

SUMMARY
The NLRP3 inflammasome senses a range of cellular disturbances, although no consensus exists regarding a common mechanism. Canonical NLRP3 activation is blocked by high extracellular K+, regardless of the activating signal. We report here that canonical NLRP3 activation leads to Ca2+ flux and increased calpain activity. Activated calpain releases a pool of Caspase-1 sequestered by the cytoskeleton to regulate NLRP3 activation. Using electrophysiological recording, we found that resting-state eukaryotic membrane potential (MP) is required for this calpain activity, and depolarization by high extracellular K+ or artificial hyperpolarization results in the inhibition of calpain. Therefore, the MP/Ca2+/calpain/ Caspase-1 axis acts as an independent regulatory mechanism for NLRP3 activity. This finding provides mechanistic insight into high K+-mediated inhibition of NLRP3 activation, and it offers an alternative model of NLRP3 inflammasome activation that does not involve K+ efflux.
Graphical Abstract
In Brief
Zhang et al. find that, in canonical NLRP inflammasome activation, calpain activity is essential for releasing caspase-1 from flightless-1 and the cytoskeleton. Membrane depolarization, such as under high extracellular K+ or hyperpolarization, impairs this activity. This work provides insight into extracellular K+ -mediated inhibition of the NLRP3 inflammasome.
INTRODUCTION
NLRP3 inflammasome activation is characterized by the diversity of its stimuli, ranging from solid particles, membrane pore inducers, to ATP (Lamkanfi and Dixit, 2014; Latz et al., 2013). ROS production, mitochondrial destabilization, and Ca2+ signaling have all been suggested to bridge upstream stimulations (Horng, 2014; Tschopp and Schroder, 2010; Zhou et al., 2011). However, whether there is a common rheostat event underlying the plethora of stimuli is under debate. In parallel, it has been suggested that K+ efflux or reduced intracellular K+ is either a prerequisite or a stand-alone self-sufficient signal for NLRP3 inflammasome activation (Muñoz-Planillo et al., 2013; Perregaux and Gabel, 1994; Pe´ trilli et al., 2007). However, critical insights integrating these two seemingly disparate types of regulations are not yet available.
Biologically, K+ flux is the key mechanism to set up a charge gradient across the plasma membrane that gives rise to the eukaryotic cell membrane potential (MP) (Wright, 2004). In stimulated excitable cells, Na+ influx depolarizes the membrane in a fraction of a millisecond that is immediately followed by equally rapid K+ efflux to restore the MP (Marks, 1997). In nonexcitable cells, including immune cells, receptor-mediated signals trigger the changes in MP more slowly. In electrochemical terms, the charge differential associated with the physiological MP sets a field strength at 200,000 V/cm (Lodish et al., 2000). While the transient change in MP is fundamental to neuronal activities, the presence of this strong electrostatic field in the resting state sets a strictly maintained platform for eukaryotic biology (Bezanilla, 2008; Cala, 1977; Daut et al., 1994; Sundelacruz et al., 2009; Zhou et al., 2015). The transitory nature in physiological MP regulation leads to the question of whether a sustained drop of intracellular K+ can be maintained long enough to serve as the intermediate for so many signals converging at NLRP3. The involvement of K+ efflux in most cases is deduced from the observation that the presence of high extracellular K+ blocks NLRP3 stimuli for IL-1β production, which also causes an experimentally sustained membrane depolarization (Belhage et al., 1993). The inhibition by extracellular K+ may indicate a potential role of the eukaryotic MP in regulating NLRP3 inflammasome activation. However, how much the MP-associated functional disruption under high extracellular K+ contributes to the loss of NLRP3 activation has not been thoroughly investigated.
We report here a scenario that the resting MP of phagocytes is an important prerequisite for NLRP3 inflammasome activation. Previous reports suggest that, under resting conditions, some Caspase-1 is sequestered in the cortical cytoskeleton (Li et al., 2008). We found that in phagocytes this pool of Caspase-1 is released by calpain by canonical triggers of NLRP3 via Ca2+ signaling. By coupling electrophysiological patch clamp with biochemical/imaging readouts, we discovered an MP-dependent, calpain-mediated mechanism to control the release of Caspase-1 from its cytoskeleton-tangled state of segregation. The Ca2+-dependent, enhanced activity of calpain relies on an undisrupted MP close to 40 mV. Either depolarization or hyperpolarization of the MP inhibits this increase, thus reducing NLRP3 activation. Therefore, the K+/MP/Ca2+/calpain-regulated sequestration state of Caspase-1 may serve as a regulatory mechanism for the general stress sensing by NLRP3, suggesting a scenario whereby NLRP3 activation can be affected by extracellular K+ other than by blockage of its cross-membrane efflux.
RESULTS
Calpain Plays an Important Role in NLRP3 Inflammasome Activation
Calpain is a large family of calcium-regulated non-endo or lysosomal proteases of about 20 members, controlling events including vesicular trafficking, protein association, motility, surface binding, and cellular metabolism (Glading et al., 2002; Sorimachi et al., 2010; Storr et al., 2011). In studying Ca2+ signaling in NLRP3 inflammasome activation, we found that, in the presence of a pan-calpain inhibitor, calpeptin, crystal-induced IL-1β production was reduced without greatly affecting tumor necrosis factor alpha (TNF-α) (Figure 1A). Another mechanistically distinct inhibitor, PD150606, had the same effect (Figure 1A).
Figure 1. Calpain Activation Plays an Important Role in NLRP3 Inflammasome Activation.

(A) BMDCs were pretreated with calpain inhibitors calpeptin (30 μM) or PD150606 (50 μM) for 30 min and then activated with different crystals for 5 hr. IL-1β and TNF-α in the supernatant were measured by ELISA.
(B) Immunoblot of activated Caspase-1 and mature IL-1β in supernatants (Sup) and all NLRP3 inflammasome components in cell lysates (Lys) of BMDCs stimulated as in (A).
(C) ELISA analysis of IL-1β production in CAST knockdown THP-1 cells stimulated with silica.
(D and E) ELISA analysis of IL-1β production in CAPN1- or CAPN2-knockdown (D) or CAPN1-knockout THP-1 cells (E, generated with 2 different small guide RNAs).
(F) Immunoblot of activated Caspase-1 and mature IL-1β in supernatants and all NLRP3 inflammasome components in cell lysates of CAPN1-deficient or wildtype (WT) THP-1 cells.
(G) CAPN1-deficient or WT THP-1 cells were treated with 3 μm biotin-coated beads for 30 min and then the cells were stained with Alexa 405-strepavidin without permeabilization to distinguish adherent or complete engulfed beads. Percentages of complete phagocytosis of 100 cells were plotted.
(H and I) BMDCs were pretreated with calpeptin or PD150606, then treated with ATP or nigericin. IL-1β secretion was measured by ELISA (H), and Caspase-1 activation and IL-1β processing were detected by immunoblotting (I).
In (A), (C)–(E), and (H), data are shown as mean ± SEM of n = 3. All results are representative of 2 or 3 independent experiments. See also Figure S1.
In a previous report, it was found that calpain was required for IL-1β and NLRP3 inflammasome component secretion in a unique mode of ATP-triggered NLRP3 activation of human macrophages (Välimäki et al., 2016), without involving the signaling chain. In our model, we wondered if it disrupted a particular juncture of NLRP3 activation. Indeed, calpeptin reduced Caspase-1 p20 and IL-1β p17 production (Figure 1B), without greatly impacting cell death (Figure S1A), mRNA or protein levels of NLRP3, pro-caspase-1, or pro-IL-1β (Figures 1B, S1B, and S1C). Calpain inhibitors also reduced crystal-induced IL-1β production in bone marrow-derived macrophages (BMDMs) and THP-1 cells (Figures S1D and S1E). As it is not feasible to reduce all calpain members at the same time, calpastatin (CAST), an endogenous inhibitor of calpain (Kawasaki et al., 1989), was knocked down by small hairpin RNA (shRNA) (Figure S1F). This treatment increased IL-1β production (Figure 1C).
Both μ-calpain and m-calpain are ubiquitously expressed. They are heterodimers composed of a large catalytic subunit (encoded by CAPN1 and CAPN2 for μ- and m-calpain, respectively) and a common regulatory subunit. shRNA knockdown of CAPN1 or CAPN2 (Figure S1G) showed that depletion of μ-calpain, but not m-calpain, was able to reduce IL-1β levels (Figure 1D). To further demonstrate the involvement of μ-calpain, we performed CRISPR-Cas9-based genomic deletion in THP-1 cells (Figure S1H). μ-calpain-deficient THP-1 cells produced reduced IL-1β levels in response to the crystals (Figure 1E). Western blot (WB) analysis also confirmed that caspase-1 activation and IL-1β maturation were blocked in μ-calpain-deficient cells, although apoptosis-associated speck-like protein containing CARD (ASC) and pro-caspase-1 protein level were upregulated (Figure 1F). It should be noted that μ-calpain deletion did not cause any reduction in phagocytosis, excluding the possibility of reduced phagocytic signaling or cell and particle contact to account for the limited IL-1β response (Figure 1G). Calpain inhibitors also strongly blocked the IL-1β production induced by ATP and nigericin (Figure 1H). Without altering the basal protein levels of NLRP3, pro-Caspase-1, and pro-IL-1β, the conversions of active Caspase-1 and IL-1β in response to ATP and nigericin were both blocked by calpeptin (Figure 1I). Therefore, the calpain activity is involved in canonical NLRP3 inflammasome activation.
Depolarization of MP by High Extracellular K+ Correlates with NLRP3 Inflammasome Inhibition
Although multiple models have been proposed regarding a common activation mechanism of NLRP3, K+ efflux is gradually moving to the focal center (Próchnicki et al., 2016). While K+ outward motion and reduced intracellular K+ have been presumed to activate an unknown intracellular sensor of ion strength, experimentally, the majority of the reports that came to this conclusion had relied on the observation that high extracellular K+ blocks NLRP3 inflammasome activation. At cell biology level, this surrogate analysis also leads to induced depolarization of the membrane, which has a profound impact on membrane ion channels and protein activities (Goldenberg and Steinberg, 2010; Kulbacka et al., 2017; Zhou et al., 2015). Therefore, in our system a question arose as to whether K+ efflux served as a conduit to transmit the calpain activation to NLRP3 signaling or extracellular K+ might block the Ca2+/ calpain activities via MP disruption.
The MP is described by the Goldman-Hodgkin-Katz equation (Figure S2A). To probe the role of MP in inflammasome activation, we elected to first use crystals to activate the NLRP3 inflammasome. Assays were performed to see if changes in MP would affect IL-1β production and to assess their qualitative association. The MP of THP-1 cells measured by patch clamp was modified by progressively increasing external K+ concentration (Figure 2A, typical step response). Initially, around −40 mV as reported (Banderali et al., 2011), the value was altered to about −20 mV in 20 mM K+, and it turned slightly positive when K+ was at 75 mM (Figure 2B). Alum, calcium pyrophosphate dihydrate (CPPD), and silica-induced release of IL-1β from THP-1 was suppressed with increasing membrane depolarization (Figure 2C). The MP of bone marrow derived dendritic cells (BMDCs) was also near −40 mV and showed a similar reversal in response to extracellular K+. At −20 mM, the MP was reduced to −20 mV; neutrality was reached between 50 and 75 mM, and the maximum potential was ~+10 mV in 130 mM K+ (Figure S2B). Coincidentally, this was accompanied by a complete loss of IL-1β production at 50 mM K+, altogether showing a slightly higher sensitivity to the MP than THP-1 cells (Figure S2C). In contrast, TNF-α and IL-6 production from BMDCs was not dramatically affected by external K+ (Figure S2D). BMDMs gave results essentially identical to BMDCs (Figure S2E). Without implicating any causality, this set of data suggests a correlation between membrane depolarization and IL-1β production triggered by canonical NLRP3 stimuli.
Figure 2. Depolarization or Hyperpolarization of Membrane Potential May Affect the Activation of NLRP3 Inflammasome.

(A) Representative membrane potential (MP) trace of a THP-1 macrophage treated with isotonically balanced salt solution containing different concentrations of K+ (mM) in whole-cell patch-clamp mode.
(B) THP-1 cell MP in response to different K+ concentrations (n = 5).
(C) THP-1 cells were treated with Al(OH)3, calcium pyrophosphate crystals (CPPD), or silica in different concentrations of extracellular K+ (5, 20, 50, 75, 100, and 130 mM, left to right). Supernatant was collected after 5 hr for IL-1β ELISA (n = 3).
(D) Representative MP trace of BMDCs treated with glyburide.
(E) BMDC MP in response to the indicated concentrations of glyburide (n = 5).
(F) ELISA analysis of IL-1β or TNF-α secreted by BMDCs stimulated with different crystals in the presence or absence of glyburide (Glb).
(G) THP-1 cells were stained with DiBAC4(5) to check the effect of CFTRinh-172 on MP. Representative images and relative fluorescence unit (RFU) plotting are shown. Each point was mean ± SEM of cells from at least 12 fields in 2 independent experiments. Scale bar, 20 μm.
(H) ELISA analysis of IL-1β and TNF-α secreted by BMDCs stimulated with different crystals in the presence or absence of CFTRinh-172.
Data are expressed as mean ± SEM (B, C, and E–H). Results are representative of at least of 3 independent experiments (C, F, and H; n = 3). See also Figures S2 and S7.
Hyperpolarization of MP Also Inhibits NLRP3 Inflammasome Activation
We wondered if IL-1β production might be increased by membrane hyperpolarization. As determined by the patch clamp, glyburide was found to drastically hyperpolarize the membrane (Figure 2D), reaching values as negative as −120 mV (Figure 2E). With an increasing concentration of glyburide, IL-1β release from BMDCs in response to crystals was progressively reduced without greatly affecting the control cytokines (Figure 2F). This finding was also seen in THP-1 and BMDMs (Figure S2F). To find more inhibitors that could cause sustained membrane hyperpolarization, DiBAC4(5), an MP indicator that yields an increased fluorescence signal upon depolarization, was used to screen a panel of ion channel regulators. CFTRinh-172 was found to induce sustained MP hyperpolarization (Figure 2G). This treatment also reduced IL-1β production in BMDCs without affecting TNF-α (Figure 2H). The inability to activate caspase-1 and produce IL-1β in the presence of CFTRinh-172 was also seen with BMDMs (Figures S2G and S2H). Since glyburide and CFTRinh-172 could target multiple types of chloride channels (Melis et al., 2014; Sheppard and Robinson, 1997), we used N-[ethoxycarbonylmethyl]-6-methoxy-quinolinium bromide (MAQE), a fluorescent chloride ion indicator (the fluorescence intensity decreases in proportion to the chloride increase in cells), to check intracellular chloride changes. Indeed, both glyburide and CFTRinh-172 increased intracellular chloride level (Figures S2I and S2J), which may explain their hyperpolarization effect. Similarly, another chloride channel inhibitor, GlyH-101, could also cause sustained membrane hyperpolarization (Figure S2K) and inhibit NLRP3 inflammasome activation (Figures S2L and S2M). Therefore, both depolarization by eliminating cross-membrane K+ ion strength differential (high extracellular K+) and hyperpolarization block IL-1β production without affecting phagocyte activation.
MP Was Maintained Close to the Resting Value for NLRP3 Inflammasome Activation
To allow extensive monitoring of MP value changes, we created two standard curves of DiBAC4(5) relative fluorescence unit (RFU) versus MP. A depolarization (~ >−40 mV) curve was produced with patch-clamp recording under varying K+, and a hyperpolarization (~ <−40 mV) one was produced with glyburide. This allowed us to obtain real-time MP readings on the fly (Figure S3A). To avoid interference from cell death and to study the role of MP on early state signaling, we shortened NLRP3 inflammasome study to 1 hr. As seen in Figure 3A, in the same period whereby Caspase-1 was processed and initial IL-1β production was detected (Figure 3C) (Hari et al., 2014), and no obvious cell death occurred as measured by lactate dehydrogenase (LDH) release (Figure S3B), silica-treated cells maintained a steady MP except for a brief depolarization that is associated with phagocytosis (silica-treated cells at 5 min) (Holevinsky and Nelson, 1995; Miles et al., 1981). When MP values converted from DiBAC4(5) intensities were plotted (Figures 3B and S3C), it became evident that a MP close to the resting value was associated with crystal-stimulated IL-1β response, at least during the early signaling cascade.
Figure 3. Maintaining MP Close to the Resting Value Is Essential for NLRP3 Inflammasome.

(A and B) Representative images of DiBAC4(5) staining (A) and corresponding calibrated MP plotting over time (B) in THP-1 cells under the indicated conditions. 100 mM [K+] and 200 μM glyburide. Results are a pool of 3 independent experiments. Original RFU result was provided in Figure S3C. Scale bar, 20 μm.
(C) Representative immunoblot analysis for Caspase-1 activation and IL-1β secretion in THP-1 cells treated with silica for 1 hr. Supernatants, Sup; lysates, Lys.
(D and E) THP-1 cells were treated with silica under the condition of high extracellular K+ (D) or inhibitor treatment (E). IL-1β secretion and intracellular K+ content measured by inductively coupled plasma optical emission spectrometry (ICP-OES) are plotted.
Similar results were obtained in 3 independent experiments (C–E). See also Figures S3 and S7.
We also directly assessed the K+ content in the treated cells following NLRP3 activation (Figures 3D and 3E). It was noted that the presence of crystal was associated with a reduction of adherent cell number (Figure S3D) and cell death (Figure S3E), resulting in the reduced total K+ content. Both changes were not correlated with depolarization or hyperpolarization. If the cell numbers were not adjusted, comparing total cell-associated K+ from the beginning to the end could lead to the false impression of K+ leakage. Figure 3D shows that, in 5 mM (permissible for K+ efflux) and 130 mM (not permissible for K+ efflux) extracellular K+ media, silica-treated cells, while following slightly different kinetics, both showed reduced total K+, likely reflecting the cell death. In the presence of silica, hyperpolarization of the membrane with glyburide and CFTRinh-172 also resulted in a similar reduction of K+ to the control (Figure 3E). This result confirmed that the loss of K+ reflected the state of cellular viability following silica treatment, rather than an indication of K+ efflux. Importantly, this loss of K+ was not correlated by IL-1β production (Figures 3D and 3E). Without excluding potential IL-1β production under a scheme of sustained K+ depletion, our results argue that NLRP3 inflammasome activation may take place without involving large quantities of K+ moving across the plasma membrane and instead suggest that an MP around −40 mV is a prerequisite for crystal-induced IL-1β production.
Resting MP Is Essential for Calpain Activity
μ-calpain is 3 logs more sensitive to Ca2+ concentration than m-calpain (Storr et al., 2011) (the prefixes refer to their activating molar concentrations of Ca2+). The intracellular Ca2+ is normally at several hundred nanomolar, which can be dramatically increased upon cellular activation (Berridge et al., 2000). Calpain is also regulated by their membrane proximity. In fact, m-calpain has been directly shown to require direct membrane binding (Leloup et al., 2010; Shao et al., 2006). Since in our system K+ efflux was not found to be an essential event for NLRP3 activation, we sought to understand whether high extracellular K+, used as a surrogate to indicate the central role of K+ efflux, actually led to calpain inhibition via Ca2+-signaling blockage or calpain inhibition via MP disruption.
To investigate how depolarization or hyperpolarization of membrane may negatively affect calpain-dependent IL-1β production, calpain activities were tested in response to increasing concentrations of K+ and glyburide, using intracellular fluorescent conversion of the standard calpain substrate CMAC as a readout. Basal levels of calpain activity in THP-1 cells were reduced in the presence of high extracellular K+, although not completely as under the calpeptin treatment. Calpain activity was strongly enhanced by silica with regular extracellular K+, while the addition of high K+ blocked this upregulation (Figure 4A). The same results were obtained from BMDCs (Figure S4A). Driving hyperpolarization by glyburide also blocked CMAC conversion (Figure 4B). Therefore, a resting-stage MP is essential for calpain activities.
Figure 4. The Resting-State MP Must Be Maintained for Optimal Calpain Activation.

(A) Calpain activity in THP-1 cells treated with silica in the presence of different concentrations of extracellular K+ was measured using calpain fluorogenic substrate CMAC. Scale bar, 20 μm.
(B) Similar to (A), calpain activity in THP-1 cells under glyburide treatment or not was measured by CMAC.
(C–E) As shown in the schematic diagram (C), calpain activity in THP-1 cells, with MP values held at −40, 0, or −100 mV by patch clamping, was measured by CMAC. Representative pictures of calpain activity at 20 min are shown in (D). Red triangle, patched cell. Corresponding bright-field images are shown above. Scale bar, 10 μm. Statistical analysis (E) by dividing CMAC intensity of patched cell over other unpatched cells in the same field is shown. n = 6 for each condition. Results are a pool of 3 independent experiments (A and B). Error bars denote SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (unpaired Student’s t test). See also Figure S4.
High K+ and glyburide could also regulate events other than the MP. To define the role of the MP in calpain activity at its most elemental form in the absence of any external treatments or stimulations, we built a hybrid system in house in which a patch clamp was combined with fluorescence microscopy (Figure 4C, schematic setup). MP differential was adjusted in both directions from the basal −40 mV via the patch electrode. This system could sustain MP changes for up to 30 min. At multiple time points, MP at the neutrality or −100 mV strongly blocked calpain activity (Figures 4D and 4E). These results therefore provide the most direct support that the resting-state MP must be maintained for optimal calpain activation.
Ca2+ Signaling Associated with NLRP3 Activators Is Not Affected by MP Disruption
Ca2+ is the critical regulator of calpain activation both at basal levels and after stimulation (Suzuki et al., 2004). To check whether disruption of MP inhibited calpain and NLRP3 inflammasome activation through blocking Ca2+ mobilization, we focused on Ca2+ flux. In our system, EGTA chelation of extracellular Ca2+ did not decrease IL-1β production (data not shown). 2-APB (an inositol triphosphate receptor blocker) inhibited IL-1β production in response to crystals, ATP, or nigericin (Figure 5A). High extracellular K+ did not affect the basal Ca2+ signal in THP-1 cells (Figure 5B). When stimulated by crystals, Ca2+ increased over time. Although the presence of high K+ slightly reduced the total Ca2+ signal, the crystal-induced increase was still evident, and the achievable maximum Ca2+ signal in each individual live cell was not affected (Figure 5C). Treating THP-1 cells with glyburide to hyperpolarize the membrane did not change the basal Ca2+ levels (Figure 5D), nor did it alter the Ca2+ increase in response to silica cumulatively or at the maximum achievable levels (Figure 5E). Although the overall Ca2+ signal in response to silica followed phases of rapid increases and slow tapering off, the Ca2+ flux in each stimulated cell displayed complex patterns (sustained, oscillating, single peak, and no response). The presence of high K+ did not significantly change the portion of each Ca2+ pattern (Figure 5F). Therefore, de- and hyperpolarization-mediated calpain inhibition and IL-1β reduction cannot be explained by a lack of Ca2+ signaling, ruling out any involvement of the voltage-gated Ca2+ channels, instead stressing the role of MP in direct regulation of calpain activities.
Figure 5. Disruption of MP Does Not Dramatically Affect Calcium Flux.

(A) BMDCs were pretreated with 50 μM 2-APB and activated with different crystals for 5 hr or ATP or nigericin for 1 hr. IL-1β in the supernatant was measured by ELISA (n = 3).
(B and C) THP-1 cells were stained with fluo-4 and medium was changed to 3 or 100 mM K+ buffer for 30 min. Ca2+ signals at basal level (B) or after silica stimulation over time (C, left) and at the maximum (C, right) were analyzed.
(D and E) THP-1 cells were stained with fluo-4 and then changed to medium with or without 200 μM glyburide for 30 min. Ca2+ signals at basal level (D) or after silica stimulation over time (E, left) and at the maximum (E, right) were analyzed.
(F) Pattern analysis of single-cell Ca2+ responses in (C) (sustained, maintaining high level for more than 3 min since induction; oscillating, with 2 or more oscillating peaks; single peak, one peak). Statistical analysis is shown on the right.
Error bars indicate SEM; ns, not significant (unpaired Student’s t test). Results are pooled from 3 independent experiments (B–F).
Calpain Activation Dissociates Caspase-1 from Motor or Structural Proteins
Inflammasome component availability, particularly Caspase-1, has become known as a regulated event. It was reported Caspase-1 is sequestered by cortical cytoskeleton and intracellular inhibitory proteins (Li et al., 2008). This may explain why an increasing number of inflammasome activators, targeting NLRP3, pyrin, NLRC4, etc., are reported to regulate the cytoskeleton via small GTPases (Man et al., 2014; Mostowy and Shenoy, 2015; Müller et al., 2009; Xu et al., 2014), which are known to regulate the state of protein association (Parsons and Adams, 2008). As the calpain is a large family of proteases (Sorimachi and Ono, 2012), we investigated whether the MP/Ca2+/calpain axis, common to NLRP3 triggers, may participate in the reversal of this sequestration.
Using ASC oligomerization as the readout, we assayed whether μ-calpain deficiency affected NLRP3 activation in general. Figure S5A shows that ASC oligomerization was not dramatically affected by μ-calpain deletion, suggesting μ-calpain regulates events downstream of ASC oligomerization. However, overall calpain blockage with calpain inhibitors reduced ASC oligomerization following the crystal (Figure S5B) as well as nigericin treatments (Figure S5C). By quantifying the number of ASC specks, we also found that pan-calpain inhibitor could decrease the percentage of ASC speck-forming cells (Figures S5D and S5E), suggesting that other calpain members participate in the signaling upstream of ASC oligomerization, a scenario we did not pursue further.
Of the many protein sequestrations reversed by the calpain family members, in one of these NLRP3 components we wished to establish a molecularly defined reversal mechanism. We next studied if the proteolytic activities of calpain were required to remove the inhibition of Caspase-1. We observed a spontaneous auto-processing of Caspase-1 and IL-1β production when pro-Caspase-1 and pro-IL-1β were co-transfected into 293FT cells (Figure 6A), as reported previously (Li et al., 2008). This common observation suggests that Caspase-1 tends to self-activate in an overexpression system and this process is likely suppressed in phagocytes. In human, Pyrin- and CARD-only proteins (POPs and COPs) provide the blockage of the spontaneous activities (de Almeida et al., 2015; Le and Harton, 2013). In mouse, an unrelated molecule, Fightless-1, is the only known protein contributing to the inhibition of Caspase-1 (Jin et al., 2013; Li et al., 2008). In the minimal 293FT system, IL-1β production as a result of dual transfection with Caspase-1 and IL-1β was blocked by an additional transfection of Flightless-1 (Figure 6A). To determine whether calpain was involved in releasing Caspase-1 from Flightless-1, protein levels of the latter and its association with Caspase-1 were analyzed. In silica-treated BMDMs, Flightless-1 level was reduced; and the reduction was blocked by calpain inhibition (Figure 6B). Similarly, the association between Flightless-1 and Caspase-1 was reduced by silica treatment, and this reduction was reverted by calpeptin (Figure 6C). Calpain inhibition had little impact on Flightless-1 mRNA level (Figure S6A).
Figure 6. Removal of Caspase-1 Inhibition by Negative Regulators Requires Calpain Activity.

(A) ELISA analysis of IL-1β secretion in 293FT cells co-transfected with the indicated amount of plasmid (μg) for 24 hr.
(B) Immunoblotting analysis of Flightless-1 protein level change in BMDMs treated with the indicated stimuli (n = 5).
(C) Caspase-1-Flightless-1 interaction in BMDMs treated with the indicated stimuli was analyzed by immunoprecipitation with Caspase-1 antibody.
(D and E) Coomassie blue staining (D, upper panel) and silver staining (E) of proteins from BMDMs immunoprecipitated by Caspase-1 antibody. Two bands at around 230 and 45 kDa (arrows) were identified as myosin-9 and two species of actin by in-gel MS (D, lower panel).
(F) MS hit numbers of Caspase-1 interaction proteins in silica alone and silica + calpeptin treatment (1 hr) were analyzed by in-gel MS. Hits are grouped per functions. Flightless-1 is marked in red.
Data are representative of 2 or 3 independent experiments (A and C–E). Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant (unpaired Student’s t test). See also Figures S5 and S6 and Table S1.
To confirm that calpain digested Flightless-1 directly, we transfected c-myc-tagged Flightless-1 in 293FT, and we found that, in the presence of calpain inhibitor, several digestion bands were diminished, as measured by either c-myc or Flightless-1 antibody (Figure S6B). We ran the Flightless-1 protein sequence with an online calpain cleavage site prediction algorithm (GPSCCD 1.0) (Liu et al., 2011). 52 potential calpain cleavage sites were predicted when the cutoff threshold was set at 0.654. Several digestion sites predicted would produce end products detected by c-myc and Flightless-1 antibodies with reciprocal sizes listed in the table (Figure S6C). Several prominent digestion bands were increased in silica-treated BMDCs, and this could be diminished by calpain inhibition (Figure S6D).
Although no COPs and POPs have been found in mouse, it is very likely that additional negative regulators are present via direct binding with Caspase-1. We first visualized total elutes from Caspase-1 co-immunoprecipitation (coIP) using Coomassie blue staining (Figure 6D). Two visible bands at about 230 and 45 kDa were decreased after silica treatment, while calpeptin blocked the reduction. They were further identified as myosin-9 (Myosin IIa) and β/γ-actin by in-gel mass spectrometry (MS) and western blotting, and control IP via caspase-3 did not show any change following silica treatment (Figures 6D and S6E). As Flightless-1 was not found, we increased the detection sensitivity with silver stain, and we visualized more bands that were decreased by silica treatment (Figure 6E).
To identify more differential Caspase-1-binding proteins that are regulated by calpain, we compared coIP proteins from silica-treated cells in the presence or absence of calpain inhibitor by in-gel MS. The species with large differences in MS hit numbers are listed (Table S1; Figure 6F). Most of the 35 species identified belong to motor, cytoskeletal, and cytoskeleton-regulatory proteins. This analysis identified Flightless-1 among MS hits, showing an intermediate hit number differential between the control and the calpeptin-treated (species 19 in Table S1). To study the overall association of Caspase-1 to actin network, BMDMs were lysed with Triton X-100 buffer. Insoluble fractions were collected by low-speed centrifugation to enrich actin network harvesting. Silica treatment reduced the amount of Caspase-1 in this fraction without affecting the actin protein levels, and this change was partially blocked by calpeptin (Figure S6F). These data collectively suggest that, in crystal-mediated NLRP3 activation, Caspase-1 release from the cytoskeleton was mediated by calpain.
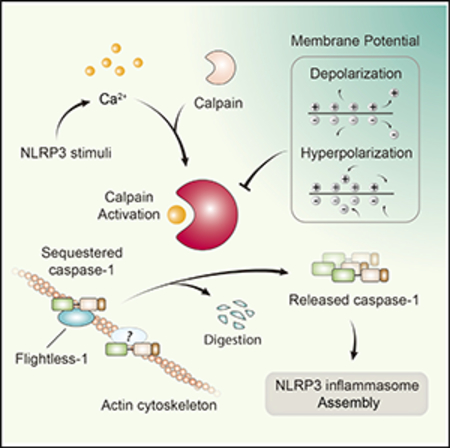
Figure 7 depicts the overall scenario that MP is essential for an active removal of endogenous Caspase-1 blockers, via Ca2+-dependent calpain activation in response to crystals.
Figure 7. Model for MP-Supported Caspase-1 Liberation by Calpain in NLRP3 Inflammasome Activation.

Crystal-induced phagocytosis initiates a calcium signal, which is the prerequisite for calpain activation. Released by the activated calpain from Flightless-1 and the cytoskeleton, the enlarged pool of Caspase-1 increases NLRP3 inflammasome oligomerization in the cytosol. Under the condition of high extracellular K+ or with the treatment of glyburide, sustained membrane depolarization or hyperpolarization impairs the ability of calpain to releasing Caspase-1 from its sequestration, inhibiting NLRP3 inflammasome activation.
The Near Resting-State MP Contributes to Non-NLRP3 Inflammasome Activation to Various Extents
To elucidate the role of MP in the general activation of the NLRP3 inflammasome, soluble stimulators were tested under MP depolarization or hyperpolarization. Although nigericin is an H+ and K+ antiporter and another NLRP3 activator gramicidin increases the general permeability for monovalent ions, they did not deviate the MP from the range required for NLRP3 activation (Figures S7A and S7B). Muñoz-Planillo et al. (2013) suggested that gramicidin plus low Na+ with a choline replacement led to membrane hyperpolarization, based on a predication that K+ outflow under gramicidin treatment without a retrograde Na+ influx would increase the charge differential. They found IL-1β production in that setting, which was used to rule out any role of MP in NLRP3 inflammasome activation (Muñoz-Planillo et al., 2013). However, upon direct measurement with DiBAC4(5), this regimen also failed to significantly alter the MP (Figure S7B). Similar to crystals, high K+, glyburide, and CFRinh-172 all inhibited IL-1β production induced by ATP, nigericin, or gramicidin, but with minimal impact on TNF-α levels (Figures S7C–S7H). Therefore, the requirement of the near resting-state MP value was also essential for these non-crystal NLRP3 activators.
While MP/Ca2+/calpain-based reversal of sequestration appeared to be important in canonical NLRP3 triggers, we wondered how much this chain of events is involved in other inflammasome activations. Degrees of Ca2+ activation are different and spatiotemporally distinct for each inflammasome complex. NLRP1b inflammasome activation by anthrax lethal toxin from Bacillus anthracis was accompanied with Ca2+ influx and relied on normal extracellular K+ (Bhatnagar et al., 1989; Shin et al., 2000; Wickliffe et al., 2008). Considering that anthrax toxin endocytosis relied on calpain (Jeong et al., 2013), here we used Val-boroPro (serine proteases Dpp8/9) inhibitor to activate NLRP1b inflammasome (Okondo et al., 2018). Similar to canonical NLRP3 inflammasome, Val-boroPro-activated NLRP1b inflammasome in BMDMs could also be inhibited by high extracellular K+, glyburide, or CFTRinh-172 (Figures S7I and S7J). Calpain inhibitors also strongly blocked the IL-1β production triggered by Val-boroPro (Figure S7K), suggesting that MP/ Ca2+/calpain-based reversal of sequestration might also regulate the NLRP1b inflammasome. Unlike the NLRP3 inflammasome, NLRC4 and AIM2 inflammasome activation was not associated with Ca2+ signaling (Horng, 2014; Lee et al., 2012). Both high extracellular K+ and glyburide showed measurable yet less dramatic impacts on poly dA:dT (AIM2 inflammasome) and flagellin (NLRC4 inflammasome) -mediated IL-1β production from BMDMs (Figures S7L and S7M). However, complete inhibition of the basal calpain activity could also impair NLRC4 and AIM2 inflammasome activation (Figure S7N). Therefore, the MP/Ca2+/calpain axis contributes to each inflammasome complex activation to different extents, which is likely correlated to the Ca2+-signaling intensity associated with these complexes.
DISCUSSION
Inflammasome components are controlled by various suppression mechanisms. Besides transcriptional regulation of NLRP3 and pro-IL-1β (Bauernfeind et al., 2009), spatial constraint or blockage is increasingly recognized as an important regulation (de Almeida et al., 2015). Caspase-1 tends to be self-activated, especially when exogenously expressed in non-phagocytes as observed in our results and other studies (Li et al., 2008). Li et al. first reported that Flightless-1, an actin-binding protein, can interact with Caspase-1 and inhibit its autoprocessing (Li et al., 2008). Pyrin, which is associated with polymerizing actin (Waite et al., 2009), mediates Caspase-1 activation in response to Rho family GTPase glucosylation by TcdB from Clostridium difficile (Xu et al., 2014). This regulation may be indirect, via sensing of actin dynamics. The targeted GTPase modifications are actually a common theme in bacterial toxin-mediated inflammatory responses (Xu et al., 2014). Bacterium-derived GTPase modifiers, such as SopE from Salmonella typhimurium (Müller et al., 2009) and YopE from Yersinia enterocolitica (Schotte et al., 2004), all mediate Caspase-1 activation. Some are individually coupled to specific types of inflammasome activation, i.e., NLRC4 in Salmonella infection (Man et al., 2014). In fact, actin depolymerization deficiency is a direct trigger of Caspase-1 (Kim et al., 2015). Therefore, it has been proposed that actin dynamic is an important upstream trigger of inflammasome and Caspase-1 activation (Müller et al., 2010). However, how this regulation is mediated in the NLRP3 inflammasome activation is less understood.
In this study, we provide evidence that, in addition to Flightless-1, several structural proteins and known calpain substrates, such as myosin-9 and vimentin (Fischer et al., 1986; Tsai et al., 2014), are also in association with Caspase-1, likely providing a redundant inhibitory network that is actively removed during NLRP3 inflammasome activation. Their release from the cytoskeleton is mediated by an MP-dependent calpain activation. All canonical NLRP3 inflammasome activators involve Ca2+ flux (Horng, 2014). Unlike the transient and tightly regulated nature of K+ efflux, Ca2+ signaling is more persistent and often oscillating, which likely explains the involvement of m-calpain that is highly sensitive to lower ranges of Ca2+ fluctuation. This provides a shared facilitator for NLRP3 inflammasome activation. We propose that, in contrast to other types of inflammasome activations in which Caspase-1/cytoskeleton association is disturbed with disparate means, the canonical NLRP3 activation is by design coupled to the Ca2+ signaling. While a pool of free Caspase-1 is available in the cytosol, which may be regulated by basal calpain activities tuned to Ca2+ fluctuation, optimal NLRP3 activation in responses to crystals, ATP, and nigericin requires the active release of cytoskeleton-associated Caspase-1 driven by the induced μ-calpain activation.
This study provides an alternative hypothesis that explains the puzzling requirement of physiological levels of extracellular K+ as the precondition for canonical NLRP3 inflammasome activation. The narrow range of the resting MP is critical to calpain activities. Persistent deviations from this range, both via high extracellular K+-induced depolarization and glyburide/CFTRinh-172-mediated hyperpolarization, cause the loss of calpain digestion and IL-1β production. With a myriad of distinct analyses, the core value of this work is to call for caution in interpreting the dependence of K+ efflux in NLRP3 inflammasome activation. This is particularly important when the methodologies used to establish such a K+ efflux model may themselves require further evaluation, i.e., whether intracellular K+ loss perceived with MS analysis may be a consequence of inconspicuous cell death, and high extracellular K+ may block cellular functions that rely on the MP. While our calpain model can provide an alternative activation mechanism impacted by high extracellular K+ in ways other than K+ efflux, it is almost certain that, given the overarching importance of MP in eukaryotic biology, activations of other inflammasome species will be regulated by this ion to various extents, which remain to be explored.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse NLRP3 | Adipogen | Cat# AG-20b-0014 |
| Anti-mouse caspase-1 p20 | Adipogen | Cat# AG-20B-0042 |
| Anti-mouse calpain 1 | Cell Signaling Technology | Cat# 2556; RRID: AB_2290836 |
| Anti-mouse calpain 2 | Cell Signaling Technology | Cat# 2539; RRID: AB_2069843 |
| Anti-β Actin | Cell Signaling Technology | Cat# 4970; RRID: AB2223172 |
| Anti-human IL-1β | Cell Signaling Technology | Cat# 12703 |
| Anti-GAPDH | Cell Signaling Technology | Cat# 2118; RRID: AB_561053 |
| Anti-mouse Myosin IIa | Cell Signaling Technology | Cat# 3403S; RRID: AB_2147297 |
| Anti-mouse Flightless-1 | Santa Cruz | Cat# SC-55583; RRID: AB_831341 |
| Anti-human Caspase-1 | Santa Cruz | Cat# SC-622; RRID: AB_2069053 |
| Anti-mouse ASC | Santa Cruz | Cat# SC-22514-R; RRID: AB_2174874 |
| Anti-mouse IL-1β antibody | R&D | Cat# AF-401-NA; RRID: AB_416684 |
| HRP-conjugated goat anti-rabbit IgG | Cell Signaling Technology | Cat# 7074s; RRID: AB_2099233 |
| HRP-conjugated horse anti-mouse IgG | Cell Signaling Technology | Cat# 7076s; RRID: AB_330924 |
| HRP-conjugated donkey anti-goat IgG | Santa Cruz | Cat# SC-2020; RRID: AB_631728 |
| All the fluorescent secondary antibodies | Jackson ImmunoResearch | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Al(OH)3 | Sigma | Cat# V900163 |
| Calcium pyrophosphate | Sigma | Cat# 401552 |
| Uric acid | Sigma | Cat# U2625 |
| ATP | Sigma | Cat# A2383 |
| PMA | Sigma | Cat# P1585 |
| Glyburide | Sigma | Cat# G2539 |
| CFTRinh-172 | Sigma | Cat# C2992 |
| Monosodium urate (MSU) crystals | (Shi et al., 2003) | N/A |
| GlyH-101 | TargetMol | Cat# T2451 |
| Calpeptin | Selleck | Cat# S7396 |
| Nigericin | Tocris | Cat# 4312 |
| PD150606 | Tocris | Cat# 1269 |
| 2-APB | Tocris | Cat# 1224 |
| E. coli 0111:B4 LPS | Sigma | Cat# L4391 |
| Naked poly(dA:dT) | Invivogen | Cat# tlrl-patn |
| Ultrapure Flagellin | Invivogen | Cat# tlrl-epstfla-5 |
| Murine GM-CSF | PeproTech | Cat# AF-315–03 |
| Murine IL-4 | PeproTech | Cat# AF-214–14 |
| DiBAC4(5) | AAT Bioquest | Cat# 21410 |
| DSS | Thermo | Cat# 21555 |
| CMAC peptidase substrate (t-BOC-Leu-Met) | Thermo | Cat# A6520 |
| MQAE fluorescent probe | Beyotime | Cat# s1082 |
| Critical Commercial Assays | ||
| LDH release detection kit | Beyotime | Cat# c0016 |
| Mouse IL-1β ELISA ready-set-go kits | eBioscience | Cat# 50–171-85 |
| Mouse TNFα ELISA ready-set-go kits | eBioscience | Cat# 50–173-31 |
| Mouse IL-6 ELISA ready-set-go kits | eBioscience | Cat# 50–112-8696 |
| Human IL-1β ELISA ready-set-go kits | eBioscience | Cat# 50–112-5196 |
| Human TNFα ELISA ready-set-go kits | eBioscience | Cat# 50–173-66 |
| Experimental Models: Cell Lines | ||
| Human: HEK293FT | Dr.Wei Guo’s Lab | N/A |
| Human: THP-1 | ATCC | ATCC: TIB-202 |
| Mouse: L929 | ATCC | ATCC: CCL-1 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 mice | The Jackson Laboratory | JAX 000664 |
| Oligonucleotides | ||
| gRNA targeting sequence: CAPN1 atcacgccggtgtactgcactgg, and tgggaccctcttccgtgatgagg. | This paper | N/A |
| qPCR primers for mouse Flightless-1: 5’-tctgcagaagctggagcac-3’ and 5’-ttggcccgagctacaatg-3’ | This paper | N/A |
| qPCR primers for mouse Nlrp3: 5’-gaattccggccttacttcaa-3’ and 5’-ggtgtgtgaagttctggttgg-3’ | This paper | N/A |
| qPCR primers for mouse II-1b: 5’-ttgacggaccccaaaagat-3’ and 5’-gaagctggatgctctcatctg-3’ | This paper | N/A |
| qPCR primers for human CAST: 5’-aaccagccacggataaagatg-3’ and 5’-agcagcggccttagattcttc-3’ | This paper | N/A |
| Non-targeting shRNA: Misson pLKO.1- puro Control Vector, TRC1.5 Negative Controls |
Sigma | SHC002 |
| Non-targeting shRNA: TRC2 Transduction Control, DNA, PLKO-PUR, TRC2 Negative Controls |
Sigma | SHC202 |
| shRNA sequence: CAST #1 | Sigma | TRCN0000073638 |
| shRNA sequence: CAST #2 | Sigma | TRCN0000073642 |
| shRNA sequence: CAPN1 | Sigma | TRCN0000003559 |
| shRNA sequence: CAPN2 | Sigma | TRCN0000003539 |
| Software and Algorithms | ||
| FlowJo_V10.0.8 | FlowJo, LLC | https://www.flowjo.com |
| Graphpad Prism 6 software | Graphpad Prism | N/A |
| Adobe Illustrator CC 2015 | Adobe | N/A |
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| Cleavage site prediction algorithm (GPS-CCD 1.0) | (Liu et al., 2011) | http://ccd.biocuckoo.org/down.php |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Yan Shi (yanshi@biomed.tsinghua.edu.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6. mice were bred and housed under specific-pathogen-free condition at Tsinghua University Animal Facilities. Male and female mice were used at 6–8 weeks old. The Animal Experiments Committee of Tsinghua University approved all of the experiments reported in this study.
Cell Lines
HEK293T cells were cultured in DMEM (GIBCO) supplemented with 10% FBS. THP-1 cells and L929 cells were cultured in cultured in complete RPMI1640 (GIBCO) supplemented with 10% FBS. THP-1 cells were differentiated with 10 ng/ml PMA for 48 hr before stimulation. All cells were cultured at 37°C and 5% CO2.
Primary Cell Cultures
Bone marrow from C57BL/6 mice was differentiated for 6 days in 20 ng/ml GM-CSF and 10 ng/ml IL-4 or in 20% L929 cell supernatant in complete RPMI medium to produce BMDCs and BMDMs respectively. On day 6, BMDCs which were grown in 24-well plate were used directly to do experiments, BMDMs were scraped from 10 cm dish and re-plated with fresh cell culture medium for overnight adhesion.
METHOD DETAILS
Cell stimulation
Cells were primed for 3 hr with Ultrapure-LPS as indicated. The subsequent NLRP3 activation was carried out for 1 hr with ATP (5 mM) or nigericin (10 μM), for 5 hr with crystals including Al(OH)3 (500 μg/ml), CPPD (400 μg/ml), MSU (400 μg/ml) and silica (400 μg/ml). In some experiments, inhibitors were added 30 minutes before the addition of stimuli. All treatments were performed in Opti-MEM medium or K+ buffer (130 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, 10 mM HEPES, and 10 mM glucose, pH 7.4; for high K+ buffer, the equal amount of NaCl was replaced by KCl).
Electrophysiology
BMDCs or PMA-treated THP-1 cells were cultured on a glass coverslip, and then transferred into an external bath solution of 130 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, 10 mM HEPES, and 10 mM glucose, pH 7.4. Borosilicate glass pipettes (Sutter Instrument Co., Novato, CA; 3–5 MU) were filled with internal solution containing 40 mM KCl, 100 mM K-Asp, 2.5 mM CaCl2, 1 mM MgCl2, 5 mM EGTA and 10 mM HEPES. Whole-cell patch clamp recordings were performed using an EPC 10 amplifier (HEKA Elektronik, Bellmore, NY) linked to a personal computer equipped with Pulse (V8.65) software. After seal formation, the membrane beneath the pipette was ruptured and the pipette solution was allowed to dialyze into the cell for 5–10 min before recording the resting MP in current-clamp mode. The different concentration of potassium solution and glyburide-containing solution were provided by a gravity-fed multichannel perfusion system with an outlet positioned in close contact to the studied cell. Data analysis was performed by using online analysis built in Pulse software (HEKA Elektronik).
MP staining
DiBAC4(5) is a translational MP dyes that redistributes within the cell membrane as MP changes. MP depolarization results in an increase in fluorescence intensity of cells at Ex/Em = 590/616nm. Conversely, hyperpolarization is indicated by a decrease in fluorescence. PMA-differentiated THP-1 cells were cultured in 35mm imaging dish (ibidi) with a concentration of 4×105 cells/ml. Cells were stained in 200 nM DiBAC4(5) in 5mM K+ buffer at 37°C for 30 min. Then the staining buffer was removed without washing and crystals (silica, Al(OH)3, CPPD) or inhibitors (glyburide or CFTRinh-172) in fresh 5mM K+ buffer containing 200nM DiBAC4(5) was added to the cells. For high K+ condition, staining buffer was replaced with 100mM K+ buffer containing 200nM DiBAC4(5). We captured 5 different fields at each time-point on fluorescent microscope, and analyzed the fluorescence intensity of all the cells by ImageJ. The relative fluorescence intensity indicates MP changes under different condition, which is normalized with the mean fluorescence intensity of non-treated cells (Laskey et al., 1992).
Calpain activity measurement
THP-1 cells or BMDCs were plated in 35 mm imaging dish (ibidi). Cells were pretreated with 3 mM or 100 mM K+ buffer, or medium with or without glyburide (200 μM) for 30 min in cell culture incubator. Cells were then moved to fluorescent microscopy with temperature control at 37°C. Half of the medium in dish was pipetted up to suspend calpain substrate BOC-LM-CMAC (50 μM) together with or without silica and then added back. After cleavage by calpain, the product emits blue-fluorescence with excitation/emission maxima ~351/430 nm. 30 min later, images were captured and CMAC fluorescence intensity in each cell was analyzed by ImageJ. For measuring calpain activity in cells under voltage control by patch clamp, the cell was patched first, CMAC was added, and then the MP was changed to hold at −40 mV, 0 mV or −100 mV. Pictures were taken at 10 min, 20 min and 30 min. CMAC fluorescence intensity in the patched cell was normalized to other unpatched cells (resting MP) in the same field.
Potassium ion measurement
PMA-differentiated 2×106 THP-1 cells in one well of 6-well plate were stimulated and briefly washed with 3 mL K+-free buffer to eliminated extracellular K+ carryover. Then the cells were extracted with 3 mL 3% HNO3 for 30 min. Intracellular K+ content was measured using inductively coupled plasma optical emission spectrometry (ICP-OES) by Chemical Analysis Center, Tsinghua University. The K+ content was plotted as % of untreated control.
Intracellular chloride ion detection
Intracellular chloride ion concentration was measured using N-[ethoxycarbonylmethyl]-6-methoxy-quinolinium bromide (MAQE), a fluorescent chloride ion indicator. MQAE with bromide ion as a counteranion has fluorescence excitation at 355 nm and emission at 460 nm. The fluorescent intensity of MQAE decreases in proportion to the chloride increase in cells. PMA-differentiated THP-1 cells were primed by 200ng/ml LPS for 3 hours. Collected cells and stained with 5mM MQAE in Kreb-HEPES buffer (20mM HEPES, 128mM NaCl, 2.5mM KCl, 2.7mM CaCl2, 1mM MgCl2 and 16mM Glucose), 37°C dark incubation for 40 mins. Cells were washed with Kreb-HEPES buffer and then treated with inhibitors at 37°C. The fluorescent intensity of cells was measured by flow cytometry at different times.
Calcium imaging
THP-1 cells or BMDCs in 35 mm imaging dishes were stained with 2 mM fluo-4 AM in HBSS-HEPS (10 mM) buffer at 37°C for 1 hr or 30 min, respectively. Then the cells were changed into 3 mM or 100 mM K+ buffer, or medium with or without glyburide for 30 min and images were acquired on an Olympus IX-73 microscope to analyze the effect on basal Ca2+ level. Then silica was added and Ca2+ flux was recorded as a time lapse for 12 min with an interval of 10 s or 20 s. The mean fluorescence intensity changes over time for individual cells were analyzed by ImageJ and normalized to the first slice (Fluo-4 F/F0). The maximum calcium level of each cell within the 12 min was selected and plotted. Results of at least 300 cells from 3 independent experiments were plotted as mean ± SEM.
Immunoblotting
The cell lysates for western blot were prepared by incubating samples in lysis buffer (1% NP-40, 50 mM Tris pH7.5, 150 mM NaCl, 5 mM EDTA, protease inhibitor cocktail from Sigma) for 20 min on ice followed by boiling in the sample buffer containing DTT. Cell culture supernatants were precipitated by the addition of 1 volume of methanol and 0.5 volumes of chloroform, then were vortexed and centrifuged for 5 min at 10,000 g. The upper phase was removed and 1 volume methanol was added to wash the interphase. This mixture was centrifuged for 15 min at 10,000 g and the protein pellet was dried and resuspended in DTT containing SDS-sample buffer. Samples were boiled and separated by 7.5%, 10% or 12% SDS-PAGE before transferring onto nitrocellulose membranes.
Immunoprecipitation
BMDMs were treated with indicated stimuli and cells were lysed in precooled lysis buffer (0.5% NP-40, 50 mM Tris pH7.5, 150 mM NaCl, 5 mM EDTA, 1 × protease inhibitor cocktail). Cell lysates were clarified by centrifugation (10000 g) at 4°C for 10 min. Pre-cleared lysates were incubated with anti-caspase-1 antibody (AG-20B-0042, 1:200) and protein A/G agarose at 4°C overnight. After extensive washing with lysis buffer, immunoprecipitates were boiled in the sample buffer and subjected to immunoblotting, Coomassie Blue staining or silver staining.
ASC oligomerization assay
For ASC oligomer cross-linking, 2.5 3 106 /well THP-1 cells were differentiated by PMA on 6 well plates and stimulated as indicated. Cells were lysed with 200 ul PBS containing 0.5% Triton X-100. Then the lysates were collected and centrifuged at 8,000 rpm for 15 min at 4°C. The soluble fraction was kept for future analysis and the Triton X-100-insoluble pellets were washed with PBS twice. The pellets were cross-linked at room temperature for 30 min in 200 ul disuccinimidyl suberate (2mM in PBS). The cross-linked pellets were spun down at 8000 rpm for 15 min and dissolved directly in SDS sample buffer for immunoblotting analysis.
Mass Spectrometry
Immunoprecipitates of Caspase-1 co-IP experiments were separated with SDS-PAGE gel and stained with Coomassie Blue. Two lanes (silica 1hr+calpeptin and silica 1hr) were cut into 8 pieces respectively and sent for in-Gel LC-MS/MS (SAMS Centre, University of Calgary). The results were searched against mouse database and all data were filtered to keep only the proteins identified with 95% confidence interval or higher. A combined Mascot search of the samples from two treatments was performed.
Fractionation of Actin
BMDMs were lysed in Triton buffer (150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 1% Triton X-100, 1 mM EDTA and protease inhibitor). Lysates were centrifuged at 15,900 g for 2 min. The insoluble low speed pellet containing bundled F-actin pool and the soluble fraction were boiled in SDS gel sample buffer for western blot.
Real-Time PCR
RNA samples were prepared using TRIzol reagent (Invitrogen) and first strand cDNA was synthesized with PrimeScript RT-PCR Kit (TAKARA). Real-time PCR was performed using 2 × RealStar Power SYBR mixture (GenStar). GAPDH was used for normalization. The primer sequences were as follows: mouse Flii, 5’-tctgcagaagctggagcac-3’ and 5’-ttggcccgagctacaatg-3’; mouse Nlrp3, 5’-gaattccggccttacttcaa-3’ and 5’-ggtgtgtgaagttctggttgg-3’; mouse Il-1b 5’-ttgacggaccccaaaagat-3’ and 5’-gaagctggatgctct catctg-3’; human CAST 5’-aaccagccacggataaagatg-3’ and 5’-agcagcggccttagattcttc-3’.
Gene knockdown in THP-1 cells
Lentiviral plasmids encoding targeting shRNA or non-targeting shRNA (SHC002 and SHC202) were purchased from Sigma and the TRC numbers were as follows: shCAST-1 TRCN0000073638, shCAST-2 TRCN0000073642, shCAPN1 TRCN0000003559, shCAPN2 TRCN0000003539. The lentivirus was produced by co-transfection of 293FT cells with lentivirus expression vector, pCMV-VSV-G, and pCMV-dR8.91 using Lipofectamine 2000. THP-1 cells were infected with lentivirus at 500 g, 32°C for 1.5 hr. THP-1 cells were selected with puromycin (2 μg/ml) 72 hr post-infection. The knockdown efficiency was analyzed by real-time PCR and/or immunoblot.
Generation of CAPN1-deficient THP-1 cells
Suitable CRISPR target sites were identified using the ‘CRISPR Design Tool’ (http://crispr.mit.edu/). The CRISPR/Cas9 expression vectors were made as previously described (Shalem et al., 2014). Two guide sequences targeting at CAPN1 were as follows: at-cacgccggtgtactgcactgg, tgggaccctcttccgtgatgagg. The lentivirus-transfected THP-1 cells were selected by puromycin (2 μg/ml). After single clonal expansion, CAPN1-deficient clones were identified by immunoblot.
Phagocytosis assay
To measure phagocytic efficiency, 3 μm polystyrene beads were coated with biotin. THP-1 cells were differentiated with PMA on clean glass slices in 24-well plate. Cells were co-cultured with coated beads, centrifuged at 900 g, 4°C for 5 min and cultured at 37°C for 30 min. Then the cells were fixed with 4% paraformaldehyde and stained with Alexa 405-strepavidin for immunofluorescent imaging. For analysis, total number of beads on each cell was calculated under bright field, the number of beads that were not engulfed were calculated under fluorescent lighting.
QUANTIFICATION AND STATISTICAL ANALYSIS
Number of experimental repeats are shown in the figure legend. All bar graphs are means with SEM. Statistical analysis was performed with Student’s t test in GraphPad Prism 6 software. P value < 0.05 was considered significant. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. ns, not significant.
Supplementary Material
Highlights.
Calpain activated by calcium is necessary for NLRP3 inflammasome activation
Calpain activity requires a relatively stable membrane potential (MP)
High K+ suppresses NLRP3 inflammasome activation via MP disruption
Activated calpain releases caspase-1 from its sequestration in cytoskeleton
ACKNOWLEDGMENTS
We thank Dr. Z.F. Jiang of Peking University for critical review of this manuscript. We thank Southern Alberta Mass Spectroscopy Center for technical assistance. Y.S. is supported by the joint Peking-Tsinghua Center for Life Sciences; the National Natural Science Foundation of China General Program (31370878); and by grants from the NIH (R01AI098995), the Natural Sciences and Engineering Research Council of Canada (RGPIN-355350/396037), and the Canadian Institutes of Health Research (MOP-119295).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.07.098.
REFERENCES
- Banderali U, Belke D, Singh A, Jayanthan A, Giles WR, and Narendran A (2011). Curcumin blocks Kv11.1 (erg) potassium current and slows proliferation in the infant acute monocytic leukemia cell line THP-1. Cell. Physiol. Biochem 28, 1169–1180. [DOI] [PubMed] [Google Scholar]
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, et al. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol 183, 787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belhage B, Hansen GH, and Schousboe A (1993). Depolarization by K+ and glutamate activates different neurotransmitter release mechanisms in GABAergic neurons: vesicular versus non-vesicular release of GABA. Neuroscience 54, 1019–1034. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, and Bootman MD (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol 1, 11–21. [DOI] [PubMed] [Google Scholar]
- Bezanilla F (2008). How membrane proteins sense voltage. Nat. Rev. Mol. Cell Biol 9, 323–332. [DOI] [PubMed] [Google Scholar]
- Bhatnagar R, Singh Y, Leppla SH, and Friedlander AM (1989). Calcium is required for the expression of anthrax lethal toxin activity in the macrophagelike cell line J774A.1. Infect. Immun 57, 2107–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cala PM (1977). Volume regulation by flounder red blood cells: the role of the membrane potential. J. Exp. Zool 199, 339–344. [DOI] [PubMed] [Google Scholar]
- Daut J, Standen NB, and Nelson MT (1994). The role of the membrane potential of endothelial and smooth muscle cells in the regulation of coronary blood flow. J. Cardiovasc. Electrophysiol 5, 154–181. [DOI] [PubMed] [Google Scholar]
- de Almeida L, Khare S, Misharin AV, Patel R, Ratsinmandresy RA, Wallin MC, Perlman H, Greaves DR, Hoffman HM, Dorfleutner A, and Stehlik C (2015). The PYRIN domain-only protein POP1 inhibits inflammasome assembly and ameliorates inflammatory disease. Immunity 43, 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer S, Vandekerckhove J, Ampe C, Traub P, and Weber K (1986). Protein-chemical identification of the major cleavage sites of the Ca2+ proteinase on murine vimentin, the mesenchymal intermediate filament protein. Biol. Chem. Hoppe Seyler 367, 1147–1152. [DOI] [PubMed] [Google Scholar]
- Glading A, Lauffenburger DA, and Wells A (2002). Cutting to the chase: calpain proteases in cell motility. Trends Cell Biol. 12, 46–54. [DOI] [PubMed] [Google Scholar]
- Goldenberg NM, and Steinberg BE (2010). Surface charge: a key determinant of protein localization and function. Cancer Res. 70, 1277–1280. [DOI] [PubMed] [Google Scholar]
- Hari A, Zhang Y, Tu Z, Detampel P, Stenner M, Ganguly A, and Shi Y (2014). Activation of NLRP3 inflammasome by crystalline structures via cell surface contact. Sci. Rep. 4, 7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holevinsky KO, and Nelson DJ (1995). Simultaneous detection of free radical release and membrane current during phagocytosis. J. Biol. Chem 270, 8328–8336. [DOI] [PubMed] [Google Scholar]
- Horng T (2014). Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 35, 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SY, Martchenko M, and Cohen SN (2013). Calpain-dependent cytoskeletal rearrangement exploited for anthrax toxin endocytosis. Proc. Natl. Acad. Sci. USA 110, E4007–E4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Yu Q, Han C, Hu X, Xu S, Wang Q, Wang J, Li N, and Cao X (2013). LRRFIP2 negatively regulates NLRP3 inflammasome activation in macrophages by promoting Flightless-I-mediated caspase-1 inhibition. Nat. Commun 4, 2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Emori Y, Imajoh-Ohmi S, Minami Y, and Suzuki K (1989). Identification and characterization of inhibitory sequences in four repeating domains of the endogenous inhibitor for calcium-dependent protease. J. Biochem 106, 274–281. [DOI] [PubMed] [Google Scholar]
- Kim ML, Chae JJ, Park YH, De Nardo D, Stirzaker RA, Ko HJ, Tye H, Cengia L, DiRago L, Metcalf D, et al. (2015). Aberrant actin depolymerization triggers the pyrin inflammasome and autoinflammatory disease that is dependent on IL-18, not IL-1β. J. Exp. Med 212, 927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulbacka J, Choromańska A, Rossowska J, Wez_gowiec J, Saczko J, and Rols MP (2017). Cell Membrane Transport Mechanisms: Ion Channels and Electrical Properties of Cell Membranes. Adv. Anat. Embryol. Cell Biol 227, 39–58. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, and Dixit VM (2014). Mechanisms and functions of inflammasomes. Cell 157, 1013–1022. [DOI] [PubMed] [Google Scholar]
- Laskey RE, Adams DJ, Cannell M, and van Breemen C (1992). Calcium entry-dependent oscillations of cytoplasmic calcium concentration in cultured endothelial cell monolayers. Proc. Natl. Acad. Sci. USA 89, 1690–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E, Xiao TS, and Stutz A (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le HT, and Harton JA (2013). Pyrin- and CARD-only proteins as regulators of NLR functions. Front. Immunol 4, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, and Chae JJ (2012). The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leloup L, Shao H, Bae YH, Deasy B, Stolz D, Roy P, and Wells A (2010). m-Calpain activation is regulated by its membrane localization and by its binding to phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem 285, 33549–33566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yin HL, and Yuan J (2008). Flightless-I regulates proinflammatory caspases by selectively modulating intracellular localization and caspase activity. J. Cell Biol 181, 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Cao J, Gao X, Ma Q, Ren J, and Xue Y (2011). GPS-CCD: a novel computational program for the prediction of calpain cleavage sites. PLoS ONE 6, e19001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, and Darnell J (2000). Intracellular ion environment and membrane electric potential Molecular Cell Biology, Fourth Edition (New York: W.H. Freeman; ). [Google Scholar]
- Man SM, Ekpenyong A, Tourlomousis P, Achouri S, Cammarota E, Hughes K, Rizzo A, Ng G, Wright JA, Cicuta P, et al. (2014). Actin polymerization as a key innate immune effector mechanism to control Salmonella infection. Proc. Natl. Acad. Sci. USA 111, 17588–17593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks AR (1997). Intracellular calcium-release channels: regulators of cell life and death. Am. J. Physiol 272, H597–H605. [DOI] [PubMed] [Google Scholar]
- Melis N, Tauc M, Cougnon M, Bendahhou S, Giuliano S, Rubera I, and Duranton C (2014). Revisiting CFTR inhibition: a comparative study of CFTRinh −172 and GlyH-101 inhibitors. Br. J. Pharmacol 171, 3716–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles PR, Bowman L, and Castranova V (1981). Transmembrane potential changes during phagocytosis in rat alveolar macrophages. J. Cell. Physiol 106, 109–117. [DOI] [PubMed] [Google Scholar]
- Mostowy S, and Shenoy AR (2015). The cytoskeleton in cell-autonomous immunity: structural determinants of host defence. Nat. Rev. Immunol 15, 559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller AJ, Hoffmann C, Galle M, Van Den Broeke A, Heikenwalder M, Falter L, Misselwitz B, Kremer M, Beyaert R, and Hardt WD (2009). The S. Typhimurium effector SopE induces caspase-1 activation in stromal cells to initiate gut inflammation. Cell Host Microbe 6, 125–136. [DOI] [PubMed] [Google Scholar]
- Müller AJ, Hoffmann C, and Hardt WD (2010). Caspase-1 activation via Rho GTPases: a common theme in mucosal infections? PLoS Pathog. 6, e1000795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, and Núñez G (2013). K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okondo MC, Rao SD, Taabazuing CY, Chui AJ, Poplawski SE, Johnson DC, and Bachovchin DA (2018). Inhibition of Dpp8/9 Activates the Nlrp1b Inflammasome. Cell Chem. Biol 25, 262–267.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M, and Adams JC (2008). Rac regulates the interaction of fascin with protein kinase C in cell migration. J. Cell Sci 121, 2805–2813. [DOI] [PubMed] [Google Scholar]
- Perregaux D, and Gabel CA (1994). Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem 269, 15195–15203. [PubMed] [Google Scholar]
- Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, and Tschopp J (2007). Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589. [DOI] [PubMed] [Google Scholar]
- Próchnicki T, Mangan MS, and Latz E (2016). Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Res. 5, F1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotte P, Denecker G, Van Den Broeke A, Vandenabeele P, Cornelis GR, and Beyaert R (2004). Targeting Rac1 by the Yersinia effector protein YopE inhibits caspase-1-mediated maturation and release of interleukin-1beta. J. Biol. Chem 279, 25134–25142. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Chou J, Baty CJ, Burke NA, Watkins SC, Stolz DB, and Wells A (2006). Spatial localization of m-calpain to the plasma membrane by phosphoinositide biphosphate binding during epidermal growth factor receptor-mediated activation. Mol. Cell. Biol 26, 5481–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, and Robinson KA (1997). Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl-channels expressed in a murine cell line. J. Physiol 503, 333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Evans JE, and Rock KL (2003). Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425, 516–521. [DOI] [PubMed] [Google Scholar]
- Shin S, Hur GH, Kim YB, Park KJ, Park YM, and Lee WS (2000). Intracellular calcium antagonist protects cultured peritoneal macrophages against anthrax lethal toxin-induced cytotoxicity. Cell Biol. Toxicol 16, 137–144. [DOI] [PubMed] [Google Scholar]
- Sorimachi H, and Ono Y (2012). Regulation and physiological roles of the calpain system in muscular disorders. Cardiovasc. Res 96, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorimachi H, Hata S, and Ono Y (2010). Expanding members and roles of the calpain superfamily and their genetically modified animals. Exp. Anim 59, 549–566. [DOI] [PubMed] [Google Scholar]
- Storr SJ, Carragher NO, Frame MC, Parr T, and Martin SG (2011). The calpain system and cancer. Nat. Rev. Cancer 11, 364–374. [DOI] [PubMed] [Google Scholar]
- Sundelacruz S, Levin M, and Kaplan DL (2009). Role of membrane potential in the regulation of cell proliferation and differentiation. Stem Cell Rev. 5, 231–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Hata S, Kawabata Y, and Sorimachi H (2004). Structure, activation, and biology of calpain. Diabetes 53 (Suppl 1), S12–S18. [DOI] [PubMed] [Google Scholar]
- Tsai JC, Lin YW, Huang CY, Lin CY, Tsai YT, Shih CM, Lee CY, Chen YH, Li CY, Chang NC, Lin FY, and Tsai CS (2014). The role of calpain-myosin 9-Rab7b pathway in mediating the expression of Toll-like receptor 4 in platelets: a novel mechanism involved in α-granules trafficking. PLoS One 9, e85833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschopp J, and Schroder K (2010). NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev.Immunol 10, 210–215. [DOI] [PubMed] [Google Scholar]
- Välimäki E, Cypryk W, Virkanen J, Nurmi K, Turunen PM, Eklund KK, Å kerman KE, Nyman TA, and Matikainen S (2016). Calpain Activity Is Essential for ATP-Driven Unconventional Vesicle-Mediated Protein Secretion and Inflammasome Activation in Human Macrophages. J. Immunol 197, 3315–3325. [DOI] [PubMed] [Google Scholar]
- Waite AL, Schaner P, Hu C, Richards N, Balci-Peynircioglu B, Hong A, Fox M, and Gumucio DL (2009). Pyrin and ASC co-localize to cellular sites that are rich in polymerizing actin. Exp. Biol. Med. (Maywood) 234, 40–52. [DOI] [PubMed] [Google Scholar]
- Wickliffe KE, Leppla SH, and Moayeri M (2008). Anthrax lethal toxininduced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell. Microbiol 10, 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SH (2004). Generation of resting membrane potential. Adv. Physiol. Educ 28, 139–142. [DOI] [PubMed] [Google Scholar]
- Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong YN, Peng X, Xi JJ, Chen S, et al. (2014). Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241. [DOI] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, and Tschopp J (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Wong CO, Cho KJ, van der Hoeven D, Liang H, Thakur DP, Luo J, Babic M, Zinsmaier KE, Zhu MX, et al. (2015). SIGNAL TRANSDUCTION. Membrane potential modulates plasma membrane phospholipid dynamics and K-Ras signaling. Science 349, 873–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.