

Abstract
Protein-protein interaction (PPI) inhibitors are a rapidly expanding class of therapeutics. Recent advances in our understanding of PPIs and success of early examples of PPI inhibitors demonstrate the feasibility of targeting PPIs. This review summarizes the techniques used for the discovery and optimization of a diverse set PPI inhibitors, focusing on the development of PPI inhibitors as new antibacterial and antiviral agents. We close with a summary of the advances responsible for making PPI inhibitors realistic targets for theraputic intervention and brief outlook of the field.
Keywords: Protein-protein interactions, Antibiotics, Antiviral, drug discovery, antibiotic resistance, small molecule inhibitors
The increasing prevalence of treatment resistant pathogens is one of the biggest challenges facing the biomedical community today. Anti-infective agents have revolutionized medicine, allowing for curative therapies that vastly reduced the morbidity and mortality of diseases caused by pathogenic bacteria, viruses and parasites. Unfortunately, the widespread overuse and misuse of these agents has led to increasing levels of resistance that threatens our ability to effectively treat not only infections, but also the use of therapies that requires prophylactic antibiotic treatment, such as surgeries. While better stewardship is critical for extending the usable life of the existing repertoire of antibacterial and antivirals agents (Goff et al., 2017), classes of agents with novel mechanisms of action are urgently needed (“The 10 x ‘20 Initiative”, 2010).
Traditionally, antibiotic development has focused primarily on inhibitors of essential bacterial enzymes for understandable reasons (Walsh, 2016). The mechanism of action was easily understood, biochemical assays existed to test for activity and substrate analog inhibitors were relatively easy to produce. Unfortunately, the supply of easily inhibited targets appears to be limited as the output of new anti-infective agents has dwindled (Ventola, 2015). Deepening our understanding of the underlying biology of pathogens will undoubtedly uncover new potential enzymatic targets, but the rate of discovery has not kept pace with the development of resistance (Ventola, 2015).
In the face of a dry anti-infective pipeline, researchers and pharmaceutic companies have begun to turn their attention to a different class of targets, protein-protein interactions (PPI) (Michelle R. Arkin & Wells, 2004; Wells & McClendon, 2007). Many essential cellular functions rely on the precise and timely recruitment of proteins, often accomplished through a PPI. Disruption of protein interfaces, either through genetic proof-of-principle studies or small molecule inhibitors can kill pathogens or render them non-virulent, making PPI inhibitors an exciting new research area in the anti-infective world. There are also plenty of PPIs with which to work. The number of cataloged PPIs varies by database, but at a minimum there are tens of thousands human PPIs and the E. coli interactions number in the thousands (Lehne & Schlitt, 2009). These counts do not include a sizeable number of host-pathogen interactions, with over 50,000 human-pathogen interactions cataloged in HPIDB (Kumar & Nanduri, 2010). While not all of these interactions are feasible targets for inhibition, a sizeable number are. We will explore examples of inhibitors that target several classes of PPI: pathogen-pathogen, host-pathogen and host-host interactions and how they might alter the treatment of infectious diseases.
Historically, PPIs were considered undruggable targets. This reputation likely stemmed from the lack of high-throughput ready screening assays as well as the thought that most PPIs are held together by large, chemically non-complex surfaces with a lack of easily druggable pockets (Spencer, 1998). While such difficult PPI targets undoubtedly exist, it is now appreciated that many PPIs use much smaller interfaces for their interaction, frequently consisting of an unstructured peptide bound to a well-defined groove (M. R. Arkin, Tang, & Wells, 2014). Furthermore, mutagenesis studies of several PPIs has revealed that surfaces contributing to the affinity of a given PPI are not evenly distributed across the entire interface. Rather, there tends to be a “hot-spot” or a small number of critical residues that anchor two proteins together (Cukuroglu, Engin, Gursoy, & Keskin, 2014). This means that a putative inhibitor would not need to displace the entirety of a given PPI, but rather only occupy the hot-spot, a more tractable problem. Recent review articles have highlighted small molecules disrupting PPIs for the treatment of oncologic targets that have reached early clinical trials, demonstrating the feasibility of the approach. Because many of these inhibitors have already been reviewed in depth (M. R. Arkin et al., 2014; Sheng, Dong, Miao, Zhang, & Wang, 2015), this review will focus on PPI inhibitors for the treatment of infectious diseases.
Antibacterial agents
ZipA-FtsZ
During bacterial cytokinesis, the cell contents must be properly partitioned between the two daughter cells and the cell wall sealed to prevent loss of cytoplasmic material or cell lysis. To accomplish this task, a ring, called the Z-ring, is formed at the site of division from the head-to-tail polymerization of the GTPase FtsZ (Adams & Errington, 2009). While the contribution that FtsZ and the Z-ring plays in generating the force required to pinch the cell membrane is debated, it is clear that FtsZ play an essential role in cytokinesis (Xiao & Goley, 2016).
To maintain contact with the cell wall throughout cytokinesis, FtsZ uses the 17 C-terminal most residues to bind to the membrane associated protein ZipA (Mosyak et al., 2000). Loss of this interaction is lethal in the gammaproteobacteria (although it is absent in other bacteria (Hale & de Boer, 1997)) likely due to the ability of ZipA to stabilize FtsZ polymers and localize them to the membrane (Kuchibhatla, Bhattacharya, & Panda, 2011). Additionally, alanine scanning mutations of the FtsZ interaction site demonstrated that the majority of the affinity between the two protein is derived from only 3 hydrophobic residues, I374, F377 and L378 (Mosyak et al., 2000). Together these data suggest that a small molecule could block the FtsZ-ZipA interaction and that an inhibitor of this PPI would have antibacterial properties.
Researchers at Wyeth Research developed a high-throughput fluorescence polarization (FP) assay to screen for inhibitors of the FtsZ-ZipA interaction. During assay development, they realized that the relatively poor affinity of the PPI (7 μM KD as determined by surface plasmon resonance) meant that a prohibitively large amount of ZipA would be required to screen an acceptable number of compounds. To circumvent this limitation, a phage display screen was conducted to identify a probe with a higher affinity to the ZipA. The resulting peptide, FtsZ-PD1, was found to have a KD of 150 nM, a 45-fold improvement and a FP high-throughput screen (HTS) of 250,000 compounds was conducted using a labeled version of the FtsZ-PD1 as a probe. This screening identified a pyridylpyrimidine inhibitor with a modest 12 μM Ki in the FP assay (Fig. 1) (Kenny et al., 2003) and several additional inhibitor scaffolds with weak activities were identified in the same screen. Crystallographic studies confirmed that the inhibitor occupied the FtsZ binding pocket on ZipA.
Figure 1.
Structure of the pyridylpyrimidine HTS hit.
Besides reducing the protein production burden, one can imagine two possible results of using a tighter binding probe for screening. First, the higher affinity peptide may serve to exclude low potency, but still active, inhibitor scaffolds that could be improved through medical chemistry efforts. The remaining hits are more likely to be active and potent against the native PPI, although reduced in number. The trade-offs associated with this approach likely depend on the size and quality of the chemical library to be screened. With a large enough library, the reduced hit rate is unlikely to be problematic and may even reduce the burden of secondary screening and hit validation. Presumably, the opposite approach could also be used; where a targeted interaction may be weakened to gain a foothold for later optimization. Secondly, the use of a higher affinity probe will introduce interaction sites not used in the wild-type interface. Ideally, a screening hit will target only the residues comprising the original interface and not the artificially introduced interactions.
Unfortunately, the pyridylpyrimidine scaffold was found to have non-specific toxicity against both bacterial and yeast cells lines which precluded further development (Rush, Grant, Mosyak, & Nicholls, 2005). A variety of techniques were tried in attempts to develop more promising inhibitor scaffolds. First, a computational scaffold hopping method was used to identify a triazolopyridazine ring structures that mimicked the pyridylpyrimidine binding pose. While a crystal structure of a triazolopyridazine inhibitor bound to ZipA was obtained, these inhibitors were quite weak (IC50 ~80 μM) and do not appear to have been pursued (Rush et al., 2005). Secondly, an additional set of weak scaffolds (IC50 >1 mM) were discovered in the initial FP screen. Remarkably in the face of this low affinity, crystal structures of these inhibitors bound to the FtsZ pocket of ZipA were obtained (Jennings et al., 2004). This led to additional medicinal chemistry optimization, as well as an attempt to merge two weak inhibitors to fill the entirety of the binding pocket (Sutherland et al., 2003). While a 10-fold reduction in IC50 was obtained, none of these compounds displayed antibacterial activity against E. coli. These compounds did inhibit the growth of E. coli strains with compromised cell membranes, leading the authors to blame poor cell penetrance for the lack of bactericidal effect. However, a subset of these compounds was found to be active against several gram-positive organisms, which lack a ZipA homolog. This suggests that observed the antibacterial activity may have been a result of off-target effects (Sutherland et al., 2003). Finally, an NMR fragment screen was conducted to identify a more cell-penetrant inhibitor scaffold. While several new inhibitor scaffolds were identified, none possessed adequate activity in the FP assay to pursued farther and the project appears to have been terminated (Tsao et al., 2006).
Despite the difficulties encountered in cell penetrance and non-specific toxicity, this early effort was a successful demonstration of the ability to identify PPI disrupting inhibitor scaffolds. We are not aware of additional work against this target, although the combination of advances in screening libraries, techniques and the ease of obtaining inhibitor bound structures suggests that potent inhibitors could yet be discovered.
Pilicides
Urinary tract infections (UTI) are one of the most prevalent infections and are responsible for nearly 10 million clinic visits per year (Schappert, 2011). Treatment of UTIs are estimated to exceed $3,500,000,000 annually (Flores-Mireles, Walker, Caparon, & Hultgren, 2015). UTIs can be caused by a wide range of pathogens, but most UTIs (65–75%) are caused by uropathogenic E. coli (UPEC) (Flores-Mireles et al., 2015). Most uncomplicated UTIs will resolve spontaneously within a week, however symptoms associated with UTIs are unpleasant (dysuria, frequency, urgency, suprapubic pain and hematuria (Bent, Nallamothu, Simel, Fihn, & Saint, 2002)). Patients are typically treated with nitrofurantoin, trimethoprim-sulfamethoxazole or fosfomycin (Gupta et al., 2011). While resistance to these commonly used antibiotics is relatively rare, resistance rates are increasing (Zowawi et al., 2015). Furthermore, treatment with broad spectrum antibiotics can lead to profound disruptions in the native microbiotia with poorly understood consequences (Dethlefsen, Huse, Sogin, & Relman, 2008). Both of these problems could be circumvented with novel therapies targeting the virulence factors of UPEC specifically, sparing the non-pathogenic members of the microbiotia.
To maintain an infection in the urinary tract despite the repeated outward flow of urine, UPEC anchor to the urothelium with the help of a variety of excreted pilus fibers (Wu, Sun, & Medina, 1996). One pilus, the type 1 pili, is capped with a FimH subunit that binds to mannose presented on urothelium glycolipids (Krogfelt, Bergmans, & Klemm, 1990). Type 1 pili also contribute towards biofilm formation and the accompanying resistance to therapy (Martinez, Mulvey, Schilling, Pinkner, & Hultgren, 2000; Wright, Seed, & Hultgren, 2007). To ascend into the kidney and cause pyelonephritis, UPEC must express P pili, terminating with PapG, which binds to kidney specific glycolipids (Lane & Mobley, 2007).
Because bacteria remain physically anchored in the urinary tract during an infection, inhibitors of FimH binding are predicted to prophylactically block UPEC binding or allow for the washout of an existing infection. Indeed, recently reported small molecule inhibitors of FimH binding, called mannocides, have activity as both prophylaxis or for treatment of established infections of murine models of UTI, demonstrating the utility of targeting UPEC pili (Cusumano et al., 2011; Han et al., 2010). While an exciting development for the treatment of UTIs, mannocide therapies disrupt only a specific pilus-ligand interaction, limiting the number of susceptible organisms.
Pilus formation and export to the bacteria cellular membrane occurs through the chaperone-usher pathway, which is widely conserved among gram-negative pathogens (Busch & Waksman, 2012). Given the need for novel antibiotics targeting gram-negative pathogens, general inhibitors of the chaperone-usher pathway could find use as therapies for a range of pathogens such as Salmonella, Yersinia, and Pseudomonas species (Nuccio & Bäumler, 2007). The chaperone-usher pilus synthesis pathway begins with the transport of pilus subunits to the periplasmic space where they are bound by a chaperone protein, PapD, that is required for proper folding and pilin assembly. The pilin subunits contain a nearly complete β-barrel, lacking only a single strand so that donation of the missing strand by the chaperone protein allows for proper folding of the subunits. Additionally, the chaperone uses a hydrophobic groove to trap and display an unstructured N-terminal extension of the pilin subunit. To add a new subunit to the growing strand, the displayed N-terminus extension replaces the chaperone strand to complete the β-barrel and allow for the release of the chaperone (Waksman & Hultgren, 2009). Loss of binding to the chaperone blocks pilus assembly and leads to the accumulation of aggregated pilin subunits (Slonim, Pinkner, Branden, & Hultgren, 1992), suggesting that small molecule inhibitors of the usher-chaperone pathway could have the same effect.
To test this hypothesis, a series of small molecules based on the core of the chaperone ligand, PapG, were synthesized. These so-called pilicides were observed to bind to PapD by SPR and NMR (Hedenstrom et al., 2005; Svensson et al., 2001). Initial biological results were promising, as the lead pilicide compound (Fig. 2A) was found to block bacterial binding to bladder epithelial cells, reduce the number of cells with developed pili and disrupt biofilm formation, albeit at relatively high inhibitor concentrations. To confirm the pilicide mechanism of action, the authors solved the structure of a pilicide bound PapD. The pilicide was found to obstruct the binding site for FimH, leading to the observed failure of pilus assembly (Pinkner et al., 2006). Further medicinal chemistry optimization of the inhibitors yielded a derivative carrying an additional benzyl functional group, with over a 16-fold improvement in potency in a biofilm formation assay (Chorell et al., 2010). In spite of these gains, even the most active pilicides to date are relatively low potency, (7 μM IC50, Fig. 2B) and have not been tested in animal models of urinary tract infections. Furthermore, recent studies have indicated that the advanced pilicides may have additional, non-pilicidal, effects on the transcriptional regulation of UPEC virulence and motility, although these experiments were conducted at high inhibitor concentrations, where off-targets effects are more likely (Greene et al., 2014).
Figure 2.
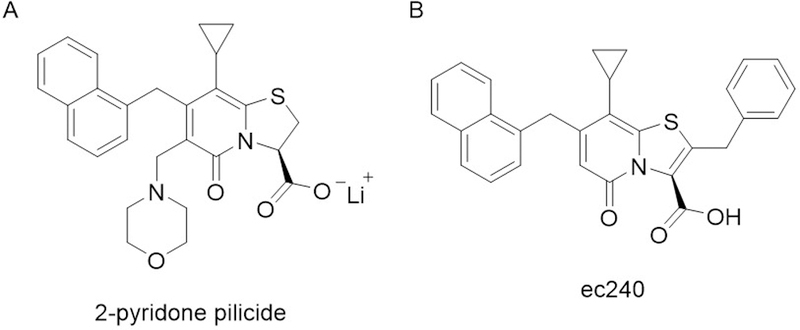
A) Scheme depicting the mechanism of chaperone-usher pilus assembly in UPEC pathogenicity B. structure of pilicides B) structure of the 2-pyridone based pilicide. C) Structure of the pilicide ec240
Despite the remaining challenges, these studies demonstrate exciting progress towards novels UTI therapies that target UPEC adhesion, both by mannosides and the pilicides. Since both classes of inhibitors target bacterial virulence rather than survival, it is possible that resistance would be slower to develop and non-virulent microbiota would be spared, both welcome attributes in a therapeutic agent.
DnaN-DnaE1
The development of effective therapies against Mycobacterium tuberculosis was a major global achievement and has made it possible to imagine that this scourge may eventually be eradicated. While the number of tuberculosis related deaths has fallen by more than 20% since 2000, it remains one of the top 10 global causes of death (Global tuberculosis report 2016, 2016). Unfortunately, there has been a remarkable increase in the extent of drug resistance in M. tuberculosis with the emergence of extensively (Jassal & Bishai, 2008) and total drug resistant strains (Udwadia, Amale, Ajbani, & Rodrigues, 2012). In 2015, over 30% of tuberculosis cases were found to be resistant to the first-line therapy rifampicin (Global tuberculosis report 2016, 2016). To continue to make strides against this pathogen, antibacterial agents acting against novel targets are needed.
To identify potential inhibitors of M. tuberculosis, Kling and coworkers identified a promising lead compound, griselimycin (GM), that was first identified at Rhône-Poulence in the 1960s (Kling et al., 2015). GM is a cyclic peptide natural product produced from Streptomyces species (Fig. 3), but the compound suffered from poor pharmacokinetics and the introduction of other active anti-tuberculosis agents. Hoping to overcome these limitations, the authors undertook a preliminary SAR study thatdemonstrate that minor derivatization of the Pro8 residue lead to both an increased stability. High doses of a cyclohexyl-derivatized GM (CGM) were found to be as active as clinical therapy isoniazid against M. tuberculosis in vitro and in an in vivo mouse lung TB model.
Figure 3.
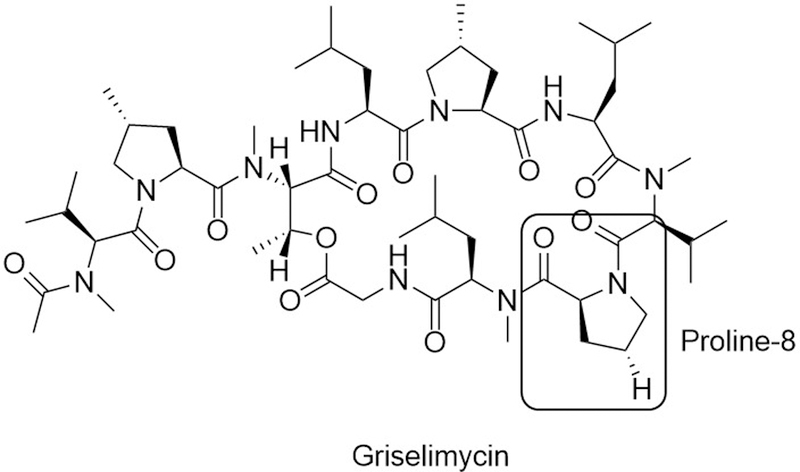
Structure of the DnaN-DnaE1 PPI inhibitor griselimycin. Methylation or cyclohexylation of the labeled hydrogen of the boxed residue, proline-8, lead to improved potency and pharmacokinetic properties.
Because the mechanism of GM was not established during the initial discovery process, resistance studies were conducted to identify the cellular target of GM. A variety of genetic approaches lead to the determination that DnaN, the DNA polymerase sliding clamp, was the target and GM binding to DnaN was confirmed by surface plasmon resonance with femtomolar dissociation constants. Both GM and CGM bind to mycoplasma DnaN with over a ~1000X higher affinity than to the E. coli homolog or the resistance associated protein GriR. The binding site of a methylated GM was determined by X-ray crystallographic studies of DnaN, which revealed the compounds block binding to DnaE1, the catalytic subunit of Pol III. The authors propose that disruption of this interaction leads to lethal failures in DNA replication and likely DNA breaks.
Intriguingly, previous screening campaigns have targeted the DnaN-DnaE interface with only modest results. An FP based chemical HTS identified a 10 μM inhibitor of the interaction, and while a crystal structure of the inhibitor bound to DnaN was obtained, no measurements of antibacterial efficacy were reported. This inhibitor contains a rhodanine, a common pan-assay interference compounds (PAINS) motif (Baell & Holloway, 2010), suggesting off-target effects would be problematic (Georgescu et al., 2008). A later fragment screening effort identified 2 possible inhibitor scaffolds (Yin et al., 2014) and while further structure activity relationship (SAR) efforts modestly improved the potency, only a 20 μM IC50 for the in vitro interaction was described (Yin et al., 2015). It seems likely that both efforts were hampered by a limited library size available for the initial screening (~30,000 and 352 compounds respectively), whereas the natural product approach taken to discover GM was agnostic to the mechanism of action and only incidentally targeted a PPI.
Antiviral PPI Inhibitors
Human Papillomavirus Ori-E1-E2 interaction
Human papillomaviruses (HPV) are double-stranded DNA viruses that infect the human epithelium and cause wart formation from the rapid growth of the epithelial tissue. One distinguishing factor of HPVs is that they are among the few known human cancer viruses, where infection of the anogenital region by high-risk strains (HPV-16 and HPV-18) can lead to the development of cervical cancer (Bosch & De Sanjosé, 2003). The increased risk of cancer results from the incorporation of the virus into the host genome, forming a provirus. To continue propagating, the host cell is then compelled to divide, simultaneously copying the provirus. This continued dysregulated replication can eventually lead to the cervical cancer (Muñoz, Castellsagué, de González, & Gissmann, 2006).
The low-risk viruses (HPV-6 and HPV-11) are rarely carcinogenic, but can lead to the development of anogenital warts. Wart removal is a painful process and, because the underlying HPV infection is not cleared, warts recur frequently. An antiviral therapy for the low-risk HPV strains could allow for the ultimate clearing of the infection and a permanent wart treatment.
Rather than reproduce as a provirus, low-risk HPV strains maintain their genomes as plasmids in infected cells. HPV relies on a host polymerase to replicate this plasmid, as the HPV genome does not encode a polymerase. Initiation of viral replication depends on the formation of ternary DNA-protein complex to recruit the polymerase to the origin of replication (Berg & Stenlund, 1997). First, the viral protein E1 binds to the origin through a DNA binding domain (DBD), which also serves as a dimerization site. A C-terminal helicase domain of E1 mediates an interaction to the transactivation domain of E2 (Chen & Stenlund, 1998). Once located at the origin, an E2 DBD domain tethers the E1-E2-DNA complex and the recruitment of additional E1 dimers leads to the formation of a hexameric structure that encircles and melts the origin DNA (Sedman & Stenlund, 1998). During this process, E2 is displaced allowing for the recruitment of a host DNA polymerase which replicates the plasmid (Conger, Liu, Kuo, Chow, & Wang, 1999). Disruption of any of these interactions is sufficient to block HPV replication.
To identify inhibitors of E1-E2-Ori complex formation, White and coworkers developed a bead-based scintillation proximity assay (SPA). A DNA substrate containing the HPV origin was radiolabeled and then incubated with E1 and E2. An SPA bead was tether to the complex by an anti-E1 antibody and this allowed for the recruitment of E1 to the DNA to be detected, which requires E1-E2 binding. Functionally, this allowed for the identification of inhibitors of the E1-E2, E1-DNA or E2-DNA interactions simultaneously, since disruption of any is sufficient to block E1 recruitment. This assay was used to screen a 140,000 compound library and identified a single lead compound with an indanedione scaffold (11 μM IC50) (Fig. 4A) (White et al., 2003). Additional chlorination of the phenyl ring through SAR studies led to the discovery of compounds with approximately 20-fold greater potency (Yoakim et al., 2003). Activity against the interaction was confirmed by ELISA and binding to E2 was confirmed by isothermal titration calorimetry (Wang et al., 2004). Unfortunately, when these compounds were tested in cellular assays, EC50 values were markedly higher than the activity in biochemical assays had suggested. Together with the fact that the inanedione class of compounds possessed relatively poor pharmacokinetic properties, this series was ultimately abandoned (White, Faucher, & Goudreau, 2010).
Figure 4.
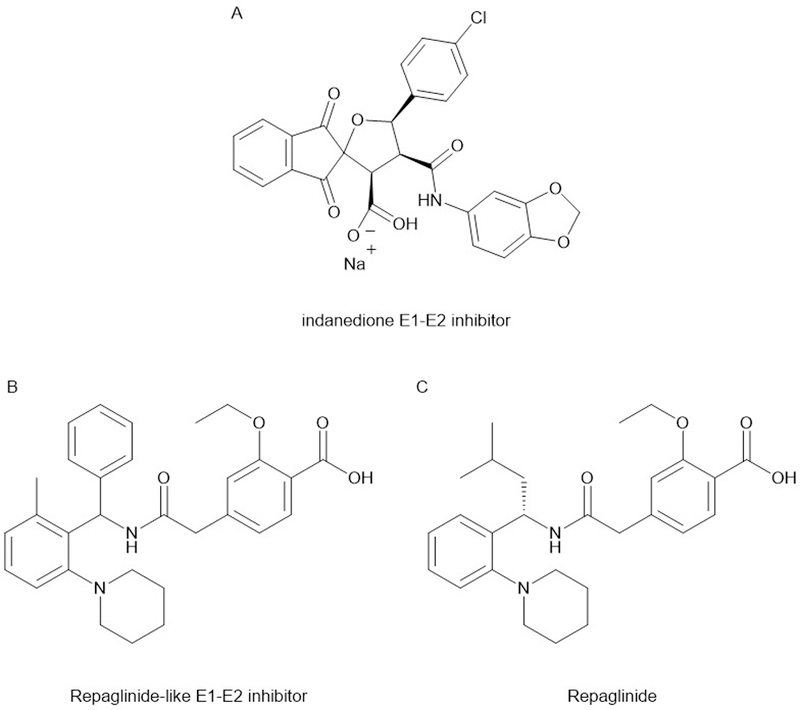
Structures of the inhibitors of the E1-E2 interface. The repaglinide like class of inhibitors was found to have undesirable hypoglycemic effects, resulting from the chemical similarity to repaglinide.
However, this well characterized set of in vitro active compounds allowed the researchers to improve their HTS assay. A tritiated version of an indanedione compound was prepared and a larger compound library was screened for compounds with the ability displace the tritiated probe. A novel inhibitor scaffold was found to both displace the tritiated probe and block the E1-E2 interaction. The scaffold showed a marked similarity to repaglinide, a diabetes drug with blood glucose lowering properties (Fig. 4B-C). Accordingly, early members of repaglinide series shared this undesirable (for an antiviral agent) property. While no crystal structure for this class was obtained, preliminary optimization SAR suggested that antiviral activity could be maintained while minimizing the glucose lowering properties. Despite these promising initial results, the introduction of an effective HPV vaccine was expected to vastly reduce the future demand for HPV therapies and the project was terminated. In this particular case, the failure to generate a clinical agent was not the due to the difficultly of targeting a PPI, but rather external market forces (White et al., 2010).
Human Immunodeficiency Virus
Prior to the development of antiretrovirus therapy, infection with human immunodeficiency virus (HIV), the causative agent of the acquired immunodeficiency syndrome (AIDS), was a death sentence. Starting in the 1990s and continuing through today, the introduction of a broad range of HIV treatments have greatly improved patient outcomes. Unfortunately, viruses resistant to every class of HIV therapy have been observed and these can lead to treatment failure (Magambo et al., 2014; Wensing et al., 2015). Using combinations of different classes of antiretrovirals greatly diminishes the risk of developing resistance, but resistance can occur because of monotherapy or if sub-therapeutic levels of antiretrovirals are maintained as a result of improper dosing. Furthermore, resistance to one member of an antiretroviral class frequently leads to cross-resistance to all members of that class, which can both reduce the number of available agents and undermine the effectiveness of pre-exposure prophylaxis (PrEP). In the face of these challenges, the development of novel classes of HIV treatment for both PrEP and therapy is needed (A Waheed & Tachedjian, 2016).
Given the limited repertoire of viral proteins, viruses rely on hijacking host proteins to carry out functions in the viral life cycle. Viruses typically rely on protein interactions with the host cellular machinery to enter or otherwise alter the normal cellular function. As such, small molecule blockades of these interactions can be used to disrupt the viral lifecycle. Two key HIV PPIs that have been targeted for disruption as antiviral therapies play vital roles in viral-host fusion and integration into the host genome.
HIV Entry Inhibitors
To access the cellular machinery required for replication, viruses must first bypass the cellular membrane. Most viruses accomplish this by binding to a cellular receptor on the outer cellular membrane, which is used as an initiation point for membrane fusion. The structure and identity of the receptor targeted varies between viruses and determines which cell types are targeted (i.e. the tropism). To target CD4+ T-cells, the gp120 subunit of the envelope protein first engages the CD4 receptor. CD4 binding causes a conformational shift that allows for binding to an additional T-cell receptor, either CXCR4 or CCR5. Once bound to both receptors, the second subunit of ENV, gp41, penetrates into the cellular membrane and begins the fusion process (Wilen, Tilton, & Doms, 2012).
There is biological support for targeting HIV fusion as a therapeutic route. In the mid-1990s, it was noted that a subset of the population lack cell surface expression of CCR5 and those lacking this receptor appear to be resistant to infection with the HIV-1 strain (Samson, Libert, Doranz, & Rucker, 1996). This is important for two principle reasons. First, this implies that blockade of the CCR5 receptor is sufficient to impede HIV infection. Secondly, inhibition of the normal function of CCR5 is likely to be tolerated with minimal adverse effects since the CCR5−/− genotype is maintained in the population.
In the early 2000s, there was much interest in developing inhibitors of CD4, CXCR4 or CCR5 receptors to block HIV entry into cells and peptide or small molecule inhibitors of gp120 binding were developed against each of these targets (Lin et al., 2003). Despite the early success of these entry inhibitors, only two have reached the clinic, primarily due to the poor bioavailability of the CD4 antagonists (Yang et al., 2005) and limited effectiveness of the CXCR4 antagonists (Doranz et al., 2001). The first clinical agent, enfuvirtide, is a peptide that binds to gp120 and blocks entry pore formation. As a peptide inhibitor, it must be dosed intravenously, is difficult to self-administer and is not recommended as first-line therapy (“Guidelines for the use…”, 2016). But because enfuvirtide targets the viral protein gp120 rather than the cellular receptor, it is active against both CCR5 and CXCR4 trophic viruses (Matthews et al., 2004).
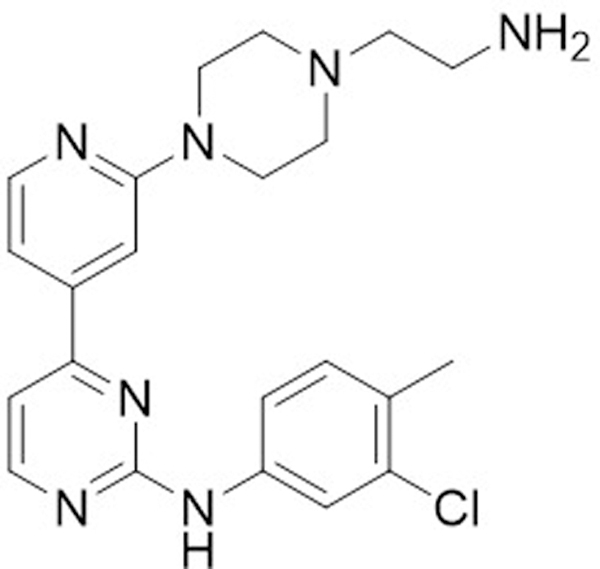
The second entry inhibitor approved to date is maraviroc. The lead that was eventually developed into maraviroc was originally identified from a Pfizer HTS. The compound (UK-107,543) was found to block binding of a radiolabeled substrate to CCR5. A heroic medicinal chemistry optimization campaign yielded maraviroc (Fig. 5), with low nanomolar IC50 values against 3 CCR5 peptide substrates. Additionally, maraviroc was found to have nanomolar antiviral activity against a variety of both lab-adapted and clinical CCR5 tropic HIV-1 strains. As expected, no activity was observed against CXCR4 tropic strains. Phase I clinical trials demonstrated that therapeutic doses could be reached safely and after a series of phase III trials demonstrated efficacy, maraviroc was approved for clinical use. Currently, maraviroc is rarely used in treatment-naive patients but is reserved for patients with treatment resistant, CCR5 specific HIV strains (“Guidelines for the use…”, 2016). Despite these limitations, the development of maraviroc represented a major accomplishment in the field of PPI inhibitors.
Figure 5.
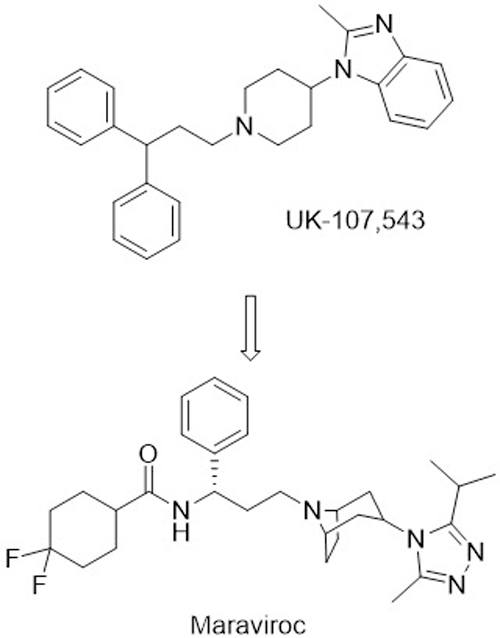
Structure of the initial screening hit for CCR5 antagonist, UK-107,543, and the optimized inhibitor, Maraviroc.
HIV Integrase inhibitors
Integration of the viral genome into the host chromosome represents another step of the HIV lifecycle that has been targeted for therapy. The validity of this target has been established by the clinical success of the integrase active site inhibitor raltegravir (Eron et al., 2013). Relatively high levels of resistance to integrase inhibitors and cross-resistance within the class has limited the utility of these therapies (Malet et al., 2014). Fortunately, integrase relies on an essential PPI where lens epithelium-derived growth factor (LEDGF/p75), tethers integrase to the DNA while stimulating the integrase activity (Poeschla, 2008). This interface is essential to carry out its function and represents a potential therapeutic target.
Several groups have conducted screens to identify small molecule inhibitors of the LEDGF/p75 interface, including both chemical and in silico screens. The first inhibitor, named D77, was found to be active in a yeast two-hybrid assay, although it proved cytotoxic to non-infected cells. This cytotoxicity likely results from the presence of a rhodanine functional group in D77 (Fig. 6A) (Du et al., 2008). Small molecules containing these functional groups are common hits in HTSs as a result of their ability to covalently modify proteins in a non-specific fashion (Baell & Holloway, 2010). Further in silico screening of clinically approved drugs identified 8 inhibitors with IC50 values in the low to mid micromolar range, although further improvements have not been reported (Hu et al., 2012). A more promising inhibitor scaffold was identified from an effort that used in silico pharmacophore screening to identify commercially available compounds. SAR studies allowed for the development of more potent inhibitors possessing a 2-(tert-butoxy) functionality, resulting in CX014442 (Fig. 6B), with 69 nM EC50 and low cytotoxicity.
Figure 6.
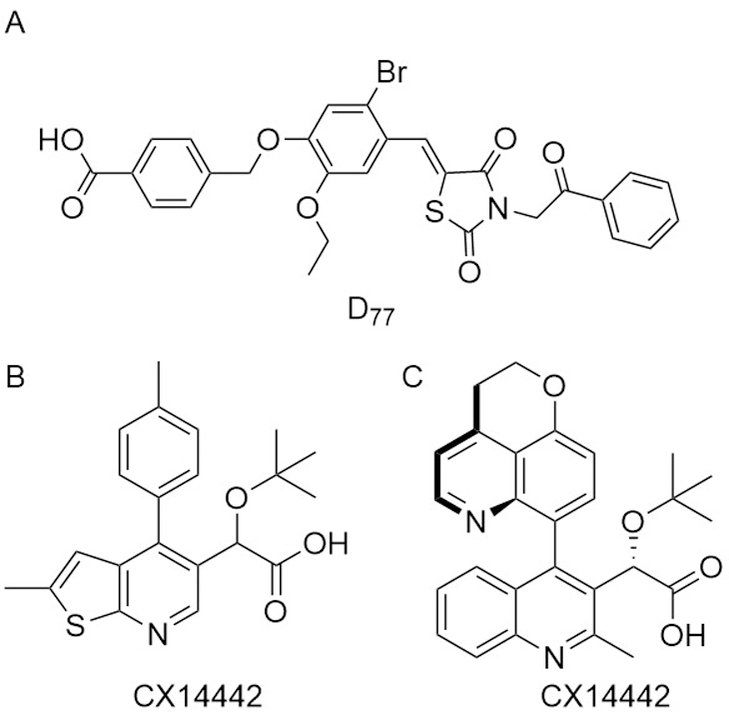
Structure of representative inhibitors of integrase/LEDGF interaction.
Another series of inhibitors of this interface was developed at Boehringer Ingelheim. Rather than specifically targeting the LEDGF/p75 interface, this group screened for inhibitors of the 3’-processing activity of HIV integrase. After successfully screening their compound collection, they noticed that the lead compound bound integrase away from the active site, leading them to name this class of inhibitors the noncatalytic site integrase inhibitors (NCINI). Optimization of this series of compounds yielded a potent inhibitor, BI 224436 (Fig. 6C).
Despite the structural similarity between the NCINI and LEDGIN inhibitors, it was not immediately clear that shared a common mechanism of action since they were discovered by different methodologies. Work by Kvaratskhelia and coworkers concluded that both inhibitors blocked LEDGF binding, albeit more potently by BI 224436, and there was substantial overlap of the inhibitor binding sites (Engelman, Kessl, & Kvaratskhelia, 2013).
During in vitro analysis, BI 224436 demonstrated antiviral activity against a range of viruses resistant to other therapeutics and had a promising animal pharmacokinetic profile, which prompted the initiation of a phase I clinical trial in 2010 (Fenwick et al., 2014). While the results from this trial have not been disclosed and no phase II trials have been started, Gilead licensed the development of the NCINI class of inhibitors in 2011 (“Boehringer license novel HIV non-catalytic integrase inhibitors to Gilead,” 2011). A paper describing the medicinal chemistry optimization of another NCINI derivative for activity against a integrase resistant mutant and improved pharmacokinetics was published in 2016, suggesting that an integrase inhibitor may yet show clinical efficacy (Fader et al., 2016).
Targeting host-host PPIs to improve infection survival:
One of the most lethal aspects of bacterial infections can be the immune response to the insult. While important for the eventual clearance of the pathogens, widespread over activation of the innate immune system and the resulting systemic inflammation can lead to multiorgan failure or even death (Marshall, 2005). Neutrophil exocytosis is a principle driver of this systemic inflammation, where neutrophils release a range of potent toxins such as reactive oxygen species and proteases (Marshall, 2005). Excessive release of these species does not improve control of the infection, but rather damages surrounding tissues (Narasaraju et al., 2011). In rats, blockade of neutrophil exocytosis was shown to reduce the extent of tissue injury (Uriarte et al., 2013) and this blockade has been proposed as a mechanism to allow for patient survival while the infection is treated by the antibiotic therapy and the immune system. Related inhibitors would also be expected to be effective for the treatment of some autoimmune conditions.
Neutrophil exocytosis inhibitors (Nexinhibs)
One of the critical regulators of neutrophil exocytosis is the interaction between JFC1 and a GTPase, Rab27a. Downregulation of either of these proteins blocks the exocytosis of neutrophil granules, but neither is known to play a role in phagosome maturation (Brzezinska et al., 2008). This raises the possibility that small molecule inhibition of the Rab27a-JFC1 interaction would lead to a targeted inhibition of the negative effects of the neutrophil granule release, while allowing for neutrophil survival and the continued clearance of the pathogens. Because this PPI a host-specific interaction, resistance is unlikely to be a problem.
A high-throughput time-resolved fluorescence resonance energy transfer (TR-FRET) was used to screen 32,000 compounds and lead compounds were confirmed by the reduction in neutrophil myeloperoxidase secretion in a cell based secondary screen. In this assay, neutrophils were pretreated with the putative nexinhibs (Neutrophil exocytosis inhibitor) and then granule release was stimulated. A decrease in the generation of H2O2 indicated that the compounds successfully inhibited the release of the myeloperoxidase containing granules. After excluding peroxide scavenging compounds, the authors concluded that compounds active in this assay must penetrate the membrane, allowing the authors to eliminate non-permeable compounds early in the hit reduction process (Johnson et al., 2016).
The most promising compound, Nexinhib 20 (Fig. 7), was found to block neutrophil degranulation in response to agonists and disrupted the Rab27a-JFC1 interaction in pulldown and ELISA assays with an IC50 of 2.6 μM in the ELISA. Nexinhib 20 treatment did not result in cell death or impair the ability of neutrophils to phagocytize, but did reduce exocytosis after neutrophil stimulation. To examine how nexinhibs function in a physiologic setting, mice were treated with nexinhib 20 prior to the induction of systemic inflammation by an intraperitoneal injection of lipopolysaccharide. The total number of neutrophils and white blood cells remained unchanged relative to an untreated control, but there was a modest, albeit significant, reduction in the plasma levels of myeloperoxidase as well as neutrophil infiltration of the kidney and liver (Johnson et al., 2016). While further in vivo testing and a mouse sepsis survival study would bolster the case for nexinhibs, this preliminary study is an exciting step.
Figure 7.
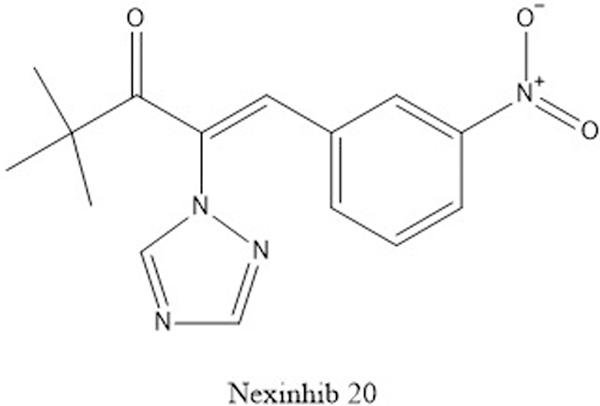
Structure of Nexinhib 20, an inhibitor of the neutrophil exocytosis.
Considerations for targeting PPIs
Historically, PPIs were considered undruggable theraputic targets. One early estimate from Pfizer suggested that while inhibitors for enzymes or GPCR targets could be discovered by high-throughput screening methods at a rate 1 per 105 compounds screen, a discovery rate of 1 per 109-1010 compounds was expected for PPI targets (Spencer, 1998). Given this discouraging prediction, it is obvious why pharmaceutical companies limited their exposure to PPIs. The prevailing view at the time was that protein interfaces consisted of interactions between two broad, mostly hydrophobic and featureless surfaces. The lack of pockets for a hit to gain an initial foothold was also thought to complicate screening.
Two major advances have made targeting PPIs more feasible. First was the realization that while many PPI match the above description, a sizeable number are much simpler. A useful system for classifying the nature and difficulty of targeting a PPI has been developed. Briefly, PPIs can be fit into one of three categories based on the complexity of the simplest member: 1) primary interfaces consist of an unstructured peptide that binds to a channel. 2) A single fold of secondary structure (α-helix or β-sheet) or 3) a lengthy stretch of protein that uses a tertiary structure (Fig. 8) (Blundell et al., 2006). While there are examples of successfully targeting each class of PPI, there is a significant bias toward the simpler interfaces (M. R. Arkin et al., 2014). Careful selection of a target, ideally guided by structural characterization of the PPI, can greatly increase the odds of success. To help calibrate expectations about a potential project, an online tool has been developed to aid in assessing the “drugability” of a given PPI (Basse et al., 2013; Basse, Betzi, Morelli, & Roche, 2016).
Figure 8.
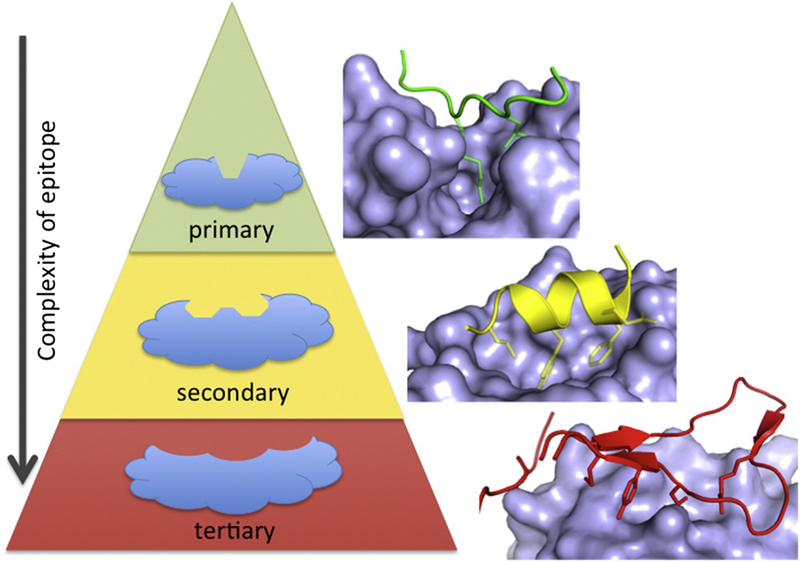
Scheme for classifying PPIs by the nature of the interaction site. Most of the successful PPI inhibitors target the simpler interactions, such as the primary or secondary ones, although there are a examples of inhibitors of tertiary interfaces. Reprinted from Chem. & Biol. 21, Arkin M.R.; Tang, Y and Wells, J.A. “Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality”, 1102–1114, 2014, with permission from Elsevier.
The second key development in the targeting of PPI has been improvements in screening methodologies. The introduction and miniaturization of PPI HTS assays and the advent of academic screening facilities have reduced the cost, and therefore the risk, of performing a chemical HTS. There has been debate regarding the suitability of existing screening libraries for targeting PPIs, as PPI inhibitors tend to be more aromatic, hydrophobic, 3-dimensional and have higher molecular weights than non-PPI inhibitors (Kuenemann, Labbe, Cerdan, & Sperandio, 2016). Armed with this information, PPI specific screening libraries have been introduced, (“iPPI Focused Libraries,” 2017; “Protein-Protein Interaction Libraries - Enamine,” 2017; Reynès et al., 2010) although more time is needed to tell if these libraries show improved hit rates. Besides chemical HTS methods, additional hit identification strategies have been successfully implemented. Some have been highlighted in this review and include in silico screening, natural product screening, rational design, scaffold hopping and fragment screening and elaboration (Sheng et al., 2015). There is unlikely to be a single best method for lead generation and different approaches may succeed where prior attempts have failed. Because hit identification methods rely on varied input libraries and individual libraries may not contain a bona fide inhibitor, trying multiple screening methods allows for the sampling of a broader range of chemical space than could be achieved by solely by increasing the library size. This is also demonstrated by several of the examples presented here, where different approaches against the same target yielded unique inhibitor scaffolds. Furthermore, the use of phenotypic screens or other assay designs allowing for the discovery of unpredicted mechanisms of action may prove fruitful (Fischer, Rossmann, & Hyvonen, 2015).
While the case studies presented here represent the successful inhibition of a range of protein interactions, most or all of these compounds will still fail to reach the clinic. It is difficult to assess the true success rate of PPI targeted drug discovery efforts as publication and survivorship bias obscures the true denominator of this calculation. However, from the examples presented here and elsewhere, it appears that small molecule disruption of many PPIs is challenging yet feasible. The development of existing inhibitors appears to be hindered by non-specific effects, poor pharmacokinetics or cell penetrance, which are challenges common to all therapeutics. As seen with the introduction of oncologic therapies targeting PPIs, once the pipeline of anti-infective PPI inhibitors expands, we have no reason to doubt that they will find increasing clinical success.
References:
- The 10 x ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. (2010). Clin Infect Dis, 50(8), 1081–1083. 10.1086/652237 [DOI] [PubMed] [Google Scholar]
- A Waheed A, & Tachedjian G (2016). Why Do We Need New Drug Classes for HIV Treatment and Prevention? Current topics in medicinal chemistry, 16(12), 1343–1349. [DOI] [PubMed] [Google Scholar]
- Adams DW, & Errington J (2009). Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nature Reviews Microbiology, 7(9), 642–653. [DOI] [PubMed] [Google Scholar]
- Arkin MR, Tang Y, & Wells JA (2014). Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol, 21(9), 1102–1114. 10.1016/j.chembiol.2014.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin MR, & Wells JA (2004). Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov, 3(4), 301–317. [DOI] [PubMed] [Google Scholar]
- Baell JB, & Holloway GA (2010). New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. Journal of medicinal chemistry, 53(7), 2719–2740. 10.1021/jm901137j [DOI] [PubMed] [Google Scholar]
- Basse MJ, Betzi S, Bourgeas R, Bouzidi S, Chetrit B, Hamon V, . . . Roche P (2013). 2P2Idb: a structural database dedicated to orthosteric modulation of protein-protein interactions. Nucleic Acids Res, 41(Database issue), D824–827. 10.1093/nar/gks1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basse MJ, Betzi S, Morelli X, & Roche P (2016). 2P2Idb v2: update of a structural database dedicated to orthosteric modulation of protein-protein interactions. Database (Oxford), 2016 10.1093/database/baw007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bent S, Nallamothu BK, Simel DL, Fihn SD, & Saint S (2002). Does this woman have an acute uncomplicated urinary tract infection? JAMA, 287(20), 2701–2710. [DOI] [PubMed] [Google Scholar]
- Berg M, & Stenlund A (1997). Functional interactions between papillomavirus E1 and E2 proteins. Journal of Virology, 71(5), 3853–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell TL, Sibanda BL, Montalvão RW, Brewerton S, Chelliah V, Worth CL, . . . Burke D (2006). Structural biology and bioinformatics in drug design: opportunities and challenges for target identification and lead discovery. Philosophical Transactions of the Royal Society of London B: Biological Sciences, 361(1467), 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehringer license novel HIV non-catalytic integrase inhibitors to Gilead (2011). Retrieved from https://www.thepharmaletter.com/article/boehringer-license-novel-hiv-non-catalytic-integrase-inhibitors-to-gilead
- Bosch FX, & De Sanjosé S (2003). Human papillomavirus and cervical cancer—burden and assessment of causality. JNCI Monographs, 2003(31), 3–13. [DOI] [PubMed] [Google Scholar]
- Brzezinska AA, Johnson JL, Munafo DB, Crozat K, Beutler B, Kiosses WB, . . . Catz SD (2008). The Rab27a effectors JFC1/Slp1 and Munc13–4 regulate exocytosis of neutrophil granules. Traffic, 9(12), 2151–2164. 10.1111/j.1600-0854.2008.00838.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A, & Waksman G (2012). Chaperone–usher pathways: diversity and pilus assembly mechanism. Philosophical Transactions of the Royal Society B: Biological Sciences, 367(1592), 1112–1122. 10.1098/rstb.2011.0206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, & Stenlund A (1998). Characterization of the DNA-binding domain of the bovine papillomavirus replication initiator E1. J Virol, 72(4), 2567–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorell E, Pinkner JS, Phan G, Edvinsson S, Buelens F, Remaut H, . . . Almqvist F (2010). Design and Synthesis of C-2 Substituted Thiazolo and Dihydrothiazolo Ring-Fused 2-Pyridones: Pilicides with Increased Antivirulence Activity. Journal of medicinal chemistry, 53(15), 5690–5695. 10.1021/jm100470t [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conger KL, Liu J-S, Kuo S-R, Chow LT, & Wang TS-F (1999). Human Papillomavirus DNA Replication: INTERACTIONS BETWEEN THE VIRAL E1 PROTEIN AND TWO SUBUNITS OF HUMAN DNA POLYMERASE α/PRIMASE. Journal of Biological Chemistry, 274(5), 2696–2705. 10.1074/jbc.274.5.2696 [DOI] [PubMed] [Google Scholar]
- Cukuroglu E, Engin HB, Gursoy A, & Keskin O (2014). Hot spots in protein–protein interfaces: Towards drug discovery. Progress in Biophysics and Molecular Biology, 116(2–3), 165–173. 10.1016/j.pbiomolbio.2014.06.003 [DOI] [PubMed] [Google Scholar]
- Cusumano CK, Pinkner JS, Han Z, Greene SE, Ford BA, Crowley JR, . . . Hultgren SJ (2011). Treatment and prevention of urinary tract infection with orally active FimH inhibitors. Sci Transl Med, 3(109), 109ra115. 10.1126/scitranslmed.3003021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, & Relman DA (2008). The Pervasive Effects of an Antibiotic on the Human Gut Microbiota, as Revealed by Deep 16S rRNA Sequencing. PLOS Biology, 6(11), e280 10.1371/journal.pbio.0060280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doranz BJ, Filion LG, Diaz-Mitoma F, Sitar DS, Sahai J, Baribaud F, . . . Doms RW (2001). Safe use of the CXCR4 inhibitor ALX40–4C in humans. AIDS Res Hum Retroviruses, 17(6), 475–486. 10.1089/08892220151126508 [DOI] [PubMed] [Google Scholar]
- Du L, Zhao Y, Chen J, Yang L, Zheng Y, Tang Y, . . . Jiang H (2008). D77, one benzoic acid derivative, functions as a novel anti-HIV-1 inhibitor targeting the interaction between integrase and cellular LEDGF/p75. Biochem Biophys Res Commun, 375(1), 139–144. 10.1016/j.bbrc.2008.07.139 [DOI] [PubMed] [Google Scholar]
- Engelman A, Kessl JJ, & Kvaratskhelia M (2013). Allosteric inhibition of HIV-1 integrase activity. Current opinion in chemical biology, 17(3), 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eron JJ, Cooper DA, Steigbigel RT, Clotet B, Gatell JM, Kumar PN, . . . Teppler H (2013). Efficacy and safety of raltegravir for treatment of HIV for 5 years in the BENCHMRK studies: final results of two randomised, placebo-controlled trials. The Lancet Infectious Diseases, 13(7), 587–596. 10.1016/S1473-3099(13)70093-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fader LD, Bailey M, Beaulieu E, Bilodeau F, Bonneau P, Bousquet Y, . . . Duan J (2016). Aligning Potency and Pharmacokinetic Properties for Pyridine-Based NCINIs. ACS medicinal chemistry letters, 7(8), 797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick C, Bailey MD, Bethell R, Bös M, Bonneau P, Cordingley M, . . . Fader LD (2014). Preclinical profile of BI 224436, a novel HIV-1 non-catalytic-site integrase inhibitor. Antimicrobial Agents and Chemotherapy, 58(6), 3233–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G, Rossmann M, & Hyvonen M (2015). Alternative modulation of protein-protein interactions by small molecules. Curr Opin Biotechnol, 35, 78–85. 10.1016/j.copbio.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Mireles AL, Walker JN, Caparon M, & Hultgren SJ (2015). Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Micro, 13(5), 269–284. doi: 10.1038/nrmicro3432 10.1038/nrmicro3432http://www.nature.com/nrmicro/journal/v13/n5/abs/nrmicro3432.html#supplementary-informationhttp://www.nature.com/nrmicro/journal/v13/n5/abs/nrmicro3432.html#supplementary-information [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu RE, Yurieva O, Kim SS, Kuriyan J, Kong XP, & O’Donnell M (2008). Structure of a small-molecule inhibitor of a DNA polymerase sliding clamp. Proc Natl Acad Sci U S A, 105(32), 11116–11121. 10.1073/pnas.0804754105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global tuberculosis report 2016. (2016). Retrieved from.
- Goff DA, Kullar R, Goldstein EJ, Gilchrist M, Nathwani D, Cheng AC, . . . Brink A (2017). A global call from five countries to collaborate in antibiotic stewardship: United we succeed, divided we might fail. The Lancet Infectious Diseases, 17(2), e56–e63. [DOI] [PubMed] [Google Scholar]
- Greene SE, Pinkner JS, Chorell E, Dodson KW, Shaffer CL, Conover MS, . . . Hultgren SJ (2014). Pilicide ec240 Disrupts Virulence Circuits in Uropathogenic Escherichia coli. mBio, 5(6). 10.1128/mBio.02038-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. (2016). Panel on Antiretroviral Guidelines for Adults and Adolescents.
- Gupta K, Hooton TM, Naber KG, Wullt B, Colgan R, Miller LG, . . . Soper DE (2011). International Clinical Practice Guidelines for the Treatment of Acute Uncomplicated Cystitis and Pyelonephritis in Women: A 2010 Update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clinical Infectious Diseases, 52(5), e103–e120. 10.1093/cid/ciq257 [DOI] [PubMed] [Google Scholar]
- Hale CA, & de Boer PA (1997). Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell, 88(2), 175–185. [DOI] [PubMed] [Google Scholar]
- Han Z, Pinkner JS, Ford B, Obermann R, Nolan W, Wildman SA, . . . Janetka JW (2010). Structure-Based Drug Design and Optimization of Mannoside Bacterial FimH Antagonists. Journal of medicinal chemistry, 53(12), 4779–4792. 10.1021/jm100438s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedenstrom M, Emtenas H, Pemberton N, Aberg V, Hultgren SJ, Pinkner JS, . . . Kihlberg J (2005). NMR studies of interactions between periplasmic chaperones from uropathogenic E. coli and pilicides that interfere with chaperone function and pilus assembly. Organic & biomolecular chemistry, 3(23), 4193–4200. 10.1039/B511857C [DOI] [PubMed] [Google Scholar]
- Hu G, Li X, Sun X, Lu W, Liu G, Huang J, . . . Tang Y (2012). Identification of old drugs as potential inhibitors of HIV-1 integrase - human LEDGF/p75 interaction via molecular docking. J Mol Model, 18(12), 4995–5003. 10.1007/s00894-012-1494-0 [DOI] [PubMed] [Google Scholar]
- iPPI Focused Libraries (2017). Retrieved from http://www.otavachemicals.com/products/targeted-libraries-and-focused-libraries/protein-protein-interaction
- Jassal M, & Bishai WR (2008). Extensively drug-resistant tuberculosis. The Lancet Infectious Diseases, 9(1), 19–30. 10.1016/S1473-3099(08)70260-3 [DOI] [PubMed] [Google Scholar]
- Jennings LD, Foreman KW, Rush TS, Tsao DH, Mosyak L, Li Y, . . . Kenny CH (2004). Design and synthesis of indolo [2, 3-a] quinolizin-7-one inhibitors of the ZipA–FtsZ interaction. Bioorganic & medicinal chemistry letters, 14(6), 1427–1431. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Ramadass M, He J, Brown SJ, Zhang J, Abgaryan L, . . . Catz SD (2016). Identification of Neutrophil Exocytosis Inhibitors (Nexinhibs), Small Molecule Inhibitors of Neutrophil Exocytosis and Inflammation: DRUGGABILITY OF THE SMALL GTPase Rab27a. J Biol Chem, 291(50), 25965–25982. 10.1074/jbc.M116.741884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny CH, Ding W, Kelleher K, Benard S, Dushin EG, Sutherland AG, . . . Ellestad G (2003). Development of a fluorescence polarization assay to screen for inhibitors of the FtsZ/ZipA interaction. Analytical biochemistry, 323(2), 224–233. [DOI] [PubMed] [Google Scholar]
- Kling A, Lukat P, Almeida DV, Bauer A, Fontaine E, Sordello S, . . . Muller R (2015). Antibiotics. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science, 348(6239), 1106–1112. 10.1126/science.aaa4690 [DOI] [PubMed] [Google Scholar]
- Krogfelt KA, Bergmans H, & Klemm P (1990). Direct evidence that the FimH protein is the mannose-specific adhesin of Escherichia coli type 1 fimbriae. Infection and immunity, 58(6), 1995–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhatla A, Bhattacharya A, & Panda D (2011). ZipA binds to FtsZ with high affinity and enhances the stability of FtsZ protofilaments. PLoS One, 6(12), e28262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuenemann MA, Labbe CM, Cerdan AH, & Sperandio O (2016). Imbalance in chemical space: How to facilitate the identification of protein-protein interaction inhibitors. Sci Rep, 6, 23815 10.1038/srep23815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, & Nanduri B (2010). HPIDB - a unified resource for host-pathogen interactions. BMC Bioinformatics, 11(6), S16 10.1186/1471-2105-11-s6-s16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane M, & Mobley H (2007). Role of P-fimbrial-mediated adherence in pyelonephritis and persistence of uropathogenic Escherichia coli (UPEC) in the mammalian kidney. Kidney international, 72(1), 19–25. [DOI] [PubMed] [Google Scholar]
- Lehne B, & Schlitt T (2009). Protein-protein interaction databases: keeping up with growing interactomes. Human Genomics, 3(3), 291 10.1186/1479-7364-3-3-291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P-F, Blair W, Wang T, Spicer T, Guo Q, Zhou N, . . . Colonno R (2003). A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proceedings of the National Academy of Sciences, 100(19), 11013–11018. 10.1073/pnas.1832214100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magambo B, Nazziwa J, Bbosa N, Gupta RK, Kaleebu P, & Parry CM (2014). The arrival of untreatable multidrug-resistant HIV-1 in sub-Saharan Africa. Aids, 28(9), 1373–1374. [DOI] [PubMed] [Google Scholar]
- Malet I, Arriaga LG, Artese A, Costa G, Parrotta L, Alcaro S, . . . Valantin M-A (2014). New raltegravir resistance pathways induce broad cross-resistance to all currently used integrase inhibitors. Journal of Antimicrobial Chemotherapy, 69(8), 2118–2122. [DOI] [PubMed] [Google Scholar]
- Marshall JC (2005). Neutrophils in the pathogenesis of sepsis. Critical care medicine, 33(12), S502–S505. [DOI] [PubMed] [Google Scholar]
- Martinez JJ, Mulvey MA, Schilling JD, Pinkner JS, & Hultgren SJ (2000). Type 1 pilus‐mediated bacterial invasion of bladder epithelial cells. The EMBO journal, 19(12), 2803–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews T, Salgo M, Greenberg M, Chung J, DeMasi R, & Bolognesi D (2004). Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat Rev Drug Discov, 3(3), 215–225. [DOI] [PubMed] [Google Scholar]
- Mosyak L, Zhang Y, Glasfeld E, Haney S, Stahl M, Seehra J, & Somers WS (2000). The bacterial cell-division protein ZipA and its interaction with an FtsZ fragment revealed by X-ray crystallography. EMBO J, 19(13), 3179–3191. 10.1093/emboj/19.13.3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz N, Castellsagué X, de González AB, & Gissmann L (2006). Chapter 1: HPV in the etiology of human cancer. Vaccine, 24, Supplement 3, S1–S10. 10.1016/j.vaccine.2006.05.115 [DOI] [PubMed] [Google Scholar]
- Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew A-A, . . . Chow VT (2011). Excessive Neutrophils and Neutrophil Extracellular Traps Contribute to Acute Lung Injury of Influenza Pneumonitis. The American Journal of Pathology, 179(1), 199–210. 10.1016/j.ajpath.2011.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuccio S-P, & Bäumler AJ (2007). Evolution of the Chaperone/Usher Assembly Pathway: Fimbrial Classification Goes Greek. Microbiology and Molecular Biology Reviews, 71(4), 551–575. 10.1128/mmbr.00014-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkner JS, Remaut H, Buelens F, Miller E, Aberg V, Pemberton N, . . . Almqvist F (2006). Rationally designed small compounds inhibit pilus biogenesis in uropathogenic bacteria. Proc Natl Acad Sci U S A, 103(47), 17897–17902. 10.1073/pnas.0606795103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poeschla EM (2008). Integrase, LEDGF/p75 and HIV replication. Cell Mol Life Sci, 65(9), 1403–1424. 10.1007/s00018-008-7540-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protein-Protein Interaction Libraries - Enamine (2017). Retrieved from http://www.enamine.net/index.php?option=com_content&task=view&id=227
- Reynès C, Host H, Camproux A-C, Laconde G, Leroux F, Mazars A, . . . Sperandio O (2010). Designing Focused Chemical Libraries Enriched in Protein-Protein Interaction Inhibitors using Machine-Learning Methods. PLOS Computational Biology, 6(3), e1000695 10.1371/journal.pcbi.1000695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush TS, Grant JA, Mosyak L, & Nicholls A (2005). A shape-based 3-D scaffold hopping method and its application to a bacterial protein− protein interaction. Journal of medicinal chemistry, 48(5), 1489–1495. [DOI] [PubMed] [Google Scholar]
- Samson M, Libert F, Doranz BJ, & Rucker J (1996). Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature, 382(6593), 722. [DOI] [PubMed] [Google Scholar]
- Schappert SM (2011). Ambulatory medical care utilization estimates for 2007. Vital and health statistics(169), 1–38. [PubMed] [Google Scholar]
- Sedman J, & Stenlund A (1998). The papillomavirus E1 protein forms a DNA-dependent hexameric complex with ATPase and DNA helicase activities. J Virol, 72(8), 6893–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng C, Dong G, Miao Z, Zhang W, & Wang W (2015). State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chemical Society Reviews, 44(22), 8238–8259. 10.1039/C5CS00252D [DOI] [PubMed] [Google Scholar]
- Slonim LN, Pinkner JS, Branden CI, & Hultgren SJ (1992). Interactive surface in the PapD chaperone cleft is conserved in pilus chaperone superfamily and essential in subunit recognition and assembly. EMBO J, 11(13), 4747–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer RW (1998). High‐throughput screening of historic collections: Observations on file size, biological targets, and file diversity. Biotechnology and bioengineering, 61(1), 61–67. [PubMed] [Google Scholar]
- Sutherland AG, Alvarez J, Ding W, Foreman K, Kenny CH, Labthavikul P, . . . Ruzin A (2003). Structure-based design of carboxybiphenylindole inhibitors of the ZipA–FtsZ interaction. Organic & biomolecular chemistry, 1(23), 4138–4140. [DOI] [PubMed] [Google Scholar]
- Svensson A, Larsson A, Emtenäs H, Hedenström M, Fex T, Hultgren SJ, . . . Kihlberg J (2001). Design and evaluation of pilicides: potential novel antibacterial agents directed against uropathogenic Escherichia coli. Chembiochem :, 2(12), 915–918. [DOI] [PubMed] [Google Scholar]
- Tsao DH, Sutherland AG, Jennings LD, Li Y, Rush TS, Alvarez JC, . . . Haney SA (2006). Discovery of novel inhibitors of the ZipA/FtsZ complex by NMR fragment screening coupled with structure-based design. Bioorganic & medicinal chemistry, 14(23), 7953–7961. [DOI] [PubMed] [Google Scholar]
- Udwadia ZF, Amale RA, Ajbani KK, & Rodrigues C (2012). Totally Drug-Resistant Tuberculosis in India. Clinical Infectious Diseases, 54(4), 579–581. 10.1093/cid/cir889 [DOI] [PubMed] [Google Scholar]
- Uriarte SM, Rane MJ, Merchant ML, Jin S, Lentsch AB, Ward RA, & McLeish KR (2013). Inhibition of Neutrophil Exocytosis Ameliorates Acute Lung Injury in Rats. Shock (Augusta, Ga.), 39(3), 286–292. 10.1097/SHK.0b013e318282c9a1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventola CL (2015). The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharmacy and Therapeutics, 40(4), 277–283. [PMC free article] [PubMed] [Google Scholar]
- Waksman G, & Hultgren SJ (2009). Structural biology of the chaperone-usher pathway of pilus biogenesis. Nat Rev Micro, 7(11), 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. a. (2016). Antibiotics : challenges, mechanisms, opportunities: Washington, DC: : ASM Press, [2016] ©2016. [Google Scholar]
- Wang Y, Coulombe R, Cameron DR, Thauvette L, Massariol M-J, Amon LM, . . . Yoakim C (2004). Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor. Journal of Biological Chemistry, 279(8), 6976–6985. [DOI] [PubMed] [Google Scholar]
- Wells JA, & McClendon CL (2007). Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature, 450(7172), 1001–1009. doi:http://www.nature.com/nature/journal/v450/n7172/suppinfo/nature06526_S1.html [DOI] [PubMed] [Google Scholar]
- Wensing AM, Calvez V, Günthard HF, Johnson VA, Paredes R, Pillay D, . . . Richman DD (2015). 2015 update of the drug resistance mutations in HIV-1. Top Antivir Med, 23(4), 132–141. [PMC free article] [PubMed] [Google Scholar]
- White PW, Faucher A-M, & Goudreau N (2010). Small molecule inhibitors of the human papillomavirus E1-E2 interaction Small-Molecule Inhibitors of Protein-Protein Interactions (pp. 61–88): Springer. [DOI] [PubMed] [Google Scholar]
- White PW, Titolo S, Brault K, Thauvette L, Pelletier A, Welchner E, . . . Yoakim C (2003). Inhibition of human papillomavirus DNA replication by small molecule antagonists of the E1-E2 protein interaction. Journal of Biological Chemistry, 278(29), 26765–26772. [DOI] [PubMed] [Google Scholar]
- Wilen CB, Tilton JC, & Doms RW (2012). HIV: cell binding and entry. Cold Spring Harb Perspect Med, 2(8). 10.1101/cshperspect.a006866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KJ, Seed PC, & Hultgren SJ (2007). Development of intracellular bacterial communities of uropathogenic Escherichia coli depends on type 1 pili. Cellular microbiology, 9(9), 2230–2241. [DOI] [PubMed] [Google Scholar]
- Wu X-R, Sun T-T, & Medina JJ (1996). In vitro binding of type 1-fimbriated Escherichia coli to uroplakins Ia and Ib: relation to urinary tract infections. Proceedings of the National Academy of Sciences, 93(18), 9630–9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, & Goley ED (2016). Redefining the roles of the FtsZ-ring in bacterial cytokinesis. Current Opinion in Microbiology, 34, 90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Zadjura L, D’arienzo C, Marino A, Santone K, Klunk L, . . . Wang T (2005). Preclinical pharmacokinetics of a novel HIV‐1 attachment inhibitor BMS‐378806 and prediction of its human pharmacokinetics. Biopharmaceutics & drug disposition, 26(9), 387–402. [DOI] [PubMed] [Google Scholar]
- Yin Z, Whittell LR, Wang Y, Jergic S, Liu M, Harry EJ, . . . Oakley AJ (2014). Discovery of lead compounds targeting the bacterial sliding clamp using a fragment-based approach. J Med Chem, 57(6), 2799–2806. 10.1021/jm500122r [DOI] [PubMed] [Google Scholar]
- Yin Z, Whittell LR, Wang Y, Jergic S, Ma C, Lewis PJ, . . . Oakley AJ (2015). Bacterial Sliding Clamp Inhibitors that Mimic the Sequential Binding Mechanism of Endogenous Linear Motifs. J Med Chem, 58(11), 4693–4702. 10.1021/acs.jmedchem.5b00232 [DOI] [PubMed] [Google Scholar]
- Yoakim C, Ogilvie WW, Goudreau N, Naud J, Hache B, O’Meara JA, . . . White PW (2003). Discovery of the first series of inhibitors of human papillomavirus type 11: inhibition of the assembly of the E1–E2–Origin DNA complex. Bioorganic & medicinal chemistry letters, 13(15), 2539–2541. [DOI] [PubMed] [Google Scholar]
- Zowawi HM, Harris PNA, Roberts MJ, Tambyah PA, Schembri MA, Pezzani MD, . . . Paterson DL (2015). The emerging threat of multidrug-resistant Gram-negative bacteria in urology. Nat Rev Urol, 12(10), 570–584. doi: 10.1038/nrurol.2015.199 10.1038/nrurol.2015.199http://www.nature.com/nrurol/journal/v12/n10/abs/nrurol.2015.199.html#supplementary-informationhttp://www.nature.com/nrurol/journal/v12/n10/abs/nrurol.2015.199.html#supplementary-information [DOI] [PubMed] [Google Scholar]