

Abstract
Significance: Iron and oxygen are intimately linked: iron is an essential nutrient utilized as a cofactor in enzymes for oxygen transport, oxidative phosphorylation, and metabolite oxidation. However, excess labile iron facilitates the formation of oxygen-derived free radicals capable of damaging biomolecules. Therefore, biological utilization of iron is a tightly regulated process. The nuclear factor (erythroid-derived 2)-like 2 (NRF2) transcription factor, which can respond to oxidative and electrophilic stress, regulates several genes involved in iron metabolism.
Recent Advances: The bulk of NRF2 transcription factor research has focused on its roles in detoxification and cancer prevention. Recent works have identified that several genes involved in heme synthesis, hemoglobin catabolism, iron storage, and iron export are under the control of NRF2. Constitutive NRF2 activation and subsequent deregulation of iron metabolism have been implicated in cancer development: NRF2-mediated upregulation of the iron storage protein ferritin or heme oxygenase 1 can lead to enhanced proliferation and therapy resistance. Of note, NRF2 activation and alterations to iron signaling in cancers may hinder efforts to induce the iron-dependent cell death process known as ferroptosis.
Critical Issues: Despite growing recognition of NRF2 as a modulator of iron signaling, exactly how iron metabolism is altered due to NRF2 activation in normal physiology and in pathologic conditions remains imprecise; moreover, the roles of NRF2-mediated iron signaling changes in disease progression are only beginning to be uncovered.
Future Directions: Further studies are necessary to connect NRF2 activation with physiological and pathological changes to iron signaling and oxidative stress.
Keywords: : NRF2, iron, oxygen, heme, cancer, ferroptosis
Introduction
Before the Great Oxygenation Event (GOE) that took place around 2.4 billion years ago, the earth was a low-oxygen stew of acidic oceans rich in dissolved ferrous iron (85). The onset of GOE was initiated when cyanobacteria and their evolutionary ancestors began utilizing photosynthesis to produce molecular oxygen (O2) by splitting water molecules. The increased O2 oxidized the ferrous iron and resulted in the rusting of the earth. Today, evidence of the oxidized iron deposits can be seen in geological deposits as banded red-colored iron minerals (20). Only following oxidation of the ferrous iron sinks could oxygen accumulate to the ∼21% atmospheric concentration seen in today's atmosphere. This profound interaction among iron, oxygen, and life persists to the present day where living organisms evolved systems to manage and exploit iron in redox chemistry.
Iron is an essential nutrient in the diet and is involved in a variety of critical intracellular processes, including DNA synthesis and cellular respiration. Iron serves as a cofactor in many enzymes involved in these processes due to its relative abundance on earth and its chemistry allowing easy participation in reduction and oxidation reactions; indeed, organisms from archaea to humans depend on iron-containing proteins (2). Despite the value of this reactivity in performing enzymatic catalysis and electron transfers, iron can also generate the highly reactive hydroxyl radical (OH•) via the Fenton reaction, which can damage lipids, proteins, and nucleic acids (29, 126, 145, 164):
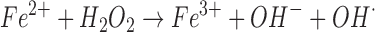 |
Aside from iron, hydrogen peroxide feeds the Fenton reaction; H2O2 can form endogenously in biology by a variety of processes ranging from metabolic by-products to immune-related oxidases (159). Thus, cells maintain an appropriately sized pool of intracellular iron to balance its beneficial catalytic functions with deleterious free radicals by carefully regulating storage of iron, export of iron, degradation of major iron-containing proteins, and recycling of iron back into biologically valuable cofactors available to redox-active proteins.
Apart from iron-mediated oxidative stress, cells must also cope with a variety of electrophilic and oxidative xenobiotic stressors. These may include metabolites from heterotrophic consumption of other organisms or inorganic and organic environmental stressors. Development of antistress systems to ameliorate these toxic exposures was likely critical in early evolutionary development. One such system is the Kelch-like ECH-associated protein 1–nuclear factor (erythroid-derived 2)-like 2 system (KEAP1-NRF2), which is theorized to have arisen around the GOE (32). In this system, homodimers of KEAP1 negatively regulate the NRF2 transcription factor by targeting it for ubiquitylation and subsequent proteasomal degradation. As expected, NRF2 protein turnover is constant and quick under unstressed conditions. Following electrophilic and oxidative stress, cysteine residues on KEAP1 are modified; this induces a conformational change in KEAP1, disrupting its interaction with NRF2 and thus preventing NRF2 degradation. Newly synthesized NRF2 accumulates, translocates to the nucleus, dimerizes with small musculoaponeurotic fibrosarcoma (MAF) proteins, and facilitates transcription of a battery of Phase II metabolism and cytoprotective genes. For example, bona fide NRF2 target genes include NADPH quinone dehydrogenase 1 (NQO1), glutamate–cysteine ligase modifier subunit (GCLM), and the peroxide-scavenging selenoproteins of the glutathione peroxidase (GPX) family (Fig. 1).
FIG. 1.

KEAP1 regulates NRF2 degradation. Under unstressed conditions, KEAP1 homodimers facilitate NRF2 ubiquitylation, which marks it for proteasomal degradation. Following NRF2 ubiquitylation, KEAP1 is recycled to bind newly synthesized NRF2. Under conditions of oxidative or electrophilic stress, key cysteine residues on KEAP1 are covalently modified, preventing it from mediating NRF2 ubiquitylation. Newly synthesized NRF2 can then accumulate and translocate to the nucleus where it dimerizes with one of the small MAF proteins to promote the transcription of cytoprotective genes. GCLM, glutamate-cysteine ligase modifier subunit; GPX, glutathione peroxidase; KEAP1, Kelch-like ECH-associated protein 1; MAF, musculoaponeurotic fibrosarcoma; NRF2, nuclear factor (erythroid-derived 2)-like 2.
Many reviews have focused on NRF2-mediated cytoprotection as well as KEAP1 and non-KEAP1-mediated NRF2 activation (34, 38, 56, 58, 86, 171). Mammalian NRF2 appears to have arisen from an ancestral NRF protein tracing back to eumetazoan lineages. Considering the ancient heritage of both NRF2 and iron regulation and their mutual crossover in the arena of intracellular stress, we sought to assess the literature to uncover relationships between NRF2 and iron.
NRF2 Was Discovered for Its Roles in Erythropoiesis
Dedicated oxygen carriers are critical for the existence of multiple phyla in the metazoan kingdom. As its name suggests, the birth of research into nuclear factor (erythroid-derived 2)-like 2 (NFE2L2, NRF2) is rooted in erythropoiesis: the production of red blood cells or erythrocytes. The core function of erythrocytes is as an oxygen carrier that delivers oxygen to tissues for use in aerobic respiration. To do so, erythrocytes are rich in the iron metalloprotein, hemoglobin, which utilizes heme-bound iron to bind and transport oxygen. To better understand the production of hemoglobin, investigators were attempting to understand the transcriptional regulation of the hemoglobin subunits, α and β. The locus control region located 5′ of the β-globin gene cluster was known to harbor binding sites for ubiquitous and erythroid-specific transcription factors, including the promiscuous AP-1 binding site. One transcription factor, nuclear factor erythroid 2 (NF-E2), was shown to bind to the AP-1 site (99) and mediate globin gene expression (102, 110, 111, 148, 149). To fully characterize all DNA-binding proteins that could interact with the NF-E2 binding site, Moi et al. utilized a cDNA expression library from K652 cells to isolate several DNA-binding proteins that bound to the AP-1/NF-E2 repeat found in the locus control region of β-globin. One of the identified proteins that could bind to this region was NRF2 (101).
Although NRF2 was discovered as part of an effort to understand globin gene regulation, investigations into NRF2 and erythropoiesis quickly diminished for two reasons. First, within 2 years of its discovery, Chan et al. generated Nrf2 knockout mice. Homozygous Nrf2 knockout (Nrf2−/−) mice showed no visible phenotypes: mice exhibited no defects in embryogenesis, were fertile, and produced normal litter sizes; most importantly, hematological markers of Nrf2−/− mice did not differ from heterozygous or wild-type mice (15). Second, any roles of NRF2 in erythropoiesis were marginalized following the seminal works by Itoh et al. that demonstrated Nrf2 was essential for orchestrating the transcriptional induction of phase II detoxification genes carrying an antioxidant response element (ARE) (51). Indeed, Nrf2 was found to regulate ARE-carrying genes. Since then, much of the NRF2 scholarship has sought to characterize its roles in detoxification and electrophilic stress mediation.
Despite a shift from erythropoiesis to toxicology, studies suggest that NRF2 target genes may still be involved in erythropoiesis. Several NRF2 target genes have been directly implicated as anabolic enzymes necessary for hematopoietic cell maturation. In the original study characterizing mouse Nrf2 as well as subsequent analyses of the β-globin locus control region, in vitro reporter assays indicated that ectopically expressed Nrf2 can increase the expression of a reporter under the control of the globin enhancer region (93, 101), indicating that Nrf2 may participate in globin transcription. More recent studies have confirmed NRF2 binding to β-globin promoter regions using chromatin immunoprecipitation (ChIP) in lymphoblastoid cells treated with 10 μM sulforaphane (SFN), an isothiocyanate found in cruciferous vegetables that is routinely used to activate NRF2. Notably, NRF2 may exhibit tissue-specific β-globin DNA binding and transcriptional induction (14). Others have extended the globin analyses to human fetal hemoglobin, which consists primarily of γ-globin, by showing that the NRF2 inducers, tert-butylhydroquinone (tBHQ, 25 μM), D3T (25 μM), and curcumin (10 μM), could induce γ-globin mRNA. Moreover, tBHQ increased the protein level, nuclear localization, and γ-globin promoter binding of NRF2, which led to increased human fetal hemoglobin (88), indicating a role for NRF2 in globin production. While globin is necessary in many organisms for oxygen transport, the globin active sites require heme moieties to bind oxygen; thus, understanding how heme synthesis can be regulated by NRF2 provides a more complete understanding of relationships between iron, oxygen, and NRF2.
NRF2 Regulates Genes Involved in Heme Biosynthesis
The profound interactions among iron, oxygen, and life are embodied in heme, a porphyrin-bound iron that is the center of many metabolic enzymes (mitochondria complexes and cytochrome P-450 enzymes), nitric oxide signaling effectors (nitric oxide synthases), oxygen storage proteins (myoglobin), and oxygen carrier proteins (hemoglobin). Of note, heme is required for oxygen transport over large distances because of poor dissolution of oxygen in aqueous solutions, including blood plasma. Thus, development of complexes capable of binding oxygen, such as heme, was critical for the evolution of large multicellular organisms; indeed, heme-bound iron accounts for ∼95% of iron within the human body (46). Heme is generated from the anabolic combinations of eight succinyl-CoA and eight glycine molecules over eight biosynthetic steps; in the final stages of heme synthesis, a single ferrous iron is inserted into a protoporphyrin to generate heme. The iron atom maintains four coordinate bonds to the rest of the heme prosthetic group, leaving two coordinate bonds available for protein interaction (via a histidine residue) and oxygen binding. The iron within heme can then bind oxygen for transportation, participation in redox reactions, or electron passaging. However, because iron can promote the formation of damaging oxygen radicals, synthesis and destruction of heme-bound iron are carefully regulated; NRF2 participates in the transcriptional regulation of heme metabolism.
Recent works have identified novel NRF2 target genes involved in heme biogeneration: ATP binding cassette subfamily B member 6 (ABCB6) and ferrochelatase (FECH). ABCB6 imports porphyrins, such as coproporphyrinogen III, from the cytosol to the mitochondria for further heme anabolism (70, 137). The ABCB6 gene also encodes the Lan blood group antigen (42). NRF2 binding to ABCB6 promoter regions was first identified in microarray analyses of airway epithelia from smokers (48). A putative NRF2-ABCB6 relationship was further characterized when knocking down NRF2 in lung-derived cell lines, A549 and BEAS-2B, and liver-derived cell line, HepG2, treated with 10 μM of sulforaphane showed ∼70% decreases in ABCB6 transcript levels. Additionally, treatment with 10 μM sulforaphane showed a fivefold induction of ABCB6 mRNA in GM12878 lymphoblastoid cells. However, the same treatment did not induce ABCB6 transcripts in bone marrow K562 cell lines, indicating that NRF2 modulation of ABCB6 may be tissue specific (14).
In addition to ABCB6, FECH is located in the mitochondria and participates in heme biosynthesis; in the last step of heme biosynthesis, ferrochelatase inserts ferrous iron into protoporphyrin IX to generate the final heme cofactor (60). Wu et al. identified ferrochelatase as an NRF2 target using a gene dose–response model. In this model, they compared gene expression profiles of Nrf2-null mice, Nrf2-wild-type mice, Keap1-knockdown mice, and hepatic Keap1-knockdown mice. After showing that these models have increasing liver Nrf2 activation, they identified a modest (<2-fold induction), but significant, induction of the Fech gene in the Keap1-knockdown and hepatic Keap1-knockdown mice relative to the Nrf2-null and Nrf2-wild-type mice (165). The role of NRF2 in mediating FECH gene expression was further evaluated in human cell lines, whereby NRF2 knockdown in sulforaphane-treated (10 μM sulforaphane for 24 h) A549, BEAS-2B, and HepG2 cells decreased FECH mRNA by 42%, 29%, and 62%, respectively (14). Consistently, treatment with 10 μM sulforaphane increased FECH mRNA expression in GM12878 lymphoblastoid cells. However, NRF2-mediated FECH induction may be tissue specific, similar to ABCB6: neither sulforaphane treatment nor KEAP1 knockdown in K562 bone marrow cells showed increased FECH expression (14). The NRF2-regulated aspects of heme anabolism are shown in Figure 2.
FIG. 2.

NRF2 regulates heme synthesis. Several genes involved in the heme biosynthetic pathway are transcription targets of NRF2 (red-colored genes). These genes include ABCB6, which transports coporphyrinogen III from the cytosol to the mitochondrial intermembrane space, and FECH, which inserts ferrous iron into the protoporphyrin ring. ABCB6, ATP binding cassette subfamily B member 6; FECH, ferrochelatase.
NRF2 Regulates Genes Involved in Heme Catabolism
While Nrf2 regulates biological utilization of oxygen and iron by facilitating the transcription of genes that incorporate iron into heme during hemoglobin anabolism, NRF2 also regulates mobilization of iron from heme during hemoglobin catabolism. Senescent red blood cells are degraded by the mononuclear phagocyte system, consisting of primarily splenic macrophages and liver Kupffer cells. Following engulfment, erythrocytes are degraded in the phagolysosome and hemoglobin is degraded by proteases to liberate the heme cofactor (10). The heme is then exported out of the lysosome into the cytosol by the lysosomal membrane-bound transporter heme-responsive gene 1 (SLC48A1, HRG1) (127, 162). Knocking down NRF2 in A549, BEAS-2B, and HepG2 cells decreases HRG1 expression up to 52%, while activation of NRF2 by either KEAP1 knockdown or treatment with 10 μM sulforaphane results in increased HRG1 mRNA expression by two- to sixfold and an increased HRG1 protein level (14). Although NRF2 activation appears to mediate HRG1 transcript and protein levels, it remains unclear to what extent heme trafficking and catabolism change.
Within macrophages, cytosolic heme from red blood cells is metabolized into ferrous iron and biliverdin by heme oxygenase (HMOX-1). Biliverdin is further metabolized, while the ferrous iron is recycled for fresh erythrocyte production. Before the discovery of Nrf2-mediated HMOX-1 regulation, Hmox-1 mRNA was known to increase following administration of phorbol esters, sodium arsenite, H2O2, ultraviolet light, heme, and sulfhydryl compounds to skin fibroblasts. The RNA synthesis inhibitor actinomycin D has been shown to inhibit Hmox-1 transcript induction following administration of many of these stimuli, thus implicating transcription as the key regulatory component for Hmox-1 induction (17). Two putative distal enhancer regions at −4 and −10 kb were known to be required for induction by several stimuli, including heme, arsenite, and H2O2. They contained stress response elements (StREs), which were critical for inducer-dependent upregulation of Hmox-1 transcription (3). As the StRE consensus sequence closely aligned with the binding site for the AP-1 groups of transcription factors, investigations focused on the well-studied Jun and Fos families of factors. However, the AP-1 site was identified to be only necessary, but not sufficient, for HMOX-1 induction by heme and cadmium; an extended AP-1 site with additional proximal three base pairs was deemed sufficient, which closely mirrored an ARE (49). Soon after, Alam et al. identified Nrf2 as the dominant mediator for Hmox-1. They showed that the expression of a dominant negative NRF2 mutant could inhibit Hmox-1 mRNA induction by multiple agents (10 μM heme, 100 μM arsenite, 10 μM cadmium, 100 μM zinc, or 50 μM tBHQ) by >85%, refuting the Jun/Fos model (4). These studies defined NRF2 as the main transcriptional regulator of HMOX-1. For a more robust discussion on the implications of NRF2 and HMOX-1 in health and disease, a comprehensive recent review has been conducted (83).
Further studies have demonstrated that a transcriptional repressor, BRCA-1-associated carboxy-terminal helicase (BACH1), can occupy the same binding sites on HMOX-1 as NRF2. Bach1 is a BTB-basic leucine zipper transcription factor that antagonizes small Maf proteins that activate transcription at NF-E2 sites, including Nrf2 (119). Notably, Bach1 is a heme sensor; upon binding heme, Bach1 is ubiquitylated and targeted for proteasomal degradation (170). It was shown that Bach1 must be displaced before Nrf2 binding to the HMOX-1 gene (129), indicating that iron-containing hemin and Nrf2 cooperate to induce HMOX-1.
While free iron generated from heme catabolism can be utilized or stored by the cell, the remaining heme catabolite, biliverdin, is metabolized to bilirubin by either biliverdin reductase A (BLVRA) or biliverdin reductase B (BLVRB). Bilirubin subsequently serves as an antioxidant free radical scavenger and can be glucuronidated for excretion. Transcription of BLVRB increased following Nrf2 activation; mice with liver-specific Keap1 knockout showed 270% increase of Blvrb mRNA (165). Human BLVRB was also shown to be an NRF2 target gene: both KEAP1 knockdown and treatment with 15 μM sulforaphane induced BLVRB transcripts one- to threefold in breast cancer MCF10A cells (1). ChIP-seq and microarray analyses were combined to demonstrate that 100 μM diethyl maleate targeted Nrf2-MafG binding to a site proximal to the Blrvb gene locus, and the Blrvb gene transcripts approximately doubled (44). Similarly, 15 μM sulforaphane enhanced transcript and protein levels of BLVRA by one- and fivefold, respectively (1). Hmox-1 and both BLVRs appear to depend on NRF2, indicating that NRF2 plays a critical role in heme catabolism; indeed, NRF2 critically mediates cross talk between these two systems: BLVRA can bind heme and transport it into the nucleus to induce Hmox-1, likely by delivering heme to the Bach1 repressor and abrogating Bach1 activity (157). Like many of the heme or erythropoietic genes associated with NRF2 activity, definitive correlation of the NRF2 role in modulation of BLVRA or BLVRB transcription with a physiological or pathological outcome is wanting.
Aside from BLVRs, NRF2 has also been implicated in the transcription of another enzyme that protects cells against oxidation by heme: alpha-1-microglobulin (A1M). The gene for A1M is alpha-1-microglobulin/bikunin precursor (AMBP); following cleavage by a protease, furin, AMBP gives rise to both A1M and bikunin, an extracellular matrix component. While the precise reactions of A1M remain unknown, A1M has cytoprotective and antioxidant effects due to its reported multifaceted role as a reductase/dehydrogenase, heme-binding and degrading protein, and free radical scavenger (114). In erythroid cells, A1M prevents intracellular oxidation and can bind heme (96, 115, 141). The proposed antioxidant mechanisms also include upregulation of its own transcription in response to hemoglobin, heme, and reactive oxygen species (ROS). These functions have been recently reviewed elsewhere (114). SFN treatment (10 μM for 8 h) of K562 chronic myeloid leukemia cells and GM12878 lymphoblastoid cells significantly increased AMBP expression by 2- and 56-fold, respectively. As expected, when KEAP1 was knocked down in K562 cells, AMBP mRNA increased more than twofold. Knocking down NRF2 in A549 lung adenocarcinoma cells and in sulforaphane-treated (10 μM) BEAS-2B bronchial epithelial cells and HepG2 hepatocellular carcinoma cells showed decreased AMBP transcript levels (14). Thus, NRF2 may mediate protection against heme- and hemoglobin-induced reactive oxygen species by promoting AMBP expression.
While NRF2 may participate in the safe destruction of heme via its induction of HRG1, HMOX-1, and the BLVRs, it may also participate in the incorporation of iron back into heme via its upregulation of genes such as FECH and ABCB6. These complementary roles could place NRF2 as a regulator at both the birth and death of iron recycling for erythrocyte production. Because iron participates in Fenton reactions, cells may utilize the redox-sensitive KEAP1-NRF2 system to ensure safe handling of the massive quantities of iron used during the generation and destruction of heme necessary for oxygen-facilitated biology. More dissection of the role of NRF2 in altering heme metabolism, quantity, or function is required in future studies. The NRF2-regulated heme homeostasis is summarized in Figure 3.
FIG. 3.

NRF2 regulates heme degradation. Putative NRF2 transcription targets are in red-colored text. (1) Aged erythrocytes, which contain hemoglobin, are endocytosed by macrophages. (2) Endocytosed erythrocytes are degraded in the phagolysosome of macrophages to liberate heme. (3) Heme is exported from the lysosome to the cytosol by HRG1, a transcriptional target of NRF2. (4) HMOX-1, a transcriptional target of NRF2, catabolizes heme into biliverdin and free iron. (5) BLVRA, a transcription target of NRF2, represses BACH1 and allows NRF2 to promote HMOX-1 expression. (6) Free iron liberated from heme is recycled for further heme synthesis, storage, or export from the cell. (7) Biliverdin is metabolized to bilirubin by BLVRA and BLVRB, which are transcription targets of NRF2. Bilirubin is then excreted to blood circulation and transported to the liver, where it is excreted as bile. (8) The NRF2 transcription target AMBP is proteolytically cleaved to A1M, which can bind heme. A1M, alpha-1-microglobulin; AMBP, alpha-1-microglobulin/bikunin precursor; BACH1, BRCA-1-associated carboxy-terminal helicase; BLVRA, biliverdin reductase A; BLVRB, biliverdin reductase B; HMOX-1, heme oxygenase; HRG1, heme-responsive gene 1; ROS, reactive oxygen species.
Other Roles of NRF2 in Erythropoiesis
Although NRF2 appears to regulate many genes involved in hemoglobin metabolism and iron utilization, Nrf2−/− mice did not show anemia (15). However, follow-up studies indicated that older Nrf2−/− mice had signs of anemia and presented with splenomegaly and spleen toxicity. Indeed, hematological analyses showed that Nrf2−/− mice had abnormal erythrocyte morphology and were more sensitive to H2O2-induced hemolysis (79). Whether these defects are due to decreased functional hemoglobin, decreased detoxification, or some combination thereof remains unclear. It is possible that alterations to red blood cell function in Nrf2−/− mice may arise from decreased antioxidant capacity in erythrocytes.
Substantial evidence indicates that accumulation of ROS is particularly deleterious to red blood cells and can lead to hemolysis (30, 33, 52). As expected, ablation of ROS-scavenging selenoproteins, such as the GPXs, can induce anemia. Red blood cell count, hemoglobin concentration, and hematocrit in mice with Cre-Lox-induced deletion of the tRNAsec gene Trsp, which normally allows synthesis of selenoproteins, were decreased to ∼60% of the control. Moreover, combined inactivation of GPX selenoproteins and Nrf2 exacerbated the anemia: the same hematopoietic parameters dropped to 30% of the control. The combined loss of selenoproteins and Nrf2 resulted in increased erythrocyte intracellular ROS levels. Thus, the battery of Nrf2 selenoprotein genes critically regulates redox homeostasis in erythrocytes and may prevent hemolytic anemia (57).
The antioxidant functions of GPX proteins are complemented by a host of other antioxidant defense systems that can be activated by NRF2. Broadly, these include the peroxiredoxin system (18, 64), the thioredoxin system, and the glutathione system. NRF2 has been shown to facilitate the transcription of enzymes involved in these defense systems, as reviewed recently (39, 173). Importantly, players in these systems such as the peroxiredoxins have been shown to be particularly efficient ROS scavengers (9, 55) capable of affecting both physiological iron homeostasis and erythrocyte health (95, 109). Excellent reviews encompassing the NRF2-peroxiredoxin system are available (35, 39, 150, 174).
Beyond its role in regulating redox stress during hematopoietic cell development, studies have also shown that NRF2 plays a role in hematopoietic stem cell (HPSC) maintenance (105). The HPSCs in Nrf2−/− mice exhibit a 65% increase in apoptosis compared with Nrf2+/+ mice, which was not associated with an increase in ROS; moreover, Nrf2 appears to balance HPSC proliferation, self-renewal, and bone marrow localization (97, 155). Nrf2-mediated Notch signaling improved HPSC function following ionizing radiation (62). Murakami et al. demonstrated the role of Nrf2 in mediating HPSC fate by using Keap1-deficient mice, which presented with almost 10% enhanced granulocyte–monocyte differentiation and compensatory decrease in erythroid and lymphoid differentiation. Keap1−/− HPSCs mimicked the nonerythroid lineage priming, while combined Nrf2/Keap1 knockout restored lineage commitment (106). Given that genes important for heme biosynthesis, such as ABCB6 and FECH, are transcription targets of NRF2, it seems surprising that NRF2 activation favors nonerythroid lineages that depend less on heme. To further complicate the role of NRF2 in erythropoiesis, some roles of NRF2 in HPSC maintenance are proposed to be independent of oxidative stress (97), while other studies indicated that NRF2 protects erythrocytes from oxidant-induced hemolysis and anemia (79). Clearly, erythropoiesis is a carefully regulated process with many players and processes, and NRF2 mediates erythropoietic processes, including cellular antioxidation, stem cell maintenance and differentiation, and transcriptional activation of heme anabolic genes. The apparently conflicting roles of NRF2 within many hematological processes require further dissection.
NRF2 Compartmentalizes Nonheme-Associated Iron
Apart from heme-bound iron, cells maintain a pool of labile (redox-active, exchangeable, and chelatable) iron to be used for biosynthetic processes such as heme or iron–sulfur cluster generation. Labile iron is very tightly regulated to a cytosolic concentration of 0.5–1.5 μM at homeostasis, which comprises <5% of total intracellular iron (12): as mentioned previously, labile iron can enable the formation of oxygen-derived free radicals, such as the highly damaging hydroxyl radical, through Fenton reaction.
The product of Fenton reaction, OH•, is a potent oxidizer that rapidly reacts with nearby proteins and lipids. Its high reactivity gives OH• a short half-life in biological systems on the order of 1 ns (67). Physiological damage from iron-associated (Fenton reaction-derived) hydroxyl radicals is extensively limited at the physiological level because of the relatively low homeostatic levels of labile iron, which are maintained by a cellular buffering system.
A central player in the iron-buffering system is the ferritin protein, an ancient intracellular iron storage protein common to all five kingdoms of life (6). Ferritin sequesters excess free iron in a protein cage that limits iron's redox switching. Human ferritin consists of 24 subunits comprising ferritin heavy chain (FTH1) and ferritin light chain (FTL) polypeptides (68). The ratio of FTH1 to FTL can vary widely by tissue or disease state (8), and the functions of each subunit also differ: FTH1 contains a ferroxidase active site, which can oxidize Fe2+ to Fe3+ for storage in the central core, while FTL is the primary stabilizer of the ferritin protein and thus can dominate the FTL/FTH1 ratio in tissues such as the liver, where long-term iron storage is necessary (152). Ferritin readily sequesters up to 4500 iron atoms into its core (87), preventing the iron from participating in Fenton reactions. Thus, increased ferritin expression can have an antioxidant effect. In times of iron deficiency, the iron reservoir within ferritin proteins can be liberated through ferritinophagy (90, 91). At the same time, translation of ferritin mRNAs is repressed by iron regulatory proteins (IRPs), which bind to iron response elements (IREs) located in the 5′ untranslated regions (UTRs). IRPs comprise IRP1 and IRP2 and their intracellular levels are responsive to the labile iron pool. High labile iron pool levels lead to destruction of IRPs through proteasomal degradation, while low labile iron stabilizes them (103, 104, 132, 158, 163). Ferritin thus serves as a reservoir to maintain an appropriately sized labile iron pool for biosynthetic processes.
While NRF2 assists with maintaining physiological iron homeostasis due to its roles in regulating heme-bound iron, NRF2 also plays a pivotal role in regulating iron homeostasis within the labile iron pools. The earliest and most striking phenotypic examples of NRF2 effects on iron storage and transportation followed the discovery that Nrf2−/− mice showed abnormally white teeth compared with Nrf2+/+ mice; this color switch was determined to be due to defective iron utilization in developing tooth enamel of Nrf2−/− mice (167). Many investigations have subsequently identified and characterized specific genes involved in management of nonheme iron. One of its most important functions in this regard is as a transcriptional activator of ferritin.
The first indication of Nrf2 as a transcription factor that modulates Fth1 and Ftl transcription arose when depletion of glutathione in rat livers was shown to increase both Ftl and Fth1 transcripts (13). Shortly after, AREs were identified within the promoter regions of murine Ftl and Fth1 genes (156, 161). However, it was unknown which transcription factors could bind to the AREs or if that binding affected transcription. The heavy chain of ferritin, Fth1, was first identified as an Nrf2 target gene when basal Fth1 mRNA levels in Nrf2-deficient mice were lower than in Nrf2-wild-type mice (74). The relationship between Nrf2 activation and Ftl was soon uncovered when Ftl transcripts were twofold higher in Nrf2+/+ compared with Nrf2-deficient mouse intestines (153). These two studies provided the first link between Nrf2 and Ftl and Fth1 transcription; however, neither study found that Nrf2-activating xenobiotics could induce Ftl or Fth1 and instead proposed that Nrf2 functioned solely to enhance basal transcription of ferritin. While chemopreventive xenobiotics such as the dithiolethiones oltipraz and 1,2-dithiole-3-thione had been known to activate Nrf2 (73) and induce ferritin (125), nobody had directly linked xenobiotic activation of Nrf2 to ferritin induction. Pietsch et al. connected these concepts by showing that both Ftl and Fth1 could be induced approximately twofold by 70 μM of various dithiolethiones only in the presence of Nrf2 in primary mouse embryo fibroblasts (123). They demonstrated using gel shift assays that Nrf2 directly bound to the known Fth1 ARE following administration of chemopreventive dithiolethiones oltipraz and 1,2-dithiole-3-thione (123). Thus, Nrf2 is responsible for basal transcription of ferritin, and xenobiotic-activated Nrf2 can induce ferritin beyond its normal levels.
While upregulation of the ferritin gene transcription by NRF2 alters iron homeostasis by increasing iron storage and decreasing labile iron, NRF2 also buffers labile iron by altering its flux in and out of the cell. NRF2 does so by modulating the expression of ferroportin (FPN1), the only known mammalian exporter of iron from the cytosol to the extracellular milieu. FPN1 exports iron from iron-absorbing enterocytes of the intestinal lining into the bloodstream, thus adjusting the amount of dietary iron entering circulation (27). Similarly, FPN1 is responsible for iron mobilization from the hemoglobin-recycling macrophages and iron-storing hepatocytes (27). As iron is only normally lost due to bleeding, menstruation, childbearing, or intestinal cell sloughing, the aforementioned processes are required to ensure adequate iron supply for cellular functions and hemoglobinization without inducing iron overload and the associated oxidative stress. The role of Nrf2 in Fpn1 regulation arose from studies on macrophages in mice. Multiple groups showed that macrophage mRNA levels of Fpn1 increased following erythrophagocytosis (22, 65). No transcription factors were identified facilitating the response to erythrophagocytosis until Marro et al. identified that the transcriptional repressor Bach1 controls Fpn1 expression. Tenfold induction of Fpn1 by heme was due to release of the heme-sensitive Bach1 repressor; subsequently, a putative ARE was identified approximately −7 kb from the FPN1 core promoter. Accordingly, treatment with sulforaphane increased Fpn1 mRNA expression more than fivefold (94). The current model proposes that Bach1 binds to the ARE until heme exposure, wherein Bach1 leaves the DNA and allows Nrf2 to bind in its place and promote Fpn1 transcription. Notably, Nrf2 activation may simply displace Bach1 without Bach1 degradation. Regardless, heme inactivates Bach1 (113), thus providing a consistent regulatory element to Nrf2 activation of both HMOX-1 and FPN1, two proteins involved in heme catabolism and iron recycling. Additional evidence points to Ftl and Fth1 regulation by Nrf2 requiring Bach1 inactivation (43).
Further studies demonstrated that several Nrf2 activators, including diethyl malate (100 μM) and sulforaphane (5 μM), can upregulate Fpn1 mRNA in murine macrophages in an NRF2-dependent iron-independent manner. The increased Fpn1 was estimated to release significantly more (5% extra) intracellular iron to the extracellular milieu. Nrf2-mediated increases in iron export from macrophages were efficient enough to counteract lipopolysaccharide-induced suppression of Fpn1 mRNA expression. This indicates that Nrf2 may regulate iron trafficking during inflammation via Fpn1 expression (37). Thus, NRF2 may reduce intracellular labile iron pools by inducing expression of the iron storage ferritin proteins, FTL and FTH1, and the iron exporter FPN1. Further studies using an in vivo model are necessary to determine whether alterations to NRF2-mediated ferroportin induction can alter inflammation or infection outcomes.
While excess iron can be sequestered in ferritin or exported by ferroportin, iron can also directly impact cellular signaling following its incorporation into a protein known as pirin (PIR). PIR directly interacts with B cell lymphoma 3-encoded (Bcl-3) to coregulate nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) transcriptional signaling. Crystal structures of PIR show it uses iron as a cofactor, suggesting PIR has an enzymatic redox function (120). PIR conformations change depending on whether the bound iron is Fe2+ or Fe3+, and the redox state of PIR iron alters its allosteric regulation of NF-κB DNA binding capabilities. It has been proposed that the NF-κB role in responding to intracellular oxidative stress may depend on the PIR redox state (80). PIR was initially identified as an NRF2 target following an investigation into differential gene expression between small airway epithelia of smokers and nonsmokers. Mobility shift assays showed direct NRF2 binding to putative AREs (48). NRF2-mediated PIR transcription was validated in HeLa cervical cancer cells: a functional ARE was identified at +281 bp downstream of the transcription start site. Additionally, NRF2 knockdown in HeLa cells decreased PIR mRNA and protein levels. NRF2 overexpression increased PIR mRNA level 30% relative to that of control, and ChIP demonstrated NRF2 binding to PIR promoter ARE (11). Although the role of NRF2 in modulating PIR levels to alter physiology or disease progression is unclear, PIR and iron may serve as a pathway linking NRF2 and NF-κB transcriptional programs.
NRF2 activation is generally expected to reduce cytosolic labile iron: ferritin can sequester iron, ferroportin can export iron, and pirin can utilize iron as a cofactor for intracellular signaling. NRF2 activation can thus enable a reduction in the intracellular iron pool, restoring homeostasis in situations of cellular iron overload and preventing oxidative stress. NRF2-mediated regulation of labile iron pool is summarized in Figure 4.
FIG. 4.

NRF2 regulates the labile iron pool. Putative NRF2 transcription targets are in red-colored font. (1) Labile iron can be exported out of the cytosol by FPN1, which is an NRF2 transcription target. (2) Labile iron can be sequestered in ferritin, which comprises FTL and FTH1 polypeptide subunits. Both FTL and FTH1 are known transcription targets of NRF2. (3) Labile iron can bind to PIR and influence NF-κB transcriptional activity. FTH1, ferritin heavy chain; FTL, ferritin light chain; FPN1, ferroportin; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cell; PIR, pirin.
Iron Homeostasis, NRF2, and Cancer
Given that iron is an essential nutrient and iron-containing proteins play diverse roles in cell cycle, DNA repair, and metabolism, many proteins involved in modulating iron homeostasis are now implicated in cancer, including the aforementioned ferroportin and ferritin proteins (92, 154). For example, glioblastoma cells were found to be particularly dependent on high ferritin levels; knocking down ferritin in vitro reduced tumor cell proliferation by sevenfold. Xenograft studies showed that shRNA-mediated FTH1 or FTL knockdown suppressed tumorigenesis of glioblastoma cells (136). Understanding how iron signaling becomes deregulated in cancer could lead to novel understanding of the tumor as well as fresh treatment avenues.
One approach is to understand how NRF2 is deregulated in cancer and how that in turn can alter iron homeostasis. Similar to many iron-modulatory proteins, NRF2 activation has also been implicated in cancer: NRF2 activation in some tumor types such as nonsmall cell lung cancer is associated with poor disease prognosis, including worse overall survival with hazard ratios of 1.75 for patients with NRF2 expression and 2.09 for patients with low or undetectable KEAP1 expression (98, 143). Current models posit that while transient NRF2 activation protects cells from deleterious insults and carcinogens, constitutive activation of NRF2 actually promotes carcinogenesis. Recent studies have highlighted this dark side to NRF2 activation in cancer (77, 124). For example, somatic mutations that activate NRF2 or inhibit KEAP1-mediated degradation of NRF2 have shown prevalence in many cancers (40, 138). Additionally, some environmental carcinogens such as arsenic are capable of constitutively activating NRF2 in nanomolar–micromolar exposure ranges (78, 122). Once activated, NRF2 can promote carcinogenesis by facilitating several classical hallmarks of cancer, including angiogenesis, metabolic reprogramming, chronic proliferation, and resistance to cell death (36, 41, 56). Additionally, one of the main consequences of constitutive NRF2 activation in tumors is its ability to upregulate cytoprotective genes that rapidly metabolize and eliminate chemotherapeutics and reduce treatment efficacy (160). While few investigations have unequivocally established relationships between dysregulated NRF2, iron, and oxygen in cancer, studies are beginning to uncover that NRF2 activation in cancer may promote the disease through its modulation of iron signaling.
Currently, the NRF2 iron-related target gene most strongly associated with cancer is HMOX-1 (107). Several studies have indicated that inhibition or elimination of HMOX-1 in NRF2-activated cells sensitizes them to chemotherapeutics (107). For example, A549 cells, a lung cancer cell line with constitutive NRF2 activation, shows HMOX-1 overexpression relative to other lung cancer cell lines H23, H127, and H460. NRF2 knockdown by RNA interference demonstrated that the high levels of HMOX-1 in A549 cells were due to its constitutive NRF2 (61). Importantly, high HMOX-1 expression decreases susceptibility to cisplatin chemotherapy; genetic or pharmacological inhibition of HMOX-1 boosts cisplatin cytotoxicity in A549 cells by 20%–50%. Mechanistically, HMOX-1 was proposed to decrease reactive oxygen species in the cells (61, 71). A similar effect was seen with a different proapoptotic compound, epigallocatechin 3-gallate (EGCG), in A549 cells. EGCG was found to upregulate HMOX-1, and following siRNA-mediated knockdown of HMOX-1, cells were significantly more susceptible to apoptosis when exposed to 50–100 μM EGCG. Intriguingly, the study also found that 100 μM of the iron chelator deferoxamine could reduce the EGCG-induced upregulation of HMOX-1, indicating a role for iron in resistance to chemotherapeutics (75). Several studies have expanded the carcinogenic roles of NRF2 and HMOX-1 beyond lung cancer. For example, NRF2-mediated HMOX-1 induction attenuated arsenic trioxide-induced cell death and ROS in glioma cells (82). NCI-H292 lung cancer cells overexpressing NRF2 and HMOX-1 showed an induction of thymidine phosphorylase and a subsequent increase in angiogenic potential as indicated by a 150%–200% increase in endothelial cell branching (151). Biallelic fumarate hydratase (FH) inactivation, which is the initiating event of hereditary leiomyomatosis and renal cell cancer (HLRCC), also activates NRF2 (116). In these cells, HMOX-1 knockdown was also shown to be synthetic lethal with FH inactivation (31). Clearly, more information on exactly how NRF2-HMOX-1 contributes to cancer is needed.
Aside from HMOX-1, oncogenic relationships between ferritin and NRF2 are emerging. Much like HMOX-1, several studies have indicated that ferritin upregulation protects cells against chemotherapeutics such as doxorubicin or carmustine. For example, siRNA-mediated FTH1 knockdown decreased the LD50 of carmustine from 100 to 40 μM in U-251 glioblastoma cells, and similar depletion of FTH1 and FTL by miR-200b lowered the IC50 of doxorubicin from 20 to <5 μM in MDA-MB-231 breast cancer cells (81, 140). Ferritin upregulation has been shown to increase resistance to tumor necrosis factor-induced cell death as well (121). Given that ferritin transcription is under the control of transcription factors beyond NRF2, such as NF-κB (72), the extent to which NRF2 specifically contributes to ferritin overexpression and associated chemotherapy resistance remains undetermined.
The roles of ferritin in cancer go beyond its role in intracellular redox stress and cell death. Recent studies on ferritin in cancer show that it can alter intracellular signaling. Schonberg et al. showed that high ferritin in glioblastoma cells induced a chronic proliferative signal by activating the promitotic Forkhead Box M1 transcription factor (FOXM1) (136). Our own group has pioneered the translation of this work into the NRF2 field through study of a hereditary cancer syndrome known as HLRCC (59). In HLRCC renal tumors, the oncometabolite fumarate constitutively activates NRF2 and inhibits IRPs (117). Thus, fumarate increases ferritin transcription and translation, leading to ferritin protein accumulation. HLRCC tumor cells subsequently rely on ferritin for FOXM1-mediated proliferation: ferritin knockdown ablated FOXM1 and abrogated cell growth (59). Whether NRF2 and FOXM1 signaling are connected through ferritin in cancers with genetic- or toxicant-induced NRF2 activation requires inquiry.
While the exact mechanism explaining how ferritin upregulation induces FOXM1 signaling is not well characterized, it could be through an iron chelation effect induced by iron-independent upregulation of ferritin. Should NRF2 activation induce intracellular iron deficiency, this could have profound implications for many biological iron-dependent carcinogenic processes, ranging from replication, DNA repair, metabolism, and iron-dependent dioxygenase activity (7, 21, 45, 84, 108, 112, 131, 134). Iron-dependent dioxygenases include the prolyl hydroxylases that negatively regulate the proangiogenic transcription factors: hypoxia-inducible factors 1α and 2α (HIF1α and HIF2α) (53). Inhibition of NRF2 has been shown to reduce HIF1α (54, 63), mirroring the activation of HIF1α seen with iron chelation treatment (53). Interestingly, upregulation of ferritin has been shown to lower available iron by an estimated 25%, decrease prolyl hydroxylase activity by ∼75%, and activate HIF1α (142). Thus, intracellular iron signaling can serve as a link between HIF and NRF2. Intriguingly, translation of HIF2α is also regulated by IRPs: akin to ferritin, HIF2α contains an IRE in its 5′ UTR that is particularly susceptible to translational inhibition by IRPs (5, 135). If the NRF2-mediated intracellular iron deficiency holds true, NRF2 activation will selectively activate HIF1α and not HIF2α, as the latter would have been repressed at the translation level. The differential activation of HIF1α versus HIF2α may have very different consequences. As was observed in renal cell carcinoma, HIF2α, but not HIF1α, confers tumorigenic potential upon inactivation of the von Hippel-Lindau tumor suppressor (128).
The interplay between transcriptional control of ferritin genes by NRF2 and translational control by IRP2 also merits exploration. KEAP1 regulation of NRF2 is mediated by a series of reactive thiols (23); different cysteines appear to react with different inducers to activate NRF2 (56, 66, 172). IRP2 also seems to show compound-dependent increases (59) or decreases (19, 175) to IRE binding, likely based on IRP2 cysteine modifications (176). Understanding the combined iron-relevant cysteine code that determines if and when ferritin protein increases could provide insight into the relationship between NRF2 and iron signaling. The roles of NRF2-mediated iron homeostasis regulation in cancer are summarized in Figure 5.
FIG. 5.

NRF2, iron, and cancer. NRF2 gain-of-function mutation (green-bordered ellipse) and KEAP1 loss-of-function mutation (red-bordered ellipse) are frequently found in cancer. These mutations lead to sustained NRF2 activation. (1) NRF2 promotes the expression of HMOX-1, which was previously shown to confer chemoresistance. (2) NRF2 promotes ferritin expression, which was shown to protect cells against TNFα-, doxorubicin-, and carmustine-induced cell death. (3) Increased ferritin level in glioblastoma cells was shown to promote FOXM1 activity and confer a proliferative phenotype. (4) In FH loss-of-function (red-bordered ellipse) HLRCC cancer cells, HMOX-1 knockdown was shown to be synthetic lethal with FH inactivation. In these cells, fumarate [Fum] inhibits KEAP1 and drives NRF2 activation. Additionally, fumarate also inhibits IRPs, which normally repress the translation of FTL and FTH1. This leads to intracellular ferritin accumulation that drives cellular proliferation through FOXM1 activation. NRF2 transcription targets are in red. FH, fumarate hydratase; FOXM1, Forkhead Box M1 transcription factor; HLRCC, hereditary leiomyomatosis and renal cell cancer; IRP, iron regulatory protein; TNF, tumor necrosis factor.
NRF2 Regulates Sensitivity to Ferroptosis, an Iron-Dependent Cell Death Mechanism
The profound interactions between oxygen and iron create a check and balance system that keeps iron-associated oxidative stress in check while allowing cells to utilize the redox properties of iron to enable living processes. When thrown out of balance, iron-induced oxidative stress may induce sufficient cellular damage to prevent biological activity and cause cell death. Indeed, free iron and its associated reactive oxygen species are important initiators and mediators of cell death, as reviewed recently (25). One of the main biomolecule targets of iron-mediated hydroxyl radicals is lipid; following abstraction of lipid hydrogen by hydroxyl radicals, the newly formed lipid radical reacts with another nearby lipid to generate a lipid peroxide and a second lipid radical. This process propagates forward, generating a series of lipid peroxides that alter membrane fluidity and membrane-bound protein mobility.
NRF2, iron, and oxygen again cross paths upon interrogation of the NRF2 role in ameliorating lipid peroxidation. Activated NRF2 protects cells against hydroperoxides by upregulating the transcription of the GPX family of proteins. GPXs utilize monomeric reduced glutathione (GSH) to eliminate peroxides, generating water and oxidized glutathione as products. Of the GPXs, glutathione peroxidase 4 (GPX4) serves as the primary neutralizer of lipid peroxides (89).
Aberrant accumulation of lipid peroxides through inhibition of GPX4 or depletion of GSH has been shown to induce ferroptosis, an iron-dependent and nonapoptotic form of cell death (168). Ferroptosis was first discovered as the mechanism by which RAS synthetic lethal compounds, erastin and RSL3, selectively killed RAS-activated cells (24). Beyond its potential exploitation in cancer chemotherapy, ferroptosis is thought to contribute to tissue ischemic reperfusion injury, acute renal failure, and neurodegeneration. Iron itself is capable of inducing lipid peroxidation, both enzymatically and nonenzymatically, although the exact role of iron in ferroptosis is unclear (26, 145). Regardless, addition of iron chelators such as deferoxamine (DFO) or knockdown of the negative regulator of ferritin translation, IRP2, inhibits ferroptosis, indicating that ferroptosis is an iron-dependent oxidative cell death (24). To date, many ferroptosis-inducing (FIN) compounds have been identified, which generally fall into two classes. Class 1 compounds (e.g., erastin) prompt ferroptosis through inhibition of SLC7A11, which is a cysteine–glutamate antiporter responsible for the import of extracellular cysteine required for GSH synthesis. Following SLC7A11 inhibition, GSH synthesis stops, GPX4 cannot eliminate lipid peroxides, and ferroptotic cell death commences. On the other hand, Class 2 FIN compounds (e.g., RSL3) directly inhibit GPX4 to induce lipid peroxide accumulation and ferroptosis (168).
NRF2 activation has long been linked to protection against cell death (58), but investigators have only just begun to dissect the role of NRF2 in ferroptotic cell death and its relationship with iron signaling. Both GPX4 and SLC7A11 are bona fide NRF2 target genes (50, 118, 133), as are the glutathione synthesis genes γ-glutamylcysteine synthetase (GCS) (153), GCLM, and glutamate–cysteine ligase catalytic subunit (GCLC) (73), so NRF2 activation was expected to protect cells against ferroptosis. Additionally, degradation of FTL and FTH1, both NRF2 target genes, was shown to enhance ferroptosis; the logical assumption follows that NRF2-mediated induction of ferritin could desensitize cells to ferroptosis (47). One of the earliest indications that NRF2 and ferroptosis were related arose from a study that showed epicatechin protects against intracerebral hemorrhage by activating NRF2. In this study, the authors also identified that epicatechin reduced gene expression of critical ferroptosis regulators such as IRP2, which is not a target gene of NRF2. It remains to be uncovered how the NRF2-mediated ferroptosis protection interacts with concurrent IRP2 downregulation and how those pathways converge with iron proteins such as FTL or FTH1 (16). Soon after, NRF2 activation via p62-mediated inhibition of KEAP1 was shown to protect hepatocellular carcinoma cells against ferroptosis (147). Specifically, NRF2-mediated induction of the iron-related target genes HMOX-1 and FTH1 protected against ferroptosis; consistently, knockdown of either HMOX-1 or FTH1 by RNA interference enhanced ferroptotic cell death in hepatocellular carcinoma cells (147). Other genes that NRF2 regulated may modify ferroptosis sensitivity: NRF2-mediated induction of metallothionein-1G (MT-1G) has been shown to protect against ferroptosis. The ferroptosis inducer sorafenib more potently reduced the Huh7 hepatocarcinoma tumor burden in mice cotreated with MT-1G shRNA. Additionally, MT-1G knockdown in hepatocellular carcinoma cells nearly doubled erastin- and sorafenib-induced lipid peroxidation (146). NRF2 activation also contributes to ferroptosis resistance of head and neck cancer cells; importantly, in vivo inhibition of the NRF2 pathway sensitized ferroptosis-resistant head and neck cancers to 50 mg/kg of artesunate, a ferroptosis inducer (130). Cancer-preventive compounds, or chemopreventives, may also protect against ferroptosis: 10 μM of the chemopreventive baicalein was shown to protect against ferroptosis by preventing NRF2 degradation and increasing GPX4 protein levels approximately threefold (166). Intriguingly, FINs appear to alter NRF2 levels themselves: the class 1 ferroptosis inducer erastin appeared to decrease NRF2 protein levels in pancreatic cancer and head and neck cancer cells (166), yet increased NRF2 levels in hepatocellular carcinoma cells (147). This mechanistic discrepancy merits further investigation to identify how different FINs affect NRF2 in different tissues. As many ferroptosis inducers concurrently activate NRF2, combining a ferroptosis inducer with an NRF2 inhibitor could provide a synergistic treatment option, although effective pharmacological NRF2 inhibitors remain elusive.
While most results thus far indicate that NRF2 plays a protective role against ferroptosis, some results indicate that pathways positively regulated by NRF2 can enhance ferroptosis. The ferroptosis inducer erastin was shown to induce expression of the NRF2 target gene HMOX-1 in HT-1080 sarcoma cells; inhibition of HMOX-1 with 10 μM zinc protoporphyrin eliminated sensitivity to erastin-induced ferroptosis (76). Given that HMOX-1 has shown both pro- and antiferroptotic activities, more evidence is needed to discern if and when NRF2-mediated upregulation of HMOX-1 promotes or inhibits ferroptosis. Importantly, changes to intracellular iron levels and localizations must be appropriately investigated in the context of NRF2 activation and ferroptosis. Furthermore, NRF2 may regulate ferroptosis in a tissue-dependent manner; many of the iron-relevant NRF2 target genes described here show cell- or tissue-specific induction. To this end, we must understand the mechanistic transcriptional grammar underlying which target genes are activated by NRF2 under different conditions and tissues.
It also remains unclear how NRF2 interacts with other regulators of ferroptosis. While some negative regulators of ferroptosis are independent of NRF2, such as IRP2 or the DNA damage repair protein Fanconi anemia complementation group D2 (FANCD2) (144), other yet to be characterized players in ferroptosis may be partially regulated by NRF2. For example, NRF2 can upregulate expression of genes in the pentose phosphate pathway, pyruvate cycling (malic enzyme), and folate metabolism (MTHFD2) (100, 165), which can generate cellular NADPH (28, 153). While NADPH abundance has been determined as a biomarker for ferroptosis sensitivity (139), no investigations to date have determined the role of NRF2 in enhancing or ameliorating ferroptosis by mediating NADPH levels. Figure 6 summarizes the regulation of ferroptosis by NRF2.
FIG. 6.

NRF2 protects cells from ferroptosis. Ferroptosis is an iron-dependent cell death inducible by FIN compounds. These compounds induce ferroptosis by directly or indirectly inhibiting GPX4 activities, causing cellular lipid peroxide accumulation and subsequent oxidative cell death. NRF2 protects cells from ferroptosis by increasing cellular glutathione production. NRF2 promotes glutathione synthesis by increasing expression of genes directly involved in the glutathione biosynthetic pathway. NRF2 also promotes the expression of SLC7A11, which increases intracellular cysteine pools, resulting in increased intracellular glutathione level. GPX4, which is the primary regulator of ferroptosis, is also an NRF2 target gene. NRF2 also regulates cellular iron availability by promoting the expression of ferritin (FTL and FTH1), which reduces the labile iron pool and thus protects cells against ferroptosis. Other NRF2 target genes, such as MT-1G and HMOX-1, have been shown to regulate ferroptosis. FIN, ferroptosis-inducing; GPX4, glutathione peroxidase 4; GSH, monomeric, reduced glutathione; MT-1G, metallothionein-1G; SLC7A11, solute carrier family 7 member 11/System xCT.
Conclusions
Since the GOE, iron and oxygen have been inexorably linked in biology. Ties between oxygen and iron are most exemplified in metabolism: aerobic organisms utilize oxygen for energy production while exploiting the redox properties of iron for oxygen transport, storage and tissue oxygenation (hemoglobin and myoglobin), oxidative phosphorylation electron shuttling (mitochondria proteins), and metabolite oxidation (cytochrome P450s). Despite the requirement for iron-mediated biology in beneficial oxidative processes, iron and oxygen can also damage cells: through the Fenton reaction, iron facilitates the production of oxygen-derived free radicals capable of damaging biomolecules. To mitigate the damaging aspects of iron and oxygen interactions, vertebrates place the bulk of their iron in the protein cofactor heme to appropriately limit the interactions of iron and oxygen to only those that benefit the organism. Similarly, most iron not bound within heme is sequestered and buffered by intracellular storage and transport systems comprising proteins such as ferritin and ferroportin, thus mitigating iron-induced oxidative damage.
The careful dance of maintaining appropriate heme levels while limiting excess free iron encompasses many carefully regulated processes under the control of the NRF2 transcription factor. As highlighted in this review, NRF2 promotes the transcription of genes involved in both the synthesis (e.g., ABCB6 and FECH) and degradation (e.g., HMOX-1 and BLVRB) of heme. However, in vivo validation of the role of NRF2 in physiological heme and iron homeostasis phenotypes remains an important knowledge gap in the field.
Interestingly, iron treatment alone (100 μM ferric citrate) cannot activate NRF2 (unpublished data). However, NRF2 mediates transcription of ferritin and ferroportin iron buffering systems (Table 1), which results in a reduced labile iron pool. Since the labile iron pool contributes to many cellular processes, a reduced labile iron pool from NRF2 activation is expected to produce a cellular phenotype. Consequently, the physiological impacts of NRF2 on iron metabolism are likely to initiate from NRF2 activation rather than from alterations to iron-mediated oxidative stress. It is also important to note that a certain species of ROS such as H2O2 also serves as an important signaling molecule (159). Emerging evidence showed that NRF2 also regulates the production of such signaling molecules through transcriptional modulation of heme-containing NAPDH oxidases (NOX) (69).
Table 1.
Iron-Related Human Genes Regulated by NRF2
| Function | Symbol | Gene name |
|---|---|---|
| Heme synthesis and O2 transport | HBB | Hemoglobin subunit beta |
| HBG1 | Hemoglobin subunit gamma 1 | |
| ABCB6 | ATP binding cassette subfamily B member 6 | |
| FECH | Ferrochelatase | |
| Heme catabolism | HRG1/SLC48A1 | Heme-responsive gene 1 |
| HMOX-1 | Heme oxygenase 1 | |
| BLVRA | Biliverdin reductase A | |
| BLVRB | Biliverdin reductase B | |
| AMBP→A1M | Alpha-1-microglobulin/bikunin precursor→Alpha-1-microglobulin | |
| Nonheme iron buffering | FTH1 | Ferritin heavy chain |
| FTL | Ferritin light chain | |
| FPN1 | Ferroportin | |
| PIR | Pirin | |
| Cancer progression | HMOX-1 | Heme oxygenase 1 |
| FTH1 | Ferritin heavy chain | |
| FTL | Ferritin light chain | |
| Ferroptosis | GPX4 | Glutathione peroxidase 4 |
| SLC7A11 | Solute carrier family 7 member 11/System xCT | |
| HMOX-1 | Heme oxygenase 1 | |
| FTH1 | Ferritin heavy chain | |
| FTL | Ferritin light chain | |
| MT-1G | Metallothionein-1G |
The importance of NRF2 in mediating interactions between iron and oxygen is becoming apparent as we identify how deregulated NRF2 facilitates iron-mediated cellular pathologies: disruptions to HMOX-1 and ferritin signaling enable cancer progression and reduce treatment efficacies. Since NRF2 can alter iron signaling to facilitate cancer and inhibit treatment, it remains critical that we understand relationships between NRF2 and iron. Indeed, considering the GOE coincided with both the onset of biological utilization of iron and NRF2 evolution, understanding whether NRF2 was eventually selected as a coping strategy for the iron-mediated oxidative toxicity that accompanies iron-facilitated biology presents an intriguing question to entertain as novel relationships among NRF2, iron, and oxygen are uncovered.
Abbreviations Used
- A1M
alpha-1-microglobulin
- ABCB6
ATP binding cassette subfamily B member 6
- AMBP
alpha-1-microglobulin/bikunin precursor
- ARE
antioxidant response element
- BACH1
BRCA-1-associated carboxy-terminal helicase
- BLVRA
biliverdin reductase A
- BLVRB
biliverdin reductase B
- ChIP
chromatin immunoprecipitation
- EGCG
epigallocatechin 3-gallate
- FECH
ferrochelatase
- FH
fumarate hydratase
- FIN
ferroptosis-inducing
- FOXM1
Forkhead Box M1 transcription factor
- FPN1
ferroportin
- FTH1
ferritin heavy chain
- FTL
ferritin light chain
- GCLM
glutamate–cysteine ligase modifier subunit
- GOE
Great Oxygenation Event
- GPX
glutathione peroxidase
- GPX4
glutathione peroxidase 4
- GSH
monomeric, reduced glutathione
- HIF1α
hypoxia-inducible factor 1α
- HLRCC
hereditary leiomyomatosis and renal cell cancer
- HMOX-1
heme oxygenase 1
- HPSC
hematopoietic stem cell
- HRG1
heme-responsive gene 1
- IRE
iron response element
- IRP
iron regulatory protein
- KEAP1
Kelch-like ECH-associated protein 1
- MT-1G
metallothionein-1G
- NF-E2
nuclear factor erythroid 2
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NRF2
nuclear factor (erythroid-derived 2)-like 2
- PIR
pirin
- ROS
reactive oxygen species
- SLC7A11
solute carrier family 7 member 11/System xCT
- StRE
stress response element
- tBHQ
tert-butylhydroquinone
- UTR
untranslated region
Acknowledgments
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1143953 (M.J.K.). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. The project is partially funded by funds from the University of Arizona College of Pharmacy and the University of Arizona Health Sciences (A.O.).
References
- 1.Agyeman AS, Chaerkady R, Shaw PG, Davidson NE, Visvanathan K, Pandey A, and Kensler TW. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res Treat 132: 175–187, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aisen P, Enns C, and Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Int J Biochem Cell Biol 33: 940–959, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Alam J, Cai J, and Smith A. Isolation and characterization of the mouse heme oxygenase-1 gene. Distal 5′ sequences are required for induction by heme or heavy metals. J Biol Chem 269: 1001–1009, 1994 [PubMed] [Google Scholar]
- 4.Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, and Cook JL. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem 274: 26071–26078, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Anderson SA, Nizzi CP, Chang Y-I, Deck KM, Schmidt PJ, Galy B, Damnernsawad A, Broman AT, Kendziorski C, Hentze MW, Fleming MD, Zhang J, and Eisenstein RS. The IRP1-HIF-2α axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab 17: 282–290, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrews SC. The Ferritin-like superfamily: evolution of the biological iron storeman from a rubrerythrin-like ancestor. Biochim Biophys Acta 1800: 691–705, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Aravind L. and Koonin EV. The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate-and iron-dependent dioxygenases. Genome Biol 2: Research0007, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arosio P, Yokota M, and Drysdale JW. Structural and immunological relationships of isoferritins in normal and malignant cells. Cancer Res 36: 1735–1739, 1976 [PubMed] [Google Scholar]
- 9.Benfeitas R, Selvaggio G, Antunes F, Coelho PM, and Salvador A. Hydrogen peroxide metabolism and sensing in human erythrocytes: a validated kinetic model and reappraisal of the role of peroxiredoxin II. Free Radic Biol Med 74: 35–49, 2014 [DOI] [PubMed] [Google Scholar]
- 10.Bratosin D, Mazurier J, Tissier J, Estaquier J, Huart J, Ameisen J, Aminoff D, and Montreuil J. Cellular and molecular mechanisms of senescent erythrocyte phagocytosis by macrophages. A review. Biochimie 80: 173–195, 1998 [DOI] [PubMed] [Google Scholar]
- 11.Brzóska K, Stępkowski TM, and Kruszewski M. Basal PIR expression in HeLa cells is driven by NRF2 via evolutionary conserved antioxidant response element. Mol Cell Biochem 389: 99–111, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cabantchik ZI. Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Front Pharmacol 5: 45, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cairo G, Tacchini L, Pogliaghi G, Anzon E, Tomasi A, and Bernelli-Zazzera A. Induction of ferritin synthesis by oxidative stress. Transcriptional and post-transcriptional regulation by expansion of the “free” iron pool. J Biol Chem 270: 700–703, 1995 [DOI] [PubMed] [Google Scholar]
- 14.Campbell MR, Karaca M, Adamski KN, Chorley BN, Wang X, and Bell DA. Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxid Med Cell Longev 2013: 120305, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan K, Lu R, Chang JC, and Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A 93: 13943–13948, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang CF, Cho S, and Wang J. (-)-Epicatechin protects hemorrhagic brain via synergistic Nrf2 pathways. Ann Clin Transl Neurol 1: 258–271, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi AM. and Alam J. Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am J Respir Cell Mol Biol 15: 9–19, 1996 [DOI] [PubMed] [Google Scholar]
- 18.Chowdhury I, Mo Y, Gao L, Kazi A, Fisher AB, and Feinstein SI. Oxidant stress stimulates expression of the human peroxiredoxin 6 gene by a transcriptional mechanism involving an antioxidant response element. Free Radic Biol Med 46: 146–153, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabon MA, Konrad C, Polverino F, Siempos II, Perez E, Mizumura K, Ghosh MC, Parameswaran H, Williams NC, Rooney KT, Chen ZH, Goldklang MP, Yuan GC, Moore SC, Demeo DL, Rouault TA, D'Armiento JM, Schon EA, Manfredi G, Quackenbush J, Mahmood A, Silverman EK, Owen CA, and Choi AM. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med 22: 163–174, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cloud P. Paleoecological significance of the banded iron-formation. Econ Geol 68: 1135–1143, 1973 [Google Scholar]
- 21.de Ungria M, Rao R, Wobken JD, Luciana M, Nelson CA, and Georgieff MK. Perinatal iron deficiency decreases cytochrome c oxidase (CytOx) activity in selected regions of neonatal rat brain. Pediatr Res 48: 169–176, 2000 [DOI] [PubMed] [Google Scholar]
- 22.Delaby C, Pilard N, Hetet G, Driss F, Grandchamp B, Beaumont C, and Canonne-Hergaux F. A physiological model to study iron recycling in macrophages. Exp Cell Res 310: 43–53, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Dinkova-Kostova AT. The role of sulfhydryl reactivity of small molecules for the activation of the KEAP1/NRF2 pathway and the heat shock response. Scientifica 2012: 606104, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, 3rd, and Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149: 1060–1072, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dixon SJ. and Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol 10: 9–17, 2014 [DOI] [PubMed] [Google Scholar]
- 26.Doll S. and Conrad M. Iron and ferroptosis: a still ill-defined liaison. IUBMB Life 69: 423–434, 2017 [DOI] [PubMed] [Google Scholar]
- 27.Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, and Andrews NC. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 1: 191–200, 2005 [DOI] [PubMed] [Google Scholar]
- 28.Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, and Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510: 298–302, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fenton H. LXXIII.—oxidation of tartaric acid in presence of iron. J Chem Soc Trans 65: 899–910, 1894 [Google Scholar]
- 30.Fibach E. and Rachmilewitz E. The role of oxidative stress in hemolytic anemia. Curr Mol Med 8: 609–619, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Frezza C, Zheng L, Folger O, Rajagopalan KN, MacKenzie ED, Jerby L, Micaroni M, Chaneton B, Adam J, and Hedley A. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 477: 225–228, 2011 [DOI] [PubMed] [Google Scholar]
- 32.Gacesa R, Dunlap WC, Barlow DJ, Laskowski RA, and Long PF. Rising levels of atmospheric oxygen and evolution of Nrf2. Sci Rep 6: 27740, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghaffari S. Oxidative stress in the regulation of normal and neoplastic hematopoiesis. Antioxid Redox Signal 10: 1923–1940, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giudice A, Arra C, and Turco MC. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. Methods Mol Biol 647: 37–74, 2010 [DOI] [PubMed] [Google Scholar]
- 35.Gorrini C, Harris IS, and Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12: 931–947, 2013 [DOI] [PubMed] [Google Scholar]
- 36.Hanahan D. and Weinberg RA. Hallmarks of cancer: the next generation. Cell 144: 646–674, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Harada N, Kanayama M, Maruyama A, Yoshida A, Tazumi K, Hosoya T, Mimura J, Toki T, Maher JM, Yamamoto M, and Itoh K. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch Biochem Biophys 508: 101–109, 2011 [DOI] [PubMed] [Google Scholar]
- 38.Harder B, Jiang T, Wu T, Tao S, de la Vega MR, Tian W, Chapman E, and Zhang DD. Molecular mechanisms of Nrf2 regulation and how these influence chemical modulation for disease intervention. Biochem Soc Trans 43: 680–686, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayes JD. and Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39: 199–218, 2014 [DOI] [PubMed] [Google Scholar]
- 40.Hayes JD. and McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci 34: 176–188, 2009 [DOI] [PubMed] [Google Scholar]
- 41.Heinäniemi M. and Levonen A-L. Role of the Keap1–Nrf2 pathway in cancer. Redox Cancer 122: 281, 2014 [DOI] [PubMed] [Google Scholar]
- 42.Helias V, Saison C, Ballif BA, Peyrard T, Takahashi J, Takahashi H, Tanaka M, Deybach J-C, Puy H, and Le Gall M. ABCB6 is dispensable for erythropoiesis and specifies the new blood group system Langereis. Nat Genet 44: 170–173, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hintze KJ, Katoh Y, Igarashi K, and Theil EC. Bach1 repression of ferritin and thioredoxin reductase1 is heme-sensitive in cells and in vitro and coordinates expression with heme oxygenase1, beta-globin, and NADP(H) quinone (oxido) reductase1. J Biol Chem 282: 34365–34371, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Hirotsu Y, Katsuoka F, Funayama R, Nagashima T, Nishida Y, Nakayama K, Engel JD, and Yamamoto M. Nrf2–MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res 40: 10228–10239, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoffbrand AV, Ganeshaguru K, Hooton JW, and Tattersall MH. Effect of iron deficiency and desferrioxamine on DNA synthesis in human cells. Br J Haematol 33: 517–526, 1976 [DOI] [PubMed] [Google Scholar]
- 46.Hooda J, Shah A, and Zhang L. Heme, an essential nutrient from dietary proteins, critically impacts diverse physiological and pathological processes. Nutrients 6: 1080–1102, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, 3rd, Kang R, and Tang D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12: 1425–1428, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hubner R, Schwartz JD, De Bishnu P, Ferris B, Omberg L, Mezey JG, Hackett NR, and Crystal RG. Coordinate control of expression of Nrf2-modulated genes in the human small airway epithelium is highly responsive to cigarette smoking. Mol Med 15: 203–219, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Inamdar NM, Ahn YI, and Alam J. The heme-responsive element of the mouse heme oxygenase-1 gene is an extended AP-1 binding site that resembles the recognition sequences for MAF and NF-E2 transcription factors. Biochem Biophys Res Commun 221: 570–576, 1996 [DOI] [PubMed] [Google Scholar]
- 50.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, and Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem 275: 16023–16029, 2000 [DOI] [PubMed] [Google Scholar]
- 51.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, and Hatayama I. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236: 313–322, 1997 [DOI] [PubMed] [Google Scholar]
- 52.Iuchi Y, Okada F, Onuma K, Onoda T, Asao H, Kobayashi M, and Fujii J. Elevated oxidative stress in erythrocytes due to a SOD1 deficiency causes anaemia and triggers autoantibody production. Biochem J 402: 219–227, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jaakkola P, Mole DR, Tian Y-M, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, and Schofield CJ. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292: 468–472, 2001 [DOI] [PubMed] [Google Scholar]
- 54.Ji X, Wang H, Zhu J, Zhu L, Pan H, Li W, Zhou Y, Cong Z, Yan F, and Chen S. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Int J Cancer 135: 574–584, 2014 [DOI] [PubMed] [Google Scholar]
- 55.Johnson RM, Ho Y-S, Yu D-Y, Kuypers FA, Ravindranath Y, and Goyette GW. The effects of disruption of genes for peroxiredoxin-2, glutathione peroxidase-1, and catalase on erythrocyte oxidative metabolism. Free Radic Biol Med 48: 519–525, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kansanen E, Kuosmanen SM, Leinonen H, and Levonen A-L. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol 1: 45–49, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawatani Y, Suzuki T, Shimizu R, Kelly VP, and Yamamoto M. Nrf2 and selenoproteins are essential for maintaining oxidative homeostasis in erythrocytes and protecting against hemolytic anemia. Blood 117: 986–996, 2011 [DOI] [PubMed] [Google Scholar]
- 58.Kensler TW, Wakabayashi N, and Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47: 89–116, 2007 [DOI] [PubMed] [Google Scholar]
- 59.Kerins MJ, Vashisht A, Liang BX-T, Duckworth SJ, Praslicka BJ, Wohlschlegel JA, and Ooi A. Fumarate mediates a chronic proliferative signal in fumarate hydratase inactivated cancer cells by increasing transcription and translation of ferritin genes. Mol Cell Biol 37: pii:, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khan AA. and Quigley JG. Control of intracellular heme levels: heme transporters and heme oxygenases. Biochim Biophys Acta 1813: 668–682, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim H-R, Kim S, Kim E-J, Park J-H, Yang S-H, Jeong E-T, Park C, Youn M-J, So H-S, and Park R. Suppression of Nrf2-driven heme oxygenase-1 enhances the chemosensitivity of lung cancer A549 cells toward cisplatin. Lung Cancer 60: 47–56, 2008 [DOI] [PubMed] [Google Scholar]
- 62.Kim JH, Thimmulappa RK, Kumar V, Cui W, Kumar S, Kombairaju P, Zhang H, Margolick J, Matsui W, Macvittie T, Malhotra SV, and Biswal S. NRF2-mediated Notch pathway activation enhances hematopoietic reconstitution following myelosuppressive radiation. J Clin Invest 124: 730–741, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim T-H, Hur E-g, Kang S-J, Kim J-A, Thapa D, Lee YM, Ku SK, Jung Y, and Kwak M-K. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Res 71: 2260–2275, 2011 [DOI] [PubMed] [Google Scholar]
- 64.Kim Y-J, Ahn J-Y, Liang P, Ip C, Zhang Y, and Park Y-M. Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: implication to tumor biology. Cancer Res 67: 546–554, 2007 [DOI] [PubMed] [Google Scholar]
- 65.Knutson MD, Vafa MR, Haile DJ, and Wessling-Resnick M. Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages. Blood 102: 4191–4197, 2003 [DOI] [PubMed] [Google Scholar]
- 66.Kobayashi M, Li L, Iwamoto N, Nakajima-Takagi Y, Kaneko H, Nakayama Y, Eguchi M, Wada Y, Kumagai Y, and Yamamoto M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol Cell Biol 29: 493–502, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kohen R. and Nyska A. Invited review: oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol Pathol 30: 620–650, 2002 [DOI] [PubMed] [Google Scholar]
- 68.Koorts AM. and Viljoen M. Ferritin and ferritin isoforms I: structure-function relationships, synthesis, degradation and secretion. Arch Physiol Biochem 113: 30–54, 2007 [DOI] [PubMed] [Google Scholar]
- 69.Kovac S, Angelova PR, Holmström KM, Zhang Y, Dinkova-Kostova AT, and Abramov AY. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta 1850: 794–801, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krishnamurthy PC, Du G, Fukuda Y, Sun D, Sampath J, Mercer KE, Wang J, Sosa-Pineda B, Murti KG, and Schuetz JD. Identification of a mammalian mitochondrial porphyrin transporter. Nature 443: 586–589, 2006 [DOI] [PubMed] [Google Scholar]
- 71.Kuroda H, Takeno M, Murakami S, Miyazawa N, Kaneko T, and Ishigatsubo Y. Inhibition of heme oxygenase-1 with an epidermal growth factor receptor inhibitor and cisplatin decreases proliferation of lung cancer A549 cells. Lung Cancer 67: 31–36, 2010 [DOI] [PubMed] [Google Scholar]
- 72.Kwak EL, Larochelle DA, Beaumont C, Torti SV, and Torti FM. Role for NF-κB in the regulation of ferritin H by tumor necrosis factor-α. J Biol Chem 270: 15285–15293, 1995 [DOI] [PubMed] [Google Scholar]
- 73.Kwak M-K, Itoh K, Yamamoto M, and Kensler TW. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol Cell Biol 22: 2883–2892, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kwak MK, Itoh K, Yamamoto M, Sutter TR, and Kensler TW. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol Med 7: 135–145, 2001 [PMC free article] [PubMed] [Google Scholar]
- 75.Kweon M-H, Adhami VM, Lee J-S, and Mukhtar H. Constitutive overexpression of Nrf2-dependent heme oxygenase-1 in A549 cells contributes to resistance to apoptosis induced by epigallocatechin 3-gallate. J Biol Chem 281: 33761–33772, 2006 [DOI] [PubMed] [Google Scholar]
- 76.Kwon M-Y, Park E, Lee S-J, and Chung SW. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 6: 24393, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lau A, Villeneuve NF, Sun Z, Wong PK, and Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res 58: 262–270, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lau A, Whitman SA, Jaramillo MC, and Zhang DD. Arsenic-mediated activation of the Nrf2-Keap1 antioxidant pathway. J Biochem Mol Toxicol 27: 99–105, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee J-M, Chan K, Kan YW, and Johnson JA. Targeted disruption of Nrf2 causes regenerative immune-mediated hemolytic anemia. Proc Natl Acad Sci U S A 101: 9751–9756, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu F, Rehmani I, Esaki S, Fu R, Chen L, de Serrano V, and Liu A. Pirin is an iron-dependent redox regulator of NF-κB. Proc Natl Acad Sci U S A 110: 9722–9727, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu X, Madhankumar A, Slagle-Webb B, Sheehan JM, Surguladze N, and Connor JR. Heavy chain ferritin siRNA delivered by cationic liposomes increases sensitivity of cancer cells to chemotherapeutic agents. Cancer Res 71: 2240–2249, 2011 [DOI] [PubMed] [Google Scholar]
- 82.Liu Y, Liang Y, Zheng T, Yang G, Zhang X, Sun Z, Shi C, and Zhao S. Inhibition of heme oxygenase-1 enhances anti-cancer effects of arsenic trioxide on glioma cells. J Neurooncol 104: 449–458, 2011 [DOI] [PubMed] [Google Scholar]
- 83.Loboda A, Damulewicz M, Pyza E, Jozkowicz A, and Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci 73: 3221, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lukianova OA. and David SS. A role for iron–sulfur clusters in DNA repair. Curr Opin Chem Biol 9: 145–151, 2005 [DOI] [PubMed] [Google Scholar]
- 85.Lyons TW, Reinhard CT, and Planavsky NJ. The rise of oxygen in Earth's early ocean and atmosphere. Nature 506: 307–315, 2014 [DOI] [PubMed] [Google Scholar]
- 86.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53: 401–426, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Macara IG, Hoy T, and Harrison PM. The formation of ferritin from apoferritin. Kinetics and mechanism of iron uptake. Biochem J 126: 151–162, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Macari ER. and Lowrey CH. Induction of human fetal hemoglobin via the NRF2 antioxidant response signaling pathway. Blood 117: 5987–5997, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Maiorino M, Bosello V, Cozza G, Roveri A, Toppo S, and Ursini F. Glutathione peroxidase-4. In: Selenium. New York, NY: Springer, 2011, pp. 181–195 [Google Scholar]
- 90.Mancias JD, Vaites LP, Nissim S, Biancur DE, Kim AJ, Wang X, Liu Y, Goessling W, Kimmelman AC, and Harper JW. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 4: e10308, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mancias JD, Wang X, Gygi SP, Harper JW, and Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509: 105–109, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manz DH, Blanchette NL, Paul BT, Torti FM, and Torti SV. Iron and cancer: recent insights. Ann N Y Acad Sci 1368: 149–161, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Marini MG, Chan K, Casula L, Kan YW, Cao A, and Moi P. hMAF, a small human transcription factor that heterodimerizes specifically with Nrf1 and Nrf2. J Biol Chem 272: 16490–16497, 1997 [DOI] [PubMed] [Google Scholar]
- 94.Marro S, Chiabrando D, Messana E, Stolte J, Turco E, Tolosano E, and Muckenthaler MU. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position −7007 of the FPN1 promoter. Haematologica 95: 1261–1268, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mattè A, De Falco L, Federti E, Levi S, Iolascon A, Federico G, Mohandas N, Janin A, Bruno M, and Leboeuf C. Peroxiredoxin-2: a novel factor involved in iron homeostasis. Am Soc Hematol, 126: 406, 2015 [Google Scholar]
- 96.Meining W. and Skerra A. The crystal structure of human α1-microglobulin reveals a potential haem-binding site. Biochem J 445: 175–182, 2012 [DOI] [PubMed] [Google Scholar]
- 97.Merchant AA, Singh A, Matsui W, and Biswal S. The redox-sensitive transcription factor Nrf2 regulates murine hematopoietic stem cell survival independently of ROS levels. Blood 118: 6572–6579, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Merikallio H, Pääkkö P, Kinnula VL, Harju T, and Soini Y. Nuclear factor erythroid-derived 2-like 2 (Nrf2) and DJ1 are prognostic factors in lung cancer. Hum Pathol 43: 577–584, 2012 [DOI] [PubMed] [Google Scholar]
- 99.Mignotte V, Wall L, Grosveld F, and Romeo P-H. Two tissue-specific factors bind the erythroid promoter of the human porphobilinogen deaminase gene. Nucleic Acids Res 17: 37–54, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, and Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22: 66–79, 2012 [DOI] [PubMed] [Google Scholar]
- 101.Moi P, Chan K, Asunis I, Cao A, and Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A 91: 9926–9930, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moi P. and Kan YW. Synergistic enhancement of globin gene expression by activator protein-1-like proteins. Proc Natl Acad Sci U S A 87: 9000–9004, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Muckenthaler MU, Galy B, and Hentze MW. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr 28: 197–213, 2008 [DOI] [PubMed] [Google Scholar]
- 104.Muckenthaler MU, Rivella S, Hentze MW, and Galy B. A red carpet for iron metabolism. Cell 168: 344–361, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Murakami S. and Motohashi H. Roles of NRF2 in cell proliferation and differentiation. Free Radic Biol Med 88: 168–178, 2015 [DOI] [PubMed] [Google Scholar]
- 106.Murakami S, Shimizu R, Romeo PH, Yamamoto M, and Motohashi H. Keap1-Nrf2 system regulates cell fate determination of hematopoietic stem cells. Genes Cells 19: 239–253, 2014 [DOI] [PubMed] [Google Scholar]
- 107.Na H-K. and Surh Y-J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic Biol Med 67: 353–365, 2014 [DOI] [PubMed] [Google Scholar]
- 108.Netz DJ, Stith CM, Stumpfig M, Kopf G, Vogel D, Genau HM, Stodola JL, Lill R, Burgers PM, and Pierik AJ. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat Chem Biol 8: 125–132, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Neumann CA, Krause DS, Carman CV, Das S, Dubey DP, Abraham JL, Bronson RT, Fujiwara Y, Orkin SH, and Van Etten RA. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 424: 561–565, 2003 [DOI] [PubMed] [Google Scholar]
- 110.Ney PA, Sorrentino BP, Lowrey CH, and Nienhuis AW. Inducibility of the HS II enhancer depends on binding of an erythroid specific nuclear protein. Nucleic Acids Res 18: 6011–6017, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ney PA, Sorrentino BP, McDonagh KT, and Nienhuis AW. Tandem AP-1-binding sites within the human beta-globin dominant control region function as an inducible enhancer in erythroid cells. Genes Dev 4: 993–1006, 1990 [DOI] [PubMed] [Google Scholar]
- 112.Oexle H, Gnaiger E, and Weiss G. Iron-dependent changes in cellular energy metabolism: influence on citric acid cycle and oxidative phosphorylation. Biochim Biophys Acta 1413: 99–107, 1999 [DOI] [PubMed] [Google Scholar]
- 113.Ogawa K, Sun J, Taketani S, Nakajima O, Nishitani C, Sassa S, Hayashi N, Yamamoto M, Shibahara S, and Fujita H. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J 20: 2835–2843, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Olsson MG, Allhorn M, Bulow L, Hansson SR, Ley D, Olsson ML, Schmidtchen A, and Akerstrom B. Pathological conditions involving extracellular hemoglobin: molecular mechanisms, clinical significance, and novel therapeutic opportunities for alpha(1)-microglobulin. Antioxid Redox Signal 17: 813–846, 2012 [DOI] [PubMed] [Google Scholar]
- 115.Olsson MG, Olofsson T, Tapper H, and Åkerström B. The lipocalin α 1-microglobulin protects erythroid K562 cells against oxidative damage induced by heme and reactive oxygen species. Free Radic Res 42: 725–736, 2008 [DOI] [PubMed] [Google Scholar]
- 116.Ooi A. and Furge KA. Fumarate hydratase inactivation in renal tumors: HIF1α, NRF2, and “cryptic targets” of transcription factors. Chin J Cancer 31: 413, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ooi A, Wong J-C, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, Min BWH, Tan M-H, Zhang Z, and Yang XJ. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 20: 511–523, 2011 [DOI] [PubMed] [Google Scholar]
- 118.Osburn WO, Wakabayashi N, Misra V, Nilles T, Biswal S, Trush MA, and Kensler TW. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch Biochem Biophys 454: 7–15, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Oyake T, Itoh K, Motohashi H, Hayashi N, Hoshino H, Nishizawa M, Yamamoto M, and Igarashi K. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol Cell Biol 16: 6083–6095, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pang H, Bartlam M, Zeng Q, Miyatake H, Hisano T, Miki K, Wong L-L, Gao GF, and Rao Z. Crystal structure of human pirin An iron-binding nuclear protein and transcription cofactor. J Biol Chem 279: 1491–1498, 2004 [DOI] [PubMed] [Google Scholar]
- 121.Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, and Beaumont C. Ferritin heavy chain upregulation by NF-κB inhibits TNFα-induced apoptosis by suppressing reactive oxygen species. Cell 119: 529–542, 2004 [DOI] [PubMed] [Google Scholar]
- 122.Pi J, Diwan BA, Sun Y, Liu J, Qu W, He Y, Styblo M, and Waalkes MP. Arsenic-induced malignant transformation of human keratinocytes: involvement of Nrf2. Free Radic Biol Med 45: 651–658, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pietsch EC, Chan JY, Torti FM, and Torti SV. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J Biol Chem 278: 2361–2369, 2003 [DOI] [PubMed] [Google Scholar]
- 124.Praslicka BJ, Kerins MJ, and Ooi A. The complex role of NRF2 in cancer: a genomic view. Curr Opin Toxicol 1: 37–45, 2016 [Google Scholar]
- 125.Primiano T, Kensler TW, Kuppusamy P, Zweier JL, and Sutter TR. Induction of hepatic heme oxygenase-1 and ferritin in rats by cancer chemopreventive dithiolethiones. Carcinogenesis 17: 2291–2296, 1996 [DOI] [PubMed] [Google Scholar]
- 126.Prousek J. Fenton chemistry in biology and medicine. Pure Appl Chem 79: 2325–2338, 2007 [Google Scholar]
- 127.Rajagopal A, Rao AU, Amigo J, Tian M, Upadhyay SK, Hall C, Uhm S, Mathew M, Fleming MD, and Paw BH. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 453: 1127–1131, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li J-L, Pugh CW, Maxwell PH, Harris AL, and Ratcliffe PJ. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol 25: 5675–5686, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Reichard JF, Motz GT, and Puga A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res 35: 7074–7086, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Roh J-L, Kim EH, Jang H, and Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol 11: 254–262, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rudolf J, Makrantoni V, Ingledew WJ, Stark MJ, and White MF. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Mol Cell 23: 801–808, 2006 [DOI] [PubMed] [Google Scholar]
- 132.Salahudeen AA, Thompson JW, Ruiz JC, Ma H-W, Kinch LN, Li Q, Grishin NV, and Bruick RK. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 326: 722–726, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Salazar M, Rojo AI, Velasco D, de Sagarra RM, and Cuadrado A. Glycogen synthase kinase-3β inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem 281: 14841–14851, 2006 [DOI] [PubMed] [Google Scholar]
- 134.Salminen A, Kauppinen A, and Kaarniranta K. 2-Oxoglutarate-dependent dioxygenases are sensors of energy metabolism, oxygen availability, and iron homeostasis: potential role in the regulation of aging process. Cell Mol Life Sci 72: 3897–3914, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sanchez M, Galy B, Muckenthaler MU, and Hentze MW. Iron-regulatory proteins limit hypoxia-inducible factor-2α expression in iron deficiency. Nat Struct Mol Biol 14: 420–426, 2007 [DOI] [PubMed] [Google Scholar]
- 136.Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, Hubert CG, Mack SC, Jarrar AM, and Karl RT. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell 28: 441–455, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schultz IJ, Chen C, Paw BH, and Hamza I. Iron and porphyrin trafficking in heme biogenesis. J Biol Chem 285: 26753–26759, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, and Hirohashi S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A 105: 13568–13573, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shimada K, Hayano M, Pagano NC, and Stockwell BR. Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem Biol 23: 225–235, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shpyleva SI, Tryndyak VP, Kovalchuk O, Starlard-Davenport A, Vasyl'F C, Beland FA, and Pogribny IP. Role of ferritin alterations in human breast cancer cells. Breast Cancer Res Treat 126: 63–71, 2011 [DOI] [PubMed] [Google Scholar]
- 141.Siebel JF, Kosinsky RL, Åkerström B, and Knipp M. Insertion of heme b into the structure of the Cys34-carbamidomethylated human lipocalin α1-microglobulin: formation of a [(heme) 2 (α1-microglobulin)] 3 complex. Chembiochem 13: 879–887, 2012 [DOI] [PubMed] [Google Scholar]
- 142.Siegert I, Schödel J, Nairz M, Schatz V, Dettmer K, Dick C, Kalucka J, Franke K, Ehrenschwender M, and Schley G. Ferritin-mediated iron sequestration stabilizes hypoxia-inducible factor-1α upon LPS activation in the presence of ample oxygen. Cell Rep 13: 2048–2055, 2015 [DOI] [PubMed] [Google Scholar]
- 143.Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, Corvalan AH, Biswal S, Swisher SG, and Bekele BN. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res 16: 3743–3753, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Song X, Xie Y, Kang R, Hou W, Sun X, Epperly MW, Greenberger JS, and Tang D. FANCD2 protects against bone marrow injury from ferroptosis. Biochem Biophys Res Commun 480: 443–449, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Stohs S. and Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med 18: 321–336, 1995 [DOI] [PubMed] [Google Scholar]
- 146.Sun X, Niu X, Chen R, He W, Chen D, Kang R, and Tang D. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 64: 488–500, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, and Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63: 173–184, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Talbot D. and Grosveld F. The 5'HS2 of the globin locus control region enhances transcription through the interaction of a multimeric complex binding at two functionally distinct NF-E2 binding sites. EMBO J 10: 1391, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Talbot D, Philipsen S, Fraser P, and Grosveld F. Detailed analysis of the site 3 region of the human beta-globin dominant control region. EMBO J 9: 2169, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, and Hayes JD. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med 88: 108–146, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tertil M, Skrzypek K, Florczyk U, Weglarczyk K, Was H, Collet G, Guichard A, Gil T, Kuzdzal J, and Jozkowicz A. Regulation and novel action of thymidine phosphorylase in non-small cell lung cancer: crosstalk with Nrf2 and HO-1. PLoS One 9: e97070, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Theil EC. Ferritin: the protein nanocage and iron biomineral in health and in disease. Inorg Chem 52: 12223–12233, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, and Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 62: 5196–5203, 2002 [PubMed] [Google Scholar]
- 154.Torti SV. and Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer 13: 342–355, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tsai JJ, Dudakov JA, Takahashi K, Shieh J-H, Velardi E, Holland AM, Singer NV, West ML, Smith OM, and Young LF. Nrf2 regulates haematopoietic stem cell function. Nat Cell Biol 15: 309–316, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tsuji Y, Ayaki H, Whitman SP, Morrow CS, Torti SV, and Torti FM. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol Cell Biol 20: 5818–5827, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tudor C, Lerner-Marmarosh N, Engelborghs Y, Gibbs PE, and Maines MD. Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem J 413: 405–416, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Vashisht AA, Zumbrennen KB, Huang X, Powers DN, Durazo A, Sun D, Bhaskaran N, Persson A, Uhlen M, and Sangfelt O. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326: 718–721, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Veal EA, Day AM, and Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell 26: 1–14, 2007 [DOI] [PubMed] [Google Scholar]
- 160.Wang X-J, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, and Wondrak GT. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29: 1235–1243, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wasserman WW. and Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci U S A 94: 5361–5366, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.White C, Yuan X, Schmidt PJ, Bresciani E, Samuel TK, Campagna D, Hall C, Bishop K, Calicchio ML, and Lapierre A. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab 17: 261–270, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wilkinson N. and Pantopoulos K. The IRP/IRE system in vivo: insights from mouse models. Fron Pharmacol 5, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Winterbourn CC. The biological chemistry of hydrogen peroxide. Methods Enzymol 528: 3–25, 2013 [DOI] [PubMed] [Google Scholar]
- 165.Wu KC, Cui JY, and Klaassen CD. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci 123: 590–600, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Xie Y, Song X, Sun X, Huang J, Zhong M, Lotze MT, Zeh HJ, Kang R, and Tang D. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem Biophys Res Commun 473: 775–780, 2016 [DOI] [PubMed] [Google Scholar]
- 167.Yanagawa T, Itoh K, Uwayama J, Shibata Y, Yamaguchi A, Sano T, Ishii T, Yoshida H, and Yamamoto M. Nrf2 deficiency causes tooth decolourization due to iron transport disorder in enamel organ. Genes Cells 9: 641–651, 2004 [DOI] [PubMed] [Google Scholar]
- 168.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, and Stockwell BR. Regulation of ferroptotic cancer cell death by GPX4. Cell 156: 317–331, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.This reference has been deleted
- 170.Zenke-Kawasaki Y, Dohi Y, Katoh Y, Ikura T, Ikura M, Asahara T, Tokunaga F, Iwai K, and Igarashi K. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol 27: 6962–6971, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev 38: 769–789, 2006 [DOI] [PubMed] [Google Scholar]
- 172.Zhang DD. and Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol 23: 8137–8151, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang M, An C, Gao Y, Leak RK, Chen J, and Zhang F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol 100: 30–47, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhu H, Santo A, and Li Y. The antioxidant enzyme peroxiredoxin and its protective role in neurological disorders. Exp Biol Med 237: 143–149, 2012 [DOI] [PubMed] [Google Scholar]
- 175.Zimmer M, Ebert BL, Neil C, Brenner K, Papaioannou I, Melas A, Tolliday N, Lamb J, Pantopoulos K, Golub T, and Iliopoulos O. Small-molecule inhibitors of HIF-2a translation link its 5′UTR iron-responsive element to oxygen sensing. Mol Cell 32: 838–848, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zumbrennen KB, Wallander ML, Romney SJ, and Leibold EA. Cysteine oxidation regulates the RNA-binding activity of iron regulatory protein 2. Mol Cell Biol 29: 2219–2229, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]