

Abstract
Stromal cell-derived factor-1α (SDF-1α) is a CXCR4-receptor agonist and dipeptidyl peptidase 4 (DPP4) substrate. SDF-1α, particularly when combined with sitagliptin to block the metabolism of SDF-1α by DPP4, stimulates proliferation of cardiac fibroblasts via the CXCR4 receptor; and this effect is greater in cells from spontaneously hypertensive (SHR) versus Wistar-Kyoto normotensive (WKY) rats. Emerging evidence indicates that ubiquitin(1–76) exists in plasma and is a potent CXCR4-receptor agonist. Therefore, we hypothesized that ubiquitin(1–76), similar to SDF-1α, should increase proliferation of cardiac fibroblasts. Contrary to our working hypothesis, ubiquitin(1–76) did not stimulate cardiac fibroblast proliferation, yet unexpectedly antagonized the pro-proliferative effects of SDF-1α combined with sitagliptin. In this regard, ubiquitin(1–76) was more potent in SHR versus WKY cells. In the presence of 6bk [selective inhibitor of insulin-degrading enzyme; an enzyme known to convert ubiquitin(1–76) to ubiquitin(1–74)], ubiquitin(1–76) no longer antagonized the pro-proliferative effects of SDF-1α/sitagliptin. Ubiquitin(1–74) also antagonized the pro-proliferative effects of SDF-1α/sitagliptin, and this effect of ubiquitin(1–74) was not blocked by 6bk and was greater than 10-fold more potent compared with ubiquitin(1–76). Neither ubiquitin(1–76) nor ubiquitin(1–74) inhibited the pro-proliferative effects of the non-CXCR4 receptor agonist neuropeptide Y (activates Y1 receptors). Cardiac fibroblasts expressed insulin-degrading enzyme mRNA, protein, and activity and converted ubiquitin(1–76) to ubiquitin(1–74). SHR fibroblasts expressed greater insulin-degrading enzyme activity. Conclusion: Extracellular ubiquitin(1–76) blocks the pro-proliferative effects of SDF-1α/sitagliptin via its conversion by insulin-degrading enzyme to ubiquitin(1–74), a potent CXCR4 antagonist. Thus insulin-degrading enzyme inhibitors, particularly when combined with DPP4 inhibitors or hypertension, could increase the risk of cardiac fibrosis.
Keywords: Cardiac Fibroblasts, Ubiquitin, SDF-1α, DPP4 Inhibitor, Insulin Degrading Enzyme
INTRODUCTION
Accumulating evidence implicates the SDF-1α/CXCR4 axis in organ fibrosis. For example, in animal models, the SDF-1α/CXCR4 axis is involved in cardiac fibrosis induced by mineralocorticoid-induced hypertension,1 genetic hypertension,2 type 1 and type 2 diabetes,3 and eNOS inhibition;4 and in injured coronary arteries the SDF-1α/CXCR4 axis stimulates neointimal hyperplasia.5,6 The SDF-1α/CXCR4 axis also participates in renal fibrosis triggered by ischemia-reperfusion injury,7 ureterial obstruction,8 and type 2 diabetes.9 Consistent with the preclinical findings, in patients there is a positive correlation between plasma SDF-1α levels and heart failure10 and all-cause mortality.11 In myocarditis patients, myocardial levels of SDF-1α are the strongest predictor of mortality and correlate with cardiac fibrosis.12
Because the SDF-1α/CXCR4 axis may be involved in cardiac fibrosis and because cardiac fibroblasts participate in cardiac fibrosis, recently we investigated the direct effects of SDF-1α on cardiac fibroblast biology.13 Our published results were the first to show that: 1) rat cardiac fibroblasts express CXCR4 receptors; 2) SDF-1α concentration-dependently increases the proliferation and hypertrophy of cardiac fibroblasts and augments collagen production by cardiac fibroblasts; 3) the effects of SDF-1α on cardiac fibroblasts are more pronounced in cardiac fibroblasts obtained from spontaneously hypertensive rats (SHR) compared with normotensive Wistar-Kyoto rats (WKY); 4) CXCR4 antagonism blocks all the effects of SDF-1α on cardiac fibroblasts; 5) SDF-1α/CXCR4 signal transduction system involves a RACK1/Gβγ/PLC/PKC signaling complex; and 6) inhibition of dipeptidyl peptidase 4 [DPP4; an enzyme that metabolizes full length SDF-1α(1–68) to the inactive truncated SDF-1α(3–68)] augments the effects of SDF-1α on cardiac fibroblasts likely by preventing its inactivation.
While investigating the role of CXCR4 receptors in cardiac fibroblasts, we became aware of literature supporting the novel concept that: 1) ubiquitin exists in the plasma at concentrations of up to 100 nmol/L; and 2) full length ubiquitin(1–76) is an agonist at CXCR4 receptors14, 15. Given the strong evidence that stimulation of CXCR4 receptors activates cardiac fibroblasts and that extracellular ubiquitin may interact with CXCR4 receptors, we hypothesized that the ubiquitin/CXCR4 axis, like the SDF-1α/CXCR4 axis, may also regulate cardiac fibroblast biology. Because the implications of this hypothesis with regard to the pathophysiology of cardiac fibrosis and heart failure are profound, we decided to investigate this concept.
METHODS
For data and for additional information on analytic methods or study materials contact Edwin K. Jackson, PhD at edj@pitt.edu.
Materials.
Chemicals were from the following sources: SDF-1α (ProSpec: Rehovot, Israel); sitagliptin (Merck: Whitehouse Station, NJ); ubiquitin(1–76) (R&D Systems: Minneapolis, MN); ubiquitin(1–74) (BostonBiochem: Cambridge, MA); neuropeptide Y(1–36) (Sigma-Aldrich: St. Louis, MO); platelet derived growth factor–BB (PDGF-BB; Sigma-Aldrich); 6bk (Phoenix Pharmaceuticals: Burlingame, CA).
Animals.
Cardiac fibroblasts were isolated from male SHR and WKY rats (approximately 12 weeks of age) obtained from Charles River Laboratories (Wilmington, MA). The University of Pittsburgh Institutional Animal Care and Use Committee approved all procedures. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (8th edition, 2011).
Culture of Cardiac Fibroblasts.
Cardiac fibroblasts from SHR and WKY were isolated, cultured, and characterized as recently described16.
Proliferation (Cell Number) Studies.
Cells were maintained in DMEM/F12 containing 10% fetal bovine serum under standard tissue culture conditions. Subconfluent cultures in 24-well plates were growth-arrested for 2 days in DMEM/F12 containing 0.4% bovine serum albumin. Next cells were placed in DMEM/F12 containing a low concentration (25 ng/mL) of PDGF-BB and then treated every day for 4 days without or with various treatments. In experiments in which cells were treated with SDF-1α, the SDF-1α was co-administered with sitagliptin (1 μmol/L). Sitagliptin was administered with SDF-1α because we have found that sitagliptin, by blocking DPP4, prevents the metabolic inactivation of SDF-1α and thus enhances its effects on proliferation of cardiac fibroblasts.13 Finally, cells were harvested and cell number quantified using a Nexcelom Cellometer Auto T4 cell counter (Nexcelom Bioscience: Lawrence, MA).
Western Blotting for Insulin Degrading Enyzme (IDE).
Western blotting for IDE was performed as previously described17. The primary antibody against IDE was a rabbit polyclonal from Abcam (Cambridge, MA, catalogue number ab32216).
Real-time PCR for IDE.
Real-time PCR for IDE was performed as previously described17. Forward and reverse primers were 5’-ttgtgactccccgtaactcc-3’ and 5’-aaggcaagatgagaccggaa-3’, respectively.
IDE Activity.
SHR and WKY cardiac fibroblasts were washed twice with phosphate-buffered saline, lysed in assay buffer with a Virsonic ultrasonic homogenizer, and then centrifuged at 10,000 g for 10 minutes at 4º C. Protein concentrations in the supernatant were measured using the Thermo Scientific Pierce BCA Protein Assay Kit, and protein concentration was adjusted to 0.625 mg protein/1 mL. Using the Anaspec SensoLyte 520 IDE Assay Kit, cellular IDE activity was determined in 50 μl of sample. As active IDE cleaves a FRET substrate, 5-carboxyfluorescein is released and its release is monitored over time by measuring fluorescence at excitation/emission of 490 nm/520 nm. IDE activity is proportional to the slope of time versus the intensity of the fluorescence signal.
Detection of Conversion of Ubiquitin(1–76) to Ubiquitin(1–74).
SHR cardiac fibroblasts were treated with ubiquitin(1–76) (1000 nmol/L) for 24 hours and with and without the IDE inhibitor 6bk (1μmol/L), and the medium was collected and lyophilized. The solid material was dissolved in 1 ml of buffer A (0.2 M NaCl, 20 mM tris, pH 7.5), and then centrifuged at 18,000 x g for 10 min. The supernatant was applied on a 3 ml Sephadex G-10 column equilibrated with buffer A, and the column was eluted by the same buffer. Fractions (0.5 ml) were manually collected and then lyophilized. The solid material was dissolved in 50 μl of buffer A containing 6 M urea. The fractions were centrifuged at 45,000 x g for 20 min. Samples were loaded onto a polar reverse phase column (Synergi polar-RP; 150 × 4.6 mm, 4 μm particle size, 80A; Phenomenex; Torrance, CA) connected to a Shimadzu (Columbia, MD) HPLC/LCMS-2010 system. Separation was conducted with a linear gradient from 20% acetonitrile to 60% acetonitrile in the presence of 0.1% formic acid at a flow rate 0.4 ml/min. The mass spectrometer was operated in the positive ion mode with selected ion monitoring that focused on ions with m/z that were diagnostic for ubiquitin(1–76) and ubiquitin(1–74) as determined by preliminary experiments with high-resolution time-of-flight (TOF) mass spectrometry (Waters Micromass Q-TOF Premier Mass Spectrometer, Waters, Milford, MA) of authentic ubiquitin(1–76) and ubiquitin(1–74). The chromatographic peak (total ion current) corresponding to the retention time of ubiquitin(1–76) and ubiquitin(1–74) (7.46 to 9.00 minutes) was scanned for multiple charged ions (M12+ to M7+): for ubiquitin(1–76) (714.80, 779.80, 857.80, 952.80, 1071.80, and 1224.80 m/z, respectively); and for ubiquitin(1–74) (705.30, 769.50, 846.20, 940.20, 1057.50, 1208.50 m/z, respectively).
Statistics.
In these protocols, different batches of SHR and WKY cardiac fibroblasts were plated on 24-well plates. On each plate, each treatment was applied to two wells (i.e., treatments were in duplicate on each plate and all 12 treatments were represented on each plate). Thus the subgroup was the plate number, and the experimental unit for statistical analysis was the average of the duplicate values on a particular plate. Therefore the reported sample size equals the number of individual/separate plates (not the number of individual wells). Data from experimental units were analysed by two-factor analysis of variance (2-Factor ANOVA). When the interaction term in the 2-factor ANOVA was significant, a Fisher’s Least Significant Difference (LSD) test was employed to explore the nature of the interaction. P<0.05 was considered statistically significant. Values are presented as mean ± SEM.
RESULTS
Ubiquitin(1–76) antagonizes the pro-proliferative effects of the CXCR4-receptor agonist SDF-1α in SHR and WKY cardiac fibroblasts.
In the absence of ubiquitin(1–76) or ubiquitin(1–74) and as previously reported by us,13 a physiological concentration (10 nmol/L) of SDF-1α (an endogenous CXCR4 receptor agonist) significantly increased the proliferation of both SHR and WKY cardiac fibroblasts (Figures 1A–1D). In contrast, even supra-physiological concentrations (10,000 nmol/L) of ubiquitin(1–76) failed to stimulate cardiac fibroblast proliferation (Figures 1A and 1B). However, physiological concentrations of ubiquitin(1–76) significantly inhibited the pro-proliferative effects of SDF-1α (Figures 1A and 1B). In SHR cardiac fibroblasts the proliferative response to SDF-1α (expressed as the percentage change in cell number) was significantly attenuated by ubiquitin(1–76) concentrations as low as 1 nmol/L, and was essentially abolished by ubiquitin(1–76) concentrations greater than or equal to 10 nmol/L (Figure 2A). Compared to SHR cardiac fibroblasts, the potency of ubiquitin(1–76) was somewhat less in WKY cardiac fibroblasts. For example, in WKY cardiac fibroblasts 100 nmol/L of ubiquitin(1–76) was required to clearly inhibit the growth response to SDF-1α (Figure 2A). Curve fitting (non-linear regression analysis) of the data in Figure 2A to the 3-parameter equation ([inhibitor] versus % inhibition; Prism GraphPad,) indicated that the IC50 of ubiquitin(1–76) was 55 nmol/L in WKY versus 4 nmol/L in SHR.
Figure 1. Ubiquitin(1–76) and ubiquitin(1–74) antagonize the pro-proliferative effects of the CXCR4-receptor agonist SDF-1α in SHR and WKY cardiac fibroblasts.

Figure illustrates the concentration-dependent effects of ubiquitin(1–76) (panels A and B) and ubiquitin (1–74) (panels C and D) on proliferation of SHR (panels A and C) and WKY (panels B and D) cardiac fibroblasts stimulated with SDF-1α (10 nmol/L). In the absence of ubiquitins, a four-day treatment with SDF-1α significantly increased proliferation (cell number) of both SHR and WKY cardiac fibroblasts. In the absence of SDF-1α, a four-day treatment with either of the two ubiquitins did not affect cardiac fibroblast proliferation in either SHR or WKY cardiac fibroblasts. However, both ubiquitin(1–76) and ubiquitin(1–74) significantly antagonized the pro-proliferative effect of SDF-1α. Values are means ± SEM.
Figure 2. Panel A: The inhibitory potency of ubiquitin(1–76) on the pro-proliferative effects of the CXCR4-receptor agonist SDF-1α is greater in SHR compared with WKY cardiac fibroblasts.
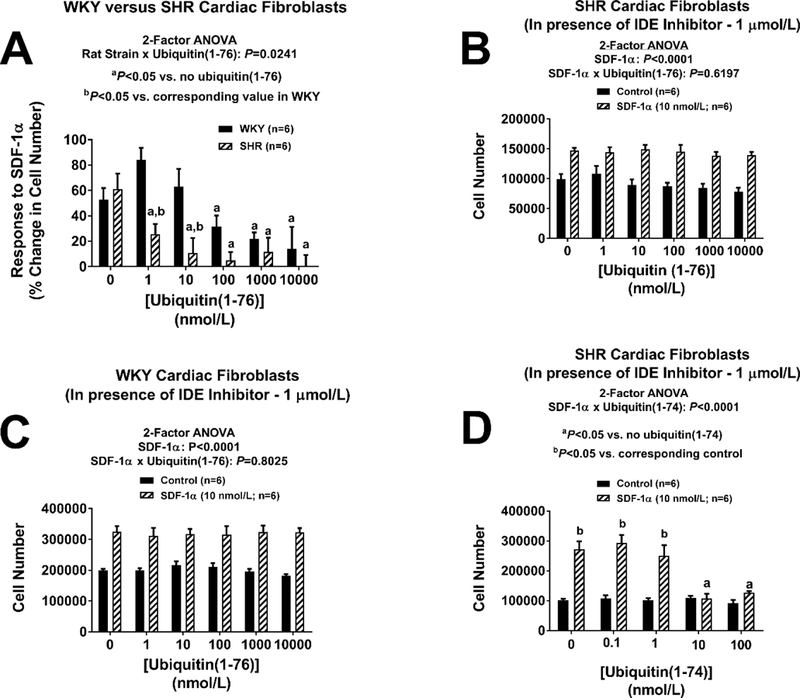
Panel A illustrates the proliferative response (expressed as the percentage change in cell number) to SDF-1α (10 nmol/L) at different concentrations of ubiquitin(1–76) (1 to 10,000 nmol/L). In SHR cardiac fibroblasts, ubiquitin(1–76) significantly inhibited the growth effects of SDF-1α at concentrations as low as 1 nmol/L. In contrast, in WKY cardiac fibroblasts a ubiquitin(1–76) concentration of 100 nmol/L was required to significantly inhibit the growth effects of SDF-1α. The calculated IC50 for ubiquitin(1–76) in SHR was 0.4 nmol/L; in contrast the IC50 for ubiquitin(1–76) in WKY was 55 nmol/L. Panels B and C: Ubiquitin(1–76) does not antagonize the pro-proliferative effects of the CXCR4-receptor agonist SDF-1α in SHR and WKY cardiac fibroblasts treated with an inhibitor of insulin-degrading enzyme. Cardiac fibroblast from SHR (panel B) and WKY (panel C) rats were treated with the insulin-degrading enzyme (IDE) inhibitor 6bk (1 μmol/L) for four days with or without SDF-1α (10 nmol/L) and with or without ubiquitin(1–76) (1 to 10,000 nmol/L). A four-day treatment with SDF-1α significantly increased proliferation (cell number) of both SHR and WKY cardiac fibroblasts, and this response was not affected by ubiquitin(1–76). Panels D: Ubiquitin(1–74) antagonizes the pro-proliferative effects of the CXCR4-receptor agonist SDF-1α in cardiac fibroblasts even in the presence of the insulin-degrading enzyme inhibitor 6bk. Cardiac fibroblasts from SHR rats were treated with the insulin-degrading enzyme (IDE) inhibitor 6bk (1 μmol/L) for four days with or without SDF-1α (10 nmol/L) and with or without ubiquitin(1–74) (1 to 100 nmol/L). A four-day treatment with SDF-1α significantly increased proliferation (cell number) of both SHR and WKY cardiac fibroblasts, and this response was inhibited by ubiquitin(1–74). In all panels, values are means ± SEM.
Ubiquitin(1–74) antagonizes the pro-proliferative effects of the CXCR4-receptor agonist SDF-1α in SHR and WKY cardiac fibroblasts.
In the absence of SDF-1α, ubiquitin(1–74) did not affect cardiac fibroblast proliferation in either SHR or WKY cardiac fibroblasts (Figures 1C and 1D). However, very low concentrations (1 nmol/L) of ubiquitin(1–74) completely inhibited the pro-proliferative effects of SDF-1α (10 nmol/L) in both SHR and WKY cardiac fibroblasts (Figures 1C and 1D). Therefore, although the inhibitory potency of ubiquitin(1–76) was greater in SHR cardiac fibroblasts, the inhibitory potency of ubiquitin(1–74) was similar in SHR versus WKY cardiac fibroblasts. Also the inhibitory potency of ubiquitin(1–74) was more than 10-fold greater than that for ubiquitin(1–76).
Ubiquitin(1–76) does not antagonize the pro-proliferative effects of the CXCR4-receptor agonist SDF-1α in SHR and WKY cardiac fibroblasts treated with an inhibitor of insulin-degrading enzyme (IDE).
Cardiac fibroblast from SHR and WKY rats were treated with the potent IDE inhibitor 6bk (1 μmol/L) and with or without SDF-1α (10 nmol/L) and with or without ubiquitin(1–76) (1 to 10,000 nmol/L). In the presence of an IDE inhibitor, SDF-1α significantly increased proliferation of both SHR and WKY cardiac fibroblasts; yet in cardiac fibroblasts treated with an IDE inhibitor, the ability of ubiquitin(1–76) to block the pro-growth effects of SDF-1α was abolished (Figures 2B and 2C). In this regard, even very high concentrations (10,000 nmol/L) of ubiquitin(1–76) were unable to attenuate growth responses to SDF-1α when IDE was inhibited (Figure 2: panels B and C). Notably, however, ubiquitin(1–74) inhibited the growth response to SDF-1α even in the presence of IDE inhibition (Figure 2D).
IDE activity is higher in cardiac fibroblasts from SHR compared with WKY cells.
The enzymatic activity of IDE was measured in SHR and WKY cardiac fibroblasts using the Anaspec SensoLyte 520 IDE Assay Kit. The performance of the assay was confirmed by assaying both recombinant IDE (large signal) and a negative control (no signal) (Figure 3A). Cardiac fibroblasts gave an IDE activity signal equivalent to the recombinant IDE positive control (Figure 3A). IDE enzymatic activity was significantly higher in SHR, compared with WKY, cardiac fibroblasts (Figures 3B, 3C, and 3D). RT-qPCR detected a high and similar expression of IDE mRNA in WKY and SHR cardiac fibroblasts (Ct: 22.48 ± 0.36 and 22.44 ± 0.06 for WKY and SHR cardiac fibroblasts, respectively; 2ΔCt relative to β-actin: 0.0013 ± 0.0002 and 0.0017 ± 0.0004 for WKY and SHR cardiac fibroblasts, respectively). Western blotting for IDE demonstrated a strong, and similar, signal for IDE expression (118 kDa) in both SHR and WKY cardiac fibroblasts (Figures 3E and 3F).
Figure 3. Panel A: Validation of the Anaspec SensoLyte® 520 insulin-degrading enzyme (IDE) assay:
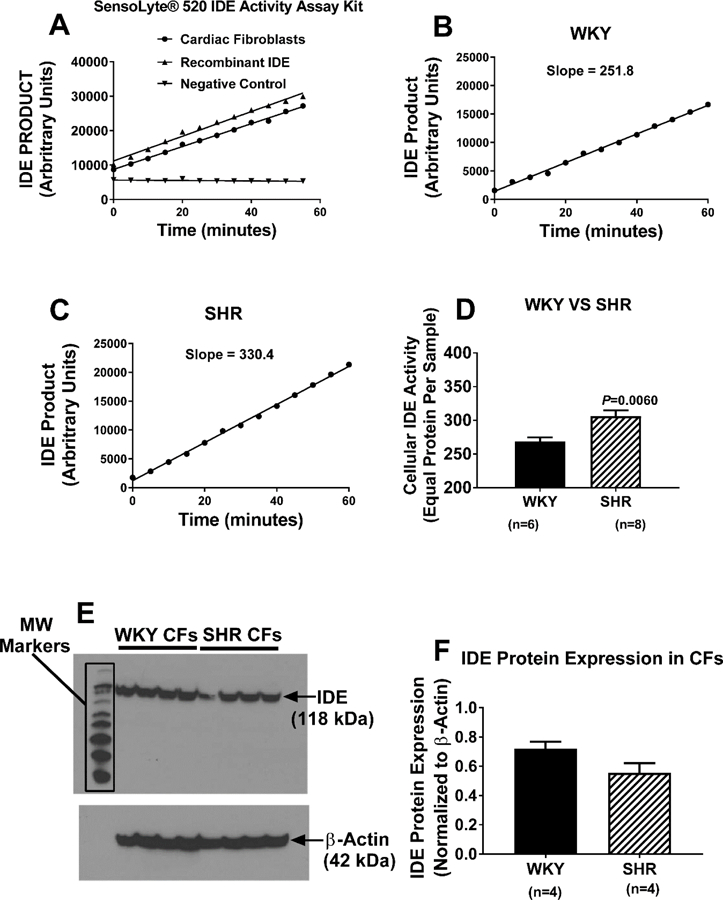
As active IDE cleaves a FRET substrate, 5-carboxyfluorescein is released, and its release is monitored over time by measuring fluorescence at excitation/emission of 490 nm/520 nm. IDE activity is proportional to the slope of the time versus product relationship. In the presence of recombinant IDE or a homogenate of cardiac fibroblasts, IDE product accumulated. In the absence of a source of IDE (negative control), there was no signal. Panels B, C, and D: IDE activity is greater in SHR compared with WKY cells. SHR and WKY cardiac fibroblasts were washed with phosphate-buffered saline, lysed in assay buffer, homogenized, and centrifuged. Cellular IDE activity was determined in 50 μl of supernatant. Panels B and C show typical results in WKY and SHR cardiac fibroblasts, respectively, and Panel D shows that the cellular IDE activity is significantly enhanced in SHR cardiac fibroblasts. Panels E and F. Both WKY and SHR cardiac fibroblasts express IDE protein. Panel E shows image of western blot for IDE in four batches of WKY and SHR cardiac fibroblast. Panel F shows the densitometry results from the image in panel E (normalized to β-actin). For all panels, values are means ± SEM.
Cardiac fibroblasts convert ubiquitin(1–76) to ubiquitin(1–74).
Figure S1 illustrates a mass spectrum generated by time-of-flight (TOF) high-mass resolution mass spectrometry using a Waters micromass Q-TOF mass spectrometer in which the collision cell was not employed so that only the non-fragmented full length peptides would be detected. The important aspects of this mass spectrum are the m/z signals corresponding to different numbers of positive charges on the full-length peptide (Mx+, where x = number of positive charges). Figure S2 illustrates a plot of the observed m/z values for each Mx+ versus the calculated m/z values from theory [assuming the signals were generated from ubiquitin(1–76)]. As shown, the plot of theoretical versus actual m/z values was linear, thus validating that the peptide used in our experiments was indeed authentic ubiquitin(1–76) uncontaminated by ubiquitin(1–74). Also, a 1-to-1 mixture of ubiquitin(1–76) and ubiquitin(1–74) was analyzed by Q-TOF mass spectrometry (Figure S3). A notable feature of this mass spectrum was the numerous closely-spaced pairs of m/z signals corresponding to different numbers of positive charges on the full-length peptides. In each doublet, the higher and lower m/z signals corresponded to ubiquitin(1–76) and ubiquitin(1–74), respectively. This mass spectrum suggested a strategy for detecting whether or not ubiquitin(1–76) can be converted to ubiquitin(1–74) by cardiac fibroblasts; i.e., by selected ion monitoring, using positive ion mass spectrometry, of medium samples collected from cardiac fibroblasts incubated with ubiquitin(1–76). Therefore, we incubated cardiac fibroblasts for 24 hours with 1000 nmol/L of highly pure ubiquitin(1–76), and after a series of sample purification steps (see Methods) subjected the samples to high-pressure liquid chromatography mass spectrometry using a Shimadzu HPLC/LCMS-2010 system operating in the positive ion mode. Although the Q-TOF mass spectrometer provided excellent high-mass resolution for identification and characterization, the Shimadzu system provided improved sensitivity for detection. The chromatographic peak (total ion current) corresponding to the retention time of ubiquitin(1–76) and ubiquitin(1–74) (retention time was the same for both peptides) was scanned for multiple charged ions (M12+ to M7+): for ubiquitin(1–76), 714.80, 779.80, 857.80, 952.80, 1071.80, and 1224.80 m/z, respectively; and for ubiquitin(1–74), 705.30, 769.50, 846.20, 940.20, 1057.50, and 1208.50 m/z, respectively. The signal for each ion under the chromatographic peak corresponding to the correct retention time was integrated and plotted. As shown in Figure 4 the expected pairs of m/z signals representing ubiquitin(1–76) and ubiquitin(1–74) were observed, with an estimated ratio of ubiquitin(1–74) to ubiquitin(1–76) of approximately 1%. This provided unequivocal evidence that cardiac fibroblasts converted some of the applied ubiquitin(1–76) to ubiquitin(1–74). This experiment was also performed in cardiac fibroblasts treated with both ubiquitin(1–76) and 6bk (IDE inhibitor). In 6bk treated cells, the signal-to-noise ratio for ubiquitin(1–74) was too low to identify or quantify the presence of ubiquitin(1–74) (Figure S4).
Figure 4. Cardiac fibroblasts convert ubiquitin(1–76) to ubiquitin(1–74).
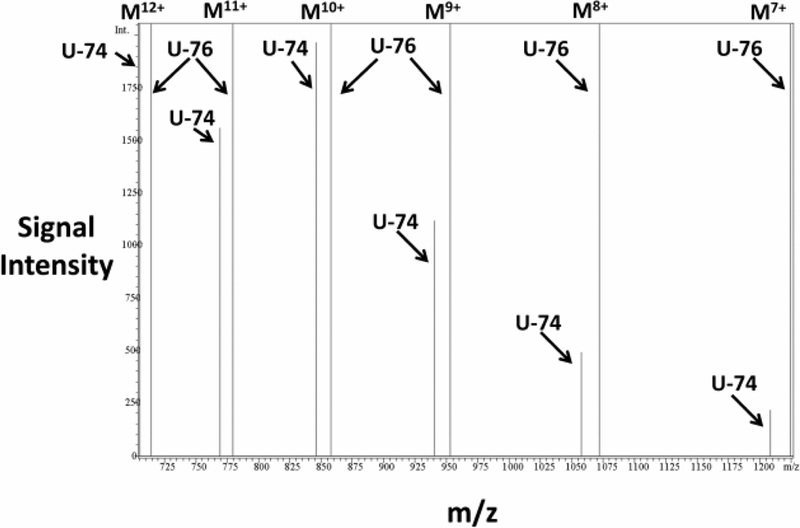
SHR cardiac fibroblasts were treated with ubiquitin(1–76) (U-76; 1000 nmol/L) for 24 hours, and the medium was collected and processed for analysis by mass spectrometry. Samples were loaded onto a polar reverse phase column connected to a Shimadzu HPLC/LCMS-2010 system. Separation was conducted with a linear gradient from 20% acetonitrile to 60% acetonitrile in the presence of 0.1% formic acid at flow rate 0.4 ml/min. The mass spectrometer was operated in the positive mode with selected ion monitoring. The chromatographic peak (total ion current) corresponding to the retention time of ubiquitin(1–76) and ubiquitin(1–74) (7.46 to 9.00 minutes) was scanned for multiple charged ions (M12+ to M7+): for ubiquitin(1–76), 714.80, 779.80, 857.80, 952.80, 1071.80, and 1224.80 m/z, respectively; for ubiquitin(1–74), 705.30, 769.50, 846.20, 940.20, 1057.50, 1208.50 m/z, respectively). The m/z versus signal intensity plot of the selected ions unequivocally identifies ubiquitin(1–74) as a product of ubiquitin(1–76).
Neither ubiquitin(1–76) nor ubiquitin(1–74) affects the pro-proliferative effects of neuropeptide Y (NPY) in cardiac fibroblasts.
As demonstrated in Figure 5 and as previously reported by us,16 treatment of cardiac fibroblasts with a physiological concentration (10 nmol/L) of NPY significantly increased proliferation of cardiac fibroblasts, and the magnitude of this effect was very similar to that observed for SDF-1α. Although the pro-growth effects of SDF-1α were abolished by both ubiquitin(1–76) (Figures 1A and 1B) and ubiquitin(1–74) (Figures 1C and 1D), ubiquitin(1–76) and ubiquitin(1–74) had no effect whatsoever on the pro-proliferative effects of NPY (Figures 5A and 5B).
Figure 5. Neither ubiquitin(1–76) nor ubiquitin(1–74) affects the pro-proliferative effects of neuropeptide Y in cardiac fibroblasts.
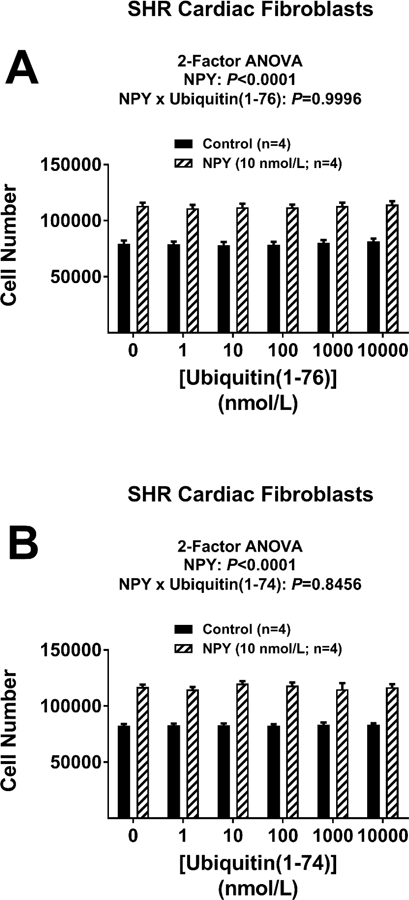
Figure illustrates the lack of effects of ubiquitin(1–76) (panel A) and ubiquitin(1–74) (panel B) (1 to 10,000 nmol/L) on cardiac fibroblasts stimulated with neuropeptide Y (NPY; 10 nmol/L). A four-day treatment with NPY significantly increased proliferation (cell number) of cardiac fibroblasts, and this effect was not attenuated by either ubiquitin(1–76) or ubiquitin(1–74). Values are means ± SEM.
DISCUSSION
Ubiquitin was identified in the early 1970s as a protein that occurs in cells across the kingdoms of life,18 and in the late 1970s and early 1980s ubiquitin was further characterized as a critical component of the ubiquitin-proteosome system.19 Nowadays, ubiquitylation is considered second only to phosphorylation as a modality of post-translational modification of proteins.15 In addition to intracellular roles, reports in the 1990s that ubiquitin exists in the extracellular compartment, including the plasma, suggested the possibility that ubiquitin might also be involved in extracellular signaling.15 This possibility was confirmed by reports that extracellular ubiquitin affects a number of different cell types, including in particular leukocytes and platelets.15 That ubiquitin is involved in extracellular signaling indicated that there likely is a cell surface ubiquitin receptor. Indeed, in 2010, Saini et al.20 made the seminal discovery that the CXCR4 receptor binds ubiquitin and in THP1 cells ubiquitin is a CXCR4 agonist.
Following the 2006 FDA approval of the DPP4 inhibitor sitagliptin for management of type 2 diabetes, we became interested in the role of DPP4 as a modulator of the activity of peptide-based extracellular signaling systems.16, 21–25 More recently, this research program motivated our investigation of the interaction of sitagliptin with the DPP4 substrate SDF-1α on cardiac fibroblast proliferation. This led to our 2017 discovery that SDF-1α, via the CXCR4 receptor, stimulates proliferation of cardiac fibroblasts and that inhibition of SDF-1α metabolism by DPP4, augments this effect.13 Following that discovery, we decided to investigate the effects of other endogenous CXCR4 agonists on cardiac fibroblast proliferation and became aware of the literature regarding ubiquitin(1–76) as a CXCR4 agonist in THP1 cells. This led to the present study.
Our working hypothesis in the current study was that ubiquitin(1–76) would stimulate cardiac fibroblast proliferation by activating CXCR4 receptors. However, our preliminary experiments in this regard were negative, thus invalidating our working hypothesis. Fortunately, in one experiment we happened to combine ubiquitin(1–76) with SDF-1α and noted that SDF-1α did not affect cardiac fibroblast proliferation in the presence of ubiquitin(1–76). To confirm that observation, in the present study we examined the concentration-dependent effects of ubiquitin(1–76) on SDF-1α-induced proliferation of cardiac fibroblasts and noted that in both SHR and WKY ubiquitin(1–76) potently inhibited the pro-proliferative effects of SDF-1α. Our next goal was to understand why ubiquitin(1–76) exerted an effect on SDF-1α signaling that was opposite of what we anticipated.
In 2013 Tripathi and coworkers published that IDE converts ubiquitin(1–76) to ubiquitin(1–74) and that ubiquitin(1–74) is a CXCR4 antagonist.14 These findings by Tripathi et al. motivated our second working hypothesis, i.e., that in cardiac fibroblasts, ubiquitin(1–76) does not activate the CXCR4 receptor but rather is metabolized by IDE expressed in cardiac fibroblasts to ubiquitin(1–74) which potently blocks CXCR4 receptors. This hypothesis has several testable predictions: 1) Ubiquitin(1–74) should be more potent that ubiquitin(1–76) with regard to inhibiting the effects of SDF-1α; 2) Cardiac fibroblasts should express IDE mRNA, protein, and activity; 3) In the presence an IDE inhibitor, ubiquitin(1–76) should no longer block the pro-proliferative actions of SDF-1α; 4) IDE inhibition should not affect the inhibitory activity of ubiquitin(1–74); 5) Cardiac fibroblasts should convert ubiquitin(1–76) to ubiquitin(1–74), and this should be blocked by inhibition of IDE; 6) Neither ubiquitin(1–76) nor ubiquitin(1–74) should affect the ability of other non-CXCR4 receptor agonists, such as NPY, to stimulate proliferation of cardiac fibroblasts. Because our experiments confirmed all of these predictions, we conclude that our second working hypothesis is correct.
As mentioned, SHR, compared with WKY, cardiac fibroblasts are more sensitive to the effects of ubiquitin(1–76). This is likely due to increased conversion of ubiquitin(1–76) to ubiquitin(1–74) in cells from genetically hypertensive animals. This conclusion is based on two observations. First, ubiquitin(1–74) blocks the effects of SDF-1α similarly in SHR and WKY cardiac fibroblasts. This rules out a difference in the affinity of SHR versus WKY CXCR4 receptors for ubiquitin(1–74) and thus focuses the explanation on differential IDE activity in SHR versus WKY cells. Second, direct measurements of IDE activity show that IDE activity is significantly higher in SHR versus WKY cardiac fibroblasts. Why IDE activity is greater in SHR versus WKY cells is unclear since the expression of IDE mRNA and protein is similar in SHR versus WKY cardiac fibroblasts. It is conceivable, however, that the cellular localization, dimerization, or post-translational modification of IDE is different in the two strains and accounts for the difference in IDE activity.
Another open question is: Why is ubiquitin(1–76) an agonist in some cell types (for example THP1 cells), but not in others (for example cardiac fibroblasts)? Conceivable this could be explained by biased agonism, post-translational modification of CXCR4 receptors, or binding partners with CXCR4 receptors that are required for ubiquitin(1–76) signaling.
Previously we showed that in cardiac fibroblasts SDF-1α/sitagliptin induced three biomarkers of cardiac fibroblast activation (i.e., proliferation, hypertrophy, and collagen production), and that antagonism of CXCR4 receptors abolished all three biomarkers of cardiac fibroblast activation.13 Thus in the current study we focused on the most important indicator of fibroblast activation, i.e., cell proliferation. Nonetheless, it would be of value to examine in future studies the interactions among SDF-1α, DPP4 inhibition, ubiquitin(1–76), and ubiquitin(1–74) on cellular hypertrophy and extracellular matrix production. Although there is strong evidence that fibroblasts are critically involved in organ fibrosis,26 in vivo studies are required to confirm whether the balance of extracellular ubiquitin(1–76) versus extracellular ubiquitin(1–74) is a determinant of the risk of organ fibrosis.
Perspectives.
An implication of this study is that IDE activity can determine whether extracellular ubiquitin is in balance an inhibitor of CXCR4-receptor activity. When IDE activity is high, the inhibitory effects of ubiquitin(1–74) would prevail; in contrast when IDE activity is low, CXCR4 receptors would not be impeded by ubiquitin(1–74), and in those cell types in which ubiquitin(1–76) is an agonist, CXCR4 receptor signaling would be augmented.
As reviewed by Brown et al.26, cardiac fibroblasts likely participate in the pathophysiology of cardiac remodeling and heart failure. Therefore, another implication of our findings is that the activity of IDE may determine patient susceptibility to cardiac remodeling and heart failure. For example, in patients with hypertension and other cardiovascular conditions associated with high levels of SDF-1α and cardiac hypertrophy, whether the levels of ubiquitin(1–74) are adequate to block the pro-proliferative effects of endogenous SDF-1α on cardiac fibroblasts could be an important determinant of whether a given patient develops heart failure.
Yet another implication of the present findings is illustrated in Figure 6. The current study shows that cardiac fibroblasts express IDE, which converts ubiquitin(1–76) to ubiquitin(1–74). And our previous study13 shows that cardiac fibroblasts express DPP4, which converts SDF-1α(1–68) to SDF-1α(3–68). SDF-1α(1–68), but not SDF-1α(3–68), is a CXCR4-receptor agonist; and ubiquitin(1–74), but not ubiquitin(1–76), is a CXCR4-receptor antagonist. DPP4 inhibitors are widely used for managing type 2 diabetes, and IDE inhibitors are being considered for the same condition.27 If IDE inhibitors are introduced to clinical medicine as anti-diabetic drugs, the present findings caution against co-administration of DPP4 inhibitors and IDE inhibitors because of the risk of increasing the levels of a CXCR4-receptor agonist while simultaneously decreasing the levels of a CXCR4-receptor antagonist. Also, our previous studies show that cardiac fibroblasts from genetically-hypertensive animals are more responsive to SDF-1α, and the present study suggests that IDE activity is greater in cardiac fibroblasts from genetically-hypertensive animals. Thus the potentially harmful interaction between DPP4 inhibitors and IDE inhibitors may be exacerbated in hypertension. Thus, viewed in a broad context, the clinical implications of our results may be profound in that the extracellular ubiquitin system could be a major determinant of heart fibrosis, and possibly fibrosis of other organ systems, in cardiovascular disease, diabetes, and hypertension.
Figure 6. Implications of the current findings.

The current findings uncover a potentially harmful interaction between DPP4 inhibitors and IDE inhibitors that may be exacerbated in hypertension.
Novelty and Significance
What Is New?
Extracellular ubiquitin(1–76) inhibits the pro-proliferative effects of the SDF-1α/CXCR4 axis in both SHR and WKY cardiac fibroblasts, but more so in SHR cardiac fibroblasts.
The mechanism by which ubiquitin(1–76) blocks the SDF-1α/CXCR4 axis involves the IDE-mediated metabolism of ubiquitin(1–76) to the CXCR4 antagonist ubiquitin(1–74).
IDE activity is greater in cardiac fibroblasts obtained from genetically-hypertensive animals.
What Is Relevant?
By governing the balance between ubiquitin(1–76) and ubiquitin(1–74), IDE activity may determine whether or not cardiac fibroblasts induce cardiac fibrosis in hypertensive patients or other patients with cardiovascular diseases and elevated levels of SDF-1α or CXCR4 receptors.
Summary.
The results of this study support the concept that ubiquitin(1–76) inhibits the pro-proliferative effects of SDF-1α on cardiac fibroblasts due to its conversion by IDE to the CXCR4 antagonist ubiquitin(1–74). This concept has major implications. One implication is that IDE activity can determine whether extracellular ubiquitin is in balance an inhibitor of CXCR4 activity. Another implication is that the activity of IDE may determine patient susceptibility to cardiac fibrosis, remodeling, and heart failure. Yet another implication relates to our previous study showing that cardiac fibroblasts express DPP4, which converts SDF-1α(1–68) to SDF-1α(3–68). SDF-1α(1–68), but not SDF-1α(3–68), is a CXCR4-receptor agonist; and ubiquitin(1–74), but not ubiquitin(1–76), is a CXCR4-receptor antagonist. DPP4 inhibitors are widely used for managing type 2 diabetes, and IDE inhibitors are being considered for the same condition. If IDE inhibitors are introduced to clinical medicine as anti-diabetic drugs, the present findings caution against co-administration of DPP4 inhibitors and IDE inhibitors because of the risk associated with increasing the levels of a CXCR4-receptor agonist while simultaneously decreasing the levels of a CXCR4-receptor antagonist. Finally, our previous studies show that cardiac fibroblasts from genetically-hypertensive animals are more responsive to SDF-1α, and the present study suggests that IDE activity is greater in cardiac fibroblasts from genetically-hypertensive animals. Thus the potentially harmful interaction between DPP4 inhibitors and IDE inhibitors may be exacerbated in hypertension.
Supplementary Material
Acknowledgments
SOURCES OF FUNDING
The work was supported by the National Institutes of Health [DK091190, HL069846, DK068575, HL109002 and DK079307].
Footnotes
Disclosures: None
Contributor Information
Edwin K. Jackson, Department of Pharmacology and Chemical Biology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania 15219
Eric Mi, Department of Pharmacology and Chemical Biology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania 15219.
Vladimir B. Ritov, Department of Pharmacology and Chemical Biology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania 15219
Delbert G. Gillespie, Department of Pharmacology and Chemical Biology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania 15219
REFERENCES
- 1.Chu P- Y, Zatta A, Kiriazis H, Chin-Dusting J, Du X- J, Marshall T, Kaye DM. CXCR4 antagonism attenuates the cardiorenal consequences of mineralocorticoid excess. Circ Heart Fail 2011;4:651–658. [DOI] [PubMed] [Google Scholar]
- 2.Shao S, Cai W, Sheng J, Yin L. Role of SDF-1 and Wnt signaling pathway in the myocardial fibrosis of hypertensive rats. Am J Transl Res 2015;7:1345–1356. [PMC free article] [PubMed] [Google Scholar]
- 3.Chu PY, Walder K, Horlock D, Williams D, Nelson E, Byrne M, Jandeleit-Dahm K, Zimmet P, Kaye DM. CXCR4 antagonism attenuates the development of diabetic cardiac fibrosis. PLoS ONE 2015;10:e0133616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kazakov A, Hall R, Jagoda P, Bachelier K, Muller-Best P, Semenov A, Lammert F, Bohm M, Laufs U. Inhibition of endothelial nitric oxide synthase induces and enhances myocardial fibrosis. Cardiovasc Res 2013;100:211–221. [DOI] [PubMed] [Google Scholar]
- 5.Schober A, Knarren S, Lietz M, Lin EA, Weber C. Crucial role of stromal cell-derived factor-1α in neointima formation after vascular injury in apolipoprotein E-deficient mice. Circulation 2003;108:2491–2497. [DOI] [PubMed] [Google Scholar]
- 6.Zernecke A, Schober A, Bot I, von Hundelshausen P, Liehn EA, Mopps B, Mericskay M, Gierschik P, Biessen EA, Weber C. SDF-1α/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells. Circ Res 2005;96:784–791. [DOI] [PubMed] [Google Scholar]
- 7.Zuk A, Gershenovich M, Ivanova Y, MacFarland RT, Fricker SP, Ledbetter S. CXCR4 antagonism as a therapeutic approach to prevent acute kidney injury. Am J Physiol Renal Physiol 2014;307:F783–797. [DOI] [PubMed] [Google Scholar]
- 8.Yuan A, Lee Y, Choi U, Moeckel G, Karihaloo A. Chemokine receptor Cxcr4 contributes to kidney fibrosis via multiple effectors. Am J Physiol Renal Physiol 2015;308:F459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sayyed SG, Hagele H, Kulkarni OP, Endlich K, Segerer S, Eulberg D, Klussmann S, Anders HJ. Podocytes produce homeostatic chemokine stromal cell-derived factor-1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia 2009;52:2445–2454. [DOI] [PubMed] [Google Scholar]
- 10.Chu PY, Mariani J, Finch S, McMullen JR, Sadoshima J, Marshall T, Kaye DM. Bone marrow-derived cells contribute to fibrosis in the chronically failing heart. Am J Pathol 2010;176:1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Subramanian S, Liu C, Aviv A, Ho JE, Courchesne P, Muntendam P, Larson MG, Cheng S, Wang TJ, Mehta NN, Levy D. Stromal cell-derived factor 1 as a biomarker of heart failure and mortality risk. Arterioscler Thromb Vasc Biol 2014;34:2100–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuern CS, Walker B, Sauter M, Schaub M, Chatterjee M, Mueller K, Rath D, Vogel S, Tegtmeyer R, Seizer P, Geisler T, Kandolf R, Lang F, Klingel K, Gawaz M, Borst O. Endomyocardial expression of SDF-1 predicts mortality in patients with suspected myocarditis. Clin Res Cardiol 2015;104:1033–1043. [DOI] [PubMed] [Google Scholar]
- 13.Jackson EK, Zhang Y, Gillespie DD, Zhu X, Cheng D, Jackson TC. SDF-1α (stromal cell-derived factor 1α) induces cardiac fibroblasts, renal microvascular smooth muscle cells, and glomerular mesangial cells to proliferate, cause hypertrophy, and produce collagen. Journal of the American Heart Association 2017;6:e007253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tripathi A, Saini V, Marchese A, Volkman BF, Tang WJ, Majetschak M. Modulation of the CXC chemokine receptor 4 agonist activity of ubiquitin through C-terminal protein modification. Biochemistry (Mosc) 2013;52:4184–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Majetschak M Extracellular ubiquitin: immune modulator and endogenous opponent of damage-associated molecular pattern molecules. J Leukoc Biol 2011;89:205–219. [DOI] [PubMed] [Google Scholar]
- 16.Zhu X, Gillespie DG, Jackson EK. NPY1–36 and PYY1–36 activate cardiac fibroblasts: an effect enhanced by genetic hypertension and inhibition of dipeptidyl peptidase 4. Am J Physiol Heart Circ Physiol 2015;309:H1528–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng D, Zhu X, Gillespie DG, Jackson EK. Role of RACK1 in the differential proliferative effects of neuropeptide Y1–36 and peptide YY1–36 in SHR vs. WKY preglomerular vascular smooth muscle cells. Am J Physiol Renal Physiol 2013;304:F770–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldstein G, Scheid M, Hammerling U, Schlesinger DH, Niall HD, Boyse EA. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proceedings of the National Academy of Sciences 1975;72:11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hershko A Ubiquitin-mediated protein degradation. J Biol Chem 1988;263:15237–15240. [PubMed] [Google Scholar]
- 20.Saini V, Marchese A, Majetschak M. CXC chemokine receptor 4 is a cell surface receptor for extracellular ubiquitin. J Biol Chem 2010;285:15566–15576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackson EK. Context-dependent effects of dipeptidyl peptidase 4 inhibitors. Curr Opin Nephrol Hypertens 2017;26:83–90. [DOI] [PubMed] [Google Scholar]
- 22.Jackson EK, Mi Z, Tofovic SP, Gillespie DG Effect of dipeptidyl peptidase 4 inhibition on arterial blood pressure is context dependent. Hypertension 2015;65:238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson EK, Kochanek SJ, Gillespie DG. Dipeptidyl peptidase IV regulates proliferation of preglomerular vascular smooth muscle and mesangial cells. Hypertension 2012;60:757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson EK, Dubinion JH, Mi Z. Effects of dipeptidyl peptidase IV inhibition on arterial blood pressure. Clin Exp Pharmacol Physiol 2008;35:29–34. [DOI] [PubMed] [Google Scholar]
- 25.Jackson EK, Zhang M, Liu W, Mi Z. Inhibition of renal dipeptidyl peptidase IV enhances peptide YY1–36-induced potentiation of angiotensin II-mediated renal vasoconstriction in spontaneously hypertensive rats. J Pharmacol Exp Ther 2007;323:431–437. [DOI] [PubMed] [Google Scholar]
- 26.Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol 2005;45:657–687. [DOI] [PubMed] [Google Scholar]
- 27.Tang W- J. Targeting Insulin-Degrading Enzyme to Treat Type 2 Diabetes Mellitus. Trends in Endocrinology & Metabolism 2016;27:24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.