

Abstract
Purpose:
The majority of ovarian carcinomas are of high-grade serous histology, which is associated with poor prognosis. Surgery and chemotherapy are the mainstay of treatment, and molecular characterization is necessary to lead the way to targeted therapeutic options. To this end, various computational methods for gene expression-based subtyping of high-grade serous ovarian carcinoma (HGSOC) have been proposed, but their overlap and robustness remain unknown.
Experimental Design:
We assess three major subtype classifiers by meta-analysis of publicly available expression data, and assess statistical criteria of subtype robustness and classifier concordance. We develop a consensus classifier that represents the subtype classifications of tumors based on the consensus of multiple methods, and outputs a confidence score. Using our compendium of expression data, we examine the possibility that a subset of tumors are unclassifiable based on currently proposed subtypes.
Results:
HGSOC subtyping classifiers exhibit moderate pairwise concordance across our data compendium (58.9%-70.9%, p < 10−5) and are associated with overall survival in a meta-analysis across datasets (p < 10−5). Current subtypes do not meet statistical criteria for robustness to re-clustering across multiple datasets (Prediction Strength < 0.6). A new subtype classifier is trained on concordantly classified samples to yield a consensus classification of patient tumors that correlates with patient age, survival, tumor purity, and lymphocyte infiltration.
Conclusion:
A new consensus ovarian subtype classifier represents the consensus of methods, and demonstrates the importance of classification approaches for cancer that do not require all tumors to be assigned to a distinct subtype.
Introduction
Ovarian carcinoma is a genomically complex disease, for which the accurate characterization of molecular subtypes is difficult but is anticipated to improve treatment and clinical outcome(1). Substantial effort has been devoted to characterize molecularly distinct subtypes of high-grade serous ovarian carcinoma (HGSOC) (Table 1). Initial large-scale efforts to classify HGSOC of the ovary did not reveal any reproducible subtypes(2). Tothill et al(3) reported four distinct HGSOC subtypes: (i) an immunoreactive expression subtype associated with infiltration of immune cells, (ii) a low stromal expression subtype with high levels of circulating CA125, (iii) a poor prognosis subtype displaying strong stromal response, correlating with extensive desmoplasia, and (iv) a mesenchymal subtype with high expression of N/P-cadherins. The Cancer Genome Atlas (TCGA) project also identified four subtypes characterized by (i) chemokine expression in the immunoreactive subtype, (ii) proliferation marker expression in the proliferative subtype, (iii) ovarian tumor marker expression in the differentiated subtype, and (iv) expression of markers suggestive of increased stromal components in the mesenchymal subtype, but did not report differences in patient survival(4). Further experimental characterization revealed an increased number of samples with infiltrating T lymphocytes for the immunoreactive subtype, whereas desmoplasia, associated with infiltrating stromal cells, was found more often for the mesenchymal subtype(5). Konecny et al.(6), independently evaluated the TCGA subtypes and also reported the presence of the four transcriptional subtypes using a de novo clustering and classification method.
Table 1:
Subtyping methodology of the algorithms reviewed.
| Citation | Probe / gene filtering for Clustering | Clustering Algorithm | Probe / gene filtering for classification | Subtype Classifier |
|---|---|---|---|---|
| Tothill/ Helland(3,9) | Probes with at least one sample expressed above 7.0, and global variance above 0.5 | Consensus k-means; diagonal LDA and kNN | Gene ranking by differentially expressed genes between groups | Linear subtype scores |
| TCGA/Verhaak(4,5) | Filter to genes that correlate above 0.7 between three platforms to unified estimate; then take top 1500 genes by median absolute deviation (MAD) | Non-negative Matrix Factorization | Filter patients by silhouette width; correlation-based feature subset selection | Single-sample Gene Set Enrichment Analysis |
| Konecny(6) | Top 2500 probes by MAD, then keep 1850 unique gene symbols | Non-negative Matrix Factorization | Prediction Analysis of Microarrays with thresholds determined by 10-fold cross validation | Nearest Centroid with Spearman’s rho |
| consensusOV | 100 Genes provided by Verhaak(5); convert the features space into binary matrix of gene-pair associations | Random Forest using unanimously classified tumors across the methods | 100 gene symbols given in Verhaak(5) | Random Forest classifier |
However, robustness and clinical relevance of these subtypes remain controversial(7). The previous subtyping efforts have assessed prognostic significance in different patient cohorts, and have taken different approaches to validate these subtypes in independent datasets. A recent review of HGSOC subtyping schemes highlighted the difficulty of comparing results of studies that used different subtyping algorithms, and that better general agreement on how molecular subtypes are defined would allow more widespread use of expression data in clinical trial design.(1)
Assessing the generalizability of subtyping algorithms is challenging as true subtype classifications remain unknown. This challenge is evident in the lack of published validation of the proposed HGSOC subtypes. Subsequent efforts have performed de novo clustering of new datasets and noted similarity in the clusters identified, but they have not reported quantitative measures such as classification accuracy or rate of concordance with previously published algorithms(8). In this article, we address these limitations by re-implementing three major subtyping methods(3,5,9) and assess between-classifier concordance and across-dataset robustness in a widely used database containing 1,770 HGSOC tumors(10), whose curation and data consistency has been independently validated(11). We show that each pair of subtype classifiers are significantly concordant, and are virtually identical for tumors classified with high certainty. However, the subtypes do not meet established standards of robustness to re-clustering(12) and only approximately one-third of tumors are classified concordantly by all three subtype classifiers. Using this core set of tumors concordantly classified by each method, we develop consensusOV, a consensus classifier that has high concordance with the three classifiers, therefore providing a standardized classification scheme for clinical applications.
Materials and Methods
Datasets
Analysis was carried out on datasets from the curatedOvarianData compendium; details of curation and of grading systems used by individual studies are described elsewhere (10). Datasets were additionally processed using the MetaGxOvarian package(13) (Supplementary Information). Analysis was restricted to datasets featuring microarray-based whole-transcriptome studies of at least 40 patients with late stage, high-grade, primary tumors of serous histology. This resulted in 15 microarray studies, providing data for 1,774 patients (Table 2). Duplicated samples identified by the doppelgangR package were removed(14). Survival analysis was performed for 13 of these datasets, which included 1,581 patients with annotated time to death or last time of follow-up.
Table 2: Compendium of gene expression datasets.
15 whole-transcriptome studies with at least 40 patients with late stage, high-grade serous histology from the curatedOvarianData compendium consisting of 1,770 patients. 13 of these datasets provided 1,581 patients with survival data. Sample size column proves the number of samples : number with survival data : number deceased (median survival in months).
| GEO(34) Accession | Sample Size | Microarray Platform | # Features |
|---|---|---|---|
| TCGA(4) | 464:452:239 (43) | Affymetrix HT HG-U133A | 12833 |
| GSE17260(35) | 43:43 22 (29) | Agilent-012391 Whole HG Oligo | 19596 |
| GSE14764(36) | 41:41:13 (30) | Affymetrix HG-U133A | 12752 |
| GSE18520(37) | 53:53:41 (21) | Affymetrix HG-U133 Plus 2.0 | 20282 |
| GSE26193(38) | 47:47:39 (34) | Affymetrix HG-U133 Plus 2.0 | 20282 |
| PMID17290060(39) | 59:59:36 (34) | Affymetrix HG-U133A | 12752 |
| GSE51088(40) | 85:84:69 (44) | Agilent-012097 Human 1A Microarray (V2) G4110B | 15299 |
| GSE13876(41) | 98:98:72 (22) | Operon human v3 ~35K 70-mer two-color oligonucleotide microarrays | 13846 |
| GSE49997(42) | 132:122:40 (23) | ABI HG Survey Microarray Version 2 | 16760 |
| E.MTAB.386(43) | 128:128:73 (30) | Illumina humanRef-8 v2.0 beadchip | 10572 |
| GSE32062(44) | 129:129:60 (40) | Agilent-014850 Whole HG 4×44K G4112F | 19596 |
| GSE9891(3) | 142:140:72 (29) | Affymetrix HG-U133 Plus 2.0 | 20282 |
| GSE26712(45) | 185:185:129 (39) | Affymetrix HG-U133A Array | 12752 |
| GSE20565(46) | 89 (0) | Affymetrix HG-U133 Plus 2.0 | 20282 |
| GSE2109 | 79 (0) | Affymetrix HG-U133Plus2 | 20282 |
Implementation of Subtype Classifiers
Subtype classifiers were re-implemented in R(15) using original data as described by Konecny(6), Verhaak(5), and Helland(9). These classifiers are based on nearest-centroids(6), subtype-specific single-sample GSEA(5), and subtype-specific linear coefficients(9), respectively. Implementations were validated by reproducing a result from each of the original publications (Supplemental File, Section ‘Reproduction of Published HGSOC Subtype Classifiers’).
Survival Analysis
Subtype calls from all included datasets were combined to generate a single Kaplan-Meier plot for each subtyping algorithm (stratified by subtype). Hazard ratios for overall survival between subtypes was estimated by Cox proportional hazards, and statistical significance was assessed by log-rank test using the survcomp R package(16). Hazard ratios were calculated using the lowest-risk subtype as the baseline group, and stratification by dataset was performed for hazard ratios and significance testing.
Prediction Strength
Prediction Strength(12) is defined as a measure of the similarity between pairwise co-memberships of a validation dataset from class labels assigned by (1) a clustering algorithm and (2) a classification algorithm trained on a training dataset (Supplementary Figure 1). The quantity is an established measure of cluster robustness with the following interpretation: a value of 0 or below indicates poor concordance, and a value of 1 indicates perfect concordance between models specified from training and validation data. Tibshirani and Walther(12), and subsequent applications of Prediction Strength(17), have considered a value of at least 0.8 to be an evidence of robust clusters. Prediction Strength was computed as implemented in the genefu Bioconductor package(18).
The tumors in each dataset were clustered de novo using our reproduced implementations of the algorithms of Konecny, TCGA/Verhaak, and Tothill (Supplemental File, Section ‘Reproduction of Subtype Clustering Methods’). Each dataset was also classified using implementation of the originally published subtype classifiers. This produced two sets of subtype labels for each sample in each validation dataset; these labels were used to compute Prediction Strength.
Concordance Analysis
For each pair of classifiers, subtypes were mapped based on the observed concordance suggested in the original studies: Subtype C2 from Tothill corresponding to Immunoreactive in TCGA/Verhaak and C1_Immunoreactive-like in Konecny; C4 corresponding to Differentiated and C2_Differentiated-like; C5 corresponding to Proliferative and C3_Proliferative-like; and C1 corresponding to Mesenchymal and C4_Mesenchymal-like. Statistical significance of pairwise concordance was assessed by Pearson’s Chi-squared test, and Cramer’s V was assessed to evaluate the strength of concordance. Two-way concordance was defined as the proportion of patients that were classified as the same mapped subtype across methods. Similarly, overall three-way concordance was defined as the proportion of tumors sharing the same mapped subtype across all three classifiers. Subtype-specific three-way concordance was defined as the number of tumors concordantly classified as that subtype by all three classifiers, divided by the number of tumors classified to that subtype by at least one method.
Filtering tumors by classification margin
Each subtype classifier outputs for each patient a real-valued score for each subtype. Marginally classifiable tumors were identified based on the difference between the top two subtype scores, denoted as the ‘margin’ value. Thus, a higher margin indicates a more confident classification. For each pair of subtype classifiers, classification concordance was assessed on both the full dataset and considering only patients classified with margins above a user-defined cutoff.
Building a consensus classifier
The consensusOV classifier was implemented using a Random Forest classifier trained on concordantly-subtyped tumors across multiple datasets. The Random Forest method has previously been used for building a multi-class consensus classifier to resolve inconsistencies among published colorectal cancer subtyping schemes(19). In order to avoid normalizing expression values across datasets, binary gene pair vectors were used as feature space, as recently applied for breast cancer subtyping(20,21). To address differences in gene expression scales due to different experimental protocols, consensusOV first standardizes genes in each dataset to the same mean and variance, and computes binary gene pairs from standardized expression values. Since the feature size of this classifier increases quadratically with respect to the size of the original gene set, we used the smallest gene set of the original subtype classifiers (the gene set of Verhaak et al.(5)), which contains 100 gene symbols. The consensusOV classifier outputs the subtype classification and a real-valued margin score to discriminate between patients that are of well-defined or indeterminate subtype. Similarly to previously published subtype classifiers, a higher margin score indicates higher confidence of classification.
Leave-one-dataset-out cross-validation
Performance of the consensus classifier for identifying concordantly classified subtypes was assessed using leave-one-dataset-out cross-validation(22). Concordant subtypes were identified to train the Random Forest classifier using 14 of the 15 datasets, and subtype predictions were tested in the remaining left-out dataset. This process was repeated for all 15 datasets. While predicting the samples in any given dataset, the training set was subsetted to contain only the concordant subtypes in other datasets.
Correlation analysis with Histopathology and Tumor Purity
Subtype calls from the Consensus Classifier were analysed for correlation with histopathology and tumor purity in the TCGA dataset. In order to best represent the most confident subtype calls, a default cutoff was used to include only the 25% of patients with the largest classification margins. Available histopathology variables included lymphocyte, monocyte, and neutrophil infiltration. Tumor purity was assessed using the ABSOLUTE algorithm(23), which estimates purity and ploidy from copy number and SNP allele frequency from SNP genotyping arrays (Synapse dataset syn3242754). Significance of associations were tested by one-way ANOVA for patient age, purity, and immune infiltration.
Research reproducibility
All results are reproducible using R/Bioconductor(24) and knitr(25) with LaTeX output at overleaf.com/read/srvqbpxpqbyz. Output of this code is provided as Supplemental File 1. Subtyping algorithms are provided by the open source consensusOV R package available from Bioconductor (http://bioconductor.org/packages/consensusOV).
Results
We performed a meta-analysis of three published subtyping algorithms for HGSOC(5,6,9) and developed a new consensus classifier to identify unambiguously classifiable tumors (Table 1). Each of these algorithms identified four distinct HGSOC subtypes with specific clinical and tumor pathology characteristics (Figure 1). We assessed the algorithms on a compendium of 15 datasets including over 1,700 HGSOC patients (Table 2) with respect to concordance, robustness, and association to patient outcome. By modifying individual algorithms to discard tumors of intermediate subtype, we found that concordance between algorithms is greatly improved.
Figure 1: Properties of Subtypes identified by Consensus Classifier.
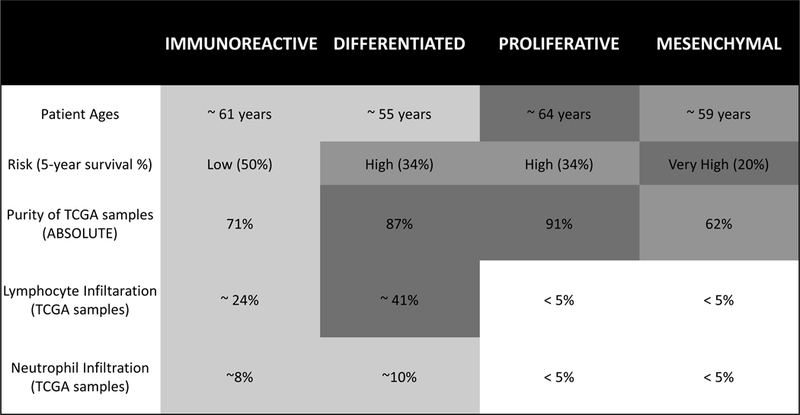
Subtype associations with patient age and overall survival were assessed across our compendium of microarray datasets; association with tumor purity and immune cell infiltration was assessed using the TCGA dataset. Tumor purity was estimated from genotyping data in TCGA; lymphocyte infiltration was based on pathology estimates from TCGA. Patient age (p < 0.001), overall survival (p < 0.005), and ABSOLUTE purity (p < 0.001) were statistically significant across subtypes. When compared to all other groups, the Immunoreactive subtype had elevated infiltration of lymphocytes (p < 0.05) and neutrophils (p < 0.10). Mean monocyte infiltration was less than 5% across all subtypes, and was excluded from this analysis. Classification was performed using default parameters, and mean values of each variable are shown.
Concordance of published classifiers
We reimplemented three published HGSOC subtype classifiers(5,6,9) (Table 1) and applied these methods to new datasets. We ensured correct implementation of classifiers by reproducing results from the original papers (Supplementary Information). When applied to independent datasets, pairwise concordance of the three methods was statistically significant (p < 10−5, Chi-square test; Figure 2A) with the highest agreement observed for Helland and Konecny subtyping schemes (70.9%), followed by Verhaak and Helland (67.4%) and Verhaak and Konecny (58.9%). Cramer’s V coefficients(26) indicated a strong association between subtypes as identified by the different algorithms (>0.5).
Figure 2: Concordance Analysis.
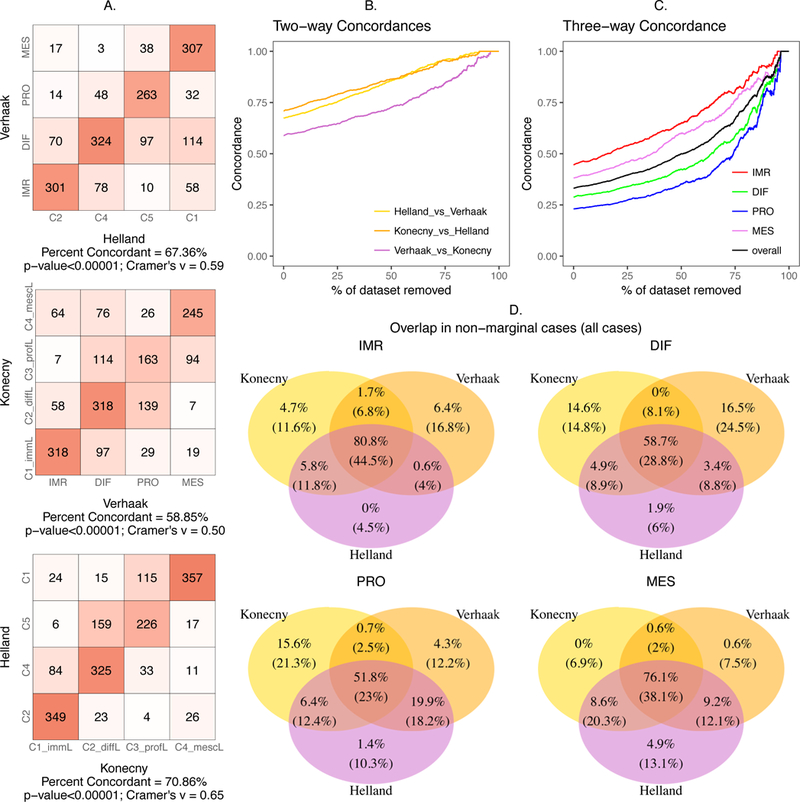
(A) Contingency table showing concordance of subtypes while comparing the methods pairwise (B) Pairwise concordance between the methods versus percentage of the dataset with samples of lower subtype margins removed, (C) three-way overall concordance between the methods and that of the individual subtypes versus percentage removed, (D) The classification of patients by three published algorithms as a Venn diagram for each of the four subtypes. Each area shows percentages of patients when all patients are classified (below, in parentheses) and after refusing to classify 75% of the most marginally classified tumors by any of the three methods (above). Thus, the numbers on the top of the three-way intersection are the concordant tumors according to the three original algorithms. Bottom numbers indicate relatively unambiguous subtype predictions by all three algorithms and which are also concordant with the others.
Tumors of intermediate subtype
The individual subtyping algorithms calculate numeric scores for each subtype, and assign each tumor to the subtype with the highest score. A tumor with a large difference or “margin” between the highest and second highest scores can be considered distinctly classifiable, whereas a tumor with two nearly equal scores could be considered of intermediate subtype. We examined the effect of modifying the individual algorithms to prevent assignment of indeterminate cases at various thresholds. For each pair of subtype classifiers, we examined the classification concordance with increasing thresholds on the margins.
For all pairs of subtype classifier, classification concordance increased as additional marginal cases are removed, approaching over 90% concordance once the majority of tumors are left unclassified (Figure 2B). Three-way concordance followed the same trend with lower overall concordance: a minimum of 23% for the proliferative subtype and maximum of 45% for the immunoreactive subtype when all tumors are classified. Restricting the concordance analysis to the top 50% of tumors by margin value resulted in an increased overlap between 35% (proliferative) and 65% (immunoreactive). At a strict threshold of where only 10% of tumors are classified, 88% of tumors overall are concordantly classified by all three published subtyping algorithms (Figure 2C). This large gain in concordance results from large reductions in both singleton calls - tumors assigned to one subtype by one algorithm, but not by the other two algorithms - and in 2-to-1 calls, tumors assigned to one subtype by two algorithms, but not by the third (Figure 2D). This indicates that tumors distinctly classifiable by a single algorithm are more likely to be concordantly classified by the other algorithms, and conversely, tumors that appear ambiguous to one algorithm are less likely to be classified in the same way by the other algorithms.
Survival Analysis
All proposed subtyping algorithms classified patients into groups that significantly differed in overall survival (Figure 3A, p < 10−5 for each subtyping algorithm, log-rank test). Comparing low-risk to high-risk subtypes for each algorithm, the hazard ratios increase from approximately 1.5 as marginal cases are removed (Figure 3B), suggesting that marginal cases may contribute to the intermediate survival profiles between subtypes.
Figure 3: Survival Analysis.
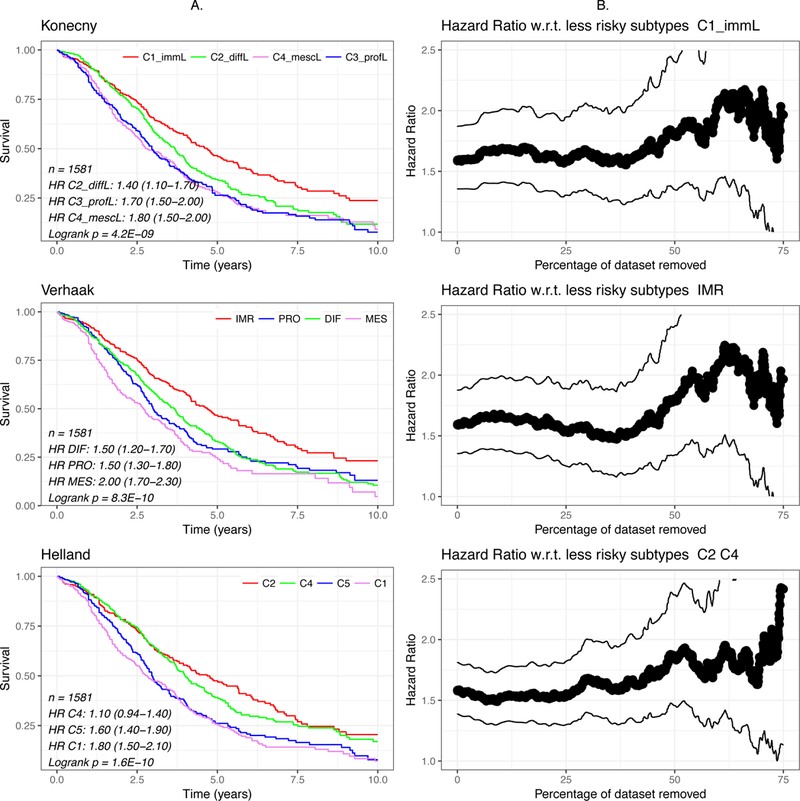
(A) Kaplan-Meier curves of subtypes of the 1581 patients with survival data under different methods. (B) Hazard ratios and 95% confidence intervals of the lowest-risk subtype (Konecny and Verhaak) or two subtypes (Helland) compared to the remaining subtypes.
Robustness of the Classifiers
Robust molecular subtyping should be replicable in multiple datasets. We performed de novo clustering in 15 independent ovarian datasets using the authors’ original gene lists and clustering methods. We compared these de novo clusters to the labels from our implementation of the published classifiers to assess robustness using the Prediction Strength (PS) statistic(12). For PS estimation, we included validation datasets with at least 100 HGSOC tumors. Overall we observed low robustness for all classifiers, with PS values under 0.6 for the three algorithms across datasets (Supplementary Figure 2), none meeting the 0.8 threshold typically indicating robust classes(12,17).
To assess whether low confidence predictions are driving the PS estimation, we re-computed the robustness of each algorithm set to classify varying fractions of the tumors with the highest margins. We used the largest dataset available, the TCGA dataset, as the validation set, and varied margin cutoffs of the Tothill and Konecny classifiers to require them to classify between 25% and 100% of the cases. From 10 random clustering runs, we report the median PS for the dataset. Clustering was performed on the full TCGA dataset and tumors of low margin values were removed subsequent to clustering and after the classifier was fully defined, in order to avoid optimistically biasing the apparent strength of clusters. We observed that the robustness of each algorithm is substantially improved by preventing them tto classify ambiguous cases. The Tothill algorithm achieved almost perfect robustness (PS = 0.96) when allowed to leave 75% of cases unclassified (Figure 4).
Figure 4: Robustness Analysis of published classifiers, by Prediction Strength.
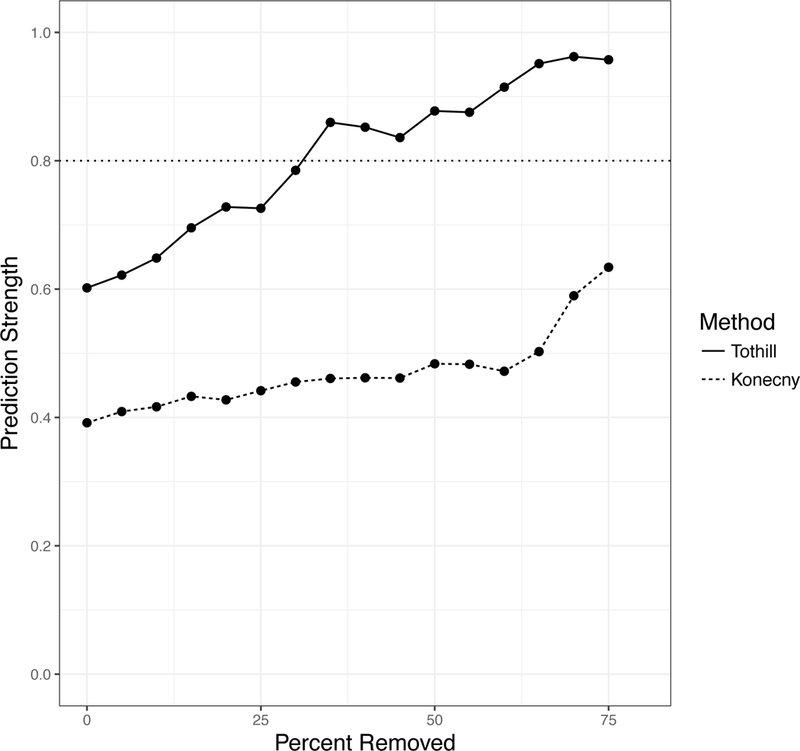
In each dataset, concordance was calculated between the published classifier and a classifier re-trained on the validation dataset. The TCGA dataset also classified using the published classifiers of Helland and Konecny (no re-training was done for the classifiers). The TCGA dataset was also clustered using the methods of Tothill and Konency (in red and blue respectively). Samples were removed from Prediction Strength calculations starting with the most ambiguous samples (with the smallest difference between the top subtype prediction and runner-up subtype prediction); the x-axis shows the percent removed before computing prediction strength. Each algorithm improves in robustness when allowed to leave ambiguous samples, that it is less certain in its classification, unclassified.
Consensus Classifier
To maximize concordance across classifiers, we developed consensusOV, a consensus subtyping scheme facilitating classification of tumors of well-defined subtypes (Figure 5). This classifier uses binary gene pairs(20,21) to support application across gene expression platforms. The consensusOV classifier exhibits overall pairwise concordance of 67 – 78% with each of the other three algorithms, when classifying all tumors; and 94% concordance with tumors that are concordantly classified by the other three algorithms (Figure 5A). The margins of consensusOV are higher for concordantly classified cases than for non-concordantly classified cases, and this difference in margins is greater than for any of the other three classifiers (Figure 6A). Accordingly, consensusOV was also most effective in identifying concordantly classified cases, although it was similar to the Konecny classifier in this respect (AUC = 0.76, Figure 6B). As expected, differences in survival of subsets identified by consensusOV are similar to those identified by previous classifiers. The highest risk subtypes are proliferative (HR=1.44, 95% CI: 1.07−1.94) and mesenchymal (HR=1.97, 95% CI: 1.46−2.67) when removing 75% of indeterminate low-margin tumors, with similar hazard ratios for the concordant cases (Figure 5B).
Figure 5: Concordance and Survival Stratification of consensusOV.
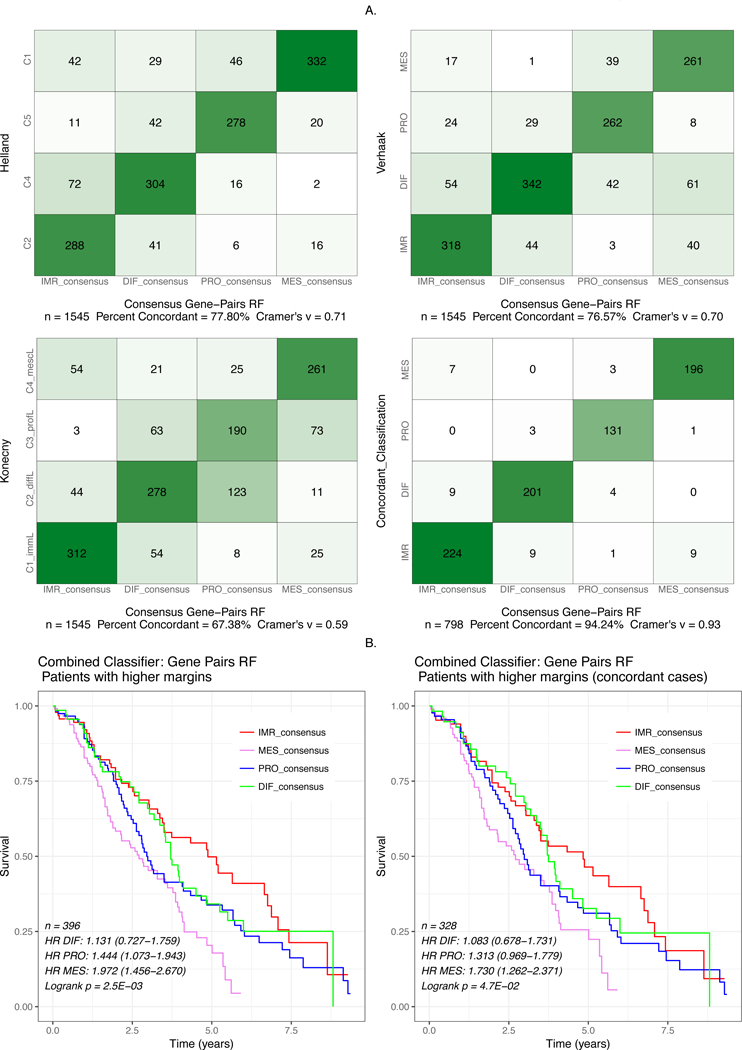
(A) Contingency plots showing concordance of subtype classification between consensusOV and the classifiers of Helland, Verhaak, Konecny. The fourth (bottom-right) plot shows the concordance between the consensus classifier and the patients concordantly classified between the three classifiers. (B) Survival curves for the pooled dataset provided by consensusOV. Classification was performed using leave-one-dataset-out validation. For the bottom two figures, classification with consensusOV was performed with the default cutoff, in which 75% of patients with the lowest margin are not classified.
Figure 6: Margin Analysis.
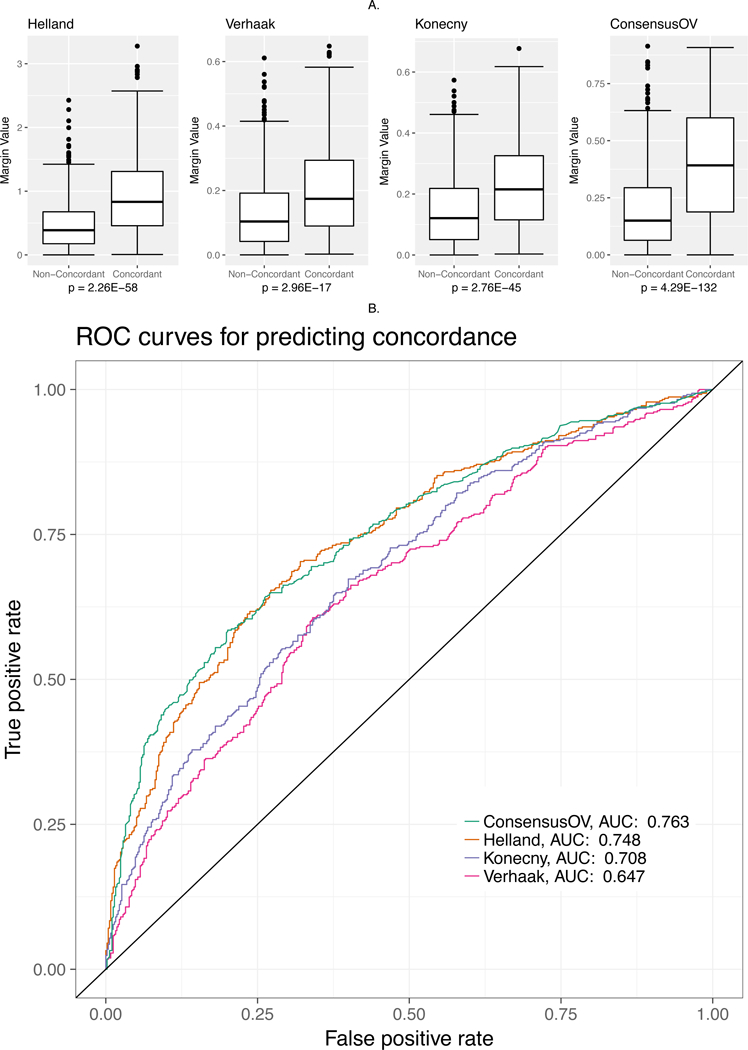
(A) Boxplots indicating the margin values assigned by each classifier to concordant and discordant cases. All statistical tests were performed using the Wilcoxon rank-sum test. (B) ROC curve for assessing the ability of margin values to discriminate between concordant and discordant cases.
Discussion
The existence of four distinct and concordant molecular subtypes of HGSOC has been reported in several studies of large patient cohorts(4–6,9), but also called into question by another effort(2) that could not identify subtypes, and by an independent validation effort that reported only two or three reproducible subtypes(27). Meanwhile, significant effort is being expended to translate these subtypes to clinical practice, for example to predict response to the angiogenesis inhibitor bevacizumab in the ICON7 trial(28,29). Our study pursues three major objectives: (1) reproduction of published subtype classification algorithms as an open-source resource; (2) evaluation of the robustness and prognostic value of each proposed subtyping scheme in independent data; and (3) consolidation of proposed subtyping schemes into a consensus algorithm.
We find that while the proposed 4-subtype classifications demonstrate significant concordance and association with patient survival, none are robust to re-training in new datasets. By modifying any of these algorithms to prevent classification of tumors of ambiguous subtype, robustness and concordance of subtyping algorithms improve dramatically. We propose a “consensus” classifier that can identify the most unambiguously classifiable tumors, although a continuous trade-off exists between classifying more tumors versus having greater confidence in those classified.
Ambiguity in tumor classification might arise from a heterogeneous admixture of different subtypes, or from a more homogeneous composition of indeterminate subtype. This distinction has implications for the therapeutic value of the proposed subtypes. Lohr et al. estimated that 90% of tumors in the TCGA HGSOC dataset are polyclonal(30) , and clonal spread of HGSOC has been directly inferred from single-nucleus sequencing(31). However, it remains unclear whether multiple clones in a tumor are consistently classifiable to the same subtype. If a tumor consists of multiple clones of different subtypes, then a subtype-specific therapy will likely lead to relapse as other clones survive and continue to grow. If this situation is common, even unambiguously classifiable tumors might be contaminated by small amounts of another subtype that could lead to relapse after subtype-specific therapy. This question could not be resolved by the current datasets, but may eventually be addressed by single-cell RNA sequencing(32) which is expected to further improve precision HGSOC molecular subtyping.
Several findings stand out in the validation of published subtyping algorithms. First, although previous studies reported inconsistent findings on whether subtypes differ by patient survival, our analysis in independent data showed clear survival differences. The 5-year survival rate for patients with different subtypes ranged from as low as 20% to as high as 50%. Second, published algorithms do not meet previously defined standards of robustness in terms of Prediction Strength, a measure of consistency between subtype classifiers trained in independent datasets. Finally, the concordance of three algorithms, established independently by different research groups from different patient cohorts, is only moderate but can be greatly improved by modifying the original algorithms to allow them to leave ambiguous tumors unclassified. In their original forms, all-way concordance of the four defined classes occurs in 23% to 45% of tumors. As the individual algorithms are modified so they are allowed to leave ambiguous cases unclassified, the minority of remaining tumors can be classified with over 90% concordance between the three algorithms. This is a novel finding of interest, because an alternate possibility was that classifiers trained on different datasets would suffer low concordance no matter how they treated uncertain tumors. This finding suggests a subset of tumors of “pure” subtype; unfortunately, such unambiguous cases account for as few as 25% of HGSOC tumors. This places important limitations on the potential for clinical application of HGSOC subtypes. The proposed alternative, consensusOV, identifies the consensus of published HGSOC subtype classifiers. By training on multiple datasets, using binary (pairwise greater-than or less-than) relationships between pairs of genes, and using a relatively small gene set, it is designed to identify robustly classifiable HGSOC tumors across gene expression platforms and datasets.
Moving forward, general agreement on how molecular subgroups of ovarian cancer are defined would facilitate the use of expression data in clinical management. (33). The present subgroups while prognostically important are not yet clinically meaningful. Much like other prognostic factors such as age, ascites, and histology, they do not alter clinical management. However, a better understanding of the biology underlying the subgroups will provide a more rational targeted treatment of those patients (perhaps first in trial) such as seen in HRD tumors with PARP inhibitors. The use of algorithms that can classify the tumor of an individual patient, while allowing some tumors to remain unclassified, along with assessment of subtype robustness in independent datasets by Prediction Strength, would move the field closer to this goal.
Supplementary Material
Statement of Translational Relevance.
High-grade serous ovarian carcinoma (HGSOC) is the fifth leading cause of cancer death in the United States and Canada. The majority of HGSOC are diagnosed as late-stage, high-grade serous ovarian carcinomas, for which prognosis is generally poor and few targeted therapies exist. Significant research effort has suggested several molecularly distinct subtypes of HGSOC, yet no consensus in the field exists and computational methods to analyze high-dimensional gene expression datasets differ across studies. Although subtypes have been shown to differ in overall survival, the lack of agreement on molecular subtype definition has been cited as a barrier to their investigation through clinical trial. In the present study, we perform an analysis of a large compendium of HGSOC transcriptomes in order to evaluate the concordance of computational methods and address the emerging consensus in the field. We develop a subtype classifier that represents the consensus of HGSOC subtypes, and show that many tumors are of intermediate or mixed subtype based on currently defined subtypes. These findings improve our understanding of the molecular basis of high-grade serous carcinoma, an important step in defining the underlying biology and identifying therapeutic targets of HGSOC.
Acknowledgements
The authors thank Brad Nelson for his feedback regarding the prognostic value of molecular subtypes in HGSOC, and Andrew Cherniak for providing ABSOLUTE purity and ploidy estimates for tumors from The Cancer Genome Atlas.
Funding
G.M. Chen was supported by the Canadian Institutes of Health Research and the Terry Fox Research Institutes. D.A.M. Gendoo was supported by the Ontario Institute for Cancer Research through funding provided by the Government of Ontario. Z. Safikhani was supported by The Cancer Research Society (Canada). B. Haibe-Kains was supported by the Gattuso-Slaight Personalized Cancer Medicine Fund at Princess Margaret Cancer Centre, the Canadian Institutes of Health Research, and the Terry Fox Research Institute. L Waldron was supported by grants from the National Cancer Institute at the National Institutes of Health (1R03CA191447–01A1 and U24CA180996). This work was part of the immunoTherapy Network supported by the Terry Fox Research Institute (Translational Research Program Grant #1060).
REFERENCES
- 1.Liu J, Matulonis UA. New strategies in ovarian cancer: translating the molecular complexity of ovarian cancer into treatment advances. Clin Cancer Res. 2014;20:5150–6. [DOI] [PubMed] [Google Scholar]
- 2.Bonome T, Levine DA, Shih J, Randonovich M, Pise-Masison CA, Bogomolniy F, et al. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008;68:5478–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14:5198–208. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verhaak RGW, Tamayo P, Yang J-Y, Hubbard D, Zhang H, Creighton CJ, et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J Clin Invest. 2013;123:517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konecny GE, Wang C, Hamidi H, Winterhoff B, Kalli KR, Dering J, et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J Natl Cancer Inst [Internet]. 2014;106 Available from: 10.1093/jnci/dju249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waldron L, Haibe-Kains B, Culhane AC, Riester M, Ding J, Wang XV, et al. Comparative meta-analysis of prognostic gene signatures for late-stage ovarian cancer. J Natl Cancer Inst [Internet]. 2014;106 Available from: 10.1093/jnci/dju049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Planey CR, Gevaert O. CoINcIDE: A framework for discovery of patient subtypes across multiple datasets. Genome Med. 2016;8:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helland Å, Anglesio MS, George J, Cowin PA, Johnstone CN, House CM, et al. Deregulation of MYCN, LIN28B and LET7 in a molecular subtype of aggressive high-grade serous ovarian cancers. PLoS One. 2011;6:e18064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ganzfried BF, Riester M, Haibe-Kains B, Risch T, Tyekucheva S, Jazic I, et al. curatedOvarianData: clinically annotated data for the ovarian cancer transcriptome. Database . 2013;2013:bat013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng X, Lu W, Liu M. Identification of homogeneous and heterogeneous variables in pooled cohort studies. Biometrics [Internet]. 2015; Available from: 10.1111/biom.12285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tibshirani R, Walther G. Cluster Validation by Prediction Strength. J Comput Graph Stat. 2005;14:511–28. [Google Scholar]
- 13.Gendoo DMA, Ratanasirigulchai N, Chen GM, Waldron L, Haibe-Kains B. MetaGxData: Breast and Ovarian Clinically Annotated Transcriptomics Datasets [Internet]. bioRxiv. 2016. [cited 2017 May 18]. page 052910 Available from: http://biorxiv.org/content/early/2016/05/12/052910.abstract [Google Scholar]
- 14.Waldron L, Riester M, Ramos M, Parmigiani G, Birrer M. The Doppelgänger Effect: Hidden Duplicates in Databases of Transcriptome Profiles. J Natl Cancer Inst [Internet]. 2016;108 Available from: 10.1093/jnci/djw146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.R Core Team. R: A Language and Environment for Statistical Computing [Internet]. Vienna, Austria: R Foundation for Statistical Computing; 2014. Available from: http://www.R-project.org [Google Scholar]
- 16.Schröder MS, Culhane AC, Quackenbush J, Haibe-Kains B. survcomp: an R/Bioconductor package for performance assessment and comparison of survival models. Bioinformatics. 2011;27:3206–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haibe-Kains B, Desmedt C, Loi S, Culhane AC, Bontempi G, Quackenbush J, et al. A three-gene model to robustly identify breast cancer molecular subtypes. J Natl Cancer Inst. 2012;104:311–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gendoo DMA, Ratanasirigulchai N, Schröder MS, Paré L, Parker JS, Prat A, et al. Genefu: an R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics. 2016;32:1097–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paquet ER, Hallett MT. Absolute assignment of breast cancer intrinsic molecular subtype. J Natl Cancer Inst. 2015;107:357. [DOI] [PubMed] [Google Scholar]
- 21.Patil P, Bachant-Winner P- O, Haibe-Kains B, Leek JT. Test set bias affects reproducibility of gene signatures. Bioinformatics [Internet]. 2015; Available from: 10.1093/bioinformatics/btv157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riester M, Wei W, Waldron L, Culhane AC, Trippa L, Oliva E, et al. Risk prediction for late-stage ovarian cancer by meta-analysis of 1525 patient samples. J Natl Cancer Inst [Internet]. 2014;106 Available from: 10.1093/jnci/dju048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. Nature Publishing Group; 2012;30:413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. Nature Publishing Group; 2015;12:115–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie Y Dynamic Documents with R and knitr, Second Edition CRC Press; 2015. [Google Scholar]
- 26.Cramér H Mathematical Methods of Statistics (PMS-9). Princeton University Press; 2016. [Google Scholar]
- 27.Way GP, Rudd J, Wang C, Hamidi H, Fridley BL, Konecny GE, et al. Comprehensive Cross-Population Analysis of High-Grade Serous Ovarian Cancer Supports No More Than Three Subtypes. G3 . 2016;6:4097–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gourley C, McCavigan A, Perren T, Paul J, Michie CO, Churchman M, et al. Molecular subgroup of high-grade serous ovarian cancer (HGSOC) as a predictor of outcome following bevacizumab. J Clin Oncol. 2014;32:5502. [Google Scholar]
- 29.Winterhoff B, Kommoss S, Oberg AL, Wang C, Riska SM, Konecny GE, et al. Bevacizumab may differentially improve survival for patients with the proliferative and mesenchymal molecular subtype of ovarian cancer. J Clin Oncol. 2014;32:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McPherson A, Roth A, Laks E, Masud T, Bashashati A, Zhang AW, et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat Genet [Internet]. 2016; Available from: 10.1038/ng.3573 [DOI] [PubMed] [Google Scholar]
- 32.Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods. 2014;11:41–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waldron L, Riester M, Birrer M. Molecular subtypes of high-grade serous ovarian cancer: the holy grail? J Natl Cancer Inst [Internet]. 2014;106 Available from: 10.1093/jnci/dju297 [DOI] [PubMed] [Google Scholar]
- 34.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshihara K, Tajima A, Yahata T, Kodama S, Fujiwara H, Suzuki M, et al. Gene expression profile for predicting survival in advanced-stage serous ovarian cancer across two independent datasets. PLoS One. 2010;5:e9615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Denkert C, Budczies J, Darb-Esfahani S, Györffy B, Sehouli J, Könsgen D, et al. A prognostic gene expression index in ovarian cancer - validation across different independent data sets. J Pathol. 2009;218:273–80. [DOI] [PubMed] [Google Scholar]
- 37.Mok SC, Bonome T, Vathipadiekal V, Bell A, Johnson ME, Wong K-K, et al. A gene signature predictive for outcome in advanced ovarian cancer identifies a survival factor: microfibril-associated glycoprotein 2. Cancer Cell. 2009;16:521–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mateescu B, Batista L, Cardon M, Gruosso T, de Feraudy Y, Mariani O, et al. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat Med. 2011;17:1627–35. [DOI] [PubMed] [Google Scholar]
- 39.Dressman HK, Berchuck A, Chan G, Zhai J, Bild A, Sayer R, et al. An integrated genomic-based approach to individualized treatment of patients with advanced-stage ovarian cancer. J Clin Oncol. 2007;25:517–25. [DOI] [PubMed] [Google Scholar]
- 40.Karlan BY, Dering J, Walsh C, Orsulic S, Lester J, Anderson LA, et al. POSTN/TGFBI-associated stromal signature predicts poor prognosis in serous epithelial ovarian cancer. Gynecol Oncol. 2014;132:334–42. [DOI] [PubMed] [Google Scholar]
- 41.Crijns APG, Fehrmann RSN, de Jong S, Gerbens F, Meersma GJ, Klip HG, et al. Survival-related profile, pathways, and transcription factors in ovarian cancer. PLoS Med. 2009;6:e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pils D, Hager G, Tong D, Aust S, Heinze G, Kohl M, et al. Validating the impact of a molecular subtype in ovarian cancer on outcomes: a study of the OVCAD Consortium. Cancer Sci. 2012;103:1334–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bentink S, Haibe-Kains B, Risch T, Fan J- B, Hirsch MS, Holton K, et al. Angiogenic mRNA and microRNA gene expression signature predicts a novel subtype of serous ovarian cancer. PLoS One. 2012;7:e30269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshihara K, Tsunoda T, Shigemizu D, Fujiwara H, Hatae M, Fujiwara H, et al. High-risk ovarian cancer based on 126-gene expression signature is uniquely characterized by downregulation of antigen presentation pathway. Clin Cancer Res. 2012;18:1374–85. [DOI] [PubMed] [Google Scholar]
- 45.Bonome T, Levine DA, Shih J, Randonovich M, Pise-Masison CA, Bogomolniy F, et al. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008;68:5478–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meyniel J-P, Cottu PH, Decraene C, Stern M-H, Couturier J, Lebigot I, et al. A genomic and transcriptomic approach for a differential diagnosis between primary and secondary ovarian carcinomas in patients with a previous history of breast cancer. BMC Cancer. 2010;10:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.