

Abstract
The molecular processes that determine the outcome of influenza virus infection in humans are multifactorial and involve a complex interplay between host, viral, and bacterial factors1. However, it is generally accepted that a strong innate immune dysregulation known as ‘cytokine storm’ contributes to the pathology of infections with 1918 H1N1 pandemic or highly pathogenic avian influenza viruses (HPAIV) of the H5N1 subtype2–4. The RNA sensor Retinoic acid-inducible gene I (RIG-I) plays an important role in sensing viral infection and initiating a signalling cascade that leads to interferon (IFN) expression5. Here we show that short aberrant RNAs (mini viral RNAs; mvRNAs), produced by the viral RNA polymerase during the replication of the viral RNA genome, bind and activate RIG-I, and lead to the expression of interferon-β. We find that erroneous polymerase activity, dysregulation of viral RNA replication, or the presence of avian-specific amino acids underlie mvRNA generation and cytokine expression in mammalian cells. By deep-sequencing RNA samples from lungs of ferrets infected with influenza viruses we show that mvRNAs are generated during infection in vivo. We propose that mvRNAs act as main agonists of RIG-I during influenza virus infection.
The negative sense viral RNA (vRNA) genome segments of influenza A viruses, as well as the complementary RNA (cRNA) replicative intermediates, contain 5′ triphosphates and partially complementary 5′ and 3′ termini that serve as the viral promoter for replication and transcription of the viral RNA genome6. RIG-I has been shown to bind and be activated by the dsRNA structure formed by the termini of influenza virus RNAs7,8. However, it remains unclear how RIG-I gains access to this dsRNA structure. Both vRNA and cRNA are assembled into ribonucleoprotein complexes (vRNP and cRNP, respectively) in which the viral RNA polymerase, a complex of the viral proteins PB1, PB2 and PA, associates with the partially complementary termini, while the rest of the RNA is bound by oligomeric nucleoprotein (NP)6 (Fig. 1a). The tight binding of the 5′ and 3′ termini of vRNA and cRNA by the RNA polymerase9 is likely to preclude an interaction with RIG-I. Moreover, it has been demonstrated that IFN expression is triggered only in a fraction of influenza virus infected cells10,11, suggesting that influenza viruses efficiently hide their genome segments during infection by replicating them in the context of RNPs11. This led to the proposal that an aberrant RNA replication product might be binding to RIG-I and triggering IFN expression12. The influenza virus polymerase is known to generate defective interfering (DI) RNAs, which are ≥178 nt long subgenomic RNAs generated during high multiplicity infections13, and small viral RNAs (svRNAs), which are 22-27 nt long and correspond to the 5′ end of vRNA segments. However, svRNAs have been shown not to be involved in the induction of antiviral cellular defences14 and DI RNAs assemble into RNP structures (Fig. 1a), as demonstrated for a 248 nt long DI RNA15, potentially precluding their interaction with RIG-I. Therefore, it remains unclear what kind of viral RNA species is recognised by RIG-I (Fig. 1a) and why different influenza virus strains trigger dramatically different levels of IFN expression2,3,16.
Figure 1. mvRNAs of influenza A virus are bound by RIG-I and induce IFN expression.
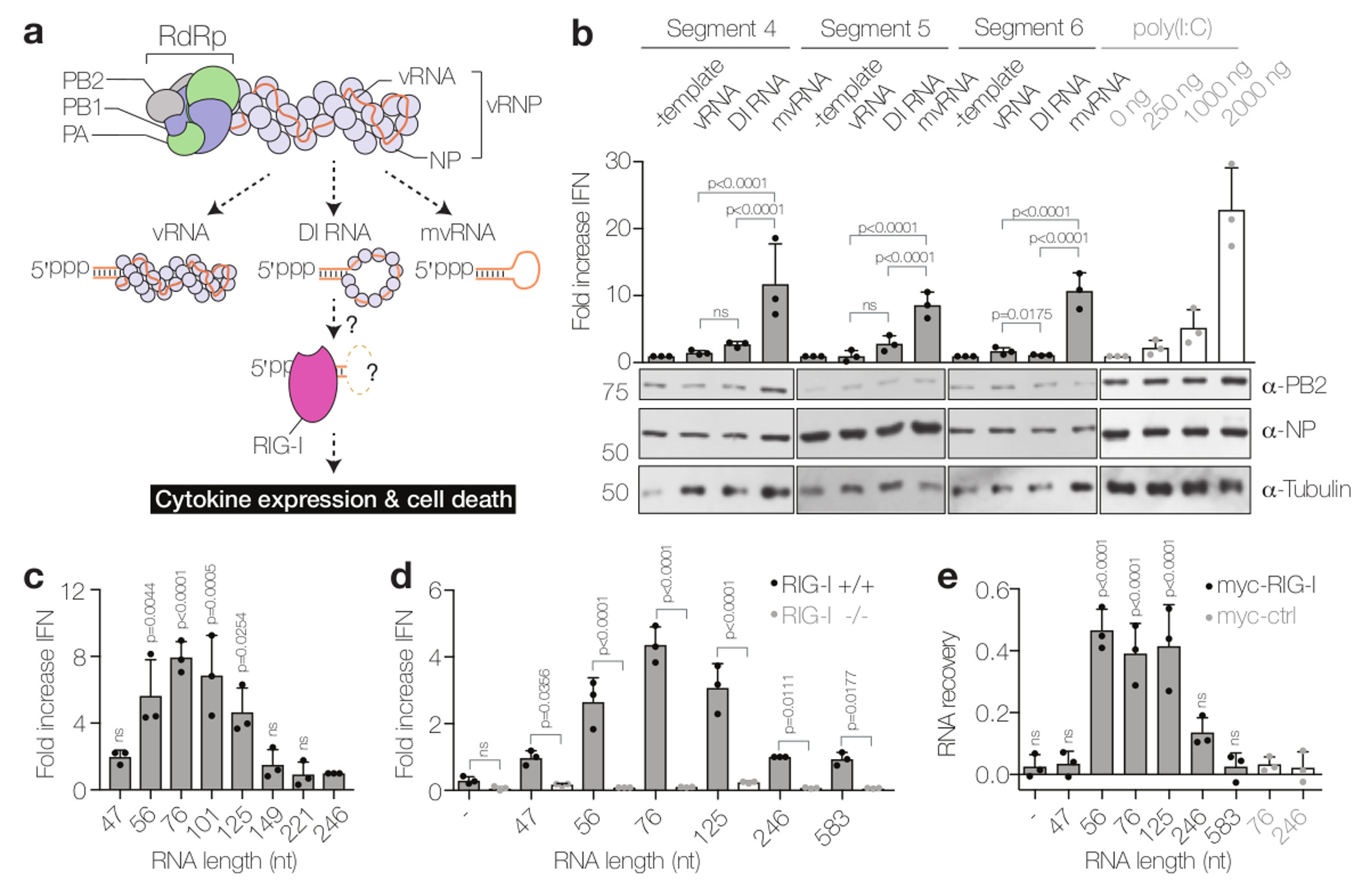
(a) Models of the influenza virus ribonucleoprotein (vRNP) complex and potential activators of RIG-I. (b) Analysis of IFN-β promoter activity induced by the replication of segment 4, 5 or 6 vRNAs, DI RNAs or mvRNAs or by the transfection of poly(I:C). PB2, NP and tubulin expression was analysed by western blot. P-values were determined using a two-sided unpaired t-test. (c) IFN-β promoter activity induced by the replication of engineered, segment 5-based, short RNAs in HEK 293T cells. P-values were determined using ANOVA with multiple testing compared to the 246 mvRNA. (d) IFN-β promoter activity induced by the replication of engineered, segment 5-based, short RNAs in wild-type (RIG-I +/+) or HEK 293 RIG-I knockout (RIG-I -/-) cells. P-values were determined as in c. (e) Binding of segment 5-based RNAs to myc-tagged RIG-I or mouse EGF control protein (myc-ctrl). P-values were determined using ANOVA with multiple testing compared to the myc-ctrl with 246 mvRNA. All graphs show standard deviation and mean of data from three (n=3) biologically independent experiments.
Engineered viral RNAs shorter than 149 nt but containing both the 5′ and 3′ termini of vRNAs can be transcribed and replicated by the viral polymerase in the absence of NP17, suggesting that they do not form canonical RNP structures. We call these short viral RNAs mvRNAs (Fig. 1a). To investigate which class of viral RNA is responsible for triggering IFN expression, we expressed a full-length segment 4 vRNA (1775 nt long) or its truncated versions, a 245 nt long DI RNA and 77 nt long mvRNA, in HEK 293T cells together with viral polymerase and NP and measured the activation of the IFN-β promoter (Fig. 1b). We found that the expression of mvRNAs induced significantly higher IFN expression than full-length vRNA or DI RNA, comparable to the levels induced by transfection of 2 μg of poly(I:C), a known activator of IFN expression18. Similar results were obtained with segment 5 and 6 vRNAs and their truncated DI RNA and mvRNA versions (Fig. 1b). To determine the optimal mvRNA length that triggers IFN-β promoter activation, we expressed 47 to 246 nt long vRNAs derived from segment 5 together with viral polymerase and NP and measured the activity of the IFN-β promoter. We found that the replication of 56 to 125 nt long mvRNAs resulted in significantly higher IFN-β promoter activity than the replication of RNAs shorter than 56 nt or longer than 125 nt (Fig. 1c and Supplementary Fig. 1a,b).
To address whether these engineered short mvRNAs triggered IFN expression via RIG-I, we co-expressed viral RNAs with polymerase and NP in HEK 293T RIG-I knockout or control cells engineered to express luciferase in response to the activation of the IFN-β promoter. We found that 56 to 125-nt long mvRNAs induced only background levels of luciferase in RIG-I knockout cells, even though the expression of RIG-I or transfection of poly(I:C) resulted in significant activation of the IFN-β promoter (Fig. 1d and Supplementary Fig. 1c). By contrast, significant levels of luciferase activity were detected in wildtype cells (Fig. 1d). mvRNAs of 56 to 125 nt induced the strongest activation of the IFN-β promoter, in agreement with the data above (Fig. 1c,d and Supplementary Fig. 1c,d). To address whether mvRNAs trigger the activation of IFN-β expression through binding to RIG-I, we immunoprecipitated myc-RIG-I from cells expressing RNAs of 47 to 583 nt. We observed that mvRNAs of 56 to 125 nt were specifically enriched in RIG-I immunoprecipitates (Fig. 1e and Supplementary Fig. 1e). No mvRNAs were detected in the myc-EGF negative control immunoprecipitates (Fig. 1e). To test if mvRNAs also activate RIG-I, we incubated purified myc-RIG-I with an in vitro transcribed 76 nt mvRNA and measured 32Pi release. We found that a triphosphorylated 76 nt mvRNA induced higher levels of ATPase activity than a dephosphorylated 76 nt mvRNA, while no ATPase activity was observed when we incubated a RIG-I mutant with the triphosphorylated 76 nt mvRNA (Supplementary Fig. 1f,g). Overall, these results demonstrate that mvRNAs longer than 47 and shorter than 125 nt are bound by RIG-I, which results in RIG-I activation and the induction of IFN-β expression. This is in agreement with findings that reconstitution of full-length influenza virus vRNPs leads to only low levels of IFN expression unless the cells are pre-treated with IFN19 and the hypothesis that aberrant replication products trigger the IFN induction cascade12.
We next asked whether mvRNAs are made during influenza virus infection. We infected HEK 293T cells with influenza A/WSN/33 (H1N1) (abbreviated as WSN) and analysed viral RNAs by RT-PCR of segment 1, RT-PCR of all segments using universal primers, or deep-sequencing of the total small RNA fraction (RNAs 17 to 200 nt in length) (Supplementary Fig. 2a,b). We found only very low levels of mvRNAs and, consistently, observed no significant IFN expression (Fig. 2a,b). We hypothesised that mvRNAs are only generated as a consequence of dysregulated viral RNA replication. To test this, we overexpressed viral RNA polymerase prior to infection to generate an imbalance between polymerase and NP levels, which is known to induce innate immune signalling20. Under this condition we found significantly higher levels of mvRNAs and IFN expression, while simultaneous overexpression of NP and polymerase reduced mvRNA and IFN production (Fig. 2a). We verified the identity of mvRNAs using gel isolation and Sanger sequencing (Supplementary Fig. 2c) as well as deep sequencing (Fig. 2b). We found that the majority of mvRNAs were derived from the PB1-, HA-, NP- and NA-encoding vRNA segments (Fig. 2c) and that mvRNAs had a size distribution with a peak around 55 to 64 nt (Fig. 2d and Supplementary Fig. 2d). In addition to mvRNAs, we also identified complementary mini viral RNAs (mcRNAs).
Figure 2. Dysregulation of RNA replication in cells infected with WSN results in the generation of mvRNAs.
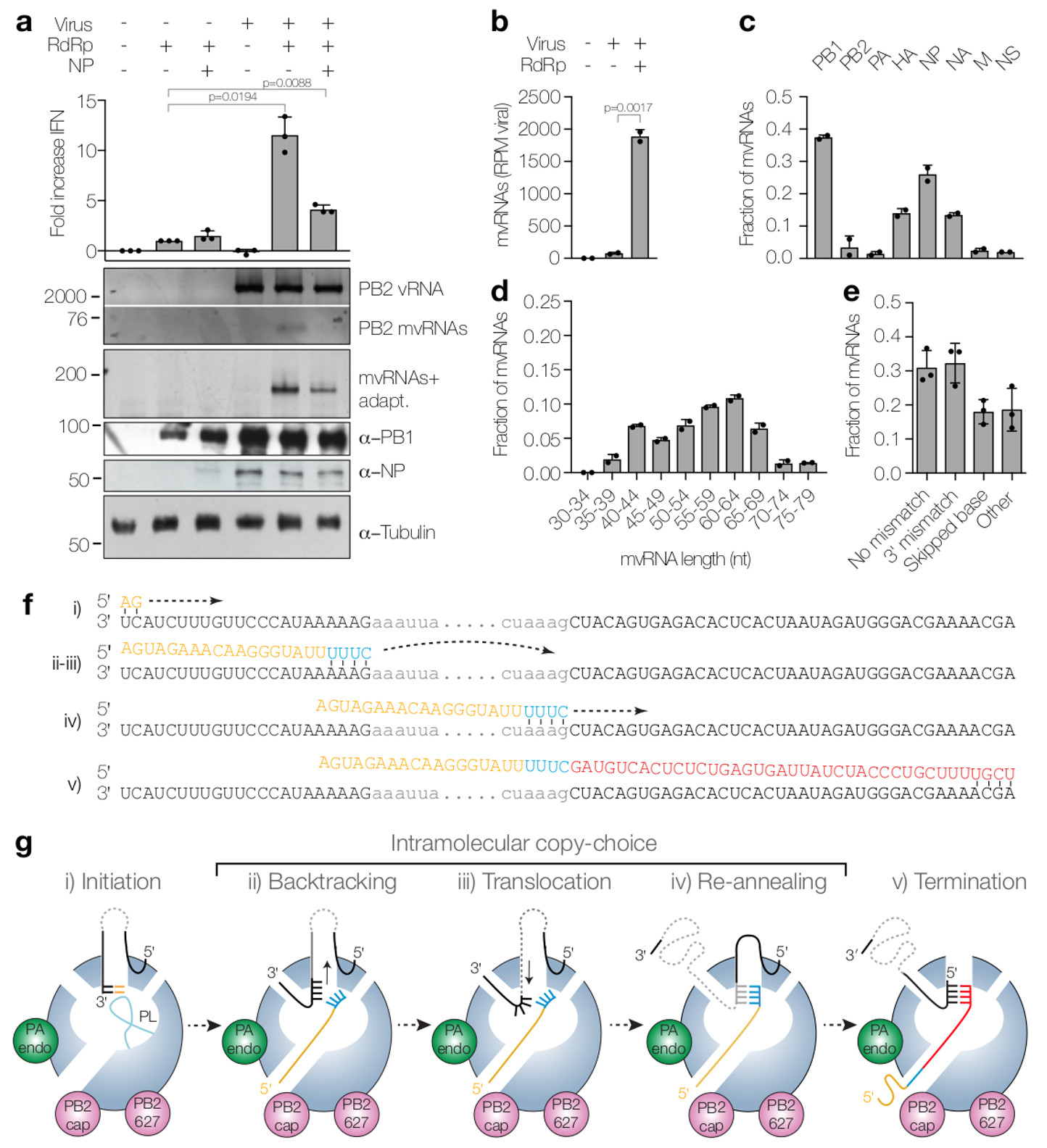
(a) Analysis of IFN-β promoter activity (graph) and steady state vRNA and mvRNA levels (top gel) in WSN infections following overexpression of viral polymerase or viral polymerase and NP. mvRNAs were also amplified with universal primers containing adapters for sequencing (mvRNAs+adapt) and analysed by PAGE (second gel). NP, PB1 and tubulin expression was analysed by western blot. P-values were determined using ANOVA compared to lane 2. (b) Quantitation of mvRNAs using deep sequencing, expressed as reads per million (RPM). P-value was determined using a two-sided unpaired t-test. (c) mvRNA distribution per genome segment. (d) Size distribution of mvRNAs. (e) mvRNA distribution per type of intramolecular copy-choice mechanism. (f) Example of mvRNA formation through an intramolecular copy-choice mechanism involving a 3′ mismatch. (g) Model of mvRNA formation by the polymerase (model adapted from6). All graphs show standard deviation and mean of data from two (n=2) (b-d) or three (n=3) (a,e) biologically independent experiments.
Analysis of mvRNA sequences suggests that mvRNAs are generated via an intramolecular copy-choice mechanism that tolerates 3' mismatches or skipped bases (Fig. 2e,f). The generation of mvRNAs can be explained by the separation of the template and nascent product RNAs by backtracking21, followed by template translocation until base pairing between template and nascent product RNA is re-established (Fig. 2f,g). This process may be induced by an imbalance between viral polymerase and NP levels (Fig. 2a and Supplementary Fig. 3a).
In humans, infection with the 1918 H1N1 pandemic virus or H5N1 HPAIV lead to strong innate immune activation2,3,16. To address whether mvRNAs could contribute to this phenomenon, we investigated the replication of a 246 nt RNA by the polymerase of these viruses. We found that the polymerases of the highly virulent A/Brevig Mission/1/18 (H1N1) (abbreviated as BM18) pandemic virus and the A/duck/Fujian/01/02 (H5N1) (abbreviated as FJ02) HPAIV generated higher levels of mvRNAs than the polymerases of WSN and A/Northern Territory/60/68 (H3N2) (abbreviated as NT60) viruses, even in the presence of high NP concentrations (Fig. 3a). No mvRNAs were observed in a control with an inactive WSN polymerase that had two point mutations in the polymerase active site (PB1a). We confirmed that the mvRNAs produced by the BM18 polymerase were similar to the WSN mvRNAs (Supplementary Fig. 3b). Isolation of total RNA from cells expressing polymerase of the BM18 or FJ02 virus and its subsequent transfection into HEK 293T cells resulted in significantly higher IFN-β promoter activity compared to when RNA from cells expressing WSN, NT60, or active site mutant WSN PB1a polymerase was transfected (Fig. 3a).
Figure 3. The PB2 polymerase subunit of highly virulent influenza A viruses promotes mvRNA synthesis.

(a) Analysis of mvRNA levels using primer extension (top gel) or RT-PCR (second gel) during the replication of a segment 5-based 246 nt RNA template by the WSN, BM18, NT60 and FJ02 polymerases, and IFN-β promoter activity induced by the transfection of total RNA isolated from these cells into reporter HEK 293T cells expressing luciferase. NP and PB1 expression was assessed by western blot. n=3 biologically independent experiments. P-values were determined using ANOVA with adjustments for multiple corrections compared to WSN. (b) Location of PB2 amino acid residues 9, 64 and 81 in the bat influenza A virus polymerase structure (PDB 4WSB). (c) Analysis of the effect of PB2 mutations on mvRNA formation using RT-PCR (top gel) and IFN-β promoter activity induced after transfection of total RNA isolated from these cells into luciferase reporter HEK 293T cells. PB2, NP and tubulin expression was analysed by western blot. P-values were determined using ANOVA with adjustments for multiple corrections compared to WSN. n=4 biologically independent experiments for all WSN mutants in top graph. n=3 biologically independent experiments for BM18 in top graph and all samples in bottom graph. (d) IFN-β promoter activity as function of PB2 mutation and mvRNA formation. Each data point was generated using the biologically independent experiments presented in c. P-values were determined using linear regression. All graphs show standard deviation and mean.
The identification of mismatches during the generation of mvRNAs (see Fig. 2e) suggests that mvRNA production might be dependent on polymerase fidelity. To investigate this further, we introduced a V43I mutation, which has been shown to confer high-fidelity on an H5N1 influenza virus polymerase22, into the PB1 subunit of the BM18 polymerase (BM18hf). We found that mvRNA levels were significantly reduced in the presence of BM18hf, with a corresponding reduction in IFN-β promoter activity (Fig. 3a). Together, the observations in Fig. 2a, Fig. 3, and Supplementary Fig. 3a suggest that dysregulation of viral RNA replication, e.g. by limiting NP availability, and replication by HPAIV polymerases in mammalian cells generates mvRNAs by employing an error-prone copy-choice mechanism, such as proposed for recombination in positive-strand RNA viruses23.
We next asked whether a particular BM18 polymerase subunit is the determinant of mvRNA production and replaced individual polymerase subunits of the BM18 polymerase with subunits of the WSN polymerase in the 246 nt RNA replication assay. We found that particularly replacement of the BM18 PB2 subunit with the WSN PB2 subunit eliminated the generation of mvRNAs (Supplementary Fig. 4a). Interestingly, the BM18 influenza PB2 subunit has been linked to the enhancement of both the kinetics and the magnitude of the host response to viral infection, leading to the induction of strong inflammatory responses in the lungs of infected mice24. To identify PB2 amino acids involved in mvRNA formation, we aligned the BM18, WSN, NT60 and FJ02 PB2 sequences and found four amino acids that distinguish the BM18 and FJ02 polymerases from the WSN and NT60 polymerases: 9 (D→N), 64 (M→T), 81 (T→M), and 661 (A→T) (Supplementary Fig. 4a). Each of these amino acids has been implicated in avian to mammalian host adaptation25 and, interestingly, all three N-terminal PB2 adaptive amino acids map to the template exit channel of the RNA polymerase (Fig. 3b)6. We generated single mutations N9D, T64M, M81T, and double mutations N9D+T64M and N9D+M81T in the PB2 subunit of the WSN polymerase and found that mutants N9D and M81T and the double mutants N9D+T64M and N9D+M81T significantly increased mvRNA formation (Fig. 3c) and IFN-β promoter activity (Fig. 3c,d). However, the levels of mvRNAs generated by these mutants did not reach the levels generated by the BM18 polymerase indicating that further amino acids contribute to mvRNA production. In line with our observations, WSN viruses that contain a PB2 N9D substitution or other PB2 mutations near the template exit channel have been reported to induce higher IFN-β expression than wild-type WSN26,27.
To address whether mvRNAs form during infection of mammalian cells, we infected A549 cells with WSN, the highly pathogenic avian strain A/Vietnam/1203/04 (H5N1) (abbreviated as VN04), and the VN04 virus with the PB1 V43I high-fidelity mutation (abbreviated as VN04hf). Infections with VN04 resulted in high levels of mvRNAs, while WSN infections produced only very low levels (Fig. 4a). Infections with VN04hf resulted in significantly reduced mvRNA levels compared to the wild-type VN04 virus. These results demonstrate that mvRNAs are formed during influenza virus infection of lung epithelial cells and that polymerase fidelity is an important determinant of mvRNA formation (Fig. 4a).
Figure 4. Levels of mvRNAs produced during infection correlate with innate immune responses.
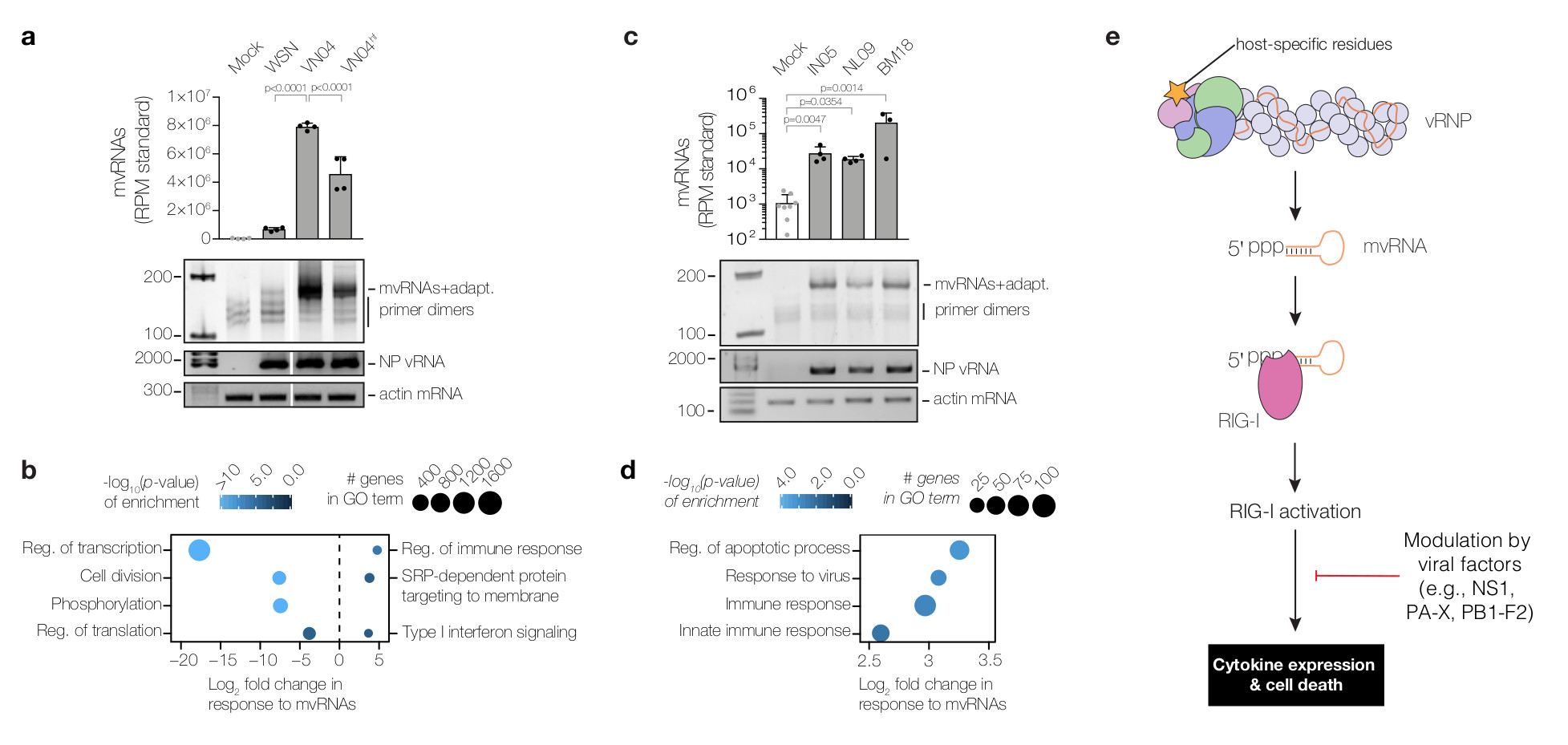
(a) Analysis of mvRNAs in A549 cells infected with WSN, VN04, VN04hf using deep sequencing or PAGE. mvRNAs were amplified using universal primers containing adapters for sequencing (mvRNAs+adapt). mvRNA counts were normalised to mvRNA and mcRNA internal standards. NP vRNA and actin mRNA levels were analysed by RT-PCR. P-values were determined using ANOVA with adjustments for multiple corrections compared to the mock. (b) Analysis of mRNAseq of infected A549 cells showing GO terms down-regulated (left) and GO terms up-regulated (right) in VN04 infection as compared to VN04hf in response to mvRNA levels. P-values were determined using a one-sample z-test (see Methods). Data are from n=4 biologically independent experiments (a,b). (c) Analysis of mvRNAs in lungs of ferrets one day after infection with IN05, NL09 or BM18 using deep sequencing or PAGE. NP vRNA and actin mRNA levels were analysed by RT-PCR. P-values were determined as in a. (d) Analysis of tissue mRNAseq showing GO terms enriched as function of mvRNA levels in lungs of ferrets infected with IN05, NL09 and BM18 influenza viruses. Data are from n=4 biologically independent experiments with separate mock samples for BM18, and IN05 and NL09 (c,d). One BM18 ferret was excluded from the analysis. P-values were determined as in b. (e) Model for the expression of cytokines in influenza virus infected cells. In a and c graphs show standard deviation and mean.
To investigate whether there is a link between mvRNA production and virus-induced innate immune responses we performed RNAseq of cells infected with VN04 and VN04hf viruses and examined which genes were differentially expressed in response to mvRNA levels. Despite significantly different mvRNA levels produced by VN04 and VN04hf, viral mRNA levels were similar (Supplementary Data 1), in agreement with previous findings that the V43I mutation has only a marginal effect on virus replication22. Gene Ontology (GO) analysis (Fig. 4b) showed that basic cellular functions were significantly compromised in VN04 infection relative to VN04hf, consistent with a greater level of cell death, which is known to exacerbate inflammation28. In addition, we observed that genes associated with innate immune responses showed a significant increase in expression in response to higher mvRNA levels (Fig. 4b). Overall, these observations are indicative of a link between erroneous polymerase activity, mvRNA synthesis, and innate immune activation and the induction of cell death. Furthermore, as VN04 exhibited a 10-fold higher lethality compared to V04hf in mice22, our data also suggest a link between mvRNA levels and virulence.
To address whether mvRNAs are produced in infection of animal models, we analysed RNA samples from ferret lungs one and three days after infection with highly pathogenic avian A/Indonesia/5/2005 (H5N1) (abbreviated as IN05), 2009 swine-origin pandemic A/Netherlands/602/2009 (H1N1) (abbreviated as NL09) or the BM18 pandemic virus29,30. mvRNAs were present in all infected lung samples one day after infection, with mvRNA levels particularly high in the BM18 infected ferret lungs (Fig. 4c, Supplementary Fig. 5). GO analysis on the ferret lung samples taken one and three days post infection, showed an up-regulation of apoptosis and innate immune responses as function of mvRNA level, independently of viral titre or the day post infection (Fig. 4d).
In summary, we identify mvRNAs, a class of influenza virus RNAs, that act as the main agonists of the pathogen recognition receptor RIG-I during influenza virus infection (Fig. 4e). mvRNAs are produced as a result of aberrant replication of the viral RNA genome by the viral RNA polymerase. Polymerase fidelity and host-specific amino acids are determinants of the ability of the viral polymerase to produce mvRNAs, which are distinct from DI RNAs and full length viral RNA segments in that they can be efficiently replicated in the absence of NP and do not form canonical RNPs17. These features of mvRNAs are likely to be critical for their preferential recognition by RIG-I over DI RNAs and full-length RNA segments. We further demonstrate that mvRNA production is linked to increased cytokine expression and cell death. Our observations thus strongly suggest that mvRNAs are a contributing factor to influenza virus virulence. We speculate that production of high levels of mvRNAs by the polymerases of the 1918 pandemic and highly pathogenic avian influenza viruses and the resulting increased innate immune activation contribute to the cytokine storm phenomenon underlying the high virulence of these viral strains. The effects of mvRNAs are likely modulated by viral factors, such as the immunomodulatory NS1 and PB1-F2 proteins12 (Fig. 4e). Further studies are required to assess mvRNA levels generated by various influenza virus strains, including seasonal strains, and their effect on virulence.
Methods
Ethics and biosafety
All work with highly pathogenic H5N1 viruses in A549 cells was conducted in the Biosafety Level-3 laboratory at the LKS Faculty of Medicine, The University of Hong Kong, under guidelines and ethics approved by the Committee on the Use of Live Animals in Teaching and Research (CULATR). Ferret experiments with IN05 and NL09 were described previously29 and conducted in the Biosafety Level-3 laboratory of the Erasmus Medical Centre in compliance with European guidelines (EU directive on animal testing 86/609/EEC) and Dutch legislation (Experiments on Animals Act, 1997), after approval by the independent animal experimentation ethical review committee of the Netherlands Vaccine Institute (permit number 200900201). Ferret experiments with BM18 were described previously30 and approved by Institutional Animal Care and Use Committee of Rocky Mountain Laboratories, National Institutes of Health, and conducted in an Association for Assessment and Accreditation of Laboratory Animal Care international-accredited facility according to the guidelines and basic principles in the United States Public Health Service Policy on Humane Care and Use of Laboratory Animals, and the Guide for the Care and Use of Laboratory Animals. Sample inactivation and shipment was performed according to standard operating procedures for the removal of specimens from high containment and approved by the Institutional Biosafety Committee.
Plasmids
Plasmids expressing the three polymerase subunits and NP of influenza A/WSN/33 (H1N1)31, A/Northern Territory/60/68 (H3N2)32, A/duck/Fujian/01/02 (H5N1)32 (all pcDNA3-based), and A/Brevig Mission/1/18 (H1N1)33 (pCAGGS-based) have been described. A PB2 E627K mutation was introduced into the A/duck/Fujian/01/02 (H5N1) PB2 subunit to enable the FJ02 polymerase to efficiently replicate vRNA in mammalian cells. Plasmids expressing mutant PB1a (D445A/D446A)34, and mutant PB2 (N9D)26, of influenza A/WSN/33 (H1N1) virus have been described previously. Full-length or internally truncated vRNAs were expressed from plasmids under the control of cellular RNA polymerase I promoter35. Luciferase reporter plasmid under the control of the IFN-β promoter (pIFΔ(-116)lucter), the β-galactosidase reporter plasmid (pJatLacZ) under the control of a constitutive promoter (β-gal), pcDNA-Myc-RIG-I expressing myc-tagged RIG-I, and pcDNA-myc-proEGF have been described previously36,37. To construct plasmids expressing mutant PB1, PB2 proteins and myc-RIG-I (myc-RIG-I mut; which contains the mutations K851A, K858A and K861A), the plasmids expressing wild-type proteins were subjected to site-directed mutagenesis using the primers listed in Supplementary Table 1.
Cells and antibodies
Human embryonic kidney HEK 293T cells were originally sourced from the ATCC, stored in the Dunn School cell bank at the University of Oxford, and mycoplasma tested, but not authenticated prior to our experiments. A549 cells were originally sourced from the ATCC and cultured at the University of Hong Kong. Cells were cultured in DMEM (Sigma-Aldrich) and 10% FCS. Western blots were performed using NP antibody GTX125989 (GeneTex), Myc antibody GTX115046 (GeneTex), RIG-I antibody GTX85488 (GeneTex), and PB2 antibody GTX125926 (GeneTex). Wild-type and RIG-I knockout HEK 293T cells expressing luciferase in response to the activation of the IFN-β promoter were described previously38.
Statistical testing
In all figures, error bars indicate standard deviation with sample sizes as indicated in figures or figure legends. Evaluation of the statistical significance between group means was performed across all experiments according to the following criteria: (i) in the case where a comparison of a single variable was made between only two groups, an unpaired t-test was used; (ii) in the case of comparisons between three of more groups of measurements derived from a single independent variable (e.g. IFN-β induction as a function of RNA length), one-way ANOVA was used and P-values were corrected for multiple comparisons using either Dunnett’s test (when a single group was taken as a reference/control to which all other groups were compared) or the Bonferroni method (when specific pairs of groups were compared to one another); (iii) in the case of comparisons between three of more groups of measurements derived from two independent variables (e.g. IFN-β induction as a function of RNA length and RIG-I expression), two-way ANOVA was used and P-values were corrected for multiple testing using the Bonferroni method; (iv) in the case of comparisons between three of more groups of log-distributed data (e.g. viral titres), measured values were first log10 transformed and then compared using one-way ANOVA, with P-values corrected for multiple comparisons by controlling the false discovery rate (FDR) to be <0.05 using the two-stage step-up method of Benjamini, Kreiger, and Yekutieli. For the evaluation of the statistical significance of the relationship between two measured values (e.g. fold increase in IFN-β induction vs. mvRNA level), linear regression analysis was used, with the P-value indicating the probability of the null hypothesis (no linear relationship), and the goodness of fit reported as r2. Statistical testing related to differential gene expression analysis is detailed below, and was performed in R; all other statistical tests were performed using GraphPad Prism.
RNP reconstitution assays and quantitative RNA analysis
RNP reconstitution assays were carried out in 24-well plates out as described previously34,39. Briefly, 0.25 μg of the plasmids pcDNA3-NP, pcDNA3-PB2, pcDNA3-PB1, pcDNA3-PA, and a pPOLI plasmid encoding full-length or truncated vRNA templates (for list of vRNA templates used see Supplementary Table 2) were transfected into HEK 293T cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Twenty-four hours post transfection, RNA was extracted using TRI Reagent (Sigma-Aldrich) and dissolved in RNase free water. For quantitative primer extensions, reverse transcription was carried out using SuperScript III reverse transcriptase (Thermo Fisher Scientific) with 32P-labelled oligonucleotides complementary to vRNA-derived RNA species and ribosomal 5S rRNA (for primers see Supplementary Table 3). cDNA synthesis was stopped with 10 μl loading dye (90% formamide, 10 mM EDTA, xylene cyanole, bromophenol blue) and 32P-labelled cDNAs generated with primer NP- were resolved by 12% denaturing PAGE (19:1 acrylamide/bis-acrylamide, 1x TBE buffer, 7 M urea). 32P-labelled cDNAs generated with primer NP-2 were resolved by 20% denaturing PAGE. The radiolabelled signals were imaged using phosphorimaging on a FLA-5000 scanner (Fuji), and analysed using AIDA (RayTek) and Prism 7 (GraphPad). In all experiments, the apparent RNA levels were background corrected using the PB1 active site mutant (PB1a) signal and normalised to the 5S rRNA control. Statistical analysis of data from at least three independent experiments was carried out using ANOVA.
RNP reconstitution assays and qualitative RNA analysis
For segment-specific qualitative RNA analysis by RT-PCR, RNA was treated with DNase (Promega) for 10 min according to the manufacturer’s instructions and reverse transcribed using SuperScript III and the PB2 primers listed in Supplementary Table 3. cDNA was amplified using Q5 polymerase (NEB) and the primers listed in Supplementary Table 3. PCR products were analysed on 1.5% agarose gels in 0.5x Tris-acetate-EDTA (TAE) buffer. For qualitative RT-PCR using universal primers, DNase treated RNA was reverse transcribed using the Lv3aa and Lv3ga primers listed in Supplementary Table 3 and Superscript III at 37 ºC for 30 min. Second strands synthesis was performed with primer Lv5 and Q5 polymerase (NEB) at 47 ºC for 10 min, followed by a further extension at 72 ºC for 3 min. The primer excess in the reactions was removed by incubating the second strand reaction with 1 U of exonuclease VII (NEB) at 37 degrees Celsius for 1 h. Following inactivation of the exonuclease at 95 ºC for 10 min, the DNA was amplified using Q5 polymerase, and primers P5 and i7 for 25 cycles. PCR products were analysed by 6% PAGE.
Luciferase-based interferon expression assays
For luciferase assays, RNP reconstitutions were performed in wild-type HEK 293T cells or HEK 293T cells engineered to express luciferase from the IFN-β promoter. RNP reconstitutions were performed in a 24-well format by transfecting 0.25 μg of the plasmids pcDNA3-NP, pcDNA3-PB2, pcDNA3-PB1, pcDNA3-PA, a pPOLI plasmid encoding full-length or truncated vRNA templates using lipofectamine2000 (Invitrogen). For RNP reconstitutions in wild-type HEK 293T cells, 100 ng of pIFΔ(-116)lucter and pJatLacZ were co-transfected with the polymerase expressing plasmids. Twenty-four hours post transfection, cells were harvested in PBS and resuspended in Reporter Lysis buffer (Promega). Luciferase activity was measured using a Luciferase Assay System (Promega) and a GloMax (Promega), and normalised using the β-galactosidase signal measured using ortho-Nitrophenyl-β-galactoside (ONPG) and a GloMax. The background was subtracted using signals obtained from cells transfected with an empty pcDNA3. Luciferase levels were corrected for viral RNA levels obtained with primer extensions and a 32P-labelled NP-2 primer (Supplementary Table 3). For total RNA transfections, 100 ng of total RNA was transfected with 100 ng of pIFΔ(-116)lucter and pJatLacZ using Lipofectamine2000. Analysis of luciferase expression was performed as described above. Statistical analysis was carried out using ANOVA.
Immunoprecipitations
For myc-RIG-I immunoprecipitations, 10 cm dishes with HEK 293T cell were transfected with 3 μg pcDNA3-NP, pcDNA3-PB2, pcDNA3-PB1, pcDNA3-PA, pcDNA-myc-RIG-I or pcDNA-myc-EGF, and a pPOLI plasmid encoding either a full-length or truncated vRNA template using Lipofectamine 2000. Twenty-four hours post transfection, the cells were harvested in cold PBS and lysed in 600 μl Tris lysis buffer (50 mM Tris-HCl, pH 8.0; 5% glycerol; 0.5% Igepal; 200 mM NaCl; 1 mM EDTA; 1 mM DTT; and 1x EDTA-free protease inhibitor (Roche)) on ice for 1 h. The lysates were cleared at 10,000 g for 5 min. Six μg of anti-myc antibody (Sigma-Aldrich) was added to 0.5 ml of cleared lysate and mixed at 4 ºC for 1.5 h. The lystate-antibody mix was bound to Dynabeads (Novex) at 4 ºC for 1.5 h, washed 3 times with IgG wash buffer (10 mM Tris-HCl pH 8.0; 150 mM NaCl; 0.1% Igepal; 1 mM PMSF; 1 mM EDTA), and finally analysed for bound RNA and protein. Statistical analysis of data from three independent experiments was carried out using ANOVA.
ATPase assay
For wild-type and mutant myc-RIG-I purification, HEK 293T cell were transfected with 5 μg pcDNA-myc-RIG-I or pcRNA-myc-RIG-I mut using Lipofectamine 2000. Twenty-four hours post transfection, the cells were harvested in cold PBS and lysed in lysis buffer (50 mM Hepes, pH 8.0; 5% glycerol; 0.5% Igepal; 200 mM NaCl; 2 mM MgCl2; 10 mM CaCl2; 1 mM DTT; 1 U/ml Micrococcal Nuclease (Thermo Scientific); and 1x EDTA-free protease inhibitor) on ice for 1 h. Three μg of anti-myc antibody was next added per 0.5 ml of cleared lysate and mixed at 4 ºC for 1.5 h. The lystate-antibody mix was bound to Protein G Mag Sepharose Xtra beads (GE Healthcare) at 4 ºC for 1.5 h, washed 6 times with 20 column volumes of RIG-I wash buffer (50 mM Hepes, pH 8.0; 200 mM NaCl; 0.1% Igepal; 5% glycerol; 1 mM PMSF; 2 mM MgCl2) at 4 ºC for 10 min, and finally myc-RIG-I was eluted from beads in 1 column volume wash buffer containing 0.5 mg/ml c-myc peptide (Pierce) for 15 min at 4 ºC. Activity assays were performed in 50 mM Hepes pH 8.0, 150 mM NaCl, 2 mM MgCl2, 5 mM DTT, and 0.1 μM [γ-32P]ATP. [γ-32P]ATP and 32Pi were resolved using PEI-cellulose TLC plates (Sigma-Aldrich) in 0.4 M KH2PO4 pH 3.4.
Cell and animal infections
HEK 293T cells were infected with influenza A/WSN/33 (H1N1) virus, free of DI RNAs, at a multiplicity of infection (MOI) of 5. RNA was extracted 5 h post infection and analysed using deep sequencing or qualitative RT-PCR. 6-wells containing A549 cells were infected with A/WSN/33 (H1N1), A/Vietnam/1203/04 (H5N1), or A/Vietnam/1203/04 (H5N1) with the V43I mutation with an MOI of 5. RNA was extracted 8 hours post infection and analysed using deep sequencing or qualitative RT-PCR. Ferret (Mustela putorius furo) lung tissue was obtained from male ferrets infected with A/Indonesia/5/2005 (H5N1) or A/Netherlands/602/2009 (H1N1)29, or female ferrets infected with A/Brevig Mission/1/1918 (H1N1)30. Ferrets were randomly assigned to groups of four before inoculation. A single ferret (lung titre = 7.6×101 log10TCID50/g) was excluded from analysis on the basis of its apparent lack of infection. Ferret RNA was isolated from lung tissue samples using Trizol (Invitrogen) and analysed using qualitative RT-PCRs and next generation mvRNA sequencing with universal primers and quantitative mRNA sequencing.
Sequence alignment and structural modelling
PB2 amino acid sequences from influenza A viruses A/WSN/33 (H1N1), A/Brevig Mission/1/18 (H1N1), A/Northern Territory/60/68 (H3N2), and A/duck/Fujian/01/02 (H5N1) were aligned using Muscle 3.0 and visualised using ESPript40. The bat influenza A virus polymerase structure (PDB 4WSB) was visualised in Pymol 1.6.
Next generation sequencing of mvRNAs using adapters
Total cell RNA from transfected or infected cells was isolated using Tri Reagent (Sigma) or Trizol (Invitrogen) according to the manufacturer’s instructions and fractionated into small (17-200 nt) and large (>200 nt) RNA fractions using an RNA Clean and Concentrator kit (Zymo Research). Next, the small RNA fraction was denatured at 70 ºC for 2 min and subsequently treated with 2 U of XRN-1 in NEB buffer 2 at 37 ºC for 15 min to deplete miRNAs. Next, XRN-1 was inactivated by adding 10 mM EDTA and incubating the reaction at 70 ºC for 10 min. Viral triphosphorylated RNAs were converted to monophosphorylated RNAs by adding 5 U of RNA 5' Pyrophosphohydrolase (RppH) and 10 mM MgCl2 and incubating the reactions at 37 ºC for 15 min. RNA was purified using an RNA Clean and Concentrator kit and libraries for deep sequencing were prepared using the NEBNext Small RNA Library Prep Kit according to the manufacturer's instructions. To ensure accurate quantitation after PCR amplification, the concentration of each library was measured by qPCR on a StepOnePlus instrument (ABI) and the number of PCR cycles used to subsequently amplify the remaining library material was calibrated so as to ensure the PCR was in the early stage of exponential amplification and to not over-cycle the PCR reactions. Amplified sequencing libraries were purified on a 6% Novex TBE PAGE according to the manufacturer’s instructions to remove primer-dimers. Paired-end sequencing (2x75bp) on an Illumina HiSeq 4000 was carried out by the Oxford Genomics Centre, Wellcome Trust Centre for Human Genetics (Oxford, UK). It is important to note that the existence of mvRNAs has likely been overlooked till now, because i) RNA isolation protocols vary in their capacity to recover small RNAs, ii) RT-PCR products from mvRNAs form a diffuse fast-migrating band on standard agarose gels that may be mistaken for primer-dimers, iii) conventional RNA deep sequencing protocols discard short library fragments, and iv) standard ligation-based deep sequencing protocols do not detect viral transcripts with a 5′-triphosphate group.
Next generation sequencing of mvRNAs using universal primers
To spike viral RNA for quantitative sequencing, 0.2 μl of 100 pM spike RNA (Supplementary Table 2) was added to 40 ng of the small RNA fraction (see above). The RNA mixture was next converted into cDNA using primers Lv3aa, Lv3ga and Lc3 and Superscript III (Invitrogen) at 37 ºC for 30 min. Second strand synthesis was performed using Q5 polymerase (NEB) and primers Lv5, Lc3a, and Lc3g at 47 ºC for 10 min, followed by a further extension at 72 ºC for 3 min. The excess of barcoded primers was removed by incubating the second strand reaction with 1 U of exonuclease VII (NEB) at 37 ºC for 1 h. The exonuclease was inactivated at 95 ºC for 10 min. Next, the DNA was amplified using Q5 polymerase, primer P5 and i7 index primers (Lexogen), and subsequently sequenced on a NextSeq 500 sequencer (Illumina).
Preparation of reference genome files for deep sequencing of mvRNAs
Prior to mapping, a reference genome file was prepared from relevant viral reference sequences in Genbank (see above). For the analysis of sequencing libraries prepared using universal influenza virus primers, the 5′ and 3′ viral promoter sequences of each segment were modified to match the degenerate universal primer sequences used in sample preparation (see Supplementary Table 3), and the sequences of the spiked-in mvRNA quantitation standards (see Supplementary Table 2) were appended to the reference genome. For WSN, VN04, and VN04hf viruses, deep-sequencing data of mRNA generated in A549 cell infections (above) was exploited to generate updated reference genome files: the mpileup and consensus commands in the bcftools software package41 were used following mapping of non-host mRNA reads to the relevant viral reference genome using STAR aligner42.
Data processing pipeline for deep sequencing of mvRNAs using universal influenza virus primers
Raw sequencing reads were first trimmed to remove sequencing adaptor sequences and reads with quality scores less than 20 using the cutadapt software package43 and the 8-nt unique molecular identifier (UMI) at the start of each read were removed from the sequence and appended to the read ID line of the FASTQ file using the extract command from the umi_tools software package44. Sequencing reads were then mapped end-to-end to the appropriate viral reference genome using the STAR aligner42, and permitting sequencing reads to have long internal deletions (i.e. an mvRNA, interpreted as splicing by STAR) with at least 16 nt anchored on either side of the deletion (--outSJfilterOverhangMin 16 16 16 16). The default settings of STAR were modified so that no alignment scoring penalty was given for an internal deletion and no preference was given to internal deletions that overlapped particular sequence motifs (--scoreGapNoncan 0 --scoreGapGCAG 0 --scoreGapATAC 0), and to ensure accurate quantitation, only the top-scoring alignment was included in the outputted BAM file (--outSAMmultNmax 1), which was sorted and indexed using samtools45. An aligned read was counted as an mvRNAs if it was anchored to the viral reference genome at the 5′ end in vRNA sense and contained an internal deletion (called as a splice junction by STAR), and the total numbers of mvRNAs and spiked-in quantitation standards were tallied using the idxstats command of samtools. mvRNA levels relative to the quantitation were then reported as number of reads per million mapped (RPM) quantitation standard. To validate the qPCR protocol used to prevent over-cycling of sequencing libraries, the quantitation was then repeated following removal of PCR duplicates, exploiting the UMI appended to each read, using the umi_tools dedup command, and counting the various mvRNA species as unique using the --spliced-is-unique option. The identities of the individual mvRNAs were then extracted from the SJ.out.tab file generated by STAR.
Data processing pipeline for deep sequencing of mvRNAs using adapter ligation
Raw sequencing reads were first trimmed using the cutadapt software package43 to remove RNA adapters, sequencing adapters, and reads with quality scores less than 20. Since adapter ligation captures both host-derived and virus-derived small RNA species, reads were first mapped end-to-end to the DASHR database of human small RNAs46 using the STAR aligner42, with spliced alignments disabled (--alignIntronMax 1). Non-human reads were outputted using the --outReadsUnmapped Fastx option, were then mapped to the appropriate viral reference genome to find mvRNAs as described above, and quantitated relative to the total number of viral reads (RPM viral) or host reads (RPM host).
Quantitative mRNA sequencing and differential gene expression analysis
Libraries for gene expression analysis were prepared using a QuantSeq 3' mRNA-Seq Library Prep Kit FWD for Illumina (Lexogen) according to the manufacturer’s instructions and sequenced on a NextSeq 500 sequencer. mRNA reads were aligned to the reference genome (CRCh38, GRCm38, or MusPutFur1.0) using the STAR read aligner42, exploiting the built-in trimming functions to remove the first 12 bases corresponding the Lexogen random primer (--clip5pNbases 12) and any contaminating poly(A) tails in the sequencing reads (--clip3pAdapterSeq AAAAAAAAAAAAAAAAAA), as well as requiring a minimum match to the reference genome of 40 bp (--outFilterMatchNmin 40). Gene counts were generated using reference genome annotations (Gencode v26 for CRCh38, and Ensembl 90 for MusPutFur1.0) using the STAR command --quantMode GeneCounts. Differential gene expression analysis was then carried out using the DEseq2 package in R47 to identify genes that were up- or down-regulated as a function of mvRNA levels, independently of viral load or titre. Specifically, the likelihood ratio test (LRT) was used to compare a full model (in which gene expression varies as a function of both viral load or titre, and mvRNA levels) to a reduced model (in which changes in gene expression are fully explained by viral load or titre alone) using analysis of deviance (ANODEV) to generate a P-value for the log-fold-change of each gene, which were adjusted for multiple testing by controlling the false discovery rate (FDR) using Independent Hypothesis Testing48 and reported as q-values. mvRNA levels were determined by deep sequencing using universal influenza virus primers, as detailed above. Viral load or titre was determined by segment 6 qRT-PCR or by using previously published values29,30. Subsequent enrichment analysis of Gene Ontology terms specifically affected by mvRNA levels was carried out using Parametric Analysis of Gene Set Enrichment49 via the GAGE package in R, with data from the above genome annotations, accessed via the biomaRt package50. Significance of enrichment for GO terms was calculated using a one-sample z-test49 in GAGE, and P-values were adjusted for multiple testing using the Benjamini-Hochberg method and were reported as q-values.
Supplementary Material
Acknowledgements
We greatly value our discussions with R. Sun and Y. Du, who independently found that mutations in the N-terminal region of PB2, near the template exit channel of the polymerase, stimulate interferon induction27. We thank G.G. Brownlee, M. Freeman and F. Vreede for plasmids, J. Rehwinkel and A. Mayer for HEK 293 RIG-I -/- cells, Y. Kawaoka for the A/Brevig Mission/1/1918 (H1N1) virus, and A. Osterhaus and T. Kuiken for A/Indonesia/5/2005 (H5N1) or A/Netherlands/602/2009 (H1N1) infected ferret tissue samples. We thank Ian Sudbery for adding spliced read functionality to the umi_tools package. We thank the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant 090532/Z/09/Z) for the generation of adapter-ligated mvRNA sequencing data. This work was supported by Wellcome Trust grant 098721/Z/12/Z, joint Wellcome Trust and Royal Society grant 206579/Z/17/Z, and a Netherlands Organization for Scientific Research (NWO) grant 825.11.029 to A.J.W.t.V; EPA Cephalosporin Junior Research Fellowship (to D.L.V.B.); support by the Intramural Research Program of NIAID, NIH, to E.d.W.; Research Grants Council of the Hong Kong Special Administrative Region, China, Project No. T11-705/14N to L.L.M.P; and Medical Research Council (MRC) programme grants MR/K000241/1 and MR/R009945/1 to E.F. and studentship to J.C.L.
Footnotes
Author Contributions
J.C.L., E.F., J.S. and A.J.W.t.V showed that subgenomic influenza virus RNAs stimulate IFN-β production and are bound by RIG-I. A.J.W.t.V. and J.C.L. found that mvRNAs are produced by influenza virus polymerases. D.L.V.B. designed sequencing strategies. D.L.V.B. and A.J.W.t.V. performed deep sequencing experiments and analyses. M.J.K., M.J.O.-M., H.F., and R.E.R. contributed reagents and protocols. E.d.W., D.v.R., and J.Y.S. provided ferret lung tissues. R.L.Y.F., H.Y. and L.L.M.P. performed A549 infections. A.J.W.t.V., D.L.V.B., J.C.L., and E.F. analysed data. A.J.W.t.V., J.C.L., D.L.V.B., and E.F. wrote the manuscript with input from co-authors.
Competing interests: Authors declare no competing interests.
Data availability statement: All sequencing data have been deposited in the NCBI Sequence Read Archive (SRA) under accession number SUB3758924. Gene expression data is available as Supplemental Data 1. Supplemental figures and tables are available in the Supplemental information file.
References
- 1.Kash JC, Taubenberger JK. The Role of Viral, Host, and Secondary Bacterial Factors in Influenza Pathogenesis. The American Journal of Pathology. 2015;185:1528–1536. doi: 10.1016/j.ajpath.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kash JC, et al. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;76:105–4. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kobasa D, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- 4.de Jong MD, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12:1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pichlmair A, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 6.te Velthuis AJW, Fodor E. Influenza virus RNA polymerase: insights into the mechanisms of viral RNA synthesis. Nat Rev Microbiol. 2016;14:479–493. doi: 10.1038/nrmicro.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kowalinski E, et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147:423–435. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 8.Lee M-K, et al. Structural features of influenza A virus panhandle RNA enabling the activation of RIG-I independently of 5'-triphosphate. Nucleic Acids Res. 2016;44:8407–8416. doi: 10.1093/nar/gkw525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robb NC, et al. Single-molecule FRET reveals the pre-initiation and initiation conformations of influenza virus promoter RNA. Nucleic Acids Res. 2016;44:10304–10315. doi: 10.1093/nar/gkw884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russell AB, Trapnell C, Bloom JD. Extreme heterogeneity of influenza virus infection in single cells. eLife. 2017 doi: 10.1101/193995. pii: e32303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Killip MJ, Jackson D, Pérez-Cidoncha M, Fodor E, Randall RE. Single-cell studies of IFN-β promoter activation by wild-type and NS1-defective influenza A viruses. J Gen Virol. 2017;98:357–363. doi: 10.1099/jgv.0.000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Killip MJ, Fodor E, Randall RE. Influenza virus activation of the interferon system. Virus Research. 2015;209:11–22. doi: 10.1016/j.virusres.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jennings PA, Finch JT, Winter G, Robertson JS. Does the higher order structure of the influenza virus ribonucleoprotein guide sequence rearrangements in influenza viral RNA? Cell. 1983;34:619–627. doi: 10.1016/0092-8674(83)90394-x. [DOI] [PubMed] [Google Scholar]
- 14.Perez JT, et al. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc Natl Acad Sci USA. 2010;107:11525–11530. doi: 10.1073/pnas.1001984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coloma R, et al. The Structure of a Biologically Active Influenza Virus Ribonucleoprotein Complex. PLoS Pathog. 2009;5:e1000491–10. doi: 10.1371/journal.ppat.1000491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, et al. Internal genes of a highly pathogenic H5N1 influenza virus determine high viral replication in myeloid cells and severe outcome of infection in mice. PLoS Pathog. 2018;14:e1006821. doi: 10.1371/journal.ppat.1006821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turrell L, Lyall JW, Tiley LS, Fodor E, Vreede FT. The role and assembly mechanism of nucleoprotein in influenza A virus ribonucleoprotein complexes. Nature Communications. 2013;4 doi: 10.1038/ncomms2589. 1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato H, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 19.Rehwinkel J, et al. RIG-I Detects Viral Genomic RNA during Negative-Strand RNA Virus Infection. Cell. 2010;140:397–408. doi: 10.1016/j.cell.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 20.Killip MJ, Smith M, Jackson D, Randall RE. Activation of the Interferon Induction Cascade by Influenza A Viruses Requires Viral RNA Synthesis and Nuclear Export. J Virol. 2014;88:3942–3952. doi: 10.1128/JVI.03109-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dulin D, et al. Backtracking behavior in viral RNA-dependent RNA polymerase provides the basis for a second initiation site. Nucleic Acids Res. 2015;43:10421–10429. doi: 10.1093/nar/gkv1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung PPH, et al. Generation and characterization of influenza A viruses with altered polymerase fidelity. Nature Communications. 2014;5:1–13. doi: 10.1038/ncomms5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woodman A, Arnold JJ, Cameron CE, Evans DJ. Biochemical and genetic analysis of the role of the viral polymerase in enterovirus recombination. Nucleic Acids Res. 2016;44:6883–6895. doi: 10.1093/nar/gkw567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forero A, et al. The 1918 Influenza Virus PB2 Protein Enhances Virulence through the Disruption of Inflammatory and Wnt-Mediated Signaling in Mice. J Virol. 2016;90:2240–2253. doi: 10.1128/JVI.02974-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miotto O, Heiny AT, Tan T, August JT, Brusic V. Identification of human-to-human transmissibility factors in PB2 proteins of influenza A by large-scale mutual information analysis. BMC Bioinformatics. 2008;9:S18–18. doi: 10.1186/1471-2105-9-S1-S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graef KM, et al. The PB2 Subunit of the Influenza Virus RNA Polymerase Affects Virulence by Interacting with the Mitochondrial Antiviral Signaling Protein and Inhibiting Expression of Beta Interferon. J Virol. 2010;84:8433–8445. doi: 10.1128/JVI.00879-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du Y, et al. Genome-wide identification of interferon-sensitive mutations enables influenza vaccine design. Science. 2018;359:290–296. doi: 10.1126/science.aan8806. [DOI] [PubMed] [Google Scholar]
- 28.Chan FK-M, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol. 2015;33:79–106. doi: 10.1146/annurev-immunol-032414-112248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van den Brand JMA, et al. Comparison of Temporal and Spatial Dynamics of Seasonal H3N2, Pandemic H1N1 and Highly Pathogenic Avian Influenza H5N1 Virus Infections in Ferrets. PLoS ONE. 2012;7:e42343–21. doi: 10.1371/journal.pone.0042343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Wit E, et al. 1918 H1N1 influenza virus replicates and induces pro-inflammatory cytokine responses in extra-respiratory tissues of ferrets. J Infect Dis. 2018 doi: 10.1093/infdis/jiy003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fodor E, et al. A Single Amino Acid Mutation in the PA Subunit of the Influenza Virus RNA Polymerase Inhibits Endonucleolytic Cleavage of Capped RNAs. J Virol. 2002;76:8989–9001. doi: 10.1128/JVI.76.18.8989-9001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kashiwagi T, Leung BW, Deng T, Chen H, Brownlee GG. The N-Terminal Region of the PA Subunit of the RNA Polymerase of Influenza A/HongKong/156/97 (H5N1) Influences Promoter Binding. PLoS ONE. 2009;4:e5473. doi: 10.1371/journal.pone.0005473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tumpey TM, et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. 2005;310:77–80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- 34.Vreede FT, Jung TE, Brownlee GG. Model Suggesting that Replication of Influenza Virus Is Regulated by Stabilization of Replicative Intermediates. J Virol. 2004;78:9568–9572. doi: 10.1128/JVI.78.17.9568-9572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fodor E, et al. Rescue of influenza A virus from recombinant DNA. J Virol. 1999;73:9679–9682. doi: 10.1128/jvi.73.11.9679-9682.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Childs KS, Andrejeva J, Randall RE, Goodbourn S. Mechanism of mda-5 Inhibition by Paramyxovirus V Proteins. J Virol. 2009;83:1465–1473. doi: 10.1128/JVI.01768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zettl M, Adrain C, Strisovsky K, Lastun V, Freeman M. Rhomboid Family Pseudoproteases Use the ER Quality Control Machinery to Regulate Intercellular Signaling. Cell. 2011;145:79–91. doi: 10.1016/j.cell.2011.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hertzog J, et al. Infection with a Brazilian isolate of Zika virus generates RIG-I stimulatory RNA and the viral NS5 protein blocks type I IFN induction and signaling. Eur J Immunol. 2018;46:509. doi: 10.1002/eji.201847483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oymans J, te Velthuis AJW. A mechanism for prime-realignment during influenza A virus replication. J Virol. 2018;92:e01773–17. doi: 10.1128/JVI.01773-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–4. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal. 2011;17:10. [Google Scholar]
- 44.Smith T, Heger A, Sudbery I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Research. 2017;27:491–499. doi: 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leung YY, et al. DASHR: database of small human noncoding RNAs. Nucleic Acids Res. 2016;44:D216–D222. doi: 10.1093/nar/gkv1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ignatiadis N, Klaus B, Zaugg JB, Huber W. Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat Methods. 2016;13:577–580. doi: 10.1038/nmeth.3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim S-Y, Volsky DJ. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. doi: 10.1186/1471-2105-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nature Protocols. 2009;4:1184–1191. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.