

Abstract
Background:
Single ventricle congenital heart disease (SV) is fatal without intervention and eventual heart failure (HF) is a major cause of morbidity and mortality. While there are no proven medical therapies for the treatment or prevention of HF in the SV population, phosphodiesterase-5 inhibitors (PDE5i), such as sildenafil, are increasingly utilized. While the pulmonary vasculature is the primary target of PDE5i therapy in patients with SV, the effects of PDE5i on the SV myocardium remain largely unknown. We sought to determine PDE5 expression and activity in the single right ventricle (RV) of SV patients relative to non-failing (NF) controls, and to determine if PDE5 impacts cardiomyocyte remodeling using a novel serum based in vitro model.
Methods and Results:
PDE5 expression (n=9 NF, n=7 SV), activity (n=8 NF, n=9 SV) and localization (n=3 SV) were determined in explanted human RV myocardium. PDE5 is expressed in SV cardiomyocytes and PDE5 protein expression and activity are increased in SV RV compared to NF RV. Isolated neonatal rat ventricular myocytes (NRVMs) were treated for 72 hours with NF or SV patient serum ± sildenafil. RT-qPCR (n=5 NF, n=12 SV) and RNAseq (n=3 NF, n=3 SV) were performed on serum-treated NRVMs and demonstrated that treatment with SV sera results in pathological gene expression changes which are attenuated with PDE5i.
Conclusions:
PDE5 is increased in failing SV myocardium and pathologic gene expression changes in SV serum-treated NRVMs are abrogated by PDE5i. These results suggest that PDE5 represents an intriguing myocardial therapeutic target in this population.
Introduction
Congenital heart defects (CHD) are the single most common type of birth defect and a leading cause of infant death in the United States1. Single ventricle heart disease (SV) is the most common form of severe CHD2 and comprises a spectrum of congenital cardiac malformations defined by severe underdevelopment of one ventricle. SV is universally fatal without intervention, and although outcomes are improving, more than 30% of SV patients die or require transplant within the first year of life and 10-year survival in this population is only 39–50%2,3. While the single ventricle can be a morphologic right ventricle (RV), left ventricle (LV) or of indeterminate morphology, single RV lesions such as hypoplastic left heart syndrome (HLHS) may be inherently at risk for heart failure presumably due to inherent limitations in the RV’s ability to tolerate increased afterload3–6. As such, eventual heart failure (HF) is a leading cause of death and indication for heart transplant in the SV population3. While surgical techniques and perioperative care for SV continue to improve, long-term survival and quality of life ultimately depend on preservation of RV function. However, the molecular mechanisms underlying failure of the single RV are poorly understood, limiting the ability to identify effective therapies. Moreover, the ability to investigate mechanisms involved in SV HF or effects of pharmacologic interventions are limited by the lack of a postnatal animal model of SV and the difficulties associated with performing in vivo research in children.
Pharmacological treatment of HF in the pediatric population is extrapolated from the adult HF experience, and treatment guidelines are based primarily on expert consensus7. In recent clinical trials, the SV population demonstrated a detrimental trend in response to carvedilol8, and a lack of benefit in preventing HF with enalapril9, medications commonly used to treat adult HF. Given the divergent response to these adult HF medications in the pediatric SV population, age-related mechanisms governing HF, as well as fundamental differences in RV versus LV failure likely exist.
While there are currently no proven medical therapies for the treatment or prevention of HF in the SV population, selective and competitive phosphodiesterase type-5 inhibitors (PDE5i), such as sildenafil, are increasingly used in children and adults with SV. However, while sildenafil has been associated with improved hemodynamics, exercise tolerance and myocardial function by echocardiography in small series of SV patients, its direct effects outside of the pulmonary vasculature remain largely unknown10–13. PDE5 expression is prominent throughout the pulmonary vasculature and in the coronary vessels, but there is virtually no detectable PDE5 expression in the normal adult myocardium14. Thus, the rationale for the use of PDE5i in the SV population is PDE5 inhibition in the pulmonary vasculature, thereby promoting vascular smooth muscle relaxation, augmenting pulmonary blood flow and secondarily SV cardiac output15. Correspondingly, PDE5i’s have been shown to lack any significant direct effects on the myocardium of normal, non-failing (NF) human and animal hearts in vitro16,17. However, recent data in both human HF patients and in animal models of HF, suggest that myocardial and cardiomyocyte-specific PDE5 expression is increased under conditions of cardiac stress, and is physiologically significant18–20. Increased myocardial PDE5 for example, has been shown to be associated with maladaptive myocardial responses, and its inhibition both globally and in cardiomyocytes specifically, can have direct beneficial myocardial effects21,22.
In the present study, we sought to determine myocardial PDE5 expression and activity in SV patients relative to NF controls, and to evaluate PDE5 localization within the myocardium. Additionally, we utilized a novel model of cardiomyocyte remodeling, in which primary cardiomyocytes are treated with SV patient sera +/− sildenafil, to assess changes in pathologic cardiac gene expression and the impact of PDE5i in vitro.
Methods
The data, analytic methods, and study materials (with the exception of patient samples, as these represent a limited resource) will be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Human Samples
All subjects gave informed consent and donated their hearts and/or blood to the Institutional Review Board-approved Investigations of Pediatric Heart Disease tissue bank at the University of Colorado, Denver. NF RV tissues were from pediatric (<18 years of age) organ donors with normal heart structure and function, whose hearts could not be placed for technical reasons (size or blood type mismatch). Samples of NF sera were obtained from control children with normal heart function. SV RV tissue and sera were from patients transplanted with SV of RV morphology who were transplanted secondary to systolic heart failure (single ventricle disease of LV or indeterminate morphology were excluded). Inclusion criteria for SV patients were: age <18 years and abnormal SV systolic function on echocardiogram. Patients that were transplanted primarily for SV lacking these defined clinical characteristics were excluded. At the time of cardiac explant, the heart tissue was rapidly dissected in the operating room, flash frozen, and stored at −80°C until further use.
PDE activity assays
Approximately 150 mg of RV myocardium was homogenized and separated into nuclear, cytosolic, and sarcoplasmic reticulum-enriched microsomal fractions by differential centrifugation. cGMP-hydrolytic activity in the cytosolic fraction was quantified at 30°C by the 2-step snake-venom method with (3H) cGMP (0.1 μmol/L) as substrate23,24. Total cGMP-hydrolytic activity was quantified by measuring activity without addition of PDE inhibitor. PDE5 activity was quantified by measuring activity in the absence and presence of 0.01 μmol/L sildenafil, a concentration that inhibits recombinant PDE5 submaximally (IC46) in order to minimize possible inhibition of other PDE families24. PDE5-specific activity was calculated by dividing the difference in activity between the absence and presence of sildenafil by the fractional inhibition of PDE5 activity at this concentration. The amount of protein used per assay were adjusted to ensure that 20% (±15%) of the total cGMP was hydrolyzed during the assay.
Western blots
Western blots were performed as described25. Protein was isolated from 10- to 25-mg frozen RV tissue in isoelectric focusing buffer at 4°C. PDE5A (Cell Signaling) and GAPDH (Santa Cruz Biotechnology) were quantified on the same blot. Blots were quantified using ImageJ (U.S. National Institutes of Health).
Immunohistochemistry and confocal microscopy
Briefly, freshly procured RV tissue was fixed in 4% paraformaldehyde at 4°C for 4–12 hours, rinsed in PBS and immersed in 30% sucrose solution for 6–12 hours at 4°C for cryoprotection. Fixed tissue was embedded and frozen in OCT media, and tissue was cryosectioned at −18 to −20°C into 6–10μm sections and mounted on microscope slides. Slides were stored at −80°C for no longer than 1 month before processing. Sections were fixed in ice cold acetone for 10 minutes, rinsed in PBS, and blocked for 1 hour in blocking buffer (2% normal goat sera, 1% BSA, and 0.1% NP-40 in PBS). Primary antibodies rabbit PDE5A (1:100, Abcam) and mouse sarcomeric α actinin (1:400, Sigma Aldrich) were incubated overnight at 4°C, and Alexa Fluor- conjugated secondary antibodies (1:1000, Alexa 555 goat anti mouse and Alexa 488 goat anti rabbit, Sigma Aldrich) were incubated for 1 hour at room temperature. Nuclei were stained with DAPI (1μM, Invitrogen) for 10 minutes at room temperature. Secondary antibody-only staining confirmed lack of nonspecific binding for all antibodies used. Slides were mounted in Prolong Diamond anti-fade mounting media (Thermo Fisher Scientific) and images were acquired on the Olympus FLUOVIEW FV1000 confocal laser scanning microscope at 60X magnification (FITC: 488nm excitation, 520nm emission; TRITC: 543nm excitation, 618nm emission; DAPI: 405nm excitation, 461nm emission).
Cell culture and serum treatment
Neonatal rat ventricular myocytes (NRVMs) were isolated from the ventricles of 1- to 2-day-old Sprague Dawley rats (Charles River) by enzymatic digestions as described26. NRVMs were treated with 2% human serum for 72 hours as described26. NRVMs were pre-incubated with 1.5μM sildenafil for 30 minutes prior to serum treatment. Efficiency of NRVM response was tested by treatment of cells with 10−5M phenylephrine (PE) for 72 hours as a positive control. All animal protocols are in accordance with PHS Animal Welfare Assurance, ID A3269–01, and approved by the University of Colorado, Denver - Animal Care and Use Committee.
RNA isolation and RT-qPCR
NRVMs were homogenized in Qiazol (Qiagen), and RNA was extracted as per manufacturer’s protocol. Human RV samples were homogenized in Qiazol, RNA was extracted using the RNeasy plus mini kit (Qiagen), and following extraction, RNA was treated with TURBO DNase (Thermo Fisher Scientific) as per manufacturer’s protocol. cDNA was synthesized by using the Verso cDNA synthesis kit (Thermo Fisher Scientific) according to manufacturer’s instructions. Gene expression was measured by RT-qPCR as described27, with Power Sybr Green PCR Master Mix (Life Technologies). Expression levels of all transcripts were normalized to 18S rRNA, and all RT-qPCR data are represented on a Log2 scale. RT-qPCR primers sequences are listed in supplemental Table S2.
RNA-seq and Transcriptome Analysis
1X150 directional mRNA sequencing was performed on an Illumina hiSEQ 4000 (HT Mode), with an average of 38–44 million mapped reads per sample. Samples were de-multiplexed and aligned to the reference genome R. norvegicus (Rnor_5.0) using gsnap28. HTSeq was used to produce gene counts for each of our 12 samples (N=3 NF serum treated NRVMs ±PDE5i and N=3 SV serum treated NRVMs ±PDE5i, from 3 different NRVM preparations)29. Counts of reads generated by HTSeq were normalized using edgeR30. While RNA-seq identified more than 20,000 genes, counts of ≥10 in any one sample were necessary for inclusion. After removal of low counts, 7,690 genes remained. Significant changes in gene expression were calculated using an ANOVA and a post-hoc Tukey with a non-stringent threshold of p<0.05. RNAseq on NRVMs was performed to generate hypotheses in regards to the human SV myocardium, therefore there was no correction for multiplicity of measured values. Differentially expressed genes of interest were validated in a cohort of human samples. Hierarchical clustering and heatmap generation were performed using R (The R Foundation). Ingenuity Pathway Analysis (IPA) and Gene Ontology (GO) categorization using PANTHER were performed to investigate molecular pathways, biological processes and toxicity functions associated with differentially regulated genes.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism (GraphPad Software), and statistical significance was set a priori at p<0.05. Normality of data was assessed, and non-normally distributed groups were Log2 transformed, after which all data were approximately normally distributed. Comparisons between 2 normally distributed groups were conducted using an unpaired t-test. Comparisons of 3 or more normally distributed groups were conducted using a 1-way ANOVA; if the overall comparison reached significance, Holm-Sidak multiple comparisons post-hoc tests were performed. Quantitative results in figures are shown as mean±SEM, and in the text as mean±SD. For log-transformed data, the mean of the log-transformed data is reported.
Results
Patient Characteristics
Aggregate characteristics for patients included in this study are listed in Table 1. A more detailed description of individual subject characteristics is included in Table S1. The NF tissue and blood group included 17 subjects with a median age of 8 years, an interquartile range (IQR) of 3.1–13.6, and was 65% male. The SV tissue and blood group had 18 subjects with a median age of 2.6 years, an IQR of 0.9–4.8, and was 67% male. As expected based on limited young donor availability, the SV group is significantly younger than the NF group (p=0.004).
Table 1.
Patient Characteristics
| Experiment | Group | No. of Subjects | % Male | Median Age (years) [IQR] | % PDE3i | % PDE5i | % Non-PDEi Inotrope* | % Digoxin | % ACEi | % β-Blocker | % Diuretic |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PDE5 Activity | NF | 8 | 62.5 | 7.7 [2.8–8.9] | 0 | 0 | 50 | 0 | 0 | 12.5 | 0 |
| SV | 9 | 55.6 | 2.8 [1–3.8] | 88.9 | 22.2 | 11.1 | 66.7 | 77.8 | 0 | 77.8 | |
| Tissue PDE5 Protein | NF | 9 | 66.7 | 7.4 [3.2–8.7] | 0 | 0 | 33.3 | 0 | 0 | 0 | 0 |
| SV | 7 | 71.4 | 2.3 [1.1–2.9] | 71.4 | 14.3 | 0 | 57.1 | 85.7 | 0 | 85.7 | |
| Tissue PDE5 Histology | NF | 1 | 100 | 2.9 [n/a] | 0 | 0 | 100 | 0 | 0 | 0 | 0 |
| SV | 3 | 100 | 2.9 [1.6–7.6] | 66.7 | 33.3 | 0 | 100 | 66.7 | 0 | 66.7 | |
| NRVM + Serum RT-PCR | NF | 5 | 40 | 11.5 [7–13.6] | 0 | 0 | 0 | 20 | 0 | 0 | 0 |
| SV | 12 | 58.3 | 2 [0.9–4.8] | 50 | 33.3 | 0 | 66.7 | 50 | 0 | 75 | |
| NRVM + Serum RNAseq | NF | 3 | 33.3 | 13.6 [12.5–14.5] | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| SV | 3 | 33.3 | 1.9 [0.9–3.2] | 100 | 66.7 | 0 | 100 | 66.7 | 0 | 66.7 | |
| Tissue RT-PCR | NF | 7 | 57.1 | 7 [2.3–7.7] | 0 | 0 | 57.1 | 0 | 0 | 14.3 | 0 |
| SV | 14 | 57.1 | 1.78 [0.8–3.6] | 71.4 | 21.4 | 0 | 64.3 | 64.3 | 0 | 78.6 | |
| All Patients | NF | 17 | 64.7 | 8.0 [3.1–13.6] | 0 | 0 | 29.4 | 5.9 | 0 | 5.9 | 0 |
| SV | 18 | 66.7 | 2.6 [0.9–4.8] | 61.1 | 27.8 | 5.6 | 66.7 | 55.6 | 0 | 77.8 |
Aggregate characteristics for all patients included in this study. ACEi = angiotensin-converting enzyme inhibitor, PDEi = phosphodiesterase inhibitor.
Non-PDEi Inotrope includes: dopamine, dobutamine, epinephrine, norepinephrine.
PDE5 Expression and Activity is Increased in Human SV Myocardium
We quantified RV PDE enzymatic activity in the cytosolic fraction of 9 RV samples from patients with SV and in 8 RV samples from normal NF pediatric donor hearts (Figure 1A, B). While total RV cytosolic PDE activity was similar between SV and NF groups [NF=48.1±8.9, SV=48.8±15.2, p=0.73], PDE5 specific activity was significantly increased in the SV group, with the average PDE5-specific cGMP hydrolyzing activity more than 3 times higher in SV patients compared to NF controls [NF=4±3.1, SV=12.4±7.4, p=0.009]. RV PDE5 protein expression was also quantified in 7 hearts from patients with SV and in 9 NF pediatric donor hearts (Figure 1C). RV PDE5 expression was significantly increased in SV relative to NF, with the mean signal intensity of PDE5 relative to the loading control GAPDH more than 8 times higher in SV myocardium compared to NF myocardium [NF=−0.28±1.3, SV=2.8±1.2, p=0.0002]. A subset of pediatric RV samples, 3 SV and 1 NF RV, were subjected to histologic staining for PDE5 localization (Figure 2, Supplementary Figure S1). We determined that PDE5 is expressed in pediatric RV myocardium and is localized primarily within cardiomyocytes as determined by co-localization with α-actinin in the z-disk region of the sarcomere.
Figure 1. PDE5 Expression and Activity is Increased in SV RV Myocardium.
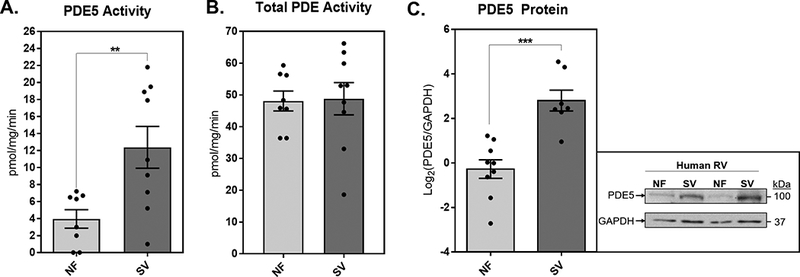
(A) PDE5 specific enzymatic activity in NF and SV. Bar equals mean±SEM, each point represents individual patient values, n=7 NF and n=13 SV RV samples; asterisk (*) denotes significant difference between groups, unpaired t-test, p<0.05. (B) Total PDE enzymatic activity in NF and SV. Bar equals mean±SEM, each point represents individual patient values, n=7 NF and n=13 SV RV samples. (C) Quantification of PDE5 protein relative to GAPDH protein in NF and SV. Quantitative data were log2 transformed, bar equals log2(mean±SEM), each point represents individual patient values, n=9 NF and n=8 SV RV samples; asterisk (*) denotes significant difference between groups, unpaired t-test, p<0.001. A representative blot of PDE5 and GAPDH protein in NF and SV RV is shown on the right.
Figure 2. PDE5 is Expressed in Pediatric SV RV Cardiomyocytes.
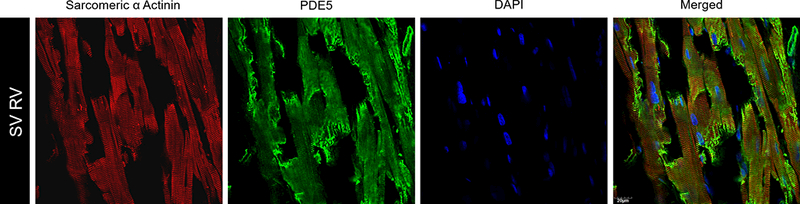
Sectioned SV tissue was stained with fluorescently labeled secondary antibodies to primary sarcomeric α actinin (red), primary PDE5 (green) and nuclei were stained with DAPI (blue). The merged confocal laser microscopy images demonstrate that PDE5 is expressed in SV cardiomyocytes. All images are at 60X magnification, scale bar = 20μM.
SV Serum Induces Pathological Gene Expression Changes in Primary Cardiomyocytes
As an in vitro model of SV-related pathological cardiomyocyte remodeling, primary cardiomyocytes (NRVMs) were treated with serum from SV patients and serum from NF controls (n=5 NF, n=12 SV in a total of 7 NRVM preparations). The α-adrenergic receptor agonist PE was used as a positive control, as it results in robust activation of the fetal gene program, a hallmark of pathologic remodeling25. As compared to NF serum treatment, SV serum treatment results in significant induction of prototypical pathological cardiac fetal genes brain natriuretic peptide (BNP) and atrial natriuretic factor (ANF), as well as a significant reduction in the ratio of α-Myosin Heavy Chain (αMHC) to β--Myosin Heavy Chain (βMHC) in NRVMs (Figure 3).
Figure 3. SV Serum Induces Pathological Gene Expression Changes in Cardiomyocytes in vitro.
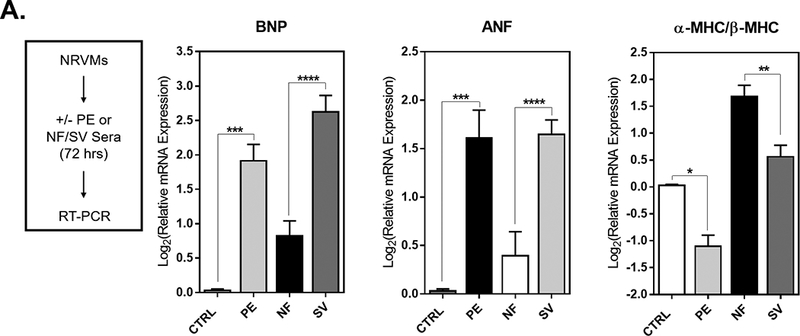
(A) NRVMs were treated with either phenylephrine (PE) or with sera from NF or SV patients for 72 hours. RNA was harvested for RT-qPCR. (B) Log2 relative mRNA expression of prototypical pathological cardiac genes (BNP, ANF, βMHC and αMHC) in NRVMs. For all groups, bar equals log2(mean±SEM), asterisk (*) denotes a significant difference between groups, one-way ANOVA, Holm-Sidak multiple comparisons (**p<0.01, ***p<0.001, ****p<0.0001), n=5 NF, n=16 SV, n=10 PE treatments in a total of 7 NRVM preparations.
PDE5 Inhibition Attenuates SV Serum-Induced Pathological Gene Expression Changes in Primary Cardiomyocytes
Additionally, we utilized the SV-serum based in vitro model of cardiomyocyte remodeling to assess the intracellular consequences of PDE5 inhibition (n=4 NF+PDE5i, n=9 SV+PDE5i in a total of 7 NRVM preparations). While PDE5i had no significant effect on untreated NRVMs or those treated with NF patient serum, PDE5i significantly attenuated SV serum-induced pathological cardiac gene expression changes (Figure 4). Namely, BNP and ANF expression were significantly attenuated in the SV+PDE5i treated group, as expression levels were not significantly different from NF+PDE5i controls and were significantly less than SV serum treatment alone. There was an increase in the ratio of α-MHC to β-MHC in the SV+PDE5i group, such that expression levels were not significantly different from NF+PDE5i controls.
Figure 4. PDE5i Attenuates SV Serum-Induced Pathological Gene Expression Changes in Cardiomyocytes in vitro.
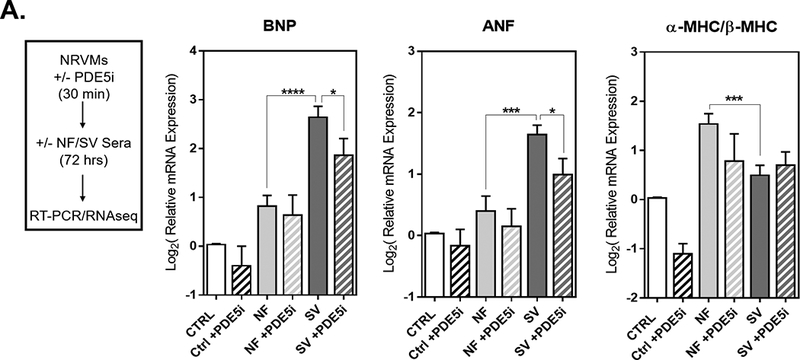
(A) NRVMs were treated with sildenafil (PDE5i), and after 30 minutes cells were treated with sera from NF or SV patients for 72 hours. RNA was harvested for RT-qPCR and RNAseq. (B) Log2 relative mRNA expression of prototypical pathological cardiac genes (BNP, ANF, βMHC and αMHC) in NRVMs. For all groups, bar equals log2(mean±SEM), asterisk (*) denotes a significant difference between groups, one-way ANOVA, Holm-Sidak multiple comparisons (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001), n=5 NF, n=16 SV, n=4 NF+PDE5i, n=14 SV+PDE5i in a total of 7 NRVM preparations.
Transcriptome Profiling Indicates SV Serum Induces Significant Transcriptional Changes and Dysregulated Cardiotoxicity Pathways in Primary Cardiomyocytes, that are Normalized by PDE5i
Next generation sequencing (RNAseq) on a subset of serum-treated NRVMs ± the PDE5i sildenafil (N=3 NF serum-treated NRVMs±PDE5i and N=3 SV serum-treated NRVMs±PDE5i, from 3 different NRVM preparations) identified more than 20,000 genes. Genes with counts <10 were removed, and further analysis was performed using the resulting 7,690 genes. Of these, 1,135 were significantly differentially expressed between NF and SV serum-treated groups (p<0.05) (Figure 5A-B). Of the 1,135 differentially expressed genes, 607 genes were significantly upregulated and 528 genes were significantly downregulated (Figure 5A). Unsupervised hierarchical clustering separated NF and SV serum-treated cells (Figure 5B). A list of all 1,135 differentially expressed genes is listed in supplemental Table S3.
Figure 5. SV Serum Induces Significant Transcriptional Changes and Dysregulated Cardiotoxicity Pathways in vitro.
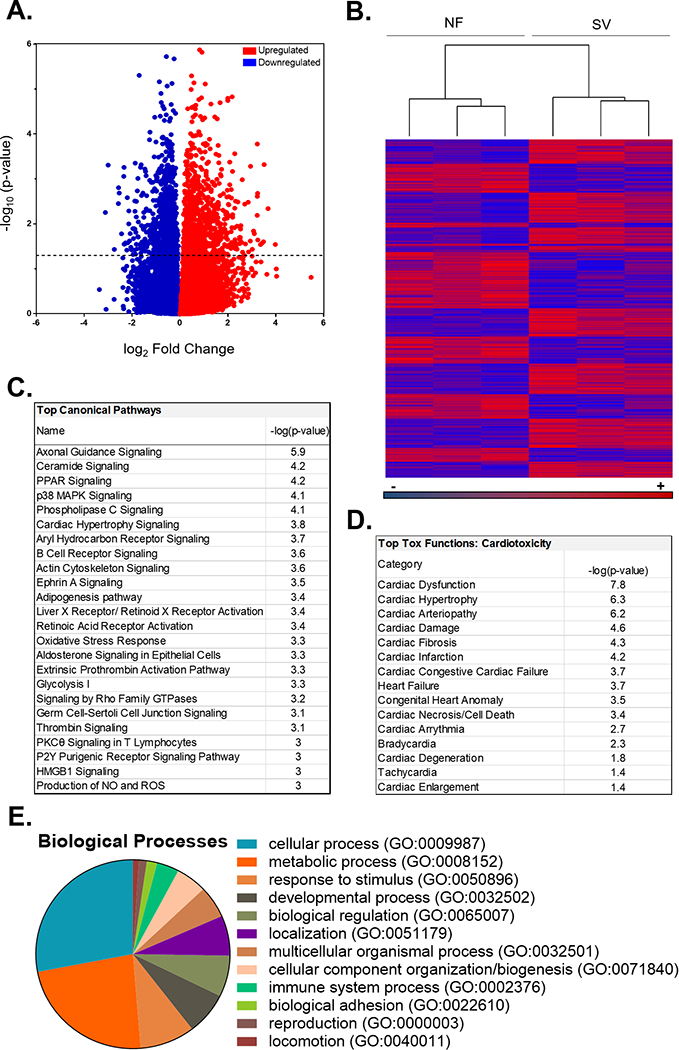
(A) Volcano plot representation of all the 20,398 transcripts detected by RNA-Seq depicted as the log2 fold-changes in expression (x-axis) and the log odds of a gene being differentially expressed (y-axis), highlighting the 1,135 transcripts that are significantly differentially expressed between NF versus SV-serum treated NRVMs (above dotted line). ANOVA, -log10(p-value)>1.3 or p<0.05). (B) Heat map of the 1,135 significantly differentially expressed transcripts (raw Fragments Per Kilobase of transcript per Million mapped reads, FPKM values). Unsupervised hierarchical clustering separated NF (n=3) and SV (n=3) serum-treated NRVM samples. (C) Significantly dysregulated canonical pathways in NF versus SV serum-treated NRVMs identified with IPA using the 1,135 transcripts that changed significantly in all 3 SV-treated cell groups. Fisher’s exact test, -log10(p-value)>2.9 or p<0.001. (D) Significantly dysregulated cardiotoxicity functions in NF versus SV serum-treated NRVMs identified with IPA using the 1,135 transcripts that changed significantly in all 3 SV-treated cell groups. Fisher’s exact test, -log10(p-value)>1.3 or p<0.05. (E) Categorization of Gene Ontology (GO) annotations for biological processes in NF versus SV serum-treated NRVMs identified with PANTHER using the 1,135 transcripts that changed significantly in all 3 SV-treated cell groups.
Pathway analysis performed using Qiagen Ingenuity Pathway Analysis (IPA) software revealed multiple dysregulated canonical pathways associated with SV serum treatment (Figure 5C). Most prominently, signaling pathways involved in: axonal guidance, ceramide, peroxisome proliferator-activated receptor (PPAR), p38 mitogen-activated protein kinase (MAPK), phospholipase C (PLC), cardiac hypertrophy, aryl hydrocarbon receptor, B cell receptor, actin cytoskeleton, and Ephrin A signaling were significantly enriched (p<0.01) in SV serum-treated NRVMs relative to NF serum-treated NRVMs. In addition to cardiac hypertrophy signaling, a more detailed IPA analysis of cardiac specific pathways revealed multiple dysregulated cardiotoxicity functions, including significant differential expression (p<0.05) of pathways related to cardiac dysfunction, arteriopathy, cardiac damage, cardiac fibrosis, infarction, heart failure, congenital heart anomalies, cardiac cell death and arrhythmias (Figure 5D).
PANTHER was used to further categorize the 1,135 differentially expressed transcripts based on implicated biological processes. The top 3 categorizations indicate a majority of the differentially expressed genes are involved in cellular processes (28%, e.g., cell communication, cell cycle and movement of cellular components), metabolic processes (23%, e.g., primary metabolism and nitrogen compound metabolism) and response to stimuli (9%, e.g., stress response and immune response) (Figure 5E).
Additionally, more than half of the 1,135 differentially expressed transcripts were normalized (p>0.05) by PDE5i, and unsupervised hierarchical clustering separated SV and SV+PDE5i serum-treated cells. (Figure 6A). Namely, 335 (55%) of the 607 significantly upregulated genes in the SV serum-treated group were no longer significantly different in the SV+PDE5i group relative to NF+PDE5i (Figure 6B), while 314 (60%) of the 528 significantly downregulated genes in the SV group were no longer significantly different in the SV+PDE5i group relative to NF+PDE5i (Figure 6C). IPA analysis of all 649 genes that were normalized with PDE5i in SV serum treated NRVMs predicted normalization of many differentially activated signaling pathways, including normalization of ceramide, p38 MAPK, PLC, cardiac hypertrophy, actin cytoskeleton, and oxidative stress response signaling (Figure 6D). IPA analysis also predicted normalization of SV serum-induced cardiotoxicity pathways, including: normalization of cardiac and ventricular hypertrophy signaling, as well as normalization of genes involved in cardiac dysfunction (Figure 7A-C). A list of all 649 genes that were normalized by PDE5i are included in supplemental Table S4.
Figure 6. PDE5i Normalizes SV Serum-Induced Transcriptional Changes in vitro.
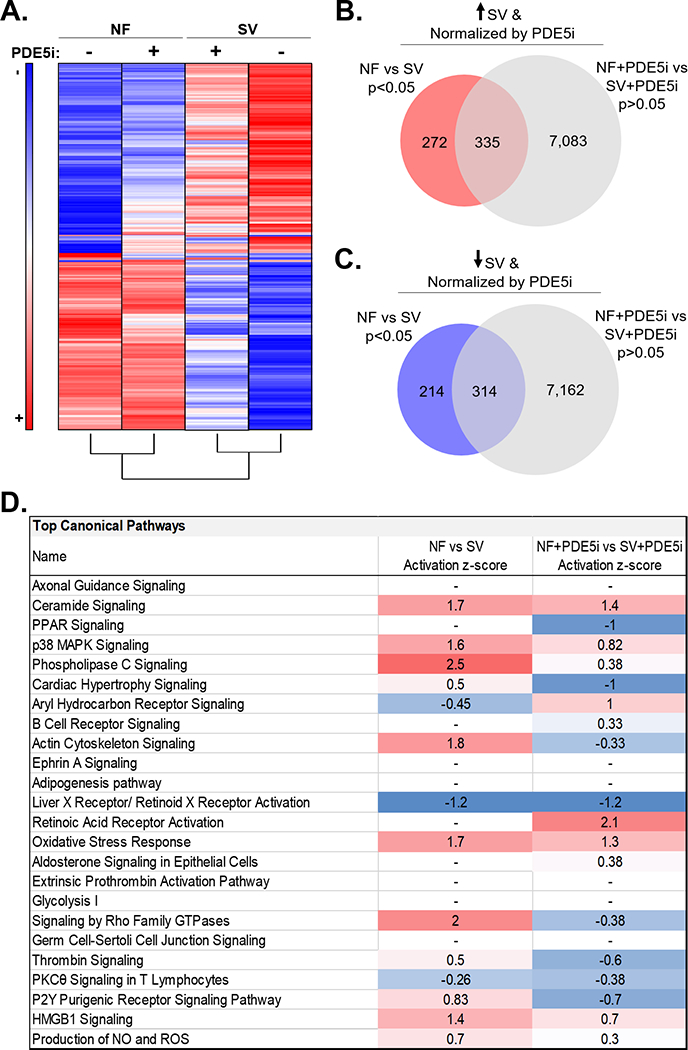
(A) Heat map summary of the 649 significantly differentially expressed transcripts that were normalized by PDE5i (average fold change relative to NF, n=3 NF, n=3 NF+PDE5i, n=3 SV, n=3 SV+PDE5i in a total of 3 NRVM preparations). Unsupervised hierarchical clustering separated NF, NF+PDE5i, SV, and SV+PDE5i serum-treated samples. (B) SV serum-induced upregulated gene expression changes were compared with gene expression changes that were not significantly different between NF+PDE5i and SV+PDE5i serum-treated groups. The Venn diagram indicates significantly upregulated genes that were normalized with PDE5i. (C) SV serum-induced downregulated gene expression changes were compared with gene expression changes that were not significantly different between NF+PDE5i and SV+PDE5i serum-treated groups. The Venn diagram indicates significantly downregulated genes that were normalized with PDE5i. (D) Activation z-scores of significantly dysregulated canonical pathways in NF versus SV serum-treated NRVMs compared to NF+PDE5i vs SV+PDE5i serum-treated NRVMs.
Figure 7. PDE5i Normalizes SV Serum-Induced Transcriptional Changes Associated with Cardiac Hypertrophy and Dysfunction in vitro.
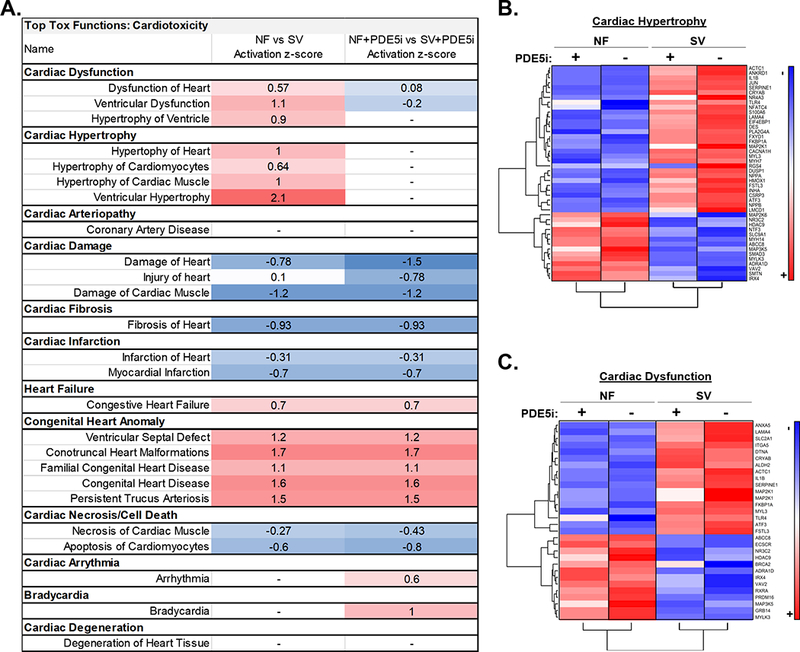
(A) Activation z-scores of significantly dysregulated cardiotoxicity pathways in NF versus SV serum-treated NRVMs compared to NF+PDE5i vs SV+PDE5i serum-treated NRVMs. (B) Heat map summary of the genes predicted to be involved in cardiac hypertrophy signaling based on IPA, that were normalized by PDE5i (average fold change relative to NF, n=3 NF, n=3 NF+PDE5i, n=3 SV, n=3 SV+PDE5i in a total of 3 NRVM preparations). (C) Heat map summary of the genes predicted to be involved in cardiac dysfunction signaling based on IPA, that were normalized by PDE5i (average fold change relative to NF, n=3 NF, n=3 NF+PDE5i, n=3 SV, n=3 SV+PDE5i in a total of 3 NRVM preparations).
Representative genes from the top 6 most significantly differentially regulated signaling pathways activated by SV serum-treatment including ceramide, p38 MAPK, PLC, cardiac hypertrophy, actin cytoskeleton, and oxidative stress response signaling (Figure S2) were chosen for further analysis. Confirmatory analysis by RT-qPCR was performed on human RV tissue for representative genes in each pathway, including (1) ceramide kinase CERK31, (2) cyclic AMP-responsive transcription factor CREB532,33, (3) non-receptor tyrosine kinase FYN34,35, (4) cardiac ankyrin repeat domain ANKRD136,37, (5) α-actinin isoform ACTN238,39, and (6) transcription factor MAFK40. RT-qPCR revealed that these representative genes were significantly upregulated in SV RV relative to NF control RV, consistent with the SV-sera NRVM treatment findings (Figure 8).
Figure 8. Representative Gene Expression Changes in SV Serum-Treated Primary Cardiomyocytes are Similar to Those in SV RV Myocardium.
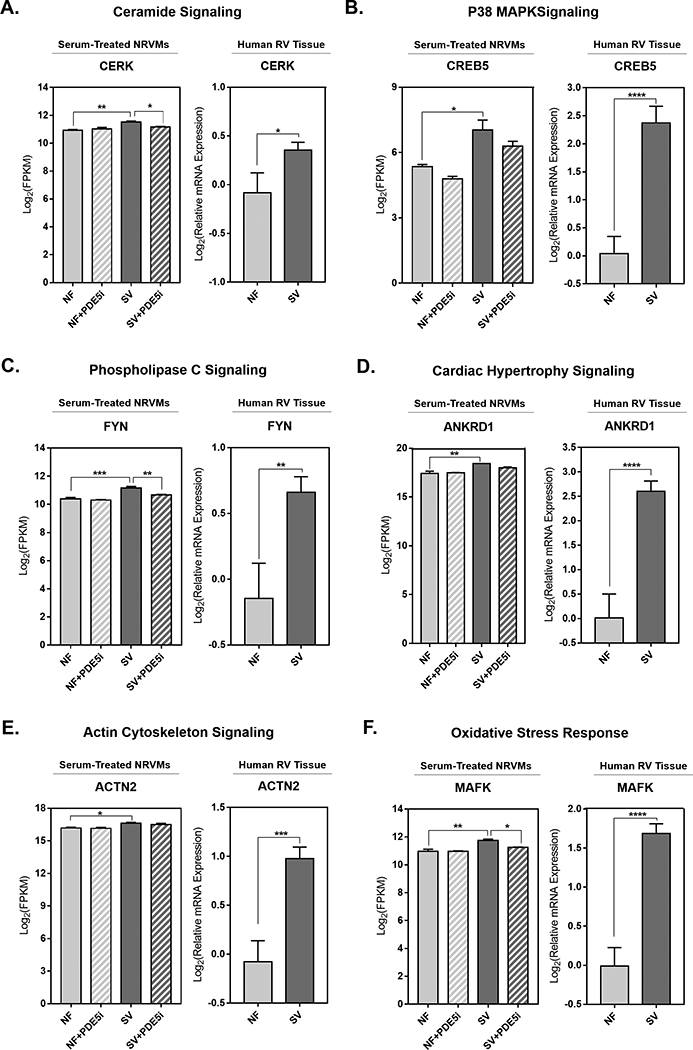
Representative genes (A) Ceramide kinase (CERK), (B) Cyclic AMP-responsive element binding protein (CREB5), (C) Proto-oncogene tyrosine-protein kinase (FYN), (D) Cardiac ankyrin repeat domain 1 (ANKRD1), (E) α-Actinin isoform ACTN2, and (F) MAF transcription factor K (MAFK) from each of the top most differentially regulated pathways (ceramide, p38 MAPK, PLC, hypertrophy, actin cytoskeleton and oxidative stress) activated by SV serum treatment in primary cardiomyocytes. For all serum-treated NRVM groups: data are depicted as log2 FPKM values, bar equals log2(mean±SEM), asterisk (*) denotes a significant difference between groups, one-way ANOVA, Holm-Sidak multiple comparisons (* p<0.05, **p<0.01, ***p<0.001), n=3 NF, n=3 NF+PDE5i, n=3 SV, n=3 SV+PDE5i in a total of 3 NRVM preparations. For all human RV tissue groups: data are depicted as log2 relative mRNA expression, bar equals log2(mean±SEM), asterisk (*) denotes a significant difference between groups, unpaired T-test (* p<0.05, **p<0.01, ***p<0.001, ****p<0.0001), n= 7 NF, n=14 SV.
Discussion
Long-term survival and quality of life in SV patients ultimately depends on preservation of RV function, particularly as more children with complex CHD are surviving into adulthood41. There are no proven therapies to treat or prevent RV failure in SV and persistence of RV dysfunction may reflect irreversible adverse remodeling that is presumably a precursor to eventual HF42. There remains a critical need to better understand the mechanisms of SV failure, and to identify clinically relevant pathways, biomarkers of disease progression, and therapeutic targets. In this study we demonstrate that myocardial PDE5 expression and activity are significantly elevated in SV patients relative to NF controls, and that PDE5 is expressed in SV cardiac myocytes. Moreover, in vitro treatment of primary cardiomyocytes with SV patient sera induces gene expression changes indicative of pathological myocardial remodeling, cardiac hypertrophy and cardiac dysfunction, which are abrogated by PDE5i. Finally, confirmatory analysis in human RV tissue revealed that gene expression changes induced by SV serum in primary cardiomyocytes are similar to those seen in SV myocardium. Together these data provide an important foundation for future mechanistic studies regarding PDE5i in SV and highlight a novel SV serum-based model that can be used to identify potential drug targets relevant to myocardial remodeling in the pediatric SV population.
PDE5 Upregulation in SV
There is mounting preclinical and clinical evidence regarding the potentially beneficial cardiac effects of PDE5i, however, their use in children with heart disease remains primarily focused on modulating pulmonary blood flow. While PDE5i therapy in failing SV patients has been associated with improved hemodynamics, exercise tolerance and cardiac function10,12,43, the direct effects of PDE5i on the SV myocardium are unknown. Given the significant increase of PDE5 mRNA20, protein and activity in SV RV and the presence of PDE5 in SV cardiomyocytes, these findings suggest that in addition to effects on the pulmonary vasculature, PDE5i may directly target the myocardium. The specific mechanisms by which PDE5i may augment cardiac function remain to be determined.
PDE5-Mediated Cardiomyocyte Remodeling
We and others have previously reported that circulating factors present in patient serum or plasma can be important paracrine modulators of cardiac gene expression26,44. Here we show that treatment of primary cardiomyocytes with SV patient serum induces changes in gene expression with similar directionality as in SV hearts45 and a pediatric rodent model of RV hypertrophy46. Paracrine effects on cardiac remodeling can be mediated by a myriad of factors present in the circulation, and the specific factors modulating SV serum-induced cardiomyocyte remodeling remain to be determined.
Transcriptome analysis of SV serum-treated primary cardiomyocytes suggests dysregulation of genes involved in various cellular and metabolic processes including a high number of genes involved in cardiac remodeling, hypertrophy and dysfunction. Pathway analysis of differentially expressed genes in SV serum-treated cardiomyocytes also suggests enrichment of several other pathways, including significant activation of: ceramide, p38 MAPK, PLC, actin cytoskeleton, and oxidative stress response signaling. Many of these dysregulated canonical pathways have known roles in cardiac hypertrophy and dysfunction. For example, ceramide signaling has been implicated in a variety of physiological functions31, and a specific species of ceramide has been shown to promote cardiomyocyte hypertrophy through generation of reactive oxygen species (ROS)47, while inhibition of ceramide signaling using a sphingosine kinase-1 inhibitor in a mouse model of hypoxia-induced pulmonary arterial hypertension resulted in reduced RV hypertrophy and cardiomyocyte death, without affecting vascular remodelling48. Additionally, PLC enzymes are important for intracellular signaling, and can be activated by non-receptor tyrosine kinases such as FYN34,35. It has been shown that cardiomyocyte hypertrophy and cardiac dysfunction can occur via stimulation of PLC, resulting in generation of potent phospholipid-derived second messenger lipid molecules that can alter cardiomyocyte signaling49,50. Moreover, the p38 MAPK signaling pathway is known as one of the key sensors of cellular stress, and signal transduction occurs via a cascade culminating in activation of key transcription factors including those in the CREB family (e.g., CREB5)33. p38 MAPK signaling has a variety of roles in the cardiovascular system including regulation of cardiomyocyte proliferation, apoptosis and hypertrophy32,51,52. Additionally, structural cytoskeletal alterations (e.g., alterations in anchor proteins such as α-actinins)53, both within cardiac myocytes and of the extracellular matrix contribute to ventricular remodeling and cardiac dysfunction38,53,54. Finally, under pathological conditions, oxidative stress in cardiomyocytes is considered a major stimulant of signal transduction, and can induce inflammatory cytokines and MAP kinases (e.g, MAFK)40,55.
Our transcriptome analysis of serum-treated primary cardiomyocytes revealed that more than half of the differentially expressed transcripts between SV-serum treated NRVMs and NF serum treated NRVMs were normalized by PDE5i, including normalization of ceramide, p38 MAPK, PLC, actin cytoskeleton, and oxidative stress signaling, as well as normalization of SV serum-induced cardiotoxicity pathways such as cardiac hypertrophy and dysfunction. Thus, these pathways may serve as potential therapeutic targets in this population and warrant future study. In summary, the in vitro model used in this study recapitulates some relevant SV myocardial gene expression patterns, poses no risk to children and provides a useful mechanistic platform to investigate PDE5, as well as other signaling pathways altered by SV physiology.
Limitations
There are important limitations of this study. The tissue bank-based aspect of this study is inherently cross-sectional, and as such, proof of mechanistic associations or knowing whether changes are pathologic or compensatory is not possible. Because of the relative rarity of SV, it is not possible for us to determine the influence of age, prior surgical procedures, or the temporal relationship of expression/activity changes in our findings. Secondary to the current widespread use of PDE inhibitors in the SV population, it is not possible to exclude all patients on a PDE5i, or to determine whether PDE5 upregulation is secondary to PDE3i (millrinone) treatment. It is important to note however, that there was no significant difference in PDE5 activity or expression between SV patients based on treatment with the PDE5i sildenafil. Because our experiments were performed using explanted RV myocardium, we acknowledge that the data are not necessarily representative of cardiomyocytes alone, and contributions of the extracellular matrix, fibroblasts and endothelial cells are unknown. Finally, we acknowledge that the unadjusted p-value (p<0.05) used in our primary cardiomyocyte RNA-seq data analysis may result in false positives. However, our goal was to identify a larger gene set to generate hypotheses regarding gene expression changes relevant in the human SV myocardium.
Conclusions
Findings from this study suggest that elevated PDE5 in failing SV myocardium may contribute to adverse myocardial remodeling, and in addition to effects on the pulmonary vasculature, PDE5i may provide direct myocardial benefit in this population. These results underscore the importance of pediatric-specific investigations and justify attempts to improve the understanding of SV-specific molecular mechanisms leading to HF.
Supplementary Material
Clinical Impact Commentary
What is new?
Phosphodiesterase-5 expression and activity are increased in the right ventricular tissue of children with single ventricle heart disease, compared to that in right ventricles of normal children.
Treatment of rat heart muscle cells (cardiomyocytes) with sera from children with single ventricle heart disease results in similar pathologic gene expression changes as are seen in single ventricle heart tissue.
Phosphodiesterase-5 inhibitor treatment results in normalization of some of the transcriptional changes seen in the single ventricle serum-treated cardiomyocytes.
What are the clinical implications?
Remodeling (pathologic hypertrophy and cardiac dysfunction) of the single ventricle heart can lead to heart failure and death or the need for heart transplant.
There are no proven therapies and a limited understanding of the mechanisms underlying single ventricle heart failure.
Elevation of phosphodiesterase-5 in heart tissue may contribute to single ventricle heart failure.
Phosphodiesterase-5 inhibitor therapy may have beneficial effects specific to the heart muscle of children with single ventricle congenital heart disease.
Treatment of primary cardiomyocytes with patient sera may represent a novel model for future studies of the mechanisms involved in single ventricle heart failure.
Acknowledgments
We would like to acknowledge the Heart Transplant Team at Children’s Hospital Colorado, especially Drs. David Campbell, Max Mitchell and James Jaggers for their assistance with obtaining explanted heart tissue; and Sam Schofield and Alix Michael for data collection and subject recruitment. We would also like to acknowledge the Pediatric Cardiovascular Research Laboratory team at the University of Colorado, Denver, including Armin Korst and Danielle Jeffrey for the dissection and transportation of fresh heart tissue. Lastly, we acknowledge Dr. Matthew Movsesian from the University of Utah, for his guidance regarding technical aspects of the PDE activity assay.
Sources of Funding
This work was supported by NIH R21 HL113846 and R01 HL126928 to Shelley D. Miyamoto, NIH R01 HL107715 to Brian L. Stauffer, the Addison Scott Memorial Fund, the Boedecker Foundation, the Nair Family and the Jack Cooper Millisor Chair in Pediatric Heart Disease. Anastacia M. Garcia was supported by NIH R01 HL126928–03S1.
Footnotes
Disclosures
Carmen C. Sucharov: Equity in miRagen, Inc., Brian L. Stauffer: Research support from Forest Laboratories, Inc. Carmen C. Sucharov, Brian L. Stauffer, Shelley D. Miyamoto are founders and scientific advisors for CoramiR Biomedical, LLC.
References
- 1.Miranović V The incidence of congenital heart disease: previous findings and perspectives. Srp Arh Celok Lek. 2016;142:243–248. [DOI] [PubMed] [Google Scholar]
- 2.Ohye RG, Schonbeck JV, Eghtesady P, Laussen PC, Pizarro C, Shrader P, Frank DU, Graham EM, Hill KD, Jacobs JP, Kanter KR, Kirsh JA, Lambert LM, Lewis AB, Ravishankar C, Tweddell JS, Williams IA, Pearson GD, Pediatric Heart Network Investigators. Cause, timing, and location of death in the Single Ventricle Reconstruction trial. J Thorac Cardiovasc Surg. 2012;144:907–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lotto AA, Hosein R, Jones TJ, Barron DJ, Brawn WJ . Outcome of the Norwood procedure in the setting of transposition of the great arteries and functional single left ventricle. Eur J Cardio-thoracic Surg. 2009;35:149–155. [DOI] [PubMed] [Google Scholar]
- 4.McGuirk SP, Winlaw DS, Langley SM, Stumper OF, de Giovanni J V, Wright JG, Brawn WJ, Barron DJ. The impact of ventricular morphology on midterm outcome following completion total cavopulmonary connection. Eur J Cardiothorac Surg. 2003;24:37–46. [DOI] [PubMed] [Google Scholar]
- 5.Kogon BE, Plattner C, Leong T, Simsic J, Kirshbom PM, Kanter KR. The bidirectional Glenn operation: A risk factor analysis for morbidity and mortality. J Thorac Cardiovasc Surg. 2008;136:1237–1242. [DOI] [PubMed] [Google Scholar]
- 6.Daebritz SH, Nollert GD, Zurakowski D, Khalil PN, Lang P, del Nido PJ, Mayer JE, Jonas RA. Results of Norwood stage I operation: comparison of hypoplastic left heart syndrome with other malformations. J Thorac Cardiovasc Surg. 2000;119:358–367. [DOI] [PubMed] [Google Scholar]
- 7.Kirk R, Dipchand AI, Rosenthal DN, Addonizio L, Burch M, Chrisant M, Dubin A, Everitt M, Gajarski R, Mertens L, Miyamoto S, Morales D, Pahl E, Shaddy R, Towbin J, Weintraub R. The International Society for Heart and Lung Transplantation Guidelines for the management of pediatric heart failure: Executive summary. J Hear Lung Transplant. 2014;33:888–909. [DOI] [PubMed] [Google Scholar]
- 8.Shaddy RE, Boucek MM, Hsu DT, Boucek RJ, Canter CE, Mahony L, Ross RD, Pahl E, Blume ED, Dodd DA, Rosenthal DN, Burr J, LaSalle B, Holubkov R, Lukas MA, Tani LY, Pediatric Carvedilol Study Group. Carvedilol for children and adolescents with heart failure: a randomized controlled trial. JAMA. 2007;298:1171–1179. [DOI] [PubMed] [Google Scholar]
- 9.Hsu DT, Zak V, Mahony L, Sleeper LA, Atz AM, Levine JC, Barker PC, Ravishankar C, McCrindle BW, Williams R V, Altmann K, Ghanayem NS, Margossian R, Chung WK, Border WL, Pearson GD, Stylianou MP, Mital S, Pediatric Heart Network Investigators. Enalapril in infants with single ventricle: results of a multicenter randomized trial. Circulation. 2010;122:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldberg DJ, French B, Szwast AL, McBride MG, Marino BS, Mirarchi N, Hanna BD, Wernovsky G, Paridon SM, Rychik J. Impact of sildenafil on echocardiographic indices of myocardial performance after the Fontan operation. Pediatr Cardiol. 2012;33:689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldberg DJ, French B, McBride MG, Marino BS, Mirarchi N, Hanna BD, Wernovsky G, Paridon SM, Rychik J. Impact of Oral Sildenafil on Exercise Performance in Children and Young Adults After the Fontan Operation: A Randomized, Double-Blind, Placebo-Controlled, Crossover Trial. Circulation. 2011;123:1185–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldberg DJ, Zak V, Goldstein BH, Chen S, Hamstra MS, Radojewski EA, Maunsell E, Mital S, Menon SC, Schumacher KR, Payne RM, Stylianou M, Kaltman JR, deVries TM, Yeager JL, Paridon SM, Pediatric Heart Network Investigators. Results of a phase I/II multi-center investigation of udenafil in adolescents after fontan palliation. Am Heart J. 2017;188:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butts RJ, Chowdhury SM, Baker GH, Bandisode V, Savage AJ, Atz AM. Effect of Sildenafil on Pressure-Volume Loop Measures of Ventricular Function in Fontan Patients. Pediatr Cardiol. 2016;37:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheitlin MD, Hutter AM, Brindis RG, Ganz P, Kaul S, Russell RO, Zusman RM. ACC/AHA expert consensus document. Use of sildenafil (Viagra) in patients with cardiovascular disease. American College of Cardiology/American Heart Association. J Am Coll Cardiol. 1999;33:273–282. [DOI] [PubMed] [Google Scholar]
- 15.Boolell M, Allen MJ, Ballard SA, Gepi-Attee S, Muirhead GJ, Naylor AM, Osterloh IH, Gingell C. Sildenafil: an orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction. Int J Impot Res. 1996;8:47–52. [PubMed] [Google Scholar]
- 16.Corbin J, Rannels S, Neal D, Chang P, Grimes K, Beasley A, Francis S. Sildenafil citrate does not affect cardiac contractility in human or dog heart. Curr Med Res Opin. 2003;19:747–752. [DOI] [PubMed] [Google Scholar]
- 17.Wallis RM, Corbin JD, Francis SH, Ellis P. Tissue distribution of phosphodiesterase families and the effects of sildenafil on tissue cyclic nucleotides, platelet function, and the contractile responses of trabeculae carneae and aortic rings in vitro. Am J Cardiol. 1999;83:3C–12C. [DOI] [PubMed] [Google Scholar]
- 18.Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, St. Aubin C, Webster L, Rebeyka IM, Ross DB, Light PE, Dyck JRB, Michelakis ED. Phosphodiesterase Type 5 Is Highly Expressed in the Hypertrophied Human Right Ventricle, and Acute Inhibition of Phosphodiesterase Type 5 Improves Contractility. Circulation. 2007;116:238–248. [DOI] [PubMed] [Google Scholar]
- 19.Zhang M, Takimoto E, Hsu S, Lee DI, Nagayama T, Danner T, Koitabashi N, Barth AS, Bedja D, Gabrielson KL, Wang Y, Kass DA. Myocardial Remodeling Is Controlled by Myocyte-Targeted Gene Regulation of Phosphodiesterase Type 5. J Am Coll Cardiol. 2010;56:2021–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakano SJ, Sucharov J, Van R, Cecil M, Nunley K, Wickers S, Karimpur-Fard A, Stauffer BL, Miyamoto SD, Sucharov CC. Cardiac Adenylyl Cyclase and Phosphodiesterase Expression Profiles Vary by Age, Disease, and Chronic Phosphodiesterase Inhibitor Treatment. J Card Fail. 2017;23:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–222. [DOI] [PubMed] [Google Scholar]
- 22.Nagayama T, Hsu S, Zhang M, Koitabashi N, Bedja D, Gabrielson KL, Takimoto E, Kass DA. Sildenafil Stops Progressive Chamber, Cellular, and Molecular Remodeling and Improves Calcium Handling and Function in Hearts With Pre-Existing Advanced Hypertrophy Caused by Pressure Overload. J Am Coll Cardiol. 2009;53:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kincaid R, Manganiello VC. Assay of cyclic nucleotide phosphodiesterase using radiolabeled and fluorescent substrates. Methods Enzymol. 1998;457–470. [DOI] [PubMed] [Google Scholar]
- 24.Vandeput F, Krall J, Ockaili R, Salloum FN, Florio V, Corbin JD, Francis SH, Kukreja RC, Movsesian MA. cGMP-Hydrolytic Activity and Its Inhibition by Sildenafil in Normal and Failing Human and Mouse Myocardium. J Pharmacol Exp Ther. 2009;330:884–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sucharov CC, Dockstader K, Nunley K, McKinsey TA, Bristow M. β-Adrenergic receptor stimulation and activation of protein kinase A protect against α1-adrenergic-mediated phosphorylation of protein kinase D and histone deacetylase 5. J Card Fail. 2011;17:592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang X, Sucharov J, Stauffer BL, Miyamoto SD, Sucharov CC. Exosomes from pediatric dilated cardiomyopathy patients modulate a pathological response in cardiomyocytes. Am J Physiol - Hear Circ Physiol. 2017;312:H818–H826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyamoto SD, Stauffer BL, Polk J, Medway A, Friedrich M, Haubold K, Peterson V, Nunley K, Nelson P, Sobus R, Stenmark KR, Sucharov CC. Gene expression and β-adrenergic signaling are altered in hypoplastic left heart syndrome. J Heart Lung Transplant. 2014;33:785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugiura M, Kono K, Liu H, Shimizugawa T, Minekura H, Spiegel S, Kohama T. Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J Biol Chem. 2002;277:23294–23300. [DOI] [PubMed] [Google Scholar]
- 32.Kumphune S, Surinkaew S, Chattipakorn SC, Chattipakorn N . Inhibition of p38 MAPK activation protects cardiac mitochondria from ischemia/reperfusion injury. Pharm Biol. 2015;53:1831–1841. [DOI] [PubMed] [Google Scholar]
- 33.Shanmugam P, Valente AJ, Prabhu SD, Venkatesan B, Yoshida T, Delafontaine P, Chandrasekar B. Angiotensin-II type 1 receptor and NOX2 mediate TCF/LEF and CREB dependent WISP1 induction and cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2011;50:928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao F, Shin HS, Rhee SG. In Vitro Tyrosine Phosphorylation of PLC-γ1 and PLC-γ2 by SRC-Family Protein Tyrosine Kinases. Biochem Biophys Res Commun. 1993;191:1028–1033. [DOI] [PubMed] [Google Scholar]
- 35.Ozdener F, Dangelmaier C, Ashby B, Kunapuli SP, Daniel JL. Activation of phospholipase Cgamma2 by tyrosine phosphorylation. Mol Pharmacol. 2002;62:672–679. [DOI] [PubMed] [Google Scholar]
- 36.Zhong L, Chiusa M, Cadar AG, Lin A, Samaras S, Davidson JM, Lim CC. Targeted inhibition of ANKRD1 disrupts sarcomeric ERK-GATA4 signal transduction and abrogates phenylephrine-induced cardiomyocyte hypertrophy. Cardiovasc Res. 2015;106:261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang N, Xie X, Wang J. Multifunctional protein: cardiac ankyrin repeat protein. J Zhejiang Univ B. 2016;17:333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su YR, Chiusa M, Brittain E, Hemnes AR, Absi TS, Lim CC, Di Salvo TG. Right Ventricular Protein Expression Profile in End-Stage Heart Failure. Pulm Circ. 2015;5:481–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ribeiro E de A, Pinotsis N, Ghisleni A, Salmazo A, Konarev PV, Kostan J, Sjöblom B, Schreiner C, Polyansky AA, Gkougkoulia EA, Holt MR, Aachmann FL, Žagrović B, Bordignon E, Pirker KF, Svergun DI, Gautel M, Djinović-Carugo K. The Structure and Regulation of Human Muscle α-Actinin. Cell. 2014;159:1447–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199–1207. [DOI] [PubMed] [Google Scholar]
- 41.Coats L, O’Connor S, Wren C, O’Sullivan J. The single-ventricle patient population: a current and future concern a population-based study in the North of England. Heart. 2014;100:1348–1353. [DOI] [PubMed] [Google Scholar]
- 42.Khairy P, Fernandes SM, Mayer JE, Triedman JK, Walsh EP, Lock JE, Landzberg MJ. Long-Term Survival, Modes of Death, and Predictors of Mortality in Patients With Fontan Surgery. Circulation. 2008;117:85–92. [DOI] [PubMed] [Google Scholar]
- 43.Goldberg DJ, Shaddy RE, Ravishankar C, Rychik J. The failing Fontan: etiology, diagnosis and management. Expert Rev Cardiovasc Ther. 2011;9:785–793. [DOI] [PubMed] [Google Scholar]
- 44.Kumar A, Kumar A, Michael P, Brabant D, Parissenti AM, Ramana C V, Xu X, Parrillo JE. Human serum from patients with septic shock activates transcription factors STAT1, IRF1, and NF-kappaB and induces apoptosis in human cardiac myocytes. J Biol Chem. 2005;280:42619–42626. [DOI] [PubMed] [Google Scholar]
- 45.Miyamoto SD, Stauffer BL, Polk J, Medway A, Friedrich M, Haubold K, Peterson V, Nunley K, Nelson P, Sobus R, Stenmark KR, Sucharov CC. Gene expression and β-adrenergic signaling are altered in hypoplastic left heart syndrome. J Heart Lung Transplant. 2014;33:785–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blakeslee WW, Demos-Davies KM, Lemon DD, Lutter KM, Cavasin MA, Payne S, Nunley K, Long CS, McKinsey TA, Miyamoto SD. Histone deacetylase adaptation in single ventricle heart disease and a young animal model of right ventricular hypertrophy. Pediatr Res. 2017;82:642–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mishra S, Chatterjee S. Lactosylceramide promotes hypertrophy through ROS generation and activation of ERK1/2 in cardiomyocytes. Glycobiology. 2014;24:518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacRitchie N, Volpert G, Al Washih M, Watson DG, Futerman AH, Kennedy S, Pyne S, Pyne NJ. Effect of the sphingosine kinase 1 selective inhibitor, PF-543 on arterial and cardiac remodelling in a hypoxic model of pulmonary arterial hypertension. Cell Signal. 2016;28:946–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singal T, Dhalla NS, Tappia PS. Norepinephrine-induced changes in gene expression of phospholipase C in cardiomyocytes. J Mol Cell Cardiol. 2006;41:126–137. [DOI] [PubMed] [Google Scholar]
- 50.Asemu G, Dhalla NS, Tappia PS. Inhibition of PLC improves postischemic recovery in isolated rat heart. AJP Hear Circ Physiol. 2004;287:H2598–H2605. [DOI] [PubMed] [Google Scholar]
- 51.Yokota T, Wang Y. p38 MAP kinases in the heart. Gene. 2016;575:369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Somvanshi RK, Qiu X, Kumar U . Isoproterenol induced hypertrophy and associated signaling pathways are modulated by Somatostatin in H9c2 cells. Int J Cardiol. 2013;167:1012–1022. [DOI] [PubMed] [Google Scholar]
- 53.Sequeira V, Nijenkamp LLA., Regan JA, van der Velden J. The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochim Biophys Acta - Biomembr. 2014;1838:700–722. [DOI] [PubMed] [Google Scholar]
- 54.Hein S, Kostin S, Heling A, Maeno Y, Schaper J. The role of the cytoskeleton in heart failure. Cardiovasc Res. 2000;45:273–278. [DOI] [PubMed] [Google Scholar]
- 55.Rababa h A, Guillory A, Mustafa R, Hijjawi T . Oxidative Stress and Cardiac Remodeling: An Updated Edge. Curr Cardiol Rev. 2018;14:53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.