

Abstract
The staphylococcal accessory regulator A (sarA) impacts the extracellular accumulation of Staphylococcus aureus virulence factors at the level of intracellular production and extracellular protease-mediated degradation. We previously used a proteomics approach that measures protein abundance of all proteoforms to demonstrate that mutation of sarA results in increased levels of extracellular proteases and assesses the impact of this on the accumulation of S. aureus exoproteins. Our previous approach was limited as it did not take into account that large, stable proteolytic products from a given protein could result in false negatives when quantified by total proteoforms. Here, our goal was to use an expanded proteomics approach utilizing a dual quantitative method for measuring abundance at both the total proteoform and full-length exoprotein levels to alleviate these false negatives and thereby provide for characterization of protease-dependent and -independent effects of sarA mutation on the S. aureus exoproteome. Proteins present in conditioned medium from overnight, stationary phase cultures of the USA300 strain LAC, an isogenic sarA mutant, and a sarA mutant unable to produce any of the known extracellular proteases (sarA/protease) were resolved using one-dimensional gel electrophoresis. Quantitative proteomic comparisons of sarA versus sarA/protease mutants identified proteins that were cleaved in a protease-dependent manner owing to mutation of sarA, and comparisons of sarA/protease mutant versus the LAC parent strain identified proteins in which abundance was altered in a sarA mutant in a protease-independent manner. Furthermore, the proteins uniquely identified by the full-length data analysis approach eliminated false negatives observed in the total proteoform analysis. This expanded approach provided for a more comprehensive analysis of the impact of mutating sarA on the S. aureus exoproteome.
Keywords: protease, proteolysis, proteomics, mass spectrometry, Staphylococcus aureus, sarA
Introduction
Staphylococcus aureus is a Gram-positive bacterial species that exists as a commensal bacterium in a significant proportion of the healthy population but remains capable of causing a diverse array of serious infections.1 Its ability to cause these infections arises from its capacity to produce an arsenal of surface-associated and extracellular virulence factors, the production of which is controlled by a complex and highly interactive regulatory network that affords the bacterium the flexibility required to survive within different microenvironments of the host.2 One component of this regulatory network is the staphylococcal accessory regulator (sarA), mutation of which has been shown to modulate the production of S. aureus virulence factors at both the transcriptional level and as a modulator of mRNA stability.3−6
In addition to its role in modulating the production of virulence factors, sarA has also been shown to serve an important post-translational role in that it enhances the accumulation of many virulence factors owing to its ability to limit protease-mediated degradation.7−9 Specifically, mutation of sarA results in the increased production of all 10 recognized S. aureus extracellular proteases (aureolysin, ScpA, SspA, SspB, and SplA-F). This has been correlated with reduced accumulation of multiple S. aureus virulence factors and reduced virulence in murine models of sepsis, catheter-associated infection, and hematogenous osteomyelitis.9−11 Moreover, a cause-and-effect relationship has been confirmed in all of these models by demonstrating that the reduced virulence of a sarA mutant can be restored to a significant extent by eliminating protease production in sarA mutants.9
One key to fully understanding the pathogenesis of S. aureus is to identify and characterize the virulence factors that contribute to its pathogenic diversity. Although the importance of many virulence factors has been demonstrated, the pathogenesis of S. aureus infections remains incompletely understood. Approximately 50% of all S. aureus proteins remain annotated as hypothetical proteins with no known function.12 It seems likely that some of these contribute to the prominence of S. aureus as a pathogen, but it is difficult to justify the studies required to experimentally address this possibility in the absence of some functional information. One approach to prioritize virulence factors for detailed in vivo studies is to identify those with reduced accumulation in a sarA mutant owing to protease-mediated degradation.
In previous work, we used a one-dimensional gel electrophoresis approach to analyze sarA-mediated S. aureus exoproteomes.9 Here, exoproteome refers to the summation of S. aureus proteins (i.e., exoproteins) present in the extracellular milieu of stationary phase cultures irrespective of whether they are actively exported or passively released from dead or dying bacterial cells. The basic principle of this published work was to resolve exoproteomes by 1D SDS-PAGE, excise the entire gel lane as a series of bands, perform in-gel trypsin digestion of proteins, and analyze the tryptic proteins by LC-MS/MS for protein identification. The presence of a given exoprotein in an exoproteome sample was determined by summation of identified tryptic peptides (i.e., spectral counts) of the exoprotein in all bands from the gel lane. This approach measures the total proteoforms of a given protein, which could be any permutation of the protein at any molecular mass (i.e., including proteolytic fragments). This previous approach was limited in two ways that could affect full characterization of the S. aureus exoproteome. First, it did not provide for the distinction of exoprotein levels regulated by sarA in a protease-independent as well as protease-dependent manner. Second, it did not take into account that large, stable proteolytic products from a given exoprotein could result in false negatives when quantifying by total proteoforms. For example, a specific exoprotein could be cleaved into a series of large fragments such that the spectral counts in the entire gel lane do not identify the protein as significantly decreased in a sarA-mediated fashion.
We have addressed these issues using an expanded exoproteomic approach, utilizing a dual quantitative method for measuring abundance at both the total proteoform and full-length exoprotein levels, to alleviate false negatives and thereby provide for characterization of protease-dependent and -independent effects of sarA mutation on the S. aureus exoproteome. Using this dual approach, we made comparisons between conditioned medium (CM) from stationary phase cultures of the USA300 S. aureus strain LAC, its isogenic sarA mutant, and its isogenic sarA mutant unable to produce any extracellular protease. The first approach is based on total spectral counts in a gel lane (i.e., measuring total proteoforms), and the second is based on spectral counts derived only from full-length exoproteins. Quantitative proteomic comparisons of sarA vs sarA/protease identified exoproteins that were cleaved in a sarA-mediated and extracellular protease-dependent manner, while comparisons of sarA/protease vs LAC identified those decreased in a sarA-mediated and extracellular protease-independent manner. Furthermore, the exoproteins uniquely identified by the full-length exoprotein data analysis approach eliminated false negatives arising from stable proteolytic products of exoproteins. Additionally, this analysis of sarA-mediated exoproteomes provided a more in-depth measurement of the exoproteins impacted by sarA in a protease-dependent vs -independent manner compared to that in our previous report.9
Materials and Methods
Bacterial Strains and Growth Conditions
The bacterial strains used in this study were fully described in our previous report.9 Briefly, they consisted of the USA300, methicillin-resistant (MRSA) strain LAC (referred to here as wild-type), its isogenic sarA mutant (sarA), and an isogenic sarA mutant unable to produce any of the 10 recognized extracellular proteases (sarA/protease). These proteases include the metalloprotease aureolysin, the cysteine proteases ScpA and SspB (also known as staphopain A and B, respectively), and the serine proteases SspA and SplA-F. With the exception of the sarA mutation, which was generated by insertion of a kanamycin-resistance cassette into the sarA open-reading frame,8 all mutations are true null mutants in which the protease genes were eliminated using either pJB38, which is a modified version of the plasmid pKOR1,13 or by replacement with an erythromycin-resistance cassette.7,14−16 All strains were maintained on tryptic soy agar (TSA) with antibiotic selection as appropriate but were grown without selection when preparing conditioned medium (CM). For exoproteome analysis, stationary phase (16 h) cultures grown in tryptic soy broth (TSB) were standardized relative to each other based on optical density and the CM prepared by centrifugation followed by filter sterilization of the clarified supernatant using 0.45 μm filters.9 Proteomics studies and statistical analysis were based on three biological replicates of each strain.
Mass Spectrometric Analysis of Proteins
An equal volume of CM from each sample was resolved by one-dimensional SDS-PAGE and visualized by Coomassie staining.17−20 Each gel lane was sliced into 24 equiv bands of 2 mm each. Gel bands were destained in 50% methanol (Fisher) containing 100 mM ammonium bicarbonate (Sigma-Aldrich). This was followed by reduction in 10 mM Tris [2-carboxyethyl] phosphine (Pierce) and alkylation in 50 mM iodoacetamide (Sigma-Aldrich). Gel slices were then dehydrated in acetonitrile (Fisher) followed by addition of 100 ng porcine sequencing-grade modified trypsin (Promega) in 100 mM ammonium bicarbonate (Sigma-Aldrich) and digestion at 37 °C for 12–16 h. Peptide products were then acidified in 0.1% formic acid (Pierce). Tryptic peptides were separated by reverse-phase Jupiter Proteo resin (Phenomenex) on a 200 × 0.075 mm column using a nanoAcquity UPLC system (Waters). Peptides were eluted using a 30 min gradient from 97:3 to 65:35 buffer A:B ratio [buffer A = 0.1% formic acid (v/v), 0.5% acetonitrile (v/v) in water; buffer B = 0.1% formic acid (v/v), 99.9% acetonitrile (v/v)]. Eluted peptides were ionized by electrospray (2.15 kV) followed by MS/MS analysis using higher-energy collisional dissociation (HCD) on an Orbitrap Fusion Tribrid mass spectrometer (Thermo) in top-speed data-dependent mode. MS data were acquired using the FTMS analyzer in profile mode at a resolution of 240,000 over a range of 375–1500 m/z. Following HCD activation, MS/MS data were acquired using the ion trap analyzer in centroid mode and normal mass range with precursor mass-dependent normalized collision energy between 28.0 and 31.0.
Proteins were identified by database search using Mascot (Matrix Science, version 2.5.1) against the USA300 S. aureus database (2653 entries, Genebank accession JTJK01000002). A decoy database (based on the reverse of the protein sequences) was used in the search to calculate the FDR for the search algorithm. The search parameters include fixed modification of carbamidomethyl on cysteine, variable modifications of acetylation on the protein N-terminus, variable modification of oxidation on methionine, a maximum of 2 missed cleavages with trypsin, parent ion tolerance of 3 ppm, and fragment ion tolerance of 0.5 Da. Scaffold (Proteome Software) was used to verify MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established with less than 1.0% false discovery by the Scaffold Local FDR algorithm. Protein identifications were accepted if they could be established with less than 1.0% false discovery and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm.21 Total spectral counts for each replicate were exported from Scaffold into Microsoft Excel for further analysis. Mass spectrometric data used for analysis is reported in Table S1. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE22 partner repository with the data set identifier PXD009614 and 10.6019/PXD009614.
Total Proteoform Data Analysis
The amount of total proteoform was identified using the total spectral counts for a given protein in the entire gel lane (i.e., in any of the 24 gel bands from a given gel lane) with spectral counts defined as assigned tandem mass spectra for a given protein.17−20,23 The spectral counts for a given protein detected in the entire gel lane were averaged across triplicate samples for each strain; this averaged spectral count value was defined as the total proteoform level of the protein in each strain. This method was the same total proteoform method employed in our earlier studies.9
Full-Length Protein Data Analysis
The amount of full-length exoprotein was measured using a 3-gel band window approach also based on spectral counting; however, this method is based on using three consecutive gel bands centered on the gel band representing the full-length protein. Specifically, spectral count data collected from wild-type was used to locate the gel band with the maximum spectral count for a given protein, which for the purposes of this study was considered the most full-length version of the protein under steady-state conditions. Spectral counts for the full-length version of that protein were determined by adding the spectral counts in that gel band with those in the gel bands immediately above and below, thus accounting for minor variations in full-length protein migration through the gel. The spectral counts observed in this 3-band window were then averaged across triplicate samples of wild-type; this averaged spectral count value was defined as the level of the steady-state, full-length version of the protein in wild-type. This information was then used to calculate spectral counts for the full-length protein in the sarA and sarA/protease mutants. For example, if the 3-band window for a specific protein was gel bands 8–10 in wild-type, then the 3-band window used for calculating the spectral counts for the full-length protein in sarA and sarA/protease mutants was also gel bands 8–10. The spectral counts observed in the 3-band window were then averaged across triplicate samples of sarA and sarA/protease strains; this averaged spectral count value was defined as the level of the steady-state, full-length version of the protein in the sarA and sarA/protease strains.
Visualizing Proteomic Data
Unsupervised hierarchical clustering for total proteoform and full-length protein analysis was used to show unique and common protein clusters between each strain (Figure S1). A clustered heat map for the total proteoform consisted of the averaged total spectral counts for significantly cleaved proteins, and the full-length protein analysis consisted of the averaged spectral counts for significantly cleaved proteins in the 3-band windows. Proteins significantly changed in abundance were defined based on p-values less than 0.05 and fold changes greater than 2. The heat maps were generated using the Euclidean distance metric, and the data were standardized by the mean and standard deviation before performing hierarchical clustering with Hierarchical Clustering Explorer (HCE, version 3.5).
Volcano plots were used to visualize the significant proteins for each comparison in the study using R studio. The y-axis consists of −log10p-values based on Student’s t test analysis using either the total proteoform or full-length protein method, and the x-axis consists of the log2 fold change. Fold changes were calculated for three different comparisons (wild-type vs sarA, sarA vs sarA/protease, and wild-type vs sarA/protease). For the total proteoform method, fold changes were calculated using the average of replicates of spectral counts from the entire gel lane. For the full-length method, fold changes were calculated using the average of replicates of spectral counts in the 3-band window. Proteins significantly changing in abundance were defined with p-values less than 0.1 and fold changes greater than 1.5 (Tables S2 and S3). Zero spectral count values were replaced with a small value of 0.001 prior to performing t tests and determining fold change.17−20,23
Proteins identified as significantly decreased in sarA compared to wild-type, sarA compared to sarA/protease, and wild-type compared to sarA/protease (top right quadrant in volcano plots) by both proteomic methods are compared with a Venn diagram created with Venny (version 2.1)24 (Figure S2 and Table S4). The Venn diagrams represent the number of shared and unique proteins identified as significant by each method. They identify proteins that are decreased in amounts as either sarA dependent, sarA-mediated protease dependent, and/or sarA-mediated protease independent.
Validation of Exoproteins Decreased in sarA-Mediated Extracellular Protease-Dependent and -Independent Mechanisms
To visualize protease-generated protein fragment profiles, we generated plots of averaged spectral count for selected proteins as a function of gel band. Immunoblots of CM were performed using commercially available antibodies for alpha toxin (Sigma-Aldrich), protein A (Spa) (Sigma-Aldrich), extracellular thermonuclease (nuc) (Toxin Technologies), and LukS (Abcam).8,25 Note that available antibodies for IsdC and ClfA lacked sufficient sensitivity for immunoblot validation; thus, quantitative parallel reaction monitoring (PRM) mass spectrometry was used to quantify levels of IsdC and ClfA.
For PRM studies, equal volumes of CM from wild-type (LAC), sarA, sarA/protease, isdC deletion, and clfA deletion strains were resolved by one-dimensional SDS-PAGE and visualized by Coomassie staining. Each gel lane was sliced into 24 equiv sections. Protein in each gel slice was digested in-gel with trypsin. Tryptic peptides were separated using a 2.5 μm Waters XSelect CSH resin on a 150 mm × 0.075 mm column using a nanoAcquity UPLC system (Waters). A 30 min chromatography gradient was used for each sample, consisting of a 4 min loading phase at 4% buffer B [0.1% formic acid (v/v), 99.9% acetonitrile (v/v)], a 14 min linear gradient from 4% buffer B to 20% buffer B, a 3 min gradient of 20%–35% buffer B, a 1 min gradient of 35%–90% buffer B, 1 min at 90% buffer B, and re-equilibration at 2% buffer B for 7 min. Eluted peptides were ionized by electrospray (2.2 kV) and analyzed by targeted MS/MS on an Orbitrap Fusion Lumos mass spectrometer (Thermo). A full-scan MS was acquired at 60,000 resolution from 375 to 1500 m/z (AGC target 4 × 105, max injection time 50 ms) followed by scheduled, targeted HCD MS/MS scans at 15,000 resolution with a 1 m/z isolation window (AGC target 1 × 105, max injection time 100 ms) and 30% collision energy.26 Scheduling windows for the short gradient were determined by injecting replicates of a subset of pooled gel slice samples prior to the full experiment. Preliminary method development was performed on unfractionated samples that were digested by filter-aided sample preparation.27
Targeted mass spectrometry files were analyzed using Skyline.28 Filter settings included precursor charge states of two or three, product ion charges of one or two, ion type inclusion of precursor, b, or y fragment ions, product ion selection from m/z > precursor to 6 ions, and autoselection of all matching transition ions. Mascot.dat files from the data-dependent runs were used for library construction. A library ion match tolerance of 0.5 m/z was used to select between three and 10 most intense product ions from filtered ion charges and types. MS/MS filtering of centroided product masses were used with a mass accuracy of 25 ppm. Explicit retention time windows of 1–2 min were used for integration. Precursor ions were used for idot product calculation, and fragment ions were used for both dot product calculation and quantitation. Transition results were exported in.csv format and analyzed using R.29 Areas for each transition (excluding precursors) were summed to generate intensity values for each peptide in each sample. Intensity values for each peptide were normalized relative to the maximum value of all samples.
Results and Discussion
Mutation of sarA Results in a Distinct Exoproteome That Is Largely Defined by the Increased Production of Extracellular Proteases
Conditioned medium (CM) samples from LAC (wild-type), sarA, and sarA/protease mutants were resolved by SDS-PAGE and visualized using Coomassie staining (Figure 1). Each of the three strains showed distinctly different molecular mass profiles of exoproteins. Visual inspection of the Coomassie-stained gels indicated that the sarA mutant produced an abundance of lower mass exoproteins relative to both wild-type and sarA/protease mutant. The sarA mutant is known to have increased levels of extracellular protease activity relative to wild-type,9,11,30 which is consistent with the observed lower mass proteins observed for the sarA mutant. The sarA/protease mutant has deletions of the ten known S. aureus extracellular proteases (aureolysin, sspA, sspB, scpA, splA-F), which appeared to partially rescue the exoprotein profile observed for the sarA mutant.
Figure 1.
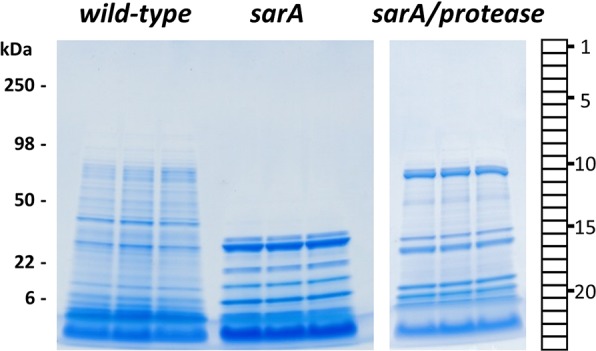
Exoproteomes from wild-type (LAC), sarA, and sarA/protease strains of S. aureus have distinct molecular masses. An equal volume of CM from each strain was resolved by one-dimensional SDS-PAGE and visualized by Coomassie staining. Strains were analyzed in biological triplicate. Each gel lane was sliced into 24 equiv sections. Protein in each gel slice was digested in-gel with trypsin and identified by high-resolution mass spectrometry.
To fully define differences in the exoproteome of these strains, each gel lane in Figure 1 was sectioned into 24 equiv bands and subjected to in-gel trypsin digestion prior to peptide analysis by high-resolution tandem mass spectrometry. A total of 18,171 unique peptides and 1,357 proteins were identified by high-resolution mass spectrometry of CM from all three strains (Table S1). Wild-type and sarA/protease mutant shared 96% of the proteins identified, indicating that these strains had similar exoproteomes in terms of total proteins present irrespective of their relative abundance. In contrast, the sarA mutant only shared 36% overlap of exoproteins identified when compared to the wild-type and sarA/protease mutant, demonstrating that the exoproteome content of sarA was distinct from the other strains. The total number of exoproteins detected in wild-type, sarA/protease, and sarA mutants were 1332, 1321, and 487, respectively. The total number of exoproteins supports the similarity of the wild-type and sarA/protease exoproteomes relative to the sarA mutant. The lower number of exoproteins detected in the sarA mutant, and the similar content of the wild-type and sarA/protease strains, support the role of sarA as a repressor of extracellular protease production. These results also suggest that exoproteins in the sarA mutant can be extensively cleaved to the point they are no longer present in the molecular mass range of the gel.
Spectral counts for each exoprotein in each gel band were used to measure the relative levels of detected exoproteins. A dual approach was used to comprehensively identify significantly cleaved exoproteins in a sarA-dependent, sarA-mediated protease-dependent and/or a sarA-mediated protease-independent fashion. The dual approach consisted of a total proteoform analysis that used total spectral counts for a given exoprotein in the entire gel lane and a full-length protein analysis that used spectral counts in a 3-gel band window centered on the gel band representing the full-length protein. Exoproteins present at different levels in pairwise comparison of strains were identified by a Student’s t test with significance defined as p < 0.05 and were visualized by hierarchical cluster heat map (Figure S1). The heat maps clearly show similarity between the CM from wild-type and sarA/protease mutant as well as differences in the sarA mutant.
Identification of Exoproteins Cleaved in a sarA-Mediated Manner through Extracellular Protease-Dependent or -Independent Mechanisms
Volcano plots were used to visualize changes in exoprotein abundance using the total proteoform and full-length protein quantitation approaches (Figure 2). Student’s t test p-values and fold changes were calculated for the total proteoform (Figure 2A–C) and for the full-length protein methods (Figure 2D–F). Three pairwise strain comparisons were analyzed to identify exoproteins cleaved in sarA-dependent (wild-type vs sarA), sarA-mediated protease-dependent (sarA/protease vs sarA), and sarA-mediated protease-independent (sarA/protease vs wild-type) fashions. The first (wild-type vs sarA mutant) was intended to identify exoproteins that were decreased in the sarA mutant relative to the parent strain irrespective of the mechanism involved (Figure 2A and D). The second (sarA/protease vs sarA) was intended to identify exoproteins cleaved by the increased production of extracelluar proteases in the sarA background (Figure 2B,E). The third (sarA/protease vs wild-type) was intended to identify exoproteins whose abundance was decreased in a sarA-mediated protease-independent manner (Figure 2C,F). In all volcano plots, exoproteins in which the abundance was reduced to a statistically significant decreased amount in the sarA mutant compared to wild-type, sarA mutant compared to the sarA/protease mutant, or wild-type compared to the sarA/protease mutant are shown as red dots in the upper right quadrant, whereas those that were present in increased amounts for the aforementioned comparisons are shown as blue circles in the upper left quadrant.
Figure 2.

Total proteoform and full-length protein data analysis methods identify exoproteins as sarA-mediated and extracellular protease dependent or independent. Volcano plots are shown using the total proteoform (A–C) and full-length protein (D–F) approaches for measuring abundance of exoproteins. Strains compared are indicated below the x-axis. Volcano plots were generated based on fold-change of protein levels using the averaged spectral counts from biological triplicates. The x-axis indicates a log2 fold-change, and the y-axis indicates −log10p-value based on Student’s t test. The horizontal line indicates a p-value < 0.1, and the vertical lines represent a fold-change >1.5. In all plots, exoproteins in which the abundance was reduced to a statistically significant degree in sarA (Figure 3A, B, D, and E) or wild-type (Figure 3C,F) are shown as red dots in the upper right quadrant, whereas those that were present in increased amounts are shown as blue circles in the upper left quadrant. Black dots indicate proteins for which differences in abundance were not statistically significant. The top left corner also indicates how many of the 10 possible extracellular proteases were detected.
The total proteoform and full-length protein methods identified 1104 and 1092 exoproteins, respectively, cleaved upon sarA deletion compared to wild-type (Figure 2A). The two methods of data analysis found 1066 of the same proteins, but the total proteoform and full-length protein methods found 38 and 26 unique exoproteins cleaved, respectively (Figure S2A). The comparison of wild-type and sarA mutant exoproteomic data also revealed a smaller number of exoproteins that were increased in levels upon mutation of sarA (Figures 2A,D, blue circles). Of the 22 exoproteins found to be present in increased levels using the total proteoform approach, nine were extracellular proteases known to be overexpressed upon deletion of sarA (Figure 2A). Of the 17 exoproteins found to be present in increased levels using the full-length protein method of data analysis, seven were extracellular proteases known to be overexpressed upon deletion of sarA (Figure 2D).
Exoproteins that were cleaved in a sarA-mediated, protease-dependent manner were identified by comparing sarA/protease and sarA mutant exoproteomic data. Exoproteins that are cleaved as a function of extracellular protease overexpression resulting from sarA deletion are highlighted in red for both the total proteoform (Figure 2B) and full-length protein (Figure 2E) methods of data analysis. The total proteoform and full-length protein methods identified 1017 and 968 sarA-mediated and protease-dependent exoproteins, respectively. The two methods of data analysis found 943 of the same proteins, but the total proteoform and full-length protein methods found 74 and 25 unique exoproteins cleaved, respectively (Figure S2B).
Exoproteins that were decreased in abundance in a sarA-mediated protease-independent manner were identified by comparing sarA/protease mutant and wild-type exoproteomic data. Exoproteins that are decreased in abundance independently of extracellular protease overexpression resulting from sarA deletion are highlighted in red for both the total proteoform (Figure 2C) and full-length protein (Figure 2F) methods of data analysis. The total proteoform and full-length protein methods identified 120 and 104 sarA-mediated protease-independent exoproteins, respectively. The two methods of data analysis found 86 of the same proteins, but the total proteoform and full-length protein methods found 34 and 18 unique exoproteins cleaved, respectively (Figure S2C).
Validation of Exoproteins Decreased in sarA-Mediated Extracellular Protease-Dependent and -Independent Mechanisms
Three well-studied exoproteins were selected for data confirmation, protein A (Spa), alpha-toxin, and thermonuclease (Nuc), all of which are implicated in the pathogenesis of S. aureus infection.31−33Figure 3A shows Spa is heavily cleaved in the sarA deletion background relative to wild-type and sarA/protease mutant. Peak levels of Spa were detected in band 12 in wild-type and sarA/protease mutant, whereas very low levels were present in the sarA mutant background. Note that the spectral counting level of Spa was not statistically different between wild-type and sarA/protease mutant. An immunoblot to Spa confirmed the spectral counting results as deletion of sarA results in a loss of the full-length Spa, whereas deletion of proteases in a sarA background recovered levels of full-length Spa (Figure 3B). These data support the extracellular presence of Spa being sarA-mediated and extracellular protease dependent. Spa was also shown to be decreased in a sarA-mediated fashion based on spectral count analysis (Figure 2A,D). The total proteoform method of data analysis found the extracellular presence of Spa to be sarA-mediated/protease-dependent and not sarA-mediated/protease-independent (Figure 2B,C). The full-length protein method agreed with the total proteoform method (Figure 2E,F). Because wild-type does produce extracellular proteases, the absence of cleaved Spa products in the sarA/protease mutant suggests that the loss of Spa in the sarA background is in fact generated via protease-mediated degradation of Spa. Taking this into account, it seems reasonable to conclude that mutation of sarA has relatively little direct transcriptional impact on the production of Spa. This is in contrast to previous reports concluding that sarA represses expression from the spa promoter.34
Figure 3.

Validation of exoproteins decreased in sarA-mediated extracellular protease-dependent and -independent mechanisms. The amounts and mass distributions of Spa (A), alpha-toxin (C), and NucB (E) were visualized by plotting the total spectral counts averaged across three biological replicates as a function of gel band. Spectral counts for the single unique NucB peptide (70-SGSEDPTVYSATSTK) were used for panel E. The vertical lines indicate the 3-band window used for data analysis in the full-length protein method. Immunoblot for Spa (B), alpha-toxin (D), and NucB (F) verified the respective spectral counting data. Shown is a representative immunoblot from biological triplicates. Controls for immunoblots are purified Spa that is known to migrate at slightly lower molecular mass relative to in vivo expressed Spa (B), purified alpha-toxin protein (D), and a control strain that does not express NucB (F). NucA is a proteolytically processed version of NucB (F).
Figure 3C shows alpha-toxin is cleaved in the sarA mutant background to a lower molecular mass species relative to wild-type and sarA/protease mutant. Similar peak levels of alpha-toxin were detected in band 15 in wild-type and sarA/protease mutant, whereas decreased levels were present at a lower molecular mass in the sarA deletion background. An immunoblot to alpha-toxin confirmed the spectral counting results as deletion of sarA produced a lower molecular mass species present at reduced amounts, whereas deletion of proteases in the sarA deletion background recovered alpha-toxin to wild-type levels (Figure 3D). Like Spa, full-length alpha toxin was restored in the sarA/protease mutant to levels comparable to those observed in wild-type, suggesting again that the primary determinant of the decreased accumulation of alpha-toxin in this sarA mutant is protease-mediated degradation rather than reduced transcription and/or mRNA stability. These data support the extracellular presence of alpha-toxin being sarA-mediated and extracellular protease dependent. Like Spa, alpha-toxin was shown to be decreased in a sarA-mediated fashion based on spectral count analysis (Figure 2A,D). The total proteoform method of data analysis found the extracellular presence of alpha-toxin to be sarA-mediated/protease-dependent and not sarA-mediated/protease-independent (Figure 2B,C). The full-length protein method agreed with the total proteoform method (Figure 2E,F).
Nuc is known to exist as a full-length protein (NucB) and in a protease-mediated processed form (NucA) under steady state wild-type conditions; accordingly, each proteoform will contain numerous overlapping tryptic peptides and therefore share many spectral counts.33 The larger molecular mass NucB does contain a single unique tryptic peptide (70-SGSEDPTVYSATSTK) that was used to determine levels of NucB independent of NucA. Analysis of spectral count data for NucB showed peak levels in band 18 under wild-type conditions, but the sarA/protease mutant induced elevated levels of NucB significantly beyond wild-type levels (Figure 3E). Deletion of sarA alone resulted in the loss of NucB. Figure 3F confirms NucB is cleaved in the sarA deletion background relative to wild-type and sarA/protease mutant but is restored beyond wild-type levels in the protease- and sarA-deficient background. These data suggest the presence of NucB is sarA-mediated and both extracellular protease-dependent and -independent. This is consistent with the spectral count data indicating that NucB was decreased in a sarA-mediated fashion (Figure 2A,D). Both the total proteoforms and full-length protein methods found the extracellular presence of NucB to be sarA-mediated/protease-dependent and sarA-mediated/protease-independent (Figure 2B,C,E,F).
Exoproteins Uniquely Identified by the Full-Length Exoprotein Data Analysis Approach Eliminate False Negatives Arising from Stable Proteolytic Products of Exoproteins
The total proteoform method of data analysis does not take into account that large, stable proteolytic products from a given exoprotein could result in false negatives when quantifying by total proteoforms. The full-length protein data analysis method used in this study provides for identification of these false negatives. Figure 4 shows two examples of this scenario for the proteins IsdC and ClfA, which are both implicated in the pathogenesis of S. aureus infection.35−37 Analysis of spectral count data for these proteins shows that sarA mutation results in the presence of abundant, large proteolytic fragments, whereas the sarA/protease mutant restores levels to wild-type (Figure 4A,B). Quantitative PRM mass spectrometry verified that the sarA mutation results in the presence of large proteolytic fragments of both IsdC and ClfA (Figure 4C,D). The total proteoform method of data analysis did not identify IsdC or ClfA as sarA-mediated and extracellular protease-dependent (Figure 2A,B), whereas the full-length protein method correctly identified both as sarA-mediated and extracellular protease dependent (Figure 2D,E). Thus, the full-length method can identify false negatives from the total proteoforms approach. Conversely, these results illustrate that, to the extent that the total proteoform approach provides for the identification of stable proteolytic fragments, it can provide information about protein production that would not be evident using the full-length protein method.
Figure 4.

Full-length protein method of data analysis identifies false negatives in the total proteoform approach. The amount and mass distribution of IsdC (A) and ClfA (B) in each strain was visualized by plotting the total spectral counts averaged across three biological replicates as a function of gel band. The vertical lines indicate the 3-band window used for data analysis in the full-length protein method. PRM mass spectrometry for IsdC (C) and ClfA (D) verifies panels A and B, respectively. Peptides used for PRM assays are indicated. Relative peptide intensity is shown for each gel section. isdC and clfA deletion strains were used as controls.
Figure 5 illustrates a more complicated example for the LukS protein that again highlights the utility of the dual method for data analysis, which in this case could be validated by Western blot data owing to the availability of an appropriate antibody. Like all of the proteins discussed above, LukS is also implicated in the pathogenesis of S. aureus infection, particularly in the context of USA300 isolates like LAC.31,38Figure 5 shows LukS to be lost in both sarA-mediated, protease-dependent and -independent pathways. Spectral count data revealed that peak levels of LukS are identified in band 15 under wild-type conditions, and deletion of proteases in a sarA background significantly increased levels of full-length LukS (Figure 5A). Interestingly, deletion of sarA alone also slightly increased levels of full-length LukS but also increased levels of multiple degradation products of LukS. Immunoblot of LukS confirmed the spectral counting results as deletion of sarA alone produced full-length and multiple cleaved versions of LukS, whereas deletion of proteases in the sarA deletion background provided for increased levels of full-length LukS (Figure 5B). LukS was shown to be increased in a sarA-mediated fashion as assessed by proteomic analysis (Figure 2A,D). The total proteoform method of data analysis found the extracellular presence of LukS to be sarA-mediated/protease-independent (Figure 2B,C). The full-length protein method correctly found the extracellular presence of LukS to be sarA-mediated/protease-dependent and sarA-mediated/protease-independent (Figure 2E,F).
Figure 5.
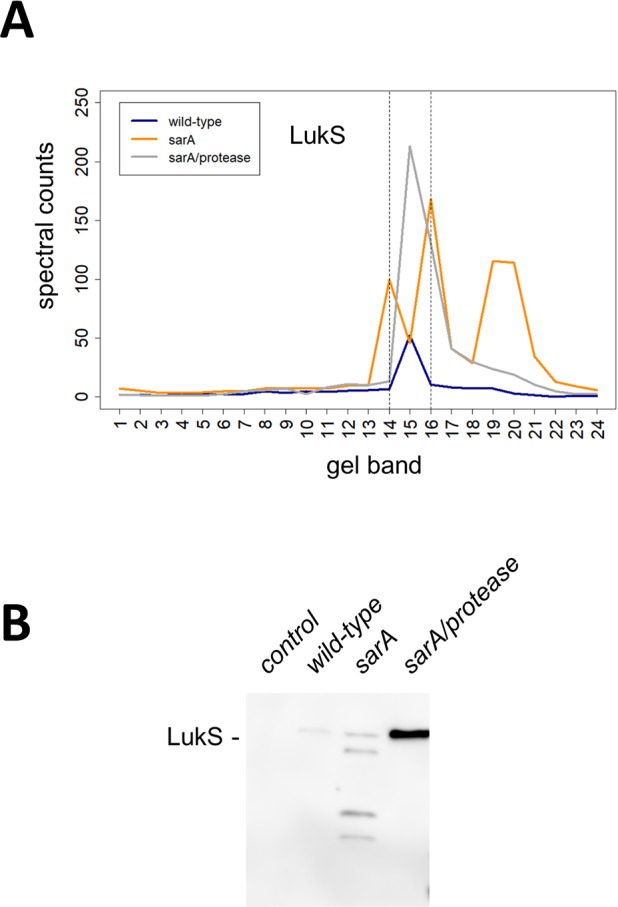
The extracellular presence of LukS is sarA-mediated and both extracellular protease dependent and independent. (A) The amount and mass distribution of LukS in each strain was visualized by plotting the total spectral counts averaged across three biological replicates as a function of gel band. The vertical lines indicate the 3-band window used for data analysis in the full-length protein method. (B) Immunoblot for LukS in each strain verifies panel A. Control is a strain that does not express LukS. Shown is a representative immunoblot from biological triplicates.
Conclusions
The sarA locus has been shown to play a key role in the pathogenesis of S. aureus.3,9,10 Exploiting this information to identify important virulence factors under the regulatory control of sarA is complicated by the fact that SarA impacts protein production at the level of both transcription and mRNA stability.3−6,46 Moreover, to some degree these regulatory functions differ in a strain-dependent manner.8 Mutation of sarA has also been shown to result in the increased production of extracellular proteases, and this has been directly correlated with reduced biofilm formation, reduced accumulation of multiple extracellular and surface-associated virulence factors, and reduced virulence in multiple animal models of infection.7,9−11,25,26 This suggests that a primary role of sarA is to limit the production of extracellular proteases. Additionally, the impact of increased protease production is apparent in diverse clinical isolates of S. aureus.10,11,47 This suggests that using a proteomics approach to fully define the impact of the increased protease production on the phenotype of clinically relevant sarA mutants has tremendous potential with respect to identifying key virulence factors that contribute to the pathogenesis of S. aureus.
The utility of using high-resolution proteomics to examine exoproteomes in pathogens and other biological systems has expanded in recent years.39,40 Several proteomic analyses of S. aureus have been performed to identify virulence factors using one-dimensional gel electrophoresis,41−44 two-dimensional gel electrophoresis,45−48 and quantitative isobaric tagging;49 however, these studies did not focus on extracellular protease activity. A recent proteomic study sought to identify the extracellular proteome in 14 clinical isolates of S. aureus.42 In addition to exploring the extracellular proteome content in these clinical isolates, the authors used a one-dimensional gel electrophoresis method to assign the mass of the extracellular protein based on migration in the gel and compared it with the predicted mass based on the amino acid sequence. This nice study revealed five percent of the proteins did not migrate in the gel as predicted but appeared to have undergone proteolytic cleavage.42 A limitation of such an approach is that steady-state levels of proteins in the wild-type scenario do not necessarily migrate as predicated on one-dimensional gels and may exist in processed states. Additionally, little mechanistic insight can be extracted from an analysis of clinical isolates as a comparative analysis of genetic variants is needed to elucidate the in vivo mechanism.
In previously reported studies, we used a one-dimensional gel electrophoresis proteomic approach that measured total proteoforms to define the impact of mutating sarA, and the consequent production of increased amounts of extracellular proteases on the LAC exoproteome.9 This approach was limited as it did not provide for the distinction of exoprotein levels regulated by sarA in a protease-independent as well as -dependent manner and did not take into account that large, stable proteolytic products from a given exoprotein could result in false negatives. In this report, we address the aforementioned issues using a dual exoproteomic approach that targets both total proteoforms and full-length exoproteins.
Application of the dual proteomic approach provided for complementary and unique information on the levels of exoproteins. Quantitative proteomic comparisons of sarA vs sarA/protease identified exoproteins that were cleaved in a sarA-mediated and extracellular protease-dependent manner, and comparisons of sarA/protease vs LAC identified those decreased in a sarA-mediated and extracellular protease-independent manner (Figure 2). The utility of these comparisons for distinguishing sarA-mediated extracellular protease-dependent and -independent mechanisms of regulation was confirmed using Spa, alpha-toxin, and NucB (Figure 3). Furthermore, IsdC, ClfA, and LukS were selected to demonstrate that exoproteins uniquely identified by the full-length data analysis approach eliminated false negatives in the total proteoforms approach (Figures 4 and 5). The current analysis of sarA-mediated exoproteomes provides a more in-depth measurement, 1042 proteins regulated in a sarA-mediated and protease-dependent manner (Figure S2B), of the exoproteins regulated by sarA compared to our previous report that identified 253 sarA-mediated and protease-dependent proteins.9
An advantage of the dual approach presented here is that one can identify unique proteins from the total proteoform method, unique proteins from the full-length protein method, and shared proteins from both methods. The unique, method-specific protein identifications help overcome false negatives, which provides for more complete and robust characterization of exoproteomes. The shared protein identifications provide for complementary data that increases rigor and confidence. Taken collectively, the dual quantification method presented provides for a more comprehensive and robust analysis of exoproteomes that can be applied to a variety of organisms beyond S. aureus.
Acknowledgments
This study was supported by the National Institutes of Health (R01AI119380, R21DA041822, P20GM121293, UL1TR000039, P20GM103625, S10OD018445, and P20GM103429). The authors acknowledge the University of Arkansas for Medical Sciences Proteomics Facility, Arkansas Children’s Research Institute Developmental Bioinformatics Core, and the Arkansas Biosciences Institute.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.8b00288.
Unsupervised hierarchical clustering of exoproteins from S. aureus wild-type (LAC), sarA, and sarA/protease mutants showing distinct profiles and total proteoform and full-length protein methods providing complementary and unique information on the presence of exoproteins (PDF)
Mass spectrometric data used to identify significant proteins (XLSX)
All significant proteins with p < 0.1 and a fold change < 1.5 (the top left quadrant of the Volcano plots from Figure 2) for each comparison in the total proteoform and full-length protein methods of analysis (XLSX)
All significant proteins with p < 0.1 and a fold change > 1.5 (the top right quadrant of the Volcano plots from Figure 2) for each comparison in the total proteoform and full-length protein methods of analysis(XLSX)
Significant proteins that were uniquely identified by both the total proteoform (total counts) and the full-length protein (sum of 3 bands) methods (XLSX)
Author Contributions
⊥ S.D.B., A.J.L., and K.E.B. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Lowy F. D. N. Engl. J. Med. 2011, 364 (21), 1987–1990. 10.1056/NEJMp1100251. [DOI] [PubMed] [Google Scholar]
- Priest N. K.; Rudkin J. K.; Feil E. J.; van den Elsen J. M. H.; Cheung A.; Peacock S. J.; Laabei M.; Lucks D. A.; Recker M.; Massey R. C. From genotype to phenotype: can systems biology be used to predict Staphylococcus aureus virulence?. Nat. Rev. Microbiol. 2012, 10, 791. 10.1038/nrmicro2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung A. L.; Nishina K. A.; Trotonda M. P.; Tamber S. The SarA protein family of Staphylococcus aureus. Int. J. Biochem. Cell Biol. 2008, 40 (3), 355–361. 10.1016/j.biocel.2007.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison J. M.; Anderson K. L.; Beenken K. E.; Smeltzer M. S.; Dunman P. M. The Staphylococcal Accessory Regulator, SarA, is an RNA-Binding Protein that Modulates the mRNA Turnover Properties of Late-Exponential and Stationary Phase Staphylococcus aureus Cells. Front. Cell. Infect. Microbiol. 2012, 2, 26. 10.3389/fcimb.2012.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts C.; Anderson K. L.; Murphy E.; Projan S. J.; Mounts W.; Hurlburt B.; Smeltzer M.; Overbeek R.; Disz T.; Dunman P. M. Characterizing the Effect of the Staphylococcus aureus Virulence Factor Regulator, SarA, on Log-Phase mRNA Half-Lives. J. Bacteriol. 2006, 188 (7), 2593–2603. 10.1128/JB.188.7.2593-2603.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterba K. M.; Mackintosh S. G.; Blevins J. S.; Hurlburt B. K.; Smeltzer M. S. Characterization of Staphylococcus aureus SarA Binding Sites. J. Bacteriol. 2003, 185 (15), 4410–4417. 10.1128/JB.185.15.4410-4417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolar S. L.; Ibarra J. A.; Rivera F. E.; Mootz J. M.; Davenport J. E.; Stevens S. M.; Horswill A. R.; Shaw L. N. Extracellular proteases are key mediators of Staphylococcus aureus virulence via the global modulation of virulence-determinant stability. MicrobiologyOpen 2013, 2 (1), 18–34. 10.1002/mbo3.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinska A. K.; Beenken K. E.; Joo H.-S.; Mrak L. N.; Griffin L. M.; Luong T. T.; Lee C. Y.; Otto M.; Shaw L. N.; Smeltzer M. S. Defining the Strain-Dependent Impact of the Staphylococcal Accessory Regulator (sarA) on the Alpha-Toxin Phenotype of Staphylococcus aureus. J. Bacteriol. 2011, 193 (12), 2948–2958. 10.1128/JB.01517-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinska A. K.; Beenken K. E.; Mrak L. N.; Spencer H. J.; Post G. R.; Skinner R. A.; Tackett A. J.; Horswill A. R.; Smeltzer M. S. sarA-mediated repression of protease production plays a key role in the pathogenesis of Staphylococcus aureus USA300 isolates. Mol. Microbiol. 2012, 86 (5), 1183–1196. 10.1111/mmi.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughran A. J.; Gaddy D.; Beenken K. E.; Meeker D. G.; Morello R.; Zhao H.; Byrum S. D.; Tackett A. J.; Cassat J. E.; Smeltzer M. S. Impact of sarA and Phenol-Soluble Modulins on the Pathogenesis of Osteomyelitis in Diverse Clinical Isolates of Staphylococcus aureus. Infect. Immun. 2016, 84 (9), 2586–2594. 10.1128/IAI.00152-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang L. H.; Cassat J. E.; Shaw L. N.; Beenken K. E.; Smeltzer M. S. Factors Contributing to the Biofilm-Deficient Phenotype of Staphylococcus aureus sarA Mutants. PLoS One 2008, 3 (10), e3361. 10.1371/journal.pone.0003361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklevitz J.; Harris L. K. Improved Annotations of 23 Differentially Expressed Hypothetical Proteins in Methicillin Resistant S.aureus. Bioinformation 2017, 13 (4), 104–110. 10.6026/97320630013104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae T.; Schneewind O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 2006, 55 (1), 58–63. 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Kavanaugh J. S.; Thoendel M.; Horswill A. R. A role for type I signal peptidase in Staphylococcus aureus quorum sensing. Mol. Microbiol. 2007, 65 (3), 780–798. 10.1111/j.1365-2958.2007.05830.x. [DOI] [PubMed] [Google Scholar]
- Reed S. B.; Wesson C. A.; Liou L. E.; Trumble W. R.; Schlievert P. M.; Bohach G. A.; Bayles K. W. Molecular Characterization of a Novel Staphylococcus aureus Serine Protease Operon. Infect. Immun. 2001, 69 (3), 1521–1527. 10.1128/IAI.69.3.1521-1527.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormann M. E.; Reichmann N. T.; Malone C. L.; Horswill A. R.; Grundling A. Proteolytic Cleavage Inactivates the Staphylococcus aureus Lipoteichoic Acid Synthase. J. Bacteriol. 2011, 193 (19), 5279–5291. 10.1128/JB.00369-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrum S.; Avaritt N. L.; Mackintosh S. G.; Munkberg J. M.; Badgwell B. D.; Cheung W. L.; Tackett A. J. A quantitative proteomic analysis of FFPE melanoma. J. Cutaneous Pathol. 2011, 38 (11), 933–936. 10.1111/j.1600-0560.2011.01761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrum S. D.; Larson S. K.; Avaritt N. L.; Moreland L. E.; Mackintosh S. G.; Cheung W. L.; Tackett A. J. Quantitative Proteomics Identifies Activation of Hallmark Pathways of Cancer in Patient Melanoma. J. Proteomics Bioinf. 2013, 6 (3), 043–050. 10.4172/jpb.1000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Q.; Byrum S. D.; Moreland L. E.; Mackintosh S. G.; Kannan A.; Lin Z.; Morgan M.; Stack B. C.; Cornelius L. A.; Tackett A. J.; Gao L. A Proteomic Study of Human Merkel Cell Carcinoma. J. Proteomics Bioinf. 2013, 6, 275–282. 10.4172/jpb.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udensi U. K.; Tackett A. J.; Byrum S.; Avaritt N. L.; Sengupta D.; Moreland L. W.; Tchounwou P. B.; Isokpehi R. D. Proteomics-Based Identification of Differentially Abundant Proteins from Human Keratinocytes Exposed to Arsenic Trioxide. J. Proteomics Bioinf. 2014, 7 (7), 166–178. 10.4172/jpb.1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii A.; Keller A.; Kolker E.; Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75 (17), 4646–58. 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- Vizcaino J. A.; Csordas A.; del-Toro N.; Dianes J. A.; Griss J.; Lavidas I.; Mayer G.; Perez-Riverol Y.; Reisinger F.; Ternent T.; Xu Q.-W.; Wang R.; Hermjakob H. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44 (Database issue), D447–D456. 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zybailov B.; Mosley A. L.; Sardiu M. E.; Coleman M. K.; Florens L.; Washburn M. P. Statistical Analysis of Membrane Proteome Expression Changes in Saccharomyces cerevisiae. J. Proteome Res. 2006, 5 (9), 2339–2347. 10.1021/pr060161n. [DOI] [PubMed] [Google Scholar]
- Oliveros J.VENNY, an interactive tool for comparing lists with Venn diagrams. citeulike-article-id:6994833.
- Weiss E. C.; Zielinska A.; Beenken K. E.; Spencer H. J.; Daily S. J.; Smeltzer M. S. Impact of sarA on Daptomycin Susceptibility of Staphylococcus aureus Biofilms In Vivo. Antimicrob. Agents Chemother. 2009, 53 (10), 4096–4102. 10.1128/AAC.00484-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson A. C.; Russell J. D.; Bailey D. J.; Westphall M. S.; Coon J. J. Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Mol. Cell. Proteomics 2012, 11 (11), 1475–1488. 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski J. R.; Zougman A.; Nagaraj N.; Mann M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359. 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; Chambers M.; Finney G. L.; Frewen B.; Kern R.; Tabb D. L.; Liebler D. C.; MacCoss M. J., Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2009, 26, ( (7), ), 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H.ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, 2016. [Google Scholar]
- Mrak L. N.; Zielinska A. K.; Beenken K. E.; Mrak I. N.; Atwood D. N.; Griffin L. M.; Lee C. Y.; Smeltzer M. S. saeRS and sarA Act Synergistically to Repress Protease Production and Promote Biofilm Formation in Staphylococcus aureus. PLoS One 2012, 7 (6), e38453. 10.1371/journal.pone.0038453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seilie E. S.; Bubeck Wardenburg J. Staphylococcus aureus pore-forming toxins: The interface of pathogen and host complexity. Semin. Cell Dev. Biol. 2017, 72, 101–116. 10.1016/j.semcdb.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Liu X.; Dou C.; Cao Z.; Liu C.; Dong S.; Fei J. Staphylococcal protein A promotes osteoclastogenesis through MAPK signaling during bone infection. J. Cell. Physiol. 2017, 232 (9), 2396–2406. 10.1002/jcp.25774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson M. E.; Nygaard T. K.; Ackermann L.; Watkins R. L.; Zurek O. W.; Pallister K. B.; Griffith S.; Kiedrowski M. R.; Flack C. E.; Kavanaugh J. S.; Kreiswirth B. N.; Horswill A. R.; Voyich J. M. Staphylococcus aureus Nuclease Is an SaeRS-Dependent Virulence Factor. Infect. Immun. 2013, 81 (4), 1316–1324. 10.1128/IAI.01242-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Stewart G. C. Regulatory Elements of the Staphylococcus aureus Protein A (Spa) Promoter. J. Bacteriol. 2004, 186 (12), 3738–3748. 10.1128/JB.186.12.3738-3748.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke S. R.; Mohamed R.; Bian L.; Routh A. F.; Kokai-Kun J. F.; Mond J. J.; Tarkowski A.; Foster S. J. The Staphylococcus aureus Surface Protein IsdA Mediates Resistance to Innate Defenses of Human Skin. Cell Host Microbe 2007, 1 (3), 199–212. 10.1016/j.chom.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Palmqvist N.; Josefsson E.; Tarkowski A. Clumping factor A-mediated virulence during Staphylococcus aureus infection is retained despite fibrinogen depletion. Microbes Infect. 2004, 6 (2), 196–201. 10.1016/j.micinf.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Sharp K. H.; Schneider S.; Cockayne A.; Paoli M. Crystal Structure of the Heme-IsdC Complex, the Central Conduit of the Isd Iron/Heme Uptake System in Staphylococcus aureus. J. Biol. Chem. 2007, 282 (14), 10625–10631. 10.1074/jbc.M700234200. [DOI] [PubMed] [Google Scholar]
- Jiang B.; Wang Y.; Feng Z.; Xu L.; Tan L.; Zhao S.; Gong Y.; Zhang C.; Luo X.; Li S.; Rao X.; Peng Y.; Xie Z.; Hu X. Panton-Valentine Leucocidin (PVL) as a Potential Indicator for Prevalence, Duration, and Severity of Staphylococcus aureus Osteomyelitis. Front. Microbiol. 2017, 8, 2355. 10.3389/fmicb.2017.02355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armengaud J.; Christie-Oleza J. A.; Clair G.; Malard V.; Duport C. Exoproteomics: exploring the world around biological systems. Expert Rev. Proteomics 2012, 9 (5), 561–575. 10.1586/epr.12.52. [DOI] [PubMed] [Google Scholar]
- Armengaud J.; Duport C. Exoproteomics of Pathogens: Analysis of Toxins and Other Virulence Factors by Proteomics. Methods Enzymol. 2017, 586, 211–227. 10.1016/bs.mie.2016.09.033. [DOI] [PubMed] [Google Scholar]
- Jones R. C.; Deck J.; Edmondson R. D.; Hart M. E. Relative Quantitative Comparisons of the Extracellular Protein Profiles of Staphylococcus aureus UAMS-1 and Its sarA, agr, and sarA agr Regulatory Mutants Using One-Dimensional Polyacrylamide Gel Electrophoresis and Nanocapillary Liquid Chromatography Coupled with Tandem Mass Spectrometry. J. Bacteriol. 2008, 190 (15), 5265–5278. 10.1128/JB.00383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. S.; Siggins M. K.; Gierula M.; Pichon B.; Turner C. E.; Lynskey N. N.; Mosavie M.; Kearns A. M.; Edwards R. J.; Sriskandan S. Identification of commonly expressed exoproteins and proteolytic cleavage events by proteomic mining of clinically relevant UK isolates of Staphylococcus aureus. Microbial Genomics 2016, 2 (2), e000049. 10.1099/mgen.0.000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreisbach A.; Hempel K.; Buist G.; Hecker M.; Becher D.; Dijl J. M. v. Profiling the surfacome of Staphylococcus aureus. Proteomics 2010, 10 (17), 3082–3096. 10.1002/pmic.201000062. [DOI] [PubMed] [Google Scholar]
- Nickerson N. N.; Prasad L.; Jacob L.; Delbaere L. T.; McGavin M. J. Activation of the SspA Serine Protease Zymogen of Staphylococcus aureus Proceeds through Unique Variations of a Trypsinogen-like Mechanism and Is Dependent on Both Autocatalytic and Metalloprotease-specific Processing. J. Biol. Chem. 2007, 282 (47), 34129–34138. 10.1074/jbc.M705672200. [DOI] [PubMed] [Google Scholar]
- Bonar E.; Wojcik I.; Wladyka B. Proteomics in studies of Staphylococcus aureus virulence. Acta Biochim Polym. 2015, 62 (3), 367–81. 10.18388/abp.2015_1083. [DOI] [PubMed] [Google Scholar]
- Le Marechal C.; Jardin J.; Jan G.; Even S.; Pulido C.; Guibert J.-M.; Hernandez D.; Francois P.; Schrenzel J.; Demon D.; Meyer E.; Berkova N.; Thiery R.; Vautor E.; Le Loir Y. Staphylococcus aureus seroproteomes discriminate ruminant isolates causing mild or severe mastitis. Veterinary Research 2011, 42 (1), 35. 10.1186/1297-9716-42-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocsfalvi G.; Cacace G.; Cuccurullo M.; Serluca G.; Sorrentino A.; Schlosser G.; Blaiotta G.; Malorni A. Proteomic analysis of exoproteins expressed by enterotoxigenic Staphylococcus aureus strains. Proteomics 2008, 8 (12), 2462–2476. 10.1002/pmic.200700965. [DOI] [PubMed] [Google Scholar]
- Scherl A.; Francois P.; Bento M.; Deshusses J. M.; Charbonnier Y.; Converset V. r.; Huyghe A.; Walter N.; Hoogland C.; Appel R. D.; Sanchez J.-C.; Zimmermann-Ivol C. G.; Corthals G. L.; Hochstrasser D. F.; Schrenzel J. Correlation of proteomic and transcriptomic profiles of Staphylococcus aureus during the post-exponential phase of growth. J. Microbiol. Methods 2005, 60 (2), 247–257. 10.1016/j.mimet.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Lin M.-H.; Li C.-C.; Shu J.-C.; Chu H.-W.; Liu C.-C.; Wu C.-C. Exoproteome Profiling Reveals the Involvement of the Foldase PrsA in the Cell Surface Properties and Pathogenesis of Staphylococcus aureus. Proteomics 2018, 18 (5–6), e1700195. 10.1002/pmic.201700195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.