

Abstract
Sparassis crispa (Hanabiratake) is a widely used medicinal mushroom in traditional Chinese medicine because it contains materials with pharmacological activity. Here, we report its 39.0-Mb genome, encoding 13,157 predicted genes, obtained using next-generation sequencing along with RNA-seq mapping data. A phylogenetic analysis by comparison with 25 other fungal genomes revealed that S. crispa diverged from Postia placenta, a brown-rot fungus, 94 million years ago. Several features specific to the genome were found, including the A-mating type locus with the predicted genes for HD1 and HD2 heterodomain transcription factors, the mitochondrial intermediate peptidase (MIP), and the B-mating type locus with seven potential pheromone receptor genes and three potential pheromone precursor genes. To evaluate the benefits of the extract and chemicals from S. crispa, we adopted two approaches: (1) characterization of carbohydrate-active enzyme (CAZyme) genes and β-glucan synthase genes and the clusters of genes for the synthesis of second metabolites, such as terpenes, indoles and polyketides, and (2) identification of estrogenic activity in its mycelial extract. Two potential β-glucan synthase genes, ScrFKS1 and ScrFKS2, corresponding to types I and II, respectively, characteristic of Agaricomycetes mushrooms, were newly identified by the search for regions homologous to the reported features of β-glucan synthase genes; both contained the characteristic transmembrane regions and the regions homologous to the catalytic domain of the yeast β-glucan synthase gene FKS1. Rapid estrogenic cell-signaling and DNA microarray-based transcriptome analyses revealed the presence of a new category of chemicals with estrogenic activity, silent estrogens, in the extract. The elucidation of the S. crispa genome and its genes will expand the potential of this organism for medicinal and pharmacological purposes.
Introduction
Sparassis crispa, alternatively known as cauliflower mushroom in English or Hanabiratake in Japanese, has been used for food and as a traditional and modern medicine1. S. crispa contains materials with pharmacological activity, such as antitumor, antiangiogenic, antimetastatic, antimicrobial, antiviral, antioxidant, antihypertensive, antidiabetic and antiallergic activities, which are attributable to different effective chemicals with low and high molecular weights such as phenolics and polyphenols, flavonoids and terpenoids2. A type of polysaccharide, β-1,3-glucan (β-glucan), which is abundantly obtained from the fruiting body of S. crispa and has polysaccharide chains linked by β-1,3-glycosidic bonds, has been highlighted because of its potential benefits, such as for prevention of cardiovascular diseases and cancer, due to its immunomodulatory or immunostimulative effects3. Immunostimulative activity of β-glucan was commercialized as a drug for the treatment of diseases4. However, β-glucan has not been confirmed to be effective for all expected diseases and symptoms, thus chemicals other than β-glucan with respective activities have been considered2.
Phytoestrogens, plant-derived chemicals with estrogenic activity, have also been considered as beneficial agents for menopausal syndromes, cardioprotection, neuroprotection and anti-carcinogenesis5. Furthermore, chemicals obtained from mushrooms have been used as sources of estrogenic chemicals and have been investigated as alternatives of synthetic estrogens because they may not cause adverse effects or unexpected side effects6.
As mushrooms are often misrepresented, it is important to identify medicinal mushrooms at the level of genomic DNA7. Here, we report the genomic structure of S. crispa (strain Scrmy26), and its genes identified by next-generation sequencing and RNA-seq-based transcriptome analysis. We further explored beneficial usages of S. crispa by two different approaches: finding new β-glucan synthase genes by genome and protein analyses, and identifying new compounds with estrogenic activity by bioassays.
Results
Genomic structure and general features
The genome of S. crispa mycelia (strain Scrmy26) was sequenced using a whole genome shotgun sequencing strategy (see Materials and Methods). A 39.0-Mb genome sequence was obtained by assembling approximately 21.3-Gbp reads (>500 × coverage; data not shown) (Table 1). This genome sequence assembly consisted of 32 contigs with an N50 length of 3.18 Mb and L50 of 5 (Fig. 1; Table S1). Based on the number of contigs along with the number of chromosomes expected for mushrooms, we expected the genome size to be close to the obtained size. In total, 13,157 protein-coding genes were predicted, characterized by an average gene length of 1,669.3 bp and average exon number of 5.7 (Table S2). The number of genes in the genome of S. crispa was comparable with that in genomes of other filamentous fungi8–14. The genes predicted formed transcripts with an average length of 1.3 kb and proteins with an average length of 147 amino acids (Table S2). Protein domains are important for the annotation of the genes and proteins identified by the genome analysis15,16. We provided here a list of protein domains predicted by the analysis of protein databases (Table S3).
Table 1.
General features of the S. crispa genome.
| Number of Contigs | 32 |
| Length of the genome assembly (Mb) | 39.0 |
| GC content (%) | 51.4 |
| Number of protein-coding genes | 13,157 |
| Average/Median gene length (bp) | 1,648.1/1,308 |
| Average/Median protein-coding sequence size (bp) | 1,326.1/1,044 |
| Average/Median number of exons per gene | 5.7/4 |
| Average/Median exon size (bp) | 233.6/137 |
| Average/Median intron size (bp) | 73.4/55 |
Figure 1.

The genomic features of S. crispa. The ideogram represents (a) contigs, (b) GC content, (c) gene numbers, (d) gene expression levels, and (e) large segmental duplications. GC content was calculated as the percentage of G + C in 20-kb non-overlapping windows. The abundances of gene transcripts were analyzed as described in Materials and Methods, and expressed in FPKM. The gene number was calculated in 20-kb non-overlapping windows, and the maximum value of the axis was 15. Gene expression levels are shown in red (FPKM > = 100), orange (FPKM > = 10), green (FPKM > 0) and black (FPKM = 0). Large segmental duplications sharing more than 90% sequence similarity are connected by orange (sequence length > = 5 kb) and grey (sequence length > = 2 kb) lines.
Recently, the genome sequence of S. latifolia was reported17, where it showed genome features very close to the data shown here, such as GC contents (51.43% vs 51.42%, our data) and the number of predicted gene models (12,471 vs 13,157), except for a quite difference in the predicted genome sizes (48.13 Mb vs 39.02 Mb). This is probably due to the depth of sequencing, as revealed in the scaffold/contig numbers (472 vs 32) and N50 values (640.83 kb vs 3,179.64 kb). Other features, such as phylogenetic analyses with other fungal genomes and for specific gene functions, did not show much difference, suggesting that reported S. latifolia and S. crispa (strain Scrmy26) are very close each other.
Comparison with other fungal genomes
The predicted proteome of S. crispa was compared with 25 other sequenced fungi (Table S4). The evolutionary history of S. crispa was examined with a phylogenetic tree (Fig. 2), which was constructed using 895 single-copy orthologous genes conserved in these 26 fungi obtained by OrthoMCL analysis (see Materials and Methods). The molecular clock analysis revealed that Postia placenta, a brown-rot fungus18,19, was evolutionarily closest fungi to S. crispa, and their divergence time was estimated to be 94 million years ago (MYA).
Figure 2.
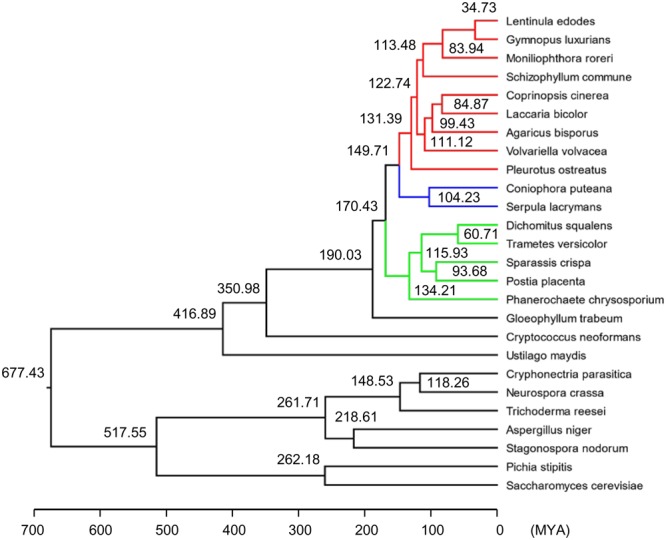
Phylogenetic tree of S. crispa with 25 other fungal species. The phylogenetic tree was constructed by the maximum likelihood method (see Materials and Methods). MYA: million years ago.
Analysis of mating type loci
Two mating type loci, A and B, were identified in the genome sequence of S. crispa on different contigs (Fig. 3; Table S5). The A-mating-type locus was identified by homology search with the genes for HD1 and HD2 homeodomain transcription factors, and the mitochondrial intermediate peptidase (MIP) of Coprinopsis cinerea and Schizophyllum commune, where relatively high protein homologies (66~67% identity) were found for MIP, but low homologies (28~34% identity) were found for HD1 and HD2. However, all three genes were found to be located in close proximity on the same contig (contig 1), and there is similarity in the arrangement of HD1 and HD2 genes (located on different strands but with the characteristic outward transcriptional direction) compared with other fungi20, suggesting them to be functional. On the other hand, we found a total of seven potential pheromone receptor genes and three potential pheromone precursor genes for the B-mating-type locus, which were mapped on contig 10 (Fig. 3; Table S5).
Figure 3.
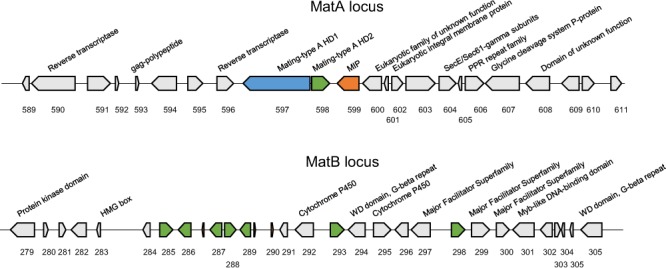
The matA and matB loci of S. crispa. The genes in the loci are shown. The matA and matB loci are positioned on contigs 1 or 10, respectively.
CAZymes and glycosyltransferases (GTs)
A total of 246 candidate carbohydrate-active enzyme genes (CAZymes) were identified in the genome of S. crispa, which included 131 glycoside hydrolases (GHs), 10 carbohydrate esterases (CEs), 61 glycosyl transferases (GTs), 4 polysaccharide lyases (PLs), 19 carbohydrate-binding modules (CBMs) and 31 auxiliary activity enzymes (AAs) (Table S6). The distribution of CAZymes in S. crispa was compared with 25 other fungi. Compared with the genomes of Agaricales, Polyporales generally had fewer CAZymes, and S. crispa had the lowest number of GHs among them (Table S6). The variety of GTs (Fig. 4; Table 2) and GHs (Fig. S1; Table 2) of S. crispa was compared with those of Phanerochaete chrysosporium21, Postia placenta18 and Lentinula edodes14. Compared with other fungi, S. crispa had the lowest number of genes in each category of CAZymes, and S. crispa, or the group containing S. crispa, had the lowest number of GT family genes of GT1, GT5, GT17, GT69 and GT90 (Table S6; Fig. 4). On the other hand, S. crispa had a low number of GH family genes (about half of that in Lentinula edodes), and thus, many GH families had the lowest numbers compared with three other fungi (Table S6; Fig. S1). Thus, S. crispa may be classified as a fungus with poorly developed carbohydrate utilization ability.
Figure 4.
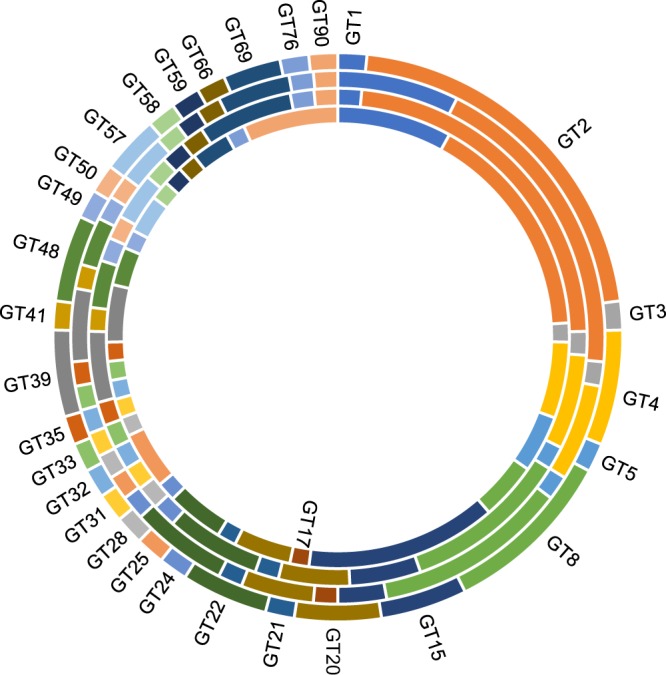
Variation of glycosyltransferases (GTs). S. crispa (the outer-most ring), Phanerochaete chrysosporium (the second ring), Postia placenta (the third ring) and Lentinula edodes (the inner-most ring).
Table 2.
CAZyme genes in fungi.
| CAZyme family | Number of genes | |||
|---|---|---|---|---|
| S. crispa | P. chrysosporium | P. placenta | L. edodes | |
| Glycosyl transferase (GT) | 61 | 68 | 65 | 75 |
| Glycoside hydrolase (GH) | 131 | 185 | 144 | 249 |
| Polysaccharide lyase (PL) | 4 | 6 | 5 | 9 |
| Carbohydrate esterase (CE) | 10 | 25 | 12 | 30 |
| Carbohydrate-binding module (CBM) | 19 | 61 | 29 | 62 |
| Auxiliary activities enzyme (AA) | 31 | 98 | 49 | 83 |
| Total | 246 | 403 | 290 | 464 |
Identification of β-glucan synthases
Among polysaccharides in the fungal cell wall, β-1,3-glucan (β-glucan) is the most prevalent and has been widely used for pharmacological purposes. β-Glucan is synthesized from uridine 5’-diphosphate (UDP)-glucose by β-glucan synthase. β-Glucan synthase is a membrane protein composed of catalytic domains and transmembrane domains. In Saccharomyces cerevisiae, there are two independent β-glucan synthase genes (FKS1 and FKS2). Identification of β-glucan synthase genes was performed by BLASTP search with reported genes (see Materials and Methods), where we found two potential β-glucan synthase genes, ScrFKS1 and ScrFKS2, which are homologous to type I and type II genes, respectively (Table S7; Fig. 5). The presence of these two different types is a common characteristic of Agaricomycetes mushrooms22. ScrFKS1 and ScrFKS2 are homologous to the previously reported type I and type II genes by 77–90% or 77–92%, respectively (Table S7), thereby indicating these to be new genes. Two regions, a region homologous to the catalytic domain of FKS123 and a β-glucan synthase-homologous region, were identified in both genes by Pfam database search, and transmembrane domains by TMHMM database search (Figs 5 and S2). Although two transmembrane domains, TM1 and TM2, are present at identical positions, they differ in that the region between TM1 and TM2 is localized outside the cell in ScrFKS1 but inside in ScrFKS2. In addition, there are several potential N-glycosylation sites, some of which are well-conserved between them (Figs 5 and S2); positions 42, 257, 441, 477/478, 912 and 1365 aa (ScrFKS1) and positions 41, 231, 456, 485, 842 and 1338 aa (ScrFKS2), which are conserved between two proteins, are also similar in a sense of the locations relative to the important domains, such as FKS1 and transmembrane domains. Some of them are conserved in another medicinal fungus, Cordyceps militaris24.
Figure 5.

Structure of β-glucan synthases. (a) Structure of ScrFKS1 (on contig 5) and ScrFKS2 (on contig 4) genes in S. crispa. The intron-exon structure is shown above, while transcriptome mapping results are shown below. Transcriptional directions are shown by arrows. (b) Structure of ScrFKS1 and ScrFKS2 proteins. The transmembrane domains predicted by TMHMM are shadowed, and their clusters are marked with TM1 and TM2. The regions homologous to FKS1 domains (FKS1) and glucan synthases (GS) predicted by Pfam are shown in orange and green, respectively. The potential N-glycosylation sites predicted by ScanProsite are shown by open triangles, and the exon-intron junctions are shown by filled triangles.
Biosynthetic gene clusters
Other than highly-polymerized structure of β-glucan, small molecules or secondary metabolites, such as terpenes, indoles and polyketides, have been known to act as bioactive compounds, and the genes responsible for their synthesis are known to form clusters15,25. A total of 15 such gene clusters were identified in the S. latifolia genome, among which the genes encoding terpene synthase for the synthesis of mono-, sesqui-, and di-terpenes, indole prenyltransferase (indole-PTase) or dimethylallyl tryptophan synthase (DMATS) for the synthesis of indole alkaloids, and type I polyketide synthases for the synthesis of aromatic and highly reduced polyketide metabolites, were respectively found17. Meanwhile, a total of 30 gene clusters were identified here in the S. crispa genome (Fig. S3), which include 15, 2, 5 and 8 genes potentially associated with the synthesis/metabolism of terpenes, indoles, polyketides and others, respectively (summarized in Table S8). Note that all these chemical categories have been known to include chemicals with estrogenic activity and that some are derived from fungi, such as zearalenone and brefeldin A26. Thus, we next tried to find chemicals with estrogenic activity in the extract of S. crispa.
Estrogenic activity associated with the mycelial extract
We then examined whether the water extract of S. crispa (SCE) contains beneficial components by screening for silent estrogens, which has been reported for an edible mushroom Agaricus blazei (A. blazei)27. A silent estrogen is a category of chemicals that exhibit estrogenic gene expression profiles but do not have any growth-stimulating activity in estrogen-responsive breast cancer MCF-7 cells, and thus may be beneficial for preventing cancer progression and as alternatives for estrogen antagonists28. SCE did not exhibit any growth-stimulating activity in MCF-7 cells (Fig. 6a), but it rapidly activated both Erk1/2 and Akt (Fig. 6c), which are markers of the non-genomic pathway of estrogen signaling29. We also examined whether the activity was retained after extraction of SCE with ethylacetate (Fig. 6b), which was the case for A. blazei30. No estrogenic activity was detected by DNA microarray-based gene expression profiling for the ethylacetate extract (R = 0.04, Fig. 6d), but SCE had profiles (R = 0.76 for SCE10 or 0.62 for SCE100) comparable with that for 17β-estradiol (E2) (R = 0.81). As SCE contained very small amounts of β-glucans (0.95% of the amount in the dry mycelium, data not shown), the chemical(s) responsible for estrogenic activity may be a water soluble chemical with a low molecular weight.
Figure 6.

Estrogenic activity of S. crispa mycelial extracts. (a) Cell proliferation assay. MCF-7 cells were treated with vehicle (DMSO), E2 (10 nM) or the extracts as indicated. Cell proliferation was examined by sulforhodamine B (SRB) assay. The ratio of cell proliferation rate in response to E2 or the extracts to that to the control (DMSO) is shown in the graphs. *p < 0.01 vs. control (Cont). (b) The extraction strategy and the yields (%) of materials (dry weight). (c) Western blot analysis of the activation of Erk1/2 and Akt. MCF-7 cells were treated with 10 nM E2 or SCE10 for the indicated times (min), and cell extracts were subjected to Western blot analysis for phosphorylated (P-) or total (T-) proteins as indicated (left). The results of three independent experiments are summarized along with statistical evaluation in the graphs (right). Significance of data compared with the controls was evaluated using p-values of *p < 0.01 or **p < 0.05. (d) The gene expression profiles for these chemicals were compared using a set of 150 estrogen-responsive genes in scatter-plot graphs. The vertical and horizontal axes indicate log2 values of the signal intensities. R-values were calculated for each graph on the basis of linear regression between two profiles.
Discussion
S. crispa is a brown-rot fungus that grows primarily on the stumps of coniferous trees, and is widely distributed throughout the North Temperate Zone2. S. crispa has been known as a source of natural products with bioactive properties, such as for nutraceutical31, medicinal32,33 and cosmetic34 applications. Among compounds found in S. crispa, β-glucan has been extensively studied as chemicals effective for neuroprotection35, cardioprotection36 and anti-inflammation37. In contrast, recent studies showed that the aromatic compounds isolated from S. crispa are effective for the treatment of cancer38 or hyperlipidemia39. Here, we used the mycelium of S. crispa (strain Scrmy26) as a source both for the genome/gene analysis and the analysis of bioactive materials to employ a consistent system for the analyses. We focused on the mycelium here because controlling the culture conditions would be easier and thus mass production and quality control could be standardized. Furthermore, S. crispa is one of the mushrooms that contain the highest phenolic contents and the highest antioxidant capacity in mycelia40.
The analysis of the S. crispa genome revealed its relatively high GC% (51.4%; Table 1) among the 26 fungal genomes compared here, while the sizes of the genome and the number of protein-coding genes are well within their variations. From the phylogenetic analysis, S. crispa was likely diverged from Postia placenta 94 MYA. Both S. crispa and P. placenta belong to brown-rot fungi, which degrade plant cell walls by ligninolysis with distinct differences from while-rot fungi including the mechanism of lignocellulose conversion18. The numbers of categories of CAZyme genes, such as GHs and GTs, are comparable between them (131 GHs for S. crispa and 144 GHs for P. placenta, or 61 GTs for S. crispa and 65 GTs for P. placenta; Table S6), suggesting similarity in their lifestyle evolutions41. As the classification of fungi has often been controversial and, several revisions have resulted in several taxonomic synonyms for P. placenta42, it will be quite informative to find the similarity as well as the differences in genome sequences between these two fungi to understand their lifestyles.
A total of 5,601 genes were annotated by Gene Ontology database, which include 1,562, 4,931 or 3,437 genes related to cellular components, molecular functions or biological processes, respectively (Fig. S4). Significant numbers of genes were found to be associated with cell/cell part/organelle/membrane (cellular component), binding/catalytic activity (molecular function), and metabolic process/cellular process (biological process). Meanwhile, a total of 6,460 genes were classified into the pathways by the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, which were further classified into the metabolic pathways (1,753 genes), and the pathways related to genetic information processing (951), environmental information processing (576), cellular processes (852), organismal systems (1,011) and diseases (1,317) (Fig. S5). A significant number of genes (465) were related to carbohydrate and glycan metabolisms, including 37 genes for glycolysis/gluconeogenesis and 48 genes for N-glycan biosynthesis, suggesting these genes to be heavily utilized for the survival of the fungus.
Polysaccharides, which account for over 90% of fungal cell walls, are predominantly β-glucans, with the interchain composed of mainly β-1,3- and 3-4% β-1,6-glucosidic linkages43. There are two identifiers for the search of β-glucan synthase (EC 2.4.1.34), Glucan_synthase (PF02364) and FKS1_dom1 (PF14288), and two GT families including β-glucan synthase, GT2 and GT48, in the CAZy database. The database search resulted in the identification of two potential genes for β-glucan synthase, ScrFKS1 and ScrFKS2 (Figs 5 and S2), which are likely the genes for type I and type II genes characteristic to Agaricomycetes mushrooms22, respectively. Although the details are not yet known, there two types may have differences in catalytic activity and function in different developmental stages22. There are significant similarities in domains and motifs between ScrFKS1 and ScrFKS2 (see Fig. 5b), where we observed conservation of transmembrane domains, the catalytic FKS1 domain and N-glycosylation sites.
Hot water extraction of S. crispa powder resulted in the yield of β-glucan to be 50.6%32, although the activity of rapid estrogenic signaling through Erk1/2 and Akt was detected in the fraction of low β-glucan contents (Fig. 6c). The same extract revealed expression profiles similar to those after the treatment with E2 (Fig. 6d), suggesting the activity to be estrogenic. However, the extract failed to promote cell proliferation (Fig. 6a), thereby categorizing the activity to be that by silent estrogens. Silent estrogens are a group of chemicals with estrogenic gene-expression profiles but lacking estrogen-induced cell proliferation28, and include chemicals such as brefeldin A30 and capsaicin44, and the mixtures of chemicals, such as the degradation product of crude oil45. While phenolics are supposed to have a main structural characteristic for estrogenic chemicals, there are a number of exceptions in the structure responsible for the activity26, enlarging the potential of activities other than the promotion of cell proliferation, and the chemicals with such a characteristic would form a quite important category of bioactive materials.
Materials and Methods
Materials
S. crispa powder was prepared from mycelia of S. crispa, strain Scrmy26, which was supplied by Intertrade Co., LTD. The genomic DNA of S. crispa was prepared with a NucleoBond buffer Set III kit (Macherey-Nagel, Düren, Germany). The DNA library for genome sequencing was constructed with 28 μg of purified DNA with the SMRTbell Template Preparation Kit 1.0 and DNA/Polymerase Binding Kit P6 (Pacific Biosciences; Menlo Park, CA).
DNA sequencing and genome assembly
The S. crispa genome was sequenced by Hokkaido System Science Co., Ltd. (Sapporo, Japan), using 16 SMRT cells on the PacBio RSII platform (Pacific Biosciences) with P6-C4 chemistry, generating 21.3 Gbp (>500 × coverage). The mean, N50, and the longest insert read length of the generated data were 9,616 bp, 13,871 bp and 52,687 bp, respectively. The obtained reads were assembled using Falcon pipeline with slight modifications to the predefined parameter set distributed by the developers46. The assembled contigs were polished using the Arrow algorithm in the GenomicConsensus software package (https://github.com/PacificBiosciences/GenomicConsensus). The genome sequence data are available in the DDBJ BioProject Database of the DNA Database of Japan (DDBJ: http://www.ddbj.nig.ac.jp/) under Accession No. BFAD01000001-BFAD01000032 (PRJDB5582).
rRNA and tRNA identification
rRNAs were identified by Barrnap (https://github.com/tseemann/barrnap), a ribosomal RNA predictor. tRNAscan-SE47 was used to detect tRNA regions and secondary structures.
Gene expression analysis
A cDNA library was prepared from the total RNA using the TruSeq Stranded mRNA Sample Prep Kit (Illumina, San Diego, CA), and subjected to sequencing on the Illumina HiSeq 2500 platform. Approximately 80 million reads were obtained and used for genome annotation and gene expression analysis. The expression level of each gene was quantified by FPKM (expected fragments per kilobase of transcript per million fragments sequenced) values using Cufflinks48 software as follows. After adapter sequences were removed from RNA-seq reads using CutAdapt49, the primary contigs of the assembly were mapped using TopHat250. Finally, the mapped reads were counted for each gene model and reported in FPKM using Cufflinks.
Protein-coding gene prediction and functional annotation
The prediction of protein-coding genes along with their functional annotation was assisted by GeneBay Inc. (Yokohama, Japan). The RNA-seq reads were first aligned to the reference genome with HISAT51, then assembled by StringTie52 to construct transcript sequences. The obtained 15,184 transcripts were used to train AUGUSTUS53 for parameters of gene models in S. crispa. AUGUSTUS was run with exon and intron hints, derived from RNA-seq mapping data, as input data. On the other hand, CodingQuarry54, which was developed to accurately predict genes in fungal genomes, was used to predict gene models using the above StringTie-derived transcript sequences. The gene models predicted by AUGUSTUS, CodingQuarry and StringTie were combined to construct 13,156 consensus gene models using EvidenceModeler55. All of the predicted gene models were functionally annotated based on the similarity to annotated genes. BLASTP56 was used to align the protein sequences to Nr and fungal taxonomic divisions of Swiss-Prot57 protein databases with an e-value < 1e-5.
Phylogenetic analysis
A total of 26 fungal species assigned to Basidiomycota or Ascomycota, including S. crispa, were used in the phylogenetic analysis. Their protein sequences were compared by BLASTP (e-value < 1e-5 and hit number < 500). The BLASTP result was then analyzed by OrthoMCL58 with default parameters to find orthologous genes, where a total of 895 single-copy orthologous genes were determined. The sequence alignments of these 895 genes were calculated by MAFFT v7.30959, and then combined into a long sequence for each species. The conserved blocks of the alignment were selected by Gblocks 0.91b60 with the default parameters. Finally, the phylogenetic tree was constructed by RAxML-8.2.961 with bootstrap 1000. The fossil calibration was done as described62 by fixing the following three points in the molecule clock analysis: the most recent common ancestor (MRCA) of Coprinopsis cinerea, Laccaria bicolor and Schizophyllum commune diverged at 122.74 MYA; the MRCA of Serpula lacrymans and Coniophora puteana diverged at 104.23 MYA; and the MRCA of Pichia stipitis, Aspergillus niger, Cryphonectria parasitica, Stagonospora nodorum and Trichoderma reesei diverged at 517.55 MYA. The divergence time of other nodes was then calculated by r8s v1.8163 with the TN algorithm, PL method and the smoothing parameter value set to 1.8.
Identification of matA and matB genes
The matA genes were identified by the similarity with the matA and MIP genes of Coprinopsis cinerea and Schizophyllum commune. The pheromone receptor genes were identified by the search with key word “pheromone receptor” in Swiss-Prot. The sequences of pheromone precursors, usually 50~60 aa in length, were searched in the ~20 kb flanking region of the pheromone receptor genes by Transdecoder (https://transdecoder.github.io/) with Pfam search (https://pfam.xfam.org/).
Protein domain analysis and gene function classification
The amino acids sequences translated from the predicted gene models were analyzed using InterProScan (5.26–65.0) to annotate functional domains by the Pfam database (https://pfam.xfam.org/)64 as well as to assign Gene Ontology (GO) categories (http://www.geneontology.org/)65. The assigned GO terms are then mapped to the plant subset of GO categories (GO Slims) using Map2Slim script in the OWLTools (https://github.com/owlcollab/owltools). Protein domains are provided in the S. crispa genome sequence database (see above). Gene function pathways were analyzed by submitting BLAST data to KEGG (http://www.genome.jp/kegg/)66 database. The Gene Ontology data were visualized by WEGO (http://wego.genomics.org.cn/)67. Biosynthetic gene clusters in the S. crispa genome were identified by antiSMASH (http://antismash.secondarymetabolites.org/)25.
Identification of CAZymes and β-glucan genes
Carbohydrate-active enzymes (CAZymes) were classified by HMM search with dbCAN HMMs 5.068 (under the default cutoff threshold) and BLASTP search of CAZy database69 (e-value < = 1e-6; the covered fraction ratio > = 0.2; the maximum hit number = 500). The BLASTP results screened with the threshold used in the study of L. edodes14 were then added to the common results to obtain the final CAZyme annotation. Meanwhile, β-glucan genes were identified by BLAST search with reported genes for type I and II genes22, and the obtained sequences were analyzed for catalytic domains and transmembrane regions by searching Pfam or TMHMM (http://www.cbs.dtu.dk/services/TMHMM/) databases.
Preparation of S. crispa mycelial extracts
The aqueous extract of S. crispa mycelia (SCE) was prepared by boiling 50 g of the S. crispa powder in 1,000 ml of Milli-Q water for 10 min. The aqueous fraction was recovered by centrifugation at 3,300 × g for 25 min and sterilized by filtration through 0.22-μm filters (Millipore; Billerica, MA). To prepare the ethylacetate fraction (SCE-EtOAc), 100 ml of 10 mg/ml SCE was mixed with 100 ml of ethylacetate, and SCE-EtOAc was recovered as the upper fraction after centrifugation at 7,000 × g for 10 min. SCE-EtOAc was then dried by evaporation. The dried material dissolved in methanol was centrifuged at 3,300 × g for 10 min at 25 °C to remove debris, and the final concentration was adjusted to 10 mg/ml.
Western blotting
MCF-7 cells were obtained from the JCRB Cell Bank (National Institute of Health Sciences, Tokyo, Japan), and cultured in RPMI 1640 (Invitrogen, Carlsbad, CA) medium supplemented with 10% fetal bovine serum (FBS) at 37 °C in 5% CO2. Western blotting was performed according to the protocol described in Dong et al.30. Phospho-Erk1/2 (P-Erk1/2) and phospho-Akt (P-Akt) were used as the primary antibodies, and rabbit antibodies against total Erk1/2 (T-Erk1/2), P-Erk1/2, total Akt (T-Akt) or P-Akt (Cell Signaling Technologies, Ipswich, MA) were used as the secondary antibodies after appropriate dilutions (1:200 to 1:1000). The antibody-antigen complexes were detected with horseradish peroxidase-coupled goat antibody against rabbit IgG (Cell Signaling Technologies) after dilution (1:3,000), and then visualized using the ECL-plus Western Blotting Detection System (Amersham Pharmacia Biotech, Arlington Heights, IL).
Cell proliferation assay
The cell proliferation assay was performed as described in Dong et al.30. MCF-7 cells were cultured in phenol red-free RPMI 1640 medium supplemented with 10% dextran-coated charcoal-treated fetal bovine serum (DCC-FBS, Invitrogen) in 24-well plates. The cells were treated with 10 nM E2 (17 β-estradiol) or the extracts under the indicated concentrations for 3 days. The cells were then fixed with 10% cold trichloroacetic acid (TCA) at 4 °C for 30 min and stained with 0.4% sulforhodamine B (SRB) dissolved in 1% acetic acid for 20 min. The bound protein was solubilized with 10 nM unbuffered Tris-base and subjected to measurement of OD490. Six independent assays were performed for each treatment and the data were analyzed by the t-test.
DNA microarray assay
MCF-7 cells were maintained in phenol red-free RPMI 1640 medium containing 10% DCC-FBS, and incubated for 3 days at 37 °C in 5% CO2. Aliquots of E2 (10 nM), SCE (10 or 100 μg/ml) or SCE-EtOAc (10 μg/ml) in 0.1% DMSO were added to the medium, and the cells were cultured for 3 days. Cells treated with 0.1% DMSO (vehicle) were used as a control. RNA preparation and cDNA labeling followed by focused oligonucleotide-DNA microarray assays were performed as described previously27,70. The values obtained were normalized and log2-transformed, and then used for correlation analysis. A coefficient of correlation between gene expression profiles was calculated based on linear regression using SPSS 12.0J (SPSS Japan; Tokyo, Japan). We here used a total of 150 genes selected as highly reproducible estrogen-responsive genes as described before27. The microarray data are available in the Gene Expression Omnibus database of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/geo/) under Accession No. GSE108549.
Electronic supplementary material
Acknowledgements
This research has been supported partly by a grant from Kyushu Sangyo University for promoting basic technologies, a grant from Intertrade Co., LTD. for the basic study of S. crispa, and a Grant-in-Aid for Basic Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We are grateful to Dr. Y. Uemura and Dr. A. Nishizawa, GeneBay Inc., for helpful discussions.
Author Contributions
R.K. and Y.F. conceived of the study. R.K., Y.F., K.K., T.N. performed the analyses. R.K. wrote manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-34415-6.
References
- 1.Chandrasekaran G, Oh DS, Shin HJ. Properties and potential applications of the culinary-medicinal cauliflower mushroom, Sparassis crispa Wulf.:Fr. (Aphyllophoromycetideae): a review. Int. J. Med. Mushrooms. 2011;13(2):177–183. doi: 10.1615/IntJMedMushr.v13.i2.100. [DOI] [PubMed] [Google Scholar]
- 2.Kimura T. Natural products and biological activity of the pharmacologically active cauliflower mushroom Sparassis crispa. Biomed. Res. Int. 2013;2013:982317. doi: 10.1155/2013/982317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barsanti L, Passarelli V, Evangelista V, Frassanito AM, Gualtieri P. Chemistry, physico-chemistry and applications linked to biological activities of β-glucans. Nat. Prod. Rep. 2011;28(3):457–466. doi: 10.1039/c0np00018c. [DOI] [PubMed] [Google Scholar]
- 4.Lemieszek M, Rzeski W. Anticancer properties of polysaccharides isolated from fungi of the Basidiomycetes class. Contemp. Oncol. (Pozn) 2012;16(4):285–289. doi: 10.5114/wo.2012.30055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rietjens IMCM, Louisse J, Beekmann K. The potential health effects of dietary phytoestrogens. Br. J. Pharmacol. 2017;174(11):1263–1280. doi: 10.1111/bph.13622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kiyama R. Estrogenic potentials of traditional Chinese medicine. Am. J. Chin. Med. 2017;45(7):1365–1399. doi: 10.1142/S0192415X17500756. [DOI] [PubMed] [Google Scholar]
- 7.Wasser SP. Medicinal mushroom science: Current perspectives, advances, evidences, and challenges. Biomed. J. 2014;37(6):345–356. doi: 10.4103/2319-4170.138318. [DOI] [PubMed] [Google Scholar]
- 8.Pel HJ, et al. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat. Biotechnol. 2007;25(2):221–231. doi: 10.1038/nbt1282. [DOI] [PubMed] [Google Scholar]
- 9.Ohm RA, et al. Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 2010;28(9):957–963. doi: 10.1038/nbt.1643. [DOI] [PubMed] [Google Scholar]
- 10.Chen S, et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012;26:913. doi: 10.1038/ncomms1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morin E, et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc. Natl. Acad. Sci. USA. 2012;109(43):17501–17506. doi: 10.1073/pnas.1206847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bao D, et al. Sequencing and comparative analysis of the straw mushroom (Volvariella volvacea) genome. PLoS One. 2013;8(3):e58294. doi: 10.1371/journal.pone.0058294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park YJ, et al. Whole genome and global gene expression analyses of the model mushroom Flammulina velutipes reveal a high capacity for lignocellulose degradation. PLoS One. 2014;9(4):e93560. doi: 10.1371/journal.pone.0093560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen L, et al. Genome Sequence of the Edible Cultivated Mushroom Lentinula edodes (Shiitake) Reveals Insights into Lignocellulose Degradation. PLoS One. 2016;11(8):e0160336. doi: 10.1371/journal.pone.0160336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar A, et al. Genome Sequencing and analyses of Two Marine Fungi from the North Sea Unraveled a Plethora of Novel Biosynthetic Gene Clusters. Sci. Rep. 2018;8(1):10187. doi: 10.1038/s41598-018-28473-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar A, et al. De Novo Assembly and Genome Analyses of the Marine-Derived Scopulariopsis brevicaulis Strain LF580 Unravels Life-Style Traits and Anticancerous Scopularide Biosynthetic Gene Cluster. PLoS One. 2015;10(10):e0140398. doi: 10.1371/journal.pone.0140398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao D, et al. De Novo Sequencing of a Sparassis latifolia Genome and Its Associated Comparative Analyses. Can. J. Infect. Dis. Med. Microbiol. 2018;2018:1857170. doi: 10.1155/2018/1857170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez D, et al. Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proc. Natl. Acad. Sci. USA. 2009;106(6):1954–1959. doi: 10.1073/pnas.0809575106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaskell J, et al. Draft genome sequence of a monokaryotic model brown-rot fungus Postia (Rhodonia) placenta SB12. Genom. Data. 2017;14:21–23. doi: 10.1016/j.gdata.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rong C, et al. Cloning of the A Mating-Type Locus from Lepista nuda and Characterization of Its Genetic Structure. Curr. Microbiol. 2015;71(6):669–677. doi: 10.1007/s00284-015-0902-y. [DOI] [PubMed] [Google Scholar]
- 21.Martinez D, et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat. Biotechnol. 2004;22(6):695–700. doi: 10.1038/nbt967. [DOI] [PubMed] [Google Scholar]
- 22.Yang YH, Kang HW, Ro HS. Cloning and Molecular Characterization of β-1,3-Glucan Synthase from Sparassis crispa. Mycobiology. 2014;42(2):167–173. doi: 10.5941/MYCO.2014.42.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okada H, et al. Multiple functional domains of the yeast l,3-β-glucan synthase subunit Fks1p revealed by quantitative phenotypic analysis of temperature-sensitive mutants. Genetics. 2010;184(4):1013–1024. doi: 10.1534/genetics.109.109892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ujita M, et al. Molecular cloning and sequence analysis of the β-1, 3-glucan synthase catalytic subunit gene from a medicinal fungus. Cordyceps militaris. Mycoscience. 2006;47(2):98–105. doi: 10.1007/S10267-005-0278-0. [DOI] [Google Scholar]
- 25.Weber T, et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43(W1):W237–W243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiyama R, Wada-Kiyama Y. Estrogenic endocrine disruptors: Molecular mechanisms of action. Environ. Int. 2015;83:11–40. doi: 10.1016/j.envint.2015.05.012/. [DOI] [PubMed] [Google Scholar]
- 27.Dong S, et al. Estrogen-like activity and dual roles in cell signaling of an Agaricus blazei Murrill mycelia-dikaryon extract. Microbiol. Res. 2012;167(4):231–237. doi: 10.1016/j.micres.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 28.Kiyama R, Zhu Y. DNA microarray-based gene expression profiling of estrogenic chemicals. Cell. Mol. Life Sci. 2014;71(11):2065–2082. doi: 10.1007/s00018-013-1544-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong S, Inoue A, Zhu Y, Tanji M, Kiyama R. Activation of rapid signaling pathways and the subsequent transcriptional regulation for the proliferation of breast cancer MCF-7 cells by the treatment with an extract of Glycyrrhiza glabra root. Food Chem. Toxicol. 2007;45(12):2470–2478. doi: 10.1016/j.fct.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 30.Dong S, et al. Brefeldin A is an estrogenic, Erk1/2-activating component in the extract of Agaricus blazei mycelia. J. Agric. Food Chem. 2013;61(1):128–136. doi: 10.1021/jf304546a. [DOI] [PubMed] [Google Scholar]
- 31.Aida FMNA, Shuhaimi M, Yazid M, Maaruf AG. Mushroom as a potential source of prebiotics: a review. Trends Food Sci. Tech. 2009;20(11-12):567–575. doi: 10.1016/j.tifs.2009.07.007. [DOI] [Google Scholar]
- 32.Ohno N, Miura NN, Nakajima M, Yadomae T. Antitumor 1,3-β-glucan from cultured fruit body of Sparassis crispa. Biol. Pharm. Bull. 2000;23(7):866–872. doi: 10.1248/bpb.23.866. [DOI] [PubMed] [Google Scholar]
- 33.Harada T, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) regulates cytokine induction by 1,3-β-D-glucan SCG in DBA/2 mice in vitro. J. Interferon Cytokine Res. 2004;24(8):478–489. doi: 10.1089/1079990041689656. [DOI] [PubMed] [Google Scholar]
- 34.Kawagishi H, et al. Novel bioactive compound from the Sparassis crispa mushroom. Biosci. Biotechnol. Biochem. 2007;71(7):1804–1806. doi: 10.1271/bbb.70192. [DOI] [PubMed] [Google Scholar]
- 35.Hu Shuang, Wang Di, Zhang Junrong, Du Mengyan, Cheng Yingkun, Liu Yan, Zhang Ning, Wang Di, Wu Yi. Mitochondria Related Pathway Is Essential for Polysaccharides Purified from Sparassis crispa Mediated Neuro-Protection against Glutamate-Induced Toxicity in Differentiated PC12 Cells. International Journal of Molecular Sciences. 2016;17(2):133. doi: 10.3390/ijms17020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong KB, et al. Hypocholesterolemic Effects of the Cauliflower Culinary-Medicinal Mushroom, Sparassis crispa (Higher Basidiomycetes), in Diet-Induced Hypercholesterolemic Rats. Int. J. Med. Mushrooms. 2015;17(10):965–975. doi: 10.1615/IntJMedMushrooms.v17.i10.60. [DOI] [PubMed] [Google Scholar]
- 37.Kim HH, et al. Sparassis crispa suppresses mast cell-mediated allergic inflammation: Role of calcium, mitogen-activated protein kinase and nuclear factor-κB. Int. J. Mol. Med. 2012;30(2):344–350. doi: 10.3892/ijmm.2012.1000. [DOI] [PubMed] [Google Scholar]
- 38.Yoshikawa K, et al. Novel phthalide compounds from Sparassis crispa (Hanabiratake), Hanabiratakelide A-C, exhibiting anti-cancer related activity. Biol. Pharm. Bull. 2010;33(8):1355–1359. doi: 10.1248/bpb.33.1355. [DOI] [PubMed] [Google Scholar]
- 39.Bang S, et al. New Aromatic Compounds from the Fruiting Body of Sparassis crispa (Wulf.) and Their Inhibitory Activities on Proprotein Convertase Subtilisin/Kexin Type 9 mRNA Expression. J. Agric. Food Chem. 2017;65(30):6152–6157. doi: 10.1021/acs.jafc.7b02657. [DOI] [PubMed] [Google Scholar]
- 40.Prasad R, Varshney VK, Harsh NS, Kumar M. Antioxidant Capacity and Total Phenolics Content of the Fruiting Bodies and Submerged Cultured Mycelia of Sixteen Higher Basidiomycetes Mushrooms from India. Int. J. Med. Mushrooms. 2015;17(10):933–941. doi: 10.1615/IntJMedMushrooms.v17.i10.30. [DOI] [PubMed] [Google Scholar]
- 41.Kohler A, et al. Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat. Genet. 2015;47(4):410–415. doi: 10.1038/ng.3223. [DOI] [PubMed] [Google Scholar]
- 42.Niemelä T, Kinnunen J, Larsson K-H, Schigel DS, Larsson E. Genus revisions and new combinations of some North European polypores. Karstenia. 2005;45(2):75–80. doi: 10.29203/ka.2005.406. [DOI] [Google Scholar]
- 43.Latgé JP. The cell wall: a carbohydrate armour for the fungal cell. Mol. Microbiol. 2007;66(2):279–290. doi: 10.1111/j.1365-2958.2007.05872.x. [DOI] [PubMed] [Google Scholar]
- 44.Zhu Y, Kiyama R. Capsaicinoids are silent estrogens, a class of estrogenic chemicals without cell-proliferation activity. Environ. Tech. Innov. 2017;7:182–193. doi: 10.1016/j.eti.2017.02.006. [DOI] [Google Scholar]
- 45.Zhu Y, Kitamura K, Maruyama A, Higashihara T, Kiyama R. Estrogenic activity of bio-degradation products of C-heavy oil revealed by gene-expression profiling using an oligo-DNA microarray system. Environ Pollut. 2012;168:10–14. doi: 10.1016/j.envpol.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Chin CS, et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods. 2016;13(12):1050–1054. doi: 10.1038/nmeth.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 50.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12(4):357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pertea M, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33(3):290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stanke M, Morgenstern B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005;33:W465–W467. doi: 10.1093/nar/gki458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Testa AC, Hane JK, Ellwood SR, Oliver RP. CodingQuarry: highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genomics. 2015;16:170. doi: 10.1186/s12864-015-1344-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haas BJ, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9(1):R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bairoch A, Boeckmann B, Ferro S, Gasteiger E. Swiss-Prot: juggling between evolution and stability. Brief. Bioinform. 2004;5(1):39–55. doi: 10.1093/bib/5.1.39. [DOI] [PubMed] [Google Scholar]
- 58.Li L, Stoeckert CJ, Jr., Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000;17(4):540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 61.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Floudas D, et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science. 2012;336(6089):1715–1719. doi: 10.1126/science.1221748. [DOI] [PubMed] [Google Scholar]
- 63.Sanderson MJ. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003;19(2):301–302. doi: 10.1093/bioinformatics/19.2.301. [DOI] [PubMed] [Google Scholar]
- 64.Finn RD, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ye J, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:W293–W297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yin Y, et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40:W445–W451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cantarel BL, et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Terasaka S, et al. Using a customized DNA microarray for expression profiling of the estrogen-responsive genes to evaluate estrogen activity among natural estrogens and industrial chemicals. Environ. Health Perspect. 2004;112(7):773–781. doi: 10.1289/ehp.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.