

Summary
Antibiotic-resistant bacterial pathogens pose an urgent healthcare threat, prompting a demand for new medicines. We report the mode of action of the natural ansamycin antibiotic kanglemycin A (KglA). KglA binds bacterial RNA polymerase at the rifampicin-binding pocket but maintains potency against RNA polymerases containing rifampicin-resistant mutations. KglA has antibiotic activity against rifampicin-resistant Gram-positive bacteria and multidrug-resistant Mycobacterium tuberculosis (MDR-M. tuberculosis). The X-ray crystal structures of KglA with the Escherichia coli RNA polymerase holoenzyme and Thermus thermophilus RNA polymerase-promoter complex reveal an altered—compared with rifampicin—conformation of KglA within the rifampicin-binding pocket. Unique deoxysugar and succinate ansa bridge substituents make additional contacts with a separate, hydrophobic pocket of RNA polymerase and preclude the formation of initial dinucleotides, respectively. Previous ansa-chain modifications in the rifamycin series have proven unsuccessful. Thus, KglA represents a key starting point for the development of a new class of ansa-chain derivatized ansamycins to tackle rifampicin resistance.
Keywords: antibiotics, RNA polymerase, kanglemycin A, rifamycin, rifampicin, antibiotic resistance, holo-enzyme crystal structure, promoter complex crystal structure, multidrug-resistant Mycobacterium tuberculosis, MDR-TB
Graphical Abstract
Highlights
-
•
Rifamycin kanglemycin A (KglA) inhibits rifampicin-resistant RNA polymerases (RNAPs)
-
•
KglA’s extra groups improve binding to RNAP and block first dinucleotide formation
-
•
KglA is effective against multidrug-resistant M. tuberculosis
Resistance to rifamycins, inhibitors of bacterial RNA polymerase used for treatment of tuberculosis, is increasing. Mosaei et al. report an analog of the rifamycins, kanglemycin A, that inhibits rifampicin-resistant RNA polymerases and is effective against multidrug-resistant M. tuberculosis, and they describe its mechanism of action.
Introduction
Antibiotic resistance is on the increase, exacerbated by the fact that, in recent years, only a few new compounds have reached the clinic. RNA polymerase (RNAP) is an essential enzyme accomplishing transcription in all living organisms. At the same time, the sequence divergence between bacterial and human RNAPs makes bacterial RNAP an excellent target for antibiotic development (Ho et al., 2009). In the last few years, details of the mode of action of a number of inhibitors of RNAP have been described (Bae et al., 2015, Degen et al., 2014, Maffioli et al., 2017, Molodtsov et al., 2015, Mukhopadhyay et al., 2008, Zhang et al., 2014a). However, most of these molecules have limited clinical utility, mainly because of their poor penetration of the bacterial cell envelope or strong binding to human plasma protein. Nevertheless, the search for new molecules that target RNAP and analysis of their mode of action is a promising approach for the development of new antibiotics, as exemplified by the now clinically used fidaxomicin (trade name Dificid) for Clostridium difficile-associated diarrhea (Tupin et al., 2010).
The most studied class of antibiotics known to inhibit bacterial RNA transcription are the rifamycins, a family of mainly semisynthetic ansamycins derived from the natural product rifamycin S, of which rifampicin (RIF) is the most widely used (Aristoff et al., 2010). RIF (and the other rifamycins) binds in a pocket (the RIF pocket) on the β subunit of RNAP in the vicinity of the catalytic site and sterically blocks propagation of the growing RNA chain through the main channel of RNAP (Campbell et al., 2001, McClure and Cech, 1978). The collision of RNA and RIF happens when the RNA reaches a length of 3–4 nucleotides, after which the RNA is released from the promoter complex as an abortive transcript. The length of the abortive RNA product can be influenced by the nature of the C3 substituent of the individual semisynthetic rifamycin. For example, rifabutin even inhibits synthesis of a trinucleotide (when reaction is started with a 5′-non-phosphorylated dinucleotide primer), whereas RIF still allows it (Artsimovitch et al., 2005).
The rifamycins are used, as part of the current combination therapy, for the first-line treatment of tuberculosis (TB), the leading cause of death by infectious disease worldwide (World Health Organization, 2017), as well as treatment for non-tuberculous mycobacterial infections (Ryu et al., 2016). One of the major problems with known rifamycins is the rapid selection for mutations in the RIF-binding pocket of RNAP that lead to resistance. Multidrug-resistant strains of Mycobacterium tuberculosis (MDR-M. tuberculosis) present a major clinical problem because these strains are resistant to RIF in addition to isoniazid. In 2016, there were 600,000 new cases with resistance to RIF, of which 490,000 had MDR-TB. With a treatment success rate of only 54%, this highlights the requirement for improved treatment options. There are ∼100 different amino acid substitutions in the RIF pocket that have been found in RIF-resistant clinical isolates of M. tuberculosis (Sandgren et al., 2009), with mutations at three positions of the RIF pocket that account for ∼70% of the total substitutions (D516, 7.4%; H526, 20%; S531, 42%; E. coli numbering).
Previous efforts to probe the structure-activity relationships of the rifamycins and, thus, to maximize their binding affinity for RNAP have shown that the minimum structural requirements essential for activity include two free hydroxyl groups at the C21 and C23 positions of the ansa chain and two polar (free hydroxyl or carbonyl) groups at the C1 and C8 positions of the napthoquinone ring. Furthermore the molecule must be able to adopt a specific conformational geometry to position these key functional groups in the correct spatial arrangement required for RNAP binding (Bacchi et al., 1998, Lancini and Zanichelli, 1977). Thus, modification of the ansa bridge (e.g., hydrogenation or epoxidation of the C16,17 or C18,19 carbon-carbon double bonds) results in decreased binding to RNAP because of conformational changes in the molecule altering the spatial arrangement of the C1, C8, C21, and C23 functional groups, thus disrupting their critical hydrogen bonds with the RIF pocket (Aristoff et al., 2010, Bacchi et al., 2008). Hence, the production of clinically used semisynthetic rifamycins (RIF, rifabutin, rifapentine, rifalazil, and rifaximin) has focused on the introduction of functionality at the C3 position to stabilize the oxidation state of the naphthoquinone core and to improve bioavailability (Floss and Yu, 2005, Perlman, 1977).
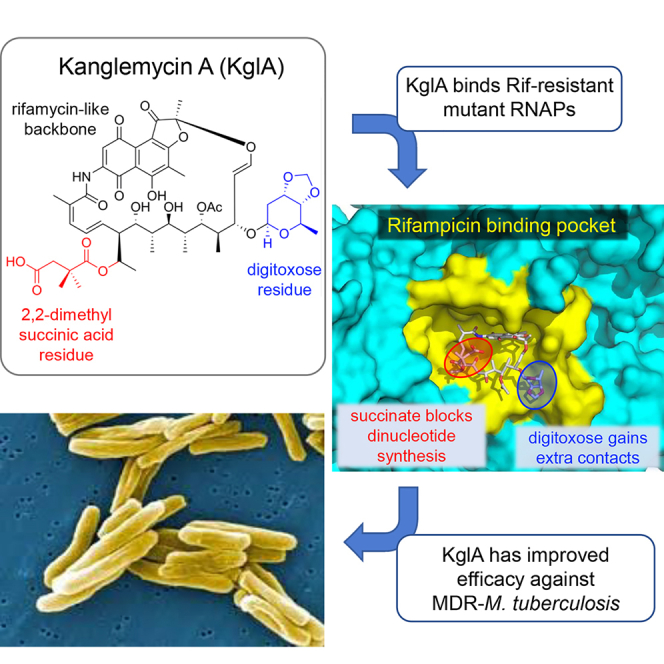
Here we isolated and, by using microbiological, biochemical, and structural biology techniques, studied an uncharacterized naturally produced antibiotic, kanglemycin A (KglA). KglA is an ansamycin antibiotic from the rifamycin family that displays two important and unusual ansa bridge modifications. We show that, despite having a mode of action similar to that of RIF, KglA has a larger binding surface with the RNAP provided by interactions of the ansa-bridge substituents and areas of RNAP that have not been previously accessible by rifamycin derivatives. As a result, KglA has an altered binding conformation compared with RIF, which makes it maintain higher activity against both RIF-resistant RNAPs and RIF-resistant bacteria.
Results
Isolation of KglA
Actinomycete bacteria have historically been an excellent source of bioactive compounds, particularly antibiotics. To identify new antibiotics targeting transcription, we screened a subset of strains from the Demuris collection of actinomycetes for their ability to inhibit cell growth and to inhibit cellular transcription. As a reporter strain, we used a previously described B. subtilis strain that has a lacZ gene fused to the promoter of the bacterial helicase helD. This promoter was selected as the one that is upregulated during the stress caused by partial inhibition of transcription in sub-inhibitory concentrations of an antibiotic (Urban et al., 2007a). In the disk assay shown in Figure 1A, when a paper disk soaked with antibiotic is placed on the X-gal-infused agar plate with a lawn of growing cells, this results in a colorimetric response at the edge of the zone of growth inhibition, observed as a blue halo (Figure 1A, left; compare the halos produced by RIF and carbenicillin, an inhibitor of cell wall synthesis). The isolate Amycolatopsis DEM30355 was of particular interest because it produced a compound (later identified as KglA, see below) that gave a blue halo at the edge of the zone of bacterial growth inhibition, indicative of transcription inhibition (Figure 1A, left). Importantly, in contrast to RIF, this new compound inhibited the RIF-resistant B. subtilis strain (βH482R; βH526R in E. coli numbering) (Figure 1A, right). We cultivated Amycolatopsis DEM30355 in a stirred tank bioreactor for 103.5 hr and extracted the secondary metabolites from the supernatant using Amberlite XAD-16 beads. Subsequent elution with methanol, extraction with ethyl acetate, and purification by multistep chromatography (STAR Methods) produced an orange crystalline compound that retained the observed biological activity against both the reporter and RIF-resistant strains (Figure 1A). Electrospray ionization-mass spectrometry (ESI-MS) revealed the mass, [M+Na]+ = 1004.3866 m/z (Figure S1A), which corresponded to the mass of the previously described KglA. 1H and 13C nuclear magnetic resonance (NMR), extensive 2D NMR analysis, and, finally, single-crystal X-ray analysis (Figures S1B–S1D; Table S1) unambiguously confirmed the structure of the isolated compound to be KglA (Figure 1B).
Figure 1.

Identification and Mode of Action of KglA
(A) Disk diffusion assay with rifampicin (RIF), carbenicillin (Cb), and kanglemycin A (KglA). Paper disks soaked with antibiotics were placed on the lawns of wild-type B. subtilis carrying the lacZ gene under the PhelD promoter, inducible during partial inhibition of transcription (left), and RIF-resistant B. subtilis (right). Note the blue halo around the growth inhibition zones in the case of transcription inhibitors.
(B) Chemical structure of KglA in comparison with RIF. The ansa chain modifications in KglA are highlighted. The semisynthetic group of RIF is shown in purple. See also Figure S1 and Table S1.
(C) Transcription in vitro by E. coli RNAP started with CpA on a linear template containing the T7A1 promoter. Runoff, termination, and abortive tri- and tetra-nucleotides are marked. Note that tetra-nucleotides migrate faster than tri-nucleotides under these electrophoretic conditions.
(D) Different ratio of the di-nucleotide-long (pppApU) and the tri-nucleotide-long (pppApUpC) abortive products in the presence of RIF and KglA. Transcription was performed in the presence of the nucleotides depicted (in the absence of the CpA primer). Note that, in the 33% gel, runoff and termination products remain in the well.
(E) The experiment was performed as in (D) but in the presence of AMP instead of ATP.
(F and G) Inhibition of wild-type and RIF-resistant E. coli RNAPs by RIF and KglA. Error bars (F) are ± SD. The brackets (G) contain ± SE and the Hill slope.
KglA is an antibiotic originally isolated from the fermentation broth of Nocardia mediterranei var. kanglensis 1741-64 (Wang et al., 1988), for which no mode of action has been previously proposed, and only limited information regarding its biological activity has been reported. It is an ansamycin antibiotic that shares similarities with rifamycins, albeit with a number of unusual structural features. Most notably, KglA differs from known rifamycins and their semisynthetic derivatives in that it contains two large ansa bridge substituents, a pendant 2,2-dimethyl succinic acid side chain at C20 and a unique sugar moiety (β-O-3,4-O,O’-methylene digitoxose) at C27 (Figure 1B). Furthermore, these modifications are unusual for bioactive natural and semisynthetic rifamycins, whose modifications are typically concentrated at the naphthoquinone core.
Mode of Action and Specificity of KglA
Activation of the helD promoter and its structural similarity to the rifamycins suggested that KglA targets bacterial RNAP. Therefore, we analyzed the effects of KglA on in vitro transcription by wild-type E. coli RNAP. KglA efficiently inhibited transcription on a linear DNA template containing the strong T7A1 promoter (Figure 1C). Inhibition of full-sized RNA products (run off or terminated) coincided with increased accumulation of short abortive tri- and tetra-nucleotide products (Figure 1C; the reactions were started with the dinucleotide primer CpA). Such a mode of action is characteristic of the rifamycins (Campbell et al., 2005, McClure and Cech, 1978), as can be seen from the pattern of inhibition of transcription by RIF (Figure 1C). In this mode of action, the antibiotic binds in the pathway of the nascent RNA, physically blocking its extension and inducing the release of short RNAs as abortive products.
Dinucleotide primer CpA that was used to start transcription does not carry a negatively charged triphosphate moiety at the 5′ end, which is present in NTPs used by RNAP in vivo. Therefore, we analyzed the effects of RIF and KglA in a native situation when transcription was started with ATP (in the absence of the CpA primer). As can be seen in Figure 1D, KglA completely blocked the formation of the triphosphorylated trinucleotide, pppApUpC, whereas RIF allowed some of its production (compare lanes 4, 5, and 6). Furthermore, KglA strongly diminished (70-fold inhibition) the formation of the first dinucleotide, pppApU, whereas RIF had only a moderate effect (3-fold inhibition) (compare lanes 7, 8, and 9). Next, to start the reaction, we used AMP, which lacks γ and β 5′ phosphates, present in ATP (Figure 1E). KglA only moderately inhibited formation of pApU (4-fold), suggesting that it is γ and, possibly, β phosphates at the 5′ end of the initiating nucleotide that clash with the additional groups of KglA (compare lanes 7, 8, and 9). However, the formation of trinucleotide pApUpC was still strongly inhibited by KglA compared with RIF (Figure 1E, lanes 4, 5, and 6). These results further suggest that extra groups and/or the mode of binding of KglA provide an additional steric and/or electrostatic (KglA has a negatively charged C20 2,2-dimethyl succinic acid side chain) hindrance for translocation of the 5′ phosphorylated dinucleotide RNA. Furthermore, KglA interference with the 5′ triphosphate moiety of the initiating nucleotide leads to inhibition even of the first phosphodiester bond formation (see structural support for these findings below).
The half maximal inhibitory concentration (IC50) values of KglA and RIF were similar against wild-type RNAP (Figures 1G and 1F). However, despite having a similar mode of action as RIF, we observed that KglA killed RIF-resistant bacteria (compare the inhibition zones in Figure 1A, right). To investigate this phenomenon, we prepared E. coli RNAPs carrying a series of RIF-resistant mutations, including βS531L, βD516V, βD516Y, βH526Y, βH526P, βH526R, and βI572F. These mutations correspond to the RIF resistance mutations in M. tuberculosis most frequently observed in clinical isolates, with βS531L, βD516V, and βH526Y accounting for 73% of all clinical RIF-resistant M. tuberculosis isolates (Gill and Garcia, 2011). Compared with RIF, KglA was much more potent against six of seven RIF-resistant RNAPs (Figures 1G and 1F); in particular, KglA was 10 times more active than RIF against the βS531L and βD516V mutants. Note that, although mutations at βH526 made RNAP insensitive to RIF (no inhibition at 2.5 mg/mL), KglA was still able to inhibit RNAPs with these mutations.
These results and in vivo data (Figure 1A) suggest that KglA binds to RNAP in the RIF-binding pocket of the β subunit but in a manner different from known rifamycins, resulting in an increased resilience to the RIF-resistant amino acid substitutions in the RIF-binding pocket of RNAP. Furthermore, the mode of action of KglA deviates from that of RIF because KglA more efficiently inhibits the early stages of RNA synthesis.
Structural Basis of RNAP Inhibition by KglA
To reveal the basis of the KglA efficacy enhancement as induced by the modifications of the ansa bridge in comparison with rifamycin, we performed a crystallographic analysis of the complexes of KglA with RNAPs. To analyze the RNAP and KglA interaction on the atomic scale, a crystal of the T. thermophilus RNAP σA holoenzyme-pyrG promoter DNA complex (Murakami et al., 2017) was used to determine the X-ray crystal structure of the RNAP-KglA complex at 3.0-Å resolution (Table 1). Additionally, to understand the mechanism of inhibition of RIF-resistant RNAP by KglA, the crystal of the E. coli βS531L RNAP σ70 holoenzyme (Molodtsov et al., 2017) was used to determine the structure of the RNAP-KglA complex at 4.1-Å resolution (Table 1). The electron density maps of both RNAPs show unambiguous densities for the compound located in the RIF-binding pockets in the β subunits (Figures 2A and 2B). The binding modes of KglA to the RNAPs are almost identical; therefore, the atomic resolution structure of T. thermophilus RNAP-KglA was used for the precise modeling of KglA in complex with E. coli RNAP.
Table 1.
Collection of Crystallographic Data and Refinement Statistics
| Complex | Eco RNAP – KglA | Tth RNAP – KglA |
|---|---|---|
| PDB code | 6CUX | 6CUU |
| Data Collection | ||
| Space group | P212121 | C2 |
| Cell dimensions | ||
| a (Å) | 188.167 | 185.221 |
| b (Å) | 204.685 | 101.315 |
| c (Å) | 311.577 | 294.279 |
| β (°) | 90 | 98.886 |
| Resolution (Å) | 50.0–4.1 | 50.0–3.0 |
| Total reflections | 608,898 | 358,319 |
| Unique reflections | 93,684 | 107,473 |
| Redundancy | 6.5 (6.2) | 3.3 (3.0) |
| Completeness (%)a | 99.9 (100.0) | 99.7 (97.1) |
| I / σa | 16.2 (1.95) | 10.2 (1.14) |
| Rsym (%)a | 7.7 (85.1) | 9.6 (61.4) |
| CC1/2a | (0.633) | (0.629) |
| Refinement | ||
| Resolution (Å) | 45.03–4.1 | 46.3–3.0 |
| Rwork | 0.2100 | 0.2012 |
| Rfree | 0.2547 | 0.2483 |
| No. of atoms | 54,867 | 28,254 |
| RMSDs | ||
| Bond length (Å) | 0.013 | 0.006 |
| Bond angles (°) | 1.776 | 1.089 |
| Clashscore | 11.47 | 5.84 |
| Ramachandran favored, % | 91.57 | 97.64 |
| Ramachandran outliers, % | 0.94 | 0.35 |
RMSD, root-mean-square deviation.
Highest-resolution shells are shown in parentheses.
Figure 2.

KglA Binding to Free and Promoter-Bound RNAP Holoenzyme
(A) Overall views of E. coli (left) and T. thermophilus (right) RNAPs with KglA bound in the RIF-binding pocket. RNAPs (gray core and orange σ) and DNA (blue) are shown as ribbon models, and KglA (red) is shown as a stick model.
(B) Electron density maps of KglA in complexes with T. thermophilus (top) and E. coli (bottom) RNAPs. Electron densities are shown as a light blue mesh, and KglA molecules are shown as stick models colored by element. The ansa bridge side chains of KglA extending from C20 and C27 are colored in yellow.
(C) A close-up view of the RIF-binding pocket of the T. thermophilus RNAP containing KglA (in red, with the side chains in yellow). RNAP is shown as a transparent surface model (light blue), KglA is shown as a stick model with the ansa bridge side chains labeled, and the β subunit residues forming the RIF-binding pocket are shown as stick models. The hydrogen bonds between KglA and β amino acid residues (colored by element) are depicted by yellow dotted lines; the βR134 involved in polar interaction with the C20 sugar of KglA is shown in blue.
(D) A side view of the RIF-binding pocket shown in (C). KglA is overlaid on RIF (gray); the side chains of the two molecules are labeled. KglA maintains a larger distance to the wall in the RIF-binding pocket than RIF.
(E) Modeling of the initiating nucleotides in the active site of the T. thermophilus RNAP (from PDB: 4Q4Z) in the presence of KglA (left) and RIF (right). The ribonucleotides (red), the βQ567 and βH999 residues coordinating the 5′ end triphosphate (blue), and KglA and RIF (gray) are shown as stick and sphere models. The side chains of the inhibitors and the positions of the initiating NTPs are indicated. Red arrows indicate the translocation directions for the corresponding substrates.
(F) Structural rearrangements in the RIF-binding pocket of E. coli βS531L RNAP triggered by KglA and RIF binding. RNAP is shown as a ribbon model in gray, the structured regions of the fork loop2 are shown in green, and the β531L residue (blue), KglA (colored by element with yellow side chains), and RIF (colored by element) are shown as stick and sphere models. The side chains of KglA and RIF are indicated. Arrows mark the clash points between the naphthalene cores of the corresponding compounds with the β531L side chain.
See also Figure S2.
The binding mode of KglA is similar to that of RIF and involves the formation of hydrogen bonds with the same amino acid residues of the RIF-binding pocket, including βQ393, βS411, and βR420 (side chain) and βF394 (main chain) (Figures 2C and S2A; the numbering is for T. thermophilus; see Figures S2C and S2D for the corresponding amino acid residues in E. coli and M. tuberculosis RNAPs). Because of a different topology of the ansa bridge of KglA (Figure S2B), the C23 hydroxyl group does not form an additional hydrogen bond with βF394, as observed in the T. thermophilus RNAP-RIF complex. However, the C27 β-O-3,4-O,O’-methylene digitoxose side chain establishes a hydrogen bond with the side chain of βR134 (Figure 2C). This KglA ansa bridge side chain inserts into a shallow pocket in the RIF-binding site and establishes additional non-polar interactions with the surrounding residues, significantly increasing the binding surface of KglA compared with RIF (Figure 2C). One of the structural consequences of the sugar side chain accommodation in the RIF-binding pocket is a rotational shift of KglA that increases the distance between the naphthalene core and the binding surface of the pocket compared with RIF (Figure 2D).
To validate the new interaction of the C27 β-O-3,4-O,O’-methylene digitoxose side chain with βR134, we constructed an E. coli RNAP carrying a mutation at the corresponding amino acid residue, βR143A. The mutation had little effect on RIF sensitivity (Figure S2E, left). However, KglA was at least an order of magnitude less effective against βR143A RNAP than against wild-type RNAP, indicating the importance of this interaction for KglA binding (Figure S2E, center). The IC50 of KglA for βR143A RNAP was still lower than for any RIF-resistant mutant RNAP, indicating that interactions of KglA within the rest of the RIF-binding pocket were still critical for its binding. Only a double mutation, βR143A/S531L, led to KglA resistance (Figure S2E, right).
The presence of additional bulky side chains on the ansa bridge of KglA has an effect on the mechanism of de novo transcription initiation. Ansamycin binding in the RIF-binding pocket inhibits transcription by clashing with the 5′ end of a nascent RNA. Modeling shows that, in the T. thermophilus RNAP-RIF complex, the triphosphate moiety of the initiating nucleoside triphosphate (NTP) guided by hydrogen bonds with side chains of two essential residues, βQ567 and βH999 (Basu et al., 2014), moderately clashes with the C3 side chain of RIF (Figure 2E, right). This clash does not strongly affect the formation of the first phosphodiester bond, likely because of flexibility of the C3 side chain. Even in the presence of RIF, there is an empty space (triphosphate retreat zone) that allows for accommodation of the 5′ triphosphate group followed by an extension of the nascent RNA to 3-mer (McClure and Cech, 1978) before a severe clash with the +1 NTP inhibits further primer extension (Figure 2E, right). In contrast, the positional shift of KglA and the orientation of the negatively charged C20 2,2-dimethyl succinic acid side chain not only do not leave space for accommodation of the 5′ triphosphate group but also interfere with the correct placement of the initiating NTPs (Figure 2E, left), which explains the strong inhibition of the initial dinucleotide synthesis (Figure 1D). The electrostatic repulsion by the C20 2,2-dimethyl succinic acid side chain of KglA can also explain the inhibition of monophosphorylated pApUpC synthesis (which is allowed by RIF; Figure 1E).
A comparative analysis of the structures of the E. coli βS531L RNAP σ70 holoenzyme in apo form and in complex with RIF and KglA reveals the mechanism of KglA’s efficiency against many RIF-resistant forms of mutant RNAPs (Figure 2F). The amino acid substitution βS531L is the most clinically important RIF-resistant mutation in bacterial RNAP because of its low structural and functional effects on the enzyme in the absence of RIF. As shown in a recent study (Molodtsov et al., 2017), RIF binding to the mutant RNAP results in a steric clash of the naphthalene core with the side chain of β531L, which triggers disordering of a constituent part of the RIF-binding pocket, the fork loop2 (Figure 2F, left versus right). This partial disordering of the RIF-binding site efficiently prevents RIF binding. The naphthalene core of KglA is almost identical to RIF; however, it is rotated in the binding site compared with RIF (Figure 2D). This positional shift allows KglA to maintain a distance to β531L, avoiding a severe clash with the residue and minimizing structural rearrangements of the fork loop2 and the binding pocket (Figure 2F, center). This fine-tuning of the core binding mode and the increased interaction surface provided by the C27 sugar side chain enable high affinity of KglA in a broad context of RIF resistance.
Kanglemycin A Has Antimicrobial Activity against MDR-M. tuberculosis and Other Gram-Positive Pathogens
The above results suggested that KglA may be effective against RIF-resistant bacteria, in particular against clinically problematic MDR-M. tuberculosis strains. Thus, we tested KglA against a clinical MDR-M. tuberculosis isolate (Beijing 08/00483E) that is fully resistant to RIF (Jamieson et al., 2014, Zhang et al., 2014b) and the other first-line antibiotics (isoniazid, ethambutol, and pyrazinamide). Beijing 08/00483E carries the most frequent RNAP amino acid change found in RIF-resistant M. tuberculosis (βS450L, M. tuberculosis numbering; βS531L, E. coli numbering; Figures S2C and S2D). As positive controls, we used the RIF-susceptible reference strain of M. tuberculosis H37Rv and a RIF-susceptible clinical isolate, Beijing 1192/015. The clinical strain Beijing 08/00483E was resistant to RIF at 1,024 times the minimum inhibitory concentration (MIC) for RIF-sensitive strains (Figures 3A and 3B). In contrast, all three strains displayed a dose-dependent response to KglA exposure (Figure 3A), demonstrating its antimicrobial activity against MDR-M. tuberculosis. KglA was statistically significantly more active against RIF-sensitive Beijing 1192/015 than RIF-resistant Beijing 08/00483E (Figures 3A and 3B).
Figure 3.

KglA Inhibits Mycobacterial RNAP and Retains Antimicrobial Activity against MDR-M. tuberculosis
(A) Dose-dependent growth inhibition of the RIF-susceptible M. tuberculosis strains H36Rv (black) and 1192/015 (red) and RIF-resistant M. tuberculosis 08/00483E (blue) by KglA (solid line, solid symbols) and RIF (dashed line, open symbols). Data presented are the mean of four independent experiments ± SD and analyzed using a Student’s t test.
(B) MIC values ± SD, as determined using the modified Gompertz function for M. tuberculosis strains H37Rv, 1192/015, and 08/00483E, which were exposed to RIF or KglA.
(C) Mode of action of KglA and RIF against M. smegmatis RNAP. The experiment was performed exactly as in Figure 1D. Note that the high radioactive background compared with Figure 1D is due to much lower activity of M. smegmatis RNAP.
(D) Inhibition of M. smegmatis RNAP by RIF and KglA.
Error bars are ± SD. Shown in brackets are the IC50 values ± SE.
As seen in Figures 3A and 3B, RIF was more active against RIF-susceptible M. tuberculosis cells than KglA. This apparently contradicted our in vitro results of very similar inhibition of E. coli wild-type RNAP by both inhibitors. It is possible that the mode of inhibition of M. tuberculosis RNAP by KglA is different from that of E. coli RNAP. Therefore, we analyzed inhibition of transcription by Mycobacterium smegmatis RNAP, which is commonly used as an experimental model for M. tuberculosis RNAP. Both RNAPs have an identical amino acid composition of the RIF-binding pocket (Figure S2C). We found that RIF and KglA inhibited RNA synthesis by M. smegmatis RNAP and E. coli RNAP in a very similar way (compare Figure 1D with Figure 3C); i.e., KglA fully blocked synthesis of the first dinucleotide (pppApU), whereas RIF partly allowed synthesis even of the trinucleotide (pppApUpC). The IC50 of KglA was nearly the same as that of RIF. Therefore, the affinity of RNAP binding cannot account for the observed difference in the activities of RIF and KglA against RIF-sensitive M. tuberculosis cells. The likely explanation of differences in activities of KglA and RIF against RIF-sensitive M. tuberculosis is the decreased ability of KglA to pass through the cell envelope.
We further tested KglA against a range of other species, including a number of clinically relevant pathogens. These included 8 Gram-positive bacteria, 10 Gram-negative bacteria, and 2 eukaryotes (Table 2). Of 10 Gram-negative pathogens, KglA was active against only Providencia rettgeri. No activity was observed toward the two strains of eukaryotic microorganisms (yeast): Candida albicans and Candida glabrata. Among the 8 Gram-positive strains, KglA was highly active (MIC, ∼1 μg/mL) against staphylococci and Streptococcus pyogenes but less active against enterococci (MIC, 64–128 μg/mL). The relatively high activity of KglA against staphylococci prompted evaluation of its activity against a large collection of methicillin-resistant Staphylococcus aureus (MRSA) and a comparison of its activity with RIF. S. aureus NCTC 6571 was re-tested as a control strain with a known MIC of RIF (EUCAST, 2018). Thirty-seven MRSA were tested in total, and 33 of these proved to be susceptible to RIF, as defined by a MIC of ≤ 0.06 μg/mL (EUCAST, 2018). KglA was substantially less active than RIF against RIF-susceptible MRSA, with MICs of KglA between 0.25 and 1 μg/mL. Conversely, for 3 of 4 RIF-resistant MRSA, MICs of KglA were 1–3 double dilutions lower than those of RIF, although still relatively high (32–128 μg/mL). In the light of our in vitro data (Figures 1C–1F and 3C), no activity against most of Gram-negative bacteria, and lower-than-RIF activity against some Gram-positive bacteria support the idea of a diminished ability of KglA to enter bacterial cells. This, however, requires further investigation coupled with further in vivo analyses to develop KglA as a future antibiotic.
Table 2.
MICs of KglA against Bacterial and Eukaryotic Pathogens
| Organism/Isolate | Strain Reference | KglA (μg/mL) | RIF (μg/mL) | |
|---|---|---|---|---|
| Gram-Negative | Acinetobacter baumannii | ATCC 19606 | >128 | – |
| Burkholderia cepacia | ATCC 25416 | >128 | – | |
| Enterobacter cloacae | NCTC 11936 | >128 | – | |
| Escherichia coli | NCTC 10418 | >128 | 16a | |
| Klebsiella pneumoniae | NCTC 9528 | >128 | – | |
| Providencia rettgeri | NCTC 7475 | 64 | – | |
| Pseudomonas aeruginosa | NCTC 10662 | >128 | – | |
| Salmonella enterica Typhimurium | NCTC 74 | >128 | – | |
| Serratia marcescens | NCTC 10211 | >128 | – | |
| Yersinia enterocolitica | NCTC 11176 | >128 | – | |
| Gram-Positive | Bacillus subtilis | NCTC 9372 | 1 | – |
| Enterococcus faecalis | NCTC 775 | 64 | – | |
| Enterococcus faecium | NCTC 7171 | 128 | – | |
| Listeria monocytogenes | NCTC 11994 | 8 | – | |
| Staphylococcus epidermidis | NCTC 11047 | 1 | – | |
| Staphylococcus aureus | NCTC 6571 | 0.25 | 0.004a | |
| Staphylococcus aureus (MRSA) | NCTC 11939 | 1 | – | |
| Streptococcus pyogenes | NCTC 8306 | 0.031 | – | |
| Yeast | Candida albicans | ATCC 90028 | >128 | – |
| Candida glabrata | NCPF 9725 | >128 | – | |
| S. aureus | Bel | 97597 | 2 | 2 |
| Bel | 97598 | 64 | >128 | |
| Fin | 37481 | 0.5 | ≤0.008 | |
| Fin | 54511 | 32 | >128 | |
| Fin | 54518 | 128 | >128 | |
| Fra | 462 | 0.5 | ≤0.008 | |
| Fra | 920 | 1 | ≤0.008 | |
| Fra | 95035 | 1 | 0.016 | |
| Ger | 131/98 (Sger 11d2) | 0.5 | 0.016 | |
| Ger | 1966/97 (Han 111c) | 0.5 | ≤0.008 | |
| Ger | 2594-1/97 (Sger 11a) | 1 | 0.016 | |
| Ger | 2594-2/97 (Sger 11b) | 0.25 | ≤0.008 | |
| EMRSA 15 | 1729/98 | 0.5 | ≤0.008 | |
| EMRSA 15 | 1758/98 | 0.5 | ≤0.008 | |
| EMRSA 15 | 14956 | 0.5 | ≤0.008 | |
| EMRSA 15 | 12484/98 | 0.5 | ≤0.008 | |
| EMRSA 15 | 14185/98 | 0.5 | ≤0.008 | |
| EMRSA 15 | 16822/98 | 0.5 | ≤0.008 | |
| EMRSA 15 | 19972/98 | 0.5 | ≤0.008 | |
| EMRSA 15 | 20460/98 | 0.5 | 0.016 | |
| EMRSA 15 | 21268/98 | 0.5 | ≤0.008 | |
| EMRSA 15 | 21698/98 | 0.5 | ≤0.008 | |
| EMRSA 15 | 2501/98 | 0.5 | ≤0.008 | |
| EMRSA 15 | 6323/98 | 0.5 | ≤0.008 | |
| EMRSA 16 | 00036/95 | 1 | 0.016 | |
| EMRSA 16 | 00998/95 | 1 | ≤0.008 | |
| EMRSA 16 | 03732/95 | 1 | 0.016 | |
| EMRSA 16 | 07121/95 | 1 | ≤0.008 | |
| EMRSA 16 | 07924/95 | 0.5 | ≤0.008 | |
| EMRSA 16 | 18200/95 | 0.5 | ≤0.008 | |
| EMRSA 16 | 18205/95 | 0.5 | ≤0.008 | |
| EMRSA 16 | 21354/95 | 1 | 0.016 | |
| EMRSA 16 | 23728/98 | 1 | ≤0.008 | |
| EMRSA 16 | 23729/98 | 1 | ≤0.008 | |
| EMRSA 16 | 32010/96 | 1 | 0.016 | |
| EMRSA 16 | 34256/96 | 1 | ≤0.008 | |
| MRSA | NCTC 11939 | 1 | ≤0.008 | |
| Staphylococcus aureus | NCTC 6571 | 0.25 | ≤0.008 |
Discussion
Easily acquired RIF resistance is a major problem in the clinical usage of rifamycins. The principal finding of our study is that KglA is much more effective against RIF-resistant RNAPs and bacteria than its commonly used relative, RIF. The structural analysis suggests that this is explained by both additional interactions and the binding mode of KglA with RNAP unique to KglA. The sugar (β-O-3,4-O,O’-methylene digitoxose) and acid (2,2-dimethyl succinic acid) moieties at C27 and C20 of KglA, respectively, establish these additional contacts and influence the overall binding mode of KglA in the pocket (Figures 2C and 2D). Compared with RIF, KglA rotates to reposition slightly away (by 1.2 Å) from the wall of the binding pocket, which may decrease the effect of resistance mutations arising in RNAP on its affinity. Thus, the increased distance between the naphthalene core along with the increased binding surface provided by the ansa-bridge side chains likely represent the mechanisms that enable KglA to maintain its inhibitory activity against many RIF-resistant bacterial RNAPs. As the result of additional interactions by the sugar moiety with the protein, only an unlikely event of two simultaneous mutations in the RIF-binding pocket can lead to KglA resistance.
KglA inhibits RNA synthesis at the stage of the first phosphodiester bond (2-mer RNA) formation, which is different from RIF, which inhibits formation of the second phosphodiester bond (3-mer RNA) (Figures 1D and 2E). Our biochemical and structural data suggest that this may happen because of the steric and/or electrostatic clash of the C20 2,2-dimethyl succinic acid side chain of KglA with γ and/or β phosphates of the +1 NTP that may disturb the conformation of initiating nucleotides and, thus, catalysis. We cannot directly compare these results with the previous study that found that some rifamycins may inhibit RNA synthesis one step earlier than RIF (Artsimovitch et al., 2005) because they used a 5′ non-phosphorylated dinucleotide to start transcription. We, however, did not observe inhibition of 5′ non-phosphorylated trinucleotide synthesis by KglA (Figure 1C), as observed for some of the rifamycins in that study (Artsimovitch et al., 2005), suggesting that inhibition of 5′ triphosphorylated dinucleotide synthesis by KglA takes place via a different mechanism.
KglA, compared with RIF, appears to show reduced penetration through the bacterial cell envelope, likely because of the presence of the polar carboxyl group at C20. For example, KglA has minimal activity against Gram-negative bacteria in comparison with RIF and displays higher MICs than RIF against most of the Gram-positive species tested. However, KglA is more effective than RIF against RIF-resistant Gram-positive bacteria. Notably, it is effective against MDR-M. tuberculosis. KglA inhibited the growth of MDR-M. tuberculosis (strain 08/00483E at a MIC of 0.854 μg/mL [850.3 nM]), which was completely resistant to RIF because of a βS450L substitution (βS531L in E. coli numbering). Finding new antibiotics for the treatment of MDR-TB is a challenge, and KglA demonstrates potential as a candidate for further development, particularly in light of the fact that the MIC of KglA is lower than the MIC values displayed for both isoniazid and pyrazinamide against drug-susceptible M. tuberculosis, and there is a wealth of information about drug toxicity and patient responses to RIF. These considerations are likely to accelerate the progression of KglA or its derivatives to the clinic for the treatment of both drug-susceptible and drug-resistant TB.
A new mode of KglA binding to RNAP may enrich the strategy of structure-guided antibiotic design against RIF-resistant pathogens such as MDR-M. tuberculosis. RIF resistance caused by a point mutation of the β subunit of RNAP is associated with either loss of important contact between a target and a compound (in the case of βS531L) or steric exclusion of a compound from its binding site (in the case of βH526Y) (Molodtsov et al., 2017). In many cases, research efforts focusing on developing new antibiotics targeting the RIF-resistant bacterial RNAP are searching for new interaction surfaces away from the RIF-binding pocket (Lin et al., 2017, Maffioli et al., 2017). The enhanced affinity of KglA for certain RIF-resistant RNAPs along with the moderate changes in its mode of binding compared with RIF (Figure 2D) indicate that the RIF-binding pocket still possesses significant potential for the development of potent antibiotics against certain RIF-resistant pathogens. Targeting the RIF-binding pocket has some advantages compared with targeting a new site. For example, development of resistance to KglA is possible via additional mutations in the RIF-binding pocket (e.g., βR143A; Figure S2E) in addition to the first RIF-resistant mutation (e.g., βS531L); however, such mutations may compromise the transcriptional integrity and further diminish the fitness of the RNAP, rendering the resulting pathogens non-viable (Song et al., 2014). Another important advantage of developing new rifamycin derivatives is that this approach exploits the well-known rifamycin scaffold and may likely overcome some obstacles common in the development of new antibiotics, such as the problems of drug permeability through bacterial membranes, human toxicity, and specificity (Aristoff et al., 2010).
The modifications in the ansa bridge are highly unusual in bioactive rifamycins, with even minor semisynthetic modifications in an ansa bridge often leading to loss of RNAP inhibition (see Introduction). Our study reveals the significant potential of increasing the affinity of the rifamycins to RNAP via ansa bridge modifications, in particular by demonstrating that C27 substitution can allow access to a previously unexplored hydrophobic pocket adjacent to the RIF-binding pocket while maintaining the critical interactions between the rifamycin backbone and the RIF-binding pocket of RNAP. Furthermore, substitution at C20 with a flexible carboxylic acid residue has revealed the potential for new drug-initiating NTP interactions in the rifamycins. Altogether, KglA provides an example of an evolved solution to RIF resistance in which these unusual ansa bridge modifications lead to improved potency against RIF-resistant RNAPs and represents an important starting point for the development of new therapeutic agents.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli BL21(DE3) | Novagen | Cat# 69450 |
| M. smegmatis mc2155 (SM07) | Mukherjee and Chatterji, 2008 | N/A |
| Mycobacterium tuberculosis H37Rv, Beijing 1192/015 and Beijing 08/00483E | PHE National Mycobacterial Reference Laboratory | N/A |
| E. coli T7 Express | New England BioLabs | Cat# C2566H |
| B. subtilis 168 amyE::PhelD-lacZ cat | Urban et al., 2007a | N/A |
| B. subtilis 168 amyE::PhelD-lacZ cat rpoBH482R | This paper | N/A |
| Enterobacter cloacae | NCTC | NCTC 11936 |
| Escherichia coli | NCTC | NCTC 10418 |
| Klebsiella pneumoniae | NCTC | NCTC 9528 |
| Providencia rettgeri | NCTC | NCTC 7475 |
| Pseudomonas aeruginosa | NCTC | NCTC 10662 |
| Salmonella typhimurium | NCTC | NCTC 74 |
| Serratia marcescens | NCTC | NCTC 10211 |
| Yersinia enterocolitica | NCTC | NCTC 11176 |
| Bacillus subtilis | NCTC | NCTC 9372 |
| Enterococcus faecalis | NCTC | NCTC 775 |
| Enterococcus faecium | NCTC | NCTC 7171 |
| Listeria monocytogenes | NCTC | NCTC 11994 |
| Staphylococcus epidermidis | NCTC | NCTC 11047 |
| Staphylococcus aureus | NCTC | NCTC 6571 |
| Staphylococcus aureus (MRSA) | NCTC | NCTC 11939 |
| Streptococcus pyogenes | NCTC | NCTC 8306 |
| Candida glabrata | NCPF | NCPF 9725 |
| Acinetobacter baumannii | ATCC | ATCC 19606 |
| Burkholderia cepacia | ATCC | ATCC 25416 |
| Candida albicans | ATCC | ATCC 90028 |
| Clinical isolates of methicillin-resistant Staphylococcus aureus (MRSA) X 36. | Public Heath England, Colindale, UK | N/A |
| T. thermophilus HB8 | ATCC | ATCC 27634 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ni-NTA agarose | QIAGEN | Cat# 30230 |
| HiTrap Q HP, 1 mL | GE Healthcare Life Sciences | Cat# 29051325 |
| HiLoad 26/600 Superdex 200 | GE Healthcare Life Sciences | Cat# 28989336 |
| HiTrap Heparin HP, 5 mL | GE Healthcare Life Sciences | Cat# 17040601 |
| HisTrap HP, 5 mL | GE Healthcare Life Sciences | Cat# 17524801 |
| Bio-Rex 70 cation exchange resin | Bio-Rad | Cat# 1425842 |
| Biotage® SNAP Ultra 100 g | Biotage | Cat# FSUL-0442-0100 |
| [alpha-P32]Uridine 5′-triphosphate (UTP) | Hartmann Analytic | Cat# SRP-810 |
| rNTP Set, 100 mM Solutions (ATP, CTP, GTP, UTP) | GE Healthcare | Cat# 27202501 |
| QuikChange II kit (Stratagene) | Agilent | Cat# 200523 |
| CpA | IBA Lifesciences | Cat# 5-0710-186 |
| RIF | Sigma | Cat# R3501 |
| Resazurin sodium salt | Sigma | Cat# R7017 |
| CMM MOD2 | James et al., 2000 | N/A |
| Middlebrook 7H9 medium | BD Difco | Cat# 271310 |
| Phosphate buffered saline, pH 7.4 | Severn Biotech | Cat# 20-74-10 |
| IsoSensitest agar | Oxoid | Cat# CM0471 |
| Amberlite XAD-16 | Sigma | Cat# XAD16 |
| Deposited Data | ||
| KglA X-ray diffraction data | This paper | CCDC: 1827601 and 1827602 |
| E. coli RNAP – KglA complex | This paper | PDB: 6CUX |
| T. thermophilus RNAP – KglA complex | This paper | PDB: 6CUU |
| E. coli RNAP | Murakami 2013 | PDB: 4YG2 |
| T. thermophilus RNAP – pyrG promoter complex | Murakami et al., 2017 | PDB: 5VOI |
| Oligonucleotides | ||
| See Table S2 for list of oligonucleotides | ||
| Recombinant DNA | ||
| pVS10(S531L) | Gill and Garcia, 2011 | N/A |
| pGEMD | Murakami, 2013 | N/A |
| pET21a-SigA | Basu et al., 2014 | N/A |
| pETLrpoB | Smith and Savery, 2005 | N/A |
| Software and Algorithms | ||
| COOT | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| HKL2000 | Otwinowski and Minor, 1997 | http://www.hkl-xray.com |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org |
| Pymol | PyMOL | https://pymol.org/2/ |
| Graphpad 7.00 | Prism | https://www.graphpad.com/ |
| SigmaPlot 11.0 | Systat Software | https://systatsoftware.com/ |
| Other | ||
| VDX48 Plate with sealant | Hampton Research | Cat# HR3-275 |
| 12 mm x 0.22 mm Siliconized circle cover slides | Hampton Research | Cat# HR3-279 |
| 18 mm Mounted CryoLoop - 20 micron | Hampton Research | Cat# HR4-973 |
| CrystalCap ALS with vial | Hampton Research | Cat# HR4-779 |
| Vivaspin sample concentrator (MWCO 10 kDa) | GE Healthcare Life Sciences | Cat# 28-9323-60 |
| Tecan Sunrise microplate absorbance reader | Tecan instruments | https://www.tecan.com/ |
| Infinite M1000 multimode plate reader | Tecan instruments | https://www.tecan.com/ |
| Isolaera ONE | Biotage | Cat# ISO-1SW |
| Jeol Lambda 500 MHz spectrometer | JEOL Ltd | N/A |
| Jeol ECS 400 MHz spectrometer | JEOL Ltd | N/A |
| Gemini X-ray Diffractometer/Atlas detector | Oxford Diffraction | N/A |
| Beam Station I19 | National Synchrotron Facility Diamond Light Source | N/A |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to Lead Contact (n.zenkin@ncl.ac.uk).
Method Details
Disk diffusion assay
Standard disc diffusion assay (Kirby-Bauer) was performed with the B. subtilis 168 amyE::PhelD-lacZ cat reporter strain or RIF-resistant B. subtilis 168 rpoBH482R strain as described (Bauer et al., 1966). Briefly, disks were loaded with to 10 μg of each drug, air-dried and placed on nutrient agar plates with embedded lawn of bacteria and x-gal (100 μg/mL). Plates were incubated overnight at 37°C and scanned.
Isolation of KglA from Amycolatopsis sp. DEM30355
The fermentation of the Amycolatiopsis sp. DEM303555 was conducted as previously described. (Kepplinger et al., 2018). One kilogram of Amberlite XAD-16 resin was washed with deionized water and then applied to 50 L of filtered culture supernatant in batch absorption. The material was eluted with 32 L of 100% methanol and concentrated under reduced pressure (2.5 L). The concentrate was adjusted to pH 6.5 and extracted eight times with an equal amount of ethyl acetate. The organic extracts were combined and evaporated to dryness to yield 22.5 g of crude extract. A sample of the crude extract (3.91 g) was subjected to silica gel chromatography (eluent = 100% ethyl acetate, Biotage SNAP 100 g cartridge). KglA-containing fractions were combined and the solvent was removed under reduced pressure to give 1.24 g of a dark colored solid. This material was subjected to a second silica gel chromatography step (eluent = linear gradient from diethyl ether to ethyl acetate. Evaporation of the KglA containing fractions yielded 806.2 mg as an orange solid.
Small molecule X-ray crystallography including absolute configuration of KglA
Slow evaporation of a methanol solution of KglA gave crystals suitable for single-crystal X-ray analysis. The molecular structure of KglA was obtained to high precision using synchrotron radiation with a very small crystal of the methanol solvate at 120 K; all H atoms were located and freely refined. The short X-ray wavelength of 0.6889 Angstroms did not permit the determination of the absolute configuration, which was subsequently established unambiguously with CuKalpha radiation and a larger crystal, giving a molecular structure with otherwise slightly lower precision. CCDC 1827601 and 1827602 contain the supplementary crystallographic data for this paper, these data can be obtained free of charge from The Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk/).
NMR analysis of KglA
All NMR spectra were recorded in CDCl3 at 10°C. 1H and 13C{1H} NMR spectra were recorded directly with a Jeol Lambda 500 MHz spectrometer (Figure S1), while 1H COSY; 45, 90 and 135 DEPT; HMQC and HMBC were recorded with a Jeol ECS 400 MHz spectrometer (not shown). Proton and carbon assignment was performed de novo from both 1D and 2D NMR experiments and was in close agreement with the literature (Wang et al., 1988, Table S1).
Purification of E. coli RNAP core enzyme and σ70, and M. smegmatis RNAP holoenzyme for transcription assays
RIF-resistant mutations were introduced in pETLRpoB plasmid (6xHis-tag on N terminus of β subunit of E. coli RNAP) (Smith and Savery, 2005) by site-directed mutagenesis using QuikChange II kit (Stratagene). Wild-type or mutant plasmids were transformed along with pACYCDuet-1_Ec_rpoZ (encoding for ω subunit) in T7 Express strain (New England Biolabs). Expression and purification of core RNAP was performed as described (Kashlev et al., 1996) (with heparin chromatography used instead of gel filtration as suggested). N-terminal 6xHis-tagged E. coli σ70 subunit was purified as described (Zenkin et al., 2007).
Cellular M. smegmatis RNAP holoenzyme with 6xHis-tag on C terminus of β’ subunit was purified as E. coli RNAP above from M. smegmatis mc2155 (SM07) (Mukherjee and Chatterji, 2008), with exception that cells were grown in MB-7H9 media supplemented with 0.05 Tween 80 and 2% glucose to late exponential phase.
In vitro transcription
One pmol of wild-type or mutant E. coli RNAP core and 3 pmols of σ70 (or one pmol of M. smegmatis holoenzyme) and 0.1 pmols of linear template containing T7A1 promoter (Julius and Yuzenkova, 2017, Roghanian et al., 2015) were mixed in 7 μL of transcription buffer (20 mM Tris HCl pH 7.9, 40 mM KCl, 10 mM MgCl2). 1 μL of DMSO containing or not containing antibiotics (concentrations specified in Figures) was added to the reaction. Transcription was initiated by the addition of 2 μL of the mixture of nucleotides in transcription buffer, containing (final concentrations): 25 μM CpA or 0.5 mM ATP or 5 mM AMP with different combinations or all of 100 μM ATP, CTP and GTP and 10 μM α-[32P]UTP (20 Ci/mmol) (Hartmann Analytic). Reactions were stopped after 10-min incubation at 37°C by the addition of formamide-containing loading buffer. Products were separated on 20% or 33% denaturing (8M urea) polyacrylamide gels, revealed by PhosphorImaging (GE Healthcare), and analyzed using ImageQuant software (GE Healthcare). Activities (a sum of run off and termination products) were normalized to the activity without antibiotic, which was taken as 100%. The average activity of at least three replicates for each concentration of antibiotic was plotted (with their standard deviations as error bars) against concentration and fitted in four parameter logistic equation by nonlinear regression using SigmaPlot. The averages and standard deviations of the three individual IC50s are reported in Figure 1E.
Crystallization of RNAP – KglA complexes
The βS531L RIF-resistant mutant E. coli RNAP σ70 holoenzyme was prepared and crystallized as previously described (Molodtsov et al., 2017). The complex of the T. thermophilus RNAP σA holoenzyme bound to pyrG promoter was prepared and crystallized as previously described (Murakami et al., 2017). To prepare complexes of RNAPs with KglA, the RNAP crystals were soaked overnight at 22°C in the respective cryoprotection solutions supplemented with 1 mM KglA. After the soaking, crystals were frozen by plunging to liquid N2.
Crystallographic data collection and X-ray crystal structure determinations
The X-ray diffraction datasets were collected at the Macromolecular Diffraction at the Cornell High Energy Synchrotron Source (MacCHESS) F1 beamline (Cornell University, Ithaca, NY) and the data were processed by HKL2000 (Otwinowski and Minor, 1997). Structures were determined by molecular replacement using the structures of the E. coli RNAP (Murakami, 2013) and the T. thermophilus RNAP – pyrG promoter complexes (Murakami et al., 2017) as corresponding initial search models. The structures were refined as the rigid body and the positional refinement with non-crystallographic symmetry and reference model structure restraints using the suite of programs Phenix (Afonine et al., 2010). The resulting electron density maps revealed clear maps corresponding to KglA that was not present in the initial search models, and the KglA model was fitted into the density maps using Coot (Emsley and Cowtan, 2004). The Table 1 contains data collection and refinement statistics for the structures of the determined RNAP – KglA complexes. The final coordinates and structure factors were deposited to the PDB with ID codes listed in Table 1.
Determination of minimum inhibitory concentrations (MICs) for M. tuberculosis
Bacterial strains and reagents
Mycobacterium tuberculosis strains: H37Rv, Beijing 1192/015 and Beijing 08/00483E (multi-drug resistant, MDR) were used in the modified resazurin microtiter assay (REMA) plate method. Beijing strains 1192/015 and 08/00483E were obtained from the PHE National Mycobacterial Reference Laboratory. Beijing 08/00483E contains a single nucleotide polymorphism (SNP) at position 761155 of C→T, which has resulted in an amino acid change, S450L (corresponding to S531L with E. coli numbering), within the RIF-binding pocket. Beijing 08/00483E is also resistant to isoniazid, ethambutol, and pyrazinamide, through mutations in known resistance genes. Other reagents used were: Middlebrook 7H9 medium (BD Difco), OADC supplemented with 0.5% glycerol (24388.295, VWR Chemicals) and 0.2% Tween 80 (P1754, Sigma), CAMR Mycobacterium Medium MOD2 (CMM MOD2) (James et al., 2000), RIF (PanReac AppliChem, cat no.A2220) and 0.02% resazurin solution (10 mg resazurin sodium salt (R7017, Sigma) in 50 mL phosphate buffered saline, pH 7.4 (Severn Biotech) containing 5% Tween 80) (Gold et al., 2016). All the antibiotics were dissolved in methanol (322415, Sigma) to a stock concentration of 10 mg/mL.
Preparation of bacterial inocula
Bacterial strains were grown in 50 mL of OADC-supplemented, Middlebrook 7H9 medium with 0.5% glycerol and 0.2% Tween 80 in vented Erlenmeyer flasks, shaken at 37°C for 5 days. Inocula for the REMA method were prepared to an OD540 of 0.5, from Middlebrook 7H9 cultures by diluting cells in CMM MOD2 medium.
Modified resazurin microtiter assay (REMA) plate method
A modification of the REMA plate method previously described by (Nateche et al., 2006) was used. A 10 mg/mL stock solution of KglA was prepared in methanol (322415, Sigma). A two-fold serial dilution of the stock solution was performed in CMM MOD2 medium. 100 μl of each KglA concentration was added to six replicate wells in a 96-well untreated flat bottomed 96-well plate (734-1554, VWR). Three of the wells remained as “drug-only” wells and the other three wells were “test wells,” which were used to test the activity of KglA. 10 μl of bacterial culture was added to the test wells to give a final OD540 of 0.05. Three further wells contained 100 μl of growth medium and 10 μl of bacterial cells as “zero-drug” controls to confirm exponential growth of the bacteria in the absence of antibiotic. The drug-only controls allowed for confirmation that there was an absence of colorimetric interference from the candidate antibiotic. Bacterial cell samples were diluted to an optical density (OD540) of 0.5 and 10 μl of the inoculum was added to each of the test wells and the zero drug controls. 10 μl of CMM MOD2 growth medium was added to the drug-only wells. The 96-well plates were incubated for 7 days at 37°C while shaking at 200 rpm. Drug activity was ascertained by adding 11 μl resazurin to every well of the 96 well-plate and incubating the plate at room temperature for 6 hours. The optical density (OD570) was measured for each well. The OD570 measurements were converted to a percentage of the zero-drug control so that data were expressed as a percentage of the growth without antibiotic.
Statistical analyses
The average OD570 for each drug concentration was fitted to a sigmoidal curve using the modified Gompertz function (y = A + Ce-eB(x - M)) and the minimum inhibitory concentration (MIC) was identified from the point of inflexion of the lower asymptote (Lambert and Pearson, 2000). This was applied to the mean of the OD570 of four biological repeats to produce a graphical representation for each condition. An unpaired, two tailed, Students t test was used to determine significance between biological repeats.
Determination of minimum inhibitory concentrations for other bacteria
MICs were determined using the agar dilution method specified by the British Society for Antimicrobial Chemotherapy against a collection of 56 isolates (Andrews, 2001). Briefly, KglA was initially dissolved in a minimal volume of dimethyl sulfoxide and diluted in water before incorporation into IsoSensitest agar (Oxoid) at a final concentration range of 128 – 0.031 μg/mL. A final inoculum of 10 000 CFU/spot was used for inoculation of plates. Cultures were incubated for 22 hours at 37 ± 0.5°C. The MIC was recorded as the lowest concentration of KglA that achieved complete inhibition of visible growth. Inhibitor-free controls were also tested, including those containing equivalent solvent concentrations. All tests were performed at least twice on separate occasions to demonstrate reproducibility.
The collection included 16 isolates acquired from the National Collection of Type Cultures (NCTC; Colindale, UK), 3 isolates from the American Type Culture Collection (ATCC; Manassas, USA), and one from the National Collection of Pathogenic Fungi (NCPF; Colindale, UK). Also included were 36 strains of methicillin-resistant Staphylococcus aureus (MRSA) kindly supplied by Public Health England (Colindale, UK). These 36 strains were isolated from clinical samples in five European countries and included representatives of the major clones responsible for outbreaks of infection.
Data and Software Availability
The atomic coordinates and structure factors have been deposited in the Protein Data Bank, https://www.pdb.org (PDB ID codes 6CUX and 6CUU). CCDC 1827601 and 1827602 contain the supplementary crystallographic data for this paper, these data can be obtained free of charge from The Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk/).
Acknowledgments
We thank the staff at the MacCHESS for support with crystallographic data collection. We also thank UK Engineering and Physical Sciences Research Council for X-ray crystallography facilities (grant EP/F03637X/1), Dr U. Baisch (Newcastle University) for measuring the CuKalpha diffraction data, Diamond Light Source for access to beamline I19 (award MT8682), Prof. W. McFarlane (Newcastle University) for NMR support, and Dr. J. Gray (Newcastle University) for HRMS analysis. We acknowledge the PHE National Mycobacterium Reference Laboratory (NMRL) for providing M. tuberculosis Beijing strains 1192/015 and 08/00483E. This work was supported by Wellcome Trust Investigator Award 102851 and Leverhulme Trust Prize PLP-2014-229 (to N.Z.), Innovate UK grants 100953 and 131143 (to N.E.E.A.), a Royal Society University Research Fellowship (to Y.Y.), NIH grant GM087350 (to K.S.M.), a Department of Health Grant in Aid and The PHE Pipeline Fund (to J.B.; the views expressed in this publication are those of the authors and not necessarily those of Public Health England or the Department of Health), and UK Medical Research Council studentship MR/N018613/1 (to J.H.).
Author Contributions
H.M., L.C., J.H., and Y.Y. performed biological experiments and analyzed the data. B.K., S.M.L., and N.E.E.A. purified KglA. V.M., Y.S., and K.S.M. performed crystallographic experiments and analyzed the structures. M.J.H., S.M.L., and C.W. performed the NMR analysis and structural assignment of KglA. W.C. performed small-molecule X-ray crystallography. C.W.M., R.E.J., and J.B. measured the MICs for the M. tuberculosis strains. J.D.P. and E.C.L.M. measured KglA MICs for other species. J.E., N.E.E.A., and N.Z. conducted the study. J.B., C.W.M., V.M., M.J.H., K.S.M., and N.Z. wrote the paper.
Declaration of Interests
N.E.E.A. is an employee of and J.E. is a scientific founder of and shareholder in Demuris Ltd. Demuris and Newcastle University have filed UK patent 1812078.2.
Published: September 20, 2018
Footnotes
Supplemental Information includes two figures and two tables and can be found with this article online at https://doi.org/10.1016/j.molcel.2018.08.028.
Contributor Information
Katsuhiko S. Murakami, Email: kum14@psu.edu.
Nikolay Zenkin, Email: n.zenkin@ncl.ac.uk.
Supplemental Information
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta. Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P.V., Mustyakimov M., Grosse-Kunstleve R.W., Moriarty N.W., Langan P., Adams P.D. Joint X-ray and neutron refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2010;66:1153–1163. doi: 10.1107/S0907444910026582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001;48(Suppl 1):5–16. doi: 10.1093/jac/48.suppl_1.5. [DOI] [PubMed] [Google Scholar]
- Aristoff P.A., Garcia G.A., Kirchhoff P.D., Showalter H.D. Rifamycins--obstacles and opportunities. Tuberculosis (Edinb.) 2010;90:94–118. doi: 10.1016/j.tube.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Artsimovitch I., Vassylyeva M.N., Svetlov D., Svetlov V., Perederina A., Igarashi N., Matsugaki N., Wakatsuki S., Tahirov T.H., Vassylyev D.G. Allosteric modulation of the RNA polymerase catalytic reaction is an essential component of transcription control by rifamycins. Cell. 2005;122:351–363. doi: 10.1016/j.cell.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Bacchi A., Pelizzi G., Nebuloni M., Ferrari P. Comprehensive study on structure-activity relationships of rifamycins: discussion of molecular and crystal structure and spectroscopic and thermochemical properties of rifamycin O. J. Med. Chem. 1998;41:2319–2332. doi: 10.1021/jm970791o. [DOI] [PubMed] [Google Scholar]
- Bacchi A., Carcelli M., Pelizzi G. Sampling rifamycin conformational variety by cruising through crystal forms: implications for polymorph screening and for biological models. New J. Chem. 2008;32:1725–1735. [Google Scholar]
- Bae B., Nayak D., Ray A., Mustaev A., Landick R., Darst S.A. CBR antimicrobials inhibit RNA polymerase via at least two bridge-helix cap-mediated effects on nucleotide addition. Proc. Natl. Acad. Sci. USA. 2015;112:E4178–E4187. doi: 10.1073/pnas.1502368112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu R.S., Warner B.A., Molodtsov V., Pupov D., Esyunina D., Fernández-Tornero C., Kulbachinskiy A., Murakami K.S. Structural basis of transcription initiation by bacterial RNA polymerase holoenzyme. J. Biol. Chem. 2014;289:24549–24559. doi: 10.1074/jbc.M114.584037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer A.W., Kirby W.M., Sherris J.C., Turck M. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 1966;45:493–496. [PubMed] [Google Scholar]
- Campbell E.A., Korzheva N., Mustaev A., Murakami K., Nair S., Goldfarb A., Darst S.A. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell. 2001;104:901–912. doi: 10.1016/s0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- Campbell E.A., Pavlova O., Zenkin N., Leon F., Irschik H., Jansen R., Severinov K., Darst S.A. Structural, functional, and genetic analysis of sorangicin inhibition of bacterial RNA polymerase. EMBO J. 2005;24:674–682. doi: 10.1038/sj.emboj.7600499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degen D., Feng Y., Zhang Y., Ebright K.Y., Ebright Y.W., Gigliotti M., Vahedian-Movahed H., Mandal S., Talaue M., Connell N. Transcription inhibition by the depsipeptide antibiotic salinamide A. eLife. 2014;3:e02451. doi: 10.7554/eLife.02451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- EUCAST (2018). European Committee on Antimicrobial Susceptibility Testing. http://www.eucast.org/clinical_breakpoints/.
- Floss H.G., Yu T.W. Rifamycin-mode of action, resistance, and biosynthesis. Chem. Rev. 2005;105:621–632. doi: 10.1021/cr030112j. [DOI] [PubMed] [Google Scholar]
- Gill S.K., Garcia G.A. Rifamycin inhibition of WT and Rif-resistant Mycobacterium tuberculosis and Escherichia coli RNA polymerases in vitro. Tuberculosis (Edinb.) 2011;91:361–369. doi: 10.1016/j.tube.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Gold B., Roberts J., Ling Y., Lopez Quezada L., Glasheen J., Ballinger E., Somersan-Karakaya S., Warrier T., Nathan C. Visualization of the Charcoal Agar Resazurin Assay for Semi-quantitative, Medium-throughput Enumeration of Mycobacteria. J. Vis. Exp. 2016;118:54690. doi: 10.3791/54690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M.X., Hudson B.P., Das K., Arnold E., Ebright R.H. Structures of RNA polymerase-antibiotic complexes. Curr. Opin. Struct. Biol. 2009;19:715–723. doi: 10.1016/j.sbi.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James B.W., Williams A., Marsh P.D. The physiology and pathogenicity of Mycobacterium tuberculosis grown under controlled conditions in a defined medium. J. Appl. Microbiol. 2000;88:669–677. doi: 10.1046/j.1365-2672.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- Jamieson F.B., Guthrie J.L., Neemuchwala A., Lastovetska O., Melano R.G., Mehaffy C. Profiling of rpoB mutations and MICs for rifampin and rifabutin in Mycobacterium tuberculosis. J. Clin. Microbiol. 2014;52:2157–2162. doi: 10.1128/JCM.00691-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius C., Yuzenkova Y. Bacterial RNA polymerase caps RNA with various cofactors and cell wall precursors. Nucleic Acids Res. 2017;45:8282–8290. doi: 10.1093/nar/gkx452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashlev M., Nudler E., Severinov K., Borukhov S., Komissarova N., Goldfarb A. Histidine-tagged RNA polymerase of Escherichia coli and transcription in solid phase. Methods Enzymol. 1996;274:326–334. doi: 10.1016/s0076-6879(96)74028-4. [DOI] [PubMed] [Google Scholar]
- Kepplinger B., Morton-Laing S., Seistrup K.H., Marrs E.C.L., Hopkins A.P., Perry J.D., Strahl H., Hall M.J., Errington J., Allenby N.E.E. Mode of Action and Heterologous Expression of the Natural Product Antibiotic Vancoresmycin. ACS Chem. Biol. 2018;13:207–214. doi: 10.1021/acschembio.7b00733. [DOI] [PubMed] [Google Scholar]
- Lambert R.J., Pearson J. Susceptibility testing: accurate and reproducible minimum inhibitory concentration (MIC) and non-inhibitory concentration (NIC) values. J. Appl. Microbiol. 2000;88:784–790. doi: 10.1046/j.1365-2672.2000.01017.x. [DOI] [PubMed] [Google Scholar]
- Lancini G., Zanichelli W. Academic Press; New York: 1977. Structure-activity relationship in rifamycins, Structure-activity relationships among the semisynthetic antibiotics edn. [Google Scholar]
- Lin W., Mandal S., Degen D., Liu Y., Ebright Y.W., Li S., Feng Y., Zhang Y., Mandal S., Jiang Y. Structural Basis of Mycobacterium tuberculosis Transcription and Transcription Inhibition. Mol. Cell. 2017;66:169–179.e8. doi: 10.1016/j.molcel.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffioli S.I., Zhang Y., Degen D., Carzaniga T., Del Gatto G., Serina S., Monciardini P., Mazzetti C., Guglierame P., Candiani G. Antibacterial Nucleoside-Analog Inhibitor of Bacterial RNA Polymerase. Cell. 2017;169:1240–1248.e23. doi: 10.1016/j.cell.2017.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure W.R., Cech C.L. On the mechanism of rifampicin inhibition of RNA synthesis. J. Biol. Chem. 1978;253:8949–8956. [PubMed] [Google Scholar]
- Molodtsov V., Fleming P.R., Eyermann C.J., Ferguson A.D., Foulk M.A., McKinney D.C., Masse C.E., Buurman E.T., Murakami K.S. X-ray crystal structures of Escherichia coli RNA polymerase with switch region binding inhibitors enable rational design of squaramides with an improved fraction unbound to human plasma protein. J. Med. Chem. 2015;58:3156–3171. doi: 10.1021/acs.jmedchem.5b00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molodtsov V., Scharf N.T., Stefan M.A., Garcia G.A., Murakami K.S. Structural basis for rifamycin resistance of bacterial RNA polymerase by the three most clinically important RpoB mutations found in Mycobacterium tuberculosis. Mol. Microbiol. 2017;103:1034–1045. doi: 10.1111/mmi.13606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee R., Chatterji D. Stationary phase induced alterations in mycobacterial RNA polymerase assembly: A cue to its phenotypic resistance towards rifampicin. Biochem. Biophys. Res. Commun. 2008;369:899–904. doi: 10.1016/j.bbrc.2008.02.118. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay J., Das K., Ismail S., Koppstein D., Jang M., Hudson B., Sarafianos S., Tuske S., Patel J., Jansen R. The RNA polymerase “switch region” is a target for inhibitors. Cell. 2008;135:295–307. doi: 10.1016/j.cell.2008.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K.S. X-ray crystal structure of Escherichia coli RNA polymerase σ70 holoenzyme. J. Biol. Chem. 2013;288:9126–9134. doi: 10.1074/jbc.M112.430900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K.S., Shin Y., Turnbough C.L., Jr., Molodtsov V. X-ray crystal structure of a reiterative transcription complex reveals an atypical RNA extension pathway. Proc. Natl. Acad. Sci. USA. 2017;114:8211–8216. doi: 10.1073/pnas.1702741114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nateche F., Martin A., Baraka S., Palomino J.C., Khaled S., Portaels F. Application of the resazurin microtitre assay for detection of multidrug resistance in Mycobacterium tuberculosis in Algiers. J. Med. Microbiol. 2006;55:857–860. doi: 10.1099/jmm.0.46513-0. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W. [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Perlman D. Academic Press; New York: 1977. Structure-activity relationships among the semisynthetic antibiotics. [Google Scholar]
- Roghanian M., Zenkin N., Yuzenkova Y. Bacterial global regulators DksA/ppGpp increase fidelity of transcription. Nucleic Acids Res. 2015;43:1529–1536. doi: 10.1093/nar/gkv003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu Y.J., Koh W.J., Daley C.L. Diagnosis and Treatment of Nontuberculous Mycobacterial Lung Disease: Clinicians’ Perspectives. Tuberc. Respir. Dis. (Seoul) 2016;79:74–84. doi: 10.4046/trd.2016.79.2.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandgren A., Strong M., Muthukrishnan P., Weiner B.K., Church G.M., Murray M.B. Tuberculosis drug resistance mutation database. PLoS Med. 2009;6:e2. doi: 10.1371/journal.pmed.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A.J., Savery N.J. RNA polymerase mutants defective in the initiation of transcription-coupled DNA repair. Nucleic Acids Res. 2005;33:755–764. doi: 10.1093/nar/gki225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song T., Park Y., Shamputa I.C., Seo S., Lee S.Y., Jeon H.S., Choi H., Lee M., Glynne R.J., Barnes S.W. Fitness costs of rifampicin resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the β′ subunit of RNA polymerase. Mol. Microbiol. 2014;91:1106–1119. doi: 10.1111/mmi.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tupin A., Gualtieri M., Leonetti J.P., Brodolin K. The transcription inhibitor lipiarmycin blocks DNA fitting into the RNA polymerase catalytic site. EMBO J. 2010;29:2527–2537. doi: 10.1038/emboj.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban A., Eckermann S., Fast B., Metzger S., Gehling M., Ziegelbauer K., Rübsamen-Waigmann H., Freiberg C. Novel whole-cell antibiotic biosensors for compound discovery. Appl. Environ. Microbiol. 2007;73:6436–6443. doi: 10.1128/AEM.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N.J., Fu Y., Yan G.H., Bao G.H., Xu C.F., He C.H. Isolation and structure of a new ansamycin antibiotic kanglemycin A from a Nocardia. J. Antibiot. (Tokyo) 1988;41:264–267. doi: 10.7164/antibiotics.41.264. [DOI] [PubMed] [Google Scholar]
- World Health Organization (2017). Global tuberculosis report. CC BY-NC- SA 3.0 IGO.
- Zenkin N., Kulbachinskiy A., Yuzenkova Y., Mustaev A., Bass I., Severinov K., Brodolin K. Region 1.2 of the RNA polymerase sigma subunit controls recognition of the -10 promoter element. EMBO J. 2007;26:955–964. doi: 10.1038/sj.emboj.7601555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Degen D., Ho M.X., Sineva E., Ebright K.Y., Ebright Y.W., Mekler V., Vahedian-Movahed H., Feng Y., Yin R. GE23077 binds to the RNA polymerase ‘i’ and ‘i+1’ sites and prevents the binding of initiating nucleotides. eLife. 2014;3:e02450. doi: 10.7554/eLife.02450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Dai F., Luo F., Zhong M., Huang Z., Hou T., Xu J. Could high-concentration rifampicin kill rifampicin-resistant M. tuberculosis? Rifampicin MIC test in rifampicin-resistant isolates from patients with osteoarticular tuberculosis. J. Orthop. Surg. Res. 2014;9:124. doi: 10.1186/s13018-014-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.