

SUMMARY
Fidaxomicin is an antibacterial drug in clinical use in treatment of Clostridium difficile diarrhea. The active ingredient of the antibacterial drug fidaxomicin, lipiarmycin A3 (Lpm), functions by inhibiting bacterial RNA polymerase (RNAP). Here, we report a cryo-EM structure of Mycobacterium tuberculosis RNAP holoenzyme in complex with Lpm at 3.5 Å resolution. The structure shows that Lpm binds at the base of the RNAP "clamp." The structure exhibits an open conformation of the RNAP clamp, suggesting that Lpm traps an open-clamp state. Single-molecule fluorescence-resonance-energy-transfer experiments confirm that Lpm traps an open-clamp state and define effects of Lpm on clamp dynamics. We suggest that Lpm inhibits transcription by trapping an open-clamp state, preventing simultaneous interaction with promoter −10 and −35 elements. The results account for the absence of cross-resistance between Lpm and other RNAP inhibitors, account for structure-activity relationships of Lpm derivatives, and enable structure-based design of improved Lpm derivatives.
Graphical Abstract
eTOC Blurb
Lin et al. report cryo-EM and single-molecule spectroscopic analyses of the antibacterial drug fidaxomicin bound to its molecular target, RNA polymerase. The results define the structure of the drug-target complex, show fidaxomicin traps the RNA polymerase clamp in an open conformational state, and enable structure-based design of improved fidaxomicin analogs.
INTRODUCTION
Fidaxomicin is an antibacterial drug in clinical use in treatment of Clostridium difficile diarrhea (Lancaster and Matthews, 2012; Venugopal and Johnson, 2012). The active pharmaceutical ingredient of fidaxomicin, lipiarmycin A3 (Lpm; Kaufmann et al., 2015; Serra et al., 2017; Fig. S1A), is a macrocyclic antibiotic with bactericidal activity against Gram-positive bacteria and efflux-deficient strains of Gram-negative bacteria (Lancaster and Matthews, 2012; Venugopal and Johnson, 2012; Srivastava et al., 2011). Lpm functions by inhibiting bacterial RNA polymerase (RNAP; Talpaert et al., 1975; Sonenshein et al., 1977; Sonenshein and Alexander, 1979). Lpm exhibits no cross-resistance with the classic RNAP inhibitor rifampin (Rif; Sonenshein et al., 1977; Ebright, 2005) and inhibits transcription initiation at an earlier step than Rif (Sonenshein et al., 1977; Ebright, 2005; Tupin et al., 2010; Artsimovitch et al., 2012), suggesting that the binding site and mechanism of Lpm differ from those of Rif. Efforts spanning more than a decade to obtain structural information on RNAP-Lpm by use of X-ray crystallography have been unsuccessful.
RESULTS AND DISCUSSION
Structure of Mtb RNAP-Lpm
Structural information on RNAP is available for three bacterial genera: the Gram-positive bacterial genus Mycobacterium (Hubin et al., 2017; Lin et al., 2017), the Gram-negative bacterial genus Escherichia (Murakami et al., 2013), and the Thermus/Deinococcus-clade bacterial genus Thermus (Zhang et al., 1999; Vassylyev et al., 2002). Lpm potently inhibits RNAP from genus Mycobacterium and genus Escherichia (IC50s = 0.3 μM and 8 μM, respectively; Talpaert et al., 1975; Kurabachew, 2008; Fig. S1B), but only poorly inhibits RNAP from genus Thermus (IC50 = 200 μM; Fig. S1B). Accordingly, efforts in our laboratory to obtain structural and functional information on RNAP-Lpm focussed on RNAP from genus Mycobacterium and genus Escherichia.
We first sought to obtain structural information on RNAP-Lpm by use of X-ray crystallography. However, we were unable to prepare crystals of RNAP-Lpm, despite numerous attempts at crystal soaking (assessing all reported crystal forms of RNAP from genus Mycobacterium and genus Escherichia) and co-crystallization (assessing crystallization conditions for all reported crystal forms of RNAP from genus Mycobacterium and genus Escherichia, and assessing >104 additional crystallization conditions by robotic screening).
We then sought to obtain structural information on RNAP-Lpm by use of cryo-EM with single-particle reconstruction (Merino and Raunser, 2017; Boland et al., 2017). Pilot experiments suggested that cryo-EM particle-orientation distributions using RNAP from genus Mycobacterium were more favorable for high-resolution structure determination than cryo-EM particle-orientation distributions using RNAP from genus Escherichia. Accordingly, our cryo-EM analysis focussed on RNAP from genus Mycobacterium.
We determined a structure of Mycobacterium tuberculosis (Mtb) RNAP σA holoenzyme in complex with Lpm by use of cryo-EM with single-particle reconstruction (Merino and Raunser, 2017; Boland et al., 2017) (Figs. 1, S1-S3). The cryo-EM density map shows unambiguous density for RNAP, including the taxon-specific, Mycobacterium-specific sequence insertion (β′MtbSI or "gate"; Lin et al., 2017), for σ conserved regions 2, 3, and 4 (σR2, σR3, and σR4), and for Lpm. The mean resolution of the structure is 3.5 Å (Fig. S1F). Local resolution ranges from ~2.5-3.8 Å in central parts of the structure, including RNAP residues close to Lpm, to ~5-6.5 Å in peripheral parts of the structure, including β′MtbSI and σR4 (Figs. 1a, S1G, S2A). Local B-factors range from ~8-100 Å2 in central regions to ~300-400 Å2 in peripheral regions (Fig. 2B). The map show clear density for backbone and sidechain atoms of RNAP and individual functional groups of Lpm (Fig. S2C-D).
Figure 1. Structure of Mtb RNAP-Lpm.

A, Density map for Mtb RNAP-Lpm (left) and Lpm (right), colored by local resolution. B, Density and atomic model for Mtb RNAP-Lpm. Gray, RNAP core other than β′MtbSI; green, β′MtbSI; yellow, σ; green mesh, density map for Lpm; cyan, Lpm; violet sphere, RNAP active-center Mg2+. C, Atomic model for Mtb RNAP-Lpm. Colors as in B. D, RNAP-Lpm interactions (residues numbered as in Mtb RNAP and, in parentheses, as in Escherichia coli RNAP). Gray ribbons, RNAP backbone; gray sticks, RNAP carbon atoms; cyan sticks, Lpm carbon atoms; red, blue, and green sticks, oxygen, nitrogen, and chlorine atoms; dashed lines, H-bonds. E, Summary of RNAP-Lpm interactions. Blue arcs, van der Waals interactions; red dashed lines, H-bonds. See also Figs. S1-S4.
Figure 2. Relationship between binding site and resistance determinant of Lpm and binding sites and resistant determinants of other RNAP inhibitors.

A, Binding positions of Lpm (cyan; Fig. 1), Rif and Sor (red; PDB: 1I6V, PDB: 1YNJ, PDB: 2A68, PDB: 2A69, PDB: 4KN4, PDB: 4KN7, PDB: 4OIR, and PDB: 5UHB), GE and PUM (dark blue; PDB: 4OIN, PDB: 4OIR, and PDB: 5X21), CBR and AAP (light blue; PDB: 4XSY, PDB: 4XSZ, PDB: 4ZH2, PDB: PDB: 4ZH3, PDB: 4ZH4, PDB: 5UHE, and PDB: 5UHF), Sal (green; PDB: 4MEX), Stl (yellow; PDB: 1ZYR), and Myx and SQ (magenta; PDB: 3DXJ, PDB: 3EQL, PDB: 4YFK, PDB: 4YFN, and PDB: 4YFX), mapped onto structure of Mtb RNAP (gray; two orthogonal views; β′MtbSI and σ omitted for clarity). Violet sphere, RNAP active-center Mg2+. B, Resistance determinants of Lpm (cyan; Fig. 2C), Rif and Sor (red; Campbell et al., 2001, 2005), GE and PUM (dark blue; Zhang et al., 2014; Maffioli et al., 2017), CBR703 and AAP (light blue; Lin et al., 2017; Feng et al., 2015; Bae et al., 2015), Sal (green; Degen et al., 2014), Stl (yellow; Tuske et al., 2005; Temiakov et al., 2005), and Myx, Cor, Rip, and SQ (magenta; Mukhopadhyay et al., 2008; Belogurov et al., 2009; Molodtsov et al., 2015) mapped onto structure of Mtb RNAP. C, Sequences and properties of E. coli Lpm-resistant mutants residues numbered as in Mtb RNAP and, in parentheses, as in Escherichia coli RNAP). D, Absence of significant cross-resistance of E. coli Lpm-resistant mutants to Rif, GE, PUM, Sal, CBR, Stl, and Myx. E, Absence of significant cross-resistance of E. coli Rif-, GE/PUM-, Sal-, CBR-, and Stl-resistant mutants to Lpm. F, Additive antibacterial activity of Lpm and Rif. [Resistance and cross-resistance levels for Lpm-, CBR-, and Stl-resistant mutants are from strains having the mutant RNAP subunit gene on a plasmid and the corresponding wild-type RNAP subunit gene on the chromosome (merodiploid strains; see Methods). Resistance and cross-resistance levels for Lpm-, CBR-, and Stl-resistant mutants are from strains having the mutant RNAP subunit gene on the chromosome and no corresponding wild-type RNAP subunit gene (non-merodipoid strains; see Methods). Resistance and cross-resistance levels are expected to be lower for merodiploid strains than for non-merodiploid strains.] See also Fig. S5.
The cryo-EM structure shows that the Lpm binding site is located at the base of the RNAP clamp (Landick, 2001; Cramer et al., 2001; Chakraborty et al., 2012) and encompasses RNAP clamp α-helices βa16α1 and β′a4α1 and RNAP switch-region (Cramer et al., 2001; Srivastava et al., 2011) switches SW2, SW3, and SW4 (Figs. 1, S3). Lpm binds to RNAP in a fully-extended conformation similar to conformations in crystal structures of Lpm alone (Fig. S3A). All five structural moieties of Lpm--isobutyryl, β-D-noviosyl, macrocycle, β-D-rhamnosyl, and homodichloro-orsellinyl--interact with RNAP (Figs. 1D-E, S1A, S3B-C). Lpm makes H-bonds with six RNAP residues: βT1096 (V1298), βK1101 (K1303), β′R84 (Q94), β′R89 (R99), β′E323 (D248), and β′R412 (R337) [residues numbered as in Mtb RNAP (UniProtKB: P9WGY9, UniProtKB: P9WGY7) and, in parentheses, as in Escherichia coli RNAP (UniProtKB: P0A8V2, UniProtKB: P0A8T7)]. Alanine substitutions of five of these RNAP residues results in Lpm-resistance, confirming the interactions are functionally relevant (Fig. S3D).
Sequence alignments show that residues of RNAP that contact Lpm are conserved in Gram-positive and Gram-negative bacterial RNAP, but are not conserved in human RNAP I, II, and III (Fig. S4A-B), accounting for the ability of Lpm to inhibit both Gram-positive and Gram-negative bacterial RNAP but not human RNAP I, II, and III (Fig. S4C).
The cryo-EM structure accounts for structure-activity relationships of Lpm analogs produced by metabolism (Babakhani et al., 2011), precursor feeding (Hochlowski et al., 1997), mutasynthesis (Xiao et al., 2011; Niu et al., 2011), and semi-synthesis (Figs. 1D-E, S3B-C, S5). The structure accounts for structure-activity relationships indicating functional importance of the Lpm isobutyryl moiety ("b1" vs. Lpm), β-D-noviosyl moiety ("c1" vs. "b2" and "a6"); macrocycle and C25 hydroxyl therein ("a5" vs. Lpm), β-D-rhamnosyl moiety and C6' methyl therein ("e1" vs. "d1" and "a7"; "a6" vs. Lpm), and homodichloro-orsellinyl moiety and C8" methyl, C3" chlorine, and C5" chlorine therein ("d1" vs. Lpm; "a2"-"a4" vs. Lpm; "a8" vs. "a6"). The observation that the Lpm C4" hydroxyl is exposed to solvent at the exterior of RNAP (Fig. S3B-C) predicts that substituents--including very large substituents--may be introduced at the C4" hydroxyl oxygen without eliminating ability to inhibit RNAP, provided the H-bond-acceptor character of the oxygen is maintained. Validating this prediction, we have prepared a Lpm analog having a large substituent, benzyl, appended at the C4" hydroxyl oxygen by semi-synthesis from Lpm and have found the analog to retain high activity in inhibiting RNAP (Fig. S5, analog "a1"; potencies within approximately an order of magnitude of that of Lpm for Mtb and S. aureus RNAP and equal to that of Lpm for Escherichia coli RNAP).
Relationship between binding site and resistance determinant of Lpm and binding sites and resistant determinants of other RNAP inhibitors
The binding site on RNAP for Lpm (cyan in Fig. 2A) does not overlap the binding sites of other RNAP inhibitors, including rifampin and sorangicin (Rif and Sor; Campbell et al., 2001, 2005); GE23077 and pseudoridimycin (GE and PUM; Zhang et al., 2014; Maffioli et al., 2017), CBR703 and D-AAP-1 (CBR and AAP; Lin et al., 2017; Feng et al., 2015; Bae et al., 2015), salinamide (Sal; Degen et al., 2014), streptolydigin (Stl; Tuske et al., 2005; Temiakov et al., 2005), and myxopyronin, corallopyronin, ripostatin, and squaramide (Myx, Cor, Rip, and SQ; Mukhopadhyay et al., 2008; Belogurov et al., 2009; Molodtsov et al., 2015) (Fig. 2A). The binding site for Lpm is close to, but does not overlap, the binding site of Myx, Cor, Rip and SQ, which bind to the RNAP switch region, contacting SW1 and the face of SW2 opposite the face contacted by Lpm. The binding site for Lpm is far from the binding sites of other RNAP inhibitors.
Amino acid substitutions conferring resistance to Lpm were identified by sequencing spontaneous Bacillus subtilis Lpm-resistant mutants (Sonenshein et al., 1977; Ebright, 2005); additional substitutions were identified by sequencing induced Lpm-resistant mutants of E. coli RNAP (Ebright, 2005); and, in this work, a full set of substitutions was identified by extending the latter method (Fig. 2B-C). Lpm-resistant substitutions are obtained at ten sites in RNAP β subunit and seven sites in RNAP β′ subunit (Fig. 2B-C). All sites of Lpm-resistant substitutions are located in the immediate vicinity of the Lpm binding site (Fig. 2B), and all high-level (≥4-fold) Lpm-resistant substitutions involve RNAP residues that make direct contact with Lpm or that contact other RNAP residues that, in turn, make direct contact with Lpm (Fig. S5A). The resistance determinant for Lpm does not overlap the resistance determinants of other RNAP inhibitors (Fig. 2B). Consistent with the absence of overlap of binding sites and resistance determinants, mutants resistant to Lpm are not cross-resistant to other RNAP inhibitors (Figs. 2D, S6B), and, reciprocally, mutants resistant to other RNAP inhibitors are not cross-resistant to Lpm (Figs. 2E, S6C). Further consistent with the absence of overlap of binding sites and resistance determinants, Lpm and Rif exhibit additive antibacterial activity when co-administered (Fig. 2F).
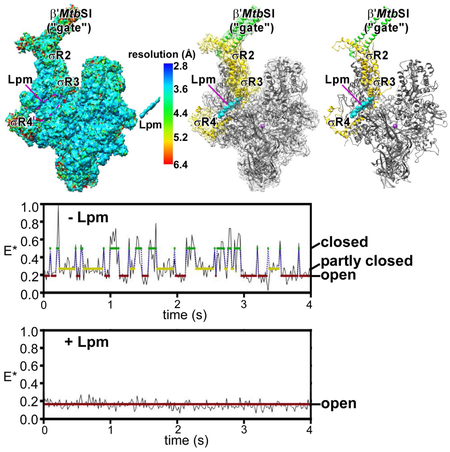
Effects of Lpm on RNAP clamp conformation: cryo-EM data
The RNAP clamp adopts different conformations--ranging from open, to partly closed, to closed--in different crystal structures (Landick, 2001; Cramer et al., 2001; Chakraborty et al., 2012; Fig. 3A). Differences in clamp conformation arise from rigid-body rotations about a hinge formed by the RNAP switch region (Landick, 2001; Cramer et al., 2001; Chakraborty et al., 2012). Importantly, differences in clamp conformation are observed even in crystal structures of RNAP from a single bacterial species, indicating that differences in clamp conformations are state differences and not species differences (Fig. 3A; Table 2).
Figure 3. Effects of Lpm on RNAP clamp conformation: cryo-EM data.

A, RNAP open (red), partly closed (yellow), and closed (green) clamp conformational states. Mtb RNAP-Lpm main mass (view as in Figs. 1-2) and, superimposed, RNAP clamps from crystal structures of T. thermophilus RNAP in different crystal lattices [T. thermophilus RNAP with open clamp (red; structure determined in this work to enable comparison of open-clamp, partly-closed-clamp, and closed-clamp states of RNAP from a single bacterial species; Table 2), T. thermophilus RNAP with partly open clamp (yellow; PDB: 5TMC), and T. thermophilus RPo (green; PDB: 4G7H)]. B, RNAP open-clamp state in Mtb RNAP-Lpm. Red, clamp in Mtb RNAP-Lpm. Other colors as in A. C, Comparisons of RNAP clamp conformation in Mtb RNAP-Lpm to RNAP clamp conformations in crystal structures of Tth RNAP with open clamp (left; Table 2), Tth RNAP with partly closed clamp (center; PDB: 5TMC), and Tth RPo with closed clamp (right; PDB: 4G7H). Each image shows Mtb RNAP-Lpm (clamp in red; view orientation and other colors as in Figs. 1-2) and, superimposed, the RNAP clamp of the comparator structure (gray). D, Stereodiagram of Lpm binding site showing conformations of RNAP structural elements SW1, SW2, SW3, SW4, βa16α1, and β'a4α1, in open-clamp (red; Mtb RNAP-Lpm), partly-closed clamp (yellow; PDB: 5TMC), and closed-clamp (green; PDB: 4G7H) states. RNAP switches. SW1, SW2, SW3, and SW4 are shown with ends that connect to the RNAP clamp as numbered circles, and ends that connect to the RNAP main mass as numbered squares. Cyan, Lpm.
Table 2.
X-ray crystallography data-collection and data-analysis statistics*
| structure | Tth RNAP core |
| PDB code | PDB: 6ASG |
| data collection | |
| data collection source | APS-19ID |
| space group | R3 |
| cell dimensions | |
| a, b, c (Å) | 280.7,280.7,184.9 |
| α, β, γ (°) | 90.0, 90.0, 120.0 |
| resolution | 50.0-3.8 (4.0-3.8) |
| number of unique reflections | 61,395 |
| Rmerge | 0.245 (0.704) |
| Rmeas | 0.275 (0.830) |
| Rpim | 0.124 (0.430) |
| highest resolution shell CC1/2 | 0.418 |
| I/σ(I) | 5.0 (1.2) |
| completeness (%) | 99.2 (92.3) |
| redundancy | 4.8 (3.2) |
| refinement | |
| resolution (Å) | 42.5-3.8 |
| number of unique reflections | 50,511 |
| number of test reflections | 2,611 |
| ssRwork/Rfree | 0.22/0.27 (0.27/0.32) |
| number of atoms | |
| protein | 23,551 |
| ligand/ion | 3 |
| root-Mean-Square(RMS) Deviations | |
| bond length (Å) | 0.003 |
| bond angle (°) | 0.658 |
| MolProbity statistics | |
| clash score | 15.2 |
| rotamer outliers (%) | 1.0 |
| Cβ outliers (%) | 0 |
| Ramachandran plot | |
| Favored (%) | 94.1 |
| Outliers (%) | 0.4 |
Data for highest-resolution shells are in parentheses.
In the cryo-EM structure of Mtb RNAP-Lpm, the clamp adopts an open conformational state, superimposable on the open state in a crystal structure of Thermus thermophilus (Tth) RNAP (Table 2), but differing by 9° from the partly closed state in a crystal structure of Tth RNAP (PDB: 5TMC), and differing by 17° from the closed state in crystal structures of catalytically competent RNAP-promoter transcription initiation complexes (Tth RPo and Mtb RPo; 4G7H and 5UH5) (Fig. 3B-C). The interactions that Lpm makes with βa16α1, β′a4α1, SW2, SW3, and SW4 require an open clamp conformation (Fig. 3D). Each of these regions is positioned to accommodate Lpm in the open state, but not in partly closed and closed states. In particular, βa16α1 and β′a4α1 would severely clash with Lpm in partly closed and closed states (Fig. 3D). We infer that Lpm, through its interactions with its binding site, induces or stabilizes an open clamp conformation.
Effects of Lpm on RNAP clamp conformation: single-molecule FRET data
RNAP clamp conformation in solution can be monitored by single-molecule fluorescence resonance energy transfer (FRET) experiments assessing distances between fluorescent probes incorporated specifically at the tips of the two walls of the RNAP active-center cleft (Chakraborty et al., 2012; Fig. 4A-C). Single-molecule FRET experiments analyzing RNAP holoenzyme in the absence of Lpm show a broad multimodal distribution of FRET efficiencies, indicative of open, partly closed, and closed states (Fig. 4D, left panel, gray curve). In contrast, analogous experiments in the presence of Lpm show a narrow unimodal peak with a FRET efficiency indicative of the open state (Fig. 4E, left panel). We conclude that Lpm traps the open-clamp state in solution. Single-molecule time traces in the absence of Lpm show static and dynamic molecules; the dynamic molecules (34%; N = 207) show transitions between three distinct FRET efficiency levels--corresponding to open-clamp, partly-closed-clamp, and closed-clamp states, with dwell times on the 100-400 ms time scale--indicating dynamic transitions between interconverting clamp states (Fig. 4D, left panel, colored curves; Fig. 4D, right panel; Fig. 4F). In contrast, analogous single-molecule time traces in the presence of Lpm show only the FRET efficiency level corresponding to the open-clamp state, indicating stable trapping of the open-clamp state (Fig. 4E, right panel; Fig 4F). Single-molecule time traces before and after addition of Lpm show a rapid (<2 s) transition from the multimodal, dynamic clamp profile of RNAP in the absence of Lpm to the unimodal, non-dynamic clamp profile of RNAP in the presence of Lpm, demonstrating rapid trapping of the open-clamp state (Fig. 4G). We conclude, based both on cryo-EM structure and on FRET, that Lpm traps the open-clamp state.
Figure 4. Effects of Lpm on RNAP clamp conformation: single-molecule FRET data.

A, Use of unnatural-amino-acid mutagenesis, Staudinger ligation with Cy3B-phosphine and Alexa647-phosphine, and total-internal-reflection fluorescence microscopy with alternating-laser excitation microscopy (TIRF-ALEX) to obtain FRET data for single molecules of RNAP having fluorescent probes at the tips of the walls of the RNAP active-center cleft (see STAR Methods). Green, fluorescence donor probe Cy3B; red, fluorescent acceptor probe Alexa647; black square, hexahistidine tag. B, Surface-immobilization of fluorescent-probe-labelled RNAP for TIRF-ALEX. C, Time trace of donor emission intensity (green) and acceptor emission intensity (red) (top) and corresponding time trace of donor-acceptor FRET efficiency (bottom). D, Single-molecule FRET data for RNAP holoenzyme in absence of Lpm. Left, histogram. Gray, all states; colors, Hidden Markov Model (HMM)-assigned open, partly closed, and closed states (red, yellow, and green). Right, time trace with HMM-assigned open, partly closed, and closed states (red, yellow, and green). E, Single-molecule FRET data for RNAP holoenzyme in presence of Lpm (histogram and time trace as in D). F, Summary of RNAP clamp angles and dwell times for open, partly closed, and closed clamp states in absence and presence of Lpm. G, Time trace for RNAP holoenzyme before and after addition of Lpm.
Strikingly, the effect of Lpm on RNAP clamp conformation is opposite the effects of Myx, Cor, and Rip on RNAP clamp conformation. Lpm, through interactions with RNAP switch-region SW2, SW3, and SW4, traps an open-clamp state (Chakraborty et al., 2012); in contrast, Myx, Cor and Rip, through interactions with SW1 and the opposite face of SW2, trap a closed-clamp state (Figs. 3-4). The different effects of these inhibitors on RNAP clamp conformation presumably underlie their different effects on isomerization of RPc to RPo [inhibition of an early step in isomerization by Lpm; inhibition of a later step in isomerization by Myx, Cor, and Rip (Tupin et al., 2010; Artsimovitch et al., 2012)].
The binding site on RNAP for Lpm overlaps the proposed binding site on RNAP for pause-inducing and termination-inducing RNA hairpins (Hein et al., 2014). It is attractive to speculate that the Lpm binding site is an "allosteric trigger" for RNAP clamp opening and that ligands of this site--including not only Lpm but also pause-inducing and termination-inducing RNA hairpins--trigger RNAP clamp opening by binding to the site.
Mechanism of transcription inhibition by Lpm
The effects of Lpm on RNAP clamp conformation suggest a model for the mechanism by which Lpm inhibits isomerization of RPc to RPo (Sonenshein and Alexander, 1979; Ebright, 2005; Tupin et al., 2010; Artsimovitch et al., 2012) in transcription initiation (Fig. 5). Because σR2 [the σ module that recognizes promoter −10 elements (Ruff et al., 2015; Zhang et al., 2012; Zuo and Steitz, 2015; Bae et al., 2015; Feng et al., 2016)] interacts with the RNAP clamp and σR4 [the σ module that recognizes promoter −35 elements (Ruff et al., 2015; Zuo and Steitz, 2015; Bae et al., 2015; Feng et al., 2016)] interacts with the main mass of RNAP, differences in RNAP clamp conformation necessarily result in differences in the spatial relationship of σR2 and σR4 (Fig. 5, left panels). We propose that, in the closed-clamp state, σR2 and σR4 can engage simultaneously with promoter −10 and −35 elements during formation of RPo (Fig. 5A, middle and right panels), but, in the open-clamp state, σR2 and σR4 cannot engage simultaneously with promoter −10 and −35 elements (Fig. 5B, middle and right panels). In particular, we propose that the σR2 "Trp wedge," which intercalates into DNA and stacks on the non-template-strand nucleotide at promoter position −12, nucleating promoter unwinding in formation of RPo (Bae et al., 2015; Feng et al., 2016), can engage DNA only in the closed-clamp state, and that the σR2 "NT-11 pocket," which captures an unstacked non-template-strand nucleotide at promoter position −11, stabilizing promoter unwinding upon formation of RPo (Zhang et al., 2012; Zuo and Steitz, 2015; Bae et al., 2015; Feng et al., 2016), can engage DNA only in the closed-clamp state (Fig. 5, middle and right panels). We suggest that the inability of the open-clamp state to engage simultaneously with −10 and −35 elements occurs during late steps in formation of RPo, when interactions of DNA downstream of the −10 element with the RNAP-active-center cleft constrain the −10 element to be near the RNAP active-center cleft, in a position unreachable by σR2 in the (Fig. 5, right panels), and possibly also occurs during earlier steps in formation of RPo, if, as proposed in Vassylyev et al., 2002 and NandyMazumdar et al., 2016, interactions of DNA downstream of the −10 element with the opposite wall of the RNAP active-center cleft constrain the −10 element to be near the RNAP active-center cleft, in a position unreachable by σR2 in the open-clamp state (Fig. 5, middle panels). The proposal that Lpm inhibits transcription initiation by trapping an open-clamp state, thereby preventing simultaneous engagement of −10 and −35 elements, is consistent with recently proposed models for transcription initiation (Feklistov et al., 2017).
Figure 5. Mechanism of transcription inhibition by Lpm.

A, RNAP holoenzyme (left), RPc (center; DNA as in Ruff et al., 2015), and RPo (right, DNA as in Zuo and Steitz, 2015; Bae et al., 2015; Feng et al., 2016) for RNAP in absence of Lpm (closed clamp). σR2 and σR4 simultaneously engage promoter −10 and −35 elements in RPc and RPo. Atomic coordinates from PDB: 5UH5. Green ribbons, σ; green surface, σR2 NT-11 pocket; yellow surfaces, σR2 Trp wedge and σR4 recognition helix; blue, DNA. Other colors as in Figs. 1-2. B, As A, but for RNAP in presence of Lpm (open clamp). σR2 and σR4 cannot simultaneously engage promoter −10 and −35 elements in RPc and RPo. Brown ribbons, σ; brown surface, σR2 NT-11 pocket; pink surface, σR2 Trp wedge and σR4 recognition helix; cyan, Lpm. Other colors as in A.
Prospect
The methods and results of this report provide a strategy to obtain structural information for any RNAP inhibitor, irrespective of species selectivity and effects on RNAP conformation. The use of cryo-EM to determine the structure of RNAP-Lpm overcame obstacles that had precluded structure determination by X-ray crystallography: (1) the species selectivity of Lpm (which only poorly inhibits Thermus RNAP; Fig. S1B) and (2) the trapping by Lpm of an RNAP open-clamp conformation (which is incompatible with known crystal lattices of non-Thermus bacterial RNAP).
The results of this report provide fundamental information on the binding site and mechanism of Lpm and provide insights for antibacterial drug development involving Lpm. The primary limitation of Lpm as an antibacterial drug is that Lpm has no systemic bioavailability and thus is ineffective against systemic infections, such as tuberculosis and S. aureus blood and lung infections (Srivastava et al., 2011; Lancaster and Matthews, 2012; Venugopal and Johnson, 2012). The chemical strategy and synthetic procedure demonstrated here for appending substituents at the Lpm C4" hydroxyl (Fig S5; "a1") provides a means to overcome this limitation. It should be possible to append substituents improving physical and pharmacokinetic properties, nanoparticle drug-delivery modules, or even other antibacterial-agent pharmacophores at the Lpm C4" hydroxyl while retaining RNAP-inhibitory activity.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for further information or reagents should be directed to Richard H. Ebright (ebright@waksman.rutgers.edu).
METHOD DETAILS
Lpm
Lpm was the kind gift of A.L. Sonenschein (Tufts University, Medford MA), was prepared by fermentation of Actinoplanes deccanensis ATCC 21983 (Parenti et al., 1975), as in Coronelli et al., 1975, or was purchased from BioAustralis or Biorbyt.
Lpm analogs: previously described Lpm analogs
Lpm analog "a4" (3"-bromo-3",5"-dideschloro-Lpm; compound 3 of Hochlowski et al., 1997) was prepared by fermentation of Dactylosporangium aurantiacum hamdenensis NRRL 18085 (Theriault et al., 1987; ARS Patent Culture Collection, Peoria IL) with precursor feeding, as in Hochlowski et al., 1997. Lpm analogs "a5" (69-1d), "a6" (70-9d), "a7" (70-10d), "a8" (70-11d), "b2" (53-11d), "c1" (54-4d), and "e1" (54-3d) were prepared by fermentation of mutants of Dactylosporangium aurantiacum hamdenensis NRRL 18085, as in Xiao et al., 2011 and Niu et al., 2011. Lpm analog "b1" (OP-1118; Babakhani et al., 2011) was purchased from Toronto Research Chemicals.
Lpm analogs: Lpm analogs "a2" (5"-deschloro-Lpm) and "d1" (desaryl-Lpm)
Lpm analogs "a2" (5"-deschloro-Lpm) and "d1" (desaryl-Lpm) were prepared by fermentation of Dactylosporangium aurantiacum hamdenensis NRRL 18085 (Theriault et al., 1987; ARS Patent Culture Collection, Peoria IL) in growth media containing limiting chloride [growth media with KCl replaced by KBr; residual chloride concentration = 40 μM, quantified using QuantiChromT Chloride Assay Kit (BioAssays Systems)]. First-stage (0.2 L) and second-stage (10 L) cultures were prepared as in Hochlowski et al., 1997, and second-stage culture broths were harvested as in Hochlowski et al., 1997. Culture broths were adjusted to pH 7 by addition of 1 ml 12 N NaOH, supplemented with 5 L acetone, shaken 1 h at 22°C, and extracted with 3×4 L ethyl acetate. The extracts were evaporated, and the resulting material (20 g) was re-dissolved in 300 ml ethyl acetate, filtered through Whatman filter paper (Grade 1; Sigma-Aldrich), and evaporated. The resulting material (14 g) was partitioned in 900 ml 1:1:1 (v/v/v) chloroform-methanol-water, and the lower phase was evaporated. The resulting material (11 g) was chromatographed on silica gel (Sigma-Aldrich; 3 cm × 30 cm; stepwise elution with 2-50% methanol in chloroform, followed by elution with 100% methanol). UV-absorbant (254 nm) fractions were collected, evaporated, and assayed for antibacterial activity by spotting on H-top-agar/LB-agar (Sambrook and Russell, 2001) plates seeded with 109 cfu E. coli D21f2tolC (Fralick and Burns-Keliher, 1994), incubating 16 h at 37, and assessing growth inhibition. Active fractions eluting at ~5% to ~10% methanol were pooled, further purified by re-chromatography on silica gel (procedures essentially as above), further purified by reversed-phase HPLC [Hitachi 7000 with L7450 detector; Supelco Discovery BIO Wide Pore C18 semi-prep column; 1:1 (v/v) acetonitrile-water isocratic elution; flow rate = 2 ml/min], and evaporated, yielding 80 mg Lpm analog "a3" [MALDI-MS m/z: calculated 1011.41 (M + Na+); found 1011.07] and 8 mg Lpm analog "a2" [MALDI-MS m/z: calculated 1045.59 (M + Na+); found 1044.60, 1046.70]. Material eluting at 100% methanol was further purified by reversed-phase HPLC (procedures essentially as above), and evaporated, yielding 0.2 mg Lpm analog "d1" [MALDI-MS m/z: calculated, 846.98 (M + Na+); found, 847.90].
Lpm analogs: Lpm analog "a1" (4"-O-benzyl-Lpm)
Lpm analog "a1" (4"-O-benzyl-Lpm) was prepared from Lpm by semi-synthesis as follows: To Lpm (Biorbyt: 3 mg; 2.8 μmol) in 50 μl anhydrous dimethylformamide (Sigma-Aldrich), was added anhydrous potassium carbonate (Sigma-Aldrich: 3 mg, 22 μmol), and the reaction mixture was stirred 30 min at 50°C and then allowed to cool to room temperature. An aliquot (10 μl) of 0.28 M benzyl bromide in anhydrous dimethylformamide [2.8 μmol; prepared by dissolving 3.4 μl benzyl bromide (Sigma-Aldrich) in 50 μl anhydrous dimethylformamide immediately before use] was added, and the reaction mixture was stirred 30 min at 50°C. The reaction mixture was evaporated to remove dimethylformamide and then re-suspended in 600 μl 100 mM monobasic sodium phosphate (ThermoFisher). Precipitated solids were collected by centrifugation, rinsed with water, re-dissolved in methanol, and purified by reversed-phase HPLC (Hitachi 2000 with L2455 detector; Phenomenex Luna C18, 100 Å, 25 mm × 4.6 mm column; phase A = water; phase B = acetonitrile; gradient = 40% B at 0 min, 100% B at 20 min; flow rate = 1 ml/min). Lpm analog "a1" eluted at 20 minutes. Yield: 0.35 mg, 10%. MALDI-MS m/z: calculated, 1171.44 (M + Na+); found 1169.64, 1171.64.
Other RNAP inhibitors
Cor A and Rip A were the kind gift of H. Irschik and R. Jansen (Helmholtz Institut, Braunschweig, Germany), Stl was the kind gift of E. Steinbrecher (Upjohn-Pharmacia, Kalamazoo, MI), and Sal A was the kind gift of W. Fenical (The Scripps Research Institute, La Jolla, CA). Myx B was prepared by synthesis, as in Ebright and Ebright, 2012; GE was prepared by fermentation of Actinomadura sp. DSMZ 13491, as in Ciciliato et al., 2004; and PUM was prepared by fermentation of Streptomyces sp. ID38640, as in Maffioli et al., 2017. Rif was purchased from Sigma-Aldrich, and CBR was purchased from Maybridge.
M. tuberculosis RNAP σA holoenzyme, RNAP core enzyme, and σA
Mtb RNAP σA holoenzyme was prepared by co-expression of genes for Mtb RNAP β' subunit, RNAP β subunit, RNAP N-terminally decahistidine-tagged α subunit, RNAP ω subunit, and N-terminally hexahistidine-tagged σA in E. coli, followed by cell lysis, polyethylenimine precipitation, ammonium sulfate precipitation, immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen), and anion-exchange chromatography on Mono Q (GE Healthcare), as in Lin et al., 2017.
For experiments in Figs. S1B, S3D, and S5, Mtb RNAP core enzyme was prepared by co-expression of genes for Mtb RNAP β' subunit, RNAP β subunit, N-terminally decahistidine-tagged RNAP α subunit, and RNAP ω subunit in E. coli[plasmids pCOLA-rpoB-rpoC, pACYC-rpoA, and pCDF-rpoZ (Banerjee et al., 2014); strain BL21(DE3) (EMD Millipore)], followed by cell lysis, polyethylenimine precipitation, ammonium sulfate precipitation, immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen), and anion-exchange chromatography on Mono Q (GE Healthcare), as in Lin et al., 2017, but using plasmid pCOLADuet-Mtb-rpoB-rpoC [prepared by replacing the NcoI-BamHI segment of plasmid pCOLA-Duet (EMD Millipore) by the NcoI-BamHI segment of a DNA fragment carrying CCATGGTG followed by Mtb rpoB codons 3-1174 followed by TAAGGATCC, prepared by PCR using plasmid pJF09 (Jacques et al., 2006; gift of S. Rodrigue, Universite de Sherbrooke, Canada), and then replacing the NdeI-MfeI segment of the resulting plasmid by the NdeI-MfeI segment of a DNA fragment carrying CATATG followed by Mtb rpoC codons 2-1316 followed by TAGCAATTG, prepared by PCR using plasmid pJF10 (Jacques et al., 2006; gift of S. Rodrigue, Universite de Sherbrooke, Canada)], or derivatives thereof constructed using site-directed mutagenesis (QuikChange Site-Directed Mutagenesis Kit; Agilent), in place of plasmid pCOLADuet-rpoB-rpoC (Banerjee et al., 2014; Lin et al., 2017).
For experiments in Figs. S1B, S3D, and S5, Mtb σA was prepared by expression of a gene for N-terminally hexahistidine-tagged Mtb σA in E. coli, followed by cell lysis, immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen), and anion-exchange chromatography on Mono Q (GE Healthcare), as in Lin et al., 2017.
E. coli RNAP σ70 holoenzyme
For experiments in Figs. S1B and S5, hexahistidine-tagged E. coli RNAP σ70 holoenzyme was prepared from E. coli strain XE54 (Tang et al., 1994) transformed with plasmid pREII-NHα (Tang et al., 1994), using culture and procedures, cell lysis, polyethylenimine precipitation, ammonium sulfate precipitation, immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen), and anion-exchange chromatography on Mono Q (GE Healthcare), as in Degen et al., 2014.
For experiments in Figs. 4C-F, fluorescent-probe-labelled, hexahistidine-tagged E. coli RNAP σ70 holoenzyme was prepared using unnatural-amino-acid mutagenesis (Chin et al., 2002) of co-expressed genes encoding RNAP β', β, α, and ω subunits and σ70, yielding an RNAP σ70 holoenzyme derivative containing 4-azido-L-phenylalanine (AzF) at position 284 of β' and position 106 of β, followed by azide-specific Staudinger ligation (Saxon and Bertozzi, 2000) to incorporate the fluorescent probes Cy3B and Alexa647 at position 284 of β' and position 106 of β, as follows (Fig. 4A). Single colonies of E. coli strain BL21(DE3) (EMD Millipore) co-transformed with plasmid pVS10-rpoB106am;rpoC284am [constructed by use of site-directed mutagenesis (QuikChange Site-Directed Mutagenesis Kit; Agilent) to replace rpoB codon 106 and rpoC codon 284 by amber codons in plasmid pVS10 (Belogurov et al., 2007)], plasmid pRSFduet-sigma (Hudson et al., 2009), and plasmid pEVOL-pAzF (Chin et al., 2002) were used to inoculate 20 ml LB broth (Sambrook and Russell, 2001) containing 100 μg/ml ampicillin, 50 μg/ml kanamycin, and 35 μg/ml chloramphenicol, and cultures were incubated 16 h at 37°C with shaking. Culture aliquots (2×10 ml) were used to inoculate LB broth (2×1 L; Sambrook and Russell, 2001) containing 2 mM AzF (Chem-Impex International), 100 μg/ml ampicillin, 50 μg/ml kanamycin, and 35 μg/ml chloramphenicol, cultures were incubated with shaking until OD600 = 0.6; L-arabinose was added to 0.2% and IPTG was added to 1 mM, and cultures were further incubated 16 h at 16°C with shaking. Cells were harvested by centrifugation (4,000 × g; 20 min at 4°C), re-suspended in 20 ml buffer A (10 mM Tris-HCl, pH 7.9, 200 mM NaCl, and 5% glycerol), and lysed using an EmulsiFlex-C5 cell disrupter (Avestin). The lysate was cleared by centrifugation (20,000 × g; 30 min at 4°C), precipitated with polyethylenimine (Sigma-Aldrich) as in Niu et al., 1996, and precipitated with ammonium sulfate as in Niu et al., 1996. The sample was dissolved in 30 ml buffer A and loaded onto a 5 ml column of Ni-NTA-agarose (Qiagen) pre-equilibrated in buffer A, and the column was washed with 50 ml buffer A containing 10 mM imidazole and eluted with 25 ml buffer A containing 200 mM imidazole. The sample was fUrther purified by anion-exchange chromatography on Mono Q (GE Healthcare; 160 ml linear gradient of 300-500 mM NaCl in 10 mM Tris-HCl, pH 7.9, 0.1 mM EDTA, and 5% glycerol; flow rate = 2 ml/min). Fractions containing AzF-derivatized hexahistidine-tagged E. coli RNAP σ70 holoenzyme were pooled, concentrated to ~1 mg/ml using 30 kDa MWCO Amicon Ultra-15 centrifugal ultrafilters (EMD Millipore), and stored in aliquots at −80°C. A reaction mixture containing 10 μM AzF-derivatized hexahistidine-tagged E. coli RNAP σ70 holoenzyme, 100 μM Alexa647-pentanoyl-ethylenediaminyl-phosphine (prepared as in Chakraborty et al., 2010, 2015), and 100 μM Cy3B-carboyl-ethylenediaminyl-phosphine (prepared as in Chakraborty et al., 2010, 2015) in 1 ml buffer B (50 mM Tris-HCl, pH 7.9, 100 mM KCl, 5% glycerol, and 2% dimethylformamide) was incubated 1 h at 15°C, incubated 16 h on ice, and subjected to 5 cycles of buffer exchange (dilution with 5 ml buffer B, followed by concentration to 0.5 ml) using 30 kDa MWCO Amicon Ultra-15 centrifugal ultrafilters (EMD Millipore). The sample was further purified by gel-filtration chromatography on HiLoad 16/60 Superdex 200 prep grade (GE Healthcare) pre-equilibrated in buffer C (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 5 mM MgCl2, 1 mM β-mercaptoethanol, and 5% glycerol) and eluted in buffer C. Fractions containing fluorescent-probe-labelled hexahistidine-tagged E. coli RNAP σ70 holoenzyme were pooled, concentrated to 1 mg/ml in buffer C using 30 kDa MWCO Amicon Ultra-15 centrifugal ultrafilters (EMD Millipore) and stored in aliquots at −80°C.
For experiments in Fig. 4G, fluorescent-probe-labelled, hexahistidine-tagged, FLAG-tagged E. coli RNAP σ70 holoenzyme was prepared using unnatural-amino-acid mutagenesis (Chin et al., 2002) of genes encoding RNAP β' and β subunits to incorporate azidophenylalanine at position 284 of β' and position 106 of β, azide-specific Staudinger ligation (Saxon and Bertozzi, 2000) to incorporate Cy3B at position 284 of the resulting β' derivative and Alexa647 at position 106 of the resulting β derivative, and in vitro reconstitution of RNAP (Tang et al., 1995) from the resulting β' and β derivatives, RNAP α and ω subunits, and σ70, as in Chakraborty et al., 2010, 2012, 2015.
Efficiencies of incorporation of fluorescent probes were determined from UV/Vis-absorbance measurements and were calculated as:
where A280 is the measured absorbance at 280 nm, ACy3B,559 is the measured absorbance at the long-wavelength absorbance maximum of Cy3B (559 nm), AAlexa,652 is the measured absorbance at the long-wavelength absorbance maximum of Alexa647 (652 nm), εP,280 is the molar extinction coefficient of RNAP σ70 holoenzyme at 280 nm (240,000 M−1 cm−1), εCy3B,280 is the molar extinction coefficient of Cy3B at 280 nm (7,350 M−1 cm−1 ), εAlexa,280 is the molar extinction coefficient of Alexa647 at 280 nm (10,400 M−1 cm−1), and εCy3B,559 is the extinction coefficient of Cy3B at its long-wavelength absorbance maximum (130,000 M−1 cm−1), and εAlexa,652 is the extinction coefficient of Alexa647 at its long-wavelength absorbance maximum (245,000 M−1 cm−1). Labelling efficiencies were ~90% for Cy3B and ~70% for Alexa647.
Specificities of incorporation of fluorescent probes were determined from the observed labelling efficiencies of (i) the labelling reaction with the AzF-derivatized hexahistidine-tagged E. coli RNAP σ70 holoenzyme and (ii) a control labelling reaction with non-AzF-derivatized hexahistidine-tagged E. coli RNAP σ70 holoenzyme, and were calculated as:
where AzF-P is AzF-derivatized hexahistidine-tagged E. coli RNAP σ70 holoenzyme, and P is non-AzF-derivatized hexahistidine-tagged E. coli RNAP σ70 holoenzyme. Labelling specificities were >90%.
S. aureus RNAP σA holoenzyme, RNAP core enzyme, and σA
S. aureus RNAP core enzyme was prepared by co-expression of genes for S. aureus RNAP β' subunit, RNAP β subunit, N-terminally decahistidine-tagged RNAP α subunit, and RNAP ω subunit in E. coli, followed by cell lysis, polyethylenimine precipitation, ammonium sulfate precipitation, immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen), and cation-exchange chromatography on HiPrep Heparin (GE Healthcare), as in Maffioli et al., 2017.
S. aureus σA was prepared by expression of a gene for N-terminally hexahistidine-tagged S. aureus σA in E. coli, followed by cell lysis, immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen), and gel-filtration chromatography on Superdex 200 (GE Healthcare), as in Maffioli et al., 2017. S. aureus RNAP σA holoenzyme was prepared by combination of S. aureus RNAP core enzyme and S. aureus σA, as in Maffioli et al., 2017.
T. thermophilus RNAP σA holoenzyme, RNAP core enzyme, and σA
T. thermophilus RNAP core enzyme was prepared from T. thermophilus strain H8 (DSM 579; DSMZ), using cell lysis, polyethylenimine precipitation, ammonium sulfate precipitation, cation-exchange chromatography on SP Sepharose FF (GE Healthcare), anion-exchange chromatography on Mono Q 10/100 GL (GE Healthcare), and cation-exchange chromatography on Mono S HR (GE Healthcare), as in Zhang et al., 2012 and Maffioli et al., 2017.
T. thermophilus σA was prepared by co-expression of a gene for N-terminally hexahistidine-tagged T. thermophilus σA in E. coli, followed by cell lysis, immobilized-metal-ion affinity chromatography on Ni-NTA agarose (Qiagen) and anion-exchange chromatography on Mono Q (GE Healthcare), as in Zhang et al., 2012.
T. thermophilus RNAP σA holoenzyme was prepared by combining T. thermophilus RNAP core enzyme and T. thermophilus σA, followed by size-exclusion chromatography on Superdex 200 (GE Healthcare), as in Zhang et al., 2014.
RNAP-inhibitory activities
Fluorescence-detected RNAP-inhibition assays with the profluorescent substrate γ-[2′-(2-benzothiazoyl)-60-hydroxybenzothiazole]-ATP (BBT-ATP; Jena Bioscience) were perfomed as in Feng et al., 2015, using 75 nM RNAP holoenzyme [Mtb RNAP σA holoenzyme (prepared by pre-incubating 75 nM Mtb RNAP core enzyme or core enzyme derivative and 300 nM Mtb σA in transcription buffer 10 min at 0°C), S. aureus RNAP σA holoenzyme, E. coli RNAP σ70 holoenzyme, or T. thermophilus RNAP σA holoenzyme] and 20 nM DNA fragment containing bacteriophage T5 N25 promoter (prepared as in Zhang et al., 2014). The transcription buffer for E. coli RNAP σ70 holoenzyme was 50 mM Tris-HCl (pH 8.0), 100 mM KCl, 10 mM MgCl2, 1 mM DTT, 10 mg/ml bovine serum albumin, and 5.5% glycerol. The transcription buffer for Mtb RNAP σA holoenzyme, S. aureus RNAP σA holoenzyme, and T. thermophilus RNAP σA holoenzyme was 40 mM Tris-HCl (pH 8.0), 75 mM NaCl, 5 mM MgCl2, 2.5 mM DTT, 5% DMSO, and 12.5% glycerol.
Radichemical RNAP-inhibition assays with HeLa nuclear extract (human RNAP I/II/III) were performed as in Degen et al., 2014.
Half-maximal inhibitory concentrations (IC50s) were calculated by non-linear regression in SigmaPlot (Systat Software).
Lpm-resistant mutants
Lpm-resistant mutants were isolated using procedures analogous to those used for isolation of Myx-resistant mutants in Mukhopadhyay et al., 2008. Mutagenesis reactions were performed using the QuikChange Site-Directed Mutagenesis Kit (Agilent) with E. coli rpoC plasmid pRL663 (Wang et al., 1995) and oligodeoxyribonucleotide forward and reverse primers corresponding to rpoC codons 1-20, 67-68, 77-81, 93-100, 245-256, 259-265, 325-355, 378-382, 393-403, 425-433, 466-481, 1319-1327, and 1347-1360, or with E. coli rpoB plasmid pRL706 (Severinov et al., 1997) and oligodeoxyribonucleotide forward and reverse primers corresponding to rpoB codons 854-857, 890-899, 914-922, and 1246-1342 (primers at 75 nM; all other components at concentrations as specified by the manufacturer). Mutagenized plasmid DNA was introduced by transformation into E. coli XL1-Blue (Agilent). Transformants (~104 cells) were applied to LB-agar (Sambrook and Russell, 2001) plates containing 200 μg/ml ampicillin, plates were incubated 16 h at 37°C, and plasmid DNA was prepared from the pooled resulting colonies. The resulting pooled mutagenized plasmid DNA was introduced by transformation into uptake-proficient, efflux-deficient E. coli strain D21f2tolC (Fralick and Burns-Keliher, 1994). Transformants (~103 cells) were applied to LB-agar plates containing 5 μg/ml Lpm (the minimal concentration that prevents colony formation by wild-type transformants), 200 μg/ml ampicillin, and 1 mM IPTG, and plates were incubated 24-48 h at 37°C. Lpm-resistant mutants were identified by the ability to form colonies on this medium, were confirmed by re-streaking on the same medium, and were demonstrated to contain plasmid-linked Lpm-resistant mutations by preparing plasmid DNA, transforming E. coli D21f2tolC with plasmid DNA, and plating transformants on the same medium. Nucleotide sequences of rpoB and rpoC were determined by Sanger sequencing (eight primers per gene).
Resistance and cross-resistance levels
Experiments in Fig. 2C and S6B-C assessing resistance and cross-resistance levels of Lpm-resistant E. coli D21f2tolC pRL706 and E. coli D21f2tolC pRL663 derivatives (preceding section) and Myx/Cor/Rip-resistant E. coli D21f2tolC pRL706 and E. coli D21f2tolC pRL663 derivatives (Mukhopadhyay et al., 2008) were performed using spiral gradient endpoint assays (Wallace and Corkill, 1989; Paton et al., 1990; Schalkowsky, 1994) on 150 mm × 4 mm exponential-gradient plates containing LB agar (Sambrook and Russell, 2001), 0.05–50 μg/ml test compound (Lpm, Rif, Myx, Cor, or Rip), 200 mg/ml ampicillin, and 1 mM IPTG. Test compounds were applied to plates using an Autoplate 4000 spiral plater (Spiral Biotech). Single colonies of transformants of E. coli D21f2tolC (Fralick and Burns-Keliher, 1994) were inoculated into 5 ml LB broth (Sambrook and Russell, 2001) containing 200 μg/ml ampicillin and incubated at 37°C with shaking until OD600 = 0.4-0.6, IPTG was added to 1 mM, and cultures were incubated 1 h at 37°C with shaking. Diluted aliquots (~1×109 cfu/ml for Fig. 2C and ~1×108 cfu/ml for Fig. S6B) were swabbed radially onto plates, and plates were incubated 16 h at 37°C. For each culture, the streak length was measured using a clear plastic template (Spiral Biotech), the test-compound concentration at the streak endpoint was calculated using the program SGE: Spiral Gradient Endpoint (v1.3; Spiral Biotech), and the minimum inhibitory concentration (MIC) was defined as the test-compound concentration at the streak endpoint.
Experiments in Fig. 2D assessing resistance and cross-resistance levels of chromosomal Lpm-resistant mutants [mutations transferred from pRL706 or pRL663 derivatives of preceding section to chromosome of E. coli D21f2tolC (Fralick and Bums-Keliher, 1994) by λ-Red-mediated recombineering (procedures essentially as in Datsenko and Wanner, 2000, but using transformation rather than electroporation)] were performed using broth microdilution assays (Clinical and Laboratory Standards Institute, 2009). Single colonies were inoculated into 5 ml LB broth (Sambrook and Russell, 2001) and incubated at 37°C with shaking until OD600 = 0.4-0.8. Diluted aliquots (~5×104 cells) in 97 μl LB broth were dispensed into wells of a 96-well plate, and were supplemented with 3 μl methanol or 3 μl of a 2-fold dilution series of Lpm (MICwild-type = 1.56 μg/ml), Rif (MICwild-type = 0.20 μg/ml), CBR (MICwild-type = 6.25 μg/ml), Sal (MICwild-type = 0.049 μg/ml), Stl (MICwild-type = 3.13 μg/ml), or Myx (MICwild-type = 0.20 μg/ml), in methanol (final concentrations = 0 and 0.006-50 μg/ml), or diluted aliquots (~1×105 cells) in 50 μl LB broth were supplemented with 50 μl LB broth or 50 μl of a 2-fold dilution series of GE (MICwild-type = 500 μg/ml) or PUM (MICwild-type = 400 μg/ml) in LB broth (final concentrations = 0 and 25-2000 μg/ml). Plates were incubated 16 h at 37°C. The MIC was defined as the lowest tested concentration that inhibited bacterial growth by ≥90%.
Experiments in Fig. 2E assessing Lpm-cross-resistance levels of Rif-, GE/PUM-, and Sal-resistant mutants were performed as described for experiments in Fig. 2D, but analyzing a panel of E. coli D21f2tolC derivatives (Degen et al., 2014; Zhang et al., 2014; Maffioli et al., 2017) comprising 4 chromosomal Rif-resistant mutants, 3 chromosomal GE/PUM-resistant mutants, and 5 chromosomal Sal-resistant mutants.
Experiments in Fig. 2E assessing Lpm-cross-resistance levels of CBR- and Stl-resistant mutants were performed essentially as described for experiments in Fig. 2D, but analyzing a panel of E. coli D21f2tolC pRL706 and E. coli D21f2tolC pRL663 derivatives (Tuske et al., 2005; Feng et al., 2015) comprising 5 CBR-resistant mutants and 5 Stl-resistant mutants. Single colonies were inoculated into 5 ml LB broth (Sambrook and Russell, 2001) containing 200 μg/ml ampicillin and incubated at 37°C with shaking until OD600 = 0.4-0.6, IPTG was added to 1 mM, and cultures were incubated 1 h at 37°C with shaking. Diluted aliquots (~5×104 cells) in 97 μl LB broth containing 200 μg/ml ampicillin and 1 mM IPTG were dispensed into wells of a 96-well plate, and were supplemented with 3 μl methanol or 3 μl of a 2-fold dilution series of CBR or Stl in methanol and further processed as described for experiments in Fig. 2D.
Checkerboard interaction assays
Antibacterial activities of combinations of Lpm and Rif were assessed in checkerboard interaction assays (White et al., 1996; Meletiadis et al., 2010). Broth-microdilution assays (procedures as described in the preceding section for determination of resistance levels of chromosomal Lpm-resistant mutants) were performed in checkerboard format, using E. coli D21f2tolC (Fralick and Burns-Keliher, 1994) and using LB broth (Sambrook and Russell, 2001) containing all pairwise combinations of: (i) Lpm at 2.0x, 1.75x, 1.5x, 1.3125x, 1.25x, 1.125x, 0.9375x, 0.75x, 0.5625x, 0.5x, 0.375x, 0.25x, and 0.1875x MICLpm and (ii) Rif at 2.0x, 1.75x, 1.5x, 1.3125x, 1.25x, 1.125x, 0.9375x, 0.75x, 0.5625x, 0.5x, 0.375x, 0.25x, and 0.1875x MICRif. Fractional inhibitory concentrations (FICs), FIC indices (FICIs), and minimum and maximum FICIs (FICImin and FICImax) were calculated as in Meletiadis et al., 2010. FICImin ≤ 0.5 was deemed indicative of super-additivity (synergism), FICImin > 0.5 and FICImax ≤ 4.0 was deemed indicative of additivity, and FICImax > 4.0 was deemed indicative of sub-additivity (antagonism) (White et al., 1996; Meletiadis et al., 2010).
Cryo-EM structure determination (M. tuberculosis RNAP-Lpm): sample preparation
Lacey carbon grids (LC300-CU-100; Electron Microscopy Sciences) were glow-discharged for 30 s using a glow-discharge cleaning system (PELCO easiGlow; Ted Pella) and mounted in the sample chamber of an EM GP grid plunger (Leica) at 18°C and relative humidity = 95%. Grids were spotted with 3.5 μl 1 μM Mtb RNAP-Lpm and 50 μM Lpm in 20 mM Tris-HCl, pH 8.0, 75 mM NaCl, 5 mM MgCl2, 5 mM dithiothreitol, and 0.1% n-octyl-β-D-glucopyranoside [prepared by pre-equilibrating 150 μl samples containing components other than n-octyl-β-D-glucopyranoside (Biosynth) 30 min at 25 °C, and adding n-octyl-β-D-glucopyranoside immediately before spotting], incubated 10 s, blotted with filter paper (Whatman Grade 541; Sigma-Aldrich) for 2.3 s, flash-frozen by plunging in liquid ethane cooled with liquid nitrogen, and stored in liquid nitrogen.
Cryo-EM structure determination (M. tuberculosis RNAP-Lpm): data collection and data reduction
Data were collected at the National Resource for Automated Molecular Microscopy of the Simons Electron Microscopy Center using a 300 keV Titan Krios (FEI/ThermoFisher) electron microscope equipped with a K2 Summit direct electron detector (Gatan) operating in counting mode and a GIF Quantum imaging filter (Gatan) with slit width of 20 eV. Data were collected semi-automatically using the software package Leginon (Suloway et al., 2005), a nominal magnification of 130,000x, a calibrated pixel size of 1.061 Å, and a dose rate of 8 electrons/pixel/s. Movies were recorded at 200 ms/frame for 10 s (50 frames total), resulting in a total radiation dose of 72.05 electrons/Å2 per movie Defocus range was varied between 1.0 μm and 2.0 μm. A total of 2,458 micrographs were recorded from two grids over 2 days.
Data were processed as summarized in Fig. S1C-E. Data processing was performed on an Ubuntu 16.04 Linux GPU workstation (Titan Computers) containing four GeForce GTX 1080 Ti graphic cards (Nvidia; Kimanius et al., 2016). Frames in individual movies were aligned using MotionCor2 (Zheng et al., 2017), and the first 35 frames per movie were merged to calculate individual micrographs.
Contrast-transfer-function estimations were performed using Gctf (Zhang, 2016), yielding defocus-range estimates ranging from 0.4 μm to 4.0 μm for individual micrographs. Initial particle picking was performed on 25 selected micrographs from the first grid (8,609 particles) using the Xmipp routine of the software package Scipion v1.1 (de la Rosa-Trevín et al., 2016), and particles were used for two-dimensional class averaging in Relion v2.0.5 (Fernandez-Leiro and Scheres, 2017). Eight distinct two-dimensional classes were selected as templates for picking 452,912 particles from 1,236 selected micrographs from the first grid and 366,594 particles from 1,025 selected micrographs from the second grid, using the Autopick routine of Relion. Two- and three-dimensional classifications were performed on the 452,912 particles from the first grid and 366,594 particles from the second grid, using Relion and using a 60 Å low-pass-filtered map calculated from the crystal structure of Mtb RPo (PDB: 5UH5; Lin et al., 2017; protein residues only) as the starting reference model for three-dimensional classification. Following identification and removal of heterogeneous particles in the two- and three-dimensional classifications, independent but similar density maps were obtained from 126,977 particles from the first grid and 97,412 particles from the second grid. Following merging of the 224,389 particles from the first and second grids, three-dimensional auto-refinement using Relion, and local angular sampling using Relion, a final density map was obtained from a subset of 68,895 particles. Gold-standard Fourier-shell-correlation analysis (FSC; Henderson et al., 2012) indicated a mean map resolution of 3.52 Å, and ResMap (Kucukelbir et al., 2014) indicated a median map resolution of 3.5 Å (Fig. S1F-G; Table 1). The initial atomic model for protein residues of Mtb RNAP-Lpm was built by manual rigid-body fitting of RNAP β', RNAP β, RNAP aI’, RNAP αII, RNAP ω, and σ segments from the crystal structure of Mtb RPo (PDB: 5UH5; Lin et al., 2017; protein residues only) into the cryo-EM density map in Coot (Emsley et al., 2010), followed by adjustment of backbone and sidechain conformations in Coot. For σR4 (residues 464-528), density was weak, suggesting high segmental flexibility; σR4 was fitted as a rigid-body segment and was not further adjusted. For the RNAP β' N and C-termini (residues 1-2 and 1282-1316), the central part of the RNAP β' trigger loop (residues 1014-1022), the RNAP β N and C-termini (residues 1-27 and 1145-1172), the RNAP αI N-terminus and C-terminal domain (residues 1-2 and 227-347), the RNAP αII N-terminus and C-terminal domain (residues 1-2 and 233-347), the RNAP ω N-terminus (residues 1-27), σR1.1 (residues 1-224), and a loop and short extended segment of the σR3-σR4 linker (residues 426-433 and 443-445), density was absent, suggesting very high segmental flexibility; these segments were not fitted. The initial atomic model for Lpm atoms of Mtb RNAP-Lpm was built by manual rigid-body fitting of a crystal structure of Lpm (CCDC 114782; Ihle et al., 2000) into the cryo-EM density map using Coot, followed by torsion-angle adjustments using Coot. Iterative cycles were performed of real-space model building in Coot followed by reciprocal-space fitting to structure-factor amplitudes and phases calculated from the cryo-EM density map in Phenix (Adams et al., 2010). The final atomic model, with map-to-model correlation of 0.83, was deposited in the Electron Microscopy Data Bank (EMDB) and Protein Data Bank (PDB) with accession codes EMDB: 4230 and PDB: 6FBV (Table 1).
Table 1.
Cryo-EM data-collection and data-analysis statistics
| structure | Mtb RNAP-Lpm | |
| EMDB and PDB codes | EMDB: 4230 PDB: 6FBV |
|
| data collection | ||
| number of grids used | 2 | |
| grid type | lacey carbon | |
| microscope/detector | Titan Krios/Gatan K2 | |
| voltage (kV) | 300 kV | |
| magnification | 130,000x | |
| recording mode | counting mode | |
| dose rate (e-/s) | 8 | |
| pixel size (Å/pix) | 1.061 | |
| total dose (e-/Å2) | 72.05 | |
| number of frames/movie | 50 | |
| total exposure time (s) | 10 | |
| defocus range (μm) | 1.0 - 2.0 | |
| data processing | grid I | grid II |
| number of micrographs | 1,238 | 1,029 |
| number of particles picked | 452,912 | 366,594 |
| number of particles after 2D/3D combined dataset | 126,977 | 97,412 |
| particles used for final map calculation | 68,895 | |
| map resolution (FSC 0.143; Å) | 3.52 | |
| map resolution (ResMap median; Å) | 3.5 | |
| structure/map fitting | ||
| resolution (Å) | 3.5 | |
| map sharpening B factor (Å2) | −109 | |
| experimental map / model correlation | 0.83 | |
| experimental map / 2Fo-Fc correlation | 0.98 | |
| average B factor (Å2) | 95.3 | |
| total number of atoms | 25,156 | |
| clash score | 13 | |
| Ramachandran plot | ||
| favored (%) | 94.5 | |
| outliers (%) | 0.03 | |
| root-mean-square (RMS) deviations | ||
| bond length (Å) | 0.006 | |
| bond angle (°) | 1.027 |
Structure factors were from cryoEM map, and 2Fo-Fc map was from final model.
Crystal structure determination (T. thermophilus RNAP core enzyme): sample preparation
Robotic crystallization trials were performed for T. thermophilus RNAP core enzyme using a Gryphon liquid handling system (Art Robbins Instruments), commercial screening solutions (Emerald Biosystems, Hampton Research, and Qiagen), and the sitting-drop vapor-diffusion technique (drop: 0.2 μl RNAP plus 0.2 μl screening solution; reservoir: 60 μl screening solution; 22°C). 900 conditions were screened. Rod-like crystals appeared under the identified crystallization conditions [0.1 M Hepes-NaOH, pH 7.5, 20 mM MgCl2, and 22% poly(acrylic acid sodium salt), 5,100 Da (Hampton Research); 22°C] within two weeks. Crystals were transferred from the sitting drop to a reservoir solution containing 20% (v/v) ethylene glycol (Sigma-Aldrich) and flash-cooled by immersing in liquid nitrogen.
Crystal structure determination (T. thermophilus core enzyme): data collection and data reduction
Diffraction data and selenium single-wavelength anomalous dispersion data were collected from cryo-cooled crystals at Argonne Photon Source beamline 19ID-D. Data were processed using HKL2000 (Otwinowski and Minor, 1997). The resolution cut-off criteria were I/σ > 1.1 and Rmerge < 1.
The structure of T. thermophilus RNAP core was solved by molecular replacement with Molrep (Vagin and Teplyakov, 1997) using PDB: 4GZY (Weixlbaumer et al., 2013) as the search model. One RNAP molecule was present in the asymmetric unit. Early-stage refinement included rigid-body refinement of RNAP, followed by rigid-body refinement of RNAP subunits, followed by rigid-body refinement of 44 RNAP domains (methods as in Zhang et al., 2012). Cycles of iterative model building with Coot (Emsley et al., 2010) and refinement with Phenix (Adams et al., 2010) were performed. Improvement of the coordinate model resulted from improvement of phasing. The final model was generated by XYZ-coordinate refinement with secondary-structure restraints in Phenix, followed by group B-factor and individual B-factor refinement in Phenix. The final model, refined to Rwork and Rfree of 0.22 and 0.27, respectively, was deposited in the PDB with accession code PDB: 6ASG (Table 2).
Single-molecule FRET: sample preparation
Observation wells were prepared essentially as described (Duchi et al., 2016, 2017; Fig. 4B): Borosilicate glass coverslips (#1.5; Menzel/ThermoFisher) were incubated in 40 ml acetone 5 min at 22°C, incubated in 40 ml 2% (v/v) Vectabond aminosilane reagent (Vector Labs) in acetone 5 min at 22°C, washed with 100 ml water at 22°C, dried under nitrogen, and bonded to CultureWell 6 mm silicone gaskets (GBL103280; Grace Bio-Labs), yielding 30 μl wells containing aminosilane-functionalized glass floors. Aliquots (20 μl ) of 30 mM methoxy-PEG succinimidyl valerate (mPEG-SVA, MW 5,000; Laysan Bio) and 0.75 mM biotinyl-PEG succinimidyl valerate (Biotin-PEG-SVA, MW 5,000; Laysan Bio) in 50 mM MOPS-NaOH, pH 7.5, were added to wells and incubated 90 min at 22°C, supernatants were removed, and wells were washed with 5×200 μl PBS (0.01 M sodium phosphate, pH. 7.4, 137 mM NaCl, and 2.7 mM KCl), yielding wells with biotin-PEG/mPEG-functionalized borosilicate glass floors. Aliquots (30 μl) of 10 μM NeutrAvidin (ThermoFisher) in 0.5xPBS then were added to wells and incubated 10 min at 22°C, supernatants were removed, and wells were washed with 3×100 μl PBS, yielding wells with NeutrAvidin-biotin-PEG/PEG-functionalized glass floors. Aliquots (30 μl) of 40 nM biotinylated anti-hexahistidine monoclonal antibody (Penta-His Biotin Conjugate; Qiagen) in buffer KG7 (40 mM Hepes-NaOH, pH 7.0, 100 mM potassium glutamate, 10 mM MgCl2, 1 mM dithiothreitol, 100 μg/ml bovine serum albumin, and 5% glycerol) then were added to wells and incubated 10 min at 22°C, supernatants were removed, and wells were washed with 3×100 μl KG7, yielding wells with (biotinylated anti-hexahistidine monoclonal antibody)-NeutrAvidin-biotin-PEG/mPEG-functionalized glass floors. For experiments in Fig. 4C-F, fluorescent-probe-labelled, hexahistidine-tagged E. coli RNAP σ70 holoenzyme was immobilized in observation wells containing (biotinylated anti-hexahistidine monoclonal antibody)-Neutravidin-biotin-PEG/mPEG-functionalized glass floors (Fig. 4B) as follows: Aliquots (30 μl) of 0.1 nM fluorescent-probe-labelled. hexahistidine-tagged E. coli RNAP σ70 holoenzyme and 0 or 20 μM (2.5×IC50) Lpm in KG7 (prepared by pre-equilibrating 50 nM fluorescent-probe-labelled. hexahistidine-tagged E. coli RNAP σ70 holoenzyme and 0 or 20 μM Lpm in KG7 10 min at 37°C, and then diluting 1:500 with 0 or 20 uM Lpm in KG7 at 37°C) were added and incubated 2-4 min at 22°C, supernatants were removed, wells were washed with 2×30 μl KG7, and 30 μl KG7 containing 2 mM Trolox (Sigma-Aldrich) and an oxygen scavenging system [12.5 μM glucose oxidase (Sigma-Aldrich), 16 nM catalase (C30; Sigma-Aldrich), and 8 mM D-glucose] at 22°C was added. Immobilization densities typically were ~30 molecules per 10 μm × 12 μm field of view. Immobilization specificities typically were >98% (assessed in control experiments omitting biotinylated anti-hexahistidine monoclonal antibody).
For experiments in Fig. 4G, fluorescent-probe-labelled, hexahistidine-tagged E. coli RNAP σ70 holoenzyme was immobilized in observation wells containing (biotinylated anti-hexahistidine monoclonal antibody)-Neutravidin-biotin-PEG/mPEG-functionalized glass floors in the absence of Lpm as described above, and 3 μl 200 μM Lpm in the same buffer was added during data acquisition [Lpm final concentration = 20 μM (2.5×IC50)].
Single-molecule FRET: data collection and data analysis
FRET experiments were performed on an objective-type total-internal-reflection (TIRF) microscope (Holden et al., 2010; Duchi et al., 2016, 2017). Light from a green laser (532 nm; Samba; Cobolt) and a red laser (635 nm; CUBE 635-30E; Coherent) was combined using a dichroic mirror, coupled into a fiberoptic cable, focused onto the rear focal plane of a 100x oil-immersion objective (numerical aperture 1.4; Olympus), and displaced off the optical axis such that the incident angle at the oil-glass interface of a stage-mounted observation chamber was greater than the critical angle, thereby creating an exponentially decaying evanescent wave. Alternating-laser excitation (ALEX; Kapanidis et al., 2004) was implemented by directly modulating the two lasers using an acousto-optical modulator (1205C; Isomet). Fluorescence emission was collected from the objective, separated from excitation light by a dichroic mirror (545 nm/650 nm; Semrock) and emission filters (545 nm LP, Chroma; and 633/25 nm notch filter, Semrock), focussed on a slit to crop the image, and then spectrally separated using a dichroic mirror (630 nm DRLP; Omega) into donor and emission channels focused side-by-side onto an electron-multiplying charge-coupled device camera (EMCCD; iXon 897; Andor Technology). A motorized x/y-scanning stage with continuous reflective-interface feedback focus (MS-2000; ASI) was used to control the sample position relative to the objective.
For experiments in Fig. 4C-F, laser powers were 3.5 mW (532 nm laser) and 0.7 mW (635 nm laser), and data were collected for 20 s using a frame rate of 1 frame per 20 ms. For experiments in Fig. 4G, laser powers were 0.5 mW (532 nm laser) and 0.15 mW (635 nm laser), and data were collected for 50 s using a frame rate of 200 ms.
Fluorescence emission intensities in donor (green) and acceptor (red) emission channels were detected using the peak-finding algorithm of the MATLAB (MathWorks) software package TwoTone-ALEX (Holden et al., 2010), as in Holden et al., 2010. Peaks detected in both emission channels (i.e., peaks for molecules containing both donor and acceptor probes) and meeting ellipticity and distance-to-nearest-neighbor thresholds (i.e., ellipticity ≤ 0.6 and distance-to-nearest-neighbor ≥ 6 pixels) were fitted with two-dimensional Gaussian functions to extract background-corrected intensity-vs.-time trajectories for donor emission intensity upon donor excitation (IDD), acceptor emission intensity upon donor excitation (IDA), and acceptor emission intensity upon acceptor excitation (IAA) (Fig. 4C, top), as described (Holden et al., 2010). Intensity-vs.-time trajectories were curated to exclude trajectories exhibiting IDD <300 or >2,000 counts or IAA <200 or >2,000 counts, trajectories exhibiting multiple-step donor or acceptor photobleaching, trajectories exhibiting donor or acceptor photobleaching in frames 1-50, and trajectories exhibiting donor or acceptor photoblinking, and to exclude portions of traces following donor or acceptor photobleaching.
Intensity-vs.-time trajectories were used to calculate trajectories of apparent donor-acceptor FRET efficiency (E*) and donor-acceptor stoichiometry (S) (Fig. 4C, bottom), as described (Kapanidis et al., 2004 and Lee et al., 2005):
E*-vs.-S plots were prepared, S values were used to distinguish species containing only donor, only acceptor, and both donor and acceptor, and E* histograms were prepared for species containing both donor and acceptor, as described (Kapanidis et al., 2004; Lee et al., 2005).
E*-vs. time trajectories that on visual inspection exhibited transitions between distinct E* states (dynamic E*-vs.-time trajectories; ~34%; N = 207) were analyzed globally to identify E* states by use of Hidden Markov Modelling (HMM) with an empirical Bayesian as implemented in the MATLAB (MathWorks) software package ebFRET (van de Meent et al., 2014), essentially as described (van de Meent et al., 2014; Duchi et al., 2016, 2017). E*-vs.-time trajectories were fitted to HMM models with two, three, four, five, or six distinct E* states; the mean scoring parameter L (lower bound per trajectory) was extracted for each model. and the model with three distinct E* states was found to best describe the data (Fig. 4D, right; L = 536, 538, 537, 537 and 537 for models with two, three, four, five, and six states). E* values from the three-state HMM model were extracted, plotted using Origin (OriginLab), and fitted to Gaussian distributions using Origin (Fig 4D, left, colored curves). The resulting histograms provide equilibrium population distributions of E* states and, for each E* state, define mean E* (Fig 4D, left, colored curves and inset).
Dwell times for E* states were extracted from E*-vs.-time trajectories exhibiting >3 transitions between E* states and were plotted as histograms in Origin. The resulting dwell-time histograms were fit with single-exponential functions, and mean dwell times were extracted (Fig. 4F).
E* values were corrected, and accurate donor-acceptor efficiencies (E) and donor-acceptor distances (R) were calculated, as follows (Lee et al., 2005).
where, C1 is "cross-talk" from leakage of donor emission into the acceptor-emission channel, C2 is "cross-talk" from direct excitation of the acceptor by the green laser, γ is the detection factor [1 in this work; determined as γ = ΔIAA/ΔIDD, where ΔIAA and ΔIDD are changes in IAA and IDD upon acceptor photobleaching (Ha et al., 1999)], E*DO is E* of the donor-only subpopulation, SAO is S of the acceptor-only subpopulation, and R0 is the Főrster parameter [60.1 Å in this work; calculated as: R0=9780(n−4κ2QDJ)1/6 Å, where n is the refractive index of the medium, κ2 is the orientation factor relating donor emission dipole and acceptor excitation dipole (approximated as 2/3, noting that all mean E values are <0.5; Wu and Brand, 1992), QD is the quantum yield of the donor in the absence of acceptor, and J is the overlap integral of donor emission spectrum and acceptor excitation spectrum].
RNAP clamp angles were determined from E values using the plot of RNAP clamp angle vs. E in Fig. 1 of Chakraborty et al., 2012.
QUANTITATION AND STATISTICAL ANALYSIS
Data for RNAP-inhibitory activities, resistance, and cross-resistance are means of at least two technical replicates
DATA AVAILABILITY
The cryo-EM map and atomic coordinates for the cryo-EM structure of Mtb RNAP-Lpm have been deposited in the EMDB and PDB with accession codes EMDB: 4230 and PDB: 6FBV. Atomic coordinates and structure factors for the crystal structure of T. thermophilus RNAP core enzyme have been deposited in the PDB with accession code PDB: 6ASG.
Supplementary Material
Highlights.
Cryo-EM structure of drug-target complex
Single-molecule spectroscopic analysis of effects of drug on target conformation
Resistance determinant of drug
Structure-activity relationships and structure-based design of drug analogs
ACKNOWLEDGEMENTS
This work was supported by a Rega Foundation award to K.D. and NIH grants GM041376, AI104660, and AI109713-01 to R.H.E. We thank W. Fenical, H. Irschik, R. Jansen, A. L. Sonenshein, and E. Steinbrecher for compounds; J. Mukhopadhyay, S. Rodrigue, and J. Swezey for strains and plasmids; APS at Argonne National Laboratory for beamline access; the Rutgers University cryo-EM facility and the National Resource for Automated Molecular Microscopy (supported by NIH grant GM103310 and Simons Foundation grant 349247) for cryo-EM access; and K. Callanan, B. Carragher, and C. Potter for discussion.
Footnotes
DECLARATION OF INTERESTS
R.H.E. has patent filings on the Lpm target (US8206898 and US8697354). The other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams P, Afonine P, Bunkóczi G, Chen V, Davis I, Echols N, Headd J, Hung L, Kapral G, Grosse-Kunstleve R, McCoy A, Moriarty N, Oeffner R, Read R, Richardson D, Richardson J, Terwilliger T, and Zwart P (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst, D 66, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnone A, Nasini G, and Cavalleri B (1987). Structure elucidation of the macrocyclic antibiotic lipiarmycin. J. Chem. Soc. Perkin Trans. I 6, 1353–1359. [Google Scholar]
- Artsimovitch I, Seddon J, and Sears P (2012). Fidaxomicin is an inhibitor of the initiation of bacterial RNA synthesis. Clin. Infect. Dis 55, S127–S131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babakhani F, Gomez A, Robert N, and Sears P (2011). Killing kinetics of fidaxomicin and its major metabolite, OP-1118, against Clostridium difficile. J. Med. Microbiol 60, 1213–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae B, Feklistov A, Lass-Napiorkowska A, Landick R, and Darst S (2015). Structure of a bacterial RNA polymerase holoenzyme open promoter complex. eLife 4, e08504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae B, Nayak D, Ray A, Mustaev A, Landick R, and Darst S (2015). CBR antimicrobials inhibit RNA polymerase via at least two bridge-helix cap-mediated effects on nucleotide addition. Proc. Natl. Acad. Sci. USA 112, E4178–E4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Rudra P, Prajapati R, Sengupta S, and Mukhopadhyay J (2014). Optimization of recombinant Mycobacterium tuberculosis RNA polymerase expression and purification. Tuberculosis 94, 397–404. [DOI] [PubMed] [Google Scholar]
- Belogurov G, Vassylyeva M, Svetlov V, Klyuyev S, Grishin N, Vassylyev D, and Artsimovitch I (2007). Structural basis for converting a general transcription factor into an operon-specific virulence regulator. Mol. Biosyst 26, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belogurov G, Vassylyeva M, Sevostyanova A, Appleman J, Xiang A, Lira R, Webber S, Klyuyev S, Nudler E, Artsimovitch I, and Vassylyev D (2009). Transcription inactivation through local refolding of the RNA polymerase structure. Nature 45, 332–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland A, Chang L, and Barford D (2017). The potential of cryo-electron microscopy for structure-based drug design. Essays Biochem. 61, 543–560. [DOI] [PubMed] [Google Scholar]
- Campbell E, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, and Darst S (2001). Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 104, 901–912. [DOI] [PubMed] [Google Scholar]
- Campbell E, Pavlova O, Zenkin N, Leon F, Irschik H, Jansen R, Severinov K, and Darst S (2005). Structural, functional, and genetic analysis of sorangicin inhibition of bacterial RNA polymerase. EMBO J. 24, 674–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Wang D, Ebright Y, and Ebright RH (2010). Azide-specific labeling of biomolecules by Staudinger-Bertozzi ligation: phosphine derivatives of fluorescent probes suitable for single-molecule fluorescence spectroscopy. Meths Enzymol. 472, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Wang D, Ebright Y, Korlann Y, Kortkhonjia E, Kim T, Chowdhury S, Wigneshweraraj S, Irschik H, Jansen R, Nixon BT, Knight J, Weiss S, and Ebright R (2012). Opening and closing of the bacterial RNA polymerase clamp. Science 337, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Mazumder A, Lin M, Hasemeyer A, Xu Q, Wang D, Ebright Y, and Ebright RH (2015). Site-specific incorporation of probes into RNA polymerase by unnatural-amino-acid mutagenesis and Staudinger-Bertozzi ligation. Meths. Mol. Biol 1276, 101–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Martin A, King D, Wang L, and Schultz P (2002). Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. USA 99, 11020–11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciciliato I, Corti E, Sarubbi E, Stefanelli S, Gastaldo L, Montanini N, Kurz M, Losi D, Marinelli F, and Selva E (2004). Antibiotics GE23077, novel inhibitors of bacterial RNA polymerase I. taxonomy, isolation and characterization. J. Antibiot 57, 210–217. [DOI] [PubMed] [Google Scholar]
- Coronelli C, White R, Lancini G, and Parenti F (1975). Lipiarmycin, a new antibiotic from Actinoplanes. II. Isolation, chemical, biological and biochemical characterization. J. Antibiot 28, 253–259. [DOI] [PubMed] [Google Scholar]
- Cramer P, Bushnell D, and Kornberg R (2001). Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science 292, 1863–1876. [DOI] [PubMed] [Google Scholar]
- Datsenko K, and Wanner B (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degen D, Feng Y, Zhang Y, Ebright KY, Ebright YW, Gigliotti M, Vahedian-Movahed H, Mandal S, Talaue M, Connell N, Arnold E, Fenical W, and Ebright RH (2014). Transcription inhibition by the depsipeptide antibiotic salinamide A. eLife 3, e02451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa-Trevín J, Quintana A, Del Cano L, Zaldívar A, Foche I, Gutiérrez J, Gómez-Blanco J, Burguet-Castell J, Cuenca-Alba J, Abrishami V, Vargas J, Otón J, Sharov G, Vilas J, Navas J, Conesa P, Kazemi M, Marabini R, Sorzano C, and Carazo J (2016). Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol 195, 93–99. [DOI] [PubMed] [Google Scholar]
- Duchi D, Bauer D, Fernandez L, Evans G, Robb N, Hwang L, Gryte K, Tomescu A, Zawadzki P, Morichaud Z, Brodolin K, and Kapanidis A (2016). RNA polymerase pausing during initial transcription. Mol. Cell 63, 939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchi D, Gryte K, Robb N, Morichaud Z, Sheppard C, Brodolin K, Wigneshweraraj S, and Kapanidis A (2017). Conformational heterogeneity and bubble dynamics in single bacterial transcription initiation complexes. Nucl. Acids Res, (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebright R (2005). RNA exit channel--target and method for inhibition of bacterial RNA polymerase. WIPO Patent Application WO/2005/001034.
- Ebright R, and Ebright Y (2012). Antibacterial agents: high-potency myxopyronin derivatives. WIPO Patent Application WO/2012/037508.
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Cryst. D 66, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feklistov A, Bae B, Hauver J, Lass-Napiorkowska A, Kalesse M, Glaus F, Altmann H, Heyduk T, Landick R, and Darst SA (2017). RNA polymerase motions during promoter melting. Science 356, 863–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Degen D, Wang X, Gigliotti M, Liu S, Zhang Y, Das D, Michalchuk T, Ebright Y, Talaue M, Connell N, and Ebright RH (2015). Structural basis of transcription inhibition by CBR hydroxamidines and CBR pyrazoles. Structure 23, 1470–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Zhang Y, and Ebright RH (2016). Structural basis of transcription activation. Science 352, 1330–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Leiro R, and Scheres S (2017). A pipeline approach to single-particle processing in RELION. Acta Cryst. D 73, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fralick J, and Burns-Keliher L (1994). Additive effect of tolC and rfa mutations on the hydrophobic barrier of the outer membrane of Escherichia coli K-12. J. Bacteriol 176, 6404–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T, Ting A, Liang J, Caldwell W, Deniz A, Chemla D, Schultz P, and Weiss S (1999). Single molecule fluorescence spectroscopy of enzyme conformational dynamics and cleavage mechanism. Proc. Natl. Acad. Sci. USA 96, 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein P, Kolb K, Windgassen T, Bellecourt M, Darst S, Mooney R, and Landick R (2014). RNA polymerase pausing and nascent-RNA structure formation are linked through clamp-domain movement. Nature Struct. Mol. Biol 21, 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R, Sali A, Baker M, Carragher B, and Devkota B (2012). Outcome of the first electron microscopy validation task force meeting. Structure 20, 2015–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochlowski J, Jackson M, Rasmussen R, Buko A, Clement J, Whittern D, and McAlpine J (1997). Production of brominated tiacumicin derivatives. J. Antibiot 50, 201–205. [PubMed] [Google Scholar]
- Holden S, Uphoff S, Hohlbein J, Yadin D, Le Reste L, Britton O, and Kapanidis A (2010). Defining the limits of single-molecule FRET resolution in TIRF microscopy. Biophys. J 99, 3102–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubin E, Fay A, Xu C, Bean J, Saecker R, Glickman M, Darst S, and Campbell E (2017). Structure and function of the mycobacterial transcription initiation complex with the essential regulator RbpA. eLife, e22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson B, Quispe J, Lara-González S, Kim Y, Berman H, Arnold E, Ebright R, and Lawson C (2009). Three-dimensional EM structure of an intact activator-dependent transcription initiation complex. Proc. Natl. Acad. Sci. U S A 106, 19830–19835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihle N, Guzei I, and Rheingold A (2000). Tiacumicin B acetone solvate. CSD Communication https://www.ccdc.cam.ac.uk/structures/Search?Ccdcid=114782.
- Jacques J, Rodrigue S, Brzezinski R, and Gaudreau L (2006). A recombinant Mycobacterium tuberculosis in vitro transcription system, FEMS Microbiol. Lett. 255, 140–147. [DOI] [PubMed] [Google Scholar]
- Jokerst R, Weeks J, Zehring W, and Greenleaf A (1989). Analysis of the gene encoding the largest subunit of RNA polymerase II in Drosophila. Mol. Gen. Genet 215, 266–275. [DOI] [PubMed] [Google Scholar]
- Kapanidis A, Lee N, Laurence T, Doose S, Margeat E, and Weiss S (2004). Fluorescence-aided molecule sorting: Analysis of structure and interactions by alternating-laser excitation of single molecules. Proc. Natl. Acad. Sci. USA 101, 8936–8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann E, Hattori H, Miyatake-Ondozabal H, and Gademann K (2015). Total synthesis of the glycosylated macrolide antibiotic fidaxomicin. Org. Lett 17, 3514–3517. [DOI] [PubMed] [Google Scholar]
- Kimanius D, Forsberg B, Scheres S, and Lindahl E (2016). Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5, e18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth F, and Tagare H (2014). Quantifying the local resolution of cryo-EM density maps. Nature Meths. 11, 63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurabachew M, Lu S, Krastel P, Schmitt E, Suresh B, Goh A, Knox J, Ma N, Jiricek J, Beer D, Cynamon M, Petersen F, Dartois V, Keller T, Dick T, and Sambandamurthy V (2008). Lipiarmycin targets RNA polymerase and has good activity against multidrug-resistant strains of Mycobacterium tuberculosis. J. Antimicrob. Chemother 62, 713–719. [DOI] [PubMed] [Google Scholar]
- Lancaster J, and Matthews S (2012). Fidaxomicin: the newest addition to the armamentarium against Clostridium difficile infections. Clin. Ther 34, 1–13. [DOI] [PubMed] [Google Scholar]
- Landick R (2001). RNA polymerase clamps down. Cell 105, 567–570. [DOI] [PubMed] [Google Scholar]
- Lane W, and Darst S (2010). Molecular evolution of multisubunit RNA polymerases: structural analysis. J. Mol. Biol 395, 686–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Kapanidis A, Wang Y, Michalet X, Mukhopadhyay J, Ebright R, and Weiss S (2005). Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys. J 88, 2939–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Mandal S, Degen D, Liu Y, Ebright YW, Li S, Feng Y, Zhang Y, Mandal S, Jiang Y, Liu S, Gigliotti M, Talaue M, Connell N, Das K, Arnold E, and Ebright RH (2017). Structural basis of Mycobacterium tuberculosis transcription and transcription inhibition. Mol Cell 66, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffioli S, Zhang Y, Degen D, Carzaniga T, Del Gatto G, Serina S, Monciardini P, Mazzetti C, Guglierame P, Candiani G, Chiriac AI, Facchetti G, Kaltofen P, Sahl H-G, Dehò G, Donadio S, and Ebright RH (2017). Antibacterial nucleoside-analog inhibitor of bacterial RNA polymerase: pseudouridimycin. Cell 169, 1240–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meletiadis J, Pournaras S, Roilides E, and Walsh TJ (2010). Defining fractional inhibitory concentration index cutoffs for additive interactions based on self-drug additive combinations, Monte Carlo simulation analysis, and in vitro-in vivo correlation data for antifungal drug combinations against Aspergillus fumigatus. Antimicrob. Agents Chemother. 54, 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino F, and Raunser S (2017). Electron cryo-microscopy as a tool for structure-based drug development. Angew. Chem. Int. Ed. Engl 56, 2846–2860. [DOI] [PubMed] [Google Scholar]
- Molodtsov V, Fleming P, Eyermann C, Ferguson A, Foulk M, McKinney D, Masse C, Buurman E, and Murakami K (2015). X-ray crystal structures of Escherichia coli RNA polymerase with switch region binding inhibitors enable rational design of squaramides with an improved fraction unbound to human plasma protein. J. Med. Chem 58, 3156–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay J, Das K, Ismail S, Koppstein D, Jang M, Hudson B, Sarafianos S, Tuske S, Patel J, Jansen R, Irschik H, Arnold E, and Ebright R (2008). The RNA polymerase "switch region" is a target for inhibitors. Cell 135, 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K (2013). The x-ray crystal structure of RNA polymerase σ70 holoenzyme. J. Biol. Chem 288, 9126–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NandyMazumdar M, Nedialkov Y, Svetlov D, Sevostyanova A, Belogurov G, and Artsimovitch I (2016). RNA polymerase gate loop guides the nontemplate DNA strand in transcription complexes. Proc. Natl. Acad. Sci. USA 113, 14994–14999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W, Kim Y, Tau G, Heyduk T, and Ebright R (1996). Transcription activation at Class II CAP-dependent promoters: two interactions between CAP and RNA polymerase. Cell 87, 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu S, Hu T, Li S, Xiao Y, Ma L, Zhang G, Zhang H, Yang X, Ju J, and Zhang C (2011). Characterization of a sugar-O-methyltransferase TiaS5 affords new tiacumicin analogues with improved antibacterial properties and reveals substrate promiscuity. ChemBioChem, 12, 1740–1748. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. Meths. Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Parenti F, Pagani H, and Beretta G (1975). Lipiarmycin, a new antibiotic from Actinoplanes. I. Description of the producer strain and fermentation studies. J. Antibiot 28, :247–252. [DOI] [PubMed] [Google Scholar]
- Paton J, Holt A, and Bywater M (1990). Measurement of MICs of antibacterial agents by spiral gradient endpoint compared with conventional dilution methods. Intl. J. Exp. Clin. Chemother 3, 31–38. [Google Scholar]
- Sambrook J, and Russell D (2001). Molecular Cloning: A Laboratory Manual (Cold Spring Harbor, NY, Cold Spring Harbor Laboratory; ). [Google Scholar]
- Saxon E, and Bertozzi C (2000). Cell surface engineering by a modified Staudinger reaction. Science 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- Ruff E, Record M, and Artsimovitch I (2015). Initial events in bacterial transcription initiation. Biomolecules 5, 1035–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalkowsky S (1994). Measures of susceptibility from a spiral gradient of drug concentrations. Adv. Exp. Med. Biol 349, 107–120. [DOI] [PubMed] [Google Scholar]
- Serra S, Malpezzi L, Bedeschi A, Fuganti C, and Fonte P (2017). Final demonstration of the co-identity of lipiarmycin A3 and tiacumicin B (fidaxomicin) through single crystal X-ray analysis. Antibiotics 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinov K, Mooney R, Darst SA, and Landick R (1997). Tethering of the large subunits of Escherichia coli RNA polymerase. J. Biol. Chem 272, 24137–24140. [DOI] [PubMed] [Google Scholar]
- Sonenshein A, and Alexander H (1979). Initiation of transcription in vitro inhibited by lipiarmycin. J. Mol. Biol 127, 55–72. [DOI] [PubMed] [Google Scholar]
- Sonenshein A, Alexander H, Rothstein D, and S., F. (1977). Lipiarmycin-resistant ribonucleic acid polymerase mutants of Bacillus subtilis. J. Bacteriol 132, 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Talaue M, Liu S, Degen D, Ebright RY, Sineva E, Chakraborty A, Druzhinin S, Chatterjee S, Mukhopadhyay J, Ebright YW, Zozula A, Shen J, Sengupta S, Niedfeldt R, Xin C, Kaneko T, Irschik H, Jansen R, Donadio S, Connell N, and Ebright RH (2011). New target for inhibition of bacterial RNA polymerase: "switch region." Curr. Opin. Microbiol 14, 532–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter C, and Carragher B (2005). Automated molecular microscopy: the new Leginon system. J. Struct. Biol 151, 41–60. [DOI] [PubMed] [Google Scholar]
- Sweetser D, Nonet M, and Young R (1987). Prokaryotic and eukaryotic RNA polymerases have homologous core subunits. Proc. Natl. Acad. Sci. USA 84, 1192–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talpaert M, Campagnari F, and Clerici L (1975). Lipiarmycin: an antibiotic inhibiting nucleic acid polymerases. Biochem. Biophys. Res. Commun 63, 328–334. [DOI] [PubMed] [Google Scholar]
- Tang H, Severinov K, Goldfarb A, Fenyo D, Chait B, and Ebright R (1994). Location, structure, and function of the target of a transcription activator protein. Genes Dev. 8, 3058–3067. [DOI] [PubMed] [Google Scholar]
- Tang H, Severinov K, Goldfarb A, and Ebright RH (1995). Rapid RNA polymerase genetics: one-day, no-column preparation of reconstituted recombinant Escherichia coli RNA polymerase. Proc. Natl. Acad. Sci. USA 92, 4902–4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temiakov D, Zenkin N, Vassylyeva M, Perederina A, Tahirov T, Kaihatsu K, Savkina M, Zorov S, Nikiforov V, Igarashi N, Matsugaki N, Wakatsuki S, Severinov K, and Vassylyev D (2005). Structural basis of transcription inhibition by antibiotic streptolydigin. Mol. Cell 19, 655–666. [DOI] [PubMed] [Google Scholar]
- Theriault R, Karwowski J, Jackson M, Girolami R, Sunga G, Vojtko C, and Coen L (1987). Tiacumicins, a novel complex of 18-membered macrolide antibiotics. I. Taxonomy, fermentation and antibacterial activity. J. Antibiot 40, 567–574. [DOI] [PubMed] [Google Scholar]
- Tupin A, Gualtieri M, Leonetti J, and Brodolin K (2010). The transcription inhibitor lipiarmycin blocks DNA fitting into the RNA polymerase catalytic site. EMBO J. 29, 2527–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuske S, Sarafianos S, Wang X, Hudson B, Sineva E, Mukhopadhyay J, Birktoft J, Leroy O, Ismail S, Clark A, Dharia C, Napoli A, Laptenko O, Lee J, Borukhov S, Ebright R, and Arnold E (2005). Inhibition of bacterial RNA polymerase by streptolydigin: stabilization of a straight-bridge-helix active-center conformation. Cell 122, 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, and Teplyakov A (1997). MOLREP: An automated program for molecular replacement. J. Appl. Cryst 30, 1022–1025. [Google Scholar]
- van de Meent J, Bronson J, Wiggins C, and Gonzalez R (2014). Empirical Bayes methods enable advanced population-level analyses of single-molecule FRET experiments. Biophys. J 106, 1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassylyev D, Sekine S, Laptenko O, Lee J, Vassylyeva M, Borukhov S, and Yokoyama S (2002). Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 Å resolution. Nature 417, 712–719. [DOI] [PubMed] [Google Scholar]
- Venugopal A, and Johnson S (2012). Fidaxomicin: a novel macrocyclic antibiotic approved for treatment of Clostridium difficile. Clin. Infect Dis 54, 568–574. [DOI] [PubMed] [Google Scholar]
- Wallace A, and Corkill J (1989). Application of the spiral plating method to study antimicrobial actio. J. Microbiol. Meths 10, 303–310. [Google Scholar]
- Wang D, Meier T, Chan C, Feng G, Lee D, and Landick R (1995). Discontinuous movements of DNA and RNA in RNA polymerase accompany formation of a paused transcription complex. Cell 81, 341–350. [DOI] [PubMed] [Google Scholar]
- Weixlbaumer A, Leon K, Landick R, and Darst S (2013). Structural basis of transcriptional pausing in bacteria. Cell 152, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R, Burgess D, Manduru M, and Bosso J (1996). Comparison of three different in vitro methods of detecting synergy: time-kill, checkerboard, and E test. Antimicrob. Agents Chemother 40, 1914–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, and Brand L (1992). Orientation factor in steady-state and time-resolved resonance energy transfer measurements. Biochem. 31, 7939–7947. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Li S, Niu S, Ma L, Zhang G, Zhang H, Zhang G, Ju J, and Zhang C (2011). Characterization of tiacumicin B biosynthetic gene cluster affording diversified tiacumicin analogues and revealing a tailoring dihalogenase. J. Am. Chem. Soc 133, 1092–1105. [DOI] [PubMed] [Google Scholar]
- Zhang G, Campbell E, Minakhin L, Richter C, Severinov K, and Darst S (1999). Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 Å resolution. Cell 98, 811–824. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Degen D, Ho M, Sineva E, Ebright K, Ebright Y, Mekler V, Vahedian-Movahed H, Feng Y, Yin R, Tuske S, Irschik H, Jansen R, Maffioli S, Donadio S, Arnold E, and Ebright RH (2014). GE23077 binds to the RNA polymerase 'i' and 'i+1' sites and prevents the binding of initiating nucleotides. eLife 3, e02450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Feng Y, Chatterjee S, Tuske S, Ho M, Arnold E, and Ebright RH (2012). Structural basis of transcription initiation. Science 338, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Palovcak E, Armache J, Verba K, Cheng Y, and Agard D (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Meths. 14, 331–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, and Steitz T (2015). Crystal structures of the E. coli transcription initiation complexes with a complete bubble. Mol. Cell 58, 534–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM map and atomic coordinates for the cryo-EM structure of Mtb RNAP-Lpm have been deposited in the EMDB and PDB with accession codes EMDB: 4230 and PDB: 6FBV. Atomic coordinates and structure factors for the crystal structure of T. thermophilus RNAP core enzyme have been deposited in the PDB with accession code PDB: 6ASG.