

Abstract
Signaling through the Hedgehog (Hh) pathway is mediated by the Patched (Ptch) family of proteins. Although the vertebrate Ptch proteins Ptch1 and Ptch2 harbor two closely related transmembrane modules related to sterol-sensing domains (SSDs), the role of these closely related receptors in the Hh pathway are not equivalent. Ptch1 is essential for development and appears to be the principal receptor mediating responses to Hh ligands, whereas Ptch2 is nonessential, and its role in Hh-signaling remains ambiguous. We hypothesized that the SSDs of the Ptch proteins function as generic modules whose protein-specific activities are determined by the adjacent cytoplasmic and luminal domains. We first showed that individual N-terminal and C-terminal halves of Ptch1 associated noncovalently to mediate ligand-dependent regulation of Hh signaling. The analogous regions of Ptch2 also interacted noncovalently but did not repress the Hh pathway. However, the SSD of Ptch2 were capable of repressing Hh signaling, as determined using chimeric proteins where the SSDs of Ptch1 were replaced by those from Ptch2. Replacement of the SSDs of Ptch1 with the analogous regions from the cholesterol transporter NPC1 failed to produce a chimeric protein capable of Hh repression. Further refinement of the specific regions in Ptch1 and Ptch2 revealed that specific cytoplasmic domains of Ptch1 were necessary but not sufficient for repression of Hh signaling and that the two principal luminal domains of Ptch1 and Ptch2 were interchangeable. These data support a model where the SSDs of the Ptch family proteins exhibit generic activities and that the adjacent cytoplasmic and luminal domains determine their protein-specific activities.
Keywords: Hedgehog signaling pathway, sonic hedgehog (SHH), transmembrane domain, membrane protein, protein chimera, transporter, NPC1, Patched-1, Patched-2, sterol-sensing domain
Introduction
Despite the centrality of the principal receptor of the Hedgehog (Hh)2 signaling pathway, Patched-1 (Ptch1), in developmental and neoplastic diseases, the structural determinants of its activities remain ill-defined. The primary sequence of Ptch1 predicts that it encodes a 12-pass transmembrane protein related to the RND class of bacterial small-molecule transporters (1–3). The best-characterized function of Ptch1 is its indirect regulation of the activity of the seven-pass, G protein–coupled protein Smoothened (Smo) (3–5). Although less well-studied, Ptch1 also modulates additional signaling cascades operating through both Smo-dependent (6–8) and Smo-independent mechanisms (9–13).
Vertebrates express a second, closely related Patched family member, Ptch2 (14–16). Despite its apparent sequence similarity to Ptch1 in regions critical for Ptch1 activity, conflicting data exist regarding the activities of Ptch2, in particular its ability to repress canonical Hh signaling and its response to Hh ligand. One group has shown that Ptch2 appears to behave similarly to Ptch1 by robustly repressing Smo-dependent Hh signaling and responding to Hh ligand (17). Other groups, however, reported only weak repression of Hh signaling (18) or no repression at all (19) for Ptch2. These apparently contradictory activities for Ptch2 lead to distinct models regarding its role in the Hh pathway. As suggested originally, Ptch2 activity may be redundant to Ptch1 in control of Hh signaling (17). These redundant activities were supported by the observation that Ptch2-deficient mice develop normally, even in tissues with normally high levels of Ptch2 expression (20), or exhibit only minor, nonlethal defects in hair follicles occurring late in adult animals (21). However, this redundancy does not appear to be reciprocal because mice lacking Ptch1 exhibit an embryonic lethal phenotype (22). These latter data imply that Ptch1 is the principal regulator of Hh signaling and that Ptch2 cannot compensate for the loss of Ptch1 activity. Indeed, only in the absence of Ptch1 were apparent roles for Ptch2 during development discernable (23). The apparent lack of Hh pathway repression activity by Ptch2 and its inability to compensate for the lack of Ptch1 suggest an alternative role for Ptch2. In this context, Ptch2 may act primarily as a ligand-dependent antagonist by sequestering Hh ligand (18, 24). In this case, Ptch2 potentially limits the range of Hh ligand activity rather than acting as a direct repressor of Smo-dependent Hh signaling. Taken together, despite the apparent structural similarities to Ptch1, the precise role of Ptch2 in the Hh signaling pathway remains ambiguous.
The cartoon in Fig. 1A illustrates the three basic modules in Ptch1 that include two luminal “loops,” the second of which (loop 2) encodes a conserved Hh ligand–binding motif (25); three large cytoplasmic domains we predicted to form intrinsically disordered protein regions; and two five-pass transmembrane regions that resemble sterol-sensing domains (SSDs) found in the family of proteins involved with cholesterol transport and homeostasis (26–28). These domains are also related to the large class of RND-containing transmembrane transporters in bacteria (for a review, see Ref. 29). Recently, the structures of the luminal and transmembrane domains for human and mouse Ptch1, alone or in complex with Shh-ligand, were solved using cryo-EM (30–32). These structures revealed that the two luminal domains and the two SSD-like regions, respectively, form distinct but closely apposed modules. These structures also provide insights into the possible means by which specific mutations found in Ptch1 in Gorlin syndrome might alter the activities of Ptch1 because at least some of the alterations arise in amino acids at the center of the interface between the two SSD-like modules.
Figure 1.

Noncovalent interaction between the N- and C-terminal halves of Ptch1. A, cartoon of Ptch1 illustrating the predicted orientation of specific regions in the membrane. B, stick diagram illustrating the specific amino acid boundaries of the Ptch1 constructs used in this assay. Amino acid numbering refers to the sequence of mPtch1. C, HEK293 cells were transfected with constructs expressing the N- or C-terminal half of Ptch1 with or without the cytoplasmic domains. i and ii, straight Western blots showing expression of the N-terminal halves (i) and HA-tagged C-terminal halves (ii) of Ptch1. iii, co-immunoprecipitation of the HA-tagged C-terminal halves with an antibody directed to the N terminus. iv, the reciprocal co-immunoprecipitation, demonstrating co-immunoprecipitation of the N-terminal halves with an anti-HA tag (C terminus) antibody.
These recent Ptch1 structures also revealed a high degree of similarity to the cholesterol transporter Niemann–Pick disease type C1 (NPC1) (33). As suggested by their highly similar sequences, the three-dimensional structure of the two transmembrane modules is predicted to be structurally similar to the analogous regions in NPC1, forming two closely apposed transmembrane domains. Likewise, the luminal regions give rise to a predicted structure similar to the luminal loops in NPC1. Despite these predicted similarities and the recent evidence that loss of NPC1 may alter cilium-dependent Hh signaling activities (34, 35), NPC1 does not appear to modulate Hh signaling directly (36). These data suggest that the transmembrane modules of Ptch1 and NPC1 may harbor distinct transport activities either because of intrinsic differences in these modules or to protein-specific activities imparted by their respective adjacent domains.
We addressed whether the transmembrane domains, referred to here as SSD1 and SSD2, of Ptch1, Ptch2, and NPC1 behave as generic transmembrane modules whose protein-specific activities are determined by the adjacent luminal and cytoplasmic domains. We showed that, despite Ptch1 and Ptch2 exhibiting distinct Smo-repressing activities, their SSD modules harbored indistinguishable activities. The Hh signaling repression activities were, however, specified in the Ptch family proteins, at least in part, by their adjacent cytoplasmic domains. We further showed that the generic activities of the SSD could not be extended to NPC1 because replacement of either of these domains in Ptch1 with the analogous regions of NPC1 generated proteins incapable of regulating the Hh signaling pathway.
Results
The N- and C-terminal halves of Ptch1 interact noncovalently
Homologs of Patched from Drosophila (dPtc) to vertebrates (Ptch) encode proteins harboring two closely related transmembrane modules known as SSDs. Co-expression of the N-terminal and C-terminal halves of dPtc recapitulated a significant amount of the activity of the intact native molecule (37). Furthermore, the two halves of dPtc could assemble noncovalently, as determined in co-immunoprecipitation assays. We first tested whether the halves of vertebrate Ptch1 also interacted noncovalently to form a functional molecule.
Vectors expressing either the N-terminal portion (Ptch1-N, aa 1–673) or the HA-tagged C-terminal half (Ptch1-CHA, aa 674–1434) of Ptch1 (Fig. 1B) were expressed in HEK293 cells. Following confirmation of their expression (Fig. 1C, i and ii), immunoprecipitation of either Ptch1-N (Fig. 1C, iii) or Ptch1-CHA (Fig. 1C, iv) co-immunoprecipitated the reciprocal half of Ptch1. Fig. 2 further illustrates that expression of the individual halves of Ptch1 had no effect on canonical Hh signaling in Ptch1-deficient MEFs. However, analogous to the reconstitution of dPtc in Drosophila, co-expression of both halves of mPtch1 potently repressed Smo-dependent Hh signaling. Furthermore, addition of N-Shh ligand reversed this repression to levels similar to those observed for the intact, full-length Ptch1 protein.
Figure 2.

The N- and C-terminal halves of Ptch1 combine to restore Smo-repressing activity. A, Ptch1-deficient MEFs were transiently transfected with an 8×Gli-luciferase construct, a constitutively expressing Renilla luciferase construct, and constructs expressing the Ptch1 halves, individually or together. Cells were serum-starved for 48 h and treated with either control or N-Shh-conditioned medium for 24 h. Neither half of Ptch1 is sufficient to repress Smo-dependent Hh signaling activity. However, when both halves are co-expressed, repression of Smo activity is restored. B, luciferase assay carried out as in A, but this time co-expressing the respective halves of Ptch1 with or without the middle loop or C terminus. Ptch1-NΔML with Ptch1-C repressed Smo-dependent Hh-signaling. However, Ptch1-CΔC with either Ptch1-N or Ptch1-NΔML did not repress the Hh pathway. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We showed previously that the cytoplasmic ML and C-terminal regions of Ptch1 mediated oligomerization of Ptch1 (38). Thus, it was determined next whether these same regions mediated the noncovalent assembly of the two halves of Ptch1. Mutants deleting the cytoplasmic regions of the Ptch1 N- and C-terminal halves, respectively (Fig. 1B), were tested by co-immunoprecipitation for their ability to associate with the reciprocal portion of Ptch1 (Fig. 1C). As lanes 4–6 in Fig. 1C, iii and iv, show, the N- and C-terminal halves of Ptch1 bound to each other regardless of the presence of either cytoplasmic domain. Similar to our previous observations, Fig. 2 shows that Ptch1-CΔML, when expressed with the C-terminal half of Ptch1 (Ptch1-C), repressed Smo-dependent Hh signaling. Deletion of the C terminus (Ptch1-CΔC), however, prevented repression of Hh signaling when expressed in the context of either Ptch1-N or Ptch1-NΔML, consistent with previous observations showing the requirement of the cytoplasmic C-terminal domain of Ptch1 for repression of Hh signaling (39). We also verified that the pairs of Ptch1 fragments were properly transported by determining whether they had been processed through the Golgi. Fig. 3, A and B, shows that P1-N, P1-NΔML, and P1-C do not appear to be processed when expressed individually because cleavage by Endo H was not protected. However, processing of P1-NΔML and, to a lesser extent, P1-N (Fig. 3B) was evident when they were co-expressed with P1-C. Likewise, processing of P1-C and P1-CΔCT was clearly evident when they were co-expressed with P1-NΔML and P1-N, respectively. Only when P1-C was co-expressed with P1-N was no processing evident for the C-terminal portion of Ptch1. The lack of processing was unrelated, however, to the ability of this pair of Ptch1 fragments to repress Hh signaling and respond to Hh ligand. Thus, the two halves of mPtch1 can interact noncovalently independently of their cytoplasmic domains to generate a functional protein that represses Smo activity and responds to Hh ligand.
Figure 3.

Posttranslational processing of the N- and C-terminal halves of Ptch1. A and B, HEK293 cells were transfected with constructs expressing the N- or C-terminal half of Ptch1, individually or together. Lysates were treated with either no enzyme, Endo H, or PNGase F for 1 h at 37 °C. Western blots were probed with an antibody directed to the first luminal loop of Ptch1 or the HA-tagged C terminus (B). Partial protection in the Endo H–treated lysates illustrates proper processing through the Golgi.
The transmembrane domains of Ptch1 and Ptch2 are equivalent functional modules
The primary structure of Ptch1 shares high sequence identity with the closely related protein Ptch2 (14–16, 40). This similarity is evident for the luminal and the transmembrane domains. However, the primary sequence of the cytoplasmic regions of Ptch1 and Ptch2 are structurally unrelated. We determined next whether the reciprocal halves of Ptch1 and Ptch2 (see Fig. 4A) could associate noncovalently and whether they gave rise to a functional regulator of canonical Hh signaling. After verification of expression of the various halves of Ptch1 and Ptch2 in HEK293 cells (Fig. 4B), the ability of these proteins to associate noncovalently was tested in co-immunoprecipitation assays. The N- and C-terminal halves of Ptch2 co-immunoprecipitated with the reciprocal C- and N-terminal portions, respectively, of Ptch1 (Fig. 4C, lanes 2, 4, and 6). However, despite their ability to form complexes, repression of Hh signaling was not apparent (Fig. 4D). The lack of activity of the heterologous complexes was consistent, however, with the failure of intact, full-length Ptch2 to repress Hh signaling in this assay. To ensure that the lack of Hh signaling repression activity of Ptch2 was not due to the presence of saturating levels of Ptch2 in the Ptch1-deficient MEFs, we deleted Ptch2 in this Ptch1-deficient line and performed a titration assay for Ptch2 activity. Fig. 5 shows that Hh signaling, although exquisitely sensitive to Ptch1 expression, is refractory to the expression of Ptch2. Only when Ptch2 was expressed at unusually high levels was a small but significant level of repression of Hh signaling observed.
Figure 4.

The N- and C-terminal halves of Ptch2 bind to but do not complement the activity of the reciprocal halves of Ptch1. A, stick diagram depicting the amino acid boundaries of the Ptch2 constructs. Amino acid numbering refers to the sequence of mPtch2. B, straight Western blots detecting transient expression of the N- and C-terminal portions of Ptch1 and Ptch2 in lysates from HEK293 cells. C, from left to right, co-immunoprecipitation of mycPtch2-C with FLAGPtch2-N (lane 2), mycPtch2-C with Ptch1-N (lane 4), and FLAGPtch2-N with Ptch1-CHA (lane 6). Both halves of Ptch2 noncovalently interact with the reciprocal halves of Ptch1. D, Ptch1-deficient MEFs were transiently transfected with an 8×Gli-luciferase construct, a constitutively expressing Renilla luciferase construct, and constructs expressing Ptch1 or Ptch2 halves individually or together. Neither half of Ptch2 is capable of complementing the respective half of Ptch1 to restore Smo-repressing activity, nor does full-length Ptch2 repress the Hh pathway. **, p < 0.01.
Figure 5.

Ptch2 does not repress Smo-dependent Hh signaling. Ptch2-deficient cells were derived from Ptch1-deficient MEFs. The ability of Ptch1 or Ptch2 to repress Hh signaling in a transient assay was assessed by titrating increasing levels of constructs expressing these two proteins. Although exquisitely sensitive to repression by Ptch1, these cells showed essentially no repression of Hh signaling when FLAGPtch2 was expressed. ***, p < 0.001.
Given the high degree of similarity between the sequences of Ptch1 and Ptch2 in their luminal and transmembrane domains, the failure of Ptch2 to repress Hh signaling appears paradoxical. Thus, we tested the Hh signaling activities of the transmembrane domains of Ptch2 in the context of the luminal and cytoplasmic regions of Ptch1. Specifically, the transmembrane modules of Ptch1 were replaced, either individually or together, with the analogous regions from Ptch2 (Fig. 6A). Potent repression of Hh signaling was observed for chimeric Ptch1 proteins harboring either individual or both transmembrane regions from Ptch2 (Fig. 6B). Thus, both transmembrane domains of Ptch2 are competent to repress Hh signaling in the context of the adjacent luminal and cytoplasmic domains of Ptch1. This activity for the SSDs of Ptch2 is observed despite their apparent inability to repress Hh signaling in the context of the adjacent domains of Ptch2.
Figure 6.

The transmembrane domains of Ptch1 and Ptch2 are equivalent functional modules. A, stick diagram illustrating the specific amino acid boundaries of the Ptch1–Ptch2 constructs used in this assay. Included below are the DNA and amino acid sequences for the boundaries of the chimeric proteins. B, Ptch1-deficient MEFs were transiently transfected with an 8×Gli-luciferase construct, a constitutively expressing Renilla luciferase construct, and constructs expressing Ptch1–Ptch2 chimeras. Cells were serum-starved for 48 h and treated with either control (pcDNA3) or N-Shh conditioned medium for 24 h. Ptch1 chimeras with either or both SSDs from Ptch2 fully repressed Smo and responded to Hh ligand, demonstrating that the SSDs in Ptch1 and Ptch2 are functionally equivalent despite the lack of repression activity of the intact Ptch2 protein. **, p < 0.01; ***, p < 0.001.
Because the SSD of Ptch2 could replace the activities of these regions of Ptch1, we next determined whether the SSDs of more distally related SSD-containing proteins acted as generic modules whose protein-specific activities were determined by the adjacent luminal and cytoplasmic domains. Thus, we replaced the SSD modules of Ptch1 with those from the cholesterol transporter NPC1 (Fig. 7A) and tested the chimeric proteins for their ability to repress Hh signaling. As Fig. 7B demonstrates, neither of the SSD regions of NPC1 mediated repression of Hh signaling, even when all but a single SSD of the chimeric protein were attributable to Ptch1.
Figure 7.

The SSDs from NPC1 cannot restore Ptch1 activity. A, stick diagram illustrating the specific amino acid boundaries of the Ptch1 and NPC1 constructs used to create chimeric Ptch1 proteins with the SSDs from NPC1. The arrow at aa 672 indicates the break point separating the two half-molecules used in C. DNA and amino acid sequences for the specific boundaries of the chimeric proteins are also shown. B, Ptch1 with individual or both SSD regions replaced with the analogous regions of NPC1 were tested in the Hh repression assay. None of the constructs tested were able to repress Smo-dependent Hh signaling, thus demonstrating that the SSDs from these distinct classes of SSD-containing proteins do not act as generic modules. C, two amino acids necessary for Ptch1 function, which are different in NPC1 (Gly495 and Asp500 in mPtch1), were mutated to those in the identical position in Ptch1. This was done in a chimeric protein encoding the N-terminal half of Ptch1 but with SSD1 replaced with that of NPC1 (PNS1-N). These were co-expressed with the WT C-terminal half of Ptch1 (Ptch1-C) or the C terminus with the second SSD replaced with that from NPC1 (PNS2-C) and tested for their ability to repress Smo-dependent Hh activity. Changing these specific amino acids was not sufficient to restore Smo repression activity. ***, p < 0.001.
We next asked whether the NPC1 and Ptch1 SSDs were fundamentally distinct or whether the NPC1 SSDs could be made functional by altering two specific amino acid residues in the third transmembrane helix of SSD1. These residues are required for Ptch1 function and, as Fig. 9 illustrates, are different in NPC1 (3, 41, 42, 32). Specifically, we targeted the Ala residue in NPC1 that is analogous to Gly495 in mPtch1 and the Asn residue in NPC1 analogous to Asp500 in mPtch1. These specific mutations in the SSD1 module of NPC1 were introduced into a chimeric protein encoding the N-terminal half of Ptch1 but with SSD1 replaced with that of NPC1 (PNS1-N, Fig. 7A).
Figure 9.
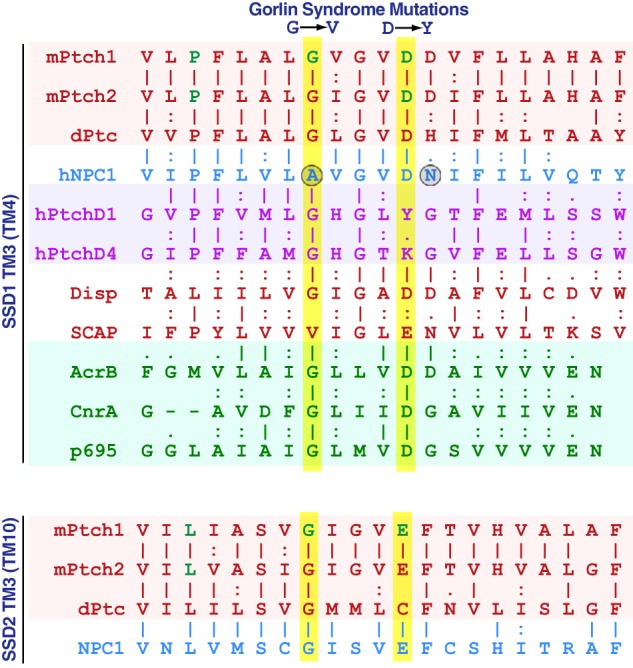
Amino acid alignment in helix 3 of the first transmembrane domain of SSD and RND proteins. Shown are the amino acid sequences for the SSD-containing proteins Ptch1, Ptch2, dPtc, NPC1, PtchD1, PtchD4, Disp, and SCAP as well as the RND proteins AcrB, CnrA, and p695. Two critical residues that are different in NPC1 and lead to Gorlin syndrome when altered in hPtch1 are indicated with circles.
Point mutants were co-expressed with the WT C-terminal half of Ptch1 (Ptch1-C) or the C terminus with the second SSD replaced with that from NPC1 (PNS2-C) and tested for their ability to repress Smo-dependent Hh-activity (Fig. 7C). Despite the requirement for these specific amino acids for Ptch1 function, the replacements in the SSD1 of NPC1 were unable to confer Smo repression activity on the NPC1 SSDs. Thus, although Ptch1 and Ptch2 are related closely enough that their SSD are interchangeable, these domains cannot be replaced by the analogous regions from NPC1, even with replacement of amino acids critical for Ptch1 activity.
The activities of the Ptch family proteins are governed by the adjacent cytoplasmic regions
The SSDs of Ptch2 were able to repress Hh signaling in the context of the adjacent domains derived from Ptch1. Given the similarity between the luminal domains of Ptch1 and Ptch2, we tested the possibility that their cytoplasmic domains governed the protein-specific activities of their SSDs. To address this possibility, the cytoplasmic C-terminal regions of Ptch1 and Ptch2 were exchanged in constructs expressing the C-terminal halves of those proteins (Fig. 8A). As Fig. 8B confirms, co-expression of the two halves of Ptch1 but not Ptch2 repressed canonical Hh signaling and responded to Hh ligand. Repression of Smo required the cytoplasmic C-terminal domain of Ptch1 because the construct deleting this region (P1-CΔCT) or replacing it with the analogous C-terminal region of Ptch2 (P1-CP2-CT) failed to repress the Hh pathway when co-expressed with the N-terminal half of Ptch1. The C terminus of Ptch1 is not sufficient, however, to confer repression activity on Ptch2 because the P2-CP1-CT protein, when expressed with the N-terminal half of Ptch2, did not suppress Hh signaling. However, when P2-CP1-CT was co-expressed with the N-terminal half of Ptch1, partial repression (40%) was consistently observed. Interestingly, when the ML region of Ptch1 (P1-NΔML) was deleted and expressed with P2-CP1-CT, potent repression (>70%) was observed. In addition, this repression was refractory to the inhibitory effects of added N-Shh ligand.
Figure 8.

The activities of the Ptch family proteins are governed, in part, by their adjacent cytoplasmic regions. A, stick diagram illustrating the specific amino acid boundaries of the Ptch1 and Ptch2 constructs. Amino acid numbering refers to the sequences of mPtch1 and mPtch2, respectively. B, specific pairs of N- and C-terminal constructs were tested in the Hh pathway repression assays. When the C-terminal half of Ptch2 containing the cytoplasmic C terminus from Ptch1 (P2-CP1-CT) was expressed with P1-N or P1-NΔML, partial or full repression, respectively, of Hh signaling was observed. However, expression of P2-CP1-CT with P2-N did not recapitulate the repression activity. Furthermore, the P1-N + P1-CΔCT and the P1-N + P1-CP2-NT combinations showed that the cytoplasmic C terminus of Ptch1 was required but not sufficient for repression activity. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Taken together, these data reveal that the cytoplasmic domains of Ptch1 play an integral role in the repression of Hh signaling activity by the Ptch family proteins Ptch1 and Ptch2. They show further that, for these related factors, the protein-specific activities of their interchangeable SSDs are determined by the adjacent cytoplasmic domains.
Discussion
Despite a fundamental role during development and its activity as a tumor suppressor, most of the basic structural aspects that give rise to the activities of Ptch1 remain nebulous. The basic structure of its transmembrane regions supports the notion that it acts as a transporter because of their similarity to other proteins harboring similar domains, such as the RND superfamily of membrane transporters and the eukaryotic cholesterol transporter NPC1 (43). Like other members in its class, Ptch1 harbors two transmembrane SSD regions, this arrangement was proposed to have arisen as the result of a gene duplication (44). These transmembrane regions also exhibit a high degree of similarity across species. As discussed in more detail below, essential amino acid residues in the transmembrane domains are conserved between proteins with SSDs as well as with the analogous regions of the RND superfamily of membrane transporters in bacteria.
The two apposing luminal domains (loop1 and loop2 in Ptch1) are also highly similar among SSD-containing proteins. In contrast, the three regions that encode the cytoplasmic domains (the N terminus, the middle loop (ML), and the C-terminal domain) are essentially unrelated between the different proteins. However, they are almost identical in the same proteins when compared between species. For example, although the N terminus, C terminus, and ML regions in Ptch1 show remarkable identity across vertebrates, the analogous regions in the otherwise closely related Ptch2 proteins are unrelated to those in Ptch1. Importantly, although we and others have demonstrated that these regions facilitate binding to factors that harbor SH3, SH2, and WW domains (38, 45–47), they appear to encode intrinsically disordered protein regions. It should be noted further that none of the recently published cryo-EM structures for Ptch1 were solved using proteins containing intact cytoplasmic domains (30–32). For Ptch1 proteins that contained segments of these cytoplasmic domains, no apparent structure for these regions was reported, further suggesting that they are disordered.
We propose that the unique sequences in the cytoplasmic regions of these SSD-containing factors lead to at least two protein-specific phenomena. First, these regions impart protein-specific complexity to the pathways through which the Hh ligands signal. This complexity arises because of the unique cohort of factors that bind to these regions. So, for example, as we and others have shown, the cytoplasmic domains of Ptch1 facilitate the noncovalent association of a number of proteins involved in distinct signaling cascades (10, 11, 45, 48). It is evident from the primary sequences of the C-terminal domains of Ptch1 and Ptch2, respectively, that the interactome of these proteins are likely distinct. Similarly, the N terminus of Ptch2 contains two uncharacterized protein interaction motifs (PPXY and YXXP, aa 11–17) that we predict bind to a unique set of WW- and SH2-containing factors. This motif is absent from the N terminus of Ptch1, although a related motif can be found in its C-terminal domain. Thus, regardless of its role in regulating Smo-dependent Hh-signaling, we expect that Ptch2 may facilitate increased complexity in the signaling cascades that respond to the Hh ligands.
Second, we demonstrated that these same cytoplasmic regions are important for intramolecular regulation of the activities of the Ptch family proteins. Specifically, we showed that, although Ptch1 but not Ptch2 repressed the canonical Hh signaling pathway, the SSDs of Ptch2 were quite capable of repressing canonical Hh signaling when present in the context of the adjacent luminal and cytoplasmic domains of Ptch1. Refinement of the domains that regulate SSD activities in Ptch1 and Ptch2 showed that the C terminus of Ptch1 was also necessary, but not sufficient, to allow the SSDs of Ptch2 to exhibit Hh pathway repression activity.
The mechanism by which the cytoplasmic domains of the Ptch family proteins control the activity of the transmembrane domains is unclear. It is possible that the cytoplasmic domains exert a conformational effect on the transmembrane regions whereby the presence or absence of the cytoplasmic domains (or interactions with another protein mediated by the cytoplasmic domains) effects changes in the functional conformation of the transmembrane domains. Alternatively, the cytoplasmic domains may mediate changes in cellular localization required for specific activities. Deletion of the C terminus of Ptch1 or replacement of this region with the C-terminal domain from Ptch2 inhibited Hh pathway repression, consistent with Kim et al. (39), who showed that the C terminus of Ptch1 is necessary for proper localization and repression of Smo.
The data in Fig. 8B also illustrate that the two principal luminal domains of Ptch1 and Ptch2 have interchangeable activities. For example, when the N-terminal half of Ptch1 with or without the ML region (P1-N and P1-NΔML, respectively) was co-expressed with the C-terminal half of Ptch2 harboring the Ptch1 C-terminus (P2-CP1-CT), repression of Hh signaling was observed. Furthermore, the ability of the combination of P1-N and P2-CP1CT to respond to ligand demonstrates that the second luminal loop of Ptch2 is capable of facilitating a response to Hh ligand. Given that this loop encodes a sequence predicted to act as a binding site for Hh ligand (25), this observation supports the previously proposed model (18) suggesting that Ptch2 may act as a ligand-dependent antagonist of Hh signaling rather than as a direct repressor of Smo activity.
The generic nature of the SSDs does not seem to extend to other members of this family. Specifically, we exchanged the SSD between the cholesterol transporter NPC1 with those from Ptch1. When tested in the Hh repression assay, even exchange of a single SSD in Ptch1 with that of NPC-1 produced a protein that failed to repress Smo. The lack of activity might have been expected because of differences in specific amino acids between Ptch1/Ptch2 and NPC-1. As Fig. 9 illustrates, one mutation in the third helix of the first SSD of Ptch1, G509V (50) (G495V in the mouse), that gives rise to Gorlin syndrome generates a Ptch1 protein with reduced repression activity in the Hh signaling pathway (3, 41). This amino acid in the analogous helix of NPC1 is an Ala residue. A similar logic applies to Asp500 which, when deleted, leads to Gorlin syndrome (32, 42). The analogous amino acid in NPC1 is an Asn and might be expected, therefore, to block Ptch1 activity in the chimeric protein. However, as Fig. 7C shows, changing these specific amino acids was insufficient to restore Smo repression activity.
The recently solved cryo-EM structures of Ptch1 (30–32) and NPC1 (33) suggest another possible explanation for why the SSDs of Ptch1 and NPC1 do not exhibit overlapping activities. Close inspection of the NPC1 structure reveals that the recently described amino acids that are critical for Ptch1 function are positioned at the bottom of the TM4 helix (TM3 of SSD1) outside of the core structure of SSD1. The position of these analogous amino acids in SSD1-TM3 of Ptch1 is distinct from NPC1, being directly at the center of the SSD1 at the interface between the two SSD-like regions. Indeed, a recent paper describing the cryo-EM structure of Ptch1 delineated the significance of the charged residues in the middle of the third helices of both SSD1 and SSD2 of Ptch1 (32). The contrasting positions of these amino acids in Ptch1 versus NPC1 suggest that the sequences surrounding this region may give rise to profoundly different three-dimensional structures. Thus, distinct activities would be expected for these SSD in Ptch1 versus NPC1. Alternatively, the nature or processing of the proteins used to generate the cryo-EM structures of this region in NPC1 or Ptch1 has induced a significant structural alteration not representative of the native proteins.
Experimental procedures
Plasmid constructs
All constructs were derived from murine Ptch1, murine Ptch2, and human NPC1. Deletion mutants and chimeric molecules were created using either convenient restriction sites combined with double-stranded oligonucleotides or gene synthesis (GenScript). The precise amino acid boundaries are indicated in each figure.
The Ptch1−/−Ptch2−/− cell line
Double Ptch1−/−Ptch2−/− MEFs were derived from Ptch1−/−MEFs (a kind gift from C. C. Hui, Hospital for Sick Children, Toronto, ON, Canada) derived from the Ptch1−/− mice harboring a lacZ gene in-frame with exon 1 of Ptch1 (22). A knockout mutation in Ptch2 was produced using the CRISPR/Cas9 system. Two gRNAs targeted against exon 3 of mPtch2 (gRNA sequences TCACCCCGCTTGACTGCTTC and GTTGATTCAGACTGCGCACC, Ref. 51) were cloned simultaneously into the pX330A-1 × 2 vector using the Golden Gate assembly system (Addgene, Ref. 52). Cells were co-transfected with 4 μg of the Cas9/gRNA vector and 200 ng PGK-Puro for selection. 48 h post-transfection, transfected cells were selected in medium containing 4.0 μg/ml puromycin. Individual colonies were isolated, and the presence of indels was verified by treating a PCR-amplified region containing exon 3 using T7 endonuclease according to manufacturer's instructions (New England Biolabs, Ref. 53). Knockout mutations in exon 3 of Ptch2 were verified by sequencing.
Western blotting and co-immunoprecipitation
HEK293 cells grown in 100-mm plates were co-transfected with 2 μg of each Ptch1 construct using 2 mg/ml polyethyleneimine at a 2:1 ratio of polyethyleneimine to DNA. Cell lysates were prepared using 0.5% NP-40 as described previously (12, 38). For straight Western blots, 50 μg of lysate was mixed with 4× SDS loading buffer (50 mm Tris (pH 6.8), 100 mm DTT, 2% SDS, 0.1% bromphenol blue, and 10% glycerol) and incubated for at least 20 min at 37 °C. Samples were separated using 10% SDS-PAGE (54) and blotted onto a nitrocellulose membrane (55). Blots were blocked in 5% skim milk and probed overnight with primary antibodies in a 3% BSA solution. The antibodies used were as follows: 1:1000 goat anti-Ptch1 (Santa Cruz Biotechnology, sc-6149), 1:1000 rabbit anti-HA (ABM, G166), 1:500 mouse anti-HA (12CA5, Developmental Studies Hybridoma Bank), 1:1000 mouse anti-myc (ABM, G019), and rabbit anti-FLAG (ABM, G188). The following day, horseradish peroxidase–linked secondary antibodies in 5% skim milk were applied for 1 h (1:5000 goat anti-rabbit IgG (Cell Signaling Technology, 7074), 1:5000 horse anti-mouse IgG (Cell Signaling Technology, 7076), or 1:5000 donkey anti-goat IgG (Santa Cruz Biotechnology, sc-2020)). Western blots were developed with Western Lightning PLUS ECL (PerkinElmer Life Sciences) and imaged using a MicroChemi 2.0 Imager (FroggaBio, Toronto, ON, Canada).
For immunoprecipitation, primary antibody was added to 250 μg of total cell lysate and incubated overnight at 4 °C in 0.5% NP-40 lysis buffer. The following day, 15 μl of protein G–agarose or protein A–agarose beads was added at 4 °C for 2 h. Beads were then spun down and washed five times in 0.5% NP-40 and then resuspended in 20 μl of 1× SDS loading buffer. Proteins were separated on 10% SDS-PAGE gels, and western blot analyses were performed as described above.
Glycosidase assay
HEK293 cells were transiently transfected, and lysates were taken in 0.5% NP-40 lysis buffer as described above. 50 μg of lysate was made up to 20 μl of volume with water and treated with either no enzyme, 500 units of Endo H (New England Biolabs), or 500 units of PNGase F (New England Biolabs) for 1 h at 37 °C. Cells were then mixed with 4× SDS loading buffer, and western blots were run as above.
Luciferase reporter assays
Shh-conditioned and control media were prepared by transiently transfecting HEK293 cells with either 5 μg of pcDNA3.1-N-Shh or 5 μg of empty pcDNA3.1, as described previously (38). Shh ligand activity was verified using Shh Light II fibroblasts that harbor an intrinsic 8×Gli-luciferase promoter as described previously (49).
To assay Ptch family protein modulation of canonical Hh signaling, 50 ng of expression plasmids encoding various Ptch1 and/or Ptch2 mutants were co-transfected with 800 ng of an 8×Gli-firefly luciferase reporter transgene and 80 ng of a constitutive Renilla luciferase transgene in either Ptch1- or Ptch1/Ptch2-deficient MEFs using FuGENE 6 (4:1 ratio, Promega). After 24 h, cells were starved in serum-free medium for 48 h. For cells that were treated with Hh ligand, conditioned medium was added after 24 h of starvation, and lysates were taken after another 24 h in serum-free-medium. Firefly and Renilla luciferase activities were determined using the Dual-Luciferase reporter assay system (Promega) according to the manufacturer's instructions. Data were analyzed by one-way analysis of variance, followed by pairwise comparison of means using a Student's t test. Data are displayed as mean ± S.E.; n = minimum of three independent experiments. Each experiment was done using biological and technical duplicates and averaged.
Author contributions
A. J. F. and P. A. H. conceptualization; A. J. F. formal analysis; A. J. F. validation; A. J. F. investigation; A. J. F. and P. A. H. visualization; A. J. F. and P. A. H. methodology; A. J. F. and P. A. H. writing-original draft; A. J. F. and P. A. H. project administration; A. J. F. and P. A. H. writing-review and editing; P. A. H. supervision; P. A. H. funding acquisition.
Acknowledgment
We thank Dr. Barbara Karten (Dalhousie University, Halifax, NS, Canada) for insights into NPC1.
This work was funded by Canadian Institutes of Health Research Grant MOP-142490 (to P. A. H.). The authors declare that they have no conflicts of interest with the contents of this article.
- Hh
- Hedgehog
- Ptch
- Patched
- RND
- resistance–nodulation–division
- SSD
- sterol-sensing domain
- aa
- amino acids
- HA
- hemagglutinin
- ML
- middle loop
- HEK
- human embryonic kidney
- MEF
- mouse embryonic fibroblast
- SH
- Src homology
- gRNA
- guide RNA
- Endo H
- endoglycosidase H
- PNGase F
- peptide:N-glycosidase F.
References
- 1. Hausmann G., von Mering C., and Basler K. (2009) The Hedgehog signaling pathway: where did it come from? PLoS Biol. 7, e1000146 10.1371/journal.pbio.1000146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tseng T. T., Gratwick K. S., Kollman J., Park D., Nies D. H., Goffeau A., and Saier M. H. Jr. (1999) The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J. Mol. Microbiol. Biotechnol. 1, 107–125 [PubMed] [Google Scholar]
- 3. Taipale J., Cooper M. K., Maiti T., and Beachy P. A. (2002) Patched acts catalytically to suppress the activity of Smoothened. Nature 418, 892–897 10.1038/nature00989 [DOI] [PubMed] [Google Scholar]
- 4. Chen Y., and Struhl G. (1998) In vivo evidence that Patched and Smoothened constitute distinct binding and transducing components of a Hedgehog receptor complex. Development. 125, 4943–4948 [DOI] [PubMed] [Google Scholar]
- 5. Ingham P. W., Nystedt S., Nakano Y., Brown W., Stark D., van den Heuvel M., and Taylor A. M. (2000) Patched represses the Hedgehog signalling pathway by promoting modification of the Smoothened protein. Curr. Biol. CB. 10, 1315–1318 10.1016/S0960-9822(00)00755-7 [DOI] [PubMed] [Google Scholar]
- 6. Riobo N. A., Haines G. M., and Emerson C. P. (2006) Protein kinase C-δ and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 66, 839–845 10.1158/0008-5472.CAN-05-2539 [DOI] [PubMed] [Google Scholar]
- 7. Chinchilla P., Xiao L., Kazanietz M. G., and Riobo N. A. (2010) Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle 9, 570–579 10.4161/cc.9.3.10591 [DOI] [PubMed] [Google Scholar]
- 8. Polizio A. H., Chinchilla P., Chen X., Manning D. R., and Riobo N. A. (2011) Sonic Hedgehog activates the GTPases Rac1 and RhoA in a Gli-independent manner through coupling of smoothened to Gi proteins. Sci. Signal. 4, pt7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barnes E. A., Kong M., Ollendorff V., and Donoghue D. J. (2001) Patched1 interacts with cyclin B1 to regulate cell cycle progression. EMBO J. 20, 2214–2223 10.1093/emboj/20.9.2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wakabayashi Y., Mao J.-H., Brown K., Girardi M., and Balmain A. (2007) Promotion of Hras-induced squamous carcinomas by a polymorphic variant of the Patched gene in FVB mice. Nature 445, 761–765 10.1038/nature05489 [DOI] [PubMed] [Google Scholar]
- 11. Klein C., Zwick A., Kissel S., Forster C. U., Pfeifer D., Follo M., Illert A. L., Decker S., Benkler T., Pahl H., Oostendorp R. A., Aumann K., Duyster J., and Dierks C. (2016) Ptch2 loss drives myeloproliferation and myeloproliferative neoplasm progression. J. Exp. Med. 213, 273–290 10.1084/jem.20150556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang H., Li Q., Moraes R. C., Lewis M. T., and Hamel P. A. (2010) Activation of Erk by sonic hedgehog independent of canonical hedgehog signalling. Int. J. Biochem. Cell Biol. 42, 1462–1471 10.1016/j.biocel.2010.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Okolowsky N., Furth P. A., and Hamel P. A. (2014) Oestrogen receptor-α regulates non-canonical Hedgehog-signalling in the mammary gland. Dev. Biol. 391, 219–229 10.1016/j.ydbio.2014.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Motoyama J., Takabatake T., Takeshima K., and Hui C. (1998) Ptch2, a second mouse Patched gene is co-expressed with Sonic hedgehog. Nat. Genet. 18, 104–106 10.1038/ng0298-104 [DOI] [PubMed] [Google Scholar]
- 15. Smyth I., Narang M. A., Evans T., Heimann C., Nakamura Y., Chenevix-Trench G., Pietsch T., Wicking C., and Wainwright B. J. (1999) Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene in basal cell carcinoma and medulloblastoma on chromosome 1p32. Hum. Mol. Genet. 8, 291–297 10.1093/hmg/8.2.291 [DOI] [PubMed] [Google Scholar]
- 16. Carpenter D., Stone D. M., Brush J., Ryan A., Armanini M., Frantz G., Rosenthal A., and de Sauvage F. J. (1998) Characterization of two patched receptors for the vertebrate hedgehog protein family. Proc. Natl. Acad. Sci. 95, 13630–13634 10.1073/pnas.95.23.13630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhulyn O., Li D., Deimling S., Vakili N. A., Mo R., Puviindran V., Chen M.-H., Chuang P.-T., Hopyan S., and Hui C.-C. (2014) A switch from low to high Shh activity regulates establishment of limb progenitors and signaling centers. Dev. Cell 10.1016/j.devcel.2014.03.002 [DOI] [PubMed] [Google Scholar]
- 18. Holtz A. M., Peterson K. A., Nishi Y., Morin S., Song J. Y., Charron F., McMahon A. P., and Allen B. L. (2013) Essential role for ligand-dependent feedback antagonism of vertebrate hedgehog signaling by PTCH1, PTCH2 and HHIP1 during neural patterning. Development. 140, 3423–3434 10.1242/dev.095083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rahnama F., Toftgård R., and Zaphiropoulos P. G. (2004) Distinct roles of PTCH2 splice variants in Hedgehog signalling. Biochem. J. 378, 325–334 10.1042/bj20031200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee Y., Miller H. L., Russell H. R., Boyd K., Curran T., and McKinnon P. J. (2006) Patched2 modulates tumorigenesis in patched1 heterozygous mice. Cancer Res. 66, 6964–6971 10.1158/0008-5472.CAN-06-0505 [DOI] [PubMed] [Google Scholar]
- 21. Nieuwenhuis E., Motoyama J., Barnfield P. C., Yoshikawa Y., Zhang X., Mo R., Crackower M. A., and Hui C. (2006) Mice with a targeted mutation of Patched2 are viable but develop alopecia and epidermal hyperplasia. Mol. Cell Biol. 26, 6609–6622 10.1128/MCB.00295-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goodrich L. V., Milenković L., Higgins K. M., and Scott M. P. (1997) Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109–1113 10.1126/science.277.5329.1109 [DOI] [PubMed] [Google Scholar]
- 23. Zhulyn O., Nieuwenhuis E., Liu Y. C., Angers S., and Hui C. (2015) Ptch2 shares overlapping functions with Ptch1 in Smo regulation and limb development. Dev. Biol. 397, 191–202 10.1016/j.ydbio.2014.10.023 [DOI] [PubMed] [Google Scholar]
- 24. Alfaro A. C., Roberts B., Kwong L., Bijlsma M. F., and Roelink H. (2014) Ptch2 mediates the Shh response in Ptch1−/− cells. Development 141, 3331–3339 10.1242/dev.110056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bosanac I., Maun H. R., Scales S. J., Wen X., Lingel A., Bazan J. F., de Sauvage F. J., Hymowitz S. G., and Lazarus R. A. (2009) The structure of SHH in complex with HHIP reveals a recognition role for the Shh pseudo active site in signaling. Nat. Struct. Mol. Biol. 16, 691–697 10.1038/nsmb.1632 [DOI] [PubMed] [Google Scholar]
- 26. Martín V., Carrillo G., Torroja C., and Guerrero I. (2001) The sterol-sensing domain of Patched protein seems to control Smoothened activity through Patched vesicular trafficking. Curr. Biol. 11, 601–607 10.1016/S0960-9822(01)00178-6 [DOI] [PubMed] [Google Scholar]
- 27. Burke R., Nellen D., Bellotto M., Hafen E., Senti K. A., Dickson B. J., and Basler K. (1999) Dispatched, a novel sterol-sensing domain protein dedicated to the release of cholesterol-modified hedgehog from signaling cells. Cell 99, 803–815 10.1016/S0092-8674(00)81677-3 [DOI] [PubMed] [Google Scholar]
- 28. Loftus S. K., Morris J. A., Carstea E. D., Gu J. Z., Cummings C., Brown A., Ellison J., Ohno K., Rosenfeld M. A., Tagle D. A., Pentchev P. G., and Pavan W. J. (1997) Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science 277, 232–235 10.1126/science.277.5323.232 [DOI] [PubMed] [Google Scholar]
- 29. Nies D. H. (2003) Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol. Rev. 27, 313–339 10.1016/S0168-6445(03)00048-2 [DOI] [PubMed] [Google Scholar]
- 30. Gong X., Qian H., Cao P., Zhao X., Zhou Q., Lei J., and Yan N. (2018) Structural basis for the recognition of Sonic Hedgehog by human Patched1. Science 10.1126/science.aas8935 [DOI] [PubMed] [Google Scholar]
- 31. Qi X., Schmiege P., Coutavas E., Wang J., and Li X. (2018) Structures of human Patched and its complex with native palmitoylated sonic hedgehog. Nature 10.1038/s41586-018-0308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y., Bulkley D. P., Roberts K. J., Xin Y., Asarnow D., Sharma A., Myers B. R., Cho W., Cheng Y., and Beachy P. A. (2018) Structural basis for cholesterol transport-like activity of the Hedgehog receptor Patched. bioRxiv 10.1101/352443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gong X., Qian H., Zhou X., Wu J., Wan T., Cao P., Huang W., Zhao X., Wang X., Wang P., Shi Y., Gao G. F., Zhou Q., and Yan N. (2016) Structural insights into the Niemann-Pick C1 (NPC1)-mediated cholesterol transfer and Ebola infection. Cell 165, 1467–1478 10.1016/j.cell.2016.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Canterini S., Dragotto J., Dardis A., Zampieri S., De Stefano M. E., Mangia F., Erickson R. P., and Fiorenza M. T. (2017) Shortened primary cilium length and dysregulated Sonic hedgehog signaling in Niemann-Pick C1 disease. Hum. Mol. Genet. 26, 2277–2289 10.1093/hmg/ddx118 [DOI] [PubMed] [Google Scholar]
- 35. Formichi P., Battisti C., De Santi M. M., Guazzo R., Tripodi S. A., Radi E., Rossi B., Tarquini E., and Federico A. (2018) Primary cilium alterations and expression changes of Patched1 proteins in Niemann-Pick type C disease. J. Cell Physiol. 233, 663–672 10.1002/jcp.25926 [DOI] [PubMed] [Google Scholar]
- 36. Incardona J. P., Gaffield W., Lange Y., Cooney A., Pentchev P. G., Liu S., Watson J. A., Kapur R. P., and Roelink H. (2000) Cyclopamine inhibition of Sonic hedgehog signal transduction is not mediated through effects on cholesterol transport. Dev. Biol. 224, 440–452 10.1006/dbio.2000.9775 [DOI] [PubMed] [Google Scholar]
- 37. Johnson R. L., Milenkovic L., and Scott M. P. (2000) In vivo functions of the Patched protein: requirement of the C terminus for target gene inactivation but not Hedgehog sequestration. Mol. Cell 6, 467–478 10.1016/S1097-2765(00)00045-9 [DOI] [PubMed] [Google Scholar]
- 38. Fleet A., Lee J. P. Y., Tamachi A., Javeed I., and Hamel P. A. (2016) Activities of the cytoplasmic domains of Patched-1 modulate but are not essential for regulation of canonical Hedgehog signaling. J. Biol. Chem. 10.1074/jbc.M116.731745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim J., Hsia E. Y., Brigui A., Plessis A., Beachy P. A., and Zheng X. (2015) The role of ciliary trafficking in Hedgehog receptor signaling. Sci. Signal. 8, ra55–ra55 10.1126/scisignal.aaa5622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zaphiropoulos P. G., Undén A. B., Rahnama F., Hollingsworth R. E., and Toftgård R. (1999) PTCH2, a novel human patched gene, undergoing alternative splicing and up-regulated in basal cell carcinomas. Cancer Res. 59, 787–792 [PubMed] [Google Scholar]
- 41. Tukachinsky H., Petrov K., Watanabe M., and Salic A. (2016) Mechanism of inhibition of the tumor suppressor Patched by Sonic Hedgehog. Proc. Natl. Acad. Sci. U.S.A. 113, E5866–E5875 10.1073/pnas.1606719113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lü Y., Zhu H., Ye W., Zhang M., He D., and Chen W. (2008) A new mutation of PTCH gene in a Chinese family with nevoid basal cell carcinoma syndrome. Chin. Med. J. (Engl.). 121, 118–121 [PubMed] [Google Scholar]
- 43. Zhong Y., Gu L. J., Sun X. G., Yang S. H., and Zhang X. H. (2014) Comprehensive analysis of patched domain-containing genes reveals a unique evolutionary pattern. Genet. Mol. Res. GMR. 13, 7318–7331 10.4238/2014.February.13.11 [DOI] [PubMed] [Google Scholar]
- 44. Saier M. H. (1994) Computer-aided analyses of transport protein sequences: gleaning evidence concerning function, structure, biogenesis, and evolution. Microbiol. Rev. 58, 71–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harvey M. C., Fleet A., Okolowsky N., and Hamel P. A. (2014) Distinct effects of the mesenchymal dysplasia variant of murine Patched-1 on canonical and non-canonical Hedgehog-signalling pathways. J. Biol. Chem. 10.1074/jbc.M113.514844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yue S., Tang L.-Y., Tang Y., Tang Y., Shen Q.-H., Ding J., Chen Y., Zhang Z., Yu T.-T., Zhang Y. E., and Cheng S. Y. (2014) Requirement of Smurf-mediated endocytosis of Patched1 in sonic hedgehog signal reception. eLife 3, e02555 10.7554/eLife.02555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang S., Zhang Z., Zhang C., Lv X., Zheng X., Chen Z., Sun L., Wang H., Zhu Y., Zhang J., Yang S., Lu Y., Sun Q., Tao Y., Liu F., et al. (2013) Activation of Smurf E3 ligase promoted by Smoothened regulates Hedgehog signaling through targeting Patched turnover. PLOS Biol. 11, e1001721 10.1371/journal.pbio.1001721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen X. L., Chinchilla P., Fombonne J., Ho L., Guix C., Keen J. H., Mehlen P., and Riobo N. A. (2014) Patched-1 proapoptotic activity is downregulated by modification of K1413 by the E3 ubiquitin-protein ligase Itchy homolog. Mol. Cell Biol. 34, 3855–3866 10.1128/MCB.00960-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sasaki H., Hui C., Nakafuku M., and Kondoh H. (1997) A binding site for Gli proteins is essential for HNF-3β floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development 124, 1313–1322 [DOI] [PubMed] [Google Scholar]
- 50. Chidambaram A., Goldstein A. M., Gailani M. R., Gerrard B., Bale S. J., DiGiovanna J. J., Bale A. E., and Dean M. (1996) Mutations in the human homologue of the Drosophila patched gene in Caucasian and African-American nevoid basal cell carcinoma syndrome patients. Cancer Res. 56, 4599–4601 [PubMed] [Google Scholar]
- 51. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sakuma T., Nishikawa A., Kume S., Chayama K., and Yamamoto T. (2014) Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci. Rep. 4, 5400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guschin D. Y., Waite A. J., Katibah G. E., Miller J. C., Holmes M. C., and Rebar E. J. (2010) A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol. 649, 247–256 10.1007/978-1-60761-753-2_15 [DOI] [PubMed] [Google Scholar]
- 54. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 55. Towbin H., Staehelin T., and Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4354 10.1073/pnas.76.9.4350 [DOI] [PMC free article] [PubMed] [Google Scholar]