

Abstract
Background
Exercise intolerance is a common clinical feature and is linked to poor prognosis in patients with heart failure (HF). Skeletal muscle dysfunction, including impaired energy metabolism in the skeletal muscle, is suspected to play a central role in this intolerance, but the underlying mechanisms remain elusive. Lysine acetylation, a recently identified post‐translational modification, has emerged as a major contributor to the derangement of mitochondrial metabolism. We thus investigated whether mitochondrial protein acetylation is associated with impaired skeletal muscle metabolism and lowered exercise capacity in both basic and clinical settings of HF.
Methods
We first conducted a global metabolomic analysis to determine whether plasma acetyl‐lysine is a determinant factor for peak oxygen uptake (peak VO2) in HF patients. We then created a murine model of HF (n = 11) or sham‐operated (n = 11) mice with or without limited exercise capacity by ligating a coronary artery, and we tested the gastrocnemius tissues by using mass spectrometry‐based acetylomics. A causative relationship between acetylation and the activity of a metabolic enzyme was confirmed in in vitro studies.
Results
The metabolomic analysis verified that acetyl‐lysine was the most relevant metabolite that was negatively correlated with peak VO2 (r = −0.81, P < 0.01). At 4 weeks post‐myocardial infarction HF, a treadmill test showed lowered work (distance × body weight) and peak VO2 in the HF mice compared with the sham‐operated mice (11 ± 1 vs. 23 ± 1 J, P < 0.01; 143 ± 5 vs. 159 ± 3 mL/kg/min, P = 0.01; respectively). As noted, the protein acetylation of gastrocnemius mitochondria was 48% greater in the HF mice than the sham‐operated mice (P = 0.047). Acetylproteomics identified the mitochondrial enzymes involved in fatty acid β‐oxidation (FAO), the tricarboxylic acid cycle, and the electron transport chain as targets of acetylation. In parallel, the FAO enzyme (β‐hydroxyacyl CoA dehydrogenase) activity and fatty acid‐driven mitochondrial respiration were reduced in the HF mice. This alteration was associated with a decreased expression of mitochondrial deacetylase, Sirtuin 3, because silencing of Sirtuin 3 in cultured skeletal muscle cells resulted in increased mitochondrial acetylation and reduced β‐hydroxyacyl CoA dehydrogenase activity.
Conclusions
Enhanced mitochondrial protein acetylation is associated with impaired FAO in skeletal muscle and reduced exercise capacity in HF. Our results indicate that lysine acetylation is a crucial mechanism underlying deranged skeletal muscle metabolism, suggesting that its modulation is a potential approach for exercise intolerance in HF.
Keywords: Heart failure, Exercise intolerance, Lysine acetylation, Skeletal muscle metabolism, Fatty acid oxidation
Introduction
Exercise intolerance, defined as dyspnea and fatigue on exertion, is a clinical hallmark of patients with heart failure (HF). As the objective assessment of exercise intolerance, a patient's peak oxygen uptake (peak VO2) is determined by cardiopulmonary exercise testing (CPX) and is well known as an independent predictor of prognosis in HF patients.1 Because exercise tolerance is not directly correlated with cardiac function and hemodynamics, the role of peripheral tissues, especially skeletal muscles, has been a focus of exercise‐tolerance research for several decades.2, 3
The intrinsic skeletal muscle abnormalities in HF include factors such as mitochondrial dysfunction,4 fibre‐type transition,5 and muscle atrophy,6 but impaired energy metabolism7 is considered the primary mechanism of the easy fatigability and exercise intolerance in HF settings.8 Indeed, our previous studies using magnetic resonance spectroscopy demonstrated that an increased accumulation of intramyocellular lipid is associated with exercise intolerance.9 This is in line with early findings of the reduced activity of diverse mitochondrial oxidative enzymes in biopsied skeletal muscle from HF patients.10 However, the exact mechanisms underlying these enzymatic alterations remain to be clarified.
Lysine acetylation, a reversible post‐translational modification, has gained much attention as a novel regulatory mechanism of mitochondrial energy metabolism.11, 12 The target proteins of acetylation include fatty acid β‐oxidation enzymes, glycolytic enzymes, the tricarboxylic acid (TCA) cycle, and the electron transport chain, resulting in the inhibition or activation of their enzyme activities.13 Of these, the acetylation of fatty acid β‐oxidation was shown to be a contributing factor to the cardiac metabolic derangements in diabetes and HF.14, 15, 16, 17 Sirtuin 3 (SIRT3), a nicotinamide adenine dinucleotide (NAD+)‐dependent deacetylase, is a major regulator of mitochondrial acetylation, and has been shown to decline in the failing heart.18, 19, 20
Although there are controversies regarding the effect of lysine acetylation on myocardial fatty acid oxidation,14, 16 several lines of evidence indicate that an increased acetylation of fatty acid β‐oxidation enzymes is linked to energy metabolic disturbances in the pathogenesis of exercise intolerance associated with HF.21, 22, 23 In addition, hyperacetylation due to decreased SIRT3 has been reported to facilitate skeletal muscle insulin resistance in diabetic animals.24, 25 We thus hypothesized that mitochondrial protein acetylation could contribute to impaired energy metabolism in the skeletal muscle that is primarily or secondarily linked to the development of exercise intolerance in HF.
Using a reverse translational approach, we first identified the circulating metabolites associated with peak VO2 in HF patients by conducting a comprehensive metabolomic analysis. We then created a murine model of HF after myocardial infarction (MI) with limited exercise capacity and skeletal muscle abnormalities, in order to determine the role of acetylation in mitochondrial fatty acid oxidation and exercise capacity.
Methods
Human studies
Ten patients with stable HF (57 ± 10 years old, New York Heart Association [NYHA] functional class II or III) and five age‐matched healthy controls (56 ± 3 years old) without underlying heart diseases were studied. HF was diagnosed on the basis of the Framingham criteria. All participants underwent CPX on an upright electromechanical bicycle ergometer (Aerobike 75XLII; CombiWellness, Tokyo) using a ramp protocol described in our previous studies.26, 27 Briefly, after 3 min of unloaded cycling, the exercise load was increased in 10 to 15 W increments every 1 min in HF patients and 25 W increments per min in the control subjects to symptom‐limited maximal work. The patients stopped the exercise when they had severe leg fatigue and/or dyspnea. Although it is possible that a different protocol could have affected the value of peak exercise capacity, this protocol was necessary for the symptomatic HF patients to achieve a fatigue‐limited exercise duration of 8–12 min based on the American Heart Association recommendation.28
Oxygen uptake was measured at rest and throughout the exercise period using a 280E Aero‐monitor (AeromonitorAE‐300S; Minato Medical Science, Osaka, Japan). Peak VO2 was defined as the maximal VO2 attained during exercise, and the anaerobic threshold was determined by the V‐slope method.29 We measured the participants' minute ventilation (VE) and carbon dioxide production (VCO2) responses throughout the exercise to calculate the VE/VCO2 slope via least squares linear regression (y = mx + b; m = slope). In echocardiographic measurements, each participant's left ventricular (LV) end‐diastolic dimension and LV end‐systolic dimension were obtained from the parasternal long‐axis view, and the LV ejection fraction was calculated based on a modification of Simpson's method.
Within 3 months after each participant's CPX was performed, blood samples were obtained from the participants after an overnight fast and were stored at −80°C until analysis. The interval between the blood sampling and the CPX was necessary to minimize the possibility that the metabolomic profile was a collective snapshot of all of the metabolic perturbations and therefore influenced by various confounding factors such as other disease states, acute illnesses, or medication use.
A 50 μL sample of plasma was mixed with 450 μL of methanol containing internal standards (20 μM of methionine sulfone and D‐camphor‐10‐sulfonic acid), and then 500 μL of chloroform was added. The mixture was centrifuged at 2,300 g for 5 min at 4°C. Subsequently, 400 μL of the upper aqueous layer was centrifugally filtered through a 5‐kDa‐cutoff ultra‐centrifugal filter unit (Ultrafree®‐MC‐PLHCC‐HMT; MilliporeSigma, Burlington, MA) at 9,100 g for 2 h at 4°C. The filtrate was dried and re‐suspended in 25 μL of Milli‐Q water for a capillary electrophoresis‐mass spectrometry (CE‐MS) analysis.
Metabolomic measurements were carried out using an Agilent CE capillary electrophoresis system equipped with an Agilent 6210 Time‐of‐Flight (TOF) MS, Agilent 1100 isocratic HPLC pump, Agilent G1603A CE‐MS adapter kit, and Agilent G1607A CE‐electrospray ionization‐MS sprayer kit (Agilent Technologies, Waldbronn, Germany) as described previously.30 For the CE‐TOFMS system control and data analysis, Agilent G2201AA ChemStation software ver. B.03.01 for CE (Agilent Technologies) was used. The CE‐TOFMS datasets were processed using the automatic integration software MasterHands (Keio University, Tsuruoka, Japan) in order to obtain peak information including the m/z, the migration time for the CE‐TOFMS measurement, and the peak area.31 Signal peaks corresponding to isotopomers, adduct ions, and other product ions of known metabolites were excluded, and the remaining peaks were annotated with putative metabolites from the database of the Human Metabolome Technologies according to their migration time and m/z values determined by CE‐TOFMS. This study was performed under protocols approved by the Medical Ethics Committee of Hokkaido University Hospital and in accord with the ethical principles in the Declaration of Helsinki (2013). Informed consent was obtained from all participants.
Experimental animals
Male C57BL6J mice (10–12 weeks old, body weight [BW] 23–25 g; CLEA Japan, Tokyo) were bred in a pathogen‐free environment and housed in an animal room under controlled conditions on a 12 h light/dark cycle maintained at 23–25°C. Normal chow containing 4.2% fat and 54.6% carbohydrate (CE‐2; CLEA Japan, Tokyo) and water were provided ad libitum. The post‐MI HF model was established by ligating the left coronary artery as described previously.32, 33 The permanent coronary occlusion model is recommended as a model of acquired dilated cardiomyopathy and HF, which usually occur 4–8 weeks post‐MI, according to a scientific statement from the American Heart Association.34 A sham operation without ligation of the coronary artery was also performed; the experiments were performed 4 weeks after the operation in both the sham (n = 11) and HF (n = 11) groups. For each surgical procedure, the mice were anaesthetised with an intraperitoneal injection of a mixture of 0.3 mg/kg of medetomidine (Dorbene® Kyoritsuseiyaku, Tokyo), 4.0 mg/kg of midazolam (Dormicum®, Astellas Pharma, Tokyo), and 5.0 mg/kg of butorphanol (Vetorphale®, Meiji Seika Kaisha, Tokyo), and the adequacy of the anaesthesia was monitored based on the disappearance of the pedal withdrawal reflex.
All procedures and animal care were approved by our institutional animal research committee and conformed to the animal care guidelines for the Care and Use of Laboratory Animals at the Hokkaido University Graduate School of Medicine. The procedures and care were also in accord with the relevant national and international guidelines and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Echocardiography, histological analysis, treadmill test, and physical activity measurement
Standard echocardiographic short‐axis and long‐axis views were obtained at the levels of the papillary muscles under light anaesthesia using tribromoethanol/amylene hydrate (avertin; 2.5% wt/vol, 8 μL/g BW, i.p.). We measured the LV endo‐diastolic diameter, LV endo‐systolic diameter, wall thickness, and LV fractional shortening from M‐mode frames at a paper speed of 50 mm/s. For histological analysis, LV tissues were fixed in 6% formaldehyde, cut into three transverse sections—the apex, middle ring, and base—and stained with Masson's trichrome. We subjected the mice to a treadmill test with a gas analysis for the measurement of indexes defining whole body exercise capacity, as described previously.35 In short, each mouse was forced to run on a motor‐driven treadmill enclosed by a metabolic chamber with constant air flow (Oxymax 2; Columbus Instruments, Columbus, OH, USA) in which the O2 and CO2 gas fractions were monitored.
After basal measurements and following a 10 min warm‐up at 6 m/min at 0° inclination, the angle was fixed at 10° and the speed was incrementally increased by 2 m/min until the mouse reached exhaustion. Exhaustion was defined as spending more than 10 s on the shocker plate without attempting to re‐engage the treadmill. Whole‐body VO2 and VCO2 were automatically acquired every 10 s by taking the difference between the inlet and output gas flows. The respiratory exchange ratio was calculated as the ratio of VCO2 to VO2. Work was defined as the product of the vertical running distance to exhaustion and BW.
Spontaneous physical activity was measured by an animal movement analysis system (ACTIMO System; Shintechno, Fukuoka, Japan) as described previously.36
Study protocol
The study protocol is illustrated in Figure S1. Briefly, 4 weeks after the MI surgery, the spontaneous physical activity of the mice was measured and then the treadmill test was conducted. Within 3 days after the measurement, echocardiography was performed. The mice were then euthanized by cervical dislocation under deep anaesthesia using additional avertin (2.5% wt/vol, total 10 μL/g BW, i.p.). The blood, heart, lungs, and hindlimb skeletal muscles including the quadriceps, gastrocnemius, and soleus were immediately collected for further analysis. To avoid direct effects of the treadmill test on the muscle acetylome analysis, we used a ≥2 day interval between the treadmill test and the tissue sampling.
Western blotting
Forty milligrams of frozen gastrocnemius was homogenized with Cell Lysis Buffer (Cell Signaling Technology, Danvers, MA, USA) supplemented with protease inhibitor cocktail (Roche, Basel, Switzerland) and deacetylase inhibitors (10 mM nicotinamide, 10 μM trichostatin A, and 10 mM sodium butylate). Mitochondrial fractions were also collected using a mitochondrial isolation kit (Thermo Scientific). Twenty micrograms of denatured proteins was subjected to 8–12% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis on a polyvinylidene difluoride membrane.
Immunoblotting was performed with antibodies against acetylated‐lysine, SIRT3 (Cell Signaling Technology), peroxisome proliferator‐activated receptor gamma coactivator 1‐α, mitochondrially encoded cytochrome c, CD36, carnitine palmitoyltransferase 1 (CPT1), CPT2, acyl‐CoA dehydrogenase (ACADL), and enoyl‐CoA hydratase and 3‐hydroxyacyl CoA dehydrogenase (Abcam, Cambridge, MA, USA). After being blocked in 5% fat‐free milk for 1 h, the membranes were probed with the primary antibodies and then incubated with the appropriate secondary antibodies (Abcam) with 5% fat‐free milk for 1 h at room temperature. Bands were detected using the enhanced chemiluminescence method.
Mass spectrometry‐based acetylome analysis
For the acetylation analysis, gastrocnemius tissues were lysed with IP buffer (50 mM Tris HCl, 150 mM NaCl, 5 mM EDTA, 0.5% NP‐40, 1% Triton‐X) supplemented with protease inhibitor cocktail (Roche, Basel, Switzerland) and the earlier‐described deacetylase inhibitors. A total of 500 μg of lysates was precleared with 40 μL of protein A/G‐agarose beads. The lysates were then incubated with acetyl‐lysine antibodies (3 μg/100 μg lysate; Cell Signaling Technology) overnight at 4°C, and 50 μL of protein A‐agarose beads was added to each sample and incubated on a rotator for 6 h at 4°C as described previously.17, 37 After 6 h, the samples were washed three times and centrifuged at 16,000 g for 5 min. The bound acetylated proteins were boiled at 100°C for 15 min in 60 μL of 1.0035× LDS buffer.
Beads were pelleted and 50% of the eluates were processed by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis using a 10% Bis‐Tris NuPAGE gel (Invitrogen, Carlsbad, CA, USA) with the MES buffer system. The mobility region was excised into 10 equal‐sized segments and subjected to in‐gel trypsin digestion. The enriched lysine‐acetylated peptides were then analysed by nano‐liquid chromatography–tandem mass spectrometry with a NanoAcquity HPLC system (Waters, Milford, MA, USA) interfaced with a ThermoFisher Q Exactive HF hybrid mass spectrometer (Thermo Scientific, Tokyo, Japan).
Mascot database search software (ver. 2.4, Matrix Science, London) was used to search for and identify lysine‐acetylated peptides. Mascot DAT files were parsed into Scaffold (Proteome software) for validation and filtering and to create a non‐redundant list per sample. Variable modifications included the acetylation of the ε‐amino group of lysine, oxidation of methionine to methionine sulfoxide, N‐terminal pyroglutamate, and the deamidation of asparagine residues; a search was also performed for carbamidomethylation of cysteine as a fixed modification. The peptide mass tolerance was set to 10 ppm for precursors and 0.02 Da for fragment ions.
The search results were filtered using a 1% protein and peptide false discovery rate with a requirement of at least two unique peptides per protein. To approximate the relative abundance of proteins within a given sample, we calculated the normalized spectral abundance factor using an in‐house program as described previously.38 A binary difference in proteins with a t‐test P‐value < 0.05 or with a 1.5‐fold or more change in the average normalized spectral abundance factor values was defined as significant.
Measurement of nicotinamide adenine dinucleotide metabolites
Approximately 50 mg of frozen gastrocnemius tissues was added to methanol (500 μL) containing internal standards (H3304‐1002; Human Metabolome Technologies, Tsuruoka, Japan), and the mixture was homogenized by a Polytron homogenizer. The lysate was further processed for the CE‐MS analysis described earlier. Nicotinamide adenine dinucleotide (NAD+) and its reduced form (NADH) were quantified using the CE‐electrospray ionization‐MS method.39
Mitochondrial respiratory capacity of the skeletal muscle
After the careful dissection of the muscle tissue, the fibre bundles were permeabilized by gentle agitation for 30 min in an ice‐cold relaxing BIOPS solution (in mmol/L: CaK2EGTA 2.77, K2EGTA 7.23, Na2ATP 5.77, MgCl2·6H2O 6.56, taurine 20, Na2‐phosphocreatine 15, imidazole 20, dithiothreitol 0.5, MES hydrate 50, pH 7.1) with saponin 50 μg/mL. After the permeabilization, the fibres were rinsed twice by agitation for 10 min in an ice‐cold respiration medium, MiR05 (in mmol/L: 248 sucrose 110, K‐lactobionate 60, EGTA 0.5, 0.1% BSA, MgCl2 3, taurine 20, KH2PO4 10, HEPES 20, pH 7.1). We measured the mitochondrial respiratory capacity with non‐fatty‐acid and fatty‐acid substrates in permeabilized gastrocnemius fibre at 37°C by using a high‐resolution respirometer (Oxygraph‐2k; Oroboros Instruments, Innsbruck, Austria) as described previously.36 All respiratory measurements were carried out in duplicate after hyperoxygenation to avoid any potential O2 limitation. Dat‐lab software (Oroboros Instruments) was used for the data acquisition and analysis.
After the addition of permeabilized fibres (approx. 1.5–3.0 mg of gastrocnemius muscle) to the chamber in the respirometer filled with 2 mL of MiR05 and the stabilization of baseline respiratory rates, the following respiratory substrates were added in the following order: (i) malate 2 mM, (ii) octanoyl‐carnitine 0.15 mM, (iii) ADP 5 mM in each step, and (iv) cytochrome c 10 μM. Mitochondrial coupling states are distinguished as leak‐state respiration (without ADP) and oxidative phosphorylation (saturating ADP). We tested the integrity of the outer mitochondrial membrane by adding cytochrome c, and the data were eliminated when the increase in the oxygen consumption rate was 10% as the sign of a damaged outer mitochondrial membrane. The respiratory rates (i.e. O2 consumption rates) are expressed as the O2 flux normalized to the muscle weight (pmoL/s/mg wet weight of muscle tissue).
Activity assay for citrate synthase and β‐hydroxyacyl CoA dehydrogenase
For citrate synthase activity measurements, frozen quadriceps were homogenized in buffer containing 50 mM Tris–HCl (pH 8.0), 1 mM EDTA, 10% glycerol (wt/vol), 0.02% Brij‐35 (wt/vol), 1 mM dithiothreitol with protease, and phosphatase inhibitor cocktails (Sigma‐Aldrich). Thirty microlitre of homogenate (1 mg/mL) was used for the analysis, and the reaction was initiated by 0.2 mM acetyl‐CoA and 0.5 mM oxaloacetate in 100 mM Tris buffer and 0.1 mM DTNB. Absorbance at 412 nm wavelength was followed for 3 min with a spectrophotometer.
β‐hydroxyacyl CoA dehydrogenase (β‐HAD) activity was assayed in whole lysates prepared from frozen tissues or cultured cells as described previously.17 First, 40 μL of homogenate (1 mg/mL) was added to a cuvette, which was then was brought to a final volume of 990 μL with 930 μL of 50 mM imidazole (pH 7.4) and 10 μL of 1.5 mM NADH. The reaction was initiated by the addition of 10 μL of 2 mM acetoacetyl‐CoA. Absorbance at 340 nm wavelength was followed for 5 min with a spectrophotometer.
Cell culture and transfection
To further examine the regulatory role of SIRT3 in fatty acid β‐oxidative activity in the skeletal muscle, we conducted cell culture studies using mouse C2C12 myoblast cell lines purchased from the American Type Culture Collection (Manassas, VA, USA). The cells were maintained in Dulbecco's Modified Eagle's Medium (4.5 g glucose/L; Sigma, St. Louis, MO, USA) containing 10% foetal bovine serum (Cell Culture Bioscience), 100 IU/mL penicillin, and 100 IU/mL streptomycin in a humidified incubator (Sanyo, Osaka, Japan) at 37°C with 5% CO2 in air.
For transfection, C2C12 myoblasts were plated in a T75 flask at a density of 1.2 × 106 cells and then transfected 24 h later with a non‐targeting pool of small interfering RNA against SIRT3 (Dharmacon, Brébières, France) using DharmaFECT reagent (Dharmacon). At 48 h after transfection, the cells were harvested and lysed with homogenization buffer containing 10 mM HEPES‐KOH (pH 7.4), 220 mM mannitol, and 0.07 M sucrose with several deacetylase inhibitors. The mitochondrial fraction was also isolated by the sequential centrifugation method. Whole cell lysates or mitochondrial fractions were then subjected to western blotting and a β‐HAD activity analysis.
Statistical analysis
Data are expressed as the mean ± SD or SEM for normally distributed variables, or the median and interquartile range for non‐normally distributed variables. The statistical analyses were performed using Student's t‐test for comparisons between two groups and a one‐way ANOVA followed by Tukey's test for more than two groups with GraphPad Prism 6 software (GraphPad, San Diego, CA, USA). A P‐value < 0.05 was considered significant. Correlations were examined by linear regression analysis using the least squares method. Significant acetylation changes were defined as a cutoff of ±1.5‐fold change in the mean values of the HF group vs. the sham group. When acetylated protein was detected only in HF mice, the protein was also defined as a significant acetylation target.
Results
The plasma acetyl‐lysine levels correlate with exercise capacity in patients with HF
The characteristics of the 10 patients with HF and five controls are summarized in Table 1. The primary aetiology of their HF was dilated cardiomyopathy (n = 4), followed by ischemic heart disease (n = 3) and hypertensive heart disease (n = 3); this distribution reflects the real‐world aetiology of HF in Japan.40 The patients' exercise capacity was reduced as evidenced by the median peak VO2 of 16.3 (interquartile range, 11.2–18.9) mL/kg/min. Our metabolomic analysis of the human plasma samples identified a total of 233 charged metabolites, in which significant increases were observed in N6‐acetyl‐lysine, arginine methylation, phospholipid metabolites, and urea cycle metabolites in the HF patients compared with controls.
Table 1.
Clinical characteristics of controls and patients with HF
| Controls (n = 5) | HF patients (n = 10) | |
|---|---|---|
| Male, n | 5 | 9 |
| Age, years | 56.6 ± 3.8 | 57.0 ± 12.3 |
| Causes of HF | ||
| ICM, n | 3 | |
| HHD, n | 2 | |
| DCM, n | 4 | |
| VHD, n | 1 | |
| NYHA classification | ||
| II, n | 6 | |
| III, n | 4 | |
| LVEF, % | 65.3 ± 5.3 | 25.0 ± 6.0* |
| BNP, pg/mL | 7.3, IQR (7.3, 35.3) | 161.3, IQR (84.9, 346.2)* |
| Cardiopulmonary exercise test | ||
| Peak VO2, mL/kg/min | 30.7 ± 7.3 | 15.6 ± 5.4* |
| AT, mL/kg/min | 15.4 ± 4.2 | 8.8 ± 1.5* |
| VE/VCO2 | 26.7, IQR (25.3, 29.5) | 33.1, IQR (29.9, 42.2)* |
| Medical therapy | ||
| ACEI/ARB, n | 0 | 9 |
| Beta‐blocker, n | 0 | 10 |
| Diuretics, n | 0 | 8 |
Data are the mean ± SD or median, IQR (first quartile, third quartile) for continuous variables. ACEI, angiotensin‐converting enzyme inhibitor; ARB, angiotensin II receptor blocker; AT, anaerobic threshold; BNP, brain natriuretic peptide; DCM, dilated cardiomyopathy; HF, heart failure; HHD, hypertensive heart disease; ICM, ischemic cardiomyopathy; IQR, interquartile range; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association; VCO2, carbon dioxide production; VE, minute ventilation; VHD, valvular heart disease; VO2, oxygen uptake.
P < 0.05 vs. controls.
Of these, N6‐acetyl‐lysine (r = −0.81), N,N‐dimethylglycine (r = −0.74), urea (r = −0.74), asymmetric dimethylarginine (r = −0.74), N‐acetylalanine (r = −0.72), choline (r = −0.66), and symmetric dimethylarginine (r = −0.62) each exhibited a strong negative correlation with peak VO2 (Figure 1A). Specifically, N6‐acetyl‐lysine had the strongest correlation with peak VO2 in all 15 subjects (r = −0.81) (Figure 1A) as well as in only the HF patients (r = −0.77) (Figure 1B,D). Consistent with the negative correlation with peak VO2, the N6‐acetyl‐lysine level tended to increase with the severity of the NYHA functional class, and the difference became significant between NYHA I and III (Figure 1C). In addition, the plasma N6‐acetyl‐lysine level was negatively correlated with the anaerobic threshold (r = −0.66) (Figure 1E) and positively correlated with VE/VCO2 (r = 0.70) (Figure 1F), and both these parameters of CPX are indispensable for determining the severity and prognosis of HF.
Figure 1.
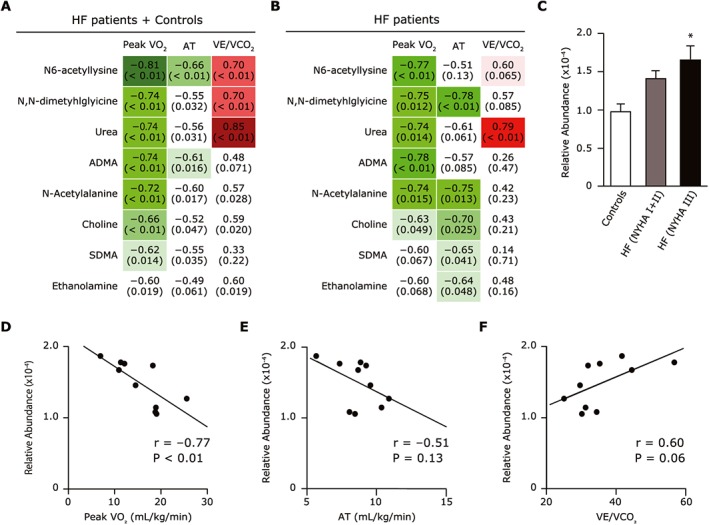
(A,B) Plasma metabolites that correlate with peak VO2, AT, or VE/VCO2 in all subjects (n = 15), i.e. the heart failure patients (HF) (n = 10) plus the normal controls (n = 5) (A), and in only the 10 HF patients (B). The upper value indicates the correlation coefficient between peak VO2, AT, or VE/VCO2 and each metabolite. The lower value in parentheses indicates the P‐value of each correlation. (C) Association between plasma acetyl‐lysine levels and NYHA functional class in the controls, the NYHA I or II HF patients, and the NYHA III HF patients. (D–F) Correlation between plasma acetyl‐lysine level and peak VO2 (D), AT (E), and VE/VCO2 (F) among the HF patients. Data are the mean ± SD. *P < 0.05 vs. controls. ADMA, asymmetric dimethylarginine; AT, anaerobic threshold; HF, heart failure; NYHA, New York Heart Association class; peak VO2, peak oxygen uptake; SDMA, symmetric dimethylarginine VE/VCO2, minute ventilation vs. carbon dioxide production.
In contrast, there was no association between the N6‐acetyl‐lysine level and the LV ejection fraction, log BNP, or thigh circumference in the HF patients, suggesting that acetylation played a specific role in exercise capacity rather than the cardiac function or congestive state, or skeletal muscle mass in HF (Figure S2). Because N‐acetyl‐lysine is a metabolite of protein lysine acetylation, this increase may reflect the hyperacetylation status, especially in skeletal muscle, which is a determinant factor of exercise capacity in HF.
Functional impairment in muscle mitochondria in a murine HF model
To examine the derangements in mitochondrial oxidative respiration in the skeletal muscle seen in HF, we used a murine model of post‐MI HF. Representative graphs of the mitochondrial respiratory capacity in permeabilized gastrocnemius tissues clearly demonstrated that the levels of complex I‐linked and complex I + II‐linked oxidative phosphorylation in the presence of octanoyl‐L‐carnitine, a fatty acid substrate, were lower in the HF mice compared with the sham‐operated mice (Figure 2A). Moreover, the β‐HAD activity was reduced in the quadriceps from the HF mice compared with the sham‐operated mice (Figure 2B). These results suggest that the acetylation of fatty acid oxidation enzymes inhibits the β‐HAD activity, and this inhibition is associated with the impaired mitochondrial oxidation that accounts for the lowered exercise tolerance in HF.
Figure 2.
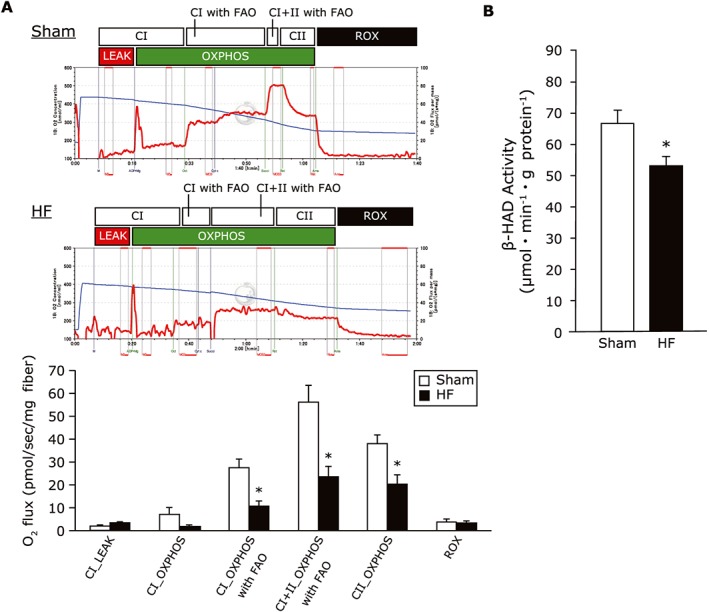
(A) Representative graphs of mitochondrial respiratory capacity with fatty acid substrates in sham and HF gastrocnemius muscles. The mitochondrial respiratory capacity at each state with octanoyl‐1‐carnitine in the gastrocnemius was lowered in the HF compared with the sham group (n = 6 each). (B) The β‐HAD enzyme activity in skeletal muscle from sham‐operated and HF mice (n = 6 each). Data are the mean ± SEM. *P < 0.05 vs. sham. CI, complex I‐linked substrates; CI + II, complex I + II‐linked substrates; FAO, fatty acid oxidation; HF, heart failure; LEAK, leak‐state respiration (non‐ADP stimulated respiration); OXPHOS, oxidative phosphorylation capacity (ADP‐stimulated respiration); ROX, residual oxygen consumption; β‐HAD, β‐hydroxyacyl CoA‐dehydrogenase.
Acetylation profile in skeletal muscle and blood in a murine model of HF
Based on the results of our study of the 10 patients with HF and five healthy controls (Figure 1), we examined the global lysine acetylation in the gastrocnemius tissues from sham‐operated and HF mice. As expected, lysine‐acetylated protein levels were higher in the mitochondrial fraction of gastrocnemius from the HF mice compared with that of the sham‐operated mice, whereas the whole muscle lysates were not significantly different between the two groups (Figure 3A,B). In conjunction with the increased mitochondrial acetylation, the protein expression of the mitochondrial deacetylase SIRT3 was decreased in the gastrocnemius from the HF mice compared with the sham group (Figure 3D). As is observed in humans, the acetylated protein levels in the murine blood samples were higher in the HF mice than the sham‐operated mice, in parallel with the increased acetylation of muscle mitochondrial proteins in the HF mice (Figure 3C). Because SIRT3 activity is dependent on NAD+ availability, we quantified the NAD+ and NAD+/NADH ratio by conducting a CE‐MS analysis. Neither the NAD+ level nor the NAD+/NADH ratio differed significantly between the HF and sham groups (Figure 3E).
Figure 3.
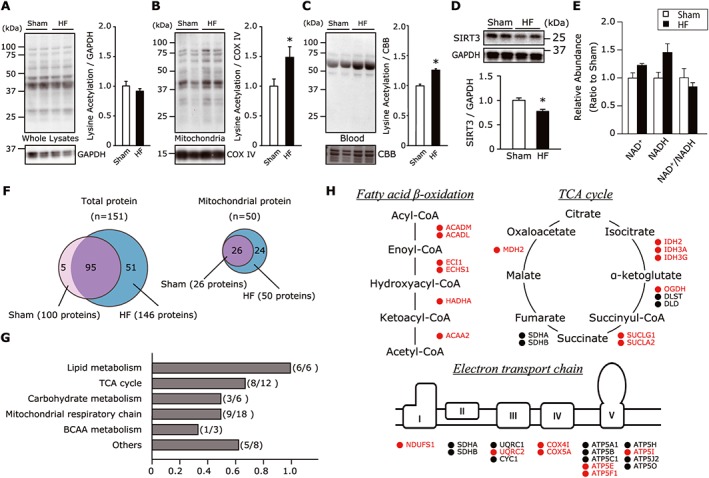
(A,B) Total protein acetylation assessed by western blotting in the whole gastrocnemius lysate (A) and its mitochondrial fraction (B) (n = 8 each). Total acetylated proteins were normalized to GAPDH in whole lysates or COX IV in the mitochondrial fraction. (C) Total protein acetylation in blood samples normalized to the non‐specific band of CBB‐stained gel from sham‐operated and HF mice (n = 6 each). (D) Protein expression of SIRT3 in gastrocnemius muscle from sham‐operated and HF mice (n = 6 each). (E) The abundance of NAD+ and NADH, and the ratio of NAD+ to NADH in skeletal muscle tissues from sham‐operated and HF mice (n = 5–6 each). Data are the mean ± SEM. *P < 0.05 vs. sham. (F) Venn diagram showing overlapping of lysine‐acetylated peptides in total and mitochondrial proteins identified in sham and HF muscles (n = 3 each). Numbers in brackets represent the total number of acetylated proteins identified in each group. A total of 151 acetylated proteins were identified, and 95 acetylated proteins overlapped between the sham and HF muscle. Of these, 50 proteins belonged to mitochondria and were all derived from HF muscle, and 26 mitochondrial proteins were shared with the sham‐operated mice, indicating that 24 proteins were unique to the HF mice. (G) Localization annotation (gene ontology) was assessed for 48 acetylpeptides that were increased significantly or at least 1.5‐fold increase in the HF mice compared with the sham‐operated mice. (H) Specific acetylation sites in the pathway map showing fatty acid β‐oxidation, TCA cycle, and electron transport chain. The acetylation status is indicated by colour coding: proteins with increased acetylation (HF/sham) are in red; proteins with decreased or unaltered acetylation are in black. ACADL, long‐chain specific acyl‐CoA dehydrogenase; BCAA, branched‐chain amino acid; CBB, Coomassie Brilliant Blue; GAPDH, glyceraldehyde‐3‐ phosphate dehydrogenase; HADHA, trifunctional enzyme subunit α; HF, heart failure; NAD+, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide (reduced form); SIRT3, Sirtuin 3; TCA, tricarboxylic acid.
To further clarify the molecular functions of acetylated proteins, we performed a mass spectroscopy‐based global analysis followed by enrichment of the acetyl lysine peptide in gastrocnemius tissues in the two groups. This approach enables the detection of low stoichiometry of acetylation and was established in previous studies.41 A total of 151 proteins were identified as having at least one portion of acetyl‐lysine residues, and 95 acetylated proteins were shared between the sham‐operated and HF mice (Figure 3F). Of these, 50 acetylated proteins were classified as mitochondrial enzymes and were all observed in HF muscle, whereas 26 of the proteins were derived from muscles of the sham‐operated animals. This indicates that 24 of the mitochondrial acetylated proteins were unique to the HF mice and that mitochondria are important targets of the post‐translational modifications in HF (Figure 3F). The hyperacetylated mitochondrial enzymes were linked to lipid metabolism, carbohydrate metabolism, the TCA cycle, branched amino acid (BCAA) metabolism, and the electron transport chain (Figure 3G). Notably, the major targets of lysine acetylation were fatty acid β‐oxidation enzymes, including long chain acyl‐CoA dehydrogenase (LCAD), enoyl‐CoA hydratase, trifunctional enzyme subunit α (HADHA), and 3‐ketoacyl‐CoA thiolase (Figure 3H).
In sharp contrast to the difference in post‐translational modification, there was no significant difference in the protein expression of peroxisome proliferator‐activated receptor gamma coactivator 1‐α, a master transcriptional regulator of mitochondrial biogenesis, between the HF mice and sham‐operated mice (Figure 4A). Consistently, the mitochondrially encoded cytochrome c protein expression and citrate synthase activity, both of which are surrogate markers for mitochondrial content, were comparable between the sham‐operated and HF mice (Figure 4B,C). Moreover, there were no significant differences in the various protein expressions related to fatty acid cellular uptake (CD36), mitochondrial transport (CPT1, CPT2), and fatty acid β‐oxidation enzymes (ACADL, EHHAD) in the gastrocnemius from the HF mice compared with the sham‐operated mice (Figure 4D–H). Taken together, these results suggest that post‐translational mechanisms are involved in the derangements in fatty acid oxidation during the development of the skeletal muscle dysfunction and exercise intolerance associated with HF.
Figure 4.
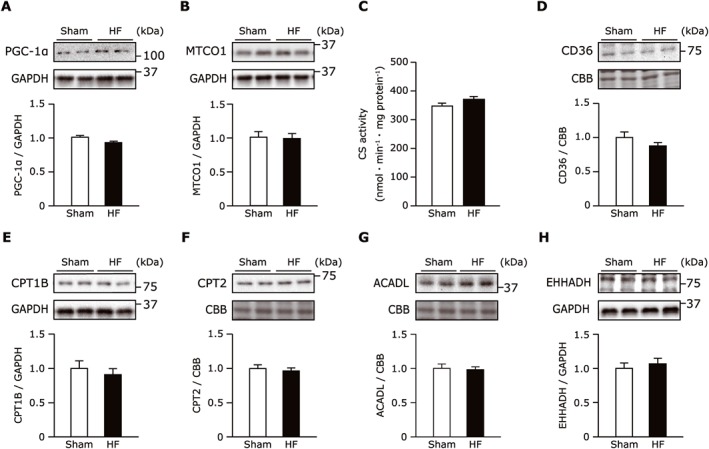
(A–H) Protein expression of PGC‐1α (A), MTCO1 (B), CD36 (D), CPT1B (E), CPT2 (F), ACADL (G), and EHHADH (H) normalized to the GAPDH or non‐specific band of CBB‐stained gel in sham and HF gastrocnemius muscles (n = 6–10 each). CS activity in the gastrocnemius muscle from the sham‐operated and the HF mice (n = 8 each). Data are the mean ± SEM. *P < 0.05 vs. sham. ACADL, long‐chain specific acyl‐CoA dehydrogenase; CBB, Coomassie Brilliant Blue; CD36, cluster of differentiation 36; CPT, carnitine palmitoyltransferase; CS, citrate synthase; EHHADH, enoyl‐CoA hydratase and 3‐hydroxyacyl CoA dehydrogenase; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; HF, heart failure; MTCO1, mitochondrially encoded cytochrome c oxidase 1; PGC‐1α, peroxisome proliferator‐activated receptor gamma coactivator 1‐α.
Post‐infarct HF mice mimic exercise intolerance in humans
Table 2 shows the characteristics of the HF mice at 4 weeks after MI. The heart weight and lung weight/BW were significantly increased in the HF mice compared with the sham‐operated mice. A representative echocardiographic image is shown in Figure 5A, indicating that the HF mice had greater LV diameters and lower LV systolic function than the sham‐operated mice (Table 2). The Masson's trichrome staining of ventricular tissues further confirmed the infarct formation (Figure 5B). The mortality rates up to 4 weeks after the operation was significantly lower in the HF mice compared with the sham group (44% vs. 0%).
Table 2.
Animal characteristics
| Sham | HF | |
|---|---|---|
| n | 11 | 11 |
| Body and organ weight | ||
| BW, g | 25.6 ± 0.2 | 25.0 ± 0.3 |
| Heart weight, mg | 116.6 ± 1.4 | 154.0 ± 7.7* |
| Lung weight/BW, mg/g | 5.4 ± 0.04 | 7.6 ± 0.09* |
| SKM weight | ||
| Quadriceps weight, mg/g | 8.0 ± 0.1 | 8.0 ± 0.2 |
| Gastrocnemius weight, mg/g | 5.5 ± 0.1 | 5.3 ± 0.1 |
| Soleus weight, mg/g | 0.4 ± 0.01 | 0.4 ± 0.01 |
| Echocardiography | ||
| Heart rate, beats/min | 688 ± 7 | 658 ± 16 |
| LV EDD, mm | 3.6 ± 0.1 | 4.9 ± 0.2* |
| LV ESD, mm | 1.5 ± 0.1 | 4.2 ± 0.2* |
| Fractional shortening, % | 56.7 ± 2.1 | 15.8 ± 1.3* |
| AWT, mm | 0.7 ± 0.02 | 0.3 ± 0.01* |
| PWT, mm | 0.8 ± 0.02 | 1.1 ± 0.06* |
| Exercise test | ||
| Run distance, m | 538.1 ± 18.2 | 245.7 ± 18.3* |
| Run time, sec | 1921 ± 33 | 1280 ± 52* |
| Peak RER | 1.2 ± 0.01 | 1.2 ± 0.01 |
Data are the mean ± SEM. AWT, anterior wall thickness; BW, body weight; HF, heart failure; LV EDD, left ventricular end‐diastolic diameter; LV ESD, left ventricular end‐systolic diameter; PWT, posterior wall thickness; RER, respiratory exchange ratio; SKM, skeletal muscle.
P < 0.05 vs. sham group.
Figure 5.
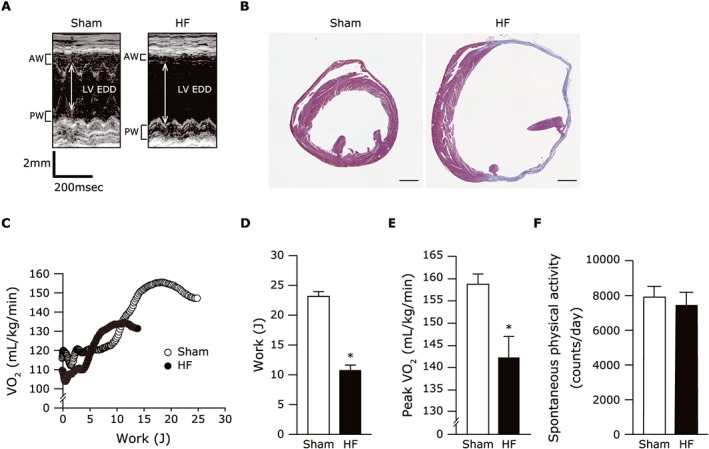
(A) Representative M‐mode echocardiographic images obtained from sham and post‐MI HF mice. (B) Representative image of the ventricular tissues from the sham‐operated and the HF mice stained by Masson's trichrome. (C) Representative graphs plotted for VO2 at each workload during exercise in sham and HF mice. (D,E) The summarized data assessed by exercise testing with expired gas analysis indicate the work (D) and peak VO2 (E) (n = 8 each). Data are the mean ± SEM. *P < 0.05 vs. sham. AW, anterior wall; EDD, end‐diastolic diameter; HF, heart failure; LV, left ventricular; peak VO2, peak oxygen uptake; PW, posterior wall.
In the CPX measurements, the peak VO2 as well as the work, run distance, and run time were significantly reduced in the HF mice compared with the sham‐operated mice at maximal effort with equivalent respiratory exchange ratios (Figure 5C–E; Table 2). In contrast, the spontaneous physical activity was not significantly different between the HF and sham groups (Figure 5F). The muscle weights including those of the quadriceps, gastrocnemius, and soleus were also comparable between the two groups (Table 2). Collectively, these results indicate that our model of post‐MI HF sufficiently mimics the exercise intolerance seen in patients with HF, possibly due to the functional alterations in skeletal muscle.
The effect of SIRT3 knockdown on the acetylation and β‐HAD activity in vitro
To further investigate whether the decrease in SIRT3 was closely associated with increased acetylation of skeletal muscle, thereby decreasing the activity of fatty acid β‐oxidation enzymes, we conducted SIRT3‐knockdown experiments in C2C12 mouse myotubes. The transfection of SIRT3 small interfering RNA in C2C12 cells reduced the SIRT3 protein expression by 70% compared with the non‐targeting pool (Figure 6A), in conjunction with a significant increase in the overall protein acetylation in the mitochondrial fraction (Figure 6B), but not in the whole muscle lysates (Figure 6A). More importantly, the β‐HAD activity was significantly reduced following SIRT3 knockdown (Figure 6C), supporting the concept that SIRT3 is a key molecule modulating the acetylation of muscle fatty acid β‐oxidation enzymes in HF.
Figure 6.
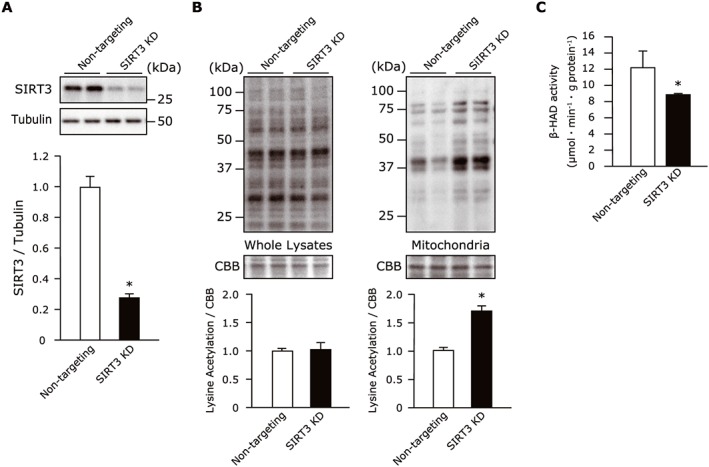
(A) The efficacy of non‐targeting pool or SIRT3 small interfering RNA on these protein levels in C2C12 cells (n = 4 each). (B) The SIRT3 knockdown with small interfering RNA increased the total protein acetylation in the mitochondrial fractions, but not in the whole cell lysates of C2C12 cells (n = 3–4 each). (C) The β‐HAD activity was reduced in SIRT3‐knockdown C2C12 cells (n = 3 each). Data are the mean ± SEM. *P < 0.05 vs. non‐targeting pool. β‐HAD, β‐hydroxyacyl CoA‐dehydrogenase; CBB, Coomassie Brilliant Blue; SIRT3, Sirtuin 3.
Discussion
The present study provides numerous novel observations. First, the mass spectroscopy‐based metabolic profiling revealed that the plasma acetyl‐lysine level was elevated in HF patients and was negatively correlated with their exercise capacities. Second, acetylation modification was enhanced in skeletal muscle mitochondria from our murine model of HF with lowered exercise capacity, which was attributed to a decreased level of a mitochondrial deacetylase, SIRT3. Third, label‐free acetylproteomics identified key metabolic pathways for acetylation, including fatty acid oxidation, the TCA cycle, BCAA metabolism, and electron transport. Specifically, the acetylation of fatty acid β‐oxidation enzymes was related to reduced activity and a further reduction in fatty acid‐derived mitochondrial oxidative capacity in the skeletal muscle. We also observed that the knockdown of SIRT3 in cultured skeletal muscle cells resulted in enhanced mitochondrial acetylation, which in turn resulted in a reduction in the enzymatic activity of fatty acid oxidation. These findings suggest that enhanced acetylation is linked to the pathogenesis of exercise intolerance in HF, at least in part through impaired fatty acid metabolism in the skeletal muscle.
Prior studies have consistently demonstrated that cardiac function does not necessarily correlate with exercise capacity in HF42 and that exercise training does reduce mortality in HF patients without any changes in cardiac function.43 These findings have raised considerable interest in skeletal muscle dysfunction as a major contributor to the pathophysiological features and symptomatology of HF.
The peak VO2 measured on CPX is a powerful prognostic indicator for HF patients, but a large clinical trial indicated that many HF patients are often unable to achieve maximal efforts due to their multiple comorbidities.43 As a result, the prognostic value of peak VO2 is suboptimal.44, 45 As an incremental risk stratification tool for HF, various biomarkers have been suggested.46 Notably, recent metabolomic analyses of plasma samples have indicated that the specific patterns of metabolites in HF patients offer substantial value for the prediction of prognosis.47, 48, 49 This corroborates the concept that metabolic impairment is pronounced not only in the heart but also in the peripheral organs.50 Until now, however, few studies have focused on metabolites associated with exercise capacity in HF.
This study is the first to show that N6 acetyl‐lysine is the most relevant metabolite that correlates with peak VO2, and that the increased levels of other metabolites such as asymmetric dimethylarginine, choline derivatives, and urea were also consistent with previous findings.47, 51 N6 acetyl‐lysine is an amino acid derivative that may be released from protein lysine acetylation, which may reflect a systemic increase in this post‐translational modification in the setting of HF. Although acetylation was originally reported to regulate gene transcription by neutralizing the positive charge of histone proteins,52 acetylation was more recently recognized as a novel regulator of mitochondrial metabolic pathways.41 Horton et al. reported that they observed a profound increase in the acetylation of mitochondrial proteins in human cardiac samples as well as murine hearts with pressure overload.22 Aside from the failing heart, we found that the muscle mitochondria were also a focus of the acetylation modification in HF. Because approx. 95% of acetyl CoA, a primary substrate for protein acetylation, is localized to the mitochondria,53 it is likely that the mitochondrion is a major targeted compartment for acetylation. It has been reported that increased acetylation of mitochondrial proteins is evident in failing or diabetic hearts.14, 22 In addition, studies of diet‐induced diabetic animals have demonstrated that mitochondrial proteins in liver and skeletal muscle were also acetylated.12, 24 Given that HF is characterized by systemic insulin resistance that involves multiple organs,50 we speculate that in HF, the acetylation of metabolic enzymes may occur in the mitochondria of organs other than skeletal muscle.
On the other hand, although it remains unclarified how much the acetyl lysine levels in human blood samples reflect acetylation alterations in the muscle mitochondrial compartment as was observed in the HF mice, we observed that the acetylated protein levels in the murine blood samples were higher in the HF mice than the sham‐operated mice, in parallel with the increased acetylation of muscle mitochondrial proteins in the HF mice. This result suggests that the levels of blood acetylated proteins may reflect, at least to some extent, those in the muscle mitochondria.
Intriguingly, our further functional profiling of acetylation revealed key fuel metabolism and energetic pathways, including fatty acid β‐oxidation, the TCA cycle, BCAA metabolism, and the electron transport chain, which are in complete agreement with the patterns reported in the failing heart.22 Alterations in mitochondrial lysine acetylation are known to be regulated by the balance between SIRT3 deacetylase activity due to NAD+ availability and/or a non‐enzymatic reaction of mitochondrial acetyl CoA to lysine.54 In the present study, the reduction in SIRT3 expression rather than a reduction in the NAD+ amount or the NAD+/NADH ratio appeared to account for the enhanced mitochondrial acetylation in the skeletal muscle from HF. However, an accurate determination of subcellular NAD+ and acetyl CoA concentrations is technically difficult at this time. A more sophisticated assay could elucidate the role of the compartmentalization of those cofactors in pathological settings.
Previous studies have shown that a decrease in SIRT3 expression is responsible for the increased lysine acetylation and promotes a shift in substrate metabolism from fatty acids toward glucose in diabetic hearts14, 55 as well as in hypertrophied hearts.16, 21, 56 This substrate alteration is thought to induce insulin resistance and therefore cardiac energetic inefficiency, leading to the development of HF.57, 58 Similar to the findings in diabetic or failing hearts, SIRT3 was also shown to regulate skeletal muscle function via alterations in mitochondrial oxidation and oxidative stress.25 Collectively, these findings lead us to speculate that the common mechanism of SIRT3‐mediated hyperacetylation between heart and skeletal muscle may predispose to energetic derangements due to alterations in substrate metabolism and hence the deterioration of exercise intolerance in HF.
In the present study, the acetylation of key fatty acid β‐oxidation enzymes was associated with a reduction in their activities and fatty acid‐derived mitochondrial oxidation. In line with this finding, early studies using muscle biopsy samples showed that a variety of mitochondrial oxidative enzymes, including β‐HAD, are decreased in patients with HF.4, 10 Our previous investigation using magnetic resonance spectroscopy revealed that intramyocellular lipid deposition was increased in HF patients, and its content was closely associated with muscle energy production and whole‐body exercise capacity.9 Similar muscular fat infiltration has also been observed in muscle dysfunction linked to muscle disuse and aging, as well as in patients with iron deficiency and patients with preserved ejection fraction (HFpEF).59, 60, 61, 62 Alternatively, it has been documented that higher exercise capacity is associated with increased fatty acid oxidation in skeletal muscle during exercise.63, 64 Combined, these observations have suggested that intrinsic deficits in fatty acid oxidative metabolism in skeletal muscle may provoke energetic deprivation in response to increased energy demand during exercise, which may be partially responsible for the reduced exercise capacity in patients with HF. However, we cannot exclude the possibility that low cardiac output is a definitive factor for the exercise capacity in HF. In addition, the causal relationship between the decreased fatty acid β‐oxidation activity and lowered exercise capacity observed in the in vivo experiments was not fully clarified in the present study.
The molecular process controlling fatty acid oxidation in the skeletal muscle has yet to be fully elucidated. Indeed, metabolic genes and related proteins were reported to remain unaltered, whereas the accumulation of intermediate metabolites was dramatically increased, in the process leading to decompensated HF.65 That finding strongly suggests that the post‐translational regulation of metabolic remodelling is an early and sensitive molecular signature of impaired energy metabolism in HF. A growing body of evidence has indicated that the acetylation of mitochondrial fatty acid oxidation enzymes can modulate their activities in a tissue‐dependent manner. For instance, Hirschey et al. demonstrated that the hyperacetylation of a key fatty acid β‐oxidation enzyme, LCAD, results in reduced enzymatic activity and hepatic lipid accumulation,12 whereas Lopaschuk and his colleagues concluded that high rates of myocardial fatty acid oxidation are attributable to hyperacetylation and the subsequent activation of fatty acid β‐oxidation enzymes.14, 17, 37
As for the effects of acetylation modification on skeletal muscle proteins, it has been consistently reported that the acetylation of a fatty acid‐related pathway negatively regulates lipid metabolism.63, 66 As evidence supporting this, our present findings demonstrated that enhanced acetylation due to SIRT3 knockdown resulted in a reduction of β‐HAD activity in skeletal muscle cells. Therefore, at least in skeletal muscle, increased acetylation via a decrement of SIRT3 has inhibitory impacts on fatty acid β‐oxidation enzymes, which in turn results in the impairment of mitochondrial oxidative capacity when fatty acids are used as a substrate. As a result, impaired fatty acid oxidation in the skeletal muscle in combination with the aforementioned insulin resistance may further exaggerate the mitochondrial energy perturbations and exercise intolerance seen in HF. Moreover, based on another study's finding that skeletal muscle from patients with mitochondrial disease exhibited enhanced ergoreflex sensitivity, we speculate that sympathetic activation and exercise intolerance due to ergoreflex hypersensitivity may also be involved in this phenomenon.67
Limitations
Given the observational nature of this study, there are several limitations that should be noted. First, although our data presented in vivo are generally solid, cause‐and‐effect relationships between acetylation and impaired fatty acid oxidation or lowered exercise capacity remain to be determined. Specifically, the lack of any evidence regarding whether hyperacetylation via SIRT3 knockdown could ultimately affect mitochondrial respiration is a major limitation of our study. In addition, off‐target effects of knockdown experiments will limit the experiments' translation into in vivo findings. We also failed to determine the correlation between acetylation and mitochondrial respiration in the skeletal muscle, due to a technical limitation. To gain direct evidence of the role of acetylation in the regulation of muscular fatty acid oxidation, further studies using skeletal muscle‐specific SIRT3‐deficient mice are necessary. Second, the number of patients in the HF group was twice the number of control subjects, due to the limited number of healthy control subjects. Our results regarding the metabolites identified herein should be verified in a future large‐scale study. Third, the upstream mechanism by which the SIRT3 amount is reduced is yet to be elucidated, but it may be linked to altered expression in response to nutrient conditions. Finally, the functional effects of mitochondrial acetylation via SIRT3 other than energy metabolism remain to be determined. In particular, as our post‐MI HF model was far from the cachectic state without any significant reduction in muscle weights, the roles of other factors such as Forkhead Box O family members and inflammatory signalling in the muscle dysfunction of HF have remained unexplored.68, 69
Conclusions
We observed that increased acetylation in skeletal muscle mitochondria was associated with reduced fatty acid oxidation and impaired exercise tolerance in HF. Novel approaches targeting the acetylation of skeletal muscle mitochondria would provide important clues for the development of an effective intervention for exercise intolerance in patients with HF.
Funding
This study was supported by grants from the Takeda Science Foundation and the Japan Ministry of Education, Science, and Culture (nos. 26350879, 15 K09115, 17 K10137).
Disclosures
The authors declare no conflicts of interest.
Supporting information
Figure S1. Schematic representation of the time schedule of the experimental protocol. SPA, spontaneous physical activity.
Figure S2. Correlations between the plasma acetyllysine level and LVEF (A), log BNP (B), and thigh circumference (C) in the 10 patients with HF.LVEF, left ventricular ejection fraction; BNP, brain natriuretic peptide.
Acknowledgements
We thank Yuki Kimura, Noriko Ikeda, and Miwako Yamane for their technical assistance.
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.70
Tsuda, M. , Fukushima, A. , Matsumoto, J. , Takada, S. , Kakutani, N. , Nambu, H. , Yamanashi, K. , Furihata, T. , Yokota, T. , Okita, K. , Kinugawa, S. , and Anzai, T. (2018) Protein acetylation in skeletal muscle mitochondria is involved in impaired fatty acid oxidation and exercise intolerance in heart failure. Journal of Cachexia, Sarcopenia and Muscle, 9: 844–859. 10.1002/jcsm.12322.
References
- 1. Mancini DM, Eisen H, Kussmaul W, Mull R, Edmunds LH Jr, Wilson JR. Value of peak exercise oxygen consumption for optimal timing of cardiac transplantation in ambulatory patients with heart failure. Circulation 1991;83:778–786. [DOI] [PubMed] [Google Scholar]
- 2. Clark AL, Poole‐Wilson PA, Coats AJ. Exercise limitation in chronic heart failure: central role of the periphery. J Am Coll Cardiol 1996;28:1092–1102. [DOI] [PubMed] [Google Scholar]
- 3. Piepoli MF, Coats AJ. The ‘skeletal muscle hypothesis in heart failure’ revised. Eur Heart J 2013;34:486–488. [DOI] [PubMed] [Google Scholar]
- 4. Drexler H, Riede U, Munzel T, Konig H, Funke E, Just H. Alterations of skeletal muscle in chronic heart failure. Circulation 1992;85:1751–1759. [DOI] [PubMed] [Google Scholar]
- 5. Sabbah HN, Hansen‐Smith F, Sharov VG, Kono T, Lesch M, Gengo PJ, Steffen RP, Levine TB, Goldstein S. Decreased proportion of type I myofibers in skeletal muscle of dogs with chronic heart failure. Circulation 1993;87:1729–1737. [DOI] [PubMed] [Google Scholar]
- 6. Fulster S, Tacke M, Sandek A, Ebner N, Tschope C, Doehner W, Anker SD, von Haehling S. Muscle wasting in patients with chronic heart failure: results from the studies investigating co‐morbidities aggravating heart failure (SICA‐HF). Eur Heart J 2013;34:512–519. [DOI] [PubMed] [Google Scholar]
- 7. Okita K, Yonezawa K, Nishijima H, Hanada A, Ohtsubo M, Kohya T, Murakami T, Kitabatake A. Skeletal muscle metabolism limits exercise capacity in patients with chronic heart failure. Circulation 1998;98:1886–1891. [DOI] [PubMed] [Google Scholar]
- 8. Kinugawa S, Takada S, Matsushima S, Okita K, Tsutsui H. Skeletal Muscle Abnormalities in Heart Failure. Int Heart J 2015;56:475–484. [DOI] [PubMed] [Google Scholar]
- 9. Hirabayashi K, Kinugawa S, Yokota T, Takada S, Fukushima A, Suga T, Takahashi M, Ono T, Morita N, Omokawa M, Harada K, Oyama‐Manabe N, Shirato H, Matsushima S, Okita K, Tsutsui H. Intramyocellular lipid is increased in the skeletal muscle of patients with dilated cardiomyopathy with lowered exercise capacity. Int J Cardiol 2014;176:1110–1112. [DOI] [PubMed] [Google Scholar]
- 10. Sullivan MJ, Green HJ, Cobb FR. Skeletal muscle biochemistry and histology in ambulatory patients with long‐term heart failure. Circulation 1990;81:518–527. [DOI] [PubMed] [Google Scholar]
- 11. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science 2010;327:1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV Jr, Alt FW, Kahn CR, Verdin E. SIRT3 regulates mitochondrial fatty‐acid oxidation by reversible enzyme deacetylation. Nature 2010;464:121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, Gibson BW. Label‐free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci U S A 2013;110:6601–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alrob OA, Sankaralingam S, Ma C, Wagg CS, Fillmore N, Jaswal JS, Sack MN, Lehner R, Gupta MP, Michelakis ED, Padwal RS, Johnstone DE, Sharma AM, Lopaschuk GD. Obesity‐induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc Res 2014;103:485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vazquez EJ, Berthiaume JM, Kamath V, Achike O, Buchanan E, Montano MM, Chandler MP, Miyagi M, Rosca MG. Mitochondrial complex I defect and increased fatty acid oxidation enhance protein lysine acetylation in the diabetic heart. Cardiovasc Res 2015;107:453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koentges C, Pfeil K, Schnick T, Wiese S, Dahlbock R, Cimolai MC, Meyer‐Steenbuck M, Cenkerova K, Hoffmann MM, Jaeger C, Odening KE, Kammerer B, Hein L, Bode C, Bugger H. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res Cardiol 2015;110:36. [DOI] [PubMed] [Google Scholar]
- 17. Fukushima A, Alrob OA, Zhang L, Wagg CS, Altamimi T, Rawat S, Rebeyka IM, Kantor PF, Lopaschuk GD. Acetylation and succinylation contribute to maturational alterations in energy metabolism in the newborn heart. Am J Physiol Heart Circ Physiol 2016;311:H347–H363. [DOI] [PubMed] [Google Scholar]
- 18. Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3‐LKB1‐AMP‐activated kinase pathway. J Biol Chem 2010;285:3133–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res 2009;105:481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pillai VB, Samant S, Sundaresan NR, Raghuraman H, Kim G, Bonner MY, Arbiser JL, Walker DI, Jones DP, Gius D, Gupta MP. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun 2015;6:6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mori J, Alrob OA, Wagg CS, Harris RA, Lopaschuk GD, Oudit GY. ANG II causes insulin resistance and induces cardiac metabolic switch and inefficiency: a critical role of PDK4. Am J Physiol Heart Circ Physiol 2013;304:H1103–H1113. [DOI] [PubMed] [Google Scholar]
- 22. Horton JL, Martin OJ, Lai L, Riley NM, Richards AL, Vega RB, Leone TC, Pagliarini DJ, Muoio DM, Bedi KC Jr, Margulies KB, Coon JJ, Kelly DP. Mitochondrial protein hyperacetylation in the failing heart. JCI insight 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karamanlidis G, Lee CF, Garcia‐Menendez L, Kolwicz SC Jr, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab 2013;18:239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jing E, O'Neill BT, Rardin MJ, Kleinridders A, Ilkeyeva OR, Ussar S, Bain JR, Lee KY, Verdin EM, Newgard CB, Gibson BW, Kahn CR. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 2013;62:3404–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, Verdin EM, Kahn CR. Sirtuin‐3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A 2011;108:14608–14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yokota T, Kinugawa S, Yamato M, Hirabayashi K, Suga T, Takada S, Harada K, Morita N, Oyama‐Manabe N, Kikuchi Y, Okita K, Tsutsui H. Systemic oxidative stress is associated with lower aerobic capacity and impaired skeletal muscle energy metabolism in patients with metabolic syndrome. Diabetes Care 2013;36:1341–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fukushima A, Kinugawa S, Homma T, Masaki Y, Furihata T, Yokota T, Matsushima S, Abe T, Suga T, Takada S, Kadoguchi T, Katsuyama R, Oba K, Okita K, Tsutsui H. Decreased serum brain‐derived neurotrophic factor levels are correlated with exercise intolerance in patients with heart failure. Int J Cardiol 2013;168:e142–e144. [DOI] [PubMed] [Google Scholar]
- 28. Balady GJ, Arena R, Sietsema K, Myers J, Coke L, Fletcher GF, Forman D, Franklin B, Guazzi M, Gulati M, Keteyian SJ, Lavie CJ, Macko R, Mancini D, Milani RV, American Heart CR. Association Exercise, C. Prevention Committee of the Council on Clinical, E. Council on, Prevention, D. Council on Peripheral Vascular, C. Interdisciplinary Council on Quality of, R. Outcomes, Clinician's Guide to cardiopulmonary exercise testing in adults: a scientific statement from the American Heart Association. Circulation 2010;122:191–225. [DOI] [PubMed] [Google Scholar]
- 29. Beaver WL, Wasserman K, Whipp BJ. A new method for detecting anaerobic threshold by gas exchange. J Appl Physiol (1985) 1986;60:2020–2027. [DOI] [PubMed] [Google Scholar]
- 30. Hirayama A, Kami K, Sugimoto M, Sugawara M, Toki N, Onozuka H, Kinoshita T, Saito N, Ochiai A, Tomita M, Esumi H, Soga T. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time‐of‐flight mass spectrometry. Cancer Res 2009;69:4918–4925. [DOI] [PubMed] [Google Scholar]
- 31. Sugimoto M, Wong DT, Hirayama A, Soga T, Tomita M. Capillary electrophoresis mass spectrometry‐based saliva metabolomics identified oral, breast and pancreatic cancer‐specific profiles. Metabolomics: official journal of the metabolomic. Society 2010;6:78–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kinugawa S, Tsutsui H, Hayashidani S, Ide T, Suematsu N, Satoh S, Utsumi H, Takeshita A. Treatment with dimethylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: role of oxidative stress. Circ Res 2000;87:392–398. [DOI] [PubMed] [Google Scholar]
- 33. Fukushima A, Kinugawa S, Takada S, Matsushima S, Sobirin MA, Ono T, Takahashi M, Suga T, Homma T, Masaki Y, Furihata T, Kadoguchi T, Yokota T, Okita K, Tsutsui H. (Pro) renin receptor in skeletal muscle is involved in the development of insulin resistance associated with postinfarct heart failure in mice. Am J Physiol Endocrinol Metab 2014;307:E503–E514. [DOI] [PubMed] [Google Scholar]
- 34. Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, Rockman HA, Kass DA, Molkentin JD, Sussman MA, Koch WJ. C.o.C.C. American Heart Association Council on Basic Cardiovascular Sciences, G. Council on Functional, B. Translational, Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res 2012;111:131–150. [DOI] [PubMed] [Google Scholar]
- 35. Takada S, Kinugawa S, Hirabayashi K, Suga T, Yokota T, Takahashi M, Fukushima A, Homma T, Ono T, Sobirin MA, Masaki Y, Mizushima W, Kadoguchi T, Okita K, Tsutsui H. Angiotensin II receptor blocker improves the lowered exercise capacity and impaired mitochondrial function of the skeletal muscle in type 2 diabetic mice. J Appl Physiol (1985) 2013;114:844–857. [DOI] [PubMed] [Google Scholar]
- 36. Takada S, Masaki Y, Kinugawa S, Matsumoto J, Furihata T, Mizushima W, Kadoguchi T, Fukushima A, Homma T, Takahashi M, Harashima S, Matsushima S, Yokota T, Tanaka S, Okita K, Tsutsui H. Dipeptidyl peptidase‐4 inhibitor improved exercise capacity and mitochondrial biogenesis in mice with heart failure via activation of glucagon‐like peptide‐1 receptor signalling. Cardiovasc Res 2016;111:338–347. [DOI] [PubMed] [Google Scholar]
- 37. Sankaralingam S, Abo Alrob O, Zhang L, Jaswal JS, Wagg CS, Fukushima A, Padwal RS, Johnstone DE, Sharma AM, Lopaschuk GD. Lowering body weight in obese mice with diastolic heart failure improves cardiac insulin sensitivity and function: implications for the obesity paradox. Diabetes 2015;64:1643–1657. [DOI] [PubMed] [Google Scholar]
- 38. Griffin NM, Yu J, Long F, Oh P, Shore S, Li Y, Koziol JA, Schnitzer JE. Label‐free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat Biotechnol 2010;28:83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Soga T, Igarashi K, Ito C, Mizobuchi K, Zimmermann HP, Tomita M. Metabolomic profiling of anionic metabolites by capillary electrophoresis mass spectrometry. Anal Chem 2009;81:6165–6174. [DOI] [PubMed] [Google Scholar]
- 40. Tsutsui H, Tsuchihashi‐Makaya M, Kinugawa S, Goto D, Takeshita A. Characteristics and outcomes of patients with heart failure in general practices and hospitals. Circ J 2007;71:449–454. [DOI] [PubMed] [Google Scholar]
- 41. Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol 2014;15:536–550. [DOI] [PubMed] [Google Scholar]
- 42. Wilson JR, Martin JL, Ferraro N. Impaired skeletal muscle nutritive flow during exercise in patients with congestive heart failure: role of cardiac pump dysfunction as determined by the effect of dobutamine. Am J Cardiol 1984;53:1308–1315. [DOI] [PubMed] [Google Scholar]
- 43. O'Connor CM, Whellan DJ, Lee KL, Keteyian SJ, Cooper LS, Ellis SJ, Leifer ES, Kraus WE, Kitzman DW, Blumenthal JA, Rendall DS, Miller NH, Fleg JL, Schulman KA, McKelvie RS, Zannad F, Pina IL. Efficacy and safety of exercise training in patients with chronic heart failure: HF‐ACTION randomized controlled trial. JAMA 2009;301:1439–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mezzani A, Corra U, Bosimini E, Giordano A, Giannuzzi P. Contribution of peak respiratory exchange ratio to peak VO2 prognostic reliability in patients with chronic heart failure and severely reduced exercise capacity. Am Heart J 2003;145:1102–1107. [DOI] [PubMed] [Google Scholar]
- 45. Nakanishi M, Takaki H, Kumasaka R, Arakawa T, Noguchi T, Sugimachi M, Goto Y. Targeting of high peak respiratory exchange ratio is safe and enhances the prognostic power of peak oxygen uptake for heart failure patients. Circ J 2014;78:2268–2275. [DOI] [PubMed] [Google Scholar]
- 46. Bayes‐Genis A, de Antonio M, Vila J, Penafiel J, Galan A, Barallat J, Zamora E, Urrutia A, Lupon J. Head‐to‐head comparison of 2 myocardial fibrosis biomarkers for long‐term heart failure risk stratification: ST2 versus galectin‐3. J Am Coll Cardiol 2014;63:158–166. [DOI] [PubMed] [Google Scholar]
- 47. Cheng ML, Wang CH, Shiao MS, Liu MH, Huang YY, Huang CY, Mao CT, Lin JF, Ho HY, Yang NI. Metabolic disturbances identified in plasma are associated with outcomes in patients with heart failure: diagnostic and prognostic value of metabolomics. J Am Coll Cardiol 2015;65:1509–1520. [DOI] [PubMed] [Google Scholar]
- 48. Zordoky BN, Sung MM, Ezekowitz J, Mandal R, Han B, Bjorndahl TC, Bouatra S, Anderson T, Oudit GY, Wishart DS, Dyck JR, Alberta H. Metabolomic fingerprint of heart failure with preserved ejection fraction. PloS one 2015;10:e0124844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ruiz M, Labarthe F, Fortier A, Bouchard B, Thompson Legault J, Bolduc V, Rigal O, Chen J, Ducharme A, Crawford PA, Tardif JC, Des Rosiers C. Circulating acylcarnitine profile in human heart failure: a surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am J Physiol Heart Circ Physiol 2017;313:H768–H781. [DOI] [PubMed] [Google Scholar]
- 50. Doehner W, Frenneaux M, Anker SD. Metabolic impairment in heart failure: the myocardial and systemic perspective. J Am Coll Cardiol 2014;64:1388–1400. [DOI] [PubMed] [Google Scholar]
- 51. Zairis MN, Patsourakos NG, Tsiaousis GZ, Theodossis Georgilas A, Melidonis A, Makrygiannis SS, Velissaris D, Batika PC, Argyrakis KS, Tzerefos SP, Prekates AA, Foussas SG. Plasma asymmetric dimethylarginine and mortality in patients with acute decompensation of chronic heart failure. Heart 2012;98:860–864. [DOI] [PubMed] [Google Scholar]
- 52. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature 1997;389:349–352. [DOI] [PubMed] [Google Scholar]
- 53. Idell‐Wenger JA, Grotyohann LW, Neely JR. Coenzyme A and carnitine distribution in normal and ischemic hearts. J Biol Chem 1978;253:4310–4318. [PubMed] [Google Scholar]
- 54. Fukushima A, Lopaschuk GD. Acetylation control of cardiac fatty acid beta‐oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochim Biophys Acta 2016;1862:2211–2220. [DOI] [PubMed] [Google Scholar]
- 55. Thapa D, Zhang M, Manning JR, Guimaraes DA, Stoner MW, O'Doherty RM, Shiva S, Scott I. Acetylation of mitochondrial proteins by GCN5L1 promotes enhanced fatty acid oxidation in the heart. Am J Physiol Heart Circ Physiol 2017;313:H265–H274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen T, Liu J, Li N, Wang S, Liu H, Li J, Zhang Y, Bu P. Mouse SIRT3 attenuates hypertrophy‐related lipid accumulation in the heart through the deacetylation of LCAD. PloS one 2015;10:e0118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev 2010;90:207–258. [DOI] [PubMed] [Google Scholar]
- 58. Fukushima A, Milner K, Gupta A, Lopaschuk GD. Myocardial energy substrate metabolism in heart failure: from pathways to therapeutic targets. Curr Pharm Des 2015;21:3654–3664. [DOI] [PubMed] [Google Scholar]
- 59. Pagano AF, Brioche T, Arc‐Chagnaud C, Demangel R, Chopard A, Py G. Short‐term disuse promotes fatty acid infiltration into skeletal muscle. J Cachexia Sarcopenia Muscle 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. St‐Jean‐Pelletier F, Pion CH, Leduc‐Gaudet JP, Sgarioto N, Zovile I, Barbat‐Artigas S, Reynaud O, Alkaterji F, Lemieux FC, Grenon A, Gaudreau P, Hepple RT, Chevalier S, Belanger M, Morais JA, Aubertin‐Leheudre M, Gouspillou G. The impact of ageing, physical activity, and pre‐frailty on skeletal muscle phenotype, mitochondrial content, and intramyocellular lipids in men. J Cachexia Sarcopenia Muscle 2017;8:213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stugiewicz M, Tkaczyszyn M, Kasztura M, Banasiak W, Ponikowski P, Jankowska EA. The influence of iron deficiency on the functioning of skeletal muscles: experimental evidence and clinical implications. Eur J Heart Fail 2016;18:762–773. [DOI] [PubMed] [Google Scholar]
- 62. Tucker WJ, Nelson MD, Beaudry RI, Halle M, Sarma S, Kitzman DW, Gerche A, Haykowksy MJ. Impact of exercise training on peak oxygen uptake and its determinants in heart failure with preserved ejection fraction. Card Fail Rev 2016;2:95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Overmyer KA, Evans CR, Qi NR, Minogue CE, Carson JJ, Chermside‐Scabbo CJ, Koch LG, Britton SL, Pagliarini DJ, Coon JJ, Burant CF. Maximal oxidative capacity during exercise is associated with skeletal muscle fuel selection and dynamic changes in mitochondrial protein acetylation. Cell Metab 2015;21:468–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Venables MC, Achten J, Jeukendrup AE. Determinants of fat oxidation during exercise in healthy men and women: a cross‐sectional study. J Appl Physiol (1985) 2005;98:160–167. [DOI] [PubMed] [Google Scholar]
- 65. Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, Vega RB, Attie AD, Muoio DM, Kelly DP. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail 2014;7:1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gao S, McMillan RP, Jacas J, Zhu Q, Li X, Kumar GK, Casals N, Hegardt FG, Robbins PD, Lopaschuk GD, Hulver MW, Butler AA. Regulation of substrate oxidation preferences in muscle by the peptide hormone adropin. Diabetes 2014;63:3242–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Giannoni A, Aimo A, Mancuso M, Piepoli MF, Orsucci D, Aquaro GD, Barison A, De Marchi D, Taddei C, Cameli M, Raglianti V, Siciliano G, Passino C, Emdin M. Autonomic, functional, skeletal muscle, and cardiac abnormalities are associated with increased ergoreflex sensitivity in mitochondrial disease. Eur J Heart Fail 2017;19:1701–1709. [DOI] [PubMed] [Google Scholar]
- 68. Bertaggia E, Coletto L, Sandri M. Posttranslational modifications control FoxO3 activity during denervation. Am J Physiol Cell Physiol 2012;302:C587–C596. [DOI] [PubMed] [Google Scholar]
- 69. Hardee JP, Counts BR, Gao S, VanderVeen BN, Fix DK, Koh HJ, Carson JA. Inflammatory signalling regulates eccentric contraction‐induced protein synthesis in cachectic skeletal muscle. J Cachexia Sarcopenia Muscle 2018;9:369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Schematic representation of the time schedule of the experimental protocol. SPA, spontaneous physical activity.
Figure S2. Correlations between the plasma acetyllysine level and LVEF (A), log BNP (B), and thigh circumference (C) in the 10 patients with HF.LVEF, left ventricular ejection fraction; BNP, brain natriuretic peptide.