

We demonstrate quantitatively controlled delivery of DNA, mRNA, and protein into primary cells using nanostraw electroporation.
Abstract
Intracellular delivery of mRNA, DNA, and other large macromolecules into cells plays an essential role in an array of biological research and clinical therapies. However, current methods yield a wide variation in the amount of material delivered, as well as limitations on the cell types and cargoes possible. Here, we demonstrate quantitatively controlled delivery into a range of primary cells and cell lines with a tight dosage distribution using a nanostraw-electroporation system (NES). In NES, cells are cultured onto track-etched membranes with protruding nanostraws that connect to the fluidic environment beneath the membrane. The tight cell-nanostraw interface focuses applied electric fields to the cell membrane, enabling low-voltage and nondamaging local poration of the cell membrane. Concurrently, the field electrophoretically injects biomolecular cargoes through the nanostraws and into the cell at the same location. We show that the amount of material delivered is precisely controlled by the applied voltage, delivery duration, and reagent concentration. NES is highly effective even for primary cell types or different cell densities, is largely cargo agnostic, and can simultaneously deliver specific ratios of different molecules. Using a simple cell culture well format, the NES delivers into >100,000 cells within 20 s with >95% cell viability, enabling facile, dosage-controlled intracellular delivery for a wide variety of biological applications.
INTRODUCTION
Delivery of exogenous biomolecules such as mRNA, DNA, and proteins through the cell membrane and into the cytoplasm has become an essential step for fundamental research and clinical applications, including induced pluripotent stem cell (iPSC) reprogramming (1–6), tissue development (7, 8), intracellular gene editing, cancer therapy, and disease pathogenesis (9–12). Various mechanisms are available for delivering these species to large numbers of cells at once, most commonly viral vectors (13, 14), chemical carriers (13, 15, 16), and bulk electroporation (BEP) (13, 17). The efficacy of each method is assessed according to the cell types, cargoes, delivery efficiencies, and cell viabilities possible. Each of these methods is effective for particular cell types and cargoes, yet general methods for delivery with high efficiency and cell viability are still active areas of research (13).
Recently, demand for more sophisticated delivery tools has emerged as researchers found that different mRNA/protein quantities, delivery timing, and relative concentrations have marked impact on iPSC development (4, 18, 19) and CRISPR editing behavior (10, 20). Traditional delivery approaches rely on bulk stochastic processes, leading to broad dosage distributions and relatively poor temporal control. To address this, more intimate interfaces between the cells and the delivery reagent have been developed, including nanochannels (21), micropipettes (22), nanotip injectors (23, 24), and single-cell electroporation systems (25). These methods control the amount of reagent delivered yet require serial injection one cell at a time, limiting the number of cells transformed.
Previously, we reported the fabrication and testing of a nanostraw-electroporation system (NES) for efficient intracellular delivery and sampling (26, 27). In this approach, a common track-etched cell culture membrane is coated with an inorganic layer to form rigid straws through the polymer membrane, connecting the top and bottom surfaces (Fig. 1 and fig. S1). The polymer on the top surface is then etched away to reveal inorganic “nanostraws” (NS), with the diameter determined by the pore size (here, 150 nm) and the height by the amount of polymer removed (here, 1.5 to 2.5 μm). Pore diameters were well controlled, with 5% size variability as determined by scanning electron microscopy (SEM). An NS density of 1 × 108 pore/cm2 was chosen to have sufficient numbers of NS per cell, while avoiding higher densities where cells merely rest on top of a bed of NS. Devices are constructed by adhering the NS membrane to a circular tube, creating a cell culture well that can easily be transported to and from an incubator and hold a number of cells equivalent to a conventional polystyrene dish of the same diameter (Fig. 1, B to D, and fig. S2). Cells cultured onto these membranes interact strongly with the NS, forming intimate cell-straw contacts (26). Cells can be cultured for weeks without adverse effects and can be readily trypsinized and removed after delivery for further use.
Fig. 1. Design and operation of the NES.
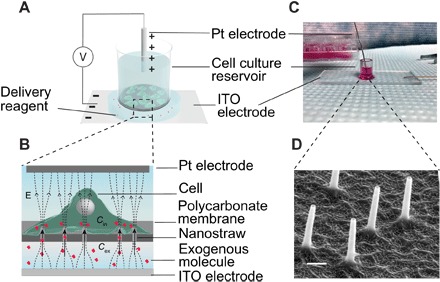
(A) Cells are cultured on the NS membrane in a well plate geometry. (B) The delivery reagent is placed under the bottom of the reservoir. An electric field is applied between the platinum and ITO electrode to deliver exogenous molecules into cells. (C) Schematic of NES delivery mechanism. The molecules beneath the NS membrane are electrophoretically injected into cells of interest through the NS. The delivered concentration (Cin) is in quadratic relationship with voltage intensity that is applied to the cells (V) and is proportional to the concentration of exogenous molecules (Cex) and the delivery duration. (D) SEM image of the NS protruding from the membrane. Scale bar, 300 nm.
Cargoes are delivered into the cells by placing the reagent in a buffer below the NS well and applying an electric field between the buffer and the cell media. Because of the tight interface formed between the cell membrane and NS, the electric field is localized on the tip of the NS, transiently permeabilizing the cell membrane near the tip. Simultaneously, the cargo is delivered through the NS at the same location, either by electrophoresis or by diffusion (Fig. 1B). By colocalizing the poration and the delivery, molecular cargoes are efficiently transported to and from the cell, with high cell viability. Previous results from NES on cell lines showed greater than 95% molecular delivery efficiency and >85% plasmid transfection with >95% cell viability (26).
Here, we apply this method for accurate dosage control for mRNA and a variety of proteins. We show that NES is a versatile and accurate delivery system for both cell lines and hard-to-transfect primary cells.
RESULTS AND DISCUSSION
Gene delivery and ratiometric control
mRNA delivery has become increasingly important to control transcription factor expression, to manipulate signaling cascades, and as a nonintegrating, “footprint-free” method for inducing protein expression (28–30). Here, we compared NS delivery to Lipofectamine (LFN), currently the most common method for mRNA, by delivery of mRNAs for two common fluorescent markers, green fluorescent protein (GFP) and mCherry (~922 nt). To make a fair comparison, we chose human embryonic kidney (HEK) 293 cells that are efficiently transfected with Lipofectamine 2000 (LFN 2000). In addition to expression analysis of a single mRNA, we demonstrate that it is possible to control the relative expression levels of these two different genes by controlling their relative concentrations in the cargo solution.
NES delivery was performed by culturing the cells onto NS for 24 hours and then placing the mRNA reagent solution on an indium tin oxide (ITO)–coated glass slide (Fig. 1, A to C). The 5-mm cell culture well was placed on top of the delivery buffer, electroporated for 20 s at 20 V, and then returned to the incubator. The positive electrode was placed above the cells to attract the negatively charged mRNA from the bottom buffer. Reversal of polarity led to no observable delivery, indicating that the mechanism is likely electrokinetic in nature. The amounts of GFP and mCherry mRNA in the buffer solution were varied between concentration ratios of 250:15.6 to 15.6:250 (Fig. 2). As a comparison, the same relative GFP/mCherry concentrations were delivered using LFN 2000 following the manufacturer’s instructions (Life Technologies). Representative images of the cells after NS delivery for each condition are shown in Fig. 2 (A and B), with histograms from flow cytometry analysis shown in Fig. 2 (C to E).
Fig. 2. Ratiometric cotransfection of GFP and mCherry mRNA by NES.
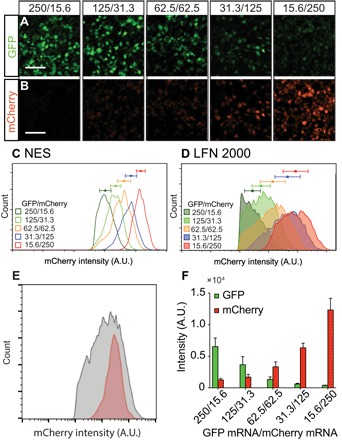
(A and B) Fluorescent microscopic images of HEK 293 cells after NES cotransfection with different concentration ratios of GFP (A) and mCherry (B) mRNA into HEK 293 cells with >70% transfection efficiency (scale bars, 500 μm). As the ratio of GFP and mCherry mRNA decreases, GFP and mCherry expression monotonically increases and decreases, respectively. (C and D) FACS histograms of mCherry expression for different GFP/mCherry mRNA delivery concentrations, showing correspondingly different expression levels using NES and LFN (error bars indicate SD of experimental triplicates). NES gives more than an order of magnitude sharper peaks than LFN [error bars indicate SD with n > 1000 in (D) and n > 5000 in (E)], indicating more uniform dosage control. A.U., arbitrary units. (E) Direct comparison of mCherry distribution for the two techniques (red, NES; gray, LFN). (F) GFP and mCherry expression levels as a function of their delivery concentrations [error bars indicate SD of experimental replicates (n = 3)].
Fluorescence-activated cell sorting (FACS) analysis of the GFP and mCherry expression following NES delivery increased with reagent concentration (Fig. 2, A and B), indicating that cytosolically active mRNA is proportional to the mRNA amount used in the delivery buffer. Transfection efficiencies were 75 to 90% with a cell viability of >90% in all cases. The dosage distribution as measured by expression was well controlled, with SDs of 50 to 70% of the mean. In comparison, LFN 2000 expression had very broad expression distributions (Fig. 2D), with SDs of 130 to 190% of the mean values. The substantial overlap in expression levels between different reagent concentrations shows that control of active mRNA in the cytoplasm was relatively poor. A direct comparison of mCherry distribution for the two techniques is shown in Fig. 2E, showing the much tighter distribution and more accurate dosage using NES delivery.
The relative expression levels of the two different mRNAs could also be controlled by varying their relative concentrations in the NES delivery buffer. Figure 2F shows the GFP and mCherry expression levels as a function of their concentrations. The expression levels for each are linear with concentration (fig. S3), although the relative brightness of mCherry was higher than that of enhanced GFP (eGFP) at the equivalent concentration. The ratio between the two species was well controlled, for example, the eGFP/mCHerry expression ratio was 6.3 ± 1.89 for the 4:1 (125:31) ratio. Note that the ratiometric amounts were still consistent even when different total amounts of reagent were used (e.g., 250:15.6 had higher total mRNA concentration than the 62.5:62.5). These results show that both the absolute quantity of reagent delivered and the ratios between reagents could be defined using the NES system.
Characteristics of NES delivery
The NES mechanism has several unique delivery characteristics relative to LFN, viruses, or BEP. Since the NES mechanism is primarily physical in nature, the method may be less cell type specific than other transfection techniques. Previous studies using the NS platform for delivery into primary macrophages (31) showed high efficacy, as did cell sampling (27) for primary cells such as human iPSC-derived cardiomyocytes (hiPSC-CMs) and astrocytes. Here, we tested whether NES could efficiently transfect five different hard-to-transfect cell types: hiPSC-CMs, human embryonic stem cells (HSCs), human fibroblasts (HFs), mouse primary glia cells (MGs), and mouse primary neuron cells (MNs).
Cells of the five types were cultured in separate NS wells with specified cell culture media for 7 days before delivery. eGFP mRNA (100 ng/ml) was delivered by NES using the standard delivery protocol developed for HEK 293 cells (20 V, 20 s). Note that no complex or proprietary transfection reagents were necessary, and the delivery buffers in each case were their specified cell culture media. Figure 3 shows that transfection of these primary cell types was highly efficient (60 to 80% for all cell types with a narrow SD in expression level). Transfection was uniform across the entire well, simultaneously transfecting 10,000 to 100,000 cells in the 5-mm wells. These cells are known to be particularly difficult to deliver into with nonviral agents, indicating that the unique NES mechanism largely avoids cell type specificity.
Fig. 3. NES eGFP mRNA transfection into different primary cell types.
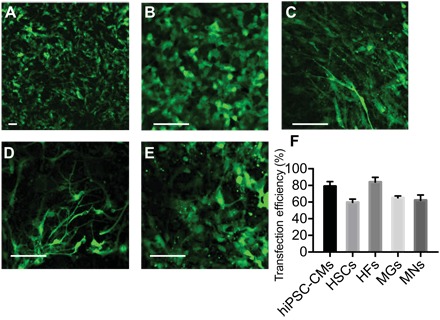
(A) hiPSC-CMs, (B) HSCs, (C) HFs, (D) MNs, and (E) MGs (scale bars, 50 μm). (F) NES transfection efficiencies of different types of difficult-to-transfect cells are more than 60% in all cases and as high as 85% [error bars indicate SD (n > 50)].
Expression via NES delivery is also expected to be faster than LFN or viral methods as the bare mRNA is injected directly into the cytoplasm, without additional endocytotic or viral unpackaging steps. To test this hypothesis, we compared eGFP expression kinetics of NES and LFN 2000 in HEK 293 cells. NES transfection was tested at both low (500 ng) and high (1500 ng) mRNA amounts using the standard delivery protocol (20 V, 20 s) and then replaced into the incubator for the prescribed time. To show that NES is not constrained to fluorescence microscopy analysis, we measured the GFP concentration at each time point by cell lysis in the well, followed by enzyme-linked immunosorbent assay (ELISA).
Detectable GFP expression was observed at 5 min after NES delivery (Fig. 4A), which indicates that the mRNA was quickly accessible for protein translation. Expression levels increased quadratically as a function of time after injection for both high and low amounts of mRNA. The reason for this functional dependence is not yet clear and may be biological in origin as no additional physical manipulation was performed. In comparison, no GFP signal was detected even 25 min after introducing LFN mixture to cells, showing much slower mRNA accessibility kinetics. The rate of mRNA availability is consistent with the observed temporal control for small-molecule delivery, which has been shown to be quite fast for the NS approach (32, 33).
Fig. 4. Characteristics of NES delivery.
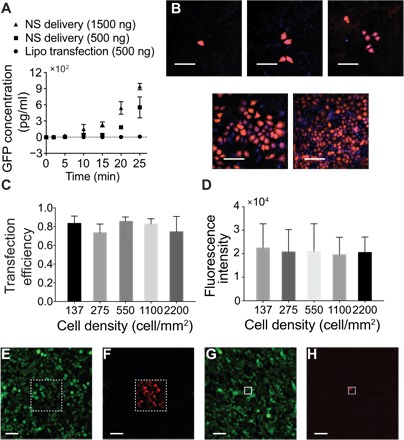
(A) mRNA expression kinetics for NES and LFN delivery. Expression is observed at 5 min for NES, indicating rapid cytosolic availability [error bars indicate SD (n = 3)]. (B) Fluorescent microscopic images show normal HEK 293 cell morphology after mRNA transfection for different cell densities (scale bars, 200 μm). (C) Transfection efficiencies for different cell culture densities were more than 75% in all cases, and no statistical significance was observed, showing that the method is consistent even for very different cell densities [error bars indicate SD of experimental replicates; P = 0.65, one-way analysis of variance (ANOVA)]. (D) Averaged mCherry expression intensity for different cell densities showed no statistically significant difference (error bars indicate SD (n = 8 to 53); P = 0.84, one-way ANOVA). (E to H) eGFP-expressing HeLa cells cultured on a patterned NS platform with square active areas of 200 μm by 200 μm [scale bars, 100 μm (E and F)] and 50 μm by 50 μm [scale bars, 100 μm (G and H)]. Only the cells cultured within the active areas with exposed NS expressed an mCherry signal (F and H) after delivery.
We also observed that NES mRNA transfection was unaffected by cell density. For many transfection methods, a tightly controlled cell density range is normally required to achieve high delivery efficiency, which can be problematic for rare cell types or patient-derived cells. To test NES performance, we cultured varying densities of HEK 293 cells ranging from 137 to 2200 cell/mm2 on the NS wells (Fig. 4, B to D). Cells spread out on the culture surface overnight, followed by NES delivery of mCherry mRNA. Expression intensity and delivery efficiency were measured fluorescently 8 hours after delivery (Fig. 4D). The average transfection efficiencies of different cell densities were all between 70 and 90% (Fig. 4C; SD, 10 to 15%). No statistically significant difference was observed (P > 0.01, one-way ANOVA), indicating that the delivery efficiency with NES is independent of cell density. No statistical difference was found between mCherry expression quantities (e.g., fluorescent intensity) of cells of different cell densities (Fig. 4D; P > 0.01, one-way ANOVA), which suggests that the dosage control characteristic is also not affected by cell density.
NES delivery can be patterned for a few cells or even a single cell without compromising cell-to-cell connectivity, which could be important for the study of cells that are sensitive to their environment and coculture conditions. As previously described (27), the active transfection regions of the NES platform could be defined by blocking the remaining NS membrane with photolithography-patterned polymers (Materials and methods). During delivery, only the cells that interface with NS in the selected regions will experience electric fields and molecular delivery, leaving cells in the blocked area unaffected. These cells should still experience the same culture environment and are free to physically and chemically communicate with neighboring cells. As a demonstration, GFP-expressing HeLa cells were cultured overnight on three different size access areas (Fig. 4, E to H, and figs. S4 and S5), of 200 μm by 200 μm, 100 μm by 100 μm, and 50 μm by 50 μm. The cells were then transfected with the cells with mCherry mRNA. As shown in Fig. 4 (E to H), cells uniformly expressed GFP, yet only the cells within the selected regions expressed mCherry. Expression efficiency within the selective areas was good: 45 of 51 cells (88%), 16 of 19 cells (84%), and 2 of 6 cells (30%) were transfected within the 200 μm by 200 μm, 100 μm by 100 μm, and 50 μm by 50 μm delivery regions, respectively. Note that, for smaller cell numbers, the statistics varied greatly because of the small (order 5) number of cells involved.
NES delivery mechanisms
Transport of biomolecules through the NS may occur by a combination of diffusive and electrokinetic mechanisms. Here, we develop a simple analytical model to predict how transport rates depend on voltage, delivery time, and reagent concentration. We treat electrophoresis as the most likely process because of the observed voltage and polarity dependence; however, we cannot completely rule out other mechanisms such as electro-osmosis. The total molecular transport per second through the NS is given by the total flux of electrokinetic and diffusive processes
| (1) |
where J is the molecular flux in moles per square meter per second. The total number of molecules delivered is then calculated from the integrated flux over time. Electrophoretic transport is taken to only occur during the period the field is on (τfield) during a pulse, which implies negligible acceleration and deceleration time that is generally reasonable for biomolecules in solution (34). Diffusion is constantly active for the total delivery period, τdiff
| (2) |
where N is the number of molecules transported, A is the NS cross-sectional area, and n is the number of NS present. Assuming ideal one-dimensional electrophoresis and diffusive steady-state transport under a uniform electric field, Eq. 2 can be expressed as
| (3) |
where μ is the electrophoretic mobility, l is the length of NS, V is the delivery voltage, C0 is the concentration of delivery reagent, and D is the diffusion coefficient. Under typical experimental conditions, the concentration of delivered reagent in cells (Ci) is much lower than C0 in the delivery buffer, such that (Ci ≪ C0) and the cell effectively acts as a molecular sink.
In addition to these traditional molecular transport mechanisms, delivery into the cell will also depend on the number and size of pores electroporated through the membrane near the NS tip. Previous studies (35) found a linear relationship between electroporation voltage and the molecular delivery, which we express by modifying the effective number of “open” NS areas, n, to be n = αV, where α is a constant that relates the number and size of electroporated holes to the applied voltage V. Equation 3 can then be rewritten as
| (4) |
This simple analysis makes several predictions about how biomolecular delivery depends on experimental conditions. The amount of material delivered should be linear with the reagent concentration C0 for both mechanisms. Delivery is linear in time, but only the time the field is applied should influence the electrophoretic mechanism. Last, electrophoretic delivery should be quadratic in voltage because of both molecular transport and electroporation being dependent on voltage.
To test this simple model, we delivered Cy5 dye–labeled DNA oligomers (Cy5-DNA, 200 nt) into HEK 293 cells by varying the reagent concentration, numbers of electric pulses, and voltage intensity and measured the amount of Cy5-DNA delivered by confocal fluorescence microscopy (Fig. 5). First, we tested whether the reagent concentration could control the delivery dosage (Fig. 5A). As the DNA concentration in the delivery buffer was increased from 1.5 to 60 μM, the oligomer concentration in cells also increased in a linear manner, consistent with the results of mRNA transfection (Fig. 2). Notably, the Cy5-DNA was homogeneously distributed for all experiments (fig. S6, A and B), suggesting that the intracellular delivery was not endocytotic. Next, we studied the dose delivery relationship with the number of electric pulses (e.g., total pulse time) (Fig. 5B). On the basis of the electrophoretic mechanism, we expect a linear relationship between total pulse time τfield and the dosage. Figure 5B shows that the Cy5-DNA concentration within the cell increased linearly as the total pulse time increases from 5 to 240 s. Cell viability was not affected even after the 240-s delivery period, consistent with our previous findings (27).
Fig. 5. Delivery of Cy5-tagged DNA oligomers and mCherry-tagged STIM1 protein into HEK 293 cells for different reagent concentration, delivery duration, and voltage.
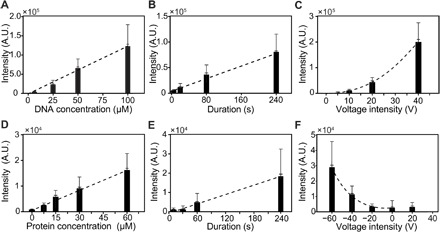
(A to C) Quantity of delivered DNA increases linearly with reagent concentration (A) and electrical pulse time duration (B). The amount increased quadratically with voltage (C), which agrees with a model where both poration and transport depend on voltage. (D to F) Similar to DNA oligomer delivery, STIM1 (stromal interaction molecule 1) protein intensity increases monotonically with protein concentration (D) or delivery duration (E) and quadratically with voltage (F). Note that the charge on STIM1 protein is positive; thus, negative fields increased the delivery rate, although some diffusive components were also observed at 0 and +20 V.
Last, we assessed the dose delivery relationship with voltage intensity. As voltage increases, the amount of Cy5-DNA delivered into cells increases as well (Fig. 5C), however, as a combination of a linear and quadratic function. This agrees with our simple model of electrophoretic transport coupled with the poration of the membrane, each of which is linear in voltage. The agreement between the theory and the measurements shows that multiple mechanisms can be effectively used to control molecular delivery quantities, with predictable amounts for given parameters.
Protein dosage control delivery
While NES allows high efficiency and controllable dosage delivery of nucleic acids (DNA and mRNA) to various cell types, it also permits delivery of proteins. As an example, we assessed whether NES could deliver the cytosolic fragment of STIM1 (residues 342 to 469) with an N-terminal 6His and mCherry tag (36, 37) into HeLa cells by adjusting concentration, duration, and voltage (Fig. 5 and fig. S7). STIM1 is an important endoplasmic reticulum membrane protein that interacts with the Orai1 calcium channel in the store-operated calcium entry pathway and should localize to the plasma membrane in the presence of Orai1. Given that the 6His-mCherry–tagged STIM1 is positively charged (Materials and methods), we applied the electric field with the negative terminal above the cell culture well except for the negative control.
The mCherry fluorescence intensity of each cell after delivery was analyzed by fluorescence imaging. The protein concentration increased in a linear manner with delivery concentrations of 7.5 to 60 μM, as well as for delivery duration from 10 to 240 s (Fig. 5, D and E). Increasing the voltage magnitude from −20 to −60 V also quadratically increased the protein delivery dosage (Fig. 5F). The quantitative protein delivery thus behaved quite similarly to the DNA oligomer delivery, suggesting that the NES delivery mechanism is largely independent of cargo identity.
STIM1 activity analysis and Cas9 ribonucleoprotein genome editing
Preservation of protein function is critical for effective intracellular protein delivery. We next tested whether STIM1 and Cas9 proteins delivered through NES retained function inside cells. The cytosolic domain of STIM1 should interact and activate the Orai1 calcium channel on the plasma membrane (36, 37). To test the binding activity of delivered STIM1 cytosolic fragment, we delivered Alexa Flour 647 maleimide–labeled STIM1 (residues 340 to 685, single cysteine at residue 512) of 10 μM to Orai1-GFP–expressing cells and plated the cells on an eight-well chamber coverslip for confocal fluorescence imaging analysis (Fig. 6 and fig. S8). Untransfected HEK 293 cells were analyzed as the negative control. For Orai1-GFP–positive cells, the GFP signal was localized on the cell membrane, showing the green circular shape of the membrane. After delivery of Alexa Flour 647–STIM1, it bound to the plasma membrane, overlapping with the GFP signal on the membrane and demonstrating proper biological localization and binding function (Fig. 6, A to D). Conversely, GFP-Orai1–negative cells show a uniform Alexa Flour 647 signal in the cytoplasm, indicating that there was no membrane binding without the presence of Orai1 (Fig. 6, E to H).
Fig. 6. STIM1 peptide interaction with the cell membrane protein Orai1 in HEK 293 cells.
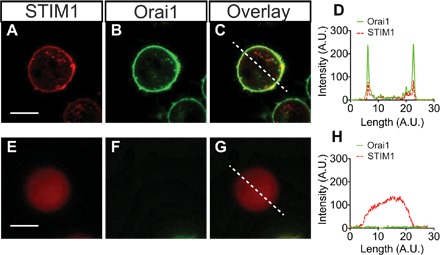
(A) GFP-tagged Orai1 is present only at the cell membrane surface. (B and C) After delivery, Alexa Fluor 647–tagged STIM1 localizes to the cell surface, with a strong overlap with Orai1 (C), indicating that specific binding has occurred. (D) A line trace through the composite image (dashed line) shows colocalization of the STIM1 and Orai1 proteins at the membrane. (E to H) In Orai1-negative cells (E), the STIM1 is uniformly distributed throughout the cytosol. Scale bar, 5 μm. (H) The composite cross section shows that STIM1 does not localize to the cell membrane without the presence of Orai1.
Since NES provides the intracellular access by opening nanosize pores on the cell membrane, the process should be largely cargo agnostic. Here, we show that NES not only delivers protein peptides but also allows intracellular delivery and gene editing of Cas9–ribonucleoproteins (RNPs). CRISPR technology (38) has had a profound impact on various research and medical applications, including identification of gene mutations of specific disease, development of disease models, and gene therapy (39). However, intracellular delivery of Cas9 RNPs for gene editing remains a challenge (40). We programmed the Cas9 RNPs to knock out PPIB, a house keeping gene that encoded an enzyme catalyzing the cis-tran isomerization and regulating protein folding and maturation. The purified recombinant Streptococcus pyogenes Cas9 (SpyCas9) protein carried two nuclear localization signal (NLS) sequences at its C terminus, followed by a GFP protein containing another NLS at C terminus (with a total molecular weight of 187 kDa). This GFP-tagged Cas9 was then incubated with in vitro–transcribed SpyCas9 single-guide RNA (sgRNA) that was engineered to uniquely target the human PPIB sequence to form the GFP-Cas9/RNA complex (RNP). This Cas9 RNP of 10 μM was delivered to HEK 293 cells by NES for 1 min. After delivery, the HEK 293 cells were resuspended and washed three times with 1× phosphate-buffered saline (PBS) before confocal microscopic imaging. The delivery efficiency was observed to be more than 90% from the GFP-tagged CRISPR complex (fig. S9, A and B). A few intense fluorescence spots were observed in cells, which is likely due to the GFP-Cas9 RNP aggregation in the cytoplasm.
Site-specific gene editing efficiency was measured by T7 endonuclease I (T7E1) assay and Sanger sequencing 48 hours after delivery. In the T7E1 assay, T7E1 cleaves DNAs at mismatched spots that can be generated by Cas9–mediated site-specific cleavage. The editing efficiency was estimated by measuring the intensity of the two cleavage products of mismatched DNA to the intact DNA strand. Figure S9C shows three bands at 550, 350, and 200 bp from DNA gel electrophoresis, indicating that the delivered GFP-Cas9 RNPs induced the mutation at the specific site in the PPIB gene. The measured genome editing efficiency was between 25 and 31% (fig. S9C). As expected, only one band at 550 bp is present in the negative control, confirming that there is no mutation at the PPIB site without the presence of Cas9 RNPs. Next, we verified the Cas9 cleavage site by Sanger sequencing. Random nucleotides are observed at the location of target site, consistent with random nucleotide insertion of nonhomologous end joining repair, indicating gene editing at the expected site (fig. S9, D and E). The estimated Cas9 editing efficiency by Sanger sequencing was 33%, which is slightly higher than the estimation from T7E1 assay. Note that the editing efficiency is lower than the delivery efficiency (>90%). This could be due to less efficient nuclear localization, protein aggregation in cells, or interference of GFP tags on the functionality of Cas9 proteins (41). This new aspect of NES offers a convenient and efficient protein delivery method to the study of intracellular protein activities and protein-based gene editing.
CONCLUSION
Delivery of exogenous mRNA, DNA, and proteins into cells is a critical step in biological research and therapeutic applications. Recent studies have demonstrated that the cytosolic quantities and/or relative ratio of mRNA and proteins has significant impact on IPSC reprograming and CRISPR editing efficiency (20), motivating the need for a refined delivery system with more precise dosage control for primary cell types. Here, we evaluated the NES approach for dosage-controlled delivery. The platform had significantly tighter dosage distributions than standard chemical delivery platforms, although some distributions in the concentrations from cell to cell still exist. Ratiometric control was also quite good, enabling specific ratios of multiple components to be delivered simultaneously. The lack of mRNA packaging or endocytosis also provided extremely fast (5 min) expression in the cytoplasm. These aspects could be quite useful for rapidly manipulating signaling networks and open up new possibilities such as delivery of DNA origami sequences for in-cell assembly.
NES delivery was also found to be effective for a variety of primary cell types with high cell viability, including primary neurons, stem cells, and cardiomyocytes. We found >95% cell viability after a single NES delivery, which enables sequential delivery over an extended time span that is often required for mRNA reprogramming. The system was fully scalable, from just a small number of cells (~5), relevant for rare cell types, to delivery of >100,000 cells at once in a single 96-well plate. The upper limit on the number of cells was simply a matter of the well size and thus could easily be increased or have multiple wells in parallel for high-throughput applications.
Mechanistically, cargo transport into the cell was found to be electrophoretic, with transport rates linearly dependent on reagent concentration and time the electric field was applied, and quadratic in voltage. Because of the physical nature of the transport, it was cargo agnostic, capable of delivering DNA, mRNA, and proteins with high efficiency, including functionally active proteins such as Cas9. No special buffers were needed for delivery, simplifying reagent preparation and cell handling.
Together, these delivery characteristics open new opportunities to control the relative levels of different proteins over time, either through dosage-controlled delivery of mRNA or by the protein itself. This capability could be particularly useful for manipulating multinode gene regulatory networks or transcription factors for iPSC reprogramming and cellular transformation.
MATERIALS AND METHODS
NS fabrication and characterization
The NS delivery platform was fabricated on a 20-μm-thick track-etched polycarbonate (PC) membrane with a pore density of 1 × 108 pore/cm2 and a pore diameter of 150 nm (GVS). A 10-nm-thick Al2O3 layer was deposited on the surface of the PC membrane including the inner side walls of the pores by atomic layer deposition (ALD) at 110°C. The top Al2O3 layer was etched by reactive ion etching (RIE) with BCl3 and Cl2 in argon [300 W, 40 sccm (standard cm3/min) of BCl3, 30 sccm of Cl2, 5 mtorr, 3 min]. Next, the PC polymer was removed by selective O2 plasma etching (100 W). The heights of NS ranged between 1.5 and 3 μm. A 4-nm layer of Au/Pd was sputter-coated on the NS platform for SEM analysis (FEI Sirion). A 3-μm-thick positive photoresist layer (MEGAPOSIT SPR 2203 i-Line photoresist, Dow) was spin-coated on the surface of the ALD-coated PC membrane (4000 rpm, 60 s) to prepare the photolitographically defined delivery regions. Following the photoresist coating, the membrane was baked at 95°C for 2 min. A square pattern of the desired size was exposed with ultraviolet light for 2 s and developed (MF-26, Shipley) for 30 s. The exposed Al2O3 surface of the square pattern was etched away by RIE. Last, O2 plasma was used to selectively etch the PC polymer to reveal the NS.
Standard NES intracellular delivery protocol
To perform intracellular delivery with NES, a total of 50,000 to 100,000 cells of interest were cultured on the NS platform within a 5-mm plastic tube for 0.5 to 7 days with the respective culture media. The recipes for each cell culture medium are listed in table S1. The cell culture devices were placed in a 48-well plate for incubation (5% CO2, 37°C). After incubation, the cell culture devices were placed on an ITO electrode with 2- to 5-μl droplets of the delivery reagent at the specified concentration. A platinum electrode (0.5 mm in diameter) was immersed in the cell culture. Square pulses of 20 Hz and pulse duration of 20 V and 200 μs were applied between the platinum electrode and ITO electrode for 20 s. The positions of the cathode and the anode were determined by the charge of the delivery molecules. For example, if the delivery molecules were charge negative, then the platinum electrode served as the anode. After delivery, the cell culture tube was returned to the 48-well plate for further incubation. When delivering fluorescently labeled molecules, the cells were washed three times with 1× PBS to remove excess molecules preceding analysis.
Cell fixation and live-cell confocal imaging
Cells were fixed with 2% glutaraldehyde and 4% paraformaldehyde (pH 7.3) for 10 min and then washed with 1× PBS. Next, the entire NS membrane with fixed cells was removed from the plastic tube and placed on a glass slide (25 mm by 75 mm) for sample mounting. A droplet of antifading mounting medium (VECTASHIELD) was applied to the cells. Last, the cells on the NS membrane were mounted between the glass slide and a 12-mm-diameter rounded glass coverslip with a thickness of 0.13 to 0.17 mm (Carolina Assistant-Brand). For live-cell imaging, cell samples were detached by trypsinization and washed three times in 1× PBS. The resuspended cells were then placed on an eight-well chamber coverslip. The cell samples were imaged by a spinning disc confocal microscope (Nikon) and were analyzed by ImageJ.
Flow cytometry analysis
After transfection by NES or LFN, the cells were incubated overnight. The cells were then removed by trypsin, washed three times in 1× PBS, and resuspended in FACS buffer (2% bovine serum albumin in PBS). Last, the GFP or mCherry intensity in each cell was analyzed by FACS analysis using an LSR II instrument (BD Biosciences).
GFP expression rate analysis
HEK 293 cells [50,000 (±5000)] were cultured on the NS platform overnight. GFP mRNA (500 or 1000 ng) was delivered to the cells on NS by the aforementioned delivery parameters (20 V, 20 Hz, and 20 s). As a control, 500 ng of GFP mRNA was delivered by LFN (Life Technologies) to the cells cultured on a 48-well plate following the manufacturer’s protocol. After NES and LFN delivery, the cells were incubated for 2, 5, 10, 15, 20, and 25 min, doubly washed with 1× PBS, and finally lysed with cell lysis buffer (Abcam). The GFP concentration in the cell lysate was determined by GFP ELISA assay (Abcam).
Protein expression
6His-mCherry–tagged STIM1 (residues 342 to 469) and single-cysteine 6His-STIM1 (residues 340 to 685, C437S, S512C) were expressed in Hi5 insect cells with the Bac-to-Bac Baculovirus Expression System (Thermo Fisher Scientific). The cells were lysed by sonication in buffer containing 20 mM Hepes and 300 mM NaCl (pH 7.5) (buffer A), and the supernatant was collected after centrifugation at 12,000g for 45 min. The supernatant was incubated with nickel–nitrilotriacetic acid beads (Qiagen) for 1 hour. The beads were washed with buffer A mixed with 50 mM imidazole, and the sample was eluted with buffer A mixed with 300 mM imidazole. The 6His-mCherry-STIM1 was then desalted with a column packed with Sephadex G-50 beads (Sigma-Aldrich) to remove imidazole.
The 6His-STIM1 was incubated with tobacco etch virus (TEV) protease overnight to cleave the 6His tag and then dialyzed into buffer containing 50 mM tris and 200 mM NaCl (pH 7.8) (buffer B). Ion exchange chromatography (HiTrap Q column, GE Healthcare) was used to obtain high purity of the protein sample. The protein was mixed with Alexa Fluor 647 C2 Maleimide dyes (Thermo Fisher Scientific) for more than 2 hours, and the free dyes were separated using a column packed with Sephadex G-50 beads (Sigma-Aldrich).
The net protein charges were positive for 6His-mCherry–tagged STIM1 that contains 51 negatively charged residues and 54 positively charged residues. Alexa Fluor 647–labeled STIM contains 42 negatively charged residues and 42 positively charged residues, which is charge neutral.
Expression and purification of Cas9
The GFP-fused recombinant SpyCas9 used in this study carries two NLS peptides between Cas9 and GFP, which is followed by another NLS at its C terminus. The Cas9 protein with an N-terminal 6His tag and maltose-binding protein was expressed in Escherichia coli Rosetta 2 cells (EMD Millipore). TEV protease was used to cleave the His tag and maltose-binding protein. The GFP-fused Cas9 was purified according to the protocols described previously (42). After purification, the GFP-fused Cas9 was stored in a protein buffer that comprised 150 mM KCl, 10% glycerol, 20 mM Hepes at pH 7.5, and 1 mM tris(2-chloroethyl) phosphate (TCEP) at −80°C.
In vitro Cas9 RNP assembly and genomic DNA extraction
The Cas9 RNP was prepared according the protocols described previously (42). The Cas9 RNP was made immediately before the experiment. To make the Cas9 RNP, purified Cas9 protein was incubated with sgRNA at a 1:1.2 molar ratio in 20 μM Hepes at pH 7.5, 150 mM KCl, 1 mM MgCl2, 10% glycerol, and 1 mM TCEP at 37°C for 10 min. Cells were incubated at 37°C for 24 to 48 hours after Cas9 RNP delivery. To extract the genomic DNA, the cells were lysed by 20 to 100 μl of QuickExtract solution (Epicentre) at 65°C for 20 min and then at 95°C for 20 min and were stored at −20°C. The concentration of genomic DNA was measured by NanoDrop (Thermo Scientific).
T7E1 assay
The T7E1 assay was performed according to the manufacturer’s instruction of T7E1 (New England BioLabs) and previous work (42). The hybridization reaction contained 200 ng of polymerase chain reaction DNA in KAPA high GC buffer and 50 mM KCl and was performed on a thermocycler with the following setting: 95°C for 10 min, 95° to 85°C at −2°C/s, 85°C for 1 min, 85° to 75°C at −2°C/s, 75°C for 1 min, 75° to 65°C at −2°C/s, 65°C for 1 min, 65° to 55°C at −2°C/s, 55°C for 1 min, 55° to 45°C at −2°C/s, 45°C for 1 min, 45° to 35°C at −2°C/s, 35°C for 1 min, and 35° to 25°C at −2°C/s, 25°C for 1 min and hold at 4°C. Buffer 2 and 5 units of T7E1 (New England BioLabs) were added to digest the reannealed DNA. After 1 hour of incubation at 37°C, the reaction was quenched with one volume of gel loading dye [50 mM tris (pH 8.5), 50 mM EDTA, 1% SDS, 50% glycerol, and 0.01% bromophenol blue] at 70°C for 10 min. The product was resolved on 1.5% agarose gel containing SYBR Gold (Life Technologies). The DNA band intensity was quantitated using ImageJ.
Supplementary Material
Acknowledgments
We thank J. Doudna for support with laboratory equipment and S. Leal-Ortiz for comments on the experimental design. Funding: This work was supported by NIH grants R01 HL133272 (to J.C.W. and H.C.), R01 HL128170 (to J.C.W.), R37 GM45374 (to R.S.L. and R.Q.), and R21 EB02533201 (to N.A.M.); NSF grant 1549696 (to Y.C.); the Knut and Alice Wallenberg Foundation (to M.H.); the Stanford Neurosciences Institute (to N.A.M.); and Bio-X Interdisciplinary Initiatives Program (to Y.C., N.A.M., and J.C.W.). Author contributions: Y.C., H.C., R.Q., M. Hjort, and N.A.M. conceived the experiments. Y.C., H.C., R.Q., and E.M. performed the experiments and analysis of experimental data. Y.C. and N.A.M. wrote the manuscript with contribution from all the authors. Competing interests: N.A.M. declares a competing interest with equity in a company related to this work. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaat8131/DC1
Fig. S1. Schematics of NS platform fabrication.
Fig. S2. Photograph of NS cell culture reservoirs.
Fig. S3. GFP and mCherry expression intensity after ratiometric cotransfection of GFP and mCherry by NES and LFN.
Fig. S4. SEM of patterned NS platform.
Fig. S5. Schematic of patterned NS fabrication.
Fig. S6. Dosage control intracellular delivery of Cy5-tagged DNA oligomers.
Fig. S7. Dosage control intracellular delivery of mCherry-tagged STIM1 protein fragment.
Fig. S8. mCherry-tagged STIM1 protein fragment interaction with cell membrane protein Orai1.
Fig. S9. GFP-tagged SpyCas9 RNP genome editing.
Table S1. Cell culture media receipts.
REFERENCES AND NOTES
- 1.Takahashi K., Yamanaka S., Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S., Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Nagamatsu G., Saito S., Kosaka T., Takubo K., Kinoshita T., Oya M., Horimoto K., Suda T., Optimal ratio of transcription factors for somatic cell reprogramming. J. Biol. Chem. 287, 36273–36282 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishimura K., Kato T., Chen C., Oinam L., Shiomitsu E., Ayakawa D., Ohtaka M., Fukuda A., Nakanishi M., Hisatake K., Manipulation of KLF4 expression generates iPSCs paused at successive stages of reprogramming. Stem Cell Reports 3, 915–929 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olariu V., Lövkvist C., Sneppen K., Nanog, Oct4 and Tet1 interplay in establishing pluripotency. Sci. Rep. 6, 25438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radzisheuskaya A., Silva J. C. R., Do all roads lead to Oct4? The emerging concepts of induced pluripotency. Trends Cell Biol. 24, 275–284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parikshak N. N., Gandal M. J., Geschwind D. H., Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet. 16, 441–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venken K. J. T., Simpson J. H., Bellen H. J., Genetic manipulation of genes and cells in the nervous system of the fruit fly. Neuron 72, 202–230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hultquist J. F., Schumann K., Woo J. M., Manganaro L., McGregor M. J., Doudna J., Simon V., Krogan N. J., Marson A., A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Rep. 17, 1438–1452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang X., Potter J., Kumar S., Zou Y., Quintanilla R., Sridharan M., Carte J., Chen W., Roark N., Ranganathan S., Ravinder N., Chesnut J. D., Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 208, 44–53 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Liu J., Gaj T., Yang Y., Wang N., Shui S., Kim S., Nagamangala Kanchiswamy C., Kim J.-S., Barbas C. F. III, Efficient delivery of nuclease proteins for genome editing in human stem cells and primary cells. Nat. Protoc. 10, 1842–1859 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Hendel A., Bak R. O., Clark J. T., Kennedy A. B., Ryan D. E., Roy S., Steinfeld I., Lunstad B. D., Kaiser R. J., Wilkens A. B., Bacchetta R., Tsalenko A., Dellinger D., Bruhn L., Porteus M. H., Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 33, 985–989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stewart M. P., Sharei A., Ding X., Sahay G., Langer R., Jensen K. F., In vitro and ex vivo strategies for intracellular delivery. Nature 538, 183–192 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Roth C. M., Sundaram S., Engineering synthetic vectors for improved DNA delivery: Insights from intracellular pathways. Annu. Rev. Biomed. Eng. 6, 397–426 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Lonn P., Kacsinta A. D., Cui X.-S., Hamil A. S., Kaulich M., Gogoi K., Dowdy S. F., Enhancing endosomal escape for intracellular delivery of macromolecular biologic therapeutics. Sci. Rep. 6, 32301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan M., Du J., Gu Z., Liang M., Hu Y., Zhang W., Priceman S., Wu L., Zhou Z. H., Liu Z., Segura T., Tang Y., Lu Y., A novel intracellular protein delivery platform based on single-protein nanocapsules. Nat. Nanotechnol. 5, 48–53 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Gehl J., Electroporation: Theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol. Scand. 177, 437–447 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Carey B. W., Markoulaki S., Hanna J. H., Faddah D. A., Buganim Y., Kim J., Ganz K., Steine E. J., Cassady J. P., Creyghton M. P., Welstead G. G., Gao Q., Jaenisch R., Reprogramming factor stoichiometry influences the epigenetic state and biological properties of induced pluripotent stem cells. Cell Stem Cell 9, 588–598 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Papapetrou E. P., Tomishima M. J., Chambers S. M., Mica Y., Reed E., Menon J., Tabar V., Mo Q., Studer L., Sadelain M., Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human iPSC induction and differentiation. Proc. Natl. Acad. Sci. U.S.A. 106, 12759–12764 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mali P., Esvelt K. M., Church G. M., Cas9 as a versatile tool for engineering biology. Nat. Methods 10, 957–963 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boukany P. E., Morss A., Liao W. C., Henslee B., Jung H., Zhang X., Yu B., Wang X., Wu Y., Li L., Gao K., Hu X., Zhao X., Hemminger O., Lu W., Lafyatis G. P., Lee L. J., Nanochannel electroporation delivers precise amounts of biomolecules into living cells. Nat. Nanotechnol. 6, 747–754 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Chow Y. T., Chen S., Wang R., Liu C., Kong C.-w., Li R. A., Han Cheng S., Sun D., Single cell transfection through precise microinjection with quantitatively controlled injection volumes. Sci. Rep. 6, 24127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shekaramiz E., Varadarajalu G., Day P. J., Wickramasinghe H. K., Integrated electrowetting nanoinjector for single cell transfection. Sci. Rep. 6, 29051 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang W., Yavari F., Minary-Jolandan M., Giraldo-Vela J. P., Safi A., McNaughton R. L., Parpoil V., Espinosa H. D., Nanofountain probe electroporation of single cells. Nano Lett. 13, 2448–2457 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ouyang M., Hill W., Lee J. H., Hur S. C., Microscale symmetrical electroporator array as a versatile molecular delivery system. Sci. Rep. 7, 44757 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie X., Xu A. M., Leal-Ortiz S., Cao Y., Garner C. C., Melosh N. A., Nanostraw-electroporation system for highly efficient intracellular delivery and transfection. ACS Nano 7, 4351–4358 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Cao Y., Hjort M., Chen H., Birey F., Leal-Ortiz S. A., Han C. M., Santiago J. G., Paşca S. P., Wu J. C., Melosh N. A., Nondestructive nanostraw intracellular sampling for longitudinal cell monitoring. Proc. Natl. Acad. Sci. U.S.A. 114, E1866–E1874 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinle H., Behring A., Schlensak C., Wendel H. P., Avci-Adali M., Concise review: Application of in vitro transcribed messenger RNA for cellular engineering and reprogramming: Progress and challenges. Stem Cells 35, 68–79 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Al-Maqtari T., Hong K. U., Vajravelu B. N., Moktar A., Cao P., Moore J. B. IV, Bolli R., Transcription factor-induced activation of cardiac gene expression in human c-kit+ cardiac progenitor cells. PLOS ONE 12, e0174242 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quabius E. S., Krupp G., Synthetic mRNAs for manipulating cellular phenotypes: An overview. N. Biotechnol. 32, 229–235 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Sander J., Schmidt S. V., Cirovic B., McGovern N., Papantonopoulou O., Hardt A.-L., Aschenbrenner A. C., Kreer C., Quast T., Xu A. M., Schmidleithner L. M., Theis H., Do Thi Huong L., Rizal Bin Sumatoh H., Lauterbach M. A. R., Schulte-Schrepping J., Günther P., Xue J., Baßler K., Ulas T., Klee K., Katzmarski N., Herresthal S., Krebs W., Martin B., Latz E., Händler K., Kraut M., Kolanus W., Beyer M., Falk C. S., Wiegmann B., Burgdorf S., Melosh N. A., Newell E. W., Ginhoux F., Schlitzer A., Schultze J. L., Cellular differentiation of human monocytes is regulated by time-dependent interleukin-4 signaling and the transcriptional regulator NCOR2. Immunity 47, 1051–1066.e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu A. M., Aalipour A., Leal-Ortiz S., Mekhdjian A. H., Xie X., Dunn A. R., Garner C. C., Melosh N. A., Quantification of nanowire penetration into living cells. Nat. Commun. 5, 3613 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu A. M., Kim S. A., Wang D. S., Aalipour A., Melosh N. A., Temporally resolved direct delivery of second messengers into cells using nanostraws. Lab Chip 16, 2434–2439 (2016). [DOI] [PubMed] [Google Scholar]
- 34.H. Berg, Random Walks in Biology (Princeton Univ. Press, 1993). [Google Scholar]
- 35.Zaharoff D. A., Henshaw J. W., Mossop B., Yuan F., Mechanistic analysis of electroporation-induced cellular uptake of macromolecules. Exp. Biol. Med. 233, 94–105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park C. Y., Hoover P. J., Mullins F. M., Bachhawat P., Covington E. D., Raunser S., Walz T., Garcia K. C., Dolmetsch R. E., Lewis R. S., STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soboloff J., Rothberg B. S., Madesh M., Gill D. L., STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 13, 549–565 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., Charpentier E., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H., La Russa M., Qi L. S., CRISPR/Cas9 in genome editing and beyond. Annu. Rev. Biochem. 85, 227–264 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Yin H., Kauffman K. J., Anderson D. G., Delivery technologies for genome editing. Nat. Rev. Drug Discov. 16, 387–399 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Zordan R. E., Beliveau B. J., Trow J. A., Craig N. L., Cormack B. P., Avoiding the ends: Internal epitope tagging of proteins using transposon Tn7. Genetics 200, 47–58 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin S., Staahl B. T., Alla R. K., Doudna J. A., Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 3, e04766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaat8131/DC1
Fig. S1. Schematics of NS platform fabrication.
Fig. S2. Photograph of NS cell culture reservoirs.
Fig. S3. GFP and mCherry expression intensity after ratiometric cotransfection of GFP and mCherry by NES and LFN.
Fig. S4. SEM of patterned NS platform.
Fig. S5. Schematic of patterned NS fabrication.
Fig. S6. Dosage control intracellular delivery of Cy5-tagged DNA oligomers.
Fig. S7. Dosage control intracellular delivery of mCherry-tagged STIM1 protein fragment.
Fig. S8. mCherry-tagged STIM1 protein fragment interaction with cell membrane protein Orai1.
Fig. S9. GFP-tagged SpyCas9 RNP genome editing.
Table S1. Cell culture media receipts.