic (see above). Linker cell clearance defects are restored to rab-35(b1013); arf-6(tm1447) double mutants by expressing wild-type arf-6 in U.l/rp cells, but not in the linker cell (Figure S6C). Furthermore, neither mutations in cnt-1 nor the dominant arf-6 allele enhance the linker cell defects of rab-35 mutants (Figure S6D). Finally, overexpression of YFP-RAB-35 in engulfing cells restores linker cell clearance to arf-6(ns388) mutants (Figure 7A). Taken together, these results suggest that RAB-35 normally functions to inactivate ARF-6 in vivo following linker cell death.
Figure 7. RAB-35 removes ARF-6 from phagolysosome membranes.
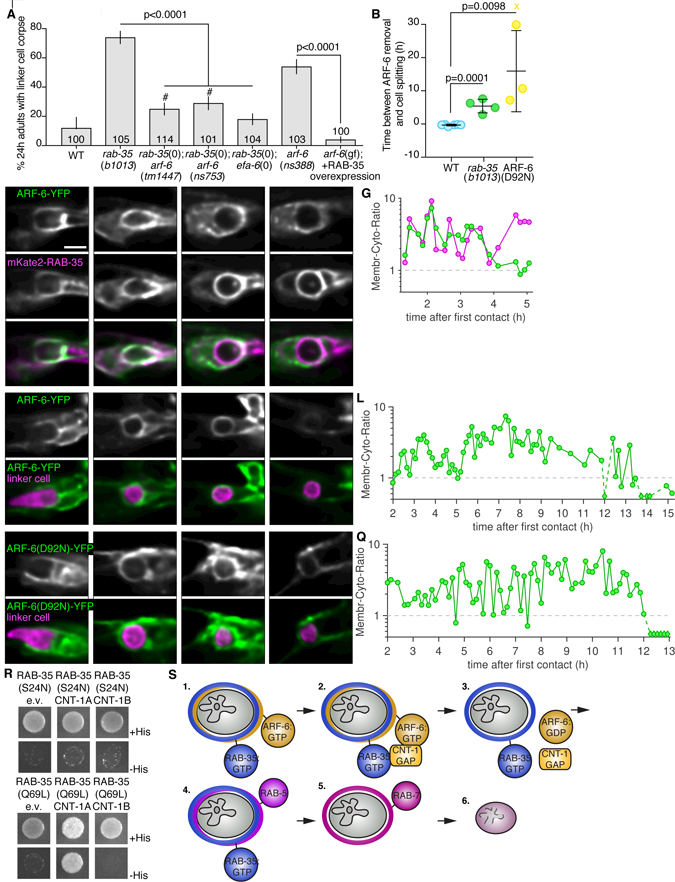
(A) Histogram details as in Figure 2b. #, significant difference compared to rab-35(b1013) and wild type. (B) Timing of ARF-6-YFP removal in relation to linker cell splitting in individual animals (circles). Bars, mean ± sd. Student’s t-test. WT, wild-type,. X, cell splitting occurred but ARF-6-YFP not removed and not included in statistical analysis. ARF-6(D92N) indicates ARF-6(D92N)-YFP imaged in otherwise WT animals. (C-F) Localization of indicated proteins with details as in Figure 3. (G) Ratio of membrane localized to cytoplasmic localized fluorescent protein with details as in Figure 3. (H-K) Same details as in (C), in rab-35(b1013) mutant. (L). Same format as in (G), in rab-35(b1013) mutant. (M-P) Same details as in (C), but with ARF-6(D92N)-YFP variant. (Q) Same format as in (G). (R) Yeast two-hybrid assay with LexA-RAB-35[S24N] (GDP) or LexA-RAB-35[Q67L] (GTP) as bait, Gal4-AD (GAD)empty vector, GAD-CNT-1A, or GAD-CNT-1B as prey. Details as in Figure 6G. (S) 1. ARF-6 and RAB-35 surround the nascent phagosome, marked with PI(4,5)P2. 2. RAB-35 recruits CNT-1. 3. CNT-1 turns off ARF-6, removing it from the phagosome membrane. RAB-35 remains on the membrane. 4. RAB-5 is recruited. 5. RAB-7 recruitment and RAB-35 loss. 6. The linker cell is degraded once the phagosome fuses with lysosomes. See also Figure S6, S7 and Table S1.
To understand how RAB-35 inhibits ARF-6, we imaged mKate2-RAB-35 and ARF-6-YFP localization together. Consistent with our other imaging studies, the two proteins initially co-localize to extending U.l/rp cell pseudopods (Figure 7C). RAB-35 then accumulates on the phagosome membrane, while ARF-6 enrichment fades (Figure 7D-G, 8/9 animals). Next, we examined ARF-6-YFP dynamics in rab-35(b1013) animals. Remarkably, we found that ARF-6-YFP remains on the membrane longer than in wild-type animals (5/6 animals; Figure 7B,H-L; note time stamps; ARF-6-YFP removed 5.4 h ± 2.0 h after cell splitting in rab-35(b1013)). Furthermore, in wild-type animals, ARF-6(D92N) remains enriched on phagosome membranes longer than ARF-6-YFP (4/4 animals; Figure 7b,M-Q; 15.9h ± 12.2h after cell splitting).
These results raise the possibility that RAB-35 promotes linker cell clearance, at least in part, by facilitating ARF-6 removal from the phagosome. Consistent with this notion, an ARF-6(D92N) gain-of-function protein is more effective in blocking linker cell clearance than ARF-6(D92N) lacking a myristoyl group addition sequence, required for membrane localization (D’Souza-Schorey and Stahl, 1995) (Figure S6C,E).
To determine whether ARF-6 localization is regulated by direct binding to RAB-35, we assessed their interactions in a yeast two-hybrid system (see Methods). While we did not detect any interactions between these two proteins, we found that RAB-35[GTP], the active form of RAB-35, binds CNT-1A (and not CNT-1B) (Shi et al., 2012) (Figure 7R). Thus, RAB-35 may recruit CNT-1A to phagosome membranes, catalyzing conversion of active ARF-6[GTP] to inactive ARF-6[GDP], consistent with its previously suggested in vitro function (Egami et al., 2011).
The RAB-35/ARF-6 module is not required for apoptotic cell clearance
Given the roles of RAB-35 and ARF-6 in the clearance of a cell dying by LCD, we wondered whether these proteins play similar roles in apoptotic cell clearance. To test this, we counted the number of persisting apoptotic cells in 3-fold embryos, after nearly all developmental cell death has taken place. In him-5 mutant control animals, we counted 0.10 ± 0.30 persisting refractile corpses on average (N=21). In arf-6(ns388) mutants, 0.47 ± 0.60 corpses were observed (N=21, p=0.0131, Student’s t-test). rab-35(b1013) mutants exhibit 0.81 ± 0.92 corpses, on average (N=21, p=0.0018, Student’s t-test). These small defects pale in comparison to defects exhibited by mutants defective in canonical apoptotic engulfment genes (e.g. ced-7; ced-10 double mutant: 16.8 ± 4.2 corpses, N = 11). Furthermore, rme-4 mutant embryos we tested have no clearance defects (ns410: 0.14 ± 0.36 apoptotic corpses, N=14; ns412: 0.13 ± 0.33 apoptotic corpses, N=24). We also examined whether rab-35 mutations exhibit genetic interactions with canonical apoptosis engulfment genes in linker cell death, but found no such interactions.
Our results, therefore, demonstrate that the RAB-35/ARF-6 module plays specific roles in dismantling the linker cell that are not shared with clearance of apoptotic cells in C. elegans.
Discussion
An engulfment and degradation pathway for non-apoptotic dying cells
Caspase-dependent apoptosis does not account for many cell death events that occur during animal development. Indeed, mice homozygous for knockout alleles of key apoptotic genes, including caspase-3, caspase-9, Apaf-1, or a triple knockout of Bax, Bak, and Bok, where intrinsic apoptosis is completely abrogated, can survive to adulthood (Honarpour et al., 2000; Ke et al., 2018; Kuida et al., 1998; Lindsten et al., 2000), a surprising observation given the prevalence of cell death in murine development. Inactivation of apoptosis genes also only weakly interferes with degenerative disease progression (Gould et al., 2006). LCD, a non-apoptotic cell death process, is prevalent in vertebrates (Kutscher and Shaham, 2017) and may account for a significant fraction of cell deaths that take place during development and in disease.
In this study, we demonstrate that the clearance machinery promoting the removal of the linker cell, which dies by LCD, differs from that used to clear apoptotic cells in C. elegans. Our studies uncover a protein network promoting linker cell engulfment and degradation in vivo during normal animal development (Figure S6F). We find that RAB-35 and ARF-6 play key early roles, controlling the sequential loading of phagosome maturation factors onto the phagosome membrane, beginning with removal of PI(4,5)P2, and continuing with recruitment of RAB-5 (Figure 7S). Our dataare consistent with a model in which RAB-35[GTP] promotes elimination of dying cells by blocking the activity of an inhibitor of the process, ARF-6[GTP]. Our studies are the first to implicate a RAB-35/ARF-6 module in the engulfment and degradation of dying cells, define in detail multiple accessory factors, identify the relevant targets (e.g. Kutscher et al. PI(4,5)P2 removal, RAB-5, RAB-7 recruitment) and, importantly, explore the functions of these proteins in vivo in a multicellular organism.
Receptor-mediated endocytosis and phagocytosis are two ways by which particles enter a cell. These processes are thought to have generally distinct molecular components, and even shared regulators, such as dynamin, appear to have different functions (De Camilli et al., 1995; Yu et al., 2006). Our findings that RAB-35 and ARF-6 control phagosome PI(4,5)P2 levels echo a similar role for these proteins in endocytosis (Brown et al., 2001; Cauvin et al., 2016; Honda et al., 1999; Shi et al., 2012), suggesting fundamental similarities between some aspects of these processes.
Although we identify the LCD corpse engulfment pathway in C. elegans, a number of studies in other systems raise the possibility that this process may be conserved. For example, in Drosophila, nurse cell death is thought to be caspase independent, and knockdown of RAB-35 results in persistent nurse cell nuclei (Timmons et al., 2016). While the molecular function of RAB-35 was not investigated here, the involvement of this protein in a non-apoptotic setting is intriguing, suggesting that the paradigms we found may also function elsewhere in non-apoptotic corpse clearance. Furthermore, concerted activities of RAB-35 and ARF-6 in other contexts have been described, including neurite outgrowth, cytokinesis, oligodendrocyte differentiation, maturation of recycling endosomes, and FcγR-mediated uptake of bacteria by macrophage (Allaire et al., 2013; Chesneau et al., 2012; Egami et al., 2011; Kobayashi and Fukuda, 2012; Miyamoto et al., 2014). In some of these cases, interactions between RAB-35 and putative ARF-6 GAPs are seen (Egami et al., 2015).
Competitive phagocytosis
In addition to unique molecular components, we found that the mechanics of linker cell engulfment are very different from engulfment of apoptotic cells. Our imaging studies reveal a process of competitive phagocytosis, where two phagocytes compete to engulf portions of the linker cell, resulting in cell splitting. RAB-35 acting through ARF-6 prevents premature activation of this engulfment process. As with the molecular constitutes of linker cell engulfment and degradation, we believe that the process of competitive phagocytosis may also be conserved. In macrophage-less mice, for example, mesenchymal cells engulf dying neighbors during digit formation, and occasionally two mesenchymal cells can be seen engulfing the same dying cell in transmission electron microscopy images (Wood et al., 2000). Consistent with this process involving different engulfment regulators, mesenchymal cells do not express the CED-7 homolog ABC1 (Wood et al., 2000). Similarly, in the developing rat cerebellum, more than one non-professional phagocyte can be seen engulfing a dying cell (Parnaik et al., 2000). A related process has been documented during germ cell development in C. elegans. Primordial germ cells extend cytoplasm-containing lobes into the adjacent endoderm, and the lobe is excised from the remaining cell, in a process reminiscent of competitive phagocytosis (Abdu et al., 2016).
The need for engulfment by more than one cell could be related to the size of the dying cell. For example, when macrophages attempt to engulf a particle that is too large, engulfment is stalled (Cannon and Swanson, 1992; Herbomel et al., 1999). Engulfment by two or more cells could solve this problem. However, in this case, cytoplasmic spillage must be avoided. The engulfment process we describe here accomplishes exactly that. Intriguingly, the interplay between ARF-6 and RAB-35 in corpse degradation is required only for the larger, nucleus-containing fragment, as the smaller, cytoplasmic-only fragment is degraded efficiently in these mutants. These results suggest that different mechanism may be required to degrade each of the cellular fragments.
A process reminiscent of competitive phagocytosis can also be seen when cultured human peripheral blood monocytes engulf a C. albicans pathogen (Rittig et al., 1998). Similarly, cultured mouse monocytes simultaneously engulf a Leishmania parasite (Rittig et al., 1998). One cell engulfs the flagellum, while the other engulfs the posterior pole. In this context, roles for RAB-35 in pathogen clearance (Egami et al., 2011; 2015; Yeo et al., 2016) are especially intriguing, and may suggest underlying mechanisms similar to linker cell clearance.
In summary, we demonstrate that engulfment and degradation of a cell that dies by LCD requires mechanics and machinery different from that used for apoptotic cell engulfment and degradation, and identify a molecular pathway governing this form of cell clearance. Given the conservation of LCD, we propose that this ancillary process may be conserved as well.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shai Shaham (shaham@rockefeller.edu).
Experimental Model and Subject Details
C. elegans
C. elegans strains were raised at 20°C on nematode growth medium (NGM) seeded with OP50 bacteria, unless otherwise indicated. Wild-type animals were of the Bristol N2 strain. All strains have one of two mutations that generate a high incidence of male progeny, him-8(e1489) IV or him-5(e1490) V. Mutants recovered by EMS mutagenesis were outcrossed five times before use. Transgenic lines were generated by injection of plasmid DNA mixes into the hermaphrodite gonad. Integrated transgenic strains were generated with UV/trioxalen treatment (Kutscher and Shaham, 2014) (Sigma, T6137) and were outcrossed at least four times before imaging experiments. Most strains also had one of three integrated linker cell markers qIs56[lag-2p::GFP] V, nsIs65[mig-24p::Venus] X, or nsIs650[mig-24p::mKate2] X. Other alleles are as follows:
LGI: rab-2(n3263), ced-1(e1735), cep-1(gk138), dpy-5(e907)
LGII: cnt-1(gk899634), cnt-1(tm2313), cdc-42(gk388), rab-7(ok511)
LGIII: rab-35(b1013), rab-35(tm2058), tbc-10(gk388086), unc-119(ed3), ttr-52(tm2003), ttr-52(tm2078), F23H11.4(gk585084), F23H11.4(gk165637), cnt-2(gm377)
LGIV: arf-6(tm1447), arf-6(ns388), arf-6(ns751[D92N]), arf-6(ns752[D92N]), arf-6(ns753), arf-6(ns760[T44N]), arf-6(ns761[T44N]), arf-6(ns762[T44N]), arf-6(ns763[I42M;P43T]), arf-6(ns765[Q67L]), arf-6(ns771[Q67L]), arf-6(ns753), arf-6(ns764), arf-6(ns767), arf-6(ns769), sand-1(or552), sand-1(ok1963), efa-6(ok3533), ced-10(n1993), ced-5(n1812), psr-1(ok714), psr-1(tm469)
LGX: rme-4(ns410), rme-4(ns412), rme-4(b1001), rme-4(tm1865), tbc-13(ok1812), piki-1(ok2346), him-4(e1267), git-1(ok1848), git-1(gk392605), gap-2(ok1001)
Yeast
The yeast strain NMY51 (Dualsystems Biotech) was maintained at 30°C on YPAD plates for yeast two-hybrid assays. Yeast were plated on SD-Leu-Trp- after transformation, and 5µl of OD600 = 0.4 of transformants were spotted on SD-Leu-Trp-His- to test for interactions.
Method Details
Forward genetic screen and artificial insemination
Young L4 hermaphrodites containing qIs56 [lag-2p::GFP] and him-5(e1490) were mutagenized with 75 mM ethylmethanesulfonate (EMS, Sigma M0880) for 4h at 20°C. Animals were synchronized twice by bleaching, and male animals were enriched by passing through a 40 µm cell strainer (BD Falcon) passively for 20 min (Lewis and Fleming, 1995). Male animals fit more readily through the mesh than hermaphrodites and were enriched to ~88%. Animals were then scored under a fluorescent dissecting microscope (Leica) for presence of a linker cell by GFP. GFP-expressing animals were isolated away from hermaphrodites for 24h to accumulate spermatids within the gonad. Mutant males were immobilized on a dried agar pad under immersion oil, and spermatids removed with a needle containing SM buffer (50 mM HEPES, pH 7, 50 mM NaCl, 25 mM KCl, 5 mM CaCl2, 1 mM MgSO4, 1 mg/ml BSA) (LaMunyon and Ward, 1994). An N2 hermaphrodite with a single row of eggs was then immobilized on the same dried agar pad and gently injected with the collected sperm through the vulva. We typically tried to insert all spermatids into a single hermaphrodite. GFP+ heterozygous progeny were collected, allowed to self fertilize, and linker cell persistence confirmed under a dissecting microscope.
Gene identification
A combination of Hawaiian Snip-SNP mapping (Wicks et al., 2001) and whole genome sequencing (Zuryn et al., 2010) was using to identify rme-4(ns410), rme-4(ns412), and arf-6(ns388) using output from galign (Shaham, 2009). rme-4 was confirmed by fosmid rescue using WRM0615bE09, and arf-6 identification was confirmed by mutant cDNA expression and CRISPR alleles.
Linker cell survival and corpse persistence assays
Linker cell death was scored as previously described (Blum et al., 2012). Briefly, unstarved gravid hermaphrodites were bleached to isolate embryos, which were allowed to hatch overnight in M9. Synchronized L1 animals were released on 9-cm NGM plates seeded with OP50 or HT115 E. coli containing the RNAi construct of interest on IPTG-RNAi plates. Male animals were isolated onto a new plate prior to the L4-to-adult transition based on full retraction of the male tail tip with rays visible under the unshed L4 cuticle. 24h later, these animals were mounted onto 2% agarose-water pads, anaesthetized in 25 mM sodium azide, and examined on an Axioplan 2 fluorescence microscope (Zeiss) with a 63x/1.4NA objective (Zeiss) and Nomarski optics. The linker cell was identified by location, morphology, and fluorescence from reporter transgenes. Cells were then scored as surviving, dead, or gone based on DIC morphology and fluorescence.
Generation of arf-6 alleles using CRISPR-Cas9 genome editing
Alleles of arf-6 were generated using the co-CRISPR-based genome editing method as previously described (Arribere et al., 2014). pDD162 was used as a vector backbone, and the following sgRNA sequences were added for each individual CRISPR attempt (D92N: 5’ CTAAAAACCTGTCTCTATCAG 3’; T44N: 5’ TCAGTGACCACAATGACGA 3’; Q67L 5’ CTTCAGGACGTCGGCGGAC 3’) to generate arf-6 targeting vectors. A dpy- sgRNA-pDD162-based vector was also generated (5’ GCTACCATAGGCACCACGAG 3’). Single-stranded repair oligos were PAGE purified and ordered from Sigma (D92N: 5’ aggaaaaaaaccaatttttccgcatttttcgcctaaaaacCTGTCTCTATtAGCaGCGTCCATCACAAAA ATGAGCGCCTGAGTTCCTGTGTAATAATGTC 3’; T44N: 5’ agCAATTCTGTACAAACTGAAGCTCGGGCAATCAGTGACCACAATTCCGAacGTGG GCTTCAATGTGGAGACTGTCACGTATAAAAATATCAAATTCAACGT 3’; Q67L 5’ ggcctaaaaacccccaaaaaccccaatttttcttcagGACGTCGGCGGACttGACAAAATTCGACCCC TCTGGCGACATTATTACACAGGAACTCAGGCGCT 3’, dpy-10(cn64): 5’ CACTTGAACTTCAATACGGCAAGATGAGAATGACTGGAAACCGTACCGCATGCGG TGCCTATGGTAGCGGAGCTTCACATGGCTTCAGACCAACAGCCTAT 3’ (Arribere et al., 2014). N2 animals were injected with the following mix: 50 ng/µl dpy-10 sgRNA, 50 ng/µl arf-6 targeting vector, 20 ng/µl dpy-10(cn64) repair oligo, 20 ng/µl arf-6 repair oligo in 1x injection buffer (20mM potassium phosphate, 3mM potassium citrate, 2% PEG, pH 7.5). F1 animals with Dpy or Rol phenotypes were picked to individual plates, indicating a CRISPR-based editing event had occurred. F1 animals were allowed to lay eggs, and then genotyped for successful co-conversion of the arf-6 locus using PCR and restriction enzyme screening or Cel1 digestion of heteroduplex DNA (Ward, 2015). Non-Rol, non-Dpy F2 animals were then singled and homozygosed for the arf-6 mutation.
Germline transformation and rescue experiments
Plasmid mixes containing the plasmid of interest, co-injection markers, and pBluescript were injected into both gonads of young adult hermaphrodites (Mello and Fire, 1995). Injected animals were singled onto NGM plates and allowed to grow for two generations. Transformed animals based on co-injection markers were picked onto single plates, and screened for stable inheritance of the extrachromosomal array. Only lines from different P0 injected hermaphrodites were considered independent. For cell-specific rescue experiments, animals expressing mCherry in either the linker cell or the U.l/rp cells were picked to a new plate at the early L4 stage, before linker cell death, to avoid bias. Isolated animals were then staged based on tail morphology under a white light microscope for appropriate linker cell scoring.
Plasmid construction
Plasmids containing lin-48p, mig-24p, or transcriptional reporters were cloned using multi-piece one-step cloning into a modified pPD95.75 backbone lacking GFP. For CRISPR-related vectors, plasmids were generated using a site-directed plasmid mutagenesis protocol on pDD162 (Dickinson et al., 2013; Liu and Naismith, 2008). Yeast vectors were generated using traditional cloning from either a pLexA-N-terminus vector or a pGAL4AD-N-terminus vector (Dualsystems Biotech), and single amino acid change variants were generated using QuikChange (Agilent).
Protein structures
The co-crystal structure of human Arf6 with the ArfGAP ASAP3 (PDB 3LVQ) was examined in Pymol (The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC.) in silico mutagenesis for D92N and I42M;P43T was performed and hydrophilic interactions tested within Pymol. Key residues noted in Figure 5A-D in ASAP3 are conserved in C. elegans CNT-1, and those for Arf6 are conserved in C. elegans ARF-6.
Long-term imaging and movie generation
Early L4 male animals were imaged in a microfluidic device described in detail in (Keil et al., 2017). Animals were fed a constant flow of NA22 bacteria in S medium, supplemented with kanamycin (50 ng/µl) to prevent bacterial overgrowth. Animals were immobilized and imaged every 8 minutes for at least 20h. Mutant animals were typically imaged for > 30h. Exposure time and light intensity was held constant across strains when the same integrated transgene was imaged. Occasionally tail development was perturbed by the flow of medium and repeated immobilization procedures, and these animals were not included in subsequent analyses.
Deconvolution
Images were cropped to a region of interest surrounding the linker cell. We measured the point-spread function (PSF) of our optical setup using red (580/605nm) and green (505/515nm) fluorescent 200nm beads (ThermoFisher Scientific). Using these PSFs, we deconvolved the cropped fluorescence z-stacks using the classic maximum likelihood estimation (CMLE) algorithm with standard parameters (refractive index of imaging medium: 1.338) in the Huygens Essential software (Huygens Essential 3.7.1) by Scientific Volume Imaging (SVI).
Image Analysis
Image analysis was performed using custom-written MATLAB R2016b (Mathworks) scripts. To overlay imaging frames, we straightened each three-dimensional image stack using a previously published algorithm (Keil et al., 2017; Peng et al., 2008) based on a manually selected worm backbone in the DIC channel. To correct for small residual animal movements during multi-channel acquisition, obvious landmarks visible in the fluorescence channels, such as the cloaca, fluorescently labeled cells in the animal’s tail, or vesicles within the U-cell descendants were then manually aligned to the DIC channel in each frame. Time-lapse movies were generated by centering all straightened, aligned images on the linker cell and cropping the entire movie to an appropriate size. Frames were removed from the movies whenever residual animal movement excessively blurred the images.
Fluorescence intensity quantification
All fluorescence intensity ratios (Figures 3,5,7, Figure S4) were quantified based on deconvolved and alignment-corrected z-stacks (see above). For each frame, we first selected the z-slice in which the linker cell or large linker cell fragment was maximally in focus in the DIC channel and used this slice for further quantification of fluorescence. In strains in which the linker cell was fluorescently labeled with the mig-24 promoter, the linker cell outline was automatically detected using Otsu thresholding. Otherwise, the LC outline was manually selected based on its DIC features and its visible delineation in the respective fluorescence channels. Additionally, using the same fluorescence channel, we manually selected a cytoplasmic area in the U.l/rp cell in the same z-slice (at least 10pixels) for each frame. Average fluorescence values were then computed both, along the linker cell outline (membrane fluorescence value) and in the selected cytoplasmic region of the U.l/rp cell, allowing us to quantify enrichment of the fluorescently tagged proteins on the linker-cell-U.l/rp-cell interface compared to the cytoplasm of the U.l/rp cell.
RNAi assay
pL4440 RNAi plasmids containing coding sequence to rme-4 or tbc-7 were isolated from the Ahringer Library (Kamath et al., 2003) (Source BioScience) and the coding sequence was confirmed by sequencing. These plasmids were grown at 37°C in HT115 bacteria, which have a plasmid containing an inducible promoter driving T7 RNA polymerase. The bacteria were plated on NGM plates that had been supplemented with the antibiotic carbomycin and IPTG. One day later, synchronized L1 animals were plated on the RNAi plates and grown for two days before assaying for linker cell corpse persistence.
Yeast two-hybrid assay
Strain and vectors used in assay were from the DUALHybrid Kit (Dualsystem Biotech). Bait cDNA was cloned into the pLexA plasmid and prey cDNA was cloned into pGAD vectors. These plasmids were cotransformed into NMY51 yeast strain using the lithium acetate method described in the DualHybrid manual. Selection was performed on SD plates lacking the amino acids leucine, tryptophan, and histidine.
Apoptotic corpse assay
We counted the number of apoptotic corpses in 3-fold embryos mounted on agar pads and examined at 63x using DIC (Nomarski optics). These corpses could be distinguished by their high refractility.
Quantification and Statistical Analysis
Statistical analysis was performed using GraphPad Prism. Statistical parameters including mean ± standard error of the proportion, mean ± SEM, and N are reported in the main text, figures and figure legends. Data is judged to be statistically significant when p < 0.05 by Fisher’s exact test, χ2-test, or student’s t-test, where appropriate.
Genetic Data Analysis
When comparing a transgenic line to the parental strain, a minimum of three independent lines was scored. Approximately 100 animals from each of these lines were examined for linker cell defects, and compared to the parental strain using a Fisher’s exact test to determine significance. For ease of presentation, we pooled the lines from a given experiment and displayed them on the relevant figure using mean ± SEM (N = 3, 4, or 5 lines). If only a subset of the three lines showed significance using a Fisher’s exact test, we indicated this on the figure and figure legend.
Data and software availability
Custom image analysis and fluorescence intensity quantification scripts are available upon request.
Supplementary Material
Highlights.
Two cells compete to engulf the C. elegans linker cell after linker cell-type death
Competitive phagocytosis and corpse clearance rely on two small GTPase networks
RAB-35 removes ARF-6 from phagosome membranes via its regulator CNT-1
ARF-6 removal promotes PI(4,5)P2 removal and subsequent phagosome maturation
Acknowledgments
We thank Zheng Zhou for sharing unpublished results; Barth Grant, Zheng Zhou, Paul Randazzo, Jacqueline Cherfils, and Shohei Mitani for reagents; Nima Tishbi for technical help; Jordan Ward for the extensive Cel1 protocol; and Shaham Lab members for discussions and critical reading of the manuscript. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This work was supported by an NIH NRSA Training Grant GM066699 to L.M.K., an HFSP postdoctoral fellowship LT000250/2013-C to W.K., an NSF grant PHY 1502151 awarded to Eric D. Siggia, and NIH grants R01NS081490, R01HD078703, and R35NS105094 to S.S.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdu Y, Maniscalco C, Heddleston JM, Chew T-L, and Nance J (2016). Developmentally programmed germ cell remodelling by endodermal cell cannibalism. Nat. Cell Biol 18, 1302–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham MC, Lu Y, and Shaham S (2007). A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Dev. Cell 12, 73–86. [DOI] [PubMed] [Google Scholar]
- Allaire PD, Marat AL, Dall’Armi C, Di Paolo G, McPherson PS, and Ritter B (2010). The Connecdenn DENN domain: a GEF for Rab35 mediating cargo-specific exit from early endosomes. Mol. Cell 37, 370–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaire PD, Seyed Sadr M, Chaineau M, Seyed Sadr E, Konefal S, Fotouhi M, Maret D, Ritter B, Del Maestro RF, and McPherson PS (2013). Interplay between Rab35 and Arf6 controls cargo recycling to coordinate cell adhesion and migration. J. Cell Sci 126, 722–731. [DOI] [PubMed] [Google Scholar]
- Andersen MH, Graversen H, Fedosov SN, Petersen TE, and Rasmussen JT (2000). Functional analyses of two cellular binding domains of bovine lactadherin. Biochemistry 39, 6200–6206. [DOI] [PubMed] [Google Scholar]
- Arribere JA, Bell RT, Fu BXH, Artiles KL, Hartman PS, and Fire AZ (2014). Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 198, 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum ES, Abraham MC, Yoshimura S, Lu Y, and Shaham S (2012). Control of nonapoptotic developmental cell death in Caenorhabditis elegans by a polyglutamine-repeat protein. Science 335, 970–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho RJ, Teruel M, Dierckman R, Anderson R, Wells A, York JD, Meyer T, and Grinstein S (2000). Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J. Cell Biol 151, 1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown FD, Rozelle AL, Yin HL, Balla T, and Donaldson JG (2001). Phosphatidylinositol 4,5-bisphosphate and Arf6-regulated membrane traffic. J. Cell Biol 154, 1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon GJ, and Swanson JA (1992). The macrophage capacity for phagocytosis. J. Cell Sci 101 (Pt 4), 907–913. [DOI] [PubMed] [Google Scholar]
- Cauvin C, Rosendale M, Gupta-Rossi N, Rocancourt M, Larraufie P, Salomon R, Perrais D, and Echard A (2016). Rab35 GTPase Triggers Switch-like Recruitment of the Lowe Syndrome Lipid Phosphatase OCRL on Newborn Endosomes. Curr. Biol 26, 120–128. [DOI] [PubMed] [Google Scholar]
- Chaineau M, Ioannou MS, and McPherson PS (2013). Rab35: GEFs, GAPs and effectors. Traffic 14, 1109–1117. [DOI] [PubMed] [Google Scholar]
- Cheng S, Wang K, Zou W, Miao R, Huang Y, Wang H, and Wang X (2015). PtdIns(4,5)P2 and PtdIns3P coordinate to regulate phagosomal sealing for apoptotic cell clearance. J. Cell Biol 210, 485–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesneau L, Dambournet D, Machicoane M, Kouranti I, Fukuda M, Goud B, and Echard A (2012). An ARF6/Rab35 GTPase Cascade for Endocytic Recycling and Successful Cytokinesis. Curr. Biol 22, 147–153. [DOI] [PubMed] [Google Scholar]
- Chu-Wang IW, and Oppenheim RW (1978). Cell death of motoneurons in the chick embryo spinal cord. I. A light and electron microscopic study of naturally occurring and induced cell loss during development. J. Comp. Neurol 177, 33–57. [DOI] [PubMed] [Google Scholar]
- D’Souza-Schorey C, and Stahl PD (1995). Myristoylation is required for the intracellular localization and endocytic function of ARF6. Exp. Cell Res 221, 153–159. [DOI] [PubMed] [Google Scholar]
- De Camilli P, Takei K, and McPherson PS (1995). The function of dynamin in endocytosis. Curr. Opin. Neurobiol 5, 559–565. [DOI] [PubMed] [Google Scholar]
- Dickinson DJ, Ward JD, Reiner DJ, and Goldstein B (2013). Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Methods 10, 1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djehiche B, Segalen J, and Chambon Y (1994). Ultrastructure of mullerian and wolffian ducts of fetal rabbit in vivo and in organ culture. Tissue Cell 26, 323–332. [DOI] [PubMed] [Google Scholar]
- Dyche WJ (1979). A comparative study of the differentiation and involution of the Mullerian duct and Wolffian duct in the male and female fetal mouse. J. Morphol 162, 175–209. [DOI] [PubMed] [Google Scholar]
- Egami Y, Fujii M, Kawai K, Ishikawa Y, Fukuda M, and Araki N (2015). Activation-Inactivation Cycling of Rab35 and ARF6 Is Required for Phagocytosis of Zymosan in RAW264 Macrophages. J. Immunol. Res 2015, 429439–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egami Y, Fukuda M, and Araki N (2011). Rab35 regulates phagosome formation through recruitment of ACAP2 in macrophages during FcγR-mediated phagocytosis. J. Cell [DOI] [PubMed] [Google Scholar]
- Ellis RE, Jacobson DM, and Horvitz HR (1991). Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics 129, 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franc NC, Heitzler P, Ezekowitz RA, and White K (1999). Requirement for croquemort in phagocytosis of apoptotic cells in Drosophila. Science 284, 1991–1994. [DOI] [PubMed] [Google Scholar]
- Fuchs Y, and Steller H (2015). Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol 16, 329–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlena RA, Lennox AL, Baker LR, Parsons TE, Weinberg SM, and Stronach BE (2015). The receptor tyrosine kinase Pvr promotes tissue closure by coordinating corpse removal and epidermal zippering. Development 142, 3403–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, and Oppenheim RW (2006). Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J. Neurosci 26, 8774– 8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P, Hu T, Zhang J, Jiang S, and Wang X (2010). Sequential action of Caenorhabditis elegans Rab GTPases regulates phagolysosome formation during apoptotic cell degradation. Proc. Natl. Acad. Sci. U.S.A 107, 18016–18021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbomel P, Thisse B, and Thisse C (1999). Ontogeny and behaviour of early macrophages in the zebrafish embryo. Development 126, 3735–3745. [DOI] [PubMed] [Google Scholar]
- Honarpour N, Du C, Richardson JA, Hammer RE, Wang X, and Herz J (2000). Adult Apaf-1-deficient mice exhibit male infertility. Dev. Biol 218, 248–258. [DOI] [PubMed] [Google Scholar]
- Honda A, Nogami M, Yokozeki T, Yamazaki M, Nakamura H, Watanabe H, Kawamoto K, Nakayama K, Morris AJ, Frohman MA, et al. (1999). Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 99, 521–532. [DOI] [PubMed] [Google Scholar]
- Ismail SA, Vetter IR, Sot B, and Wittinghofer A (2010). The structure of an Arf-ArfGAP complex reveals a Ca2+ regulatory mechanism. Cell 141, 812–821. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237. [DOI] [PubMed] [Google Scholar]
- Ke FFS, Vanyai HK, Cowan AD, Delbridge ARD, Whitehead L, Grabow S, Czabotar PE, Voss AK, and Strasser A (2018). Embryogenesis and Adult Life in the Absence of Intrinsic Apoptosis Effectors BAX, BAK, and BOK. Cell 173, 1217–1230. [DOI] [PubMed] [Google Scholar]
- Keil W, Kutscher LM, Shaham S, and Siggia ED (2017). Long-Term High-Resolution Imaging of Developing C. elegans Larvae with Microfluidics. Dev. Cell 40, 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchen JM, and Ravichandran KS (2010). Identification of two evolutionarily conserved genes regulating processing of engulfed apoptotic cells. Nature 464, 778– 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchen JM, Doukoumetzidis K, Almendinger J, Stergiou L, Tosello-Trampont A, Sifri CD, Hengartner MO, and Ravichandran KS (2008). A pathway for phagosome maturation during engulfment of apoptotic cells. Nat. Cell Biol 10, 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinet MJ, Malin JA, Abraham MC, Blum ES, Silverman MR, Lu Y, and Shaham S (2016). HSF-1 activates the ubiquitin proteasome system to promote non-apoptotic developmental cell death in C. elegans. Elife 5, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, and Fukuda M (2012). Rab35 regulates Arf6 activity through centaurin-β2 (ACAP2) during neurite outgrowth. J. Cell Sci 125, 2235–2243. [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, and Flavell RA (1998). Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94, 325–337. [DOI] [PubMed] [Google Scholar]
- Kutscher LM, and Shaham S (2014). Forward and reverse mutagenesis in C. elegans. WormBook 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutscher LM, and Shaham S (2017). Non-apoptotic cell death in animal development. Cell Death Differ 24, 1326–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMunyon CW, and Ward S (1994). Assessing the viability of mutant and manipulated sperm by artificial insemination of Caenorhabditis elegans. Genetics 138, 689–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JA, and Fleming JT (1995). Basic culture methods. Methods Cell Biol 48, 3– 29. [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong W-X, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. (2000). The Combined Functions of Proapoptotic Bcl-2 Family Members Bak and Bax Are Essential for Normal Development of Multiple Tissues. Mol. Cell 6, 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, and Naismith JH (2008). An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 8, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu N, and Zhou Z (2012). Membrane trafficking and phagosome maturation during the clearance of apoptotic cells. Int. Rev. Cell Mol. Biol 293, 269–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maat-Schieman MLC, Dorsman JC, Smoor MA, Siesling S, Van Duinen SG, Verschuuren JJGM, Dunnen Den, J.T., Van Ommen G-JB, and Roos RAC (1999). Distribution of Inclusions in Neuronal Nuclei and Dystrophic Neurites in Huntington Disease Brain. J. Neuropath. Exp. Neurol 58, 129–137. [DOI] [PubMed] [Google Scholar]
- Macia E, Chabre M, and Franco M (2001). Specificities for the small G proteins ARF1 and ARF6 of the guanine nucleotide exchange factors ARNO and EFA6. J. Biol. Chem 276, 24925–24930. [DOI] [PubMed] [Google Scholar]
- Macia E, Luton F, Partisani M, Cherfils J, Chardin P, and Franco M (2004). The GDP-bound form of Arf6 is located at the plasma membrane. J. Cell Sci 117, 2389– 2398. [DOI] [PubMed] [Google Scholar]
- Malin JA, Kinet MJ, Abraham MC, Blum ES, and Shaham S (2016). Transcriptional control of non-apoptotic developmental cell death in C. elegans. Cell Death Differ 23, 1985–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangahas PM, Yu X, Miller KG, and Zhou Z (2008). The small GTPase Rab2 functions in the removal of apoptotic cells in Caenorhabditis elegans. J. Cell Biol 180, 357–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello C, and Fire A (1995). DNA transformation. Methods Cell Biol 48, 451–482. [PubMed] [Google Scholar]
- Miyamoto Y, Yamamori N, Torii T, Tanoue A, and Yamauchi J (2014). Rab35, acting through ACAP2 switching off Arf6, negatively regulates oligodendrocyte differentiation and myelination. Mol. Biol. Cell 25, 1532–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-Y, and Kim I-S (2017). Engulfment signals and the phagocytic machinery for apoptotic cell clearance. Exp. Mol. Med 49, e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnaik R, Raff MC, and Scholes J (2000). Differences between the clearance of apoptotic cells by professional and non-professional phagocytes. Curr. Biol 10, 857– 860. [DOI] [PubMed] [Google Scholar]
- Peng H, Long F, Liu X, Kim SK, and Myers EW (2008). Straightening Caenorhabditis elegans images. Bioinformatics 24, 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon IKH, Lucas CD, Rossi AG, and Ravichandran KS (2014). Apoptotic cell clearance: basic biology and therapeutic potential. Nat. Rev. Immunol 14, 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JM, Donahoe PK, Ito Y, and Hendren WH (1977). Programmed cell death in the Müllerian duct induced by Müllerian inhibiti ng substance. Am. J. Anat 149, 353– 375. [DOI] [PubMed] [Google Scholar]
- Rittig MG, Burmester GR, and Krause A (1998). Coiling phagocytosis: when the zipper jams, the cup is deformed. Trends Microbiol 6, 384–388. [DOI] [PubMed] [Google Scholar]
- Sato M, Sato K, Liou W, Pant S, Harada A, and Grant BD (2008). Regulation of endocytic recycling by C. elegans Rab35 and its regulator RME-4, a coated-pit protein. Embo J 27, 1183–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham S (2009). galign: a tool for rapid genome polymorphism discovery. PLoS ONE 4, e7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi A, Liu O, Koenig S, Banerjee R, Chen CC-H, Eimer S, and Grant BD (2012). RAB-10-GTPase-mediated regulation of endosomal phosphatidylinositol-4,5-bisphosphate. Proc. Natl. Acad. Sci. U.S.A 109, e2306–e2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Albertson DG, and Thomson JN (1980). The Caenorhabditis elegans male: postembryonic development of nongonadal structures. Dev. Biol 78, 542–576. [DOI] [PubMed] [Google Scholar]
- Timmons AK, Mondragon AA, Schenkel CE, Yalonetskaya A, Taylor JD, Moynihan KE, Etchegaray JI, Meehan TL, and McCall K (2016). Phagocytosis genes nonautonomously promote developmental cell death in the Drosophila ovary. Proc. Natl. Acad. Sci. U.S.A 113, e1246–e1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosello-Trampont AC, Brugnera E, and Ravichandran KS (2001). Evidence for a conserved role for CRKII and Rac in engulfment of apoptotic cells. J. Biol. Chem 276, 13797–13802. [DOI] [PubMed] [Google Scholar]
- Venegas V, and Zhou Z (2007). Two alternative mechanisms that regulate the presentation of apoptotic cell engulfment signal in Caenorhabditis elegans. Mol. Biol. Cell 18, 3180–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JD (2015). Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics 199, 363–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicks SR, Yeh RT, Gish WR, Waterston RH, and Plasterk RH (2001). Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat. Genet 28, 160–164. [DOI] [PubMed] [Google Scholar]
- Wood W, Turmaine M, Weber R, Camp V, Maki RA, McKercher SR, and Martin P (2000). Mesenchymal cells engulf and clear apoptotic footplate cells in macrophageless PU.1 null mouse embryos. Development 127, 5245–5252. [DOI] [PubMed] [Google Scholar]
- Yeo JC, Wall AA, Luo L, and Stow JL (2016). Sequential recruitment of Rab GTPases during early stages of phagocytosis. Cell Logist 6, e1140615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Lu N, and Zhou Z (2008). Phagocytic receptor CED-1 initiates a signaling pathway for degrading engulfed apoptotic cells. PLoS Biol 6, e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Odera S, Chuang C-H, Lu N, and Zhou Z (2006). C. elegans Dynamin mediates the signaling of phagocytic receptor CED-1 for the engulfment and degradation of apoptotic cells. Dev. Cell 10, 743–757. [DOI] [PubMed] [Google Scholar]
- Zuryn S, Le Gras S, Jamet K, and Jarriault S (2010). A strategy for direct mapping and identification of mutations by whole-genome sequencing. Genetics 186, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.