

Cellular senescence, characterized by an irreversible cell-cycle arrest1 accompanied by a distinctive secretory phenotype2, can be induced through a variety of intracellular and extracellular factors. Senescent cells expressing the cell cycle inhibitory protein p16INK4A, have been found to actively drive naturally occurring age-related tissue deterioration3,4 and contribute to several aging-associated diseases, including atherosclerosis5 and osteoarthritis6. Various markers of senescence have been observed in patients suffering from neurodegenerative diseases7-9, however, a role for senescent cells in the etiology of these pathologies is unknown. Here we show a causal link between the accumulation of senescent cells and cognition-associated neuronal loss. We found that the MAPT P301S PS19 mouse model of tau-dependent neurodegenerative disease10 accumulates p16Ink4a-positive senescent astrocytes and microglia. Clearance of these cells as they arise using INK-ATTAC transgenic mice prevented gliosis, hyper-phosphorylation of both soluble and insoluble tau leading to neurofibrillary tangle (NFT) deposition, and degeneration of cortical and hippocampal neurons to preserved cognitive function. Lastly, pharmacological intervention with a first generation senolytic modulated tau aggregation. Collectively, these results demonstrate that senescent cells play a role in tau-mediated disease initiation and progression; suggesting that targeting senescent cells may provide a therapeutic avenue for treating these pathologies.
Senescent cells accumulate with aging and have been shown to contribute to tissue dysfunction11, although their role in neurodegenerative disease has remained elusive. To address this key open question, we selected the tau MAPT P301S PS19 (hereafter PS19) transgenic mouse line that expresses high levels of mutant human tau specifically in neurons under the regulation of the mouse prion promoter10. The model is characterized by gliosis, neurofibrillary tangle (NFT) deposition, neurodegeneration, and loss of cognitive function. Pathology typically initiates in the hippocampus and radiates outwards to the neocortex10. First, we performed RT-qPCR for p16Ink4a on isolated hippocampi and cortices from Wildtype and PS19 littermates. p16Ink4a expression was significantly increased beginning at 4 months of age in the hippocampus and at 6 months in the cortex (Fig. 1a), which precedes the onset of NFT deposition10. Importantly, increased p16Ink4a expression correlated with expression of widely established senescence markers (Extended Data Fig. 1), indicating that senescent cells accumulate at sites of pathology in the PS19 model.
Figure 1. Senescent astrocytes and microglia that accumulate in brains of P301S (MAPT) PS19 mice can be removed using the INK-ATTAC transgene.
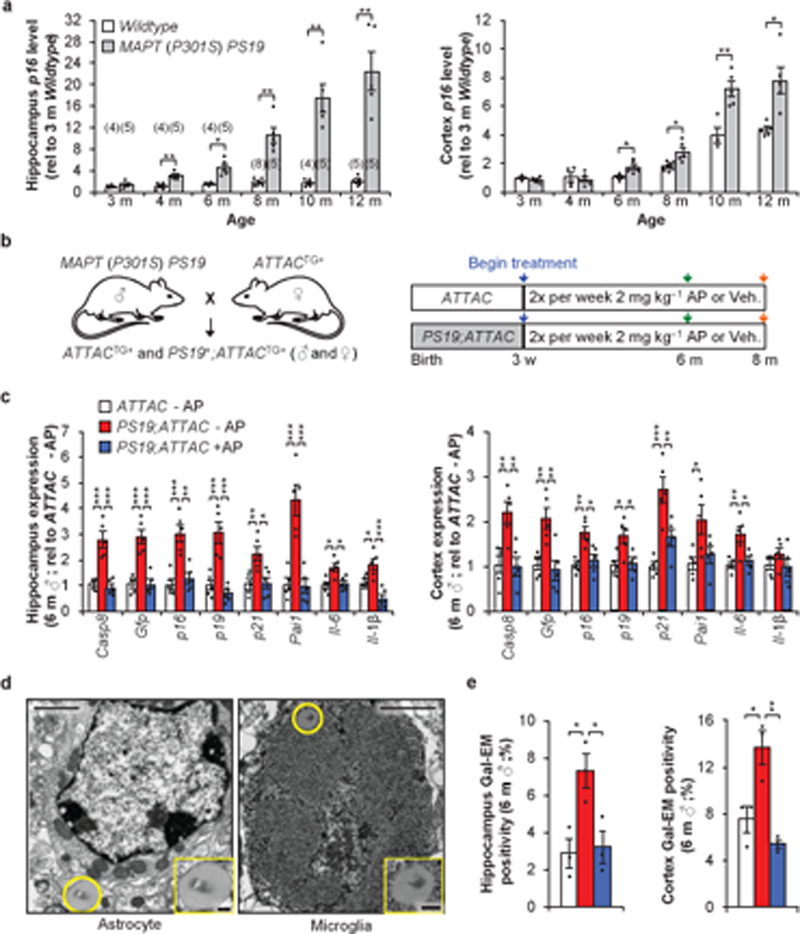
a, RT-qPCR analysis for p16Ink4a expression in hippocampus (left) and cortex (right) from Wildtype and P301S (MAPT) PS19 mice (animal numbers for each column are indicated in parentheses of hippocampus graph, 2 independent experiments; normalized to 3 m Wildtype group). b, Study design for clearance of senescent cells in PS19;ATTAC mice. Abbreviations: AP – AP20187; Veh. – vehicle. c, Expression of senescence markers from 6-month-old male hippocampus (left) and cortex (cortex) either vehicle (–AP) or AP20187 (+AP) treated assessed by RT-qPCR (n = 5 mice per group; normalized to ATTAC –AP group). p21 is also known as Cdkn1a; Pai1 is also known as Serpine1. d, Electron micrograph showing an X-Gal-positive astrocyte (left) and microglia (right) following SA-β-Gal staining from a 6-month-old vehicle-treated PS19;ATTAC male. e, Quantification of cells containing X-Gal crystals in the hippocampus (left) or cortex (right; n = 3 male mice per group, 2 independent experiments). Legend is as in (c). Scale bars, 1 μm (d) and 200 nm (d, insets). Data are mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001 (unpaired two-sided t-tests with Welch’s correction (a) and one-way ANOVA with Tukey’s multiple comparisons test (c and e)). Exact P values can be found in the accompanying source data file.
To investigate the role of senescent cells in disease development, we crossed the INK-ATTAC transgene (hereafter ATTAC) to the PS19 strain to eliminate p16Ink4a-expressing senescent cells through biweekly administration of AP20187 (hereafter AP)3,4 from weaning age (Fig. 1b). Hippocampi and cortices isolated from 6-month-old vehicle administered PS19;ATTAC mice displayed an elevated level of the ATTAC transgene as measured by Casp8 and GFP (Fig. 1c and Extended Data Fig. 2). Senescence indicators, including the cell cycle regulators p16Ink4a, p19Arf, p21Cip1/Waf1 and the pro-inflammatory genes Pai1 (also called Serpine1), Il-6, and Il-1β, were also elevated (Fig. 1c, Extended Data Fig. 2). AP administration in PS19;ATTAC mice maintained the expression of these genes at a level comparable to control mice (Fig. 1c, Extended Data Fig. 2). Importantly, AP treatment of ATTAC mice lacking the PS19 transgene had no impact on the expression of these markers (Extended Data Fig. 2). Thus, AP administration effectively and selectively cleared senescent cells in the hippocampus and cortex of PS19;ATTAC mice.
To understand the mechanistic contribution of senescence to tau–mediated pathology, we sought to identify the specific cell types that were becoming senescent. Firstly, we stained cortices and hippocampi from 6-month-old vehicle-treated ATTAC and PS19;ATTAC and AP-treated PS19;ATTAC mice for senescence-associated-β-galactosidase (SA-β-Gal)12 and screened for cells that contained X-Gal crystals by transmission electron microscopy (TEM)13. We found that cells that clearly and morphologically resembled astrocytes or microglia contained X-Gal crystals, irrespectively of the mouse group (Fig. 1d). In contrast, no crystals were found in any clearly identifiable neurons (Extended Data Fig. 3). Vehicle-treated PS19;ATTAC mice had nearly double the number of cells containing X-Gal crystals in both the hippocampus and cortex (Fig. 1e), whereas AP-treated PS19;ATTAC mice had a similar incidence of X-Gal crystals as control mice (Fig. 1e). To validate that senescence was impacting astrocytes and microglia, we performed FACS on 6-month-old Wildtype and PS19 mice (Extended Data Fig. 4a). Isolated astrocytes and microglia had increased expression of senescence-associated genes, including p16Ink4a (Extended Data Fig. 4b, c). A similar induction was not observed in oligodendrocytes or neuron-enriched CD56+ cells (Extended Data Fig. 4d, e and Extended Data Fig. 5), supporting the conclusion that senescence occurs in astrocytes and microglia of PS19 mice. To verify that AP administration selectively targeted senescent cells, we made in vitro cultures of primary microglia and astrocytes isolated from ATTAC mice. These cultures were not sensitive to AP-mediated elimination in the absence of senescence-inducing stimuli (Extended Data Fig. 6). Furthermore, short-term AP administration did not promote excessive cellular death (Extended Data Fig. 7a) or increased proliferation of microglia with extended treatment of ATTAC transgenic mice in vivo (Extended Data Fig. 7b).
PS19 mice present progressive gliosis with disease progression10. To assess if AP administration impacted this process, RT-qPCR was performed on 6-month-old hippocampi for markers of astrocytes (GFAP and S100β) and microglia (Cd11b). Vehicle-treated PS19;ATTAC mice had an ~2–3 fold induction in these markers, whereas AP-treated PS19;ATTAC mice expressed these markers at a similar level to control mice (Extended Data Fig. 8a, b). Immunohistochemistry (IHC) for GFAP and Iba1 confirmed these observations (Extended Data Fig. 8c, d). Taken together, these results suggest that both gliosis and glial cell senescence in the PS19 mouse model are effectively eliminated upon the administration of AP in ATTAC mice.
A distinguishing characteristic of PS19 mice is the development of aggregates consisting of hyperphosphorylated tau protein by 6 months of age10. To assess if tau aggregation was impacted with senescence clearance, we probed for the levels of soluble total and phosphorylated tau (Ser202/Thr205) in addition to the level of insoluble phosphorylated tau in vehicle-treated PS19;ATTAC and AP-treated ATTAC and PS19;ATTAC mice. As expected10, vehicle-treated PS19;ATTAC mice displayed increased soluble total and phosphorylated tau and insoluble phosphorylated tau (Fig. 2a, Extended Data Fig. 9a, b). AP-treated PS19;ATTAC mice showed identical levels of soluble total tau protein to vehicle-treated PS19;ATTAC mice (Fig. 2a), indicating that tau over-expression from the transgene was maintained. Surprisingly, AP treatment of PS19;ATTAC mice significantly reduced the amount of phosphorylated tau in both the soluble and insoluble fraction (Fig. 2a, Extended Data Fig. 9b). IHC staining for phospho-tau modifications at S202/T205, T231, and S396 confirmed that senescent cell clearance attenuated tau phosphorylation at a number of residues relevant for tau aggregation (Fig. 2b, Extended Data Fig. 9c). Furthermore, thioflavin S staining of 8-month-old mice from these same groups revealed that NFT deposition in the dentate gyrus, the site of neurogenesis in the hippocampus traditionally associated with memory formation and cognition14, was substantially reduced when senescent cells were removed (Fig. 2c). Collectively, these results indicate that senescent cell accumulation promotes the formation of hyperphosphorylated tau aggregates.
Figure 2. Senescent cells promote insoluble tau aggregates.
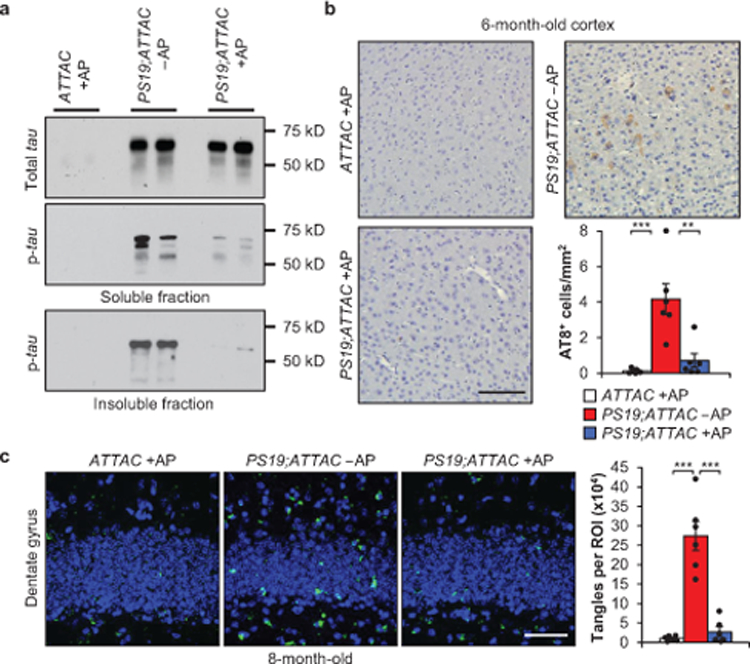
a, Representative western blot (≥ 3 independent experiments) analysis of 6-month-old whole brain for soluble tau (top), soluble phosphorylated tau (S202/T205; middle), and insoluble phosphorylated tau (S202/T205; bottom). b, Immunostaining and quantification of cortex sections for phosphorylated tau (S202/T205) protein aggregates (n = 6 mice per group, 3 independent experiments). c, Thioflavin S staining and quantification for neurofibrillary tangles located within the dentate gyrus (n = 6 mice per group, 2 independent experiments; normalized to ATTAC +AP group). Key is as indicated in b. Scale bars, 100 μm (b) and 50 μm (c). Data are mean ± s.e.m. **P < 0.01; ***P < 0.001 (one-way ANOVA with Tukey’s multiple comparisons test). Exact P values can be found in the accompanying source data file. For gel source data, see Supplementary Figure 1.
PS19 mice show neurodegeneration by 8 months of age10. As NFT deposition was attenuated with AP treatment in both the cortex and hippocampus of PS19;ATTAC mice, we performed assessments for degeneration in these areas. Overt brain size of vehicle-treated PS19;ATTAC mice was reduced compared to both ATTAC and AP-treated PS19;ATTAC mice (Fig. 3a). In addition, we observed localized neurodegeneration in the dentate gyrus of the hippocampus through Nissl staining in vehicle-treated PS19;ATTAC mice (Fig. 3b). AP administration prevented thinning of the dentate gyrus and increased neuron density. Sequential coronal sectioning and NeuN staining revealed that the dentate gyrus was significantly reduced in vehicle-treated PS19;ATTAC mice (Fig. 3c), further demonstrating that senescent cells promote neurodegeneration in PS19 mice.
Figure 3. Senescent cells drive neurodegenerative disease.
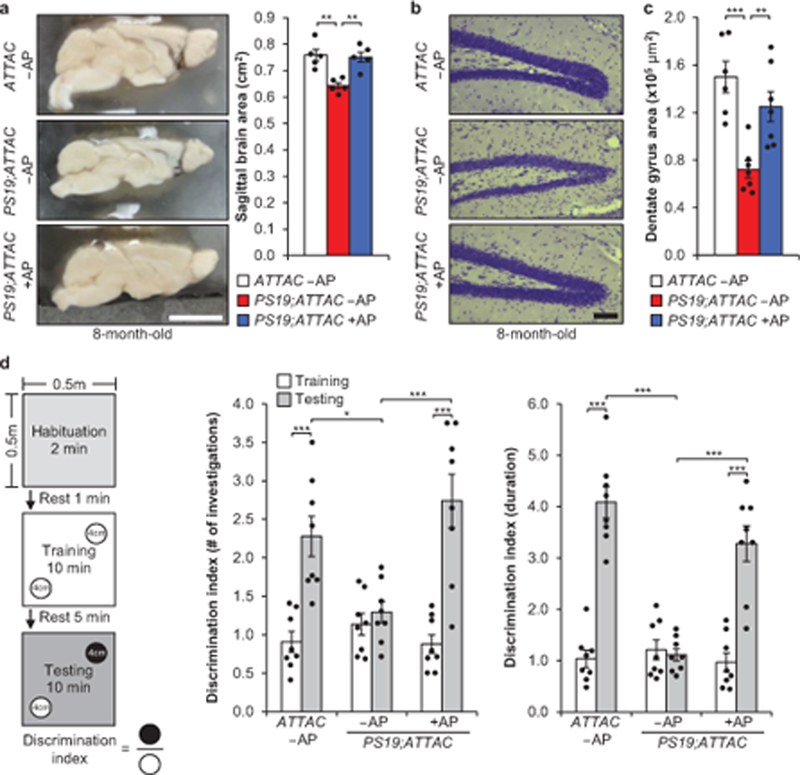
a, Sagittal midline brain area of 8-month-old mice (n = 5 males per group, 2 independent experiments). b, Nissl stains of the dentate gyrus from 8-month-old mice. c, Average area of the dentate gyrus (measuring the pyramidal neuron layer) from serial, coronal NeuN-stained free-floating sections (n = 6 ATTAC –AP and n = 7 PS19;ATTAC –AP and PS19;ATTAC +AP mice, 2 independent experiments). d, Novel object recognition experiment setup and average ratio for the number of investigations and duration of those investigations (n = 8 female mice per group). Scale bars, 0.5 cm (a) and 100 μm (b). Data are mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with Tukey’s multiple comparisons test (a and c) and two-way ANOVA with Tukey’s multiple comparisons test (d)). Exact P values can be found in the accompanying source data file.
To test whether this improved cognitive function, we performed novel scent discrimination assessments to test for changes in short-term memory (see experimental setup Fig. 3d)15. Whereas AP-treated ATTAC mice were more inquisitive to the novel scent during the testing phase, vehicle-treated PS19;ATTAC mice were not (Fig. 3d). In contrast, AP-treated PS19;ATTAC mice behaved nearly identically to control mice, indicating that senescent cell elimination mitigated the short-term memory loss observed in vehicle-treated PS19;ATTAC mice. Importantly, the overall distance traveled by mice in all groups was unchanged (data not shown) and similar results were obtained with novel object discrimination tests using the same setup using visual cues instead of scents (Extended Data Fig. 10). Thus, these results demonstrate that senescent cells drive neurodegeneration and loss of cognition in PS19 mice.
Lastly, we tested whether pharmacological elimination of senescent cells with the senolytic ABT263 (navitoclax)5,6,16,17 exhibited similar impacts to our genetic interventions in PS19 mice. Recent work has demonstrated a therapeutic effect in orthotopically implanted glioblastomas with peripheral administration pf ABT26318. WT and PS19 mice were treated with a repeating schedule of ABT263 beginning at weaning age until the mice reached 6 months of age. Importantly, this treatment prevented the upregulation of senescence-associated genes (Fig. 4a) and attenuated tau phosphorylation in PS19 mice (Fig. 4b), indicating that senolytic interventions can recapitulate key observations from transgenic mouse models of senescent cell ablation.
Figure 4.
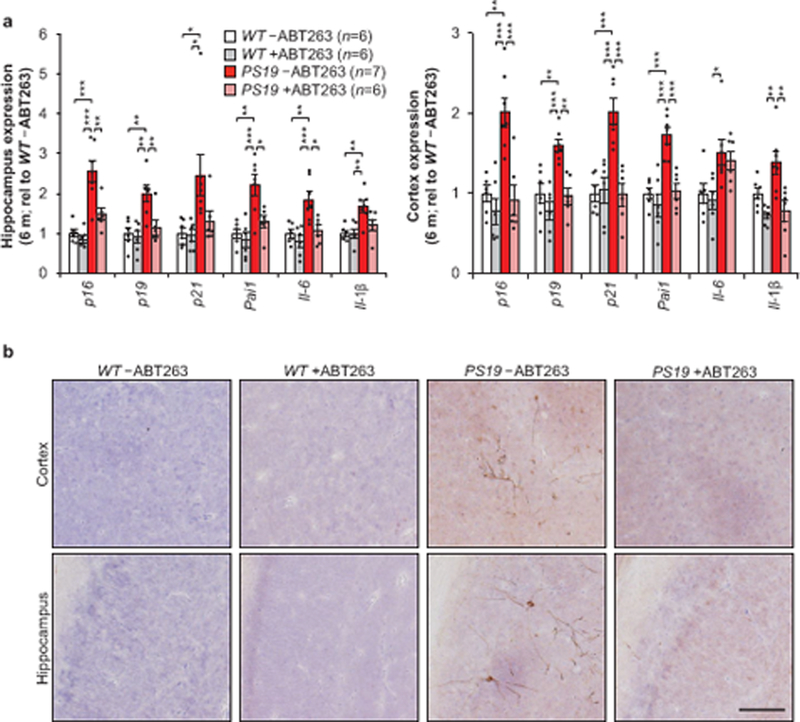
ABT263 can modulate senescent cells and attenuate tau phosphorylation. a, Expression of senescence markers from 6-month-old hippocampus (left) and cortex (cortex) either vehicle (–ABT263) or ABT263 (+ABT263) treated assessed by RT-qPCR (animal numbers indicated in the legend; normalized to WT –ABT263 group). p21 is also known as Cdkn1a; Pai1 is also known as Serpine1. b, Representative immunostaining of cortex (top) and hippocampal (bottom) sections for phosphorylated tau (S202/T205) protein aggregates (n = 4 mice per group, 2 independent experiments). Scale bar, 100µm. Data are mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with Tukey’s multiple comparisons test). Exact P values can be found in the accompanying source data file.
The mechanistic contribution of cells with features reminiscent of senescence to the pathophysiology of neurodegenerative diseases has been a common question in recent years7–9,19–21. Furthermore, recent work has suggested that senescent cells may contribute to Parkinson’s disease pathology in both mice and humans22. Here we show that continuous clearance of p16Ink4a-expressing senescent cells prior to disease onset in a model of aggressive tauopathy has a significant impact on various aspects of disease progression including gliosis, NFT formation, neurodegeneration, and cognitive decline. Remarkably, senescent cell clearance has a significant impact on the accumulation of phosphorylated tau protein in both the soluble and insoluble fractions. The amount of total soluble tau was unchanged in AP-treated PS19;ATTAC mice (Fig. 2a), indicating that the aberrant hyper-phosphorylation of tau protein and subsequent aggregation into NFTs is mediated by extracellular signaling from p16Ink4a-expressing senescent glial cells. The molecular mechanisms that senescent astrocytes and microglia exploit to promote pathological conversion of tau into NFTs within neurons require additional investigation. The absence of neurodegeneration in AP-treated mice (Fig. 3) demonstrates that attenuated disease severity is not due to clearance of neurons harboring NFTs. However, it is important to leave open the possibility that other neurodegenerative disease models may exhibit senescence-associated alterations in cell types not observed in the present study. Regardless, it is likely that intervention in senescent cell accumulation in these models would also reduce disease severity based on our observations. As this study was designed to prevent senescent cells from accumulating to determine how this impacts disease, future studies of senolysis in established disease models will be necessary to determine the utility of senolytic strategies to translate into the clinic to stall or perhaps revert disease. As senescent cells exhibit a unique and identifiable SASP, exploiting this phenotype may serve as a possible therapeutic avenue to attenuate many tau-dependent pathologies. Our observation that p16Ink4a expression increases prior to NFT aggregation further supports the now commonly held belief that early intervention in these diseases is essential to provide more favorable impacts to patients.
Methods
Mouse strains and drug treatment
MAPT P301S PS19 (PS19) mice were purchased from The Jackson Laboratory (stock #008169) and bred to C57BL/6 for three generations. C57BL/6 ATTAC transgenic mice are as described3,4. Male PS19 mice were bred to ATTAC females to generate cohorts of ATTAC and PS19;ATTAC mice. All mice were on a pure C57BL/6 genetic background. Mice from this cohort were randomly assigned to receive AP20187 (AP; B/B homodimerizer; Clontech) or vehicle twice a week beginning at weaning age (3 weeks). Dosing of AP was 2.0 mg kg–1 body weight. 6-month-old short-term AP pulse treated animals (Extended Data Fig. 6a) received a dose of 10 mg kg–1 body weight for 5 consecutive days prior to tissue collection. Senolytic intervention was performed in C57BL/6 WT and PS19 animals. At weaning, mice were assigned to receive either ABT263 (Cayman, 923564–51-6) or vehicle (Phosal 50 PG, Lipoid NC0130871 – 60%; PEG400, Sigma 91893 – 30%, EtOH – 10%). ABT263 was administered by oral gavage at a dose of 50 mg kg–1 body on a repeating regiment of five consecutive days of treatment followed by 16 days of rest. Animals were housed in a 12h L/D cycle environment in pathogen-free barrier conditions as described in detail3. Compliance with relevant ethical regulations and all animal procedures were reviewed and approved by the Mayo Clinic Institutional Animal Care and Use Committee.
Statistical analysis
Prism software was used for all statistical analysis. A student’s two-tailed unpaired t-test with Welch’s correction was used in Fig. 1a and Extended Data Fig. 4b – e; two-way ANOVA with Tukey’s multiple comparisons test was used for Fig. 3d and Extended Data Fig. 10; and one-way ANOVA with Tukey’s multiple comparisons test was used in all other figures. For consistency in these comparisons, the following denotes significance in all figures: *P < 0.05, **P < 0.01, ***P < 0.001. We note that no power calculations were used. Sample sizes are based on previously published experiments where differences were observed. No samples were excluded. Investigators were blinded to allocation during experiments and outcomes assessment, except for rare instances where blinding was not possible. All source data and exact P values (if applicable) for every figure are included in the supporting information that accompanies the paper.
Senescence-associated β-galactosidase transmission electron microscopy (Gal-TEM)
Detection of X-Gal crystals by transmission electron microscopy (TEM) after senescence-associated β-galactosidase (SA-β-Gal) staining was performed as described3,5 with the following alterations to accommodate central nervous tissue. Mice were transcardially perfused with ice-cold Dulbecco’s phosphate-buffered saline (DPBS; pH 7.4) until fluid runoff was clear. This was followed by perfusion with 4% paraformaldehyde (PFA) for 10 minutes at a rate of 3 ml per minute, and then ice-cold DPBS was perfused again for 2 minutes at the same rate to remove the remaining fixative. Brains were then isolated and the hippocampus and cortex were dissected out. A 1 mm x 1 mm piece from the CA1 and M1 region, respectively, was then incubated in SA-β-Gal staining solution (Cell Signaling) at 37°C for 6 h (hippocampus) or 18 h (cortex). The samples were placed in Trump’s fixative overnight at 4°C before being processed for routine transmission electron microscopy (dehydration by xylene-alcohol series, osmium tetroxide staining, and Epon resin embedding). Images were acquired and quantified using a Jeol 1400+ electron microscope with 80 kV acceleration voltage. Two grids from each tissue were produced, and >100 cells were scanned per grid at a magnification of 20,000x to detect X-Gal crystal containing cells. On average, half of all cells examined were neurons. Cells with one or more crystals and the total number of cells were counted. Cells containing crystals were imaged and independently assessed for distinguishing morphology. To define cell type, the following criteria were applied: 1) astrocytes – circular nucleus with spattered electron density pattern; 2) microglia – abnormally shaped nucleus with a much darker, often phagosome containing cytoplasm; and 3) neuron – large circular nucleus with less electron density and periodically denoted by an offshooting axon. Only cells with morphology consistent of astrocytes or microglia were clearly X-gal crystal positive.
Western blotting for soluble and sarkosyl insoluble proteins
Half brains were weighed and homogenized in 5X volume of Buffer I (50 mM Tris base [pH 7.4], 50mM NaCl, 1mM EDTA, 1mM PMSF, 1X Halt™ Protease and Phosphatase Inhibitor Cocktail [Thermo]). 250ul of the homogenate was then added to an equal volume of Buffer S (50 mM Tris [pH 8.0], 274 mM NaCl, 5 mM KCl, 1 mM PMSF, 1X Halt™ Protease and Phosphatase Inhibitor Cocktail [Thermo]) and ultracentrifuged at 150,000g for 15 minutes at 4°C. The supernatant (S1-soluble protein fraction) was transferred to a new tube and the pellet homogenized in 3x volume of sucrose buffer (10 mM Tris [pH7.4], 0.8M NaCl, 10% sucrose, 1mM EGTA, 1 mM PMSF) before being ultracentrifuged at 150,000g for 15 minutes at 4°C. The pellet was discarded and the supernatant incubated with sarkosyl (Sodium lauroyl sarcosinate) at a final concentration of 1% for 1 hour at 37°C. Following incubation, the samples were ultracentrifuged at 150,000g for 30 minutes at 4°C. The supernatant was discarded and the pellet was re-suspended in 25ul Buffer F (10 mM Tris [pH8.0], 1mM EDTA) to get the insoluble protein fraction (S2). Equal parts of 2x laemmli buffer (Bio-Rad) containing 5% β-mercaptoethanol was added to each fraction (S1 and S2) and boiled at 100°C for 15 minutes to prepare the sarkosyl soluble and insoluble protein lysates. For total protein lysate, 90ul of the homogenate (half brain in 5X volume Buffer I) was added to 110ul of Buffer T (2% SDS, 50 mM Tris [pH7.4], 274 mM NaCl, 5 mM KCl, 5mM EDTA, 1% Triton-X-100, 1 mM PMSF, X Halt™ Protease and Phosphatase Inhibitor Cocktail [Thermo]). The samples were then sonicated and centrifuged at 16,000g for 15 minutes at 4°C to remove debris. The supernatant was removed and added to equal parts 2x laemmli buffer with 5% β-mercaptoethanol and boiled at 100°C for 15 minutes to prepare the total protein lysate. Western blotting was performed as previously described23. Blots were probed with antibodies for total tau (ThermoFisher; MN1000, 1:5000) and phospho-tau S202/T205 (ThermoFisher; MN1020, 1:1000). Ponceau S staining was performed to normalize lysate loading for the total and S1 fraction lysates. Quantification was performed using ImageJ as described3.
Quantitative RT-PCR
RNA extraction, cDNA synthesis, and RT-qPCR analysis were performed on hippocampi and cortical samples from mouse brains as previously described24. Primers used to amplify Casp8, GFP, p16Ink4a, p19Arf, p21, Pai1, Il-6, Il-1b and Cd11b were as previously described3,5,24. The following additional primers were used: GFAP forward 5’-CCTTCTGACACGGATTTGGT-3’, reverse 5’-TAAGCTAGCCCTGGACATCG-3’; S100β forward 5’-CCGGAGTACTGGTGGAAGAC-3’, reverse 5’-GGACACTGAAGCCAGAGAGG-3’; Aqp4 forward 5’-TGAGCTCCACATCAGGACAG-3’, reverse 5’-TCCAGCTCGATCTTTTGGAC-3’; Cx3cr1 forward 5’-GTTCCAAAGGCCACAATGTC-3’, reverse 5’-TGAGTGACTGGCACTTCCTG-3’; Olig forward 5’-CCCCAGGGATGATCTAAGC-3’, reverse 5’-CAGAGCCAGGTTCTCCTCC-3’; NeFL forward 5’-AGGCCATCTTGACATTGAGG-3’, reverse 5’-GCAGAATGCAGACATTAGCG-3’; TBP forward 5’-GGCCTCTCAGAAGCATCACTA-3’, reverse 5’-GCCAAGCCCTGAGCATAA-3’. Expression for all experiments was normalized first to TBP.
Immunohistochemistry and immunofluorescence staining
Mice were transcardially perfused as described above. Brains were stored in 4% PFA overnight at 4°C and then cryoprotected by incubating in a 30% sucrose solution for 48 hours at 4°C. Samples were sectioned into 30 μM thick coronal sections and stored in antifreeze solution (300g Sucrose, 300 mL Ethylene Glycol, 500 mL PBS) at −20°C. Nissl staining (Bregma −2.1 to −2.4 mm), thioflavin S staining (Bregma −1.4 to −1.6 mm), and phospho-tau S202/S205 (ThermoFisher, MN1020; 1:500), phospho-tau T231 (ThermoFisher, MN1040; 1:500), phospho-tau S396 (Abcam, 109390; 1:500), and Gfap (Dako, Z0334; 1:500) and Iba1 (Novus, NB100–1028; 1:100) IHC staining (Bregma 1.6 to 1.0 mm and Lateral 2.0 to 2.7 mm) was performed on free-floating sections as described25–27. NeuN staining (EMD, MAB377; 1:200) of five sections (between Bregma −1.3 to −2.5) to measure dentate gyrus area was performed as previously described28. For cellular proliferation assays, animals were injected with EdU (Carbosynth, NE08701; 75mg/kg) intraperitoneally 24 hours prior to sacrifice. Imaging of EdU positive cells (Lateral 0.75 to 1.25 mm) was performed following manufacturer’s instructions (Invitrogen - Click-iT™ EdU Alexa Fluor™ 488 Imaging Kit, C10337). Iba1 (Wako, 019–19741; 1:500) immunofluorescent staining and Iba1/EdU colocalization assessments were performed as previously described28. TUNEL staining (Lateral 0.75 to 1.25 mm) was performed according to the manufacturer’s instructions (Roche In Situ Cell Death Detection Kit, Fluorescein: 11684795910). Thioflavin S, EdU/Iba1 colocalization, and in vivo TUNEL stained images were acquired on a Zeiss LSM 780 confocal system using multi-track configuration.
Single cell preparation and FACS
Dissociation of cerebral tissue was performed using the Adult Brain Dissociation kit from Miltenyi (MACS, 130–107-677) according to the manufacturer’s instructions. Samples were then incubated with a viability dye, LIVE/DEAD Aqua (Invitrogen, L34966; 1:250) followed by incubation with Cd11b eFluor 450 (eBioscience, 48–0112-80, 1:100), Cd45 APC eFluor 780 (eBioscience, 47–0451-82; 1:200), Glast1 PE (Miltenyi Biotec, 130–095-821; 1:100), O1 AF 700 (R&D Systems, FAB1327N-100UG; 1:100), and Cd56 APC (R&D Systems, FAB7820A; 1:100). These samples were then sorted using a FACSAria IIu SORP (BD Biosciences), with gating parameters created using FACSDiva 8.0.1 (BD Biosciences). A precise gating strategy was used to maximize the purification of each isolated cell population. Briefly, populations were isolated first by a negative report of the viability dye indicating the cell is viable (Extended Data Figure 4), followed by a positive report of the desired marker, then negative reports of the other labels used. This strategy allowed for live cells containing only the desired marker to be sorted, while eliminating dead cells. Cells were sorted directly into lysis buffer and RNA was extracted with RNeasy Micro kits according to manufacturer’s instruction (Qiagen, cat #: 74004). cDNA synthesis and RT-qPCR analysis were performed as described above.
Novel object recognition
Novel object recognition testing was performed as previously described15. Briefly, mice from each cohort were acclimated to a 50 cm x 50 cm testing environment for a period of two minutes. After acclimation, the mice were removed, the testing area was cleaned with 70% EtOH, and two identical scented candles were placed in either corner of the testing area approximately 5 cm from either wall. Mice were reintroduced, and the ratio of both the number of visits and time spent at each candle was recorded for a period of ten minutes. Recording was performed from above (Panasonic WV-CP294) and all video files were analyzed with TopScan Version 3.00 (Clever Sys Inc.). Afterwards, the mice were removed, the testing area cleaned with 70% EtOH, and one candle was replaced with a novel scent. The mice were reintroduced and the number of visits and total time per candle was recorded as before. Testing also was performed with visual stimuli by placing identical toy brick towers at either corner and then replacing with a different toy brick tower in the testing phase using the same experimental paradigm monitoring for the number of investigations.
In vitro astrocyte and microglia culture
Astrocyte and microglia primary cultures were prepared in tandem from mixed glial cultures as previously described29. C57BL/6 WT and ATTAC pups (p0-p3) were sacrificed, and the cerebellum was discarded. The remaining tissue had its meninges removed using forceps and a dissection scope. Cleaned cerebral tissue was placed in chilled Earle’s Balanced Salt Solution with HEPES (EBSH) (NaCl [120 mM], NaH2PO4 [10 mM], KCl [2.5 mM], C6H12O6 [20 mM], HEPES [20 mM], NaHCO3 [10 mM], BSA [0.3%], H2O) until the remaining animals were sacrificed, and then animals were pooled together based on genotype (3–4 brains/group). The tissue was minced using a razor blade and dissociated by shaking in a 0.025% Trypsin/EBSH solution at 37°C for 30 minutes. FBS and MgSO4 (3.82%)/ DNAse I (1mg/ml) were added, and the sample was placed on ice for 5 minutes to halt trypsinization. Samples were mixed and centrifuged at 200g at 25°C for 5 minutes. The supernatant was discarded, and the remaining pellet was resuspended in EBSH. Tissue was triturated using a 1 ml pipet to completely dissociate the sample and allowed to settle for 5 minutes to remove large debris. Samples were then transferred to clean tubes and underlayed with a 4% BSA/EBSH solution. The tissue was then centrifuged at 100g at 25°C for 8 minutes. Cells were counted using trypan blue and a hemocytometer and plated on a PDL-coated T75 dish (7–10 million cells/flask) with glial cell culture media (GCM) consisting of DMEM with 10% FBS, C3H3NaO3 (1mM), Pen/Strep (500 ug/ml), and InvivoGen Primocin. Cultures were grown for 14 days (37°C ambient O2) with media changes every 4 days. Microglia were isolated as previously described30 using the EasySep Mouse CD11b Positive Selection Kit from Stem Cell (cat #: 18970). Microglia were collected and plated on 10-well glass slides (5,000 cells/well) and cultured for 6 days in GCM with LADMAC-conditioned media (20%, generously provided by the Howe Laboratory) before further experimentation. This conditioned media aids in the proliferation and maintenance of microglia cultures through the secretion of M-CSF by the LADMAC cells31. Microglia were allowed to proliferate for 6 days prior to experimentation. The mixed glial culture flow-through from the EasySep CD11b kit was replated in GCM on a PDL-coated T75 dish (10 million cells/flask). These cultures then underwent purification for astrocytes as previously described29. After 48 hours, flasks were placed on an orbital shaker and agitated at 200 rpm for two 24-hour periods with media refreshed once during and after the shaking. Flasks were then exposed to GCM containing liposomal clodronate (Clodrosome, 8909; 100 µg/ml) for 72 hours to remove any remaining microglia from the culture. The liposomal clodronate media was then removed and culture plates washed prior to further experimentation.
Microglia activation and TUNEL staining
Microglia samples were exposed to media containing IFNy (R&D Systems, 285-IF; 200ng/ml), LPS (Sigma, L2654; 100ng/ml) or a combination of both for a period of 24 hours to induce an inflammatory response32. Cells were then processed for immunofluorescence to determine inflammation state as previously described33. Anti-Cd11b antibody (BioRad, MCA711G; 1:500) and goat-anti-rat AlexaFluor 594 (Invitrogen, A-11007; 1:500) staining was counterstained with DAPI (Invitrogen, D1306; 1:1000). To assess AP-mediated cell clearance specificity, activated or basal microglia were exposed to AP20187 (Clontech, 635059; 10nM or 100nM) for a period of 24 hours. TUNEL staining was then performed according to manufacturer’s instructions (Roche In Situ Cell Death Detection Kit, Fluorescein: 11684795910). All imaging was performed using an Olympus BX53 Fluorescence microscope and DP80 digital camera. Analysis was performed using the Fiji distribution of ImageJ (version 1.51n)34. To obtain a TUNEL positive percentage, a region of interest was defined using Li Auto Thresholding of the DAPI channel, and the colocalization percentage was calculated using the colocalization threshold plugin bound by that region.
Incucyte tracking of basal and activated astrocytes
To track basal and activated astrocytic response to AP, astrocytes were plated in a 48 well culture plate (10,000 cells/well) and placed into the IncuCyte S3 Live-Cell Analysis System. The IncuCyte System is a time-lapse imaging system that records cell culture changes through photographic capture of the culture well within the incubator. Cultures were acclimated to the system for a period of 6 hours, then exposed to media containing IFNy (R&D Systems, 285-IF; 200ng/ml), LPS (Sigma, L2654; 100ng/ml) or a combination of both for a period of 24 hours to induce an inflammatory response35. Cells were also plated on 10-well slides and processed in tandem for immunofluorescence staining to verify activation status with anti-Gfap (DAKO, Z0334; 1:500) and counterstained with DAPI (Invitrogen, D1306; 1:1000). After activation, astrocytes were exposed to AP20187 (Clontech, 635059; 10nM or 100nM) for a period of 24 hours. The IncuCyte captured phase images of each culture well were taken every 30 minutes over this period using the following settings: (Segmentation Adjustment: 0.8, Hole Fill: 450, Adjust size (pixels): −1, Minimum area (uM2): 0.1). Phase confluency difference was calculated by subtracting the final phase confluency of each image from its initial value.
Extended Data
Extended Data Figure 1. Senescent cells accumulate in PS19 mice.
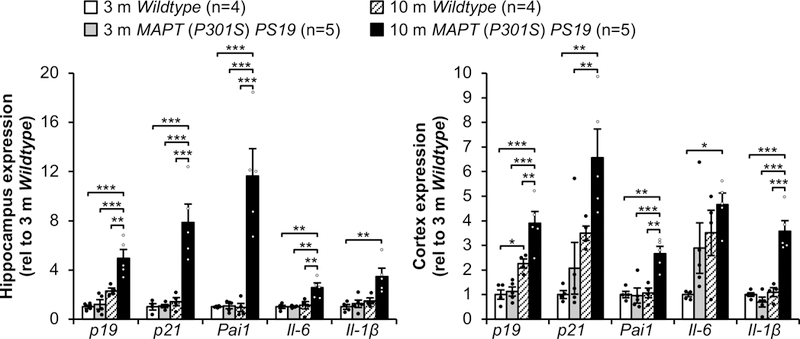
RT-qPCR analysis for senescence-associated genes in hippocampi (left) and cortices (right) of 3- and 10-month-old male mice (animal numbers indicated in the legend, 2 independent experiments; normalized to 3 m Wildtype group). Data are mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with Tukey’s multiple comparisons test). Exact P values can be found in the accompanying source data file.
Extended Data Figure 2. AP-mediated clearance selectively removes senescent cells that accumulate in brains of PS19;ATTAC mice.
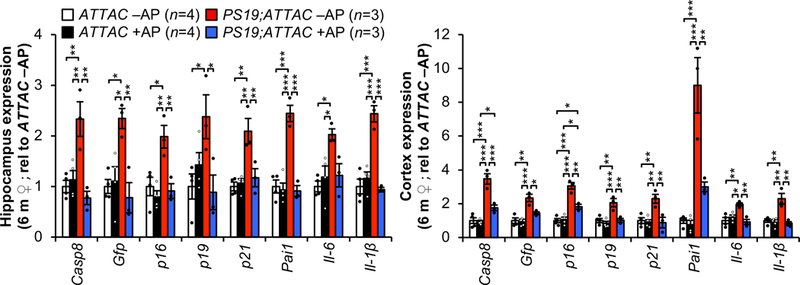
Expression of senescence markers from 6-month-old female hippocampus (left) and cortex (cortex) either vehicle (–AP) or AP20187 (+AP) treated assessed by RT-qPCR (animal numbers indicated in the legend; normalized to ATTAC –AP group). p21 is also known as Cdkn1a; Pai1 is also known as Serpine1. Data are mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with Tukey’s multiple comparisons test). Exact P values can be found in the accompanying source data file.
Extended Data Figure 3. Neurons do not exhibit X-Gal crystals by Gal-TEM.

Representative electron microscopy image of neurons after SA-β-Gal staining from a 6-month-old vehicle-treated PS19;ATTAC male mouse (n = 3 male mice, 2 independent experiments). The image has been artificially colored to denote individual cell bodies. Scale bar: 10 µm.
Extended Data Figure 4. Increased senescence-associated gene expression is observed in astrocytes and microglia isolated from PS19 mice.
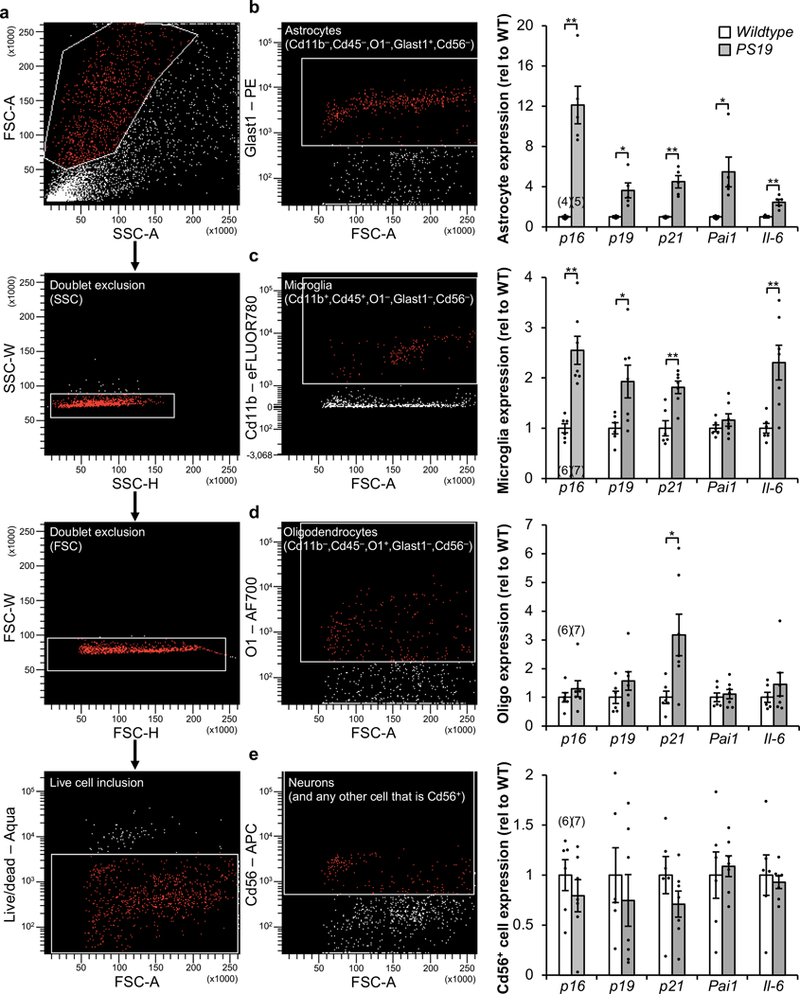
a–e, Gating strategy (a) for FACS isolation of living astrocytes (b), microglia (c), oligodendrocytes (d), and neuron-enriched Cd56+ cells (e) from cortices from 6-month-old WT and PS19 mice. b, Astrocyte (Cd11b–,Cd45–,O1–,GLAST+,Cd56–) fraction (left) and RT-qPCR analysis (right). c, Microglia (Cd11b+,Cd45+,O1,GLAST+,Cd56–) fraction (left) and RT-qPCR analysis (right). d, Oligodendrocyte (Cd11b–,Cd45–,O1+,GLAST–,Cd56–) fraction (left) and RT-qPCR analysis (right). e, Neuron-enriched Cd56+ (Cd11b–,Cd45–,O1–,GLAST–,Cd56+) fraction (left) and RT-qPCR analysis (right). p21 is also known as Cdkn1a; Pai1 is also known as Serpine1. Individual numbers of independent animal cell population isolations are indicated in the parentheses above p16Ink4a columns (2 independent experiments). Data are mean ± s.e.m. *P < 0.05; **P < 0.01 (unpaired two-sided t-tests with Welch’s correction). Exact P values can be found in the accompanying source data file.
Extended Data Figure 5. Cell identity verification of cell populations isolated by FACS.
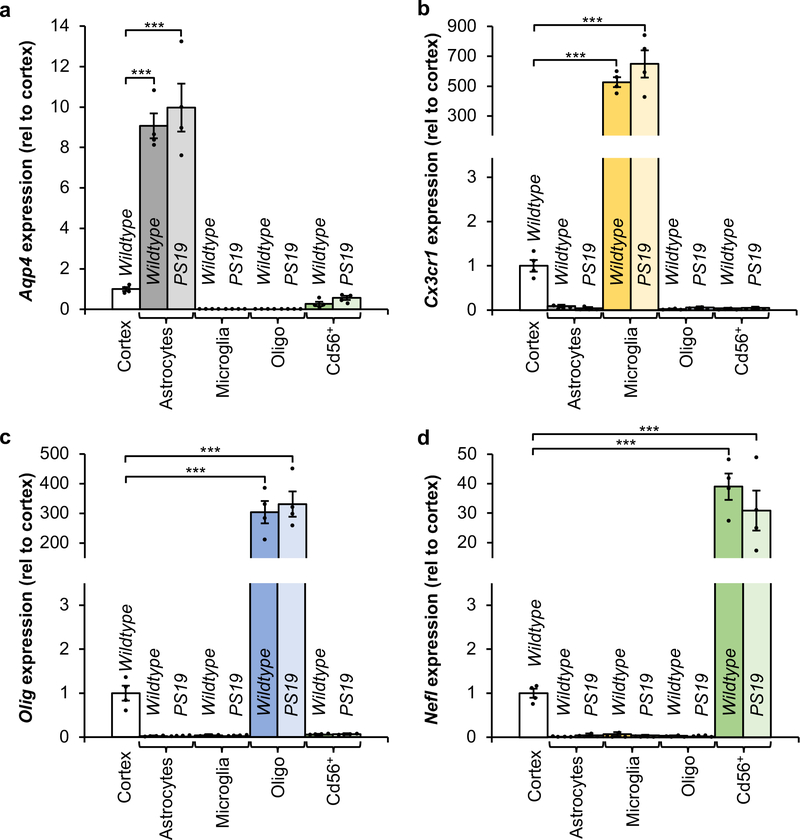
a–d, RT-qPCR analysis for cell identity markers from cell populations isolated from 6-month-old Wildtype and PS19 mice for Aqp4 expression enriched in astrocytes (a), Cx3cr1 expression enriched in microglia (b), Olig expression enriched in oligodendrocytes (c), and Nefl expression enriched in neurons (d). Expression is normalized to intact cortices of 6-month-old Wildtype mice (n = 4 biologically independent cell isolations for each group, 2 independent experiments). Data are mean ± s.e.m. ***P < 0.001 (one-way ANOVA with Tukey’s multiple comparisons test). Exact P values can be found in the accompanying source data file.
Extended Data Figure 6. AP administration does not precociously eliminate non-senescent glial cells isolated from ATTAC mice.
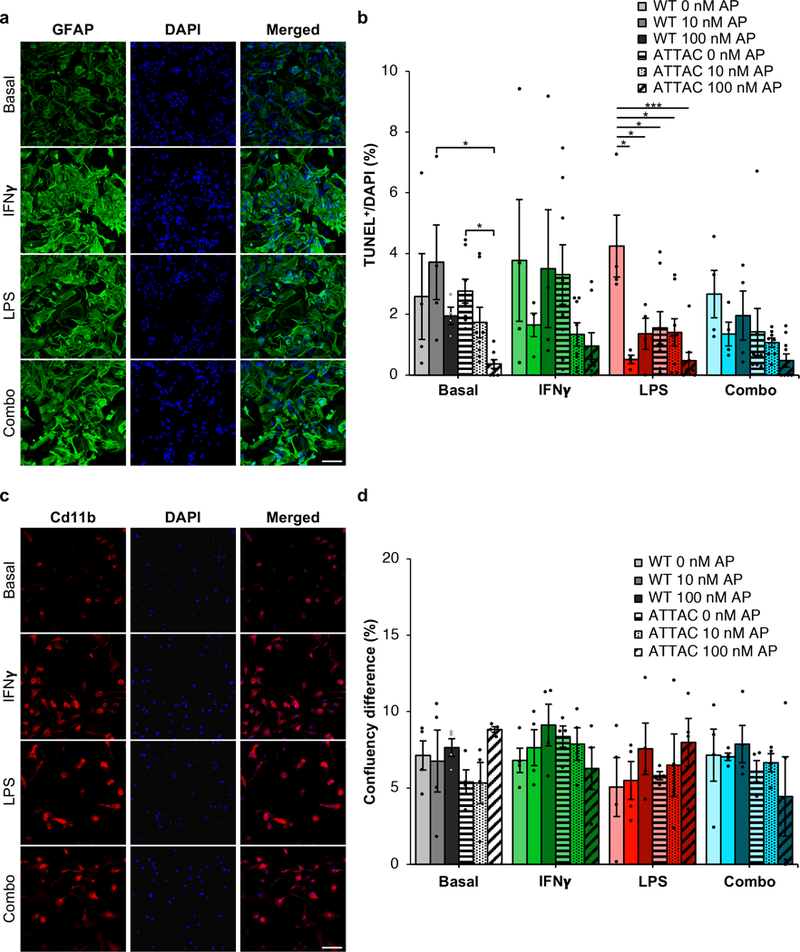
a, Cd11b staining of primary microglia treated with IFNγ (200 ng/ml), LPS (100 ng/ml), or a combination of both (n = 3 biologically independent samples). b, Quantification of TUNEL positive bodies in basal or activated microglia (n = 4 WT and 8 ATTAC cultures for each treatment group, 2 independent experiments). c, GFAP staining of primary astrocytes treated with IFNγ, LPS, or a combination of both as described in (a) (n = 3 biologically independent samples). d, Quantification of confluency change over 24 hours in basal or activated astrocytes (n = 4 biologically independent cultures of each genotype and treatment). Scale Bars, 100 μm (a and c). Data are mean ± s.e.m. *P < 0.05; ***P < 0.001 (one-way ANOVA with Tukey’s multiple comparisons test (b and d)). Exact P values can be found in the accompanying source data file.
Extended Data Figure 7. AP-administration does not broadly eliminate cells or increase proliferation of microglia.
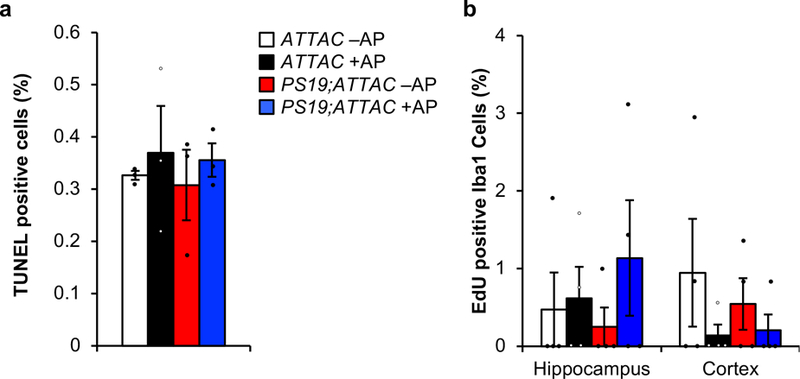
a, Quantification of TUNEL positive bodies (as a percentage of all cells) at the transition between the CA2 and CA3 within the hippocampus after a short-term AP administration in 6-month-old mice (n = 3 mice per genotype and treatment group). b, Quantification of Iba1/EdU double positive cells in hippocampus and cortex from 6-month-old mice administered AP beginning at weaning age (n = 4 mice per genotype and treatment group). Data are mean ± s.e.m. We note that no comparison is statistically significant (one-way ANOVA with Tukey’s multiple comparisons test). Exact P values can be found in the accompanying source data file.
Extended Data Figure 8. Senescent cells promote gliosis.
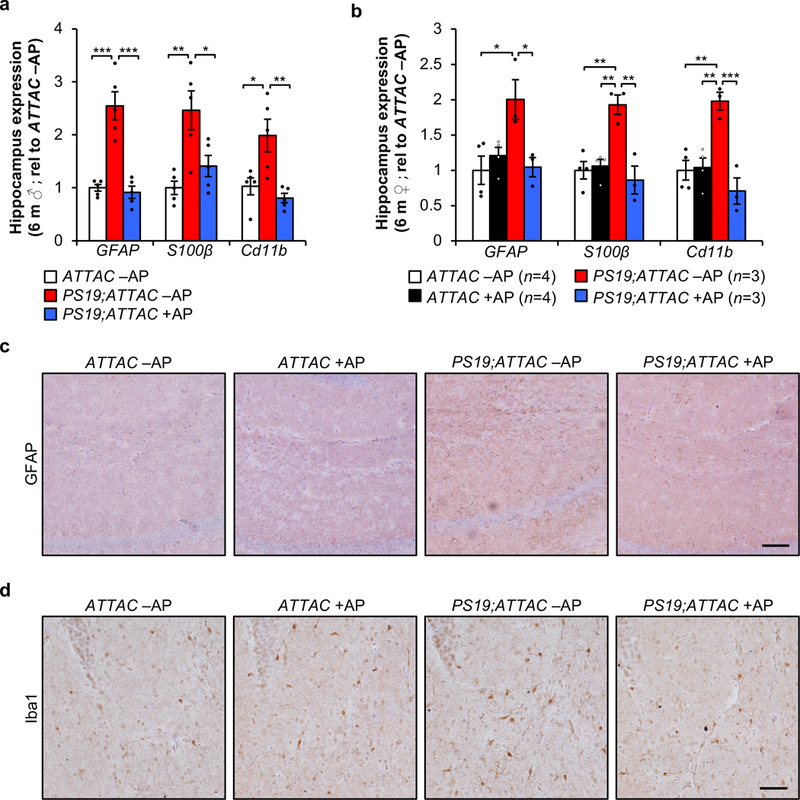
a, RT-qPCR analysis for Gfap, S100β, and Cd11b in hippocampi of 6-month-old male mice (n = 5 mice per group; normalized to ATTAC –AP group). b, RT-qPCR analysis as in (a) in hippocampi of 6-month-old female mice (animal number indicated in legend; normalized to ATTAC –AP group). c, Representative Gfap IHC staining in the hippocampus of 6-month-old vehicle and AP-treated ATTAC and PS19;ATTAC female mice (n = 4 mice per group, 2 independent experiments). d, Representative Iba1 staining in the hippocampus of 6-month-old vehicle and AP-treated ATTAC and PS19;ATTAC female mice (n = 4 mice per group, 2 independent experiments). Scale bar, 100 µm (c) and 50 µm (d). Data are mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with Tukey’s multiple comparisons test). Exact P values can be found in the accompanying source data file.
Extended Data Figure 9. AP treatment attenuates tau phosphorylation.
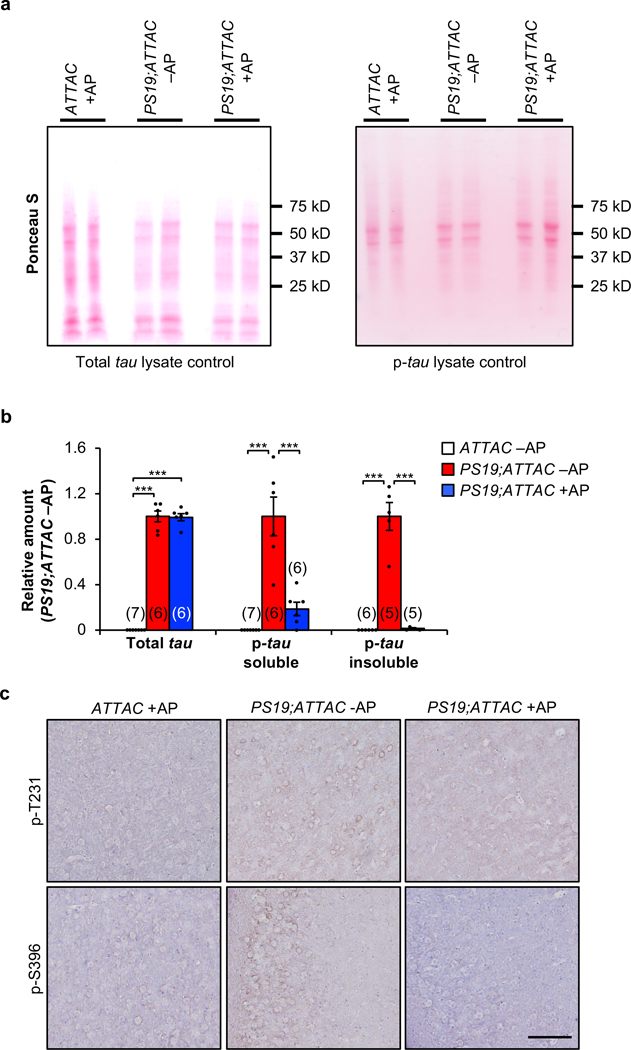
a, Ponceau S loading controls for western blot lysates of 6-month-old whole brain total-tau (left) and phosphorylated tau (S202/T205; right) shown in Figure 3a. b, Quantification of westerns blot analysis from 6-month-old whole brain for soluble tau (left), soluble phosphorylated tau (S202/T205; middle), and insoluble phosphorylated tau (S202/T205; right). Biologically independent animal numbers are indicated, data are from ≥ 3 independent experiments. c, Immunostaining of 6-month-old cortex for phosphorylated tau protein at T231 (top) and S396 (bottom; n = 4 mice per group, 2 independent experiments). Scale bar, 100µm. Data are mean ± s.e.m. ***P < 0.001 (one-way ANOVA with Tukey’s multiple comparisons test). Exact P values can be found in the accompanying source data file.
Extended Data Figure 10. Vision-based novel object discrimination remains intact in AP-treated PS19;ATTAC mice.
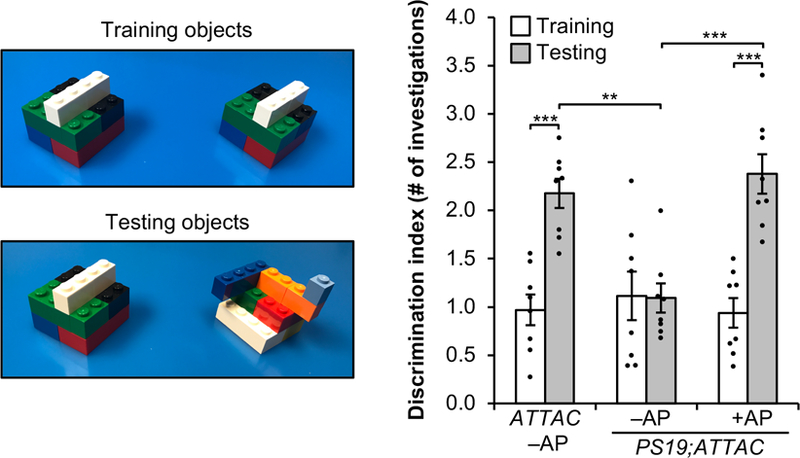
Objects used for novel object recognition during the training and testing phase for visual discrimination (left) and the average ratio for the number of investigations (right, n = 8 female mice per group). Data are mean ± s.e.m. **P < 0.01; ***P < 0.001 (two-way ANOVA with Tukey’s multiple comparisons test). Exact P values can be found in the accompanying source data file.
Supplementary Material
Acknowledgements
The authors thank Chang Hoon Cho for his numerous contributions to the experiments; the laboratory of Charles Howe and specifically Miranda Standiford for her help with the microglia and astrocyte cultures; Michael Poeschla for his assistance on the phospho-tau IHC; Grace Nelson for genotyping and animal support; Bennett Childs for his input and assistance in Gal-TEM; the Mayo Clinic Microscopy and Cell Analysis Core and staff for flow cytometry and TEM assistance; the Mayo Clinic Medical Genome Facility Gene Expression Core for RT-qPCR instrumentation; and Ron Petersen and Charles Howe for helpful feedback on this manuscript. This work was supported by the Ellison Medical Foundation, the Glenn Foundation for Medical Research, the National Institutes of Health (R01AG053229), the Mayo Clinic Children’s Research Center and the Alzheimer’s Disease Research Center of Mayo Clinic (all to D.J.B.).
Footnotes
Data availability
All source data and exact P values (if applicable) for every figure are included in the supporting information that accompanies the paper.
References
- 1.Hayflick L & Moorhead PS The serial cultivation of human diploid cell strains. Exp Cell Res 25, 585–621 (1961). [DOI] [PubMed] [Google Scholar]
- 2.Coppe JP et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6, 2853–2868 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker DJ et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker DJ et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Childs BG et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354, 472–477 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeon OH et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 23, 775–781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhat R et al. Astrocyte senescence as a component of Alzheimer’s disease. PLoS One 7, e45069 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan FC, Hutchison ER, Eitan E & Mattson MP Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology 15, 643–660 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo XG, Ding JQ & Chen SD Microglia in the aging brain: relevance to neurodegeneration. Mol Neurodegener 5, 12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshiyama Y et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Childs BG et al. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov, doi: 10.1038/nrd.2017.116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dimri GP et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 92, 9363–9367 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stollewerk A, Klambt C & Cantera R Electron microscopic analysis of Drosophila midline glia during embryogenesis and larval development using beta-galactosidase expression as endogenous cell marker. Microsc Res Tech 35, 294–306 (1996). [DOI] [PubMed] [Google Scholar]
- 14.Ming GL & Song H Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buenz EJ et al. Apoptosis of hippocampal pyramidal neurons is virus independent in a mouse model of acute neurovirulent picornavirus infection. Am J Pathol 175, 668–684 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang J et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22, 78–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu Y et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karpel-Massler G et al. Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat Commun 8, 1067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flanary BE, Sammons NW, Nguyen C, Walker D & Streit WJ Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res 10, 61–74 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Salminen A et al. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur J Neurosci 34, 3–11 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Streit WJ, Braak H, Xue QS & Bechmann I Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol 118, 475–485 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chinta SJ et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep 22, 930–940 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kasper LH et al. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol Cell Biol 19, 764–776 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker DJ et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol 10, 825–836 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parent JM, von dem Bussche N & Lowenstein DH Prolonged seizures recruit caudal subventricular zone glial progenitors into the injured hippocampus. Hippocampus 16, 321–328 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Ly PT, Cai F & Song W Detection of neuritic plaques in Alzheimer’s disease mouse model. J Vis Exp, doi: 10.3791/2831 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oh KJ et al. Staging of Alzheimer’s pathology in triple transgenic mice: a light and electron microscopic analysis. Int J Alzheimers Dis 2010, doi: 10.4061/2010/780102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Z et al. Age-related decline in BubR1 impairs adult hippocampal neurogenesis. Aging Cell 16, 598–601 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumamaru H et al. Liposomal clodronate selectively eliminates microglia from primary astrocyte cultures. J Neuroinflammation 9, 116, doi: 10.1186/1742-2094-9-116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon R et al. A simple magnetic separation method for high-yield isolation of pure primary microglia. J Neurosci Methods 194, 287–296 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsay HJ et al. Amyloid beta peptide-mediated neurotoxicity is attenuated by the proliferating microglia more potently than by the quiescent phenotype. J Biomed Sci 20, 78 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chao CC, Hu S, Molitor TW, Shaskan EG & Peterson PK Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol 149, 2736–2741 (1992). [PubMed] [Google Scholar]
- 33.Wang G et al. Apoptosis and proinflammatory cytokine responses of primary mouse microglia and astrocytes induced by human H1N1 and avian H5N1 influenza viruses. Cell Mol Immunol 5, 113–120 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chung IY & Benveniste EN Tumor necrosis factor-alpha production by astrocytes. Induction by lipopolysaccharide, IFN-gamma, and IL-1 beta. J Immunol 144, 2999–3007 (1990). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.