

Abstract
Background
Respiratory fungal exposure is known to be associated with severe allergic lung inflammation. Airway epithelium is an essential controller of allergic inflammation. An innate immune recognition receptor, nucleotide-binding domain, leucine-rich-containing family, pyrin-domain-containing-3 (NLRP3) inflammasome, and phosphoinositide 3 kinase (PI3K)-δ in airway epithelium are involved in various inflammatory processes.
Objectives
We investigated the role of NLRP3 inflammasome in fungi-induced allergic lung inflammation and examined the regulatory mechanism of NLRP3 inflammasome, focusing on PI3K-δ in airway epithelium.
Methods
We used two in vivo models induced by exposure to Aspergillus fumigatus (Af) and Alternaria alternata (Aa), as well as an Af-exposed in vitro system. We also checked NLRP3 expression in lung tissues from patients with allergic bronchopulmonary aspergillosis (ABPA).
Results
Assembly/activation of NLRP3 inflammasome was increased in the lung of Af-exposed mice. Elevation of NLRP3 inflammasome assembly/activation was observed in Af-stimulated murine and human epithelial cells. Similarly, pulmonary expression of NLRP3 in patients with ABPA was increased. Importantly, neutralisation of NLRP3 inflammasome derived IL-1β alleviated pathophysiological features of Af-induced allergic inflammation. Furthermore, PI3K-δ blockade improved Af-induced allergic inflammation through modulation of NLRP3 inflammasome, especially in epithelial cells. This modulatory role of PI3K-δ was mediated through the regulation of mitochondrial reactive oxygen species (mtROS) generation. NLRP3 inflammasome was also implicated in Aa-induced eosinophilic allergic inflammation, which was improved by PI3K-δ blockade.
Conclusion
These findings demonstrate that fungi-induced assembly/activation of NLRP3 inflammasome in airway epithelium may be modulated by PI3K-δ, which is mediated partly through the regulation of mtROS generation. Inhibition of PI3K-δ may have potential for treating fungi-induced severe allergic lung inflammation.
Keywords: airway epithelium, allergic lung disease, innate immunity, aspergillus lung disease, asthma mechanisms, oxidative stress
Key messages.
What is the key question?
Phosphoinositide 3-kinase (PI3K)-δ isoform has been shown as an important mediator of allergic lung inflammation, including the fungi-induced form; however, a role of PI3K-δ in the modulation of innate immune response against fungal allergens has not been reported.
What is the bottom line?
PI3K-δ is critically implicated in fungi-induced allergic lung inflammation through regulation of nucleotide-binding domain, leucine-rich-containing family, pyrin-domain-containing-3 (NLRP3) inflammasome assembly/activation, which is mediated partly through the regulation of mitochondrial reactive oxygen species generation, particularly in the airway epithelium.
Why read on?
Inhibition of PI3K-δ signalling may have potential for treating fungi-induced severe allergic lung inflammation in humans.
Introduction
Bronchial asthma is recognised as a heterogeneous clinical syndrome, in which complex interplay between innate and adaptive immune response is central to airway inflammation.1 Among many cells, airway epithelium which expresses various pattern recognition receptors (PRRs)2 is the first line of defence on encountering allergens and an essential controller of immune responses.3 Particularly, the nucleotide-binding domain, leucine-rich repeat containing receptors (NLRs) represent one important PRR family in the intracellular compartment.4
Several members of NLRs are involved in the activation of caspase-1 through assembling cytosolic multiprotein complexes called inflammasomes, leading to cleavage of the proinflammatory IL-1 family of cytokines, such as pro-IL-1β into its bioactive form, IL-1β. Nucleotide-binding domain, leucine-rich-containing family, pyrin-domain-containing-3 (NLRP3) inflammasome consisting of NLRP3, apoptosis-associated speck-like protein containing a carboxy-terminal caspase-recruitment domain (ASC), and caspase-1 is the most characterised inflammasome.5 NLRP3 inflammasome has been reported to be implicated in diverse human disorders.6–8 Assembly/activation of NLRP3 inflammasome is induced through several intermediate factors.5 Specifically, functional association between NLRP3 inflammasome and intracellular organelles, including endoplasmic reticulum (ER) and mitochondria, is a key regulatory mechanism of NLRP3 inflammasome activation, partly via release of mitochondrial reactive oxygen species (mtROS).9 10 Recently, we showed that NLRP3 inflammasome activation by mtROS in airway epithelial cells (EpCs) is important in allergic lung inflammation.11
Fungal exposure has been recognised as a precipitating factor for development and exacerbation of asthma.12 According to a previous study, over 50% of patients with severe asthma were sensitised to one or more fungi.13 Among various species, Aspergillus fumigatus (Af) and Alternaria alternata (Aa) are common airborne fungi implicated in severe asthma.12 14 Particularly, the impact of Af on asthma severity has been well characterised15 and it is also involved in allergic bronchopulmonary aspergillosis (ABPA), a representative allergic bronchopulmonary mycosis which is assumed as an extreme spectrum of allergic response against fungi.12 14 16 In patients with fungi-associated allergic lung inflammation, conventional treatment using inhaled corticosteroid (CS) seems to be inadequate and other treatment modalities such as systemic CS and antifungal agents have been suggested.17 Nevertheless, limited information exists on the mechanisms of how fungi are linked to the allergic inflammation.
Although NLRP3 inflammasome is vital in anti-fungal immune response,18 the role of NLRP3 inflammasome in fungus-induced allergic lung inflammation remains poorly understood. Contradictory findings exist regarding the involvement of NLRP3 inflammasome in allergic lung inflammation.8 11 19 20 Given the involvement of NLRP3 inflammasome in many pathological conditions, controlling NLRP3 inflammasome may offer beneficial effects in fungi-induced allergic lung disorders. We previously demonstrated that activation of phosphoinositide 3-kinase (PI3K)-δ in lung EpCs contributes to allergic lung inflammation through the control of inflammatory mediators.21 In addition, PI3K-δ in airway EpCs was shown to be involved in fungi-induced allergic lung inflammation through modulating ER stress,22 which has been known as a regulator of NLRP3 inflammasome assembly/activation.10 23
In this study, the role of NLRP3 inflammasome in fungi-induced allergic lung inflammation was evaluated using two murine models induced by Af and Aa. We also examined whether PI3K-δ influences NLRP3 inflammasome assembly/activation in fungi-induced allergic lung inflammation, especially in airway EpCs using Af-stimulated primary cultured EpCs. Lastly, we checked expression of NLRP3 in lung tissues from patients with ABPA to verify the involvement of NLRP3 inflammasome in fungi-induced human allergic lung disorder.
Methods
Detailed methods are provided in the online supplement.
thoraxjnl-2017-210326supp013.pdf (460.1KB, pdf)
Animals and experimental protocol
Female C57BL/6 mice, 8–10 weeks of age, were used. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Chonbuk National University and were performed in accordance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. For the generation of fungi-induced allergic lung inflammation murine models, mice were exposed to Af or Aa extract (Greer Laboratories, Lenoir, North Carolina, USA) as previously described.22 24
Cell culture and treatment
Primary cultured murine tracheal EpCs were isolated under sterile conditions and treated with IC87114, a selective inhibitor of PI3K-δ, or PI3K-δ-specific siRNA as described previously.22 25Normal human bronchial epithelial (NHBE) cells were purchased from Lonza (Walkersville, Maryland, USA).
Immunofluorescence staining for NLRP3, caspase-1 and ASC and cytoplasmic localisation of mtROS
Immunofluorescence staining and cytoplasmic localisation of mtROS were performed as previously described.22
Western blot analysis
Protein levels were determined as previously described.22
Immunoprecipitation
Co-immunoprecipitation (IP) of NLRP3 and interacting proteins was performed using Dynabeads protein G (ThermoFisher Scientific Inc., Waltham, Massachusetts, USA) according to the manufacturer’s instructions.
Trichloroacetic acid-mediated protein precipitation
Trichloroacetic acid (TCA)-mediated protein precipitation of bronchoalveolar lavage (BAL) fluids was performed as described elsewhere.26
ASC oligomerisation assay
Cross-linking of ASC oligomers using disuccinimidyl suberate cross-linker was performed as previously described elsewhere.27
Determination of airway responsiveness to methacholine
Invasive measurement was performed as previously described.22
Statistics
We used SPSS software (version 18.0, SPSS, Chicago, Illinois, USA). Data are expressed as mean±SEM. Statistical comparisons were performed using one-way ANOVA followed by the Scheffe’s test. Significant differences between two groups (analyses between control and Af-exposed groups and analyses between Af-exposed wild type and Af-exposed p110δ knockout (KO) mice) were determined using unpaired t test. A value of P<0.05 was considered statistically significant.
Results
NLRP3 inflammasome is implicated in Af-induced allergic lung inflammation
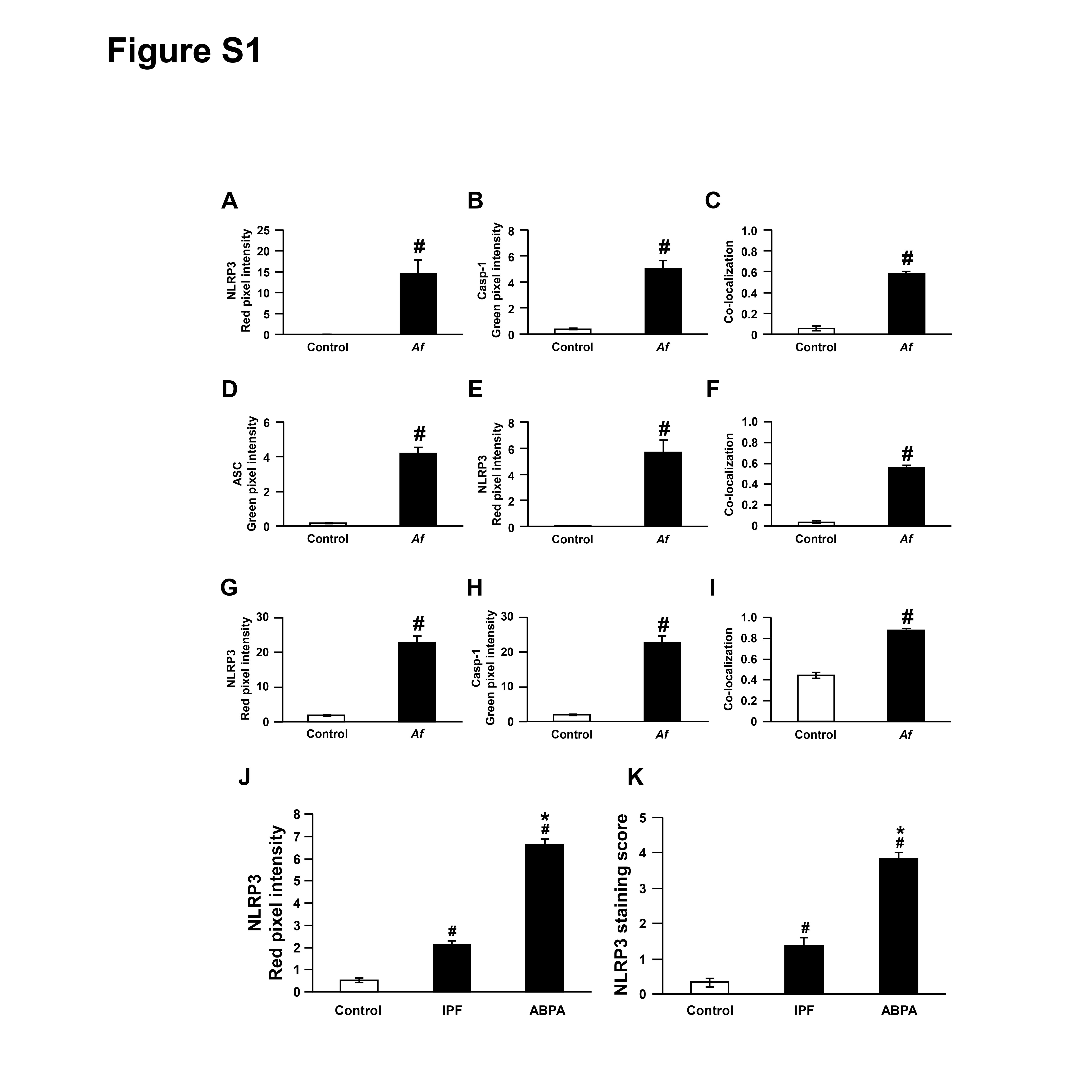
First, we determined expression of NLRP3 inflammasome components and IL-1β in Af-sensitised/challenged mice. Immunofluorescence intensities of NLRP3 and caspase-1 in BAL cells were remarkably increased in Af-exposed mice (figure 1A and online supplementary figure S1A, B). We observed the co-localisation of NLRP3 and caspase-1, predominantly in the cytoplasmic area (figure 1A and online supplementary figure S1C). Immunofluorescence intensities of ASC and NLRP3 and their co-localisation were also increased, especially in cytoplasm, in BAL cells (figure 1B and online supplementary figure S1D-F). Furthermore, IL-1β was elevated in BAL fluids (figure 1C). Similarly, immunofluorescence intensities of NLRP3 and caspase-1 and their co-localisation were increased, particularly in epithelial cell layers, in lung tissues (figure 1D and online supplementary figure S1G-I). IL-1β was also increased in lung tissues (figure 1E).
Figure 1.

Nucleotide-binding domain, leucine-rich-containing family, pyrin-domain-containing-3 (NLRP3) inflammasome is involved in Aspergillus fumigatus (Af)-induced allergic lung inflammation. (A and B) Representative confocal images of bronchoalveolar lavage (BAL) cells show the localisation of NLRP3 (red), caspase-1 (Casp-1, green) and apoptosis-associated speck-like protein containing a carboxy-terminal caspase-recruitment domain (ASC; green) from saline-exposed (control) or Af-exposed (Af) mice. 4',6-Diamidino-2-phenylindole (DAPI) stain was used for nuclear localisation. Bars indicate 20 µm. (C) Representative immunoblots and densitometric analyses of IL-1β in BAL fluids from saline-exposed (control) and Af-exposed (Af) mice. Bars represent mean±SEM from six mice per group. # P<0.05 versus control. (D) Representative confocal images show the localisation of NLRP3 (red) and Casp-1 (green) in lung tissues from saline-exposed (control) and Af-exposed (Af) mice. Bars indicate 50 µm. (E) Representative immunoblots and densitometric analyses of IL-1β in lung tissues from saline-exposed (control) and Af-exposed (Af) mice. Bars represent mean±SEM from six mice per group. # P<0.05 versus control. (F) Representative confocal image for NLRP3 in lung tissues from a healthy control, a patient with idiopathic pulmonary fibrosis (IPF; disease control), and a patient with allergic bronchopulmonary aspergillosis (ABPA), respectively. Bars indicate 50 µm. (G) Representative immunohistochemical staining of NLRP3 in lung tissues of a healthy person, a patient with IPF and a patient with ABPA. Brown-stained cells were considered to express the NLRP3 protein. Bars indicate 20 µm.
thoraxjnl-2017-210326supp001.jpg (1.2MB, jpg)
{kind=link}
NLRP3 is increased in lung tissues from patients with ABPA
Aspergillus-related disorders encompass a spectrum of diseases.28 Notably, ABPA represents the most severe type of T helper type 2 (Th2)-mediated allergic response against aspergillus species.29 We evaluated immunofluorescence and immunohistochemical analyses for NLRP3 in lung tissues from patients with ABPA, patients with idiopathic pulmonary fibrosis (IPF), and healthy controls, because NLRP3 expression is a limiting factor in NLRP3 inflammasome activation.30 Immunofluorescence intensity of NLRP3 was increased in patients with ABPA or IPF. However, increase of NLRP3 in patients with ABPA was significantly higher than in patients with IPF (figure 1F and online supplementary figure S1J). Immunohistochemical analyses also showed that immunoreactive NLRP3 was increased in patients with ABPA or IPF. However, immunoreactive NLRP3 was significantly increased in patients with ABPA (figure 1G and online supplementary figure S1K). These data indicate that Af exposure may be related to the NLRP3 inflammasome activation in human lung disorder, though we cannot rule out the possibility of the involvement of other factors.
Af leads to NLRP3 inflammasome activation in tracheal EpCs
Airway epithelium is the first contact site with Af and a key modulator of innate immune response against Af.31 We checked expression of NLRP3 inflammasome components in Af-stimulated tracheal EpCs. Immunofluorescence intensities of NLRP3 and caspase-1 were increased after Af stimulation (figure 2A and online supplementary figure S2A-C). Moreover, cytoplasmic co-localisation of NLRP3 and caspase-1 was increased (figure 2A and online supplementary figure S2A, D). Similarly, immunofluorescence intensities of ASC and NLRP3 and their cytoplasmic co-localisation were increased in these cells (figure 2B and online supplementary figure S2E-H).
Figure 2.

Stimulation with Aspergillus fumigatus (Af) leads to nucleotide-binding domain, leucine-rich-containing family, pyrin-domain-containing-3 (NLRP3) inflammasome assembly in tracheal epithelial cells (EpCs) and neutralisation of IL-1β improves Af-induced allergic lung inflammation. (A and B) Representative confocal images of tracheal EpCs show the localisation of NLRP3 (red), caspase-1 (Casp-1, green) and apoptosis-associated speck-like protein containing a carboxy-terminal caspase-recruitment domain (ASC; green) in normal (control) and Af-stimulated (Af) cells. 4',6-Diamidino-2-phenylindole (DAPI) stain was used for nuclear localisation. Bars indicate 20 µm. (C) Enzyme immunoassays of IL-1β in bronchoalveolar lavage (BAL) fluids from saline-exposed mice administered drug vehicle (SAL+VEH), Af-exposed mice administered drug vehicle (Af+VEH), or Af-exposed mice administered MCC 950 (Af+MCC). Bars represent mean±SEM from six mice per group. # P<0.05 versus SAL+VEH; * P<0.05 versus Af+VEH. Right upper inset image demonstrates the representative immunoblot of IL-1β in BAL fluids. (D) Airway responsiveness assessed by invasive measurements in saline-exposed mice administered isotype antibody (Ab) (SAL+ISO Ab), Af-exposed mice administered isotype Ab (Af+ISO Ab), or Af-exposed mice administered anti-IL-1β neutralising Ab (Af+IL-1β Ab). Bars represent mean±SEM from six mice per group. # P<0.05 versus SAL+ISO Ab; *P<0.05 versus Af+ISO Ab. (E) Cellular changes in BAL fluids. Bars represent mean±SEM from five mice per group. # P<0.05 versus SAL+ISO Ab; *P<0.05 versus Af+ISO Ab. (F–H) Representative H&E stained sections of the lung from SAL+ISO Ab (F), Af+ISO Ab (G), and Af+IL-1β Ab (H). Bars indicate 50 µm.
thoraxjnl-2017-210326supp002.jpg (1.4MB, jpg)
{kind=link}
IL-1β inhibition improves Af-induced allergic lung inflammation
To investigate whether NLRP3 inflammasome-derived IL-1β is implicated in Af-induced allergic lung inflammation, we evaluated the effects of IL-1β blockade on features of the disease. First, we determined the effect of MCC 950, a selective inhibitor of NLRP3 inflammasome,32 on IL-1β in BAL fluids. Results showed that Af-induced increase of IL-1β was significantly lowered by MCC 950 (figure 2C), indicating that NLRP3 inflammasome plays a key role in IL-1β generation in Af-exposed mice. Notably, IL-1β neutralising antibody (Ab) treatment improved respiratory system resistance (Rrs) (figure 2D). Increased numbers of total cells, lymphocytes, and eosinophils in BAL fluids were lowered by IL-1β neutralising Ab (figure 2E). Furthermore, Af-exposed mice administered IL-1β neutralising Ab showed reduction in the infiltrations of inflammatory cells into the peribronchiolar and perivascular regions (figure 2F-H).
PI3K-δ blockade ameliorates Af-induced allergic lung inflammation through regulation of NLRP3 inflammasome assembly/activation
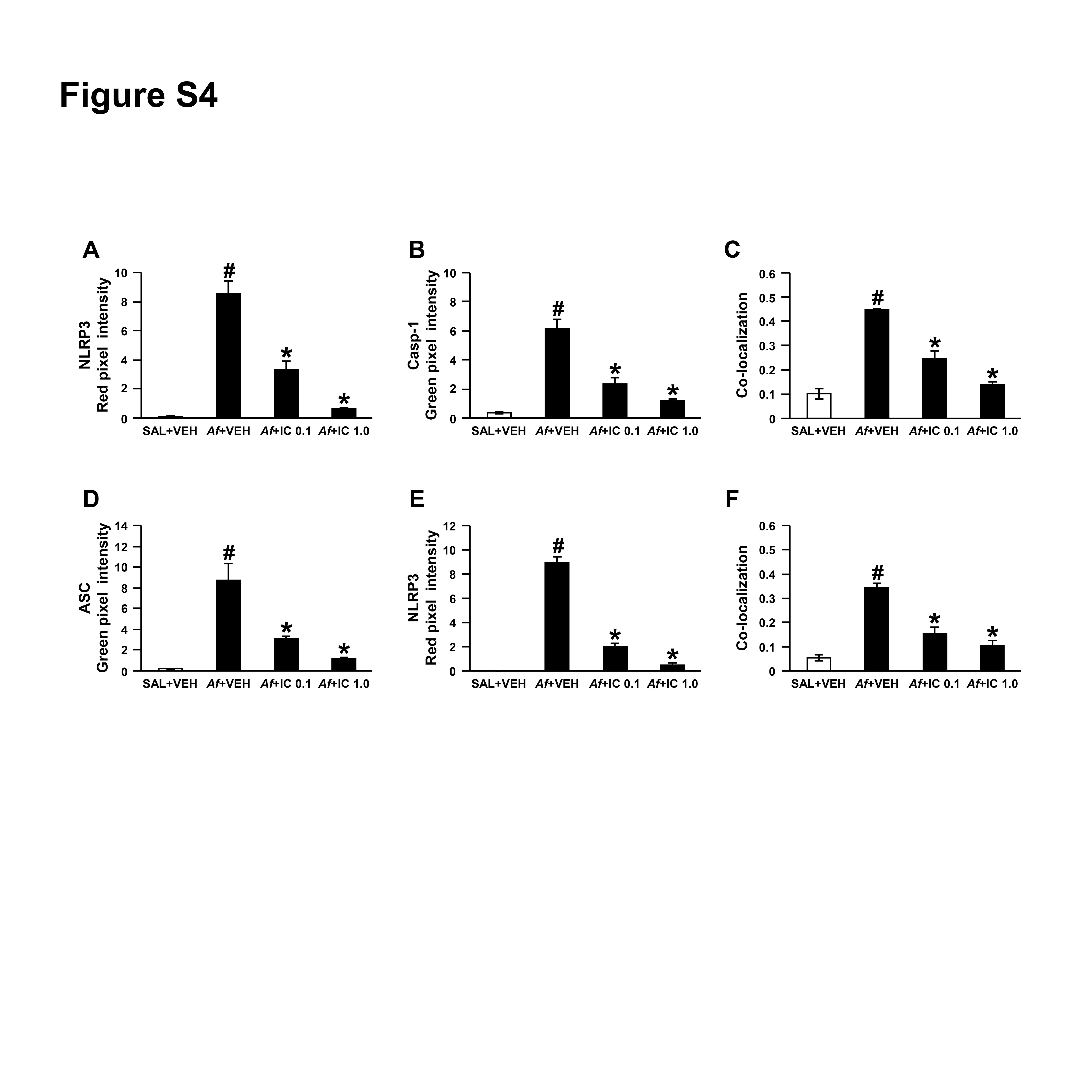
First, through assessing the therapeutic effects of IC87114, a selective inhibitor of PI3K-δ, in Af-sensitised/challenged mice, we observed that PI3K-δ activity contributes to overall activities of class I PI3Ks in the lung and plays a key role in Af-induced allergic lung inflammation (online supplementary figure S3). Then, we determined the effect of PI3K-δ blockade on NLRP3 inflammasome assembly/activation in vivo. Af-induced increases in immunofluorescence intensities of NLRP3, caspase-1, and ASC and their cytoplasmic co-localisations in BAL cells were notably reduced by IC87114 (figure 3A, B and online supplementary figure S4). Viability assay of BAL cells after intratracheal administration of IC87114 revealed that there was no significant toxic effect of the inhibitor on cells (online supplementary figure S5). IP assay also showed that Af-induced increase of NLRP3 co-precipitated with caspase-1 or ASC in lung tissues was reduced by IC87114 (figure 3C). Furthermore, Af-induced increases of NLRP3 and caspase-1 p10 (an autoprocessed cleaved form of caspase-1) in lung tissues were significantly reduced by IC87114 (figure 3D, E). Increased levels of IL-1β in lung tissues and BAL fluids were also lowered by IC87114 (figure 3F-H). To further determine whether PI3K-δ affects NLRP3 inflammasome, KO mice without the catalytic subunit of PI3K-δ (p110δ) were used. Whereas Af exposure led to the increase of p110δ in wild type (WT) mice, p110δ expression was absent in lung tissues of Af-exposed p110δ KO mice. In addition, the extent of the infiltration of numerous inflammatory cells into the peribronchial and perivascular regions was much lower in Af-exposed p110δ KO mice (figure 3I). Furthermore, p110δ KO mice after Af exposure demonstrated the lower levels of NLRP3 and cleaved caspase-1 in lung tissues (figure 3J, K). In addition, Af-exposed p110δ KO mice had lower levels of IL-1β in lung tissues and BAL fluids (figure 3L, M).
Figure 3.

Inhibition of phosphoinositide 3-kinase (PI3K)-δ improves Aspergillus fumigatus (Af)-induced allergic lung inflammation through regulation of nucleotide-binding domain, leucine-rich-containing family, pyrin-domain-containing-3 (NLRP3) inflammasome assembly/activation. (A and B) Representative confocal images of bronchoalveolar lavage (BAL) cells show the localisation of NLRP3 (red), caspase-1 (Casp-1; green) and apoptosis-associated speck-like protein containing a carboxy-terminal caspase-recruitment domain (ASC; green) from saline-exposed mice administered drug vehicle (SAL+VEH), Af-exposed mice administered drug vehicle (Af+VEH), Af-exposed mice administered 0.1 mg/kg IC87114 (Af+IC 0.1) or Af-exposed mice administered 1.0 mg/kg IC87114 (Af+IC 1.0). 4',6-Diamidino-2-phenylindole (DAPI) stain was used for nuclear localisation. Bars indicate 20 µm. (C) Representative immunoblots of Casp-1 or ASC co-precipitated (IP) with NLRP3 in lung tissues. (D–F) Representative immunoblots and densitometric analyses of NLRP3 (D), Casp-1 (E) and IL-1β (F) in lung tissues. (G and H) Enzyme immunoassay (G) and representative immunoblots and densitometric analyses (H) of IL-1β in BAL fluids. Bars represent mean±SEM from six mice per group. # P<0.05 versus SAL+VEH; *P<0.05 versus Af+VEH. (I) Representative immunoblots and densitometric analyses of p110δ in lung tissues and representative H&E stained sections of the lung from Af-exposed mice with p110δ KO (p110δ–/–) or WT. Bars indicate 50 µm. (J–M) Representative immunoblots and densitometric analyses of NLRP3 (J), Casp-1 (K) and IL-1β (L) in lung tissues, and IL-1β in BAL fluids (M) from Af-exposed mice with p110δ–/– or WT. Bars represent mean±SEM from seven mice per group. # P<0.05 versus WT+Af.
thoraxjnl-2017-210326supp003.jpg (1.9MB, jpg)
{kind=link}
thoraxjnl-2017-210326supp004.jpg (1.1MB, jpg)
{kind=link}
thoraxjnl-2017-210326supp005.jpg (1,006.7KB, jpg)
{kind=link}
Af-induced assembly/activation of NLRP3 inflammasome in bronchial EpCs is regulated by PI3K-δ
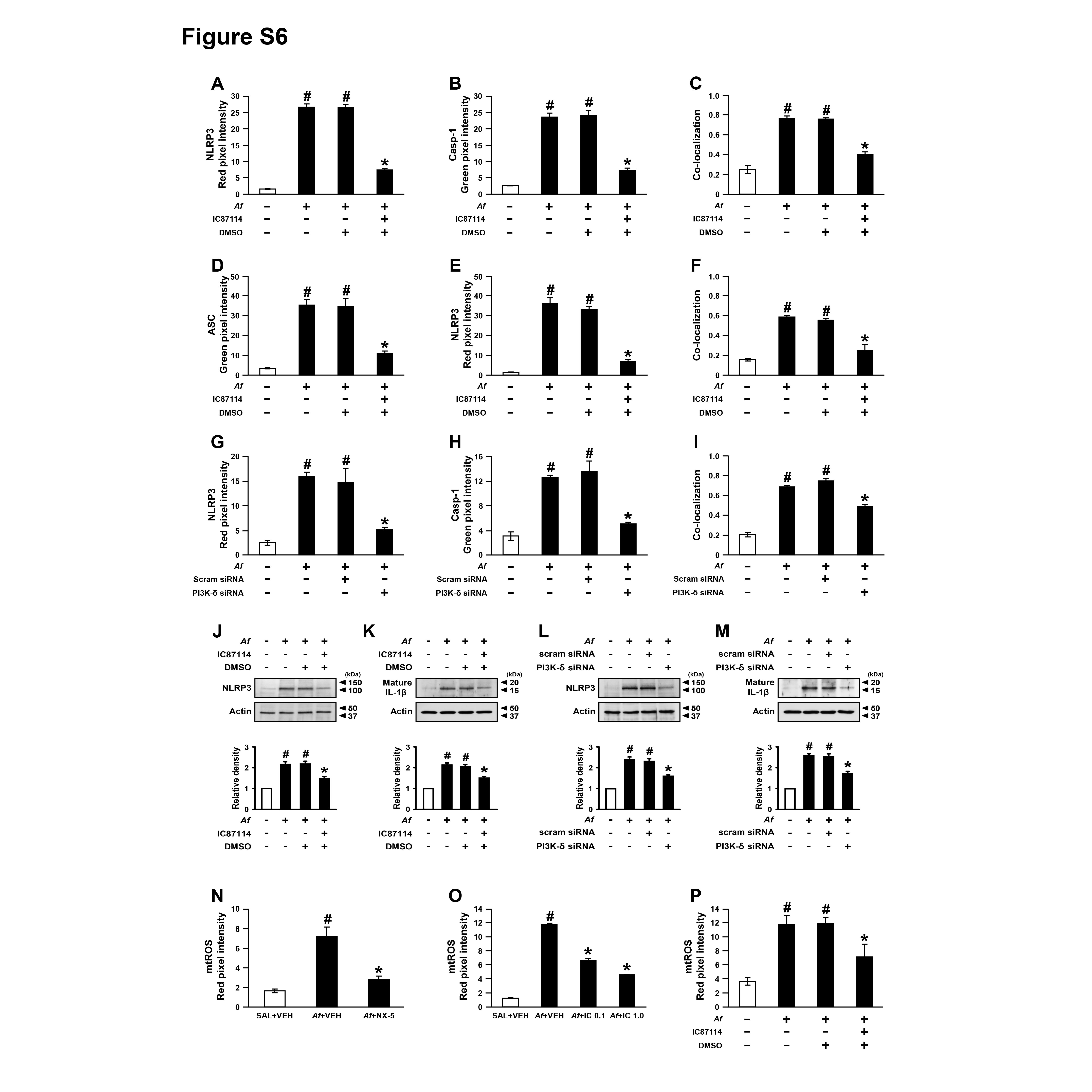
Then, we examined whether PI3K-δ modulates NLRP3 inflammasome in airway EpCs. IC87114 substantially limited Af-induced increases in immunofluorescence intensities of NLRP3, caspase-1 and ASC and their cytoplasmic co-localisations in EpCs (figure 4A, B and online supplementary figure S6A-F). We also examined the formation of ASC oligomerisation, a key process in NLRP3 inflammasome assembly/activation,32 in these cells and IC87114 substantially lowered Af-induced formation of ASC monomers and high order complexes (figure 4C). Furthermore, when PI3K-δ in EpCs was knocked down, Af-induced expression of NLRP3 and caspase-1 and their cytoplasmic co-localisation were substantially lowered (figure 4D and online supplementary figure S6G-I). Supporting these data, Af-induced increases of NLRP3 and IL-1β in cell lysates were reduced by IC87114 or PI3K-δ siRNA (online supplementary figure S6J-M). Meanwhile, ROS generation acts as one of the major upstream signals for NLRP3 inflammasome activation.9 Mitochondrion is a major source of intracellular ROS, thereby being involved in NLRP3 inflammasome activation.9 33 To investigate the role of mtROS in Af-induced allergic lung inflammation, we determined the effects of a potent mtROS scavenger, NecroX-522, in Af-exposed mice. Fluorescence intensity of mtROS was increased in cytoplasm of BAL cells, mainly macrophages and eosinophils, from Af-exposed mice (figure 4E, F and online supplementary figure S6N). NecroX-5 reduced Af-induced increase in fluorescence intensity of mtROS. Notably, NecroX-5 dramatically reduced the Af-induced increase of IL-1β (figure 4G). Moreover, NecroX-5 ameliorated Af-induced allergic lung inflammation and airway hyper-responsiveness (AHR) (figure 4H-L). Furthermore, Af-induced mtROS generation was decreased by IC87114 in both BAL cells and EpCs (figure 4M, N and online supplementary figure S6O, P).
Figure 4.

Aspergillus fumigatus (Af)-induced nucleotide-binding domain, leucine-rich-containing family, pyrin-domain-containing-3 (NLRP3) inflammasome assembly/activation is regulated by phosphoinositide 3-kinase (PI3K)-δ in epithelial cells (EpCs). (A and B) Representative confocal images of tracheal EpCs show the localisation of NLRP3 (red), caspase-1 (Casp-1; green) and apoptosis-associated speck-like protein containing a carboxy-terminal caspase-recruitment domain (ASC; green) in the control (no treatment), Af-stimulated cells (Af), Af-stimulated cells administered drug vehicle (Af+VEH), and Af-stimulated cells administered IC87114 (Af+IC). 4',6-Diamidino-2-phenylindole (DAPI) stain was used for nuclear localisation. Bars indicate 20 µm. (C) Representative immunoblot of ASC monomers and high order complexes in cross-linked cytosolic pellets from Af-stimulated EpCs in the presence and absence of IC87114. (D) Representative confocal images of tracheal EpCs show the localisation of NLRP3 (red) and Casp-1 (green) in the control (no treatment), Af-stimulated cells (Af), Af-stimulated cells administered scrambled siRNA (Af+Scram), or Af-stimulated cells administered PI3K-δ siRNA (Af+PI3K-δ). Bars indicate 20 µm. (E) Representative confocal images show the localisation of mitochondrial ROS (mtROS) in bronchoalveolar lavage (BAL) cells from saline-exposed mice administered drug vehicle (SAL+VEH), Af-exposed mice administered drug vehicle (Af+VEH), or Af-exposed mice administered NecroX-5 (Af+NX-5). Bars indicate 20 µm. (F) Cellular changes in BAL fluids of Af-exposed mice. Arrows and arrowheads indicate macrophages and eosinophils, respectively. Bars indicate 20 µm. (G) Representative immunoblots and densitometric analyses of IL-1β in lung tissues. Bars represent mean±SEM from seven mice per group. #P<0.05 versus SAL+VEH; *P < 0.05 versus Af+VEH. (H) Airway responsiveness assessed by invasive measurements from SAL+VEH, Af+VEH or Af+NX-5. Bars represent mean±SEM from six mice per group. (I) Cellular changes in bronchoalveolar lavage fluids. Bars represent mean±SEM from six mice per group. #P<0.05 versus SAL+VEH; *P < 0.05 versus Af+VEH. (J–L) Representative H&E stained sections of the lung from SAL+VEH (J), Af+VEH (K) and Af+NX-5 (L). Bars indicate 50 µm. (M) Representative confocal images show the localisation of mtROS in BAL cells from saline-exposed mice administered drug vehicle (SAL+VEH), Af-exposed mice administered drug vehicle (Af+VEH), Af-exposed mice administered 0.1 mg/kg IC87114 (Af+IC 0.1) or Af-exposed mice administered 1.0 mg/kg IC87114 (Af+IC 1.0). Bars indicate 20 µm. (N) Representative confocal images of tracheal EpCs show the localisation of mtROS in the control, Af, Af+VEH and Af+IC. Bars indicate 10 µm.
thoraxjnl-2017-210326supp006.jpg (1.5MB, jpg)
{kind=link}
Af-stimulated NHBE cells replicate the in vitro murine studies
To investigate whether PI3K-δ also modulates NLRP3 inflammasome in human epithelial cells, we checked expression of NLRP3 inflammasome components and their co-localisations in Af-stimulated NHBE cells. IC87114 significantly lowered the Af-induced increases in immunofluorescence intensities of NLRP3, caspase-1 and ASC and their cytoplasmic co-localisations in Af-stimulated NHBE cells (figure 5A, B and online supplementary figure S7A-F). Furthermore, IC87114 lowered the Af-induced ASC-complex formation (figure 5C). In the same vein, increased levels of NLRP3 and IL-1β in Af-stimulated NHBE cells were reduced by IC87114 (figure 5D, E). Similarly, when PI3K-δ was knocked down, increased levels of NLRP3 and IL-1β in Af-stimulated NHBE cells were significantly reduced (figure 5F, G). Consistent with these observations, Af-induced increased immunofluorescence intensities of NLRP3 and caspase-1 and their cytoplasmic co-localisation were substantially lowered by PI3K-δ siRNA (figure 5H and online supplementary figure S7G-I). These data suggest that PI3K-δ also regulates Af-induced assembly/activation of NLRP3 inflammasome in NHBE cells. Additionally, Af-induced generation of mtROS were significantly lowered by IC87114 in NHBE cells (figure 5I and online supplementary figure S7J).
Figure 5.

Phosphoinositide 3-kinase (PI3K)-δ regulates Aspergillus fumigatus (Af)-induced nucleotide-binding domain, leucine-rich-containing family, pyrin-domain-containing-3 (NLRP3) inflammasome assembly/activation in normal human bronchial epithelial (NHBE) cells. (A and B) Representative confocal images of NHBE cells show the localisation of NLRP3 (red), caspase-1 (Casp-1; green) and apoptosis-associated speck-like protein containing a carboxy-terminal caspase-recruitment domain (ASC; green) in the control (no treatment), Af-stimulated cells (Af), Af-stimulated cells administered drug vehicle (Af+VEH), or Af-stimulated cells administered IC87114 (Af+IC). 4',6-Diamidino-2-phenylindole (DAPI) stain was used for nuclear localisation. Bars indicate 20 µm. (C) Representative immunoblot of ASC monomers and high order complexes in cross-linked cytosolic pellets from Af-stimulated NHBE cells in the presence and absence of IC87114. (D and E) Representative immunoblots and densitometric analyses of NLRP3 (D) and IL-1β (E) after stimulation with Af in NHBE cells in the presence or absence of IC87114. Bars represent mean±SEM from three independent experiments. # P<0.05 versus control; *P<0.05 versus cells stimulated with Af alone. (F and G) Representative immunoblots and densitometric analyses of NLRP3 (F) and IL-1β (G) after stimulation with Af in NHBE cells transfected with either non-targeting siRNA (NT siRNA) or PI3K-δ siRNA. Bars represent mean±SEM from three independent experiments. # P<0.05 versus control; *P<0.05 versus cells stimulated with Af transfected with NT siRNA. (H) Representative confocal images of NHBE cells show the localisation of NLRP3 (red) and Casp-1 (green) in the control (no treatment), Af-stimulated cells (Af), Af-stimulated cells administered NT siRNA (Af+NT), or Af-stimulated cells administered PI3K-δ siRNA (Af+PI3K-δ). Bars indicate 20 µm. (I) Representative confocal images of NHBE cells show the localisation of mitochondrial reactive oxygen species (mtROS) in the control, Af, Af+VEH or Af+IC. Bars indicate 20 µm.
thoraxjnl-2017-210326supp007.jpg (1.3MB, jpg)
{kind=link}
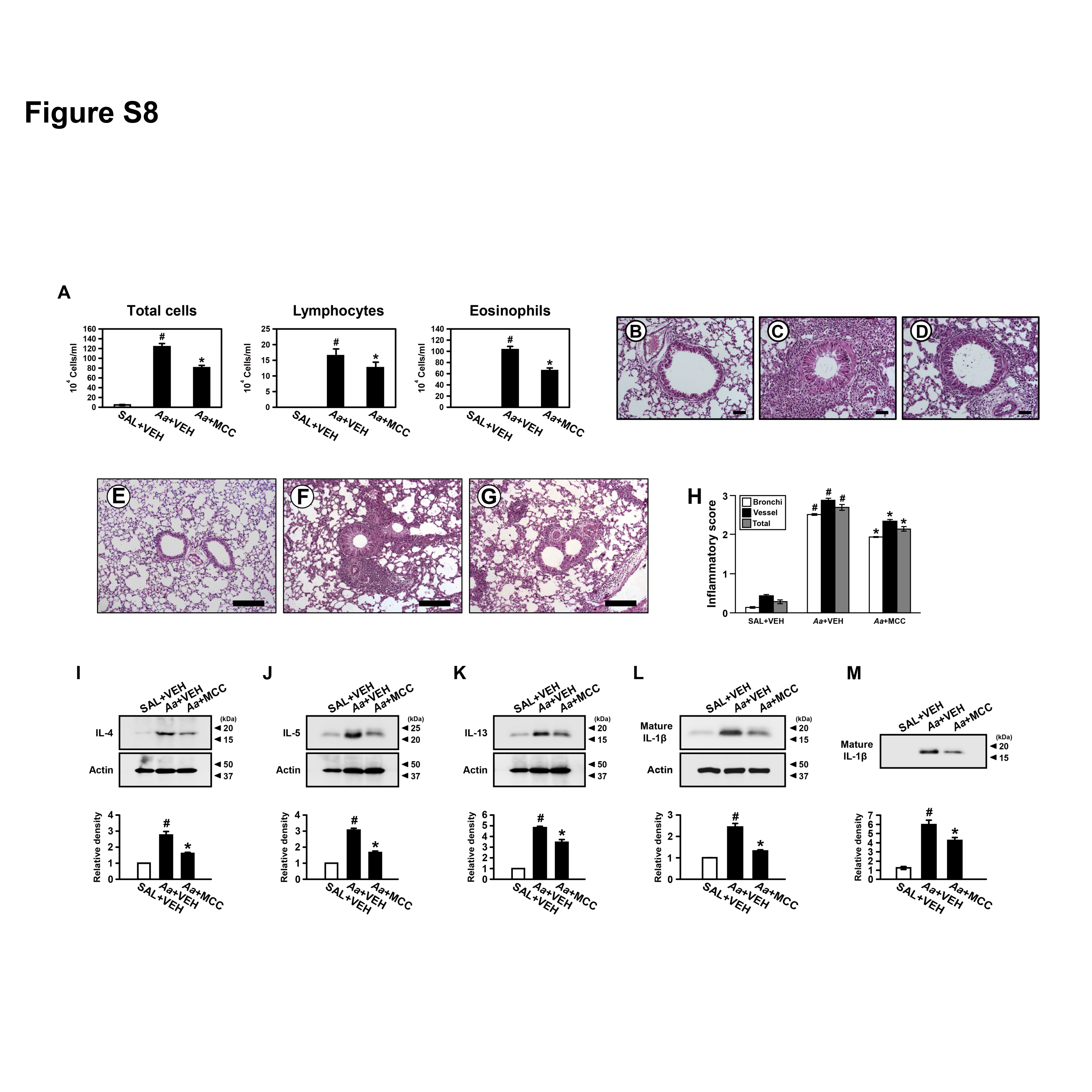
PI3K-δ regulates NLRP3 inflammasome in Aa-induced allergic lung inflammation
To clarify the involvement of NLRP3 inflammasome in fungi-induced allergic lung inflammation, we evaluated the effects of MCC 950 on allergic lung inflammation induced by Aa. Treatment with MCC 950 significantly reduced Aa-induced increases of eosinophil-dominant inflammatory cells infiltration, Th2 cytokines and IL-1β in the lung of mice (online supplementary figure S8). We then explored whether PI3K-δ contributes to Aa-induced allergic lung inflammation, and if PI3K-δ regulates NLRP3 inflammasome activation. Data showed that Aa-induced increases of airway inflammatory cells infiltration and pulmonary Th2 cytokines were significantly reduced by treatment with IC87114. Interestingly, Aa-induced increases of IL-1β in lung tissues and BAL fluids were significantly lowered by IC87114 (online supplementary figure S9).
thoraxjnl-2017-210326supp008.jpg (1.9MB, jpg)
{kind=link}
thoraxjnl-2017-210326supp009.jpg (2.1MB, jpg)
{kind=link}
CS-resistant NLRP3 inflammasome contributes to Af-induced allergic lung inflammation
CS resistance is a common feature in severe allergic lung inflammation.34 Recently, we showed that the current Af-induced model represents a CS-resistant severe phenotype.22 Therefore, we examined whether or not CS can influence NLRP3 inflammasome activation in the present two murine models. Data showed that dexamethasone did not improve the Af-induced increases of AHR, eosinophil-dominant inflammatory cells infiltration, and Th2 cytokines in the lung of mice (online supplementary figure S10A-I). In contrast, dexamethasone substantially improved the Aa-induced increases of AHR, eosinophil-dominant inflammatory cell infiltration, and Th2 cytokines in the lung (online supplementary figure S10J-Q). Interestingly, increases of IL-1β in lung tissues and BAL fluids from Af-exposed mice were not lowered by dexamethasone. In contrast, increases of IL-1β in lung tissues and BAL fluids from Aa-exposed mice were significantly reduced by dexamethasone (online supplementary figure S11A-D). Similarly, dexamethasone treatment failed to lower the Af-induced increases of NLRP3 and caspase-1 in lung tissues. However, Aa-induced increases of NLRP3 and caspase-1 were significantly reduced by dexamethasone (online supplementary figure S11E-H).
thoraxjnl-2017-210326supp010.jpg (1.6MB, jpg)
{kind=link}
thoraxjnl-2017-210326supp011.jpg (1.3MB, jpg)
{kind=link}
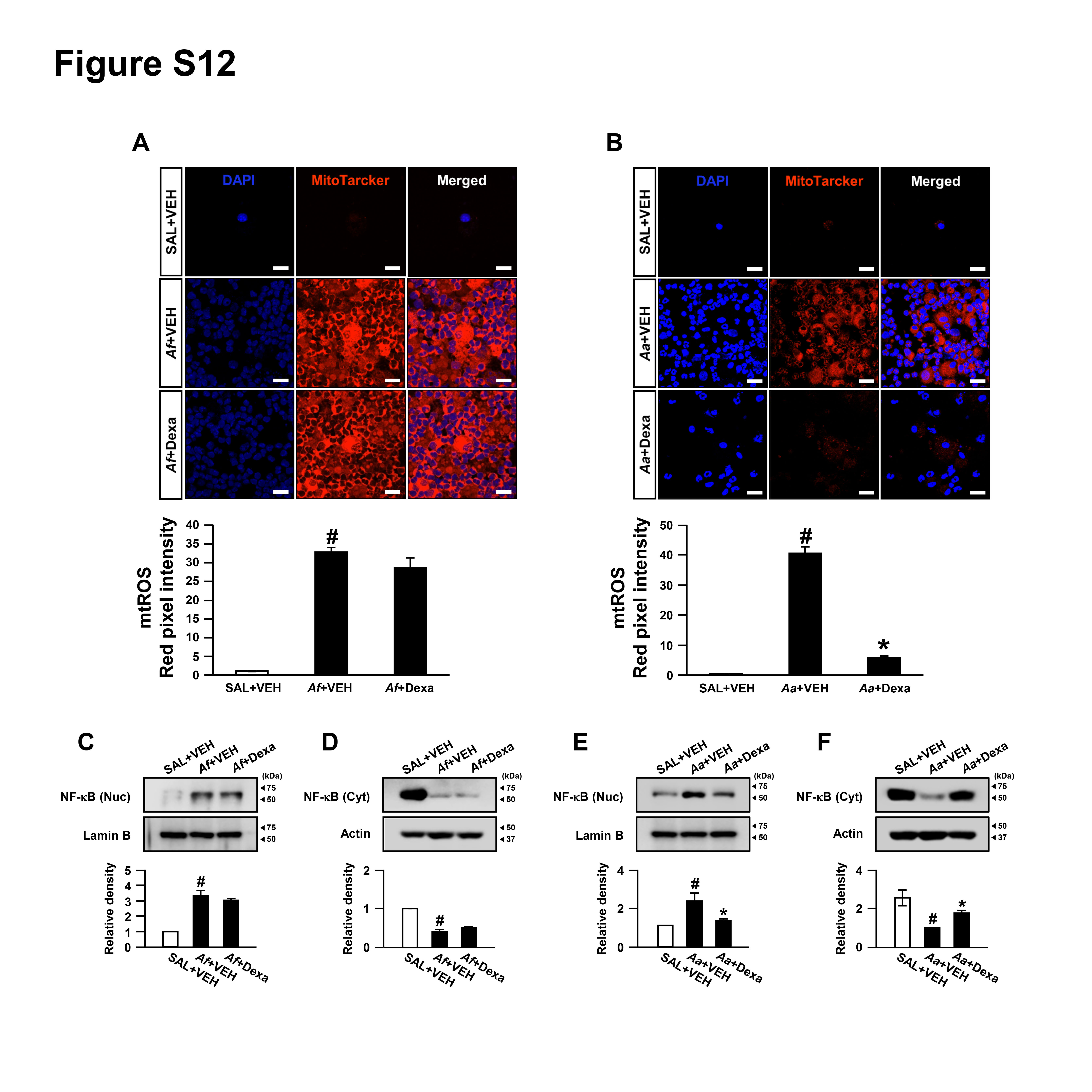
Effects of dexamethasone on mtROS generation and nuclear translocation of nuclear factor-κB in lungs of two different murine models
Oxidative stress is closely involved in the development of CS resistance in intractable pulmonary disorders, including severe asthma and chronic obstructive pulmonary disease.34 In addition, generation of mtROS is implicated in CS-resistant inflammation in the lung.22 To investigate the molecular mechanisms which underlie the different responses to CS between the two mouse models, we determined mtROS and the related activation of nuclear factor (NF)-κB in the lung. Fluorescence intensity of mtROS was increased in cytoplasm of BAL cells from Aa-exposed mice, as seen in that of Af-exposed mice. Dexamethasone significantly reduced the Aa-induced generation of mtROS in these cells. However, dexamethasone failed to reduce the Af-induced generation of mtROS (online supplementary figure S12A, B). Furthermore, levels of NF-κB p65 in nuclear protein extracts of lung tissues were significantly increased after exposure to Af or Aa, but levels of NF-κB p65 in cytosolic protein extracts of lung tissues were remarkably decreased after exposure to Af and Aa. Interestingly, while dexamethasone significantly reduced the Aa-induced increases in the nuclear translocation of NF-κB p65, it failed to lower the Af-induced increases in NF-κB p65 in nuclear protein extracts (online supplementary figure S12C-F). These findings suggest that the difference in the ability of dexamethasone to control inflammation in the two mouse models is in part dependent on whether CS can regulate fungi-induced generation of mtROS.
thoraxjnl-2017-210326supp012.jpg (1.9MB, jpg)
{kind=link}
Discussion
In this study, assembly/activation of NLRP3 inflammasome was increased in the lung of Af-exposed mice. Elevation of NLRP3 inflammasome assembly/activation was also observed in Af-stimulated murine and human EpCs. Similarly, expression of NLRP3 was increased in lung tissue from patients with ABPA. Importantly, neutralisation of NLRP3-derived IL-1β alleviated various features of Af-induced allergic inflammation. Furthermore, inhibition of PI3K-δ improved Af-induced allergic inflammation through modulating NLRP3 inflammasome assembly/activation, especially in EpCs. This modulatory role of PI3K-δ was mediated through the regulation of mtROS generation. NLRP3 inflammasome was also implicated in Aa-induced allergic lung inflammation, which was improved by PI3K-δ blockade. These findings demonstrate for the first time that fungi-induced assembly/activation of NLRP3 inflammasome in bronchial EpCs may be modulated by PI3K-δ, which is mediated partly through the regulation of mtROS generation.
Cytokine imbalance is important in chronic lung disorders.35 Particularly, IL-1β, which activates many inflammatory genes implicated in allergic inflammation,36 is increased in asthmatic airways, and blockade of IL-1β ameliorated allergic lung inflammation in a murine model.37 NLRP3 inflammasome has been known to be implicated in allergic inflammatory processes as well as antifungal defence through IL-1β.8 18 Nevertheless, the role of NLRP3 inflammasome in allergic lung inflammation is still unclear. While several reports favour the critical involvement of NLRP3 inflammasome in allergic lung inflammation,8 11 conflicting data also exist.19 20 However, given that upregulation of NLRP3 inflammasome is observed in patients with asthma having neutrophilic inflammation,38 the role of NLRP3 inflammasome in allergic inflammation might be different according to underlying biological processes. In our study, Af exposure resulted in the increases of NLRP3, caspase-1 and IL-1β, and physical associations between NLRP3 inflammasome components in lung tissues. It was supported by the observation that immunofluorescence intensities of NLRP3 components and their cytoplasmic co-localisations were increased in BAL cells. Increases in immunofluorescence intensities and co-localisation of NLRP3 and caspase-1 were also observed in lung tissues. Especially, NLRP3 expression is increased in lung tissues from patients with ABPA, one proposed asthma endotype characterised by severe allergic inflammation associated with Af.39 Furthermore, Af stimulation led to the increases in immunofluorescence intensities and their cytoplasmic co-localisations of NLRP3 inflammasome components as well as ASC oligomerisation in murine and human EpCs. Notably, selective inhibition of NLRP3 inflammasome led to the decrease of IL-1β in the lung of Af-exposed mice and neutralisation of IL-1β improved numerous features of Af-induced allergic lung inflammation. Similarly, NLRP3 inflammasome blockade ameliorated Aa-induced increases of IL-1β and allergic inflammation in the lung. Taken together, NLRP3 inflammasome may play a pivotal role in fungi-induced allergic lung inflammation.
NLRP3 inflammasome senses and reacts to diverse stimuli through various mechanisms.5 However, scarce information exists on the regulatory mechanism of NLRP3 inflammasome activation against fungal exposure, particularly in allergic inflammation. PI3K-δ plays a key role in regulation of immune processes through activating immune cells and trafficking inflammatory cells.40 PI3K-δ is also activated in response to fungal exposure and plays a role in the regulation of ER stress, thereby being crucially implicated in fungal allergic inflammation.22 ER stress is one of the triggers of NLRP3 inflammasome activation.10 23 In this study, PI3K-δ inhibition attenuated Af-induced airway inflammatory cell infiltrations, AHR and elevations of pulmonary Th2 cytokines and serum total and Af-specific IgE, highlighting the pivotal role of PI3K-δ in Af-induced allergic inflammation. Importantly, Af-induced increases of NLRP3, active caspase-1 and IL-1β in the lung were significantly reduced by IC87114. Furthermore, Af-exposed p110δ KO mice showed the lower levels of NLRP3, caspase-1 and IL-1β in the lung compared with Af-exposed WT mice. The regulatory role of PI3K-δ on NLRP3 inflammasome was further verified by the IP assay. Af-induced increases in the immunofluorescence intensities of NLRP3 inflammasome components and their cytoplasmic co-localisations in BAL cells were also reduced by IC87114. In Af-stimulated murine and human EpCs, Af-induced increased ASC oligomerisation was reduced by IC87114. Moreover, Af-induced increases in the immunofluorescence intensities and cytoplasmic co-localisations of NLRP3 inflammasome components in EpCs were reduced by IC87114 or PI3K-δ siRNA. Af-induced increases of NLRP3 and IL-1β were also reduced by IC87114 or PI3K-δ siRNA in these cells. In Aa-induced allergic inflammation, IC87114 attenuated the Aa-induced increases of inflammatory cell infiltrations, Th2 cytokines and IL-1β in the lung. These findings imply that PI3K-δ is critically implicated in fungi-induced allergic lung inflammation through regulation of NLRP3 inflammasome in airway epithelium.
Oxidative stress is one of the most characteristic features of chronic airway disorders41 and mtROS are a common molecular platform in NLRP3 inflammasome activation.5 9 33 42 Since most of the intracellular ROS originate from mitochondria, we examined roles of mtROS in this study. Fluorescence intensity of mtROS was increased in the cytoplasm of BAL cells, particularly macrophages and eosinophils, from Af-exposed and Aa-exposed mice. A potent mtROS scavenger alleviated Af-induced mtROS generation in these cells. Importantly, the mtROS scavenger reduced the Af-induced increase in IL-1β, and that improved various features of allergic lung inflammation in Af-exposed mice. Recently, we reported that mtROS generation in airway epithelium is fundamental in allergic inflammation11 and PI3K-δ regulates mtROS production in airway EpCs under fungal exposure.22 Likewise, in the present study, Af-induced increases in the fluorescence intensity of mtROS were lowered by IC87114 in murine and human EpCs as well as BAL cells from Af-exposed mice. These findings suggest that PI3K-δ modulates NLRP3 inflammasome, partly through inducing mtROS in airway epithelium, thereby contributing to fungi-induced allergic inflammation.
CS is the most powerful anti-inflammatory agent available for immune-mediated disorders. However, 5~10% of patients with asthma have severe disorders accounting for more than 50% of the total healthcare cost, and CS resistance is a common feature for severe asthma.34 43 Fungi-associated asthma endotype may present more severe disease with frequent exacerbations and eosinophilic inflammation.39 Among fungi implicated in severe asthma, Af can tolerate body temperature and grow in the airways, thereby colonising and inducing potent immune responses. Therefore, it has been suggested that the most severe form of allergic fungal disease frequently involves Af.44 In the current study, whereas dexamethasone improved various features of Aa-induced allergic lung inflammation, it failed to alleviate Af-induced inflammatory cell infiltration, AHR and elevations of Th2 cytokines in the lung. Dexamethasone also reduced Aa-induced elevations of NLRP3, caspase-1 and IL-1β in the lung. However, dexamethasone failed to reduce Af-induced elevations of NLRP3, caspase-1 and IL-1β. Additionally, the difference in the ability of dexamethasone to control inflammation in the two mouse models may be partly dependent on whether CS can regulate fungi-induced generation of mtROS. In fact, oxidative stress is closely involved in the development of CS resistance in severe asthma and chronic obstructive pulmonary disease.34 Moreover, generation of mtROS is closely implicated in CS-resistant inflammation in the lung.22 Further, NLRP3 inflammasome has been suggested to be involved in severe allergic lung inflammation, particularly in neutrophilic inflammation11 38 45 that is generally regarded as an important phenotype for severe asthma. Moreover, NLRP3 activation has been proposed to be associated with increased CS resistance in certain leukaemia cells.46 Therefore, pulmonary mtROS generation and the related activation of NLRP3 inflammasome may be linked to fungi-induced CS resistance associated with specific fungal species including Af.
In summary, we demonstrated that NLRP3 inflammasome is involved in fungi-induced allergic lung inflammation and may be linked to CS resistance in Af-induced allergic inflammation. Furthermore, PI3K-δ is critically implicated in fungi-induced allergic lung inflammation through regulation of NLRP3 inflammasome assembly/activation in airway epithelium, which is mediated partly through the regulation of mtROS generation. This suggests that inhibition of PI3K-δ may have potential for treating fungi-induced severe allergic lung inflammation in humans.
Footnotes
JSJ, KBL and SRK contributed equally.
Contributors: JSJ interpreted data and wrote the manuscript. KBL, DIK and HJP conducted experiments and performed analysis. H-KL, HJK, SHC, NK and SHK reviewed and edited the manuscript. SRK and YCL designed research, interpreted data, and edited the manuscript.
Funding: This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (NRF-2014R1A2A1A01002823; Y.C.L.).
Competing interests: None declared.
Ethics approval: Institutional Review Board of the Biomedical Research Institute of Chonbuk National University Hospital (IRB file No. 2013-11-007-001).
Provenance and peer review: Not commissioned; externally peer reviewed.
Presented at: Some of our data have been previously presented in the form of an abstract at an international scientific meeting (the 22nd congress of the Asia Pacific Society of Respirology, 2017).
References
- 1. Holgate ST. Innate and adaptive immune responses in asthma. Nat Med 2012;18:673–83. 10.1038/nm.2731 [DOI] [PubMed] [Google Scholar]
- 2. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010;140:805–20. 10.1016/j.cell.2010.01.022 [DOI] [PubMed] [Google Scholar]
- 3. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol 2015;16:45–56. 10.1038/ni.3049 [DOI] [PubMed] [Google Scholar]
- 4. Kanneganti TD, Lamkanfi M, Núñez G. Intracellular NOD-like receptors in host defense and disease. Immunity 2007;27:549–59. 10.1016/j.immuni.2007.10.002 [DOI] [PubMed] [Google Scholar]
- 5. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015;21:677–87. 10.1038/nm.3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schroder K, Tschopp J. The inflammasomes. Cell 2010;140:821–32. 10.1016/j.cell.2010.01.040 [DOI] [PubMed] [Google Scholar]
- 7. Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol 2012;13:325–32. 10.1038/ni.2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Besnard AG, Guillou N, Tschopp J, et al. . NLRP3 inflammasome is required in murine asthma in the absence of aluminum adjuvant. Allergy 2011;66:1047–57. 10.1111/j.1398-9995.2011.02586.x [DOI] [PubMed] [Google Scholar]
- 9. Zhou R, Yazdi AS, Menu P, et al. . A role for mitochondria in NLRP3 inflammasome activation. Nature 2011;469:221–5. 10.1038/nature09663 [DOI] [PubMed] [Google Scholar]
- 10. Bronner DN, Abuaita BH, Chen X, et al. . Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity 2015;43:451–62. 10.1016/j.immuni.2015.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim SR, Kim DI, Kim SH, et al. . NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis 2014;5:e1498 10.1038/cddis.2014.460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Denning DW, O’Driscoll BR, Hogaboam CM, et al. . The link between fungi and severe asthma: a summary of the evidence. Eur Respir J 2006;27:615–26. 10.1183/09031936.06.00074705 [DOI] [PubMed] [Google Scholar]
- 13. O’Driscoll BR, Powell G, Chew F, et al. . Comparison of skin prick tests with specific serum immunoglobulin E in the diagnosis of fungal sensitization in patients with severe asthma. Clin Exp Allergy 2009;39:1677–83. 10.1111/j.1365-2222.2009.03339.x [DOI] [PubMed] [Google Scholar]
- 14. Knutsen AP, Bush RK, Demain JG, et al. . Fungi and allergic lower respiratory tract diseases. J Allergy Clin Immunol 2012;129:280–91. 10.1016/j.jaci.2011.12.970 [DOI] [PubMed] [Google Scholar]
- 15. Fairs A, Agbetile J, Hargadon B, et al. . IgE sensitization to Aspergillus fumigatus is associated with reduced lung function in asthma. Am J Respir Crit Care Med 2010;182:1362–8. 10.1164/rccm.201001-0087OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Denning DW, Pashley C, Hartl D, et al. . Fungal allergy in asthma-state of the art and research needs. Clin Transl Allergy 2014;4:14 10.1186/2045-7022-4-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Denning DW, O’Driscoll BR, Powell G, et al. . Randomized controlled trial of oral antifungal treatment for severe asthma with fungal sensitization: The Fungal Asthma Sensitization Trial (FAST) study. Am J Respir Crit Care Med 2009;179:11–18. 10.1164/rccm.200805-737OC [DOI] [PubMed] [Google Scholar]
- 18. Gross O, Poeck H, Bscheider M, et al. . Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 2009;459:433–6. 10.1038/nature07965 [DOI] [PubMed] [Google Scholar]
- 19. Kool M, Willart MA, van Nimwegen M, et al. . An unexpected role for uric acid as an inducer of T helper 2 cell immunity to inhaled antigens and inflammatory mediator of allergic asthma. Immunity 2011;34:527–40. 10.1016/j.immuni.2011.03.015 [DOI] [PubMed] [Google Scholar]
- 20. Allen IC, Jania CM, Wilson JE, et al. . Analysis of NLRP3 in the development of allergic airway disease in mice. J Immunol 2012;188:2884–93. 10.4049/jimmunol.1102488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim SR, Lee KS, Park HS, et al. . HIF-1α inhibition ameliorates an allergic airway disease via VEGF suppression in bronchial epithelium. Eur J Immunol 2010;40:2858–69. 10.1002/eji.200939948 [DOI] [PubMed] [Google Scholar]
- 22. Lee KS, Jeong JS, Kim SR, et al. . Phosphoinositide 3-kinase-δ regulates fungus-induced allergic lung inflammation through endoplasmic reticulum stress. Thorax 2016;71:52–63. 10.1136/thoraxjnl-2015-207096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Menu P, Mayor A, Zhou R, et al. . ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis 2012;3:e261 10.1038/cddis.2011.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oczypok EA, Milutinovic PS, Alcorn JF, et al. . Pulmonary receptor for advanced glycation end-products promotes asthma pathogenesis through IL-33 and accumulation of group 2 innate lymphoid cells. J Allergy Clin Immunol 2015;136:747–56. 10.1016/j.jaci.2015.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kwak YG, Song CH, Yi HK, et al. . Involvement of PTEN in airway hyperresponsiveness and inflammation in bronchial asthma. J Clin Invest 2003;111:1083–92. 10.1172/JCI16440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Francesco EM, Lappano R, Santolla MF, et al. . HIF-1α/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res 2013;15:R64 10.1186/bcr3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lopez-Castejon G, Luheshi NM, Compan V, et al. . Deubiquitinases regulate the activity of caspase-1 and interleukin-1β secretion via assembly of the inflammasome. J Biol Chem 2013;288:2721–33. 10.1074/jbc.M112.422238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Segal BH. Aspergillosis. N Engl J Med 2009;360:1870–84. 10.1056/NEJMra0808853 [DOI] [PubMed] [Google Scholar]
- 29. Stevens DA, Moss RB, Kurup VP, et al. . Allergic bronchopulmonary aspergillosis in cystic fibrosis--state of the art: Cystic Fibrosis Foundation Consensus Conference. Clin Infect Dis 2003;37 Suppl 3(Suppl 3):S225–S264. 10.1086/376525 [DOI] [PubMed] [Google Scholar]
- 30. Bauernfeind FG, Horvath G, Stutz A, et al. . Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009;183:787–91. 10.4049/jimmunol.0901363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Osherov N. Interaction of the pathogenic mold Aspergillus fumigatus with lung epithelial cells. Front Microbiol 2012;3:346 10.3389/fmicb.2012.00346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Coll RC, Robertson AA, Chae JJ, et al. . A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015;21:248–55. 10.1038/nm.3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakahira K, Haspel JA, Rathinam VA, et al. . Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011;12:222–30. 10.1038/ni.1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol 2013;131:636–45. 10.1016/j.jaci.2012.12.1564 [DOI] [PubMed] [Google Scholar]
- 35. Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest 2008;118:3546–56. 10.1172/JCI36130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 2009;27:519–50. 10.1146/annurev.immunol.021908.132612 [DOI] [PubMed] [Google Scholar]
- 37. Johnson VJ, Yucesoy B, Luster MI. Prevention of IL-1 signaling attenuates airway hyperresponsiveness and inflammation in a murine model of toluene diisocyanate-induced asthma. J Allergy Clin Immunol 2005;116:851–8. 10.1016/j.jaci.2005.07.008 [DOI] [PubMed] [Google Scholar]
- 38. Simpson JL, Phipps S, Baines KJ, et al. . Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur Respir J 2014;43:1067–76. 10.1183/09031936.00105013 [DOI] [PubMed] [Google Scholar]
- 39. Lötvall J, Akdis CA, Bacharier LB, et al. . Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol 2011;127:355–60. 10.1016/j.jaci.2010.11.037 [DOI] [PubMed] [Google Scholar]
- 40. Park SJ, Min KH, Lee YC. Phosphoinositide 3-kinase delta inhibitor as a novel therapeutic agent in asthma. Respirology 2008;13:764–71. 10.1111/j.1440-1843.2008.01369.x [DOI] [PubMed] [Google Scholar]
- 41. Ciencewicki J, Trivedi S, Kleeberger SR. Oxidants and the pathogenesis of lung diseases. J Allergy Clin Immunol 2008;122:456–68. 10.1016/j.jaci.2008.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dostert C, Pétrilli V, Van Bruggen R, et al. . Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008;320:674–7. 10.1126/science.1156995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chung KF, Wenzel SE, Brozek JL, et al. . International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J 2014;43:343–73. 10.1183/09031936.00202013 [DOI] [PubMed] [Google Scholar]
- 44. Agarwal R, Gupta D. Severe asthma and fungi: current evidence. Med Mycol 2011;49(Suppl 1):S150–7. 10.3109/13693786.2010.504752 [DOI] [PubMed] [Google Scholar]
- 45. Kim HY, Lee HJ, Chang YJ, et al. . Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med 2014;20:54–61. 10.1038/nm.3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Paugh SW, Bonten EJ, Savic D, et al. . NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nat Genet 2015;47:607–14. 10.1038/ng.3283 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
thoraxjnl-2017-210326supp013.pdf (460.1KB, pdf)
thoraxjnl-2017-210326supp001.jpg (1.2MB, jpg)
thoraxjnl-2017-210326supp002.jpg (1.4MB, jpg)
thoraxjnl-2017-210326supp003.jpg (1.9MB, jpg)
thoraxjnl-2017-210326supp004.jpg (1.1MB, jpg)
thoraxjnl-2017-210326supp005.jpg (1,006.7KB, jpg)
thoraxjnl-2017-210326supp006.jpg (1.5MB, jpg)
thoraxjnl-2017-210326supp007.jpg (1.3MB, jpg)
thoraxjnl-2017-210326supp008.jpg (1.9MB, jpg)
thoraxjnl-2017-210326supp009.jpg (2.1MB, jpg)
thoraxjnl-2017-210326supp010.jpg (1.6MB, jpg)
thoraxjnl-2017-210326supp011.jpg (1.3MB, jpg)
thoraxjnl-2017-210326supp012.jpg (1.9MB, jpg)