

ABSTRACT
During severe septic shock and/or severe acute respiratory distress syndrome (ARDS) patients present with a limited cardio-ventilatory reserve (low cardiac output and blood pressure, low mixed venous saturation, increased lactate, low PaO2/FiO2 ratio, etc.), especially when elderly patients or co-morbidities are considered. Rescue therapies (low dose steroids, adding vasopressin to noradrenaline, proning, almitrine, NO, extracorporeal membrane oxygenation, etc.) are complex. Fever, above 38.5–39.5°C, increases both the ventilatory (high respiratory drive: large tidal volume, high respiratory rate) and the metabolic (increased O2 consumption) demands, further limiting the cardio-ventilatory reserve. Some data (case reports, uncontrolled trial, small randomized prospective trials) suggest that control of elevated body temperature (“fever control”) leading to normothermia (35.5–37°C) will lower both the ventilatory and metabolic demands: fever control should simplify critical care management when limited cardio-ventilatory reserve is at stake. Usually fever control is generated by a combination of general anesthesia (“analgo-sedation”, light total intravenous anesthesia), antipyretics and cooling. However general anesthesia suppresses spontaneous ventilation, making the management more complex. At variance, alpha-2 agonists (clonidine, dexmedetomidine) administered immediately following tracheal intubation and controlled mandatory ventilation, with prior optimization of volemia and atrio-ventricular conduction, will reduce metabolic demand and facilitate normothermia. Furthermore, after a rigorous control of systemic acidosis, alpha-2 agonists will allow for accelerated emergence without delirium, early spontaneous ventilation, improved cardiac output and micro-circulation, lowered vasopressor requirements and inflammation. Rigorous prospective randomized trials are needed in subsets of patients with a high fever and spiraling toward refractory septic shock and/or presenting with severe ARDS.
KEYWORDS: septic shock, acute respiratory distress syndrome, spontaneous breathing, high PEEP, fever control, hyperthermia, hypothermia, sympathetic system, clonidine, dexmedetomidine
Abbreviations and glossary
- ALI
acute lung injury (200<P/F<300)
- APRV-SV
airway pressure release ventilation-spontaneous ventilation
- ARDS
Acute respiratory distress syndrome
- CCU
critical care unit
- CMV
controlled mandatory ventilation
- ECMO
extracorporeal membrane oxygenation
- EIT
electrical impedance tomography
- FEV1
forced expiratory volume measured over 1s (Tiffeneau-Pinelli index)
- FRC
functional residual capacity
- HR
heart rate
- 5HT
serotonin
- 5HT1A
subtype of serotonin receptor
- IL6, IL10
pro- and anti-inflammatory cytokines
- LPS
lipopolysaccharide evoking “sterile inflammation”
- NA
noradrenaline
- NO
nitric oxide
- PaO2/FiO2 = P/F
index of oxygenation
- Paracetamol
acetaminophen
- Pendel-luft
lung dys-coordination
- PEEP
positive end-expiratory pressure
- PCT
procalcitonin
- P-SILI
patient-self inflicted lung injury (ventilation-induced lung injury as opposed to ventilator-induced self injury)
- RASS
Richmond agitation sedation scale
- RR
respiratory rate
- RRT
renal replacement therapy
- TIVA
total intravenous anesthesia
- VA/Q
ventilation/perfusion ratio
- VO2
O2 consumption
- Vt
tidal volume
I. Introduction
Manipulation of temperature in the critical care unit (CCU) has been proposed early (“artificial hibernation” [1]. However, as early as 1954, manipulation of temperature was to be considered in only ≈10% of the patients, those presenting with the most severe combat injuries [2]. This overview focuses on these very sick patients presenting with an elevated body temperature. Indeed, induced-normothermia (control of elevated body temperature, “fever control” [3]) is not straightforward. Given the focus on severe cardioventilatory distress in the setting of septic shock and severe acute respiratory distress syndrome (ARDS) and recent reviews [4–9], pros and cons for respecting elevated body temperature are briefly discussed.
A). Restriction of the topic
The present hypothesis restricts itself to fever control and will not consider spontaneous hypothermia or induced-hypothermia. Nevertheless, the literature on spontaneous and induced-hypothermia has to be overviewed.
The US infectious diseases society defines hyperthermia as temperature ≥38.3°C [7]. Hypothermia is defined <36°C [7], <35.5°C) [10] or <35°C [11]. Mild hypothermia (≈34–32°C), in the post-cardiac arrest setting, avoids any after-drop and ventricular arrhythmias. In the setting of traumatic brain injury, temperature is sometimes lowered to ≈33–35.5°C [12]. Conversely, in the cardiac arrest setting, the upper limit ≤37.6°C is considered [7]. Only fever control or evoked-normothermia will be considered in the following discussion, defined as 35.5–37.5°C. These are the lowest and highest temperatures observed during the day-night cycle in any healthy non-exercising or sleeping human. This range allows one to skip the coagulation and immunological pathologies [11] associated with deeper hypothermia (≤35°C).
In hospitalized patients, ≈75% of the elevated body temperature is linked to systemic inflammation in the setting of sepsis. Malignancy, tissue ischemia, drug reactions account for the remainder. Neurogenic and endocrinology-evoked (thyroid, adrenal medulla) fevers are uncommon [8].
B). Beneficial effects of hyperthermia
A negative relationship exists between body temperature and mortality (n = 10 834 patients) [13]: hyperthermic (>38°C) and hypothermic (<36°C) patients present a 22 and 47% mortality respectively [13]. High temperature>39.0°C was associated with reduced mortality in septic patients, outside septic shock. Moderate fever (37.5–39.4°C) associated with sepsis may be associated with an increased survival (quoted from [8]. Similarly, moderate fever (37.5–38.4°C) is beneficial in septic patients [7]. A temperature>39.5°C is detrimental in non-septic patients: high peak temperature were associated with increased mortality in non-infectious fever [14].
Presumably, elevated body temperature decreases the bacterial/viral load [7]. Increased temperature may enhance the ability of the immune system to eliminate invading micro-organisms [15]. As elevated body temperature comes at a metabolic cost, a teleological conclusion is that its persistence across a broad range of species provides circumstantial evidence that the response has evolutionary advantage: the immune system would have evolved to function optimally when elevated body temperature does not reach too high levels [14]. However arguments based on evolution do not necessarily apply to humans under CCU conditions, and supported beyond the limits of physiological homeostasis [14]: in the presence of antibiotics and other therapeutics, the harms of the febrile response may outweigh its benefits [16]. Therefore, elevated body temperature may either be a protective response (i.e. a mechanism that the physician ought not to interfere with) or a marker of severity or a risk factor implying active therapy i.e. fever control [6].
C). Effects of hypothermia on the outcome of sepsis
In the setting of septic shock, thirty three per cent of the patients presenting for inclusion in a trial of control of elevated body temperature were excluded for absence of fever [3]. Some ten per cent of septic patients present hypothermia and increased mortality (quoted from [5]). Presumably, the spontaneous absence of elevated body temperature characterizes the most severely ill patients with shock and/or without metabolic resource to produce heat [3]. Hypothermia (<35.5°C; 9% of the patients) is associated with a higher incidence of confusion, higher lactate (not significant: ns), higher incidence of shock (94% of hypothermic patients vs. 61% of febrile patients), low filling pressure, higher mortality (mortality in hypothermia group: 62% vs. febrile group: 26%) [10]. Increased mortality is observed in elderly patients presenting with community-acquired pneumonia and absence of elevated body temperature as opposed to patients developing fever (quoted from [8]).
Does spontaneous or evoked-hypothermia worsen the outcome [15]? In rats developing lipopolysaccharide (“sterile inflammation” evoked by LPS) or E Coli-induced shock, the outcome is better in the hypothermic group as opposed to the fever group, if hypothermia develops in a colder environment for both groups [15]: therefore spontaneous hypothermia may be a protective response in a subset of septic shock patients. This hypothesis [17,18] posits that elevated body temperature is the first phase of sepsis followed by reduced body temperature observed when the animal is getting sicker, the only possibility in the wild: mortality may be a consequence of a more severe sepsis. The severity of sepsis and the occurrence of spontaneous hypothermia does not necessarily imply that spontaneous hypothermia occurs immediately before death, septic shock, ARDS or use of antipyretics [19]. In addition, the situation of a very sick animal in the wild is not necessarily identical with the situation of a very sick patient under maximal support in the CCU [14]. Therefore, the patient presenting with septic shock and spontaneous hypothermia does not necessarily need active rewarming, irrespective of his clinical situation. Moreover, multiple organ failure is postulated to be a temporary adaptive, protective, mechanism in the setting of septic shock [20]. As in the setting of myocardial stunning, the hypothesis is that the metabolic requirements are reduced temporarily: reduced mitochondrial activity may enhance cell survival. Therefore, the thermometabolic adaptation hypothesis holds that spontaneous hypothermia should be allowed to run, within limits, if lowered O2 consumption (VO2) is matched by a lowered O2 delivery [21]. Outcome should be assessed prospectively [21] in well defined subsets [22] (Figure 1; figure 2 in reference [7]: a control hypothermic septic shock group where spontaneous hypothermia is let to run within limits as opposed to a treated hypothermic septic shock group where evoked-normothermia is re-established through active rewarming [21].
Figure 1.
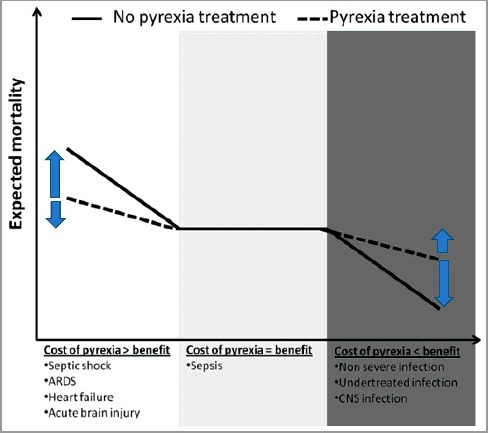
A niche for normothermia in the critical care setting? Suggested impact of fever control on outcome according to clinical context: obviously fever control is to be restricted to niches (cost of pyrexia>benefit : left part of schema) in which the benefit is superior to the cost for the patient : severe septic shock, severe ARDS, circulatory insufficiency. Abbreviations: ARDS: acute respiratory distress syndrome; CNS: central nervous system. Reused from Doyle, Crit Care, 2016, 20: 303 [7] under Creative Commons attribution license 4.0.
The peripheral shut-down observed in the hypothermic septic shock patients needs discussion: if therapy achieves only adequate cardiac output and systemic pressure, improved ventilation, improved general homeostasis using renal replacement therapy (RRT), in addition to anti-infectious strategy, adequate transfer of heat from core to periphery and vice-versa will occur, at best, very slowly: then, the metabolic issue is the limiting factor. This brings up restoring micro-circulation [23,24] immediately after restoring cardiac output. Within the mechanisms of heat production (shivering and non-shivering thermogenesis) and dissipation, only shivering, vasoconstriction and vasodilation may be considered in the CCU. After normalization of cardio-ventilatory distress, vasodilation will enhance heat dissipation from core to periphery in the setting of elevated body temperature and septic shock: this will lower body temperature. Conversely, reopening of unperfused capillaries (i.e. vasodilation) will presumably allow the transfer of an external, therapeutic, heat source to the core in the setting of spontaneous hypothermia and septic shock, allowing faster rewarming.
D). Evoking fever control?
Shivering occurs when core temperature is ≈1.5°C below the activation threshold1. of cold defense effectors (quoted from [7]). In the septic setting (baseline temperature ≈38.7°C), external cooling evokes shivering: VO2 increased (+57%) [22]. «Counter-warming» with an air circulating blanket suppresses shivering evoked by hypothermia in the setting of brain injury/cardiac arrest hypothermia (VO2 : −16%) [12]. In the setting of subarachnoid hemorrhage, traumatic brain injury and strict normothermia, water-circulating blankets (−1.12°C.h-1) appear superior to air-circulating blanket (−0.15°C.h-1) or conventional cooling (iced saline combined to surface cooling : 0.06°C.h-1) [25].
Antipyretics (non steroidal anti-inflammatory drugs, paracetamol = acetaminophen) are administered to the most severe patients unable to tolerate elevated body temperature [5]. In septic patients, ibuprofen lowered temperature (38°C to 36.7°C), ventilation (≈−2 L.min-1), heart rate (HR≈115 to 100 bpmin) and lactate (−17%) [26]. Antipyretics increased 28-day mortality in the septic group [8].
This overview focuses on control of elevated body temperature in the setting of limited cardioventilatory reserve (elderly patients, co-morbidities): septic shock and severe acute respiratory distress syndrome (ARDS) presenting with elevated body temperature. A rationale for using alpha-2 adrenergic agonists (“alpha-2 agonists”) to achieve fever control while controlling for shivering is delineated.
II. Fever control in the setting of septic shock
For didactic purposes, septic shock and acute respiratory distress syndrome will be discussed respectively as pure circulatory vs. pure ventilatory disorders, with evident oversimplification.
A). Present knowledge
In healthy volunteers, injection of endotoxin to healthy volunteers increase both energy expenditure and temperature [27]. Furthermore, administration of noradrenaline increases VO2 by +15% [28]. Thus, therapy adds to injury, reinforcing the need of fever control when limited cardio-ventilatory reserve is present.
By contrast, cooling febrile CCU patients reduces VO2 by ≈8–10% per °C [14]. In paralyzed sedated patients under controlled mechanical ventilation, surface cooling (39.4±0.8 °C to 37.0±0.5) decreases VO2 (359±65 to 295±57 mL.min-1; aggregated data: −18%) [29]. In one mild shivering patient, lowering temperature to 36.2°C reduces VO2 by −39%. VCO2 is lowered (303±43 to 243±37 mL.min-1:−20%). This is relevant when low driving pressure [30] and permissive hypercapnia [31] are considered in the setting of ARDS (“protective” ventilation). Cardiac output, O2 extraction ratio2. and HR decrease (respectively: 8.4±3.2 L.min-1 to 6.5±1.8 L.min-1, 28±7 to 23±5% and 119±21 to 102±14 bpmin). Mixed venous SO2 increases from 68±8 to 71±6% (ns). Therefore, mechanical ventilation, paralysis, and cooling (from 40 to 36° C) could reduce V02 in febrile, critically ill patients by as much as 190 mL/min, lowering metabolic demand by 47%: the manipulation of VO2 is a salvage therapy in severe shock or hypoxemic respiratory failure, e.g. for febrile, mechanically ventilated patients who do not respond to sedation and antipyretics [29]. In the setting of synchronized intermittent mandatory ventilation (n = 8) or pressure support (n = 10), external cooling lowered minute ventilation (39.1 to 37°C: 14.7±1 to 13.1 ±0.9 L.min-1: −11%) and decreased energy expenditure (−12%; sedation: morphine+midazolam to Ramsay 3–4) [32]. In this trial [32], only subsets of « septic episode » or severe hypoxia were studied [32].
1). Outcome
After sedation, intubation, mechanical ventilation, patients presenting with early septic shock were randomized to external cooling within 2 h after enrollment (n = 101, 36.5–37.5°C for 48 h) vs. no external cooling (n = 99) [3]. Paracetamol was seldom used (n = 2 patients*2) [33]: only external cooling was used, avoiding almost entirely a possible confounding factor (pharmacology: heat production reduction as opposed to external cooling only). The sample is biased as, respectively, 70 and ≈50% of the included patients presented an infection related to pneumonia and moderate ARDS, irrespective of group assignment to fever control vs. no external cooling [3]. Nevertheless, the strength of this trial [3] is its high degree of homogeneity both with respect to recruitment of septic shock patients with elevated body temperature and therapeutics. In this trial [3], ≈33% of the patients were excluded for absence of elevated body temperature in the setting of septic shock [3].
Cooling was short (48 h) to allow one assessing the evolution of the initial infection and early detection of nosocomial infection [3]. Fourteen days mortality was lowered (34 vs. 19%: −16%) [3]. Evoked normothermia is supposed not to increase infection [3,11]. Nevertheless, a) incidence of acquired infection by day 14 was higher in the normothermia group. b) a late mortality, upon hospital discharge, was observed in the fever control group [3]: 24% vs. 14%. The immuno-suppressive response develops after the initial phase of severe sepsis, depending on the severity and duration of the septic phase. Therefore, taken together, fever control may present opposite effects which may or may not cancel each other: decreased risk of CCU-acquired infection by accelerating the rate of recovery of the initial septic shock vs. increased secondary immuno-depression [5]. A post-hoc analysis [34] showed that the reduced mortality was related to lowered temperature <38.4°C but not to lowered HR. Reduced vasopressor requirement was not associated with lowered HR [34].
This favorable outcome observed in a subset of septic shock patients presenting with elevated body temperature [3] contrasts with worsened outcome when all critical care patients with elevated body temperature are randomized to paracetamol and external cooling (aggressive group) vs. no treatment until temperature reached >40°C (permissive group). Interim analysis showed higher mortality in the aggressive group (44 vs. 38 patients and 7 deaths vs. 1 respectively; p = 0.06) [35]. Thus, elevated temperature may be more important for the localized tissue response than for the systemic or whole-body immune responses (Atkins quoted from [18]. Therefore, the conventional wisdom of beneficial effect of hyperthermia may not only be broken down to severe vs. non-severe hyperthermia but also to local vs. systemic phenomenons. A niche [3] approach, favoring external cooling in the setting of elevated temperature, is to be favored if fever control is to improve CCU outcome. However, suppression of heat production is another option to achieve fever control, as delineated earlier [36].
2). Vasopressor requirement
A decrease in vasopressor requirement was significant by 12 h (−50%) but not by the 48 h interval, more pronounced in those patients having the highest baseline vasopressor doses [3]. The authors delineate a limitation: the fever control group presented slightly lower noradrenaline requirement at inclusion (0.50 μg.kg-1.min-1 vs. 0.65). Early shock reversal was more common with external cooling, which prevented early death. This reduction in vasopressor requirement [3] during normothermia has been previously observed [37]:
-
a)
a pressor effect was observed in 80% of the patients [38]. An early report (1961) observed a 50% survival in the setting of refractory septic shock (« irreversible septic shock ») treated with 1961's state-of-the-art combined to hypothermia (32°C; 1–30 days, usually 3 days; no control group). A recovery from confusion/coma evoked by sepsis was observed in 94% of the patients [38].
-
b)
moribund patients with sepsis+ARDS presented a reduction in vasopressor requirements in the setting of hypothermia (32–35°C) [39].
The vasopressor sparing effect of lowered temperature is observed following corticosteroids administration or continuous hemofiltration. This vasopressor sparing effect linked to hypothermia is presumably opposed to the mechanism linked to hyperthermia: capillary dilatation, vascular stasis, and extravasation in the interstitium [8].
To sum up, fever control should be considered in septic shock with increasing dose of vasopressors [5]: the sicker patients are the ones to benefit the most from artificial hibernation [2], external cooling [3,35], and alpha-2 agonists in the setting of septic shock [40]. Nevertheless, although septic patients with high temperature require fever control to reduce VO2 and catabolism, this reduction in temperature is not yet proven as harmful or not [37]. As the evidence [3] is limited to one large trial, independent validation is required.
B). Alpha-2 agonists to modify the management of septic shock?
1). Working hypothesis
In our hands, in the setting of acute cardio-ventilatory distress and septic shock, obtaining fever control in febrile patients combines within an “analytical” bundle, i.e. state-of-the-art treatment combined with alpha-2 agonists: a) paracetamol, external cooling. b) normalization of volemia up to an absence of response to rigorous [41] passive leg raising and minimized changes in the diameter of the vena cava evoked by ventilation. c) administration of a vasopressor to rise the systemic pressure according to systemic perfusion (urine output, micro-circulation, etc.) and co-morbidities (hypertension, coronary disease, etc.). Vasopressor is thought to be administered early when diastolic pressure is low [42]. High systemic perfusion pressure may be needed early in the sicker patients [23]. d) after normalization of volemia, auriculo-ventricular conduction and blood pressure, and immediately following tracheal intubation, an alpha-2 agonist is administered (infusion of clonidine 1 μg.kg-1.h-1 or dexmedetomidine 0.75 μg.kg-1.h-13. to −3<RASS<−1)4..
Several questions remain: first, should the hypothermic patients presenting with septic shock be actively warmed up to 35.5°C [21]? second, is it beneficial to actively rewarm the hypothermic septic shock patients up to 35.5°C, under alpha-2 agonists, to improve micro-circulation? Presumably, following alpha-2 agonists, the adequacy of the cardiac output (superior vena cava S02>70–75%, CO2 gap <5–6 mm Hg, adequate trend in lactate toward <2mM) combines to adequate micro-circulation. This will allow transferring active heating from periphery to the core. c) conversely, may antipyretics be used alone, without alpha-2 agonists, to evoke fever control? Alpha-2 agonists will provide cooperative sedation without depression of the respiratory generator [43] and without emergence delirium [44–46]. In addition, alpha-2 agonists normalize a dysfunctional vasomotor and skin sympathetic nervous hyperactivity: this will presumably improve micro-circulation and favor heat transfer from core to periphery, evoking fever control.
2). Pharmacology
Application of serotonin (5HT) in the hypothalamus activates heat loss mechanisms whereas application of noradrenaline increases heat conservation and production (quoted from [47]): serotonin and noradrenaline are considered as thermogenic vs. thermolytic, respectively [10], depending on the considered agonist or antagonist. The alpha-2 adrenoceptors may be located on a cholinergic-serotoninergic pathway [48]. The activation threshold of cold defense effectors is lowered by alpha-2 agonists [49]. The hypothermic effect of clonidine is increased following lesioning the ventral noradrenergic bundle which projects to the hypothalamus [50]. Clonidine produces hypothermia either by reducing heat production and/or by enhancing heat loss (cutaneous vasodilation) through hypothalamic alpha-2 adrenoceptors.
In animals, alpha-2 agonists reduce muscle tremor [51] (Figure 2; figure 2 in reference [52]), explaining reduced VO2 [53]. In healthy semi-recumbent volunteers, alpha-2 agonists reduce temperature (−0.7°C with dexmedetomidine high dose: 2 μg.kg-1) and VO2 (dexmedetomidine high-dose: −18%; clonidine 5 μg.kg-1 p.o.: −13%) [54,55]. Clonidine [56], dexmedetomidine (0.4 ng.mL-1) and meperidine lower shivering threshold (−0.7 and −1.2°C respectively). The combination is additive (−2.0°C) [57]. Dexmedetomidine produces marked and linear decrease in the vasoconstriction and shivering thresholds (quoted from [9].
Figure 2.

Raw electromyogram (EMG) from quadriceps femoris muscle. The rat is chronically spinalized to suppress descending control. This will allow one to assess absence or presence of peripherally-evoked tremor. Note the increase in EMG after administration of 5-hydroxy-tryptophan (5HTP) and the reduction after systemic administration of an alpha-2 agonist, clonidine. Phasic bursting persists after clonidine, as observed in another setting [58]. The observation of reduced muscular activity is key to understand reduced VO2 and enlarged cardio-ventilatory reserve, after alpha-2 agonists. Reused from Tremblay, Neuropharmacology, 1986, 25, 41–46 [41] under Elsevier reuse free agreement (Dec, 06, 2017) with thanks.
3). Applied pharmacology
Alpha-2 agonists affect minimally baseline VO2 in resting volunteers (≈−15% [53–55]) but suppress increased metabolic activity (≈−50 to −80% depending of the amplitude of shivering or of the increased VO2 [53,59]). Clonidine low dose (75 μg i.v.) suppresses post-operative shivering [56]: a large reduction in VO2 was observed only during post-operative shivering itself. VO2 reduction was not observed in non-shivering patients [53], in line with the data observed in resting volunteers. By contrast, during withdrawal from remifentanil-propofol sedation and weaning from the respirator in trauma patients, a large reduction of VO2 (−41%) was observed following high dose clonidine bolus [59] (Figure 3). In some patients, when weaning from the ventilator was combined to opioid and/or alcohol withdrawal, only very high doses of alpha-2 agonists achieved VO2 control (up to 2700 μg≈38 μg.kg-1 of clonidine) (figure 2 in [59]).
Figure 3.
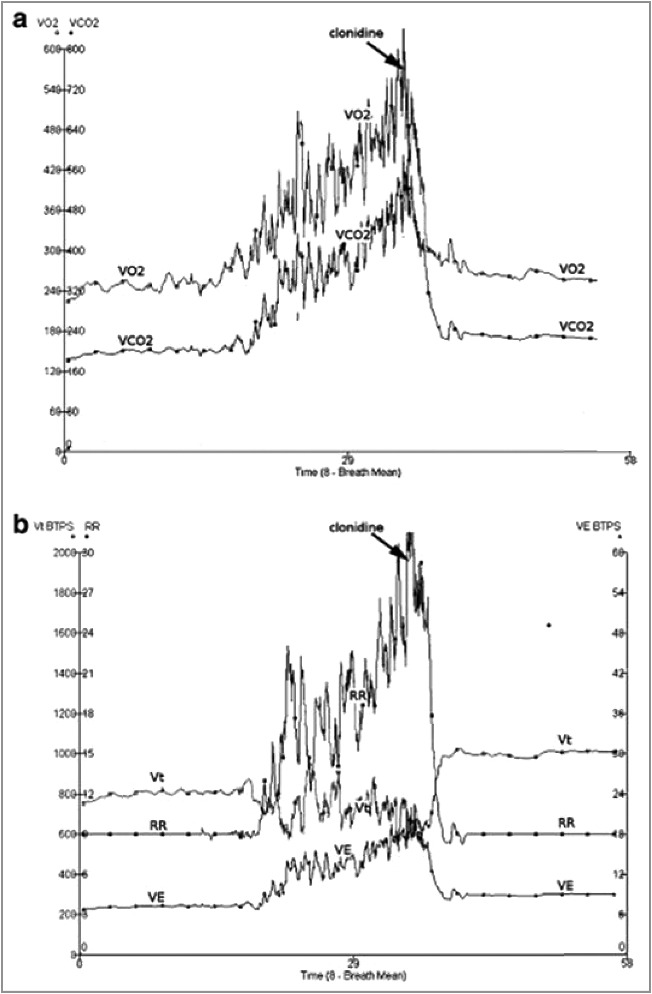
Reduced oxygen consumption following administration of an alpha-2 agonist (clonidine (900 μg i.v.) during withdrawal from remifentanil-propofol sedation, while depth of sedation decreased (Ramsay score: from ≈3.5 under remifentanil-propofol to ≈2.2 after clonidine). Abbreviations: VO2: O2 consumption; VCO2: CO2 production; RR: respiratory rate; VE: minute ventilation. Note a) the normalized tachypnea and increased Vt after clonidine relative to pre-interruption of remifentanil-propofol administration b) some patients need very high dose of clonidine (2700 μg, i.e. 1.6 μg.kg-1.h-1) to lower VO2. These very high requirements of alpha-2 agonists fit with similar observation in a minority of patients in which sympathetic activity has been increased for an extended period of time, or in patients presenting with addiction and/or delirium tremens or Gayet-Wernicke (Pichot and Quintin, unpublished data). Reused from Liatsi, Intens Care Med, 2009, 35, 271–81 [45] with paid agreement from Springer (Dec, 7, 2017).
Presumably, alpha-2 agonists reset the activation threshold of cold defense effectors, evoke adaptation to a lower external temperature, suppress or minimize shivering and generate vasodilation. To our knowledge, heat transfer from core to periphery or from periphery to core is poorly described in the setting of septic shock with, respectively, elevated body temperature or spontaneous hypothermia. Suppressed shivering with an alpha-2 agonist is the opposite to the vasoconstriction and shivering evoked by surface cooling performed without “cooperative” sedation or without general anesthesia. The current practice is to use light general anesthesia (light “total intravenous anesthesia”: TIVA; “analgo-sedation”) to decrease the activation threshold of cold defense effectors for shivering: this is considered as the most efficient way to prevent shivering and achieve VO2 reduction [7]. Given the ventilatory, cognitive, immuno-suppressive [60] effects and complexity of administration of general anesthesia, our present hypothesis is at variance with the present clinical practice: we suggest not to use “analgo-sedation” with hypnotics and opioid analgesics i.e. not to use light total intravenous anesthesia5.. Minimal sedation and well preserved ventilation are key: for clinical applicability, therapeutic hypothermia will be induced in patients handled on a ward [57] or an intermediate care unit. Although patients presenting with septic shock or ARDS are handled in a CCU, suppressed vasoconstriction and shivering should be achieved simply: the activation threshold may be modified while maintaining spontaneous ventilation and « cooperative » sedation with alpha-2 agonists6..
4). Reduced vasopressor requirements?
In addition to reduced VO2 and reduced shivering, may alpha-2 agonists be of help in the setting of septic shock? In the setting of septic shock, following alpha-2 agonists, a reduction of the dose of noradrenaline was observed clinically [24,61], experimentally [24,62,63], and, post-hoc, in the setting of sepsis [64]. Outside the setting of sepsis and septic shock, administration of alpha-2 agonists in the anesthesia or CCU setting leads to reduced vasopressor [65–69] or isoproterenol [70] requirements. In the setting of neonate [24] or terminal adult [71] septic shock, an alpha-2 agonist lowers noradrenaline requirements by 90% over 13 h [24] and 25% over 6 h and 80% over 48 h [71] respectively, irrespective of unabated elevated body temperature [71]. At first glance, such observations are thoroughly counter-intuitive: how a centrally acting anti-hypertensive agent can reduce pressor requirements in vasodilated septic patients? Adrenergic receptor sensitivity to endogenous catecholamines undergoes major changes during sleep, rest, or exercise [72–74] or under pathological circumstances [75]. In resting healthy volunteers, clonidine (2 μg.kg-1 i.v. over 10 min) lowers plasma noradrenaline concentration, increases lymphocytes beta-adrenergic receptor density, and decreases the affinity of the beta receptor [76]. The kinetics of the changes is compatible with the observed changes in circulatory variables and plasma noradrenaline concentration. Presumably, a rapid externalization of the receptors occurs immediately after a reduction in sympathetic nervous activity. But the phenomenon is not observed in vitro [76]. Thus, the simplest explanation for the increased noradrenaline requirement during septic shock and the lowered noradrenaline requirement following administration of alpha-2 agonists is a switch from “hyper-innervation desensitivity” (septic shock without alpha-2 agonist: high endogenous NA plasma concentration [77]) to “denervation hypersensitivity” (septic shock with alpha-2 agonist: lowered endogenous NA plasma concentration i.e. normalized sympathetic nervous activity back toward baseline levels) [61,62,71]. As septic shock involves many factors, other explanations are possible (NO, etc.) [78,79]: a) lowered temperature leading to vasoconstriction: this does not fit with the vasodilation generated by alpha-2 agonists b) normalization of an impaired micro-circulation (see below), leading to normalized local pH back toward normal, increased sensitivity to noradrenaline and reduced extravasation. Given the reduced requirement of vasopressor observed during hypothermia [38] or normothermia [3] in the absence of administration of alpha-2 agonists and the reduction in vasopressors requirement following administration of alpha-2 agonists [24,71], a combination of fever control and alpha-2 agonist may help in refractory septic shock.
Two issues are relevant:
-
a)
Micro-circulation. In the setting of septic shock, cardiac output should be increased7. back to adequacy, defined as the output needed to match VO2. This combines pre-load and afterload manipulations of the right [80] and left ventricles, right [81] and left coronary perfusion pressures, and adjustments of rhythm, contractility, and compliance. Nevertheless, achieving adequate cardiac output does not necessarily translate into achieving adequate micro-circulation [23], i.e. mixed venous saturation or superior vena cava saturation >70–75%8. [82], CO2 gap <5–6 mmHg [83–85], lactate trending <2mM and excellent capillary refill. Few drugs achieved popularity to improve the micro-circulation [86]. Fast suppression of peripheral mottling is observed when alpha-2 agonists are administered in the setting of septic shock treated with very high dose noradrenaline (Quintin, unpublished data). Evidence-based demonstration is required.
-
b)
Renal replacement. In the setting of major acidosis and early multiple organ failure, renal replacement therapy is used early, to correct the systemic acidosis. Fever control, reduced vasopressor requirement and normalized systemic acidosis [37,87] will be achieved more rapidly, when the RRT circuit is not actively warmed-up.
To sum up, fever control reduces vasopressor requirement [3,37,38]. Therefore, fever control in patients presenting with high noradrenaline dose [5] when limited cardio-ventilatory reserve is observed, especially in the setting of “refractory”9. septic shock. Combined to fever control, the use of alpha-2 agonist may reduce the vasopressor requirements. A prospective randomized clinical trial e.g. in the setting of refractory10. [88–90] septic shock should document the hypothesis [24,61,62,71,78,79].
III). Fever control in the setting of severe ARDS?
A). Present knowledge
Data are conflicting: high fever (≥39.5°) is associated with the lowest mortality in the setting of ARDS [91]: presumably most of these patients did not suffer from a limited cardio-ventilatory reserve. By contrast, elevated body temperature>38°C in the setting of mild ARDS (200<P/F<300) is associated with increased duration of ventilation [16]. As stated with respect to septic shock [10], hypothermia<36.0°C is associated with increased mortality in the setting of mild ARDS [16,91]. However, fever is also associated with delayed weaning [16]. Therefore, aggressive fever control should be considered in mild ARDS [16]: indeed, reducing the metabolic and ventilatory demand should be among the most important of unproven rules that guides management especially in the initial stage of ARDS [92].
In the setting of severe ARDS (P/F<100 on PEEP = 5 cm H20, bilateral opacities not explained by cardiac failure or fluid overload [93]), evoked hypothermia or fever control are associated with improved gas exchange (Table 1; table 1 in reference [94]):
-
1)
Experimentally, evoked hypothermia (33°C) increases PaO2, lowers lung edema formation, and alveolar hemorrhage when exposed to high pressure controlled ventilation [95].
-
2)
Following evoked hypothermia (34°C), increased PaO2 was observed. PaO2 fell when temperature returned to 37°C. In patients presenting with ARDS (P/F ≤50, n = 8) following trauma, blood loss, and resuscitation, hypothermia was used as an alternative to extracorporeal membrane oxygenation (ECMO; survival in the hypothermia group without ECMO group: 87%) [96].
-
3)
Following bronchospasm, evoked hypothermia (34.5°C, 5 days), sedation and paralysis are associated with increased PaO2 (P/F = 100 to 225–400), improved radiologic findings and recovery [97].
-
4)
Under sedation and paralysis, evoked hypothermia (33–35°C) is associated with decreased intrapulmonary shunt and VO2 and increased arterio-venous difference and O2 extraction ratio [98]: evoked hypothermia may be detrimental by decreasing O2 transport more than O2 consumption: a disequilibrium may occur11..
-
5)
Given refractory hypoxia12. (P/F = 54) and a low cardiac output, high frequency ventilation combined to evoked hypothermia (33°C, 10 days, positive end-expiratory pressure: PEEP = 30 cm H20, pancuronium-fentanyl, steep Trendelenburg position) was associated with recovery [99].
-
6)
Under general anesthesia (propofol-phenoperidine), hypothermia (32–34°C, 11 days) led to permissive hypercapnia (PaCO2≈145 mm Hg), improved P/F from a low 58 and recovery. Evoked hypothermia may be used as a “bridge” during transfer to ECMO [100].
-
7)
Moribund septic ARDS patients (P/F = 47–101 on PEEP≈10–11 cm H20; expected mortality: 100%) were allocated to state-of-the-art vs. state-of-the-art+evoked hypothermia (32–35°C, ≈70 h, discontinued if P/F >90) [39]. Under evoked hypothermia, the intrapulmonary shunt was reduced (42 to 32%) and PaO2/PAO2 increased (0.15 to 0.27). Lowered VO2 was observed during the first 6 h (243 to 217 mL.min-1, ns). O2 extraction was increased (26% to 30%), as observed earlier [98]. Death was 100 and 66% respectively in the state-of-the-art and state-of-the-art +evoked hypothermia groups [39].
-
8)
Mild evoked hypothermia (34.8 °C) in septic shock patients undergoing high-flow continuous hemofiltration reduces VO2 and venous admixture (30% to 18%), increases PaO2 and mixed venous PO2 with unchanged outcome [37]. Under continuous hemofiltration, evoked hypothermia (30.5°C, 7 days) is associated with reduced minute ventilation (30 to 6 L.min-1), increased PaCO2 (38 to 54 mm Hg) and increased P/F (118 to 235). This allows one to reduce PEEP (30 to 10 cmH20) [87]. Presumably, permissive hypercapnia allows lung rest and recovery [87].
-
9)
As P/F = 109 further worsened, very low Vt (≤4 mL.kg-1) and lowered plateau pressure (23 cm H2O to 18) were achieved using evoked hypothermia (35–36°C) and mild permissive hypercapnia (50–68 mm Hg) [101]. This temperature was selected to avoid coagulation and infection problems.
-
10)
In severe ARDS (P/F≈40–50, n = 2) refractory to veno-venous ECMO and proning (n = 1), evoked hypothermia (32–34°C, 24 h) improved oxygenation (P/F≈50 to 119 and 40 to 69 respectively). Survival occurred in one patient [94].
-
11)
PEEP (from zero to 20 cm H20) lowers cardiac output and shunt, increases P/F (calculated with FiO2 = 0.7 from table 1 in reference [102]: ≈90 to ≈115) and underestimates lung injury [103]: shunt changes as a function of cardiac output without change in ventilation/perfusion (VA/Q) ratio (figures 1 and 3 in reference [104]. Under veno-venous ECMO, in the setting of severe septic shock and ARDS requiring very high noradrenaline dose, lowering temperature (37 to 34°C) increases SaO2 from 82 to 94% [105]. Evoked hypothermia reduces cardiac output and increases the ratio ECMO flow/cardiac output: pulmonary flow and intrapulmonary shunt are reduced. Thus, an imbalance between ECMO flow and cardiac output improves oxygenation [105].
-
12)
A retrospective study of evoked hypothermia (32–34°C, ≈30 h) in the cardiac arrest setting observed lower PaCO2 and Vt, increased P/F with similar PEEP, increased compliance. The increased P/F was maintained after rewarming [106].
-
13)
Mild evoked hypothermia (34–36°C, 48 h) was generated prospectively in ARDS patients (P/F<150, n = 8) under paralysis. When historical controls are compared, the mortality is lowered (mild hypothermia: 25%; controls: 75%). More ventilator- and CCU-free days, higher P/F upon day 3 (from 86 to 255) were observed [107].
-
14)
A large (n = 101) trial on fever control in septic shock was conducted primarily on patients with moderate ARDS (50% of the patients; P/F: 153–165) [3]. The 14d mortality decrease is most relevant to the present problem. Nevertheless, no prospective randomized clinical trial addressed fever control in the setting of severe ARDS.
Table 1.
Literature search on hypothermia in the setting of ARDS. Retyped from Hayek, J Intens Care Med, 2017, 1–5 [94] with paid agreement from SAGE (Dec, 8, 2017).
| Authors | Year | n | Age | TH Temp (°C) | TH Length (days) | Etiology | Paralysis | Tidal volume | Other therapies |
|---|---|---|---|---|---|---|---|---|---|
| Flachs | 1977 | >8 | 34 | Trauma, blood loss, ressuscitation | Yes | None | |||
| Hurst | 1985 | 1 | 32 | 33 | 10 | Trauma, massive transfusion | Yes | HFV | None |
| Browning | 1992 | 1 | 20 | 30 | 5 | Status asthmaticus | Yes | Halothane | |
| Wetterberg | 1992 | 1 | 20 | 32-4 | 11 | Pneumonia | PCV, peakP=40 cm H20 | Isotonic buffering, prostacyclin | |
| Villar | 1993 | 9 | 40±16 | 32-5 | 2.9±0.6 | Sepsis, trauma, pneumonia | Yes | TPN, Swan Ganz catheter | |
| Moonka | 1996 | 1 | 48 | 30.5 | 7 | Trauma, massive transfusion | Yes | Hemodialysis | |
| Matsuno | 1997 | 10 | 35 | Respiratory failure secondary to infection | None | ||||
| Erikson | 1998 | 2 | 41 | 35 | 2 | Lung transplantation : rejection vs. infection | isotonic buffering, steroids | ||
| 55 | 32 | 4 | Lung transplantation : rejection vs. infection | isotonic buffering, steroids | |||||
| Duan | 2011 | 1 | 29 | 35-36 | 6 | Aspiration | Y | 4 mL.kg-1 | NO, inhaled prostaglandin, oral PDI |
| Varon | 2012 | 4 | 32 | 1 | not available | none |
Abbreviations: HFV: high frequency ventilation; NO: nitric oxide; PCV: pressure controlled ventilation; PDI: phosphodiesterase inhibitor; TH: therapeutic hypothermia; TPN; total parenteral nutrition.
B). Alpha-2 agonists to modify the management of severe ARDS?
Time-wise, severe ARDS undergoes 3 phases. First phase: acute cardio-ventilatory distress upon admission to the CCU [108] (“shock state”), until its correction. Controlled mandatory ventilation, with paralysis, is appropriate in the setting of «Friday night ventilation» [108] to address acute cardio-ventilatory distress immediately when the patient reaches the CCU. However, the intensivist should keep in mind that the goal is to make the patient self-sufficient, ventilation-wise, as early as possible: the short time-window to skip CCU-acquired diseases implies literarily running against time. Second phase: early severe ARDS. Early ARDS is to be managed under spontaneous ventilation-pressure support as soon as the situation has improved [109], our present hypothesis. Third phase: late ARDS after 3–5 days, not considered here.
1). Working hypothesis: a new bundle?
As delineated [110–112], after correction of one factor after the other factor generating the acute cardio-ventilatory distress [108], the present hypothesis is that spontaneous ventilation combined to alpha-2 agonists helps in the setting of severe diffuse ARDS. An “analytical” bundle separates the factors involved in the ARDS conundrum, using an itemized, step by step, process:
-
a)
Position: upright position improves oxygenation [113,114] especially when severe ARDS is considered (Figure 4; panel B in electronic supplement [115]), as delineated [111,116]. Intra-abdominal pressure is to be lowered.
-
b)
Temperature: in the setting of elevated body temperature, fever control reduces VO2 [29]. As alpha-2 agonists lower VO2 [53–55], an additive effect between fever control [3,29], alpha-2 agonists [54] and noradrenaline requirements [28], lowered in the presence of alpha-2 agonists, is a possibility. As proposed in septic shock [3] and ARDS [107], fever control should be carried away for 48–72 h, depending on the cardio-ventilatory status, and the micro-circulation.
-
c)
Acidosis: adequate cardiac output implies correction of a “low PvO2 effect” [109], assessment of a possible patent foramen ovale [117] and improvement of right ventricular function [81,118]. Adequate cardiac output will improve micro-circulation [23], normalize systemic pH and suppress the systemic acidotic drive on the respiratory generator. In the setting of ARDS [119], a pH>7.23–29 is required to use spontaneous ventilation (synchronized intermittent mandatory ventilation). Accordingly, pH = 7.29 appears as a cut-off point [120] if spontaneous ventilation is to be used early in the setting of ARDS. By contrast, severe acidosis generates a high RR, making spontaneous ventilation impossible [121].
-
d)
Permissive hypercapnia: lowered temperature lowers PaCO2, driving pressure, Vt [101] and respiratory rate (RR). Following alpha-2 agonist, an observation gathered in healthy volunteers [54] of lowered Vt and lowered sensitivity to CO2 may apply to mild permissive hypercapnia in the CCU. This would improve right ventricular function [122] and minimize patient self-inflicted lung injury [123] (ventilation13.-induced lung injury).
-
e)
High FiO2: a degree of permissive hypoxemia (SaO2≈88–92%) is acceptable in the setting of controlled mandatory ventilation (CMV) [124] and paralysis. By contrast, under spontaneous ventilation, heightened SaO2 at or above 96% lowers the hypoxic respiratory drive and RR [125]. Thus, in the setting of severe diffuse ARDS, firstly, immediately upon admission, the hypoxic patient is administered immediately high FiO2 = 0.8–1.0 and high PEEP, with acceptable plateau pressure. Secondly, all the other factors are to be improved, one by one, according to the equation (Vt, RR = f(temperature, pH, PaCO2, PaO2)), during the stabilization of the cardio-ventilatory distress. Thirdly, hypoxia is handled specifically, using high FiO2 and high PEEP: FiO2 is lowered to 0.4 as quickly as possible. Then PEEP is lowered from high levels (15–24 cm H20) to acceptable levels (≤10 cm H20), as soon as possible. The objective of the analytical bundle is to isolate low PaO2 as the only variable left evoking high respiratory drive. Therefore, the issue is not to fully re-open the whole lung with very high PEEP [126,127], curing P/F at once (“open lung strategy”) [126–128]. Under spontaneous ventilation-high PEEP-low pressure support, the objective is only to recruit a “penumbra area” [116] and increase P/F≈50 to P/F≈150–200. Schematically, the issue is to move from the worst case scenario (FiO2 = 1, PEEP = 24, P/F = 50) to an acceptable scenario (FiO2 = 0.4, PEEP = 10, P/F = 150): after having used the above mentioned bundle (combined if appropriate with veno-venous ECMO), the sole stimulus on the respiratory generator is hypoxemia. Then, given improved overall clinical status, early switch to continuous non-invasive ventilation should be considered immediately following correction of temperature, systemic pH, and acceptable permissive hypercapnia.
-
f)
High PEEP [129] resets the chest wall and diaphragm [130–132], lowers RR [133], minimize inflammation [134] and improves oxygenation over 12–72h [135–138]. While “focal” [139] ARDS requires low PEEP [139], diffuse ARDS requires higher PEEP [129,140,141].
-
g)
Spontaneous ventilation: Airway Pressure Release ventilation (APRV+SV) [142,143]) is under trial (BIRDS:clinicaltrials.gov). Based on our practice, we use inspiratory assistance [144] i.e. Pressure Support [110,138,145]. Pressure support reduces ventilatory demand outside the setting of ARDS [146–148]. If selected in the setting of early severe ARDS, after control of the acute cardio-ventilatory distress, pressure support should be set rigorously: high peak flow rate [148,149] to handle the high respiratory drive [150] and lower the work of breathing, low inspiratory trigger, low expiratory trigger [151] given a restrictive lung, 100% automatic tube compensation [152] to lower work of breathing, low pressure support [138,145,153] to avoid increasing Vt. In this respect, the “Smart Care” software [154] (Drager Evita XL4/infinity V500) works surprisingly well in severe “focal” ARDS [138] and lowers pressure support and Vt, as early as possible (“inverted settings” [155]), despite high PEEP levels.
-
h)
Cooperative sedation evoked by alpha-2 agonists is compatible with spontaneous ventilation [43,54,156]. After optimized volemia and A-V conduction, clonidine or dexmedetomidine infusion are administered immediately following tracheal intubation (respectively: 1–2 μg.kg-1.h-1 [157,158]; 0.75–1.5 μg.kg-1.h-1 [68] to −3<RASS<0)14.. After correction of systemic acidosis (pH>7.30, CO2 gap<5–6 mm Hg, lactate trending <2mM), paralysis is withheld as soon as possible, usually in our hands within ≈3–12h: the patient regulates his Vt (≤4–6 mL.kg-1) and RR (≤20–25 breaths per min : bpmin) by himself, under pressure support, as early as possible Presumably, a patient presenting with early multiple organ failure and major acidosis will need a longer stabilization of the acute cardio-ventilatory distress (extrapulmonary ARDS) than a patient presenting single organ failure (pulmonary ARDS). Following extubation, cooperative sedation will allow one to provide continuous non-invasive ventilation [159–161] with PEEP≤10 cm H20.
-
i)
“Rescue therapy” (controlled mandatory ventilation with low driving pressure [30] or low Vt, paralysis [162], proning [163], veno-venous ECMO, NO, inhaled prostacyclin, almitrine, etc.) should be considered if this bundle is inapplicable in the considered patient.
Figure 4.
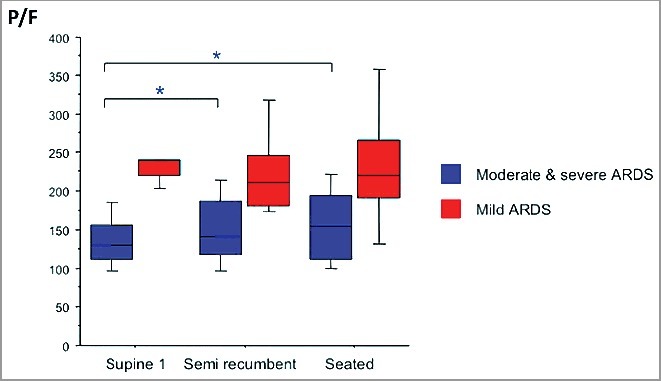
Effect of seated position on PaO2/FiO2 (P/F) in moderate and severe ARDS. P/F increased from ≈125 to ≈155 in moderate and severe ARDS. Four epochs were studied: supine1, semi-recumbent, seated, supine 2 (not shown). Blue boxes represent moderate & severe ARDS defined as PaO2/FiO2 = P/F<200. Note the statistically significant, and clinically relevant, changes for moderate and severe ARDS when seated is compared to supine. Modified from Dellamonica, Intens Care Med, 2013, 39 : 1121–7 (figure in electronic supplement, panel B) under Springer reuse free agreement (Dec, 7, 2017) with thanks.
2). Spontaneous ventilation vs. controlled mandatory ventilation
Presently, the state-of-the-art combines deep sedation [164], paralysis [162] and proning [163] throughout acute cardioventilatory distress and early ARDS. The result [163] is remarkable, halving mortality (prone: 24 vs. supine: 41%, ≈17 vs. 19 days of invasive ventilation respectively, circa 4–5 days of general anesthesia and paralysis; Guerin, personal communication). The use of general anesthesia and paralysis [162,164] is based on suppressing ventilator-patient asynchrony [165] and lowering VO2 for 48 h after admission to the CCU. Rescue therapies are used without paying attention to lowering VO2 in the setting of limited cardio-ventilatory reserve. As there is no randomized trial showing a benefit of CMV+paralysis+general anesthesia vs. normothermia+spontaneous ventilation+cooperative sedation, these papers [162–164] offer no appropriate control15., with respect to the present hypothesis. The weight of these data [162,163] rests on absence of knowledge and existing clinical practice rather than on full evidence-based medicine with appropriate control group. As the management of severe ARDS requires deep sedation [164], the dogma of deep sedation combined to paralysis is reinforced when hypothermia (34.5°C) is considered [97]. Such successful management [97] is part and parcel of advances within previous clinical management. Nevertheless, the present advancement in physiopathology and pharmacology allows one to propose an alternative bundle without general anesthesia and paralysis.
The ARDS is a pathophysiological conundrum (temperature, pH, PCO2, Vt, RR, inflammation, “wet” lung, etc.). Adding therapeutic complexity to this conundrum is unwise. General anesthesia, paralysis and controlled mandatory ventilation are followed by reduced cardiac output. In turn, lowered cardiac output leads to lowered P/F [103,109]. Then iterative echocardiography, pre-emptive volume loading [166,167], vasopressors, inotropes, etc. are needed. Schematically, the intensivist has two options: general anesthesia, paralysis, controlled mandatory ventilation, etc., then controlling for the deterioration evoked by this combination. Conversely, the intensivist should concentrate on improving low PaO2, the hallmark of ARDS, by shortening the course of general anesthesia, paralysis, controlled mandatory ventilation. From the data delineated in this review, a therapeutic shift (VO2 control, early spontaneous ventilation-high PEEP-low pressure support, cooperative sedation) is now possible, after swift and itemized control of the factors evoking the acute cardio-ventilatory distress, as delineated above.
3). Alpha-2 agonists
a). Pharmacology
Briefly [157,111], alpha-2 agonists evoke: i) ataraxia (slow-wave sleep [168,169], “arousable” [170] or “cooperative sedation” [171] and minimized emergence delirium [44–46]. In addition, a degree of analgesia [172] and analgognosia [173] is observed following alpha-2 agonist administration. Deducing sedative properties from circulatory properties, or vice-versa, is irrelevant: circulatory and cognitive effects of alpha-2 agonists are anatomically un-related [174], being located respectively in the lower brain stem vs. the forebrain. The only common link is the alpha-2 adrenergic receptor distributed widely in the central nervous system and the autonomic nervous system: this wide distribution explains the multiple, protean (“pleiotropic”), effects of the alpha-2 agonists. ii) absence of depression of the respiratory generator [43,156], increased number of ventilator-free days [175,176]. iii) increased peak expiratory flow rate and forced expiratory volume (FEV1) in asthmatic patients [177]. This is relevant in the setting of early ARDS: an intrinsic PEEP as high as 7 cm H20 is observed [178]. iv) reduced intrapulmonary shunt [179,180]. v) reduction in pulmonary hypertension evoked by normalizing the sympathetic hyperactivity [181]. vi) increased left ventricular diastolic compliance [182] and systolic performance [183–186]. vii) improved micro-circulation [187,188] and lactate clearance [189], reduced vasopressor requirement in the setting of septic shock [24,71] and sepsis [64]. viii) reduced microvascular permeability [190]. ix) increased diuresis in the setting of ascites [191–193], cardiac failure [186] and critical care [67]. x) reduced intra-abdominal pressure [194]. xi) lowered PCT [24] and pro-inflammatory IL6 [195], increased anti-inflammatory IL10 [196].
In semi-recumbent healthy volunteers [54], dexmedetomidine high-dose (2 μg.kg-1.h-1) reduces minute ventilation. This affects predominantly Vt (−33%) as opposed to RR. PaCO2 increased ≈+4 mmHg. The combination of lowered minute ventilation and increased systemic CO2 indicates lowered sensitivity to hypercapnia16. [54]. Under alpha-2 agonist, during CO2 rebreathing (PaCO2 = 55 mm Hg), minute ventilation was lowered (−43% from baseline; ≈−46% vs. placebo [calculated from figure 4 in [54]]. pH and PaO2 were unchanged. The point is whether increased set-point to CO2, lowered slope, lowered Vt, absence of acidosis (figure 6 in [54], reduced temperature [54] and VO2 (−17% [54]; −13% [55]) present relevance when early severe ARDS is considered. After controlling all confounding factors, will alpha-2 agonists allow one combining fever control, permissive hypercapnia and high PEEP-low pressure support and achieving low Vt and RR? Following an alpha-2 agonist, in healthy volunteers, lowered Vt and unchanged RR are observed [54]. By contrast, in the setting of weaning from the ventilator and alcohol/opiates withdrawal, Vt is unchanged and RR lowered [59]. Indeed, lowering RR has relevance in the setting of ARDS [125,150,197]. In our hands, despite a PaCO2≈45–55 mm Hg in ARDS patients, no hyperpnoea nor tachypnea were observed under alpha-2 agonists and spontaneous ventilation-low pressure support, following control of temperature and systemic acidosis (Quintin, unpublished observations). Given the present hypothesis, recent data [120,150,198] require an itemized discussion in order to carve out a niche for alpha-2 agonists:
b). High Vt in early ARDS
One key issue addresses the possibility to handle tachypnea and hyperpnoea in the setting of acute cardio-ventilatory distress occurring during early severe acute respiratory distress syndrome.
High Vt:
-
i)
Experimentally, inspiratory resistive breathing increases alveolar-capillary membrane permeability, deteriorates P/F and ventilatory mechanics, evoking inflammation [199].
-
ii)
Exercise: Intense exercise increases red cells and proteins concentrations in the broncho-alveolar lavage [200]: this requires analysis, bearing in mind patient's-self-inflicted lung injury (P-SILI) [123]. However, marathoners pulls Vt>25 mL.kg-1 for hours without major untoward effects [201].
-
iii)
early ARDS: in the early phase of ARDS under non-invasive ventilation (NIV) [150], the sicker patients (P/F = 122 ; range = 98–191) generated a large Vt = 10.6 mL.kg-1 (range = 9.6–12) and required intubation. However neither pH (success: 7.41 vs. failure: 7.45) nor PaCO2 (no intubation required: PaCO2 = 36; intubation required: PaCO2 = 32 mm Hg) allows one partitioning hyperpnoea along the equation (Vt, RR = f(pH, PaCO2, etc.)).
-
iv)
late ARDS: rapid shallow breathing (RR = 27–32 bpmin; decreased Vt: 243–335 mL, increased respiratory drive [P0.1 = 4.9–6.6 cmH2O], intrinsic PEEP = 2–5 cmH2O) was observed in the chronic phase of lung injury (≈14 days) [197]. This represents muscular weakness or impending fatigue, at variance with early ARDS.
Taken together these data suggest to look for P-SILI in the early phase of ARDS in order to suppress it. The altered geometry of the chest wall (including the diaphragm), lowered functional residual capacity (FRC) [130], atelectasis [202], lung water [203], inflammation, etc. imposes generating large end-inspiratory transpulmonary pressure to re-open the alveoli. This generates a large Vt and harms the lung. Strong patient's inspiratory efforts are observed even with pressure support as low as 7 cm H20 [150]: Vt cannot be controlled noninvasively despite very low pressure support [204]. The limiting factor is not the ventilator's set up but the patient himself (hyperpnoea: “soif d'air”) which generates P-SILI.
The first move to handle a large Vt is an appropriate resetting of the geometry of the chest wall. Experimentally, under spontaneous ventilation, a) high PEEP (17 cm H20 vs. ≈6 cm H20) reduces the curvature of the diaphragm, reduces the generation of negative transpulmonary pressure and lung dyscoordination [132] b) expiratory diaphragmatic mechanics is improved [131]. Clinically, a) spontaneous ventilation improves rib cage mechanism [130] b) increasing PEEP (0 to 10 cm H20) increases the expiratory time and lowers respiratory rate at constant Vt: the Hering-Breuer reflex acts within 1 breath17. [133]. The RR is lowered more in patients with impaired compliance, as opposed to patients without impaired compliance [133]. To sum up, high PEEP increases the expiratory time [133], restores the functional residual capacity and cures hypoxemia over 12–72 h [135,136].
Pulmonary edema. Strong inspiratory efforts maximize the possibility of pulmonary edema (figure 1 in reference [205]. This mimics hydrostatic post-obstructive pulmonary edema [206]. In figure 1 of [205], when spontaneous ventilation is added to mechanical breath18., the hydrostatic transvascular pressure19. increases from +2 to +28 mm Hg. However, the reasoning [205] accounts for only one side of the phenomenon: the lymphatic transmural pressure gradient is positive under spontaneous ventilation throughout the ventilatory cycle, at variance to what is observed under controlled mechanical ventilation [207]. Thus, under spontaneous ventilation, lymph resorption may counteract transalveolar water leakage: this is an unsolved issue. Pharmacologically, an alpha-adrenergic blocker, phentolamine, restores alveolar liquid clearance following hemorrhagic shock [208]. Therefore, given the ARDS lung, the question of lung water being lowered by a combination of gentle spontaneous ventilation and normalized sympathetic activity is to be studied.
At first glance, a large Vt in the setting of early ARDS [150] favors [205] controlled mandatory ventilation and paralysis. Only mild spontaneous efforts can be tolerated to recruit the collapsed lung [198,209]. During the early acute cardio-ventilatory distress, acidosis, hypercapnia, hypoxemia, pain, elevated body temperature, pulmonary inflammation evoke large Vt. Thus, each factor should be handled separately during control of the acute cardioventilatory distress. The equation (Vt, RR = f(temperature, pH, PaCO2, PaO2)) should be re-written: (Vt, RR = f(temperature [VO2], pH [lactate, mixed venous SO2, CO2 gap, metabo-reflex20.], PaCO2, PaO2, inflammation, “cooperative” sedation)). To separate these various items, several questions need analysis:
-
i)
Differences between severe vs. mild or moderate ARDS: The mechanisms set in motion between mild and moderate ARDS are identical [145]: the issue is not the severity of lung injury itself but the ability of spontaneous breathing first to generate enough end-inspiratory transpulmonary pressure21. to open collapsed units and second the settings of PEEP and/or expiratory time to maintain the collapsed units open at end-expiration. This will determine whether spontaneous breathing is protective or harmful [145]. We presume this holds true in the setting of severe ARDS.
-
ii)
Lowering PS level [155] is presumably not sufficient [204] in order not to increase an already enlarged Vt evoked by the severity of the disease [150].
-
iii)
Cooperative sedation may be the pharmacological adjunct of a physiology-based management. Our observations [137,138,210] are at odd with [150,204]: combined to fever control, pH and PaCO2 control and alpha-2 agonists, high PEEP-low pressure support [145,153,155] allow one to observe a low Vt, acceptable RR, no signs of ventilatory fatigue (anxiety, nasal flaring, sternal notch retraction, use of accessory muscles and thoraco-abdominal dyscoordination). The analytical management delineated above should minimize the duration of controlled mandatory ventilation with paralysis, away from a prescription which entails an arbitrary length: 24- 48 h [162,211].
c). High transpulmonary pressure generated by pressure support
Spontaneous ventilation-pressure support may be harmful even beyond the period of early acute cardioventilatory distress [150]. Spontaneous ventilatory efforts make pleural pressure more negative and increases transpulmonary pressure at identical airway pressure [198,212]. Thus, “naïve” [213] use of spontaneous ventilation-pressure support generates a high transalveolar pressure: pressure support = 18 cm H2O evokes transpulmonary pressure = 51 cm H20 [213]. Transpulmonary pressure and Vt reach excessive levels despite low airway pressure when attention is not paid to the Vt delivered under pressure support [214]. This could be a consequence of either a high pressure support (ventilator-induced lung injury) or high ventilatory demand (P-SILI), or both.
d). Lung dyscoordination
Under CMV and paralysis in an ARDS patient, simultaneous inflation of lung regions occurring at different inflation rates is observed. By contrast, upon spontaneous ventilation, inflation of dependent lung region in the lowest position (dependent lung region, usually at the base of the lung in the erect patient or the dorsal part of the lung in the supine patient) occured. This inflation was greater than with controlled breaths [215]: this is a positive finding. Furthermore, the early inflation of the dependent lung region was accompanied by simultaneous transient deflation of non-dependent region: during early inspiration, gas moves from non-dependent regions to dependent regions causing lung dys-coordination (“pendel-luft”; figure 1 and table 1 in reference [198]. Thus, asynchronies develop within the lung under spontaneous ventilation [198]. However this observation occurred in one acidotic patient (pH = 7.28; PaCO2 = 46 mm Hg; Base excess≈−8; Vt = 532 mL). Presumably, acidosis increases the inspiratory effort and generates P-SILI. To hold true, the reasoning [198] needs replication in a large group of non-acidotic patients.
When atelectasis occurs, lung dyscoordination [198] is of further concern (figure 1 in reference [209]: an atelectatic lung (“focal” ARDS [139] does not behave as an healthy homogeneous lung (“fluid-like behavior”). The diaphragmatic contraction is concentrated asymmetrically in dependent lung regions (“solid-like behavior”): a pendelluft is possible. Furthermore, the overstretch reinforces inflammation. This view holds for “focal” or patchy [139] ARDS. It is unknown whether this view hold true when diffuse [139] ARDS is considered. Furthermore, this view does not account for the modification of the geometry of the diaphragm: first, in the supine position, the human diaphragm subsides with paralysis [216]. Second, under spontaneous breathing and high PEEP, the geometry of the pig diaphragm is restored [132], generating lower transpulmonary pressure.
The current state-of-the-art [162–164] claims curing all the problems, at once, by combining general anesthesia, paralysis and proning: paralysis suppresses patient-to-the-ventilator asynchronies, intrapulmonary overstretch, pendelluft and inflammation. However, to skip CCU-acquired diseases, the point is that the trachea of the patient should be extubated as early as possible. There are no data addressing the duration of controlled mandatory ventilation with paralysis to obviate the tachypnea and hyperpnea observed during the acute cardio-ventilatory distress: 24 h [211]? 48 h [162] ? 4–5 days [163]? This requires a specify study, opposing the analytical management delineated above vs. state-of-the-art management. Meanwhile, CCU-acquired diseases creep in: we observed repeatedly ARDS patients left several weeks under paralysis (Quintin, unpublished data). Rather than resorting to a black box (general anesthesia, CMV, paralysis, proning) to solve the ARDS conundrum, physiopathological analysis will help sorting out, one by one, the factors involved, as early as possible during the management of the acute cardio-ventilatory distress.
e). Awake ECMO: Spontaneous ventilation in the setting of early severe ARDS?
Spontaneous ventilation on ECMO prevent barotrauma, volutrauma, circulatory impact of sedation, diaphragmatic dysfunction, ventilator-acquired pneumonia and sepsis [120]. Therefore, cooperative awake patients presenting with early severe ARDS under veno-venous ECMO were maintained under spontaneous ventilation with minimal sedation (27% of ARDS patients; 6 out of 8 extubated before ECMO but 50% re-intubated during the whole course of ECMO lasting 11 days, 38% of ECMO days spent on spontaneous ventilation; survival : 75%) [120]. Gas flow was increased to ≈84% of CO2 production, aiming for near-apnea when the whole VCO2 production is cleared out by ECMO. This suppresses the respiratory drive generated by CO2, as proposed above (§ III B 1). Nevertheless, at identical VO2, only 50% of the ARDS patients lowered their RR<25 bpmin and reduced pressure esophageal swings. Thus, dyspnea persists in half of the ARDS-ECMO patients, despite suppressed CO2 drive: another mechanism comes into play, e.g. inflammation22..
The patients which sustain spontaneous ventilation under ECMO are less acidotic and present lower out-of-hospital referral, duration of ventilation before ECMO (1 vs. 3 days), higher P/F (86 vs. 76), higher pH (7.29 vs. 7.23), similar PaCO2≈61–2 mm Hg, less non-aerated tissue (43 vs. 56%) and better cough ability [120]. When compared to the non-ARDS patients (transplant, acute decompensation of chronic obstructive pulmonary disease), the ARDS patients under spontaneous ventilation with ECMO present a lower CO2 tension measured at entry of the oxygenator [120]. A putative explanation for this surprising result is that the ARDS patients present worse peripheral micro-circulation and/or inadequate systemic circulatory improvement, when compared to chronic obstructive pulmonary disease or transplant patients. Nevertheless, this paper reports a major breakthrough [120], at variance with deep sedation [164], 48 h paralysis [162], and proning [163].
These data [120] show that the equation (Vt, RR = f(temperature, pH, PaCO2, PO2)) does not hold in most awake ARDS patients. Dyspnea is unrelated to CO2 drive, as CO2 removal is nearly complete. Another possibility would be dyspnea related to a huge degree of inflammation present during early ARDS [120]. Thus, the equation should be: (Vt, RR = f(temperature, systemic and local pH i.e. metabo-reflex, PaCO2, PaO2, inflammation, cooperative sedation))? As the feasibility of the awake+veno-venous ECMO+spontaneous ventilation technique appears limited when severe ARDS with another organ dysfunction is considered [120], our hypothesis [110,112] needs reformulation:
-
i)
physiology: could fever control be of help to lower Vt and RR? Each factor involved in generating tachypnea or hypercapnea should be improved, one factor after the other: the conundrum is to be dismembered. The data [120] suggest that the patients sustaining spontaneous ventilation under ECMO are either less sick or more adequately managed with respect to microcirculation. Presumably, a higher systemic pH, improved cardiac output and microcirculation would increase the feasibility of awake-ECMO-spontaneous ventilation.
-
ii)
pharmacology: Inflammation is prominent is this setting [120]. Should a short course of steroids considered? Conversely, alpha-2 agonists lower inflammation [24,195,196]. This anti-inflammatory property may add up to other properties (see III, B, 3, a).
To sum up, after itemized correction of the acute cardio-ventilatory distress [108], the combination of fever control, upright position, rigorous control of pH, PaCO2, micro-circulation, spontaneous ventilation with low pressure support (with or without veno-venous ECMO) and administration of alpha-2 agonists should isolate hypoxemia as the only variable addressed with high FiO2 [125] lowered as early as possible), and high PEEP-low pressure support. Low Vt and RR should be achieved to allow for spontaneous ventilation, as early as possible, in the setting of early severe ARDS in order to cause no self-inflicted harm to the lung.
IV). Perspectives
The suggestion for using fever control in the setting of septic shock and ARDS is substantiated by preliminary findings [3,71,137,138,217] and provides a rationale to set up randomized prospective trials [218] combining pharmacological treatment to a physiological management in the setting of limited cardio-ventilatory reserve, septic shock and/or early severe ARDS. Two issues deserve comments: a) may the intensivist improve his therapeutic approach to major CCU illness? b) does major, prolonged, CCU illness fits within a larger overall disease?
A). Extending the analysis?
The 2018 approach combines analytic diagnosis with analytic therapeutics: fever control ((3) and present hypothesis); improved macro [81,103,109] and micro-circulation [23,24] to handle circulatory shock, peripheral shut-down, systemic and local acidosis; controlled mandatory ventilation with paralysis and proning (or present hypothesis: spontaneous ventilation-high PEEP-low pressure support [111,155]) to lower the work of breathing and increase ventilator-to-patient synchrony; anti-infectious strategy, etc. Nevertheless, we surmise that this organ-targeted approach (heart, lung, kidney) should extend the analysis to other systems:
-
1)
cognition: minimized cognitive dysfunction following alpha-2 agonists [44,219].
-
2)
sympathetic activity: the normalization of the sympathetic hyperactivity toward baseline levels may normalize more quickly the micro-circulation (see above) and inflammation. Alpha-2 agonists increase the concentration of the anti-inflammatory IL10 [196] and lower pro-inflammatory cytokines [195]. This raises 2 questions. Firstly, the issue of the anti-inflammatory vs. the pro-inflammatory effects of general anesthesia (propofol, benzodiazepine and opioid analgesics [220] is complex [60]. The anti-inflammatory effects of anesthetics may occur in the setting of a pre-existing low inflammatory state (e.g. brief general anesthesia in the setting of minor surgery performed on an healthy patient). By contrast, anti-inflammatory effects of general anesthetics are at best unproven when they are administered, firstly for days/weeks in the CCU, secondly in the setting of a high inflammatory state generated by septic shock or ARDS. Conversely, the alpha-2 agonists may lower pro-inflammatory cytokines and increase anti-inflammatory cytokines as a consequence of normalized sympathetic activity, back toward baseline activity.
-
3)
inflammation: the influence of inflammation on the circulatory response to sepsis is underappreciated, e.g. when it comes to HR. For example, in the setting of experimental sepsis, tachycardia is independent of the sympathetic nervous system but mediated by inflammatory mediators [221]. In septic patients, the effect of ibuprofen in lowering HR (≈115 to 100 bpmin) [26] may be a consequence of lowered inflammation or of lowered temperature.
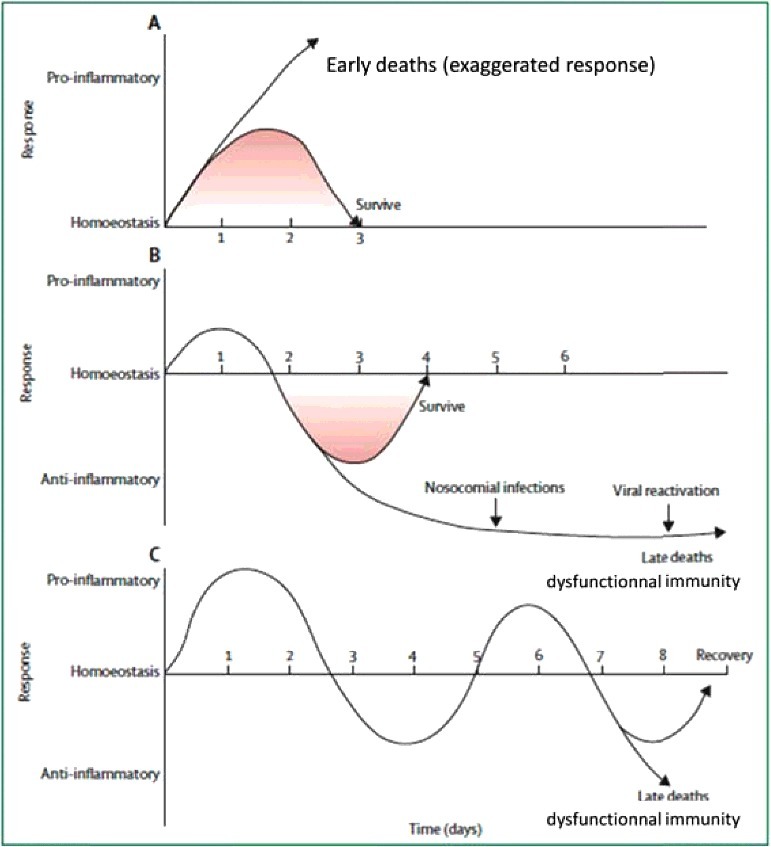
In septic shock, intertwined pro- and anti-inflammatory mechanisms lead to immunoparalysis [222]. Extreme pro-inflammatory activity is associated with a cytokine “storm”, hypotension refractory to high doses of noradrenaline and early death [88–90]. Extreme anti-inflammatory response is associated with a dysfunctional innate immune system and death (Figures 5 and 6; figure 1B in [223]; figure 2 in [224]). The sympathetic system is an adaptive system with complex anti- [225,226] and pro- [227] inflammatory effects under physiological conditions. By contrast, under major prolonged pathophysiological condition, this adaptive system becomes a mal-adaptive system23.. The massive release of noradrenaline presumably upsets the fine tuning of adrenergic receptors with functional consequences on immuno-competent cells (macrophages, natural killer and dendritic cells, neutrophil, eosinophil, basophil cells, T cells, natural killer T Cells). Secondly, acute activation of the sympathetic system inhibits the innate immune system [228,229]. Conversely, inhibited sympathetic activity may possibly evoke anti-anti-inflammatory effects: could this generate pro-inflammatory effects? Thirdly, the sympathetic system may present different actions in the acute (septic shock, etc.) vs. chronic (auto-immune diseases, cancer) settings. Thus a manipulation of the sympathetic system in the setting of septic shock is not straightforward.
Figure 5.
Inflammatory responses in sepsis. Immune responses in sepsis are determined by many factors including pathogen virulence, size of the bacterial inoculum, comorbidities, etc.
A) Although both pro-inflammatory and anti-inflammatory responses begin rapidly after sepsis, the initial response in previously healthy patients with severe sepsis is typified with an overwhelming hyperinflammatory phase (fever, hyperdynamic circulation, shock). During the early phase, deaths are generally caused by circulatory collapse, metabolic derangements, and multiple organ failure. Although no specific anti-inflammatory therapies have improved survival in large phase III trials, short-acting anti-inflammatory or anticytokine therapies offer a theoretical benefit. B) Many patients who develop sepsis are elderly with co-morbidities that impair immune response. When these individual develop sepsis, a blunted hyperinflammatory phase is common. Patients develop impaired immunity and an anti-inflammatory state;. Immuno adjuvant therapy offers promise in this setting. C) A third theoretical immunological response to sepsis is characterized by cycling between first hyperinflammatory then hypoinflammatory states. The development of a new secondary infection lead to a repeat hyperinflammatory response and may recover or re-enter the hypoinflammatory phase. Patients may die in either state. There is less evidence for this theory. The longer the sepsis carries on, the more likely a patient is to develop profound immunodepression. Presumably figures 5,6 and 7 look up at phenomenons occurring simultaneously with respect to metabolism and the immune system. This raises the question: should normalized sympathetic activity, normothermia and immuno-stimulation be combined? Reused from Hotchkiss et al, Lancet Infect Dis, 2013, 13, 260–8 223under Elsevier free reuse agreement (Dec, 6, 2017) with thanks.
Figure 6.
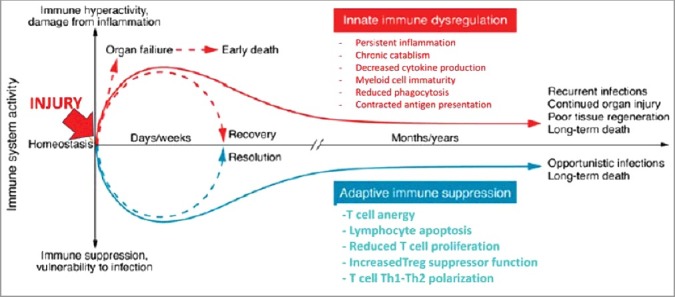
Immune dysregulation in sepsis. New insights into immune dysregulation were achieved using samples from deceased septic patients and severely injured trauma patients. An enduring inflammatory state driven by an upregulated innate and a suppressed adaptative immunity culminates in persistent organ injury and death. Accordingly, 80% of patients presented unresolved septic foci observed during a post mortem study (Torgensen 2009 quoted from [223]. The unabated initial inflammatory process contributes to organ failure and early mortality. This maladative syndrome is improved by newer “analytic” management. However, considering that the vast majority of sepsis survivors are elderly patients with multiple comorbidities, the short-term gain in survival have been merely pushed back by months or years. The widespread consensus is that persistent derangement in innate and adaptive immune systems are the main culprits driving long-term mortality. This raises the question: does normalized sympathetic hyperactivity would in turn normalize a dysfunctional innate immune system? Reused from Delano, J Clin Investigation, 2016, 126, 23–31 [224] under American Society of Clinical Investigation paid agreement (Dec, 6, 2017).
In the setting of experimental sepsis, depletion (reserpine, 6-0HDA) of noradrenaline neurons [230,231] and normalization toward baseline activity of sympathetic activity with an alpha-2 agonist, dexmedetomidine [232], improve survival24.. In healthy resting volunteers, inhibition of sympathetic activity with clonidine increases the density of beta-receptors on mononuclear cells and lowers the affinity of those beta receptors [76]: upregulation of adrenergic receptors [62,63,71,217] may occur on immunocompetent cells not only for beta receptors but for alpha-2 receptors as well. In addition, radiation increases the bacteria-induced inflammatory cytokines of the innate immune system; a ganglionic blocker, chlorisondamine, suppresses the phenomenon25. [233]: the implication is that the sympathetic system modifies the activity of the innate immune system, explaining, partially, the immuno-paralysis observed in the acute setting (septic shock). Clinically, administration of alpha-2 agonists lowers mortality in various subsets of CCU patients [40,64,234,235]. In the setting of septic shock or major surgery, normalization of sympathetic hyperactivity toward baseline levels by alpha-2 agonists has anti-inflammatory effects [195,217] including in the setting of septic shock ((233), Quintin, unpublished data)). Apparently, in our hands, the net overall effect of alpha-2 agonists is anti-inflammatory26.. Thus, the possibility of normalizing the sympathetic hyperactivity back towards baseline levels to optimize a dysfunctional innate immune system, inflammation, immuno-paralysis and outcome in the acute setting of septic shock or ARDS should be studied.
B). Enlarging the approach?
A “general alarm reaction” occurs in rats exposed for an extended period to various deleterious agents [236]: the description of a general alarm reaction matches major systemic inflammatory response. In the CCU, prolonged severe illness [224] may be considered as an expression of this general alarm reaction. In this respect, septic shock or ARDS are associated with a prolonged sympathetic hyperactivity which generates a maladaptive syndrome (dysfunctional response to shock and trauma: “reaction oscillante post-aggressive”: Figure 7; figure 1 in reference [237]. Normalization of this maladaptive syndrome was proposed to handle major surgery [1] and restricted to a subset of very severe combat casualties [2]. These early observations in the most severe combat casualties [2] match the improved survival in the most severe septic patients treated with dexmedetomidine [40], administered to normalize sympathetic activity back toward baseline levels. This maladaptive syndrome [237] occurs simultaneously with the immuno-paralysis [222] observed in the setting of septic shock (Figures 5 and 6). Nevertheless, a direct causality between sympathetic hyperactivity and innate immuno-dysfunction requires documentation. Early on [1], normalization of this maladaptive syndrome was achieved with neuroleptics (artificial hibernation, “protection neurovegetative” using “lytic cocktail”) [1]. Nowadays, alpha-2 agonists present relevance as they specifically normalize the sympathetic hyperactivity back toward baseline levels and achieve fever control more easily. They may, as well, engage the maladaptive syndrome, the increased inflammation [195,217] and the immuno-paralysis observed in septic shock and prolonged CCU stay.
Figure 7.

Dysfonctionnal response following shock or trauma (“reaction oscillante post aggressive”). Modified from Laborit, Therapie, 1951, 6, 207–10 [237] with free permission of Therapie and Societe Française de Pharmacologie et Therapeutique (Dec, 8, 2017) with thanks. Excessive catabolism may lead to prolonged, exaggerated disorders, multiple organ failure and death (“reaction dysharmonique”: dysfunctionnal response. The author [237] questioned the excessive use of sympathomimetic drugs. This led him to use of phenothiazines (“lytic cocktails”: chlorpromazine, promethazine, meperidine) to evoke hypothermia (“artificial hibernation”) [1]. Do alpha-2 agonists represent a tool more specific than phenothiazines to normalize early the sympathetic hyperactivity back to baseline levels?
Conclusion
Alpha-2 agonists allow one to evoke fever control without shivering, respiratory depression and cognitive dysfunction, at variance with general anesthesia/deep conventional sedation. Volemia and auriculo-ventricular conduction are to be optimized before early administration of alpha-2 agonists as first-line sedative agents [157,238] immediately following endotracheal intubation. Combining fever control to state-of-the-art therapy and alpha-2 agonists-evoked cooperative sedation may be worth considering when limited cardio-ventilatory reserve is observed in septic shock and severe ARDS with elevated body temperature. Evidence-based demonstrations are required.
Disclosure of potential conflicts of interest
L Quintin holds US Patents 8 703 697, April 22 2014: Method for treating early severe diffuse acute respiratory distress syndrome and 8 846 606 B2, September 30, 2014: Method and drug composition for treating septic shock hypotension. The other authors declare no conflict of interest.
Biographies

F Petitjeans, MD, OF5, trained with Ecole du Service de Santé des Armées, Lyon, physician associated with the Foreign Legion (Castelnaudary), Hôpitaux d'Instruction des Armées Paris-Val de Grâce Percy and Begin, Centre Hospitalier Français des Armées, Djibouti, Head of Critical Care and Anesthesiology, Hôpital d'Instruction des Armées Desgenettes, Lyon, France.

S Leroy, MD, PhD, trained at the Assistance Publique-Hôpitaux de Paris (resident in immunology, pediatrics and pediatric critical care) and at Centre for Statistics in Medicine, University of Oxford, UK; physician and clinical scientist in Epidemiology and Biostatistics, Teaching Hospital Avicennes, Paris-Bobigny, Founder and Head of Episcience Ltd.

C Pichot, MD, trained with Teaching Hospital and School of Medicine, Caen, France (Anesthesiology/Critical Care); Head of Critical Care, Louis Pasteur Hospital, Dole, France.

Alain Geloen, Research Director at Centre National de la Recherche Scientifique, member of Cardiovascular, Metabolism, Diabetologia and Nutrition (CarMeN) Laboratory (UMR INSERM 1060, INSA de Lyon). He completed his Ph.D. at Laval University (Québec, Canada) and his undergraduate studies at U of Lyon, France. Biologist of the XXIXth French expedition in Antarctica, his interests cover physiological studies of energy regulation in homeotherms, development of adipose tissues and applications of nanomaterials in cell and animal physiology, published in more than 115 articles in leading peer-review physiology journals.

M Ghignone, MD, FRCPC, FCCP, MBA, ABMS (Anesthesiology/Critical Care), trained with Torino, Ottawa and Manitoba Schools of Medicine, Assistance Publique-Hôpitaux de Paris (Hôpital H Mondor, Créteil), former Associate Professor, Director of Cardiac Anesthesiology, U of Pittsburgh and Texas Tech University, Lubbock, TX, former Chair of Anesthesiology, Critical Care and Pain Clinic Division, JFK North County Hospital WPalm Beach, Fl, presently Clinical Professor, School of Natural and Health Sciences Barry University and Associate Clinical Professor of Surgery, Division of Anesthesiology, Nova Southeastern University.

L Quintin, MD, PhD, trained with U of Western Brittany and Lyon, Assistance Publique-Hôpitaux de Paris (Hôpital H Mondor, Créteil), Royal Victoria Hospital, McGill University, Montreal, former Visiting full Professor, Hospital of the University of Pennsylvania, Philadelphia, Senior Investigator (retired), Centre National de la Recherche Scientifique.
Notes
Thermophysiology underwent revamping: In place of a single integrator with multiple intputs and outputs, the model includes as many integrators as there are thermoregulatory responses. These integrators are postulated to be represented at many levels of the central nervous system, with each level facilitated or inhibited by levels above and below. This arrangement achieves finer control over body temperature [239,240].
O2 consumption/O2 transport (rest: 0.25–0.35) [241].
The dose proposed here is tentative: should the alpha-2 agonist be administered to achieve a) adequate sedation to allow for nursing (−3<RASS<0) b) or the lowest vasopressor requirement (see below) or improved micro-circulation as assessed from capillary refill, lactate<2mM and CO2 gap<5–6 mm Hg ? The speed of the administration of the alpha-2 agonist in the setting of septic shock is important. Obviously when volume is adequate, administration of dexmedetomidine or clonidine, respectively at 1.5 and 2 μg.kg-1.h-1, from endotracheal intubation onwards is the simplest alternative after withdrawal of conventional sedation. When volemia or auriculo-ventricular (A-V) conduction is inadequate, volemia and/or A-V conduction should be restored. Then the alpha-2 agonist is administered and the speed of administration increased step-wise (0.25 μg.kg-1.h-1 for 3 h, then iterative echocardiography and passive leg raising and volume loading if appropriate, then 0.5 μg.kg-1.h-1 for 3 h with again echocardiography, passive leg raising and volume loading, etc. ; up to maximal dose).
If the alpha-2 agonist does not allow one to achieve -3<RASS<1, neuroleptics may be added [157,242]. If amnesia is a concern, during a brief period of paralysis needed to handle an acute cardio-ventilatory distress or to control high respiratory rate (RR), bolus(es) of midazolam may be considered (very low dose midazolam bolus as “rescue sedation” [44] rather than continuous infusion≤1 mg.h-1). Midazolam is withheld as soon as paralysis is stopped.
We suggest to use analgo-sedation evoked by alpha-2 agonists, and if needed, non-opioid analgesics [243].
Further options are neuroleptics [1,244] or meperidine-buspirone (5HT1A agonist) [47]. In addition to centrally acting agents, dantrolene, acting peripherally, reduces the peripheral gain of shivering, evokes little central thermoregulatory inhibition and lowers VO2 by 39% [245]. Chlorpromazine (RP 4560) lowers temperature centrally and inhibits centrally shivering more potently than peripheral ganglionic blocker or other peripheral sympatholytics, as a consequence of vasodilation and reduced cellular metabolism [246]. However even if chlorpromazine was the single most effective agent on shivering (1954), the effect of chlorpromazine is variable and unpredictable. Chlorpromazine was administered within a “lytic cocktail” (chlorpromazine, promethazine, meperidine) to evoke hypothermia via vasodilation without shivering (“artificial hibernation”). If shivering occurs under chlorpromazine, cooling should be stopped and chlorpromazine re-injected .
The hyperkinetic state is a different matter.
Increased mixed venous saturation >80–85% is a different matter implying an hyperkinetic state or major peripheral shutdown.
NA (range)=1.22–3 μg.kg-1.min-1 [88]; cut off point >2.33 μg.kg-1.min-1 [≈10 mg.h-1/70 kg]: 62% mortality within 48 h [89]; 100% mortality in the high NA requirement group ; 90% mortality when NA >1 μg.kg-1.min-1 [4.2 mg.h-1/70 kg] quoted from [42]
Helsinki declaration, §37 : Unproven Interventions in Clinical Practice. In the treatment of an individual patient, where proven interventions do not exist or other known interventions have been ineffective, the physician, after seeking expert advice, with informed consent from the patient or a legally authorised representative, may use an unproven intervention if in the physician's judgement it offers hope of saving life, re-establishing health or alleviating suffering. This intervention should subsequently be made the object of research, designed to evaluate its safety and efficacy. In all cases, new information must be recorded and, where appropriate, made publicly available [247].
Some reports do not show consistently reduced VO2 in the setting of ARDS and hypothermia [39,98]. Overall, there are few clear-cut data [29] delineating decreased O2 extraction.
Refractory hypoxia is defined as: a) P/F <50 with FiO2 >0.8, PEEP >15 cm H20, Vt <6 mL.kg-1, Pplat <30 cm H20 for >3h [248]. b) P/F <70–87, PEEP >10 cm H2O for >12–24 h [249].
Ventilation-induced lung injury is patient self inflicted lung injury (P-SILI: tachypnea, hyperpnoea, lung dys-coordination: pendel-luft) generated by a high respiratory drive (P-SILI) [123] during acute cardio-ventilatory distress. This is at odd from ventilator-induced lung injury (VILI: patient-ventilator dys-synchrony, volutrauma, barotrauma) [250]: high Vt under controlled mandatory ventilation, with or without paralysis.
If needed, neuroleptics may be added (§ II B 1) to achieve -3<RASS<0 .
The appropriate design would be spontaneous ventilation as delineated above after full correction of the acute cardio-ventilatory distress vs. controlled mandatory ventilation, paralysis and general anesthesia, i.e. testing the present hypothesis vs. state-of-the-art.
As observed during slow-wave sleep. During slow-wave sleep, PaCO2 should increase by 5–7 mm Hg to evoke an increase in minute ventilation ≈20 L.min-1 [251].
This contributes to explain why some patients improve almost immediately under high FiO2-high PEEP-low pressure support [210].
Presumably under assist-control mode [205].
From intracapillary pressure to interstitial fluid: Pcap-Ppl
Metabolic reflex originating in skeletal muscle « activated when blood flow to contracting muscles is insufficient to warrant both O2 delivery and metabolite washout » [252]
Heightened respiratory drive has been reported in late ARDS, not considered here. Briefly, first, ≈ 14 days within the course of ARDS, the patients present with rapid, shallow breathing [197], as opposed to hyperpnea and tachypnea in the setting of early ARDS [150]. Accordingly, after ≈ 28 days of ECMO, a high CO2 drive (high P0.1, high tidal volume, large esophageal pressure swings, etc.) is still present [255].
Young, well-trained athletes, aware of their physiological limits and engaging into ultra-long mountain races contrast with elderly patients exposed to a CCU environment for weeks : this last situation imposes general exhaustion of integrative systems (sympathetic and immune systems) and target organs (heart, lung, kidney, etc.).
A proinflammatory effect of alpha-2 receptors and an anti-inflammatory effect of an alpha-2 antagonist, yohimbine, has been shown [256,257]: the interpretation is complex.
Chlorisondamine may act either on the parasympathetic ganglia or on nicotinic receptors on immune cells.
A complex arrangement is postulated. Either there are two sets of sympathetic nerves, one pro and one anti-inflammatory. Conversely, the same sympathetic nerve may present different actions according to circumstances (e.g acute vs. chronic).
Note added in proof
With respect to the discussion in pp 14-sq, the observation of lowered hyperventilation following administration of dexmedetomidine (Saito, JA Clin Report, 2017, 3: 22), helps explain our observations regarding the absence of tachypnea and hyperpnea in the setting of early severe diffuse ARDS treated with high PEEP-low pressure support and no ventilator-to-the-patient dysynchrony.
Acknowledgments
R McAllen, Howard Florey Institute, Melbourne, JM Pequignot, Physiology, U of Lyon, and C Guerin, Critical Care, Hôpital de la Croix Rousse, Lyon are thanked for enlightening comments on the interaction between the sympathetic and immune systems, respiratory physiology and clinical pathophysiology. Springer, Elsevier and Therapie/Société Française de Pharmacologie et Thérapeutique are thanked for the reproduction, free of charge, of their figures.
References
- [1].Laborit H, Huguenard P. Technique actuelle de l'hibernation artificielle. Presse Med. 1952;60:1455–1456. PMID:13027061 [PubMed] [Google Scholar]
- [2].Chippaux C, Carayon A, Rouffilange F, et al. Artificial hibernation in war surgery: with reference to its use in Indochina. Presse Med. 1954;62:504–506. L'hibernation artificielle en chirurgie de guerre: d'apres son emploi actuel en Indochine. PMID:13166963 [PubMed] [Google Scholar]
- [3].Schortgen F, Clabault K, Katsahian S, et al. Fever control using external cooling in septic shock: a randomized controlled trial. Am J Respir Crit Care Med. 2012;185:1088–1095. doi: 10.1164/rccm.201110-1820OC. PMID:22366046 [DOI] [PubMed] [Google Scholar]
- [4].Varon J, Acosta P. Therapeutic hypothermia: past, present, and future. Chest. 2008;133:1267–1274. doi: 10.1378/chest.07-2190. PMID:18460529 [DOI] [PubMed] [Google Scholar]
- [5].Schortgen F. Fever in sepsis. Minerva Anestesiol. 2012;78:1254–1264. PMID:22772856 [PubMed] [Google Scholar]
- [6].Taccone FS, Saxena M, Schortgen F. What's new with fever control in the ICU. Intensive Care Med. 2014;40:1147–1150. doi: 10.1007/s00134-014-3277-9. PMID:24691575 [DOI] [PubMed] [Google Scholar]
- [7].Doyle JF, Schortgen F. Should we treat pyrexia? And how do we do it? Crit Care. 2016;20:303. doi: 10.1186/s13054-016-1467-2. PMID:27716372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Walter EJ, Hanna-Jumma S, Carraretto M, et al. The pathophysiological basis and consequences of fever. Crit Care. 2016;20:200. doi: 10.1186/s13054-016-1375-5. PMID:27411542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sessler DI. Temperature monitoring and perioperative thermoregulation. Anesthesiology. 2008;109:318–338. doi: 10.1097/ALN.0b013e31817f6d76. PMID:18648241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Clemmer TP, Fisher CJ, Jr., Bone RC, et al. Hypothermia in the sepsis syndrome and clinical outcome. The Methylprednisolone Severe Sepsis Study Group. Crit Care Med. 1992;20:1395–1401. doi: 10.1097/00003246-199210000-00006. PMID:1395659 [DOI] [PubMed] [Google Scholar]
- [11].Polderman KH. Application of therapeutic hypothermia in the intensive care unit. Opportunities and pitfalls of a promising treatment modality–Part 2: Practical aspects and side effects. Intensive Care Med. 2004;30:757–769. doi: 10.1007/s00134-003-2151-y. PMID:14767590 [DOI] [PubMed] [Google Scholar]
- [12].Badjatia N, Strongilis E, Prescutti M, et al. Metabolic benefits of surface counter warming during therapeutic temperature modulation. Crit Care Med. 2009;37:1893–1897. doi: 10.1097/CCM.0b013e31819fffd3. PMID:19384208 [DOI] [PubMed] [Google Scholar]
- [13].Rumbus Z, Matics R, Hegyi P, et al. Fever is associated with reduced, hypothermia with increased mortality in septic patients: a meta-analysis of clinical trials. PLoS One. 2017;12:e0170152. doi: 10.1371/journal.pone.0170152. PMID:28081244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Young PJ, Saxena M. Fever management in intensive care patients with infections. Crit Care. 2014;18:206. doi: 10.1186/cc13773. PMID:25029624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liu E, Lewis K, Al-Saffar H, et al. Naturally occurring hypothermia is more advantageous than fever in severe forms of lipopolysaccharide- and Escherichia coli-induced systemic inflammation. Am J Physiol Regul Integr Comp Physiol. 2012;302:R1372–R1383. doi: 10.1152/ajpregu.00023.2012. PMID:22513748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Netzer G, Dowdy DW, Harrington T, et al. Fever is associated with delayed ventilator liberation in acute lung injury. Ann Am Thorac Soc. 2013;10:608–615. doi: 10.1513/AnnalsATS.201303-052OC. PMID:24024608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12:123–137. doi: 10.1016/S0149-7634(88)80004-6. PMID:3050629 [DOI] [PubMed] [Google Scholar]
- [18].Romanovsky AA, Szekely M. Fever and hypothermia: two adaptive thermoregulatory responses to systemic inflammation. Med Hypotheses. 1998;50:219–226. doi: 10.1016/S0306-9877(98)90022-6. PMID:9578327 [DOI] [PubMed] [Google Scholar]
- [19].Fonseca MT, Rodrigues AC, Cezar LC, et al. Spontaneous hypothermia in human sepsis is a transient, self-limiting, and nonterminal response. J Appl Physiol (1985). 2016;120:1394–1401. doi: 10.1152/japplphysiol.00004.2016. PMID:26989218 [DOI] [PubMed] [Google Scholar]
- [20].Singer M, De Santis V, Vitale D, et al. Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet. 2004;364:545–548. doi: 10.1016/S0140-6736(04)16815-3. PMID:15302200 [DOI] [PubMed] [Google Scholar]
- [21].Steiner AA. Reduced oxygen utilization in septic shock: disorder or adaptation? Temperature. 2015;2:447–448. doi: 10.1080/23328940.2014.996484. PMID:27227060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shah NG, Cowan MJ, Pickering E, et al. Nonpharmacologic approach to minimizing shivering during surface cooling: a proof of principle study. J Crit Care. 2012;27:746 e1–8. doi: 10.1016/j.jcrc.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].De Backer D, Donadello K, Taccone FS, et al. Microcirculatory alterations: potential mechanisms and implications for therapy. Ann Intensive Care. 2011;1:27. doi: 10.1186/2110-5820-1-27. PMID:21906380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Geloen A, Pichot C, Leroy S, et al. Pressor response to noradrenaline in the setting of septic shock: anything new under the sun-dexmedetomidine, clonidine? A minireview. Biomed Res Int. 2015;2015:863715. doi: 10.1155/2015/863715. PMID:26783533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hoedemaekers CW, Ezzahti M, Gerritsen A, et al. Comparison of cooling methods to induce and maintain normo- and hypothermia in intensive care unit patients: a prospective intervention study. Crit Care. 2007;11:R91. doi: 10.1186/cc6104. PMID:17718920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bernard GR, Wheeler AP, Russell JA, et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N Engl J Med. 1997;336:912–918. doi: 10.1056/NEJM199703273361303. PMID:9070471 [DOI] [PubMed] [Google Scholar]
- [27].Michaeli B, Martinez A, Revelly JP, et al. Effects of endotoxin on lactate metabolism in humans. Crit Care. 2012;16:R139. doi: 10.1186/cc11444. PMID:22839504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ensinger H, Weichel T, Lindner KH, et al. Effects of norepinephrine, epinephrine, and dopamine infusions on oxygen consumption in volunteers. Crit Care Med. 1993;21:1502–1508. doi: 10.1097/00003246-199310000-00018. PMID:8403959 [DOI] [PubMed] [Google Scholar]
- [29].Manthous CA, Hall JB, Olson D, et al. Effect of cooling on oxygen consumption in febrile critically ill patients. Am J Respir Crit Care Med. 1995;151:10–14. doi: 10.1164/ajrccm.151.1.7812538. PMID:7812538 [DOI] [PubMed] [Google Scholar]
- [30].Amato MB, Meade MO, Slutsky AS, et al. Driving pressure and survival in the acute respiratory distress syndrome. N Engl J Med. 2015;372:747–755. doi: 10.1056/NEJMsa1410639. PMID:25693014 [DOI] [PubMed] [Google Scholar]
- [31].Darioli R, Perret C. Mechanical controlled hypoventilation in status asthmaticus. Am Rev Respir Dis. 1984;129:385–387. PMID:6703497 [DOI] [PubMed] [Google Scholar]
- [32].Poblete B, Romand JA, Pichard C, et al. Metabolic effects of i.v. propacetamol, metamizol or external cooling in critically ill febrile sedated patients. Br J Anaesth. 1997;78:123–127. doi: 10.1093/bja/78.2.123. PMID:9068325 [DOI] [PubMed] [Google Scholar]
- [33].Schortgen F, Brochard L. Reply: Does cooling really improve outcomes in patients with septic shock? Am J Respir Crit Care Med. 2013;187:1275. doi: 10.1164/rccm.201212-2163LE. PMID:23725627 [DOI] [PubMed] [Google Scholar]
- [34].Schortgen F, Charles-Nelson A, Bouadma L, et al. Respective impact of lowering body temperature and heart rate on mortality in septic shock: mediation analysis of a randomized trial. Intensive Care Med. 2015;41:1800–1808. doi: 10.1007/s00134-015-3987-7. PMID:26202042 [DOI] [PubMed] [Google Scholar]
- [35].Schulman CI, Namias N, Doherty J, et al. The effect of antipyretic therapy upon outcomes in critically ill patients: a randomized, prospective study. Surg Infect (Larchmt). 2005;6:369–375. doi: 10.1089/sur.2005.6.369. PMID:16433601 [DOI] [PubMed] [Google Scholar]
- [36].Harden LM, Kent S, Pittman QJ, et al. Fever and sickness behavior: Friend or foe? Brain Behav Immun. 2015;50:322–333. doi: 10.1016/j.bbi.2015.07.012. PMID:26187566 [DOI] [PubMed] [Google Scholar]
- [37].Matamis D, Tsagourias M, Koletsos K, et al. Influence of continuous haemofiltration-related hypothermia on haemodynamic variables and gas exchange in septic patients. Intensive Care Med. 1994;20:431–436. doi: 10.1007/BF01710654. PMID:7798448 [DOI] [PubMed] [Google Scholar]
- [38].Blair E, Buxton RW, Cowley RA, et al. The use of hypothermia in septic shock. JAMA. 1961;178:916–919. doi: 10.1001/jama.1961.73040480005008b. PMID:13869732 [DOI] [PubMed] [Google Scholar]
- [39].Villar J, Slutsky AS. Effects of induced hypothermia in patients with septic adult respiratory distress syndrome. Resuscitation. 1993;26:183–192. doi: 10.1016/0300-9572(93)90178-S. PMID:8290813 [DOI] [PubMed] [Google Scholar]
- [40].Kawazoe Y, Miyamoto K, Morimoto T, et al. Effect of dexmedetomidine on mortality and ventilator-free days in patients requiring mechanical ventilation with sepsis: a randomized clinical trial. JAMA. 2017;317:1321–1328. doi: 10.1001/jama.2017.2088. PMID:28322414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Monnet X, Teboul JL. Passive leg raising. Intensive Care Med. 2008;34:659–663. doi: 10.1007/s00134-008-0994-y. PMID:18214429 [DOI] [PubMed] [Google Scholar]
- [42].Hamzaoui O, Scheeren TWL, Teboul JL. Norepinephrine in septic shock: when and how much? Curr Opin Crit Care. 2017.; doi: 10.1097/MCC.0000000000000418. PMID:28509668 [DOI] [PubMed] [Google Scholar]
- [43].Voituron N, Hilaire G, Quintin L. Dexmedetomidine and clonidine induce long-lasting activation of the respiratory rhythm generator of neonatal mice: possible implication for critical care. Respir Physiol Neurobiol. 2012;180:132–140. doi: 10.1016/j.resp.2011.11.003. PMID:22108092 [DOI] [PubMed] [Google Scholar]
- [44].Riker RR, Shehabi Y, Bokesch PM, et al. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA. 2009;301:489–499. doi: 10.1001/jama.2009.56. PMID:19188334 [DOI] [PubMed] [Google Scholar]
- [45].Pandharipande PP, Pun B, Herr DI, et al. Effect of sedation with dexmedetomidine vs lorazepam on acute brain dysfunction in mechanically ventilated patients: the MENDS randomized controlled trial. J Am Med Assoc. 2007;298:2644–2653. doi: 10.1001/jama.298.22.2644. [DOI] [PubMed] [Google Scholar]
- [46].Maldonado JR, Wysong A, van der Starre PJ, et al. Dexmedetomidine and the reduction of postoperative delirium after cardiac surgery. Psychosomatics. 2009;50:206–217. doi: 10.1176/appi.psy.50.3.206. PMID:19567759 [DOI] [PubMed] [Google Scholar]
- [47].Mokhtarani M, Mahgoub AN, Morioka N, et al. Buspirone and meperidine synergistically reduce the shivering threshold. Anesth Analg. 2001;93:1233–1239. doi: 10.1097/00000539-200111000-00038. PMID:11682404 [DOI] [PubMed] [Google Scholar]
- [48].Lin MT, Shian LR, Leu SY. Clonidine-induced hypothermia: possible involvement of cholinergic and serotonergic mechanisms. Naunyn Schmiedebergs Arch Pharmacol. 1984;326:124–128. doi: 10.1007/BF00517308. PMID:6472490 [DOI] [PubMed] [Google Scholar]
- [49].Myers RD, Beleslin DB, Rezvani AH. Hypothermia: role of alpha 1- and alpha 2-noradrenergic receptors in the hypothalamus of the cat. Pharmacol Biochem Behav. 1987;26:373–379. doi: 10.1016/0091-3057(87)90132-8. PMID:3033698 [DOI] [PubMed] [Google Scholar]
- [50].Zis AP, Fibiger HC. Functional evidence for postsynaptic supersensitivity of central noradrenergic receptors after denervation. Nature. 1975;256:659–660. doi: 10.1038/256659a0. PMID:1153000 [DOI] [PubMed] [Google Scholar]
- [51].Jerussi TP, Capacchione JF, Benvenga MJ. Reversal of opioid-induced muscular rigidity in rats: evidence for alpha-2 adrenergic involvement. Pharmacol Biochem Behav. 1987;28:283–289. doi: 10.1016/0091-3057(87)90226-7. PMID:2891145 [DOI] [PubMed] [Google Scholar]
- [52].Tremblay LE, Bedard PJ. Effect of clonidine on motoneuron excitability in spinalised rats. Neuropharmacology. 1986;25:41–46. doi: 10.1016/0028-3908(86)90056-0. PMID:3005904 [DOI] [PubMed] [Google Scholar]
- [53].Quintin L, Viale JP, Annat G, et al. Oxygen uptake after major abdominal surgery: effect of clonidine. Anesthesiology. 1991;74:236–241. doi: 10.1097/00000542-199102000-00008. PMID:1990899 [DOI] [PubMed] [Google Scholar]
- [54].Belleville JP, Ward DS, Bloor BC, et al. Effects of intravenous dexmedetomidine in humans. I. Sedation, ventilation, and metabolic rate. Anesthesiology. 1992;77:1125–1133. doi: 10.1097/00000542-199212000-00013. PMID:1361310 [DOI] [PubMed] [Google Scholar]
- [55].Takahashi H, Nishikawa T, Mizutani T, et al. Oral clonidine premedication decreases energy expenditure in human volunteers. Can J Anaesth. 1997;44:268–272. doi: 10.1007/BF03015364. PMID:9067045 [DOI] [PubMed] [Google Scholar]
- [56].Delaunay L, Bonnet F, Liu N, et al. Clonidine comparably decreases the thermoregulatory thresholds for vasoconstriction and shivering in humans. Anesthesiology. 1993;79:470–474. doi: 10.1097/00000542-199309000-00009. PMID:8363071 [DOI] [PubMed] [Google Scholar]
- [57].Doufas AG, Lin CM, Suleman MI, et al. Dexmedetomidine and meperidine additively reduce the shivering threshold in humans. Stroke. 2003;34:1218–1223. doi: 10.1161/01.STR.0000068787.76670.A4. PMID:12690216 [DOI] [PubMed] [Google Scholar]
- [58].Saunier CF, Akaoka H, de La Chapelle B, et al. Activation of brain noradrenergic neurons during recovery from halothane anesthesia: persistence of phasic activation after clonidine. Anesthesiology. 1993;79:1072–1082. doi: 10.1097/00000542-199311000-00026. PMID:8238984 [DOI] [PubMed] [Google Scholar]
- [59].Liatsi D, Tsapas B, Pampori S, et al. Respiratory, metabolic and hemodynamic effects of clonidine in ventilated patients presenting with withdrawal syndrome. Intensive Care Med. 2009;35:275–281. doi: 10.1007/s00134-008-1251-0. PMID:18709354 [DOI] [PubMed] [Google Scholar]
- [60].Cruz FF, Rocco PR, Pelosi P. Anti-inflammatory properties of anesthetic agents. Crit Care. 2017;21:67. doi: 10.1186/s13054-017-1645-x. PMID:28320449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pichot C, Geloen A, Ghignone M, et al. Alpha-2 agonists to reduce vasopressor requirements in septic shock? Med Hypotheses. 2010;75:652–656. doi: 10.1016/j.mehy.2010.08.010. PMID:20817367 [DOI] [PubMed] [Google Scholar]
- [62].Geloen A, Chapelier K, Cividjian A, et al. Clonidine and dexmedetomidine increase the pressor response to norepinephrine in experimental sepsis: a pilot study. Crit Care Med. 2013;41:e431–e438. doi: 10.1097/CCM.0b013e3182986248. PMID:23963131 [DOI] [PubMed] [Google Scholar]
- [63].Lankadeva YR, Booth LC, Kosaka J, et al. Clonidine restores pressor responsiveness to phenylephrine and angiotensin II in ovine sepsis. Crit Care Med. 2015;43:e221–e229. doi: 10.1097/CCM.0000000000000963. PMID:25860204 [DOI] [PubMed] [Google Scholar]
- [64].Pandharipande PP, Sanders RD, Girard TD, et al. Effect of dexmedetomidine versus lorazepam on outcome in patients with sepsis: an a priori-designed analysis of the MENDS randomized controlled trial. Critical Care. 2010;14:R38. doi: 10.1186/cc8916. PMID:20233428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Nishikawa T, Kimura T, Taguchi N, et al. Oral clonidine preanesthetic medication augments the pressor responses to intravenous ephedrine in awake or anesthetized patients. Anesthesiology. 1991;74:705–710. doi: 10.1097/00000542-199104000-00014. PMID:2008952 [DOI] [PubMed] [Google Scholar]
- [66].Parlow J, Sagnard P, Begou G, et al. The effects of clonidine on sensitivity to phenylephrine and nitroprusside in patients with essential hypertension recovering from surgery. Anesth Analg. 1999;88:1239–1243. doi: 10.1213/00000539-199906000-00010. PMID:10357325 [DOI] [PubMed] [Google Scholar]
- [67].Herr DL, Sum-Ping ST, England M. ICU sedation after coronary artery bypass graft surgery: dexmedetomidine-based versus propofol-based sedation regimens. J Cardiothorac Vasc Anesth. 2003;17:576–584. doi: 10.1016/S1053-0770(03)00200-3. PMID:14579210 [DOI] [PubMed] [Google Scholar]
- [68].Venn RM, Newman PJ, Grounds RM. A phase II study to evaluate the efficacy of dexmedetomidine for sedation in the medical intensive care unit. Intensive Care Med. 2003;29:201–207. doi: 10.1007/s00134-002-1579-9. PMID:12594584 [DOI] [PubMed] [Google Scholar]
- [69].Lam F, Ransom C, Gosett J, et al. Safety and efficacy of dexmedetomidine in children with heart failure. Pediatr Cardiol. 2012;33:1067–1077. [DOI] [PubMed] [Google Scholar]
- [70].Quintin L, Bouilloc X, Butin E, et al. Clonidine for aortic surgery in hypertensive patients: a double blind controlled randomized study. Anesth Analg. 1996;83:687–695. doi: 10.1213/00000539-199610000-00005. PMID:8831304 [DOI] [PubMed] [Google Scholar]
- [71].Pichot C, Mathern P, Khettab F, et al. Increased pressor response to noradrenaline during septic shock following clonidine? Anaesth Intensive Care. 2010;38:784–785. PMID:20715756 [PubMed] [Google Scholar]
- [72].Butler J, Kelly JG, O'Malley O, et al. Beta-adrenoceptor adaptation to acute exercise. J Physiol (Lond). 1983;344:113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ohman EM, Butler J, Kelly J, et al. Beta-adrenoceptor adaptation to endurance training. J Cardiovasc Pharmacol. 1987;10:728–731. doi: 10.1097/00005344-198712000-00018. PMID:2450245 [DOI] [PubMed] [Google Scholar]
- [74].Schultz KD, Fritschka E, Kribben A, et al. Alpha 2- and beta 2-adrenoceptor downregulation in marathon runners. J Hypertens Suppl. 1989;7:S48–S49. [PubMed] [Google Scholar]
- [75].Robertson D, Goldberg MR, Hollister AS, et al. Clonidine raises blood pressure in severe idiopathic orthostatic hypotension. Am J Med. 1983;74:193–200. doi: 10.1016/0002-9343(83)90607-1. PMID:6824002 [DOI] [PubMed] [Google Scholar]
- [76].Zoukos Y, Thomaides T, Pavitt DV, et al. Upregulation of beta adrenoceptors on circulating mononuclear cells after reduction of central sympathetic outflow by clonidine in normal subjects. Clin Auton Res. 1992;2:165–170. doi: 10.1007/BF01818957. PMID:1323363 [DOI] [PubMed] [Google Scholar]
- [77].Benedict CR, Rose JA. Arterial norepinephrine changes in patients with septic shock. Circ Shock. 1992;38:165–172. PMID:1292880 [PubMed] [Google Scholar]
- [78].Lakandeva Y, Booth L, Kosaka J, et al. Clonidine restores pressor responsiveness to phenylephrine and angiotensin II in ovine sepsis. Crit Care Med. 2015;in press. [DOI] [PubMed] [Google Scholar]
- [79].Julien C, Orea V, Quintin L, et al. Renal sympathetic nerve activity and vascular reactivity to phenylephrine after lipopolysaccharide administration in conscious rats. Physiol Rep. 2017;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Vieillard-Baron A. Is right ventricular function the one that matters in ARDS patients? Definitely yes. Intensive Care Med. 2009;35:4–6. doi: 10.1007/s00134-008-1308-0. PMID:18839136 [DOI] [PubMed] [Google Scholar]
- [81].Prewitt RM, Ghignone M. Treatment of right ventricular dysfunction in acute respiratory failure. Crit Care Med. 1983;11:346–352. doi: 10.1097/00003246-198305000-00005. PMID:6340963 [DOI] [PubMed] [Google Scholar]
- [82].Marini JJ, Gattinoni L. Ventilatory management of acute respiratory distress syndrome: a consensus of two. Crit Care Med. 2004;32:250–255. doi: 10.1097/01.CCM.0000104946.66723.A8. PMID:14707588 [DOI] [PubMed] [Google Scholar]
- [83].Silbert BI, Litton E, Ho KM. Central venous-to-arterial carbon dioxide gradient as a marker of occult tissue hypoperfusion after major surgery. Anaesth Intensive Care. 2015;43:628–634. PMID:26310414 [DOI] [PubMed] [Google Scholar]
- [84].Vallee F, Vallet B, Mathe O, et al. Central venous-to-arterial carbon dioxide difference: an additional target for goal-directed therapy in septic shock? Intensive Care Med. 2008;34:2218–2225. doi: 10.1007/s00134-008-1199-0. PMID:18607565 [DOI] [PubMed] [Google Scholar]
- [85].Viale JP. The venous-arterial partial pressure of carbon dioxide as a new monitoring of circulatory disorder: no so simple. J Clin Monit Comput. 2016;30:757–760. doi: 10.1007/s10877-016-9872-2. PMID:27038162 [DOI] [PubMed] [Google Scholar]
- [86].Correa TD, Filho RR, Assuncao MS, et al. Vasodilators in septic shock resuscitation: a clinical perspective. Shock. 2017;47:269–275. doi: 10.1097/SHK.0000000000000777. PMID:27787407 [DOI] [PubMed] [Google Scholar]
- [87].Moonka R, Gentilello L. Hypothermia induced by continuous arteriovenous hemofiltration as a treatment for adult respiratory distress syndrome: a case report. J Trauma. 1996;40:1026–1028. doi: 10.1097/00005373-199606000-00031. PMID:8656459 [DOI] [PubMed] [Google Scholar]
- [88].Vieillard-Baron A, Caille V, Charron C, et al. Reversal of refractory septic shock with drotrecogin alpha (activated). Intensive Care Med. 2009;35:1204–1209. doi: 10.1007/s00134-009-1538-9. PMID:19529911 [DOI] [PubMed] [Google Scholar]
- [89].Conrad M, Perez P, Thivilier C, et al. Early prediction of norepinephrine dependency and refractory septic shock with a multimodal approach of vascular failure. J Crit Care. 2015;in press. doi: 10.1016/j.jcrc.2015.03.029. PMID:25900257 [DOI] [PubMed] [Google Scholar]
- [90].Dargent A, Nguyen M, Fournel I, et al. Vasopressor cumulative dose requirement and risk of early death during septic shock: an analysis from the episs cohort. Shock. 2017; doi: 10.1097/SHK.0000000000001022. [DOI] [PubMed] [Google Scholar]
- [91].Schell-Chaple HM, Puntillo KA, Matthay MA, et al. Body temperature and mortality in patients with acute respiratory distress syndrome. Am J Crit Care. 2015;24:15–23. doi: 10.4037/ajcc2015320. PMID:25554550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Marini JJ. Unproven clinical evidence in mechanical ventilation. Curr Opin Crit Care. 2012;18:1–7. doi: 10.1097/MCC.0b013e32834ef425. PMID:22157253 [DOI] [PubMed] [Google Scholar]
- [93].Ranieri VM, Rubenfeld GD, Thompson BT, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307:2526–2533. PMID:22797452 [DOI] [PubMed] [Google Scholar]
- [94].Hayek AJ, White HD, Ghamande S, et al. Is therapeutic hypothermia for acute respiratory distress syndrome the future? J Intensive Care Med. 2017:885066617701117. [DOI] [PubMed] [Google Scholar]
- [95].Suzuki S, Hotchkiss JR, Takahashi T, et al. Effect of core body temperature on ventilator-induced lung injury. Crit Care Med. 2004;32:144–149. doi: 10.1097/01.CCM.0000098857.14923.44. PMID:14707573 [DOI] [PubMed] [Google Scholar]
- [96].Flachs J, Bookallil M, Clarke B. Extracorporeal oxygenation or hypothermia in respiratory failure. Lancet. 1977;1:489–490. doi: 10.1016/S0140-6736(77)91982-1. PMID:65598 [DOI] [PubMed] [Google Scholar]
- [97].Gilston A. A hypothermic regime for acute respiratory failure. Intensive Care Med. 1983;9:37–39. doi: 10.1007/BF01693705. PMID:6833627 [DOI] [PubMed] [Google Scholar]
- [98].D'Empaire G, Wilson RS, Hotchkiss R. The effect of induced hypothermia on oxygen uptake, oxygen transport and pulmonary shunt in ARDS. Am Rev Respir Dis. 1985;131:A134. [Google Scholar]
- [99].Hurst JM, deHaven CB, Branson R, et al. Combined use of high-frequency jet ventilation and induced hypothermia in the treatment of refractory respiratory failure. Crit Care Med. 1985;13:771–772. doi: 10.1097/00003246-198509000-00019. PMID:3928260 [DOI] [PubMed] [Google Scholar]
- [100].Wetterberg T, Steen S. Combined use of hypothermia and buffering in the treatment of critical respiratory failure. Acta Anaesthesiol Scand. 1992;36:490–492. doi: 10.1111/j.1399-6576.1992.tb03504.x. PMID:1632175 [DOI] [PubMed] [Google Scholar]
- [101].Duan M, Berra L, Kumar A, et al. Use of hypothermia to allow low-tidal-volume ventilation in a patient with ARDS. Respir Care. 2011;56:1956–1958. doi: 10.4187/respcare.01211. PMID:21682985 [DOI] [PubMed] [Google Scholar]
- [102].Hemmer M, Suter PM. Treatment of cardiac and renal effects of PEEP with dopamine in patients with acute respiratory failure. Anesthesiology. 1979;50:399–403. doi: 10.1097/00000542-197905000-00005. PMID:378029 [DOI] [PubMed] [Google Scholar]
- [103].Repesse X, Aubry A, Vieillard-Baron A. On the complexity of scoring acute respiratory distress syndrome: do not forget hemodynamics! J Thorac Dis. 2016;8:E758–E764. doi: 10.21037/jtd.2016.07.54. PMID:27618840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Dantzker DR, Lynch JP, Weg JG. Depression of cardiac output is a mechanism of shunt reduction in the therapy of acute respiratory failure. Chest. 1980;77:636–642. doi: 10.1378/chest.77.5.636. PMID:6988180 [DOI] [PubMed] [Google Scholar]
- [105].Kimmoun A, Vanhuyse F, Levy B. Improving blood oxygenation during venovenous ECMO for ARDS. Intensive Care Med. 2013;39:1161–1162. doi: 10.1007/s00134-013-2903-2. PMID:23584469 [DOI] [PubMed] [Google Scholar]
- [106].Karnatovskaia LV, Festic E, Freeman WD, et al. Effect of therapeutic hypothermia on gas exchange and respiratory mechanics: a retrospective cohort study. Ther Hypothermia Temp Manag. 2014;4:88–95. doi: 10.1089/ther.2014.0004. PMID:24840620 [DOI] [PubMed] [Google Scholar]
- [107].Slack DF, Corwin DS, Shah NG, et al. Pilot feasibility study of therapeutic hypothermia for moderate to severe acute respiratory distress syndrome. Crit Care Med. 2017;in press. doi: 10.1097/CCM.0000000000002338. PMID:28406814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Gattinoni L, Carlesso E, Brazzi L, et al. Friday night ventilation: a safety starting tool kit for mechanically ventilated patients. Minerva Anestesiol. 2014;80:1046–1057. PMID:24847737 [PubMed] [Google Scholar]
- [109].Page B, Vieillard-Baron A, Beauchet A, et al. Low stretch ventilation strategy in acute respiratory distress syndrome: eight years of clinical experience in a single center. Crit Care Med. 2003;31:765–769. doi: 10.1097/01.CCM.0000055402.68581.DC. PMID:12626981 [DOI] [PubMed] [Google Scholar]
- [110].Pichot C, Petitjeans F, Ghignone M, et al. Is there a place for pressure support ventilation and high end-expiratory pressure combined to alpha-2 agonists early in severe diffuse acute respiratory distress syndrome? Med Hypotheses. 2013;80:732–737. doi: 10.1016/j.mehy.2013.02.023. PMID:23561575 [DOI] [PubMed] [Google Scholar]
- [111].Petitjeans F, Pichot C, Ghignone M, et al. Early severe acute respiratory distress syndrome: What's going on? Part II: controlled vs. spontaneous ventilation? Anaesthesiol Intensive Ther. 2016;48:339–351. doi: 10.5603/AIT.2016.0057. PMID:28000205 [DOI] [PubMed] [Google Scholar]
- [112].Pichot C, Petitjeans F, Ghignone G, et al. Commentary: spontaneous ventilation in the setting of early severe stabilized ARDS: heresy? Austin J Pulm Respir Med. 2016;3:1046 ( 1–8.). [Google Scholar]
- [113].Burns SM, Egloff MB, Ryan B, et al. Effect of body position on spontaneous respiratory rate and tidal volume in patients with obesity, abdominal distension and ascites. Am J Crit Care. 1994;3:102–106. PMID:8167771 [PubMed] [Google Scholar]
- [114].Richard JC, Maggiore SM, Mancebo J, et al. Effects of vertical positioning on gas exchange and lung volumes in acute respiratory distress syndrome. Intensive Care Med. 2006;32:1623–1626. doi: 10.1007/s00134-006-0299-y. PMID:16896856 [DOI] [PubMed] [Google Scholar]
- [115].Dellamonica J, Lerolle N, Sargentini C, et al. Effect of different seated positions on lung volume and oxygenation in acute respiratory distress syndrome. Intensive Care Med. 2013;39:1121–1127. doi: 10.1007/s00134-013-2827-x. PMID:23344832 [DOI] [PubMed] [Google Scholar]
- [116].Petitjeans F, Pichot C, Ghignone M, et al. Early severe acute respiratory distress syndrome: What's going on? Part I: pathophysiology. Anaesthesiol Intensive Ther. 2016;48:314–338. doi: 10.5603/AIT.2016.0056. PMID:28000204 [DOI] [PubMed] [Google Scholar]
- [117].Mekontso Dessap A, Boissier F, Leon R, et al. Prevalence and prognosis of shunting across patent foramen ovale during acute respiratory distress syndrome. Crit Care Med. 2010;38:1786–1792. doi: 10.1097/CCM.0b013e3181eaa9c8. PMID:20601861 [DOI] [PubMed] [Google Scholar]
- [118].Jardin F, Farcot JC, Boisante L, et al. Influence of positive end-expiratory pressure on left ventricular performance. N Engl J Med. 1981;304:387–392. doi: 10.1056/NEJM198102123040703. PMID:7005679 [DOI] [PubMed] [Google Scholar]
- [119].Hickling KG, Henderson SJ, Jackson R. Low mortality associated with low volume pressure limited ventilation with permissive hypercapnia in severe adult respiratory distress syndrome. Intensive Care Med. 1990;16:372–377. doi: 10.1007/BF01735174. PMID:2246418 [DOI] [PubMed] [Google Scholar]
- [120].Crotti S, Bottino N, Ruggeri GM, et al. Spontaneous breathing during extracorporeal membrane oxygenation in acute respiratory failure. Anesthesiology. 2017;126:678–687. doi: 10.1097/ALN.0000000000001546. PMID:28212205 [DOI] [PubMed] [Google Scholar]
- [121].Pichot C, Petitjeans F, Ghignone G, et al. Spontaneous ventilation-high PEEP upon severe ARDS: an erratum to further the analysis. Med Hypotheses. 2013;81:966–967. doi: 10.1016/j.mehy.2013.07.031. PMID:23993471 [DOI] [PubMed] [Google Scholar]
- [122].Mekontso DA, Charron C, Devaquet J, et al. Impact of acute hypercapnia and augmented positive end-expiratory pressure on right ventricle function in severe acute respiratory distress syndrome. Intensive Care Med. 2009;35:1850–1858. doi: 10.1007/s00134-009-1569-2. PMID:19652953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Brochard L, Slutsky A, Pesenti A. Mechanical ventilation to minimize progression of lung injury in acute respiratory failure. Am J Respir Crit Care Med. 2017;195:438–442. doi: 10.1164/rccm.201605-1081CP. PMID:27626833 [DOI] [PubMed] [Google Scholar]
- [124].Aggarwal NR, Brower RG. Targeting normoxemia in acute respiratory distress syndrome may cause worse short-term outcomes because of oxygen toxicity. Ann Am Thorac Soc. 2014;11:1449–1453. doi: 10.1513/AnnalsATS.201407-297PS. PMID:25314313 [DOI] [PubMed] [Google Scholar]
- [125].Pesenti A, Rossi N, Calori A, et al. Effects of short-term oxygenation changes on acute lung injury patients undergoing pressure support ventilation. Chest. 1993;103:1185–1189. doi: 10.1378/chest.103.4.1185. PMID:8131462 [DOI] [PubMed] [Google Scholar]
- [126].Kirby RR, Downs JB, Civetta JM, et al. High level positive end expiratory pressure (PEEP) in acute respiratory insufficiency. Chest. 1975;67:156–163. doi: 10.1378/chest.67.2.156. PMID:1090420 [DOI] [PubMed] [Google Scholar]
- [127].Borges JB, Carvalho CR, Amato MB. Lung recruitment in patients with ARDS. N Engl J Med. 2006;355:319–320. doi: 10.1056/NEJMc061434. PMID:16855277 [DOI] [PubMed] [Google Scholar]
- [128].Lachmann B. Open up the lung and keep the lung open. Intensive Care Med. 1992;18:319–321. doi: 10.1007/BF01694358. PMID:1469157 [DOI] [PubMed] [Google Scholar]
- [129].Guerin C. The preventive role of higher PEEP in treating severely hypoxemic ARDS. Minerva Anestesiol. 2011;77:835–845. PMID:21730932 [PubMed] [Google Scholar]
- [130].Hedenstierna G, Edmark L. The effects of anesthesia and muscle paralysis on the respiratory system. Intensive Care Med. 2005;31:1327–1335. doi: 10.1007/s00134-005-2761-7. PMID:16132894 [DOI] [PubMed] [Google Scholar]
- [131].Pellegrini M, Hedenstierna G, Roneus A, et al. The diaphragm acts as a brake during expiration to prevent lung collapse. Am J Respir Crit Care Med. 2016. [DOI] [PubMed] [Google Scholar]
- [132].Yoshida T, Roldan R, Beraldo MA, et al. Spontaneous effort during mechanical ventilation: maximal injury with less positive end-expiratory pressure. Crit Care Med. 2016;44:e678–e688. doi: 10.1097/CCM.0000000000001649. PMID:27002273 [DOI] [PubMed] [Google Scholar]
- [133].Haberthur C, Guttmann J. Short-term effects of positive end-expiratory pressure on breathing pattern: an interventional study in adult intensive care patients. Crit Care. 2005;9:R407–R415. doi: 10.1186/cc3735. PMID:16137354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Ranieri VM, Suter PM, Tortorella C, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA. 1999;282:54–61. doi: 10.1001/jama.282.1.54. PMID:10404912 [DOI] [PubMed] [Google Scholar]
- [135].Villar J, Perez-Mendez L, Lopez J, et al. An early PEEP/FIO2 trial identifies different degrees of lung injury in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2007;176:795–804. doi: 10.1164/rccm.200610-1534OC. PMID:17585106 [DOI] [PubMed] [Google Scholar]
- [136].Talmor D, Sarge T, Malhotra A, et al. Mechanical ventilation guided by esophageal pressure in acute lung injury. N Engl J Med. 2008;359:2095–2104. doi: 10.1056/NEJMoa0708638. PMID:19001507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Pichot C, Picoche A, Saboya-Steinbach M, et al. Combination of clonidine sedation and spontaneous breathing-pressure support upon acute respiratory distress syndrome: a feasability study in four patients. Acta Anaesthesiol Belg. 2012;63:127–133. PMID:23397665 [PubMed] [Google Scholar]
- [138].Galland C, Ferrand FX, Cividjian A, et al. Swift recovery of severe hypoxemic pneumonia upon morbid obesity. Acta Anaesthesiol Belg. 2014;65:109–117. PMID:25470892 [PubMed] [Google Scholar]
- [139].Rouby JJ, Puybasset L, Cluzel P, et al. Regional distribution of gas and tissue in acute respiratory distress syndrome. II. Physiological correlations and definition of an ARDS Severity Score. CT Scan ARDS Study Group. Intensive Care Med. 2000;26:1046–1056. doi: 10.1007/s001340051317. PMID:11030160 [DOI] [PubMed] [Google Scholar]
- [140].Oba Y, Thameem DM, Zaza T. High levels of PEEP may improve survival in acute respiratory distress syndrome: a meta-analysis. Respir Med. 2009;103:1174–1181. doi: 10.1016/j.rmed.2009.02.008. PMID:19269800 [DOI] [PubMed] [Google Scholar]
- [141].Cressoni M, Chiumello D, Algieri I, et al. Opening pressures and atelectrauma in acute respiratory distress syndrome. Intensive Care Med. 2017.; doi: 10.1007/s00134-017-4754-8. [DOI] [PubMed] [Google Scholar]
- [142].Putensen C, Zech S, Wrigge H, et al. Long-term effects of spontaneous breathing during ventilatory support in patients with acute lung injury. Am J Respir Crit Care Med. 2001;164:43–49. doi: 10.1164/ajrccm.164.1.2001078. PMID:11435237 [DOI] [PubMed] [Google Scholar]
- [143].Richard JC, Lyazidi A, Akoumianaki E, et al. Potentially harmful effects of inspiratory synchronization during pressure preset ventilation. Intensive Care Med. 2013;39:2003–2010. doi: 10.1007/s00134-013-3032-7. PMID:23928898 [DOI] [PubMed] [Google Scholar]
- [144].Hansen J, Wendt M, Lawin P. Inspiratory flow assistance. Anaesthesist. 1984;33:428–432. PMID:6388405 [PubMed] [Google Scholar]
- [145].Guldner A, Pelosi P, Gama de Abreu M. Spontaneous breathing in mild and moderate versus severe acute respiratory distress syndrome. Curr Opin Crit Care. 2014;20:69–76. doi: 10.1097/MCC.0000000000000055. PMID:24335656 [DOI] [PubMed] [Google Scholar]
- [146].Viale JP, Annat G, Bouffard Y, et al. Oxygen cost of breathing in postoperative patients: pressure support ventilation versus continuous positive airway pressure. Chest. 1988;93:506–509. doi: 10.1378/chest.93.3.506. PMID:3125013 [DOI] [PubMed] [Google Scholar]
- [147].Brochard L, Harf A, Lorino H, et al. Inspiratory pressure support prevents diaphragmatic fatigue during weaning from mechanical ventilation. Am Rev Respir Dis. 1989;139:513–521. doi: 10.1164/ajrccm/139.2.513. PMID:2643905 [DOI] [PubMed] [Google Scholar]
- [148].Chiumello D, Pelosi P, Croci M, et al. The effects of pressurization rate on breathing pattern, work of breathing, gas exchange and patient comfort in pressure support ventilation. Eur Respir J. 2001;18:107–114. doi: 10.1183/09031936.01.00083901. PMID:11510780 [DOI] [PubMed] [Google Scholar]
- [149].Bonmarchand G, Chevron V, Menard JF, et al. Effects of pressure ramp slope values on the work of breathing during pressure support ventilation in restrictive patients. Crit Care Med. 1999;27:715–722. doi: 10.1097/00003246-199904000-00023. PMID:10321660 [DOI] [PubMed] [Google Scholar]
- [150].Carteaux G, Millan-Guilarte T, De Prost N, et al. Failure of noninvasive ventilation for de novo acute hypoxemic respiratory failure: role of tidal volume. Crit Care Med. 2016;44:282–290. doi: 10.1097/CCM.0000000000001379. PMID:26584191 [DOI] [PubMed] [Google Scholar]
- [151].Mauri T, Bellani G, Grasselli G, et al. Patient-ventilator interaction in ARDS patients with extremely low compliance undergoing ECMO: a novel approach based on diaphragm electrical activity. Intensive Care Med. 2013;39:282–291. doi: 10.1007/s00134-012-2755-1. PMID:23196419 [DOI] [PubMed] [Google Scholar]
- [152].Wrigge H, Zinserling J, Hering R, et al. Cardiorespiratory effects of automatic tube compensation during airway pressure release ventilation in patients with acute lung injury. Anesthesiology. 2001;95:382–389. doi: 10.1097/00000542-200108000-00020. PMID:11506110 [DOI] [PubMed] [Google Scholar]
- [153].Anjos CF, Schettino GP, Park M, et al. A randomized trial of noninvasive positive end expiratory pressure in patients with acquired immune deficiency syndrome and hypoxemic respiratory failure. Respir Care. 2012;57:211–220. PMID:21762561 [DOI] [PubMed] [Google Scholar]
- [154].Dojat M, Harf A, Touchard D, et al. Clinical evaluation of a computer-controlled pressure support mode. Am J Respir Crit Care Med. 2000;161:1161–1166. doi: 10.1164/ajrccm.161.4.9904064. PMID:10764306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Petitjeans F, Quintin L. Non-invasive Failure in de novo acute hypoxemic respiratory failure: high positive end-expiratory pressure-low PS, i.e. “inverted” settings? Crit Care Med. 2016; in press. doi: 10.1097/CCM.0000000000001967. PMID:27755097 [DOI] [PubMed] [Google Scholar]
- [156].Bailey PL, Sperry RJ, Johnson GK, et al. Respiratory effects of clonidine alone and combined with morphine, in humans. Anesthesiology. 1991;74:43–48. doi: 10.1097/00000542-199101000-00008. PMID:1898841 [DOI] [PubMed] [Google Scholar]
- [157].Pichot C, Ghignone M, Quintin L. Dexmedetomidine and clonidine: from second- to first-line sedative agents in the critical care setting? J Intensive Care Med. 2012;27:219–237. doi: 10.1177/0885066610396815. PMID:21525113 [DOI] [PubMed] [Google Scholar]
- [158].Sauder P, Andreoletti M, Cambonie G, et al. Sedation and analgesia in intensive care. French Critical Care Society. Ann Fr Anesth Reanim. 2008;27:541–551. doi: 10.1016/j.annfar.2008.04.021. PMID:18579339 [DOI] [PubMed] [Google Scholar]
- [159].Akada S, Takeda S, Yoshida Y, et al. The efficacy of dexmedetomidine in patients with noninvasive ventilation: a preliminary study. Anesth Analg. 2008;107:167–170. doi: 10.1213/ane.0b013e3181732dc2. PMID:18635484 [DOI] [PubMed] [Google Scholar]
- [160].Huang Z, Chen YS, Yang ZL, et al. Dexmedetomidine versus midazolam for the sedation of patients with non-invasive ventilation failure. Intern Med. 2012;51:2299–2305. doi: 10.2169/internalmedicine.51.7810. PMID:22975538 [DOI] [PubMed] [Google Scholar]
- [161].Tian X, Li H, Ji Z, et al. Application of dexmedetomidine sedation in the treatment of continuous state of asthma: a case report. Zhonghua Wei Zhong Bing Ji JIU Yi Xue. 2014;26:598. PMID:25124915 [PubMed] [Google Scholar]
- [162].Papazian L, Forel JM, Gacouin A, et al. Neuromuscular blockers in early acute respiratory distress syndrome. N Engl J Med. 2010;363:1107–1116. doi: 10.1056/NEJMoa1005372. PMID:20843245 [DOI] [PubMed] [Google Scholar]
- [163].Guerin C, Reignier J, Richard JC, et al. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med. 2013;368:2159–2168. doi: 10.1056/NEJMoa1214103. PMID:23688302 [DOI] [PubMed] [Google Scholar]
- [164].Chanques G, Jaber S, Jung B, et al. Sédation-analgésie en réanimation de l'adulte. Encyclopedie Medico-Chirurgicale, Anesthésie Reanimation. 2013;10:1–12. [Google Scholar]
- [165].Slutsky AS. Neuromuscular blocking agents in ARDS. N Engl J Med. 2010;363:1176–1180. doi: 10.1056/NEJMe1007136. PMID:20843254 [DOI] [PubMed] [Google Scholar]
- [166].Quintin L, Whalley DG, Wynands JE, et al. Oxygen-high dose fentanyl-droperidol anesthesia for aortocoronary bypass surgery. Anesthesia Analgesia. 1981;60:412–416. doi: 10.1213/00000539-198106000-00008. PMID:6972181 [DOI] [PubMed] [Google Scholar]
- [167].Quenot JP, Binquet C, Pavon A. Cardiovascular collapse: lack of understanding or failure to anticipate heart-lung interaction? Reanimation (Paris). 2012;21:710–714. doi: 10.1007/s13546-012-0523-4. [DOI] [Google Scholar]
- [168].Miyazaki S, Uchida S, Mukai J, et al. Clonidine effects on all-night human sleep: opposite action of low- and medium-dose clonidine on human NREM-REM sleep proportion. Psychiatry Clin Neurosci. 2004;58:138–144. doi: 10.1111/j.1440-1819.2003.01207.x. [DOI] [PubMed] [Google Scholar]
- [169].Huupponen E, Maksimow A, Lapinlampi P, et al. Electroencephalogram spindle activity during dexmedetomidine sedation and physiological sleep. Acta Anaesthesiol Scand. 2008;52:289–294. doi: 10.1111/j.1399-6576.2007.01537.x. PMID:18005372 [DOI] [PubMed] [Google Scholar]
- [170].Maze M. Dexmedetomidine: a view point. Drugs. 2000;59:269. doi: 10.2165/00003495-200059020-00013. [DOI] [Google Scholar]
- [171].Davies DS, Wing LMH, Reid JL, et al. Pharmacokinetics and concentration-effect relationships of intravenous and oral clonidine. Clin Pharmacol Ther. 1977;21:593–601. doi: 10.1002/cpt1977215593. PMID:870272 [DOI] [PubMed] [Google Scholar]
- [172].Hood DD, Mallak KA, Eisenach JC, et al. Interaction between intrathecal neostigmine and epidural clonidine in human volunteers. Anesthesiology. 1996;85:315–325. doi: 10.1097/00000542-199608000-00013. PMID:8712447 [DOI] [PubMed] [Google Scholar]
- [173].Kauppila T, Kemppainen P, Tanila H, et al. Effect of systemic medetomidine, an alpha-2 adrenoceptor agonist, on experimental pain in humans. Anesthesiology. 1991;74:3–8. doi: 10.1097/00000542-199101000-00002. PMID:1670912 [DOI] [PubMed] [Google Scholar]
- [174].Lorez HP, Kiss D, Da Prada M, et al. Effects of clonidine on the rate of noradrenaline turnover in discrete areas of the rat central nervous system. Naunyn-Schmiedeberg's Arch Pharmacol. 1983;323:307–314. doi: 10.1007/BF00512468. [DOI] [PubMed] [Google Scholar]
- [175].Ruokonen E, Parviainen I, Jakob SM, et al. Dexmedetomidine versus propofol/midazolam for long-term sedation during mechanical ventilation. Intensive Care Med. 2009;35:282–290. doi: 10.1007/s00134-008-1296-0. PMID:18795253 [DOI] [PubMed] [Google Scholar]
- [176].Jakob SM, Ruokonen E, Grounds RM, et al. Dexmedetomidine vs midazolam or propofol for sedation during prolonged mechanical ventilation: two randomized controlled trials. JAMA. 2012;307:1151–1160. doi: 10.1001/jama.2012.304. PMID:22436955 [DOI] [PubMed] [Google Scholar]
- [177].Lindgren BR, Ekstrom T, Andersson RGG. The effects of inhaled clonidine in patients with asthma. Am Rev Respir Dis. 1986;134:266–269. [DOI] [PubMed] [Google Scholar]
- [178].Koutsoukou A, Armaganidis A, Stavrakaki-Kallergi C, et al. Expiratory flow limitation and intrinsic positive end-expiratory pressure at zero positive end-expiratory pressure in patients with adult respiratory distress syndrome. Am J Respir Crit Care Med. 2000;161:1590–1596. doi: 10.1164/ajrccm.161.5.9904109. PMID:10806160 [DOI] [PubMed] [Google Scholar]
- [179].Kernan S, Rehman S, Meyer T, et al. Effects of dexmedetomidine on oxygenation during one-lung ventilation for thoracic surgery in adults. J Minimal Access Surg. 2011;7:227–231. doi: 10.4103/0972-9941.85645. PMID:22022111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Huang SQ, Zhang J, Zhang XX, et al. Can dexmedetomidine improve arterial oxygenation and intrapulmonary shunt during one-lung ventilation in adults undergoing thoracic surgery? A meta-analysis of randomized, placebo-controlled trials. Chin Med J (Engl). 2017;130:1707–1714. doi: 10.4103/0366-6999.209891. PMID:28685722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Ghignone M, Calvillo O, Quintin L, et al. Clonidine: a new pulmonary vasodilator. Crit Care Med. 1986;14:344. doi: 10.1097/00003246-198604000-00073. [DOI] [Google Scholar]
- [182].Stefanadis C, Manolis A, Dernellis J, et al. Acute effect of clonidine on left ventricular pressure-volume relation in hypertensive patients with diastolic heart dysfunction. J Hum Hypertens. 2001;15:635–642. doi: 10.1038/sj.jhh.1001243. PMID:11550110 [DOI] [PubMed] [Google Scholar]
- [183].Giles TD, Iteld BJ, Mautner RK, et al. Short-term effects of intravenous clonidine in congestive heart failure. Clin Pharmacol Ther. 1981;30:724–728. doi: 10.1038/clpt.1981.229. PMID:7307422 [DOI] [PubMed] [Google Scholar]
- [184].Hermiller JB, Magorien RD, Leithe ME, et al. Clonidine in congestive heart failure: a vasodilator with negative inotropic effects. Am J Cardiol. 1983;51:791–795. doi: 10.1016/S0002-9149(83)80135-0. PMID:6829440 [DOI] [PubMed] [Google Scholar]
- [185].Olivari MT, Levine TB, Cohn JN. Acute hemodynamic and hormonal effects of central vs. peripheral sympathetic inhibition in patients with congestive heart failure. J Cardiovasc Pharmacol. 1986;8:973–978. doi: 10.1097/00005344-198609000-00014. PMID:2429099 [DOI] [PubMed] [Google Scholar]
- [186].Manolis AJ, Olympios C, Sifaki M, et al. Suppressing sympathetic activation in congestive heart failure. A new therapeutic strategy. Hypertension. 1995;26:719–724. doi: 10.1161/01.HYP.26.5.719. PMID:7591009 [DOI] [PubMed] [Google Scholar]
- [187].De Kock M, Laterre PF, Van Obbergh L, et al. The effects of intraoperative intravenous clonidine on fluid requirements, hemodynamic variables, and support during liver transplantation: a prospective, randomized study. Anesth Analg. 1998;86:468–476. doi: 10.1213/00000539-199803000-00003. PMID:9495395 [DOI] [PubMed] [Google Scholar]
- [188].Kulka PJ, Tryba M, Reimer T, et al. Clonidine prevents tissue-malperfusion during extracorporal circulation. Anesth Analg. 1996;82:482–487. [Google Scholar]
- [189].Miyamoto K, Nakashima T, Shima N, et al. Effect of dexmedetomidine on lactate clearance in patients with septic shock: a sub-analysis of a multicenter randomized controlled trial. Shock. 2017. doi: 10.1097/SHK.0000000000001055. PMID:29117063 [DOI] [PubMed] [Google Scholar]
- [190].Schmidt K, Hernekamp JF, Philipsenburg C, et al. Time-dependent effect of clonidine on microvascular permeability during endotoxemia. Microvasc Res. 2015;101:111–117. doi: 10.1016/j.mvr.2015.07.002. PMID:26177515 [DOI] [PubMed] [Google Scholar]
- [191].Esler M, Dudley F, Jennings G, et al. Increased sympathetic nervous activity and the effects of its inhibition with clonidine in alcoholic cirrhosis. Ann Intern Med. 1992;116:446–455. doi: 10.7326/0003-4819-116-6-446. PMID:1739234 [DOI] [PubMed] [Google Scholar]
- [192].Singhal S, Baikati KK, Jabbour II, et al. Management of refractory ascites. Am J Ther. 2012;19:121–132. doi: 10.1097/MJT.0b013e3181ff7a8b. PMID:21192246 [DOI] [PubMed] [Google Scholar]
- [193].Lenaerts A, Codden T, Meunier JC, et al. Effects of clonidine on diuretic response in ascitic patients with cirrhosis and activation of sympathetic nervous system. Hepatology. 2006;44:844–849. doi: 10.1002/hep.21355. PMID:17006921 [DOI] [PubMed] [Google Scholar]
- [194].Tasdogan M, Memis D, Sut N, et al. Results of a pilot study on the effects of propofol and dexmedetomidine on inflammatory responses and intraabdominal pressure in severe sepsis. J Clin Anesth. 2009;21:394–400. doi: 10.1016/j.jclinane.2008.10.010. PMID:19833271 [DOI] [PubMed] [Google Scholar]
- [195].von Dossow V, Baehr N, Moshirzadeh M, et al. Clonidine attenuated early proinflammatory response in T-cell subsets after cardiac surgery. Anesth Analg. 2006;103:809–814. doi: 10.1213/01.ane.0000237308.28739.d8. PMID:17000786 [DOI] [PubMed] [Google Scholar]
- [196].Xu B, Makris A, Thornton C, et al. Antihypertensive drugs clonidine, diazoxide, hydralazine and furosemide regulate the production of cytokines by placentas and peripheral blood mononuclear cells in normal pregnancy. J Hypertens. 2006;24:915–922. doi: 10.1097/01.hjh.0000222762.84605.03. PMID:16612254 [DOI] [PubMed] [Google Scholar]
- [197].Kallet RH, Hemphill JC, Dicker RA, et al. The spontaneous breathing pattern and work of breathing of patients with acute respiratory distress syndrome and acute lung injury. Respir Care. 2007;52:989–995. PMID:17650353 [PubMed] [Google Scholar]
- [198].Yoshida T, Torsani V, Gomes S, et al. Spontaneous effort causes occult pendelluft during mechanical ventilation. Am J Respir Crit Care Med. 2013;188:1420–1427. doi: 10.1164/rccm.201303-0539OC. PMID:24199628 [DOI] [PubMed] [Google Scholar]
- [199].Toumpanakis D, Kastis GA, Zacharatos P, et al. Inspiratory resistive breathing induces acute lung injury. Am J Respir Crit Care Med. 2010;182:1129–1136. doi: 10.1164/rccm.201001-0116OC. PMID:20622034 [DOI] [PubMed] [Google Scholar]
- [200].Hopkins SR, Schoene RB, Henderson WR, et al. Intense exercise impairs the integrity of the pulmonary blood-gas barrier in elite athletes. Am J Respir Crit Care Med. 1997;155:1090–1094. doi: 10.1164/ajrccm.155.3.9116992. PMID:9116992 [DOI] [PubMed] [Google Scholar]
- [201].Marini JJ. Lessons learned: the conditional importance of high positive end-expiratory pressure in acute respiratory distress syndrome. Crit Care Med. 2006;34:1540–1542. doi: 10.1097/01.CCM.0000216194.15882.CC. PMID:16633248 [DOI] [PubMed] [Google Scholar]
- [202].Gattinoni L, Pesenti A. The concept of “baby lung”. Intensive Care Med. 2005;31:776–784. doi: 10.1007/s00134-005-2627-z. PMID:15812622 [DOI] [PubMed] [Google Scholar]
- [203].Jozwiak M, Silva S, Persichini R, et al. Extravascular lung water is an independent prognostic factor in patients with acute respiratory distress syndrome. Crit Care Med. 2013;41:472–480. doi: 10.1097/CCM.0b013e31826ab377. PMID:23263578 [DOI] [PubMed] [Google Scholar]
- [204].Carteaux G, de Prost N, Razazi K, et al. The authors reply. Crit Care Med. 2016;44:e1154. doi: 10.1097/CCM.0000000000001995. PMID:27755098 [DOI] [PubMed] [Google Scholar]
- [205].Yoshida T, Fujino Y, Amato M, et al. Spontaneous breathing during mechanical ventilation: risk, mechanisms, and management. Am J Respir Crit Care Med. 2017;in press. doi: 10.1164/rccm.201604-0748CP. [DOI] [PubMed] [Google Scholar]
- [206].Fremont RD, Kallet RH, Matthay MA, et al. Postobstructive pulmonary edema: a case for hydrostatic mechanisms. Chest. 2007;131:1742–1746. doi: 10.1378/chest.06-2934. PMID:17413051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Moriondo A, Mukenge S, Negrini D. Transmural pressure in rat initial subpleural lymphatics during spontaneous or mechanical ventilation. Am J Physiol Heart Circ Physiol. 2005;289:H263–H269. doi: 10.1152/ajpheart.00060.2005. PMID:15833809 [DOI] [PubMed] [Google Scholar]
- [208].Laffon M, Lu LN, Modelska K, et al. alpha-adrenergic blockade restores normal fluid transport capacity of alveolar epithelium after hemorrhagic shock. Am J Physiol. 1999;277:L760–L768. PMID:10516217 [DOI] [PubMed] [Google Scholar]
- [209].Hraiech S, Yoshida T, Papazian L. Balancing neuromuscular blockade versus preserved muscle activity. Curr Opin Crit Care. 2015;21:26–33. doi: 10.1097/MCC.0000000000000175. PMID:25517889 [DOI] [PubMed] [Google Scholar]
- [210].Pichot C, Petitjeans F, Ghignone M, et al. Swift recovery of severe acute hypoxemic respiratory failure under non-invasive ventilation. Anaesthesiol Intensive Ther. 2015;47:138–142. doi: 10.5603/AIT.a2014.0053. PMID:25338517 [DOI] [PubMed] [Google Scholar]
- [211].Yoshida T, Papazian L. When to promote spontaneous respiratory activity in acute respiratory distress patients? Anesthesiology. 2014;120:1313–1315. doi: 10.1097/ALN.0000000000000260. PMID:24722176 [DOI] [PubMed] [Google Scholar]
- [212].Rittayamai N, Brochard L. Recent advances in mechanical ventilation in patients with acute respiratory distress syndrome. Eur Respir Rev. 2015;24:132–140. doi: 10.1183/09059180.00012414. PMID:25726563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Freebairn R, Hickling KG. Spontaneous breathing during mechanical ventilation in ARDS. Crit Care Shock. 2005;8:61–66. [Google Scholar]
- [214].Leray V, Bourdin G, Flandreau G, et al. A case of pneumomediastinum in a patient with acute respiratory distress syndrome on pressure support ventilation. Respir Care. 2010;55:770–773. PMID:20507662 [PubMed] [Google Scholar]
- [215].Putensen C, Mutz NJ, Putensen-Himmer G, et al. Spontaneous breathing during ventilatory support improves ventilation-perfusion distributions in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 1999;159:1241–1248. doi: 10.1164/ajrccm.159.4.9806077. PMID:10194172 [DOI] [PubMed] [Google Scholar]
- [216].Froese AB, Bryan AC. Effects of anesthesia and paralysis on diaphragmatic mechanics in man. Anesthesiology. 1974;41:242–255. doi: 10.1097/00000542-197409000-00006. PMID:4604401 [DOI] [PubMed] [Google Scholar]
- [217].Leroy S, Aladin L, Laplace C, et al. Introduction of a centrally anti-hypertensive, clonidine, reduces noradrenaline requirements in septic shock caused by necrotizing enterocolitis Am J Emerg Med. 2017;35:e3–377. doi: 10.1016/j.ajem.2016.08.027. [DOI] [PubMed] [Google Scholar]
- [218].Longrois D, Quintin L. Dexmedetomidine: Superiority trials needed? Anaesth Crit Care Pain Med. 2016;35:237–238. doi: 10.1016/j.accpm.2016.03.001. PMID:26972484 [DOI] [PubMed] [Google Scholar]
- [219].Pandharipande PP, Sanders RD, Girard T. Comparison of sedation with dexmedetomidine versus lorazepam in septic ICU patients. Critical Care. 2008;12:P275. doi: 10.1186/cc6496. [DOI] [Google Scholar]
- [220].Cantazucene J. Nouvelles recherches sur le mode de destruction des vibrions dans l'organisme. Annales de l'Institut Pasteur. 1898;12:273–300. [Google Scholar]
- [221].Takayama K, Yuhki K, Ono K, et al. Thromboxane A2 and prostaglandin F2alpha mediate inflammatory tachycardia. Nat Med. 2005;11:562–566. doi: 10.1038/nm1231. PMID:15834430 [DOI] [PubMed] [Google Scholar]
- [222].Hamers L, Kox M, Pickkers P. Sepsis-induced immunoparalysis: mechanisms, markers, and treatment options. Minerva Anestesiol. 2015;81:426–439. PMID:24878876 [PubMed] [Google Scholar]
- [223].Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013;13:260–268. doi: 10.1016/S1473-3099(13)70001-X. PMID:23427891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. 2016;126:23–31. doi: 10.1172/JCI82224. PMID:26727230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Martelli D, McKinley MJ, McAllen RM. The cholinergic anti-inflammatory pathway: a critical review. Auton Neurosci. 2014;182:65–69. doi: 10.1016/j.autneu.2013.12.007. [DOI] [PubMed] [Google Scholar]
- [226].Martelli D, Yao ST, McKinley MJ, et al. Reflex control of inflammation by sympathetic nerves, not the vagus. J Physiol. 2014;592:1677–1686. doi: 10.1113/jphysiol.2013.268573. PMID:24421357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Bellinger DL, Lorton D. Autonomic regulation of cellular immune function. Auton Neurosci. 2014;182:15–41. doi: 10.1016/j.autneu.2014.01.006. [DOI] [PubMed] [Google Scholar]
- [228].Kox M, Stoffels M, Smeekens SP, et al. The influence of concentration/meditation on autonomic nervous system activity and the innate immune response: a case study. Psychosom Med. 2012;74:489–494. doi: 10.1097/PSY.0b013e3182583c6d. PMID:22685240 [DOI] [PubMed] [Google Scholar]
- [229].Kox M, van Eijk LT, Zwaag J, et al. Voluntary activation of the sympathetic nervous system and attenuation of the innate immune response in humans. Proc Natl Acad Sci USA. 2014;111:7379–7384. doi: 10.1073/pnas.1322174111. PMID:24799686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Smith IM, Kennedy LR, RegnÇ-Karlsson MH, et al. Adrenergic mechanisms in infection. III: alpha and beta-receptor blocking agents in treatment. Am J Clin Nutr. 1977;30:1285–1288. doi: 10.1093/ajcn/30.8.1285. PMID:18930 [DOI] [PubMed] [Google Scholar]
- [231].Seeley EJ, Barry SS, Narala S, et al. Noradrenergic neurons regulate monocyte trafficking and mortality during gram-negative peritonitis in mice. J Immunol. 2013;190:4717–4724. doi: 10.4049/jimmunol.1300027. PMID:23543756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Taniguchi T, Kidani Y, Kanakura H, et al. Effects of dexmedetomidine on mortality rate and inflamatory response to endotoxin-induced shock in rats. Crit Care Med. 2004;32:1322–1326. doi: 10.1097/01.CCM.0000128579.84228.2A. PMID:15187514 [DOI] [PubMed] [Google Scholar]
- [233].Pecaut MJ, Mehrotra S, Luo-Owen X, et al. Chlorisondamine, a sympathetic ganglionic blocker, moderates the effects of whole-body irradiation (WBI) on early host defense to a live bacterial challenge. Immunol Lett. 2015;167:103–115. doi: 10.1016/j.imlet.2015.07.008. PMID:26235133 [DOI] [PubMed] [Google Scholar]
- [234].Gregorakos L, Kerezoudi E, Dimopoulos G, et al. Management of blood pressure instability in severe tetanus: the use of clonidine. Intensive Care Med. 1997;23:893–895. doi: 10.1007/s001340050428. PMID:9310809 [DOI] [PubMed] [Google Scholar]
- [235].Moritz RD, Machado FO, Pinto EP, et al. [Evaluate the clonidine use for sedoanalgesia in intensive care unit patients under prolonged mechanical ventilation]. Revista Brasileira de terapia intensiva. 2008;20:24–30. Avaliacao do uso da clonidina para sedoanalgesia de pacientes sob ventilacao mecanica prolongada, internados em unidade de terapia intensiva. doi: 10.1590/S0103-507X2008000100004. PMID:25306944 [DOI] [PubMed] [Google Scholar]
- [236].Selye H. A syndrome produced by diverse nocuous agents. Nature. 1936;138:32. doi: 10.1038/138032a0. [DOI] [PubMed] [Google Scholar]
- [237].Laborit H. La notion de “reaction oscillante post agressive”. Therapie. 1951;6:207–210. PMID:14922295 [PubMed] [Google Scholar]
- [238].Cann C, Melia D. Sedation in intensive care: should we reach for dexmedetomidine sooner? Br J Hosp Med (Lond). 2017;78:598. doi: 10.12968/hmed.2017.78.10.598. PMID:29019730 [DOI] [PubMed] [Google Scholar]
- [239].Satinoff E. Neural organization and evolution of thermal regulation in mammals. Science. 1978;201:16–22. doi: 10.1126/science.351802. PMID:351802 [DOI] [PubMed] [Google Scholar]
- [240].Romanovsky AA. Thermoregulation: some concepts have changed. Functional architecture of the thermoregulatory system. Am J Physiol Regul Integr Comp Physiol. 2007;292:R37–R46. doi: 10.1152/ajpregu.00668.2006. PMID:17008453 [DOI] [PubMed] [Google Scholar]
- [241].Dantzker DR, Foresman B, Gutierrez G. Oxygen supply and utilization relationships. A reevaluation. Am Rev Respir Dis. 1991;143:675–679. doi: 10.1164/ajrccm/143.3.675. PMID:2001082 [DOI] [PubMed] [Google Scholar]
- [242].Shehabi Y, Botha JA, Ernest D, et al. Clinical application, the use of dexmedetomidine in intensive care sedation. Crit Care Shock. 2010;13:40–50. [Google Scholar]
- [243].Pichot C, Longrois D, Ghignone M, et al. Dexmédetomidine et clonidine: revue de leurs propriétés pharmacodynamiques en vue de définir la place des agonistes alpha-2 adrénergiques dans la sédation en réanimation. Ann Fr Anesth Reanim. 2012;31:876–896. doi: 10.1016/j.annfar.2012.07.018. PMID:23089375 [DOI] [PubMed] [Google Scholar]
- [244].Little DM., Jr. Hypothermia. Anesthesiology. 1959;20:842–877. doi: 10.1097/00000542-195911000-00016. PMID:13856089 [DOI] [PubMed] [Google Scholar]
- [245].Lin CM, Neeru S, Doufas AG, et al. Dantrolene reduces the threshold and gain for shivering. Anesth Analg. 2004;98:1318–1324. table of contents. doi: 10.1213/01.ANE.0000108968.21212.D7. PMID:15105208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Dundee JW, Mesham PR, Scott WE. Chlorpromazine and the production of hypothermia. Anaesthesia. 1954;9:296–302. doi: 10.1111/j.1365-2044.1954.tb01927.x. PMID:13207699 [DOI] [PubMed] [Google Scholar]
- [247].World Medical A. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–2194. doi: 10.1001/jama.2013.281053. PMID:24141714 [DOI] [PubMed] [Google Scholar]
- [248].Pham T, Richard JC, Brochard L. Veno-venous extracorporeal support to treat acute respiratory distress syndrome: rationale and clinical objectives Reanimation. 2013;22:S577–S583. doi: 10.1007/s13546-014-0872-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Villar J, Kacmarek RM. What is new in refractory hypoxemia? Intensive Care Med. 2013;39:1207–1210. doi: 10.1007/s00134-013-2905-0. PMID:23575611 [DOI] [PubMed] [Google Scholar]
- [250].Gattinoni L. Ventilation-induced lung injury exists in spontaneously breathing patients with acute respiratory failure: We are not sure. Intensive Care Med. 2017;43:256–258. doi: 10.1007/s00134-016-4483-4. PMID:28074229 [DOI] [PubMed] [Google Scholar]
- [251].Reed DJ, Kellogg RH. Changes in respiratory response to CO2 during natural sleep at sea level and at altitude. J Appl Physiol. 1958;13:325–330. doi: 10.1152/jappl.1958.13.3.325. PMID:13587410 [DOI] [PubMed] [Google Scholar]
- [252].Nobrega AC, O'Leary D, Silva BM, et al. Neural regulation of cardiovascular response to exercise: role of central command and peripheral afferents. Biomed Res Int. 2014;2014:478965. doi: 10.1155/2014/478965. PMID:24818143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Grasso S, Terragni P, Birocco A, et al. ECMO criteria for influenza A (H1N1)-associated ARDS: role of transpulmonary pressure. Intensive Care Med. 2012;38:395–403. doi: 10.1007/s00134-012-2490-7. PMID:22323077 [DOI] [PubMed] [Google Scholar]
- [254].Colebatch HJ, Greaves IA, Ng CK. Exponential analysis of elastic recoil and aging in healthy males and females. J Appl Physiol Respir Environ Exerc Physiol. 1979;47:683–691. PMID:511674 [DOI] [PubMed] [Google Scholar]
- [255].Mauri T, Grasselli G, Suriano G, et al. Control of respiratory drive and effort in extracorporeal membrane oxygenation patients recovering from severe acute respiratory distress syndrome. Anesthesiology. 2016;125:159–167. doi: 10.1097/ALN.0000000000001103. PMID:26999639 [DOI] [PubMed] [Google Scholar]
- [256].Zhou M, Yang S, Koo DJ, et al. The role of Kupffer cell alpha(2)-adrenoceptors in norepinephrine-induced TNF-alpha production. Biochim Biophys Acta. 2001;1537:49–57. doi: 10.1016/S0925-4439(01)00055-2. PMID:11476962 [DOI] [PubMed] [Google Scholar]
- [257].Miksa M, Das P, Zhou M, et al. Pivotal role of the alpha(2A)-adrenoceptor in producing inflammation and organ injury in a rat model of sepsis. PLoS One. 2009;4:e5504. doi: 10.1371/journal.pone.0005504. PMID:19430535 [DOI] [PMC free article] [PubMed] [Google Scholar]