

Abstract
The development of new chemical entities against the major diseases caused by parasites is highly desired. A library of thirty diamines analogs following a minimalist approach and supported by chemoinformatics tools have been prepared and evaluated against apicomplexan parasites. Different member of the series of N,N′−disubstituted aliphatic diamines shown in vitro activities at submicromolar concentrations and high levels of selectivity against Toxoplasma gondii and in chloroquine-sensitive and resistant-strains of Plasmodium falciparum. In order to demonstrate the importance of the secondary amines, ten N,N,N′,N′−tetrasubstituted aliphatic diamines derivatives were synthesized being considerably less active than their disubstituted counterpart. Theoretical studies were performed to establish the electronic factors that govern the activity of the compounds.
Keywords: N,N′-disubstituted diamines; Polyamines; Anti-apicomplexa; NTDs; Cheminformatics
1. Introduction
The phylum Apicomplexa includes intracellular protozoan pathogens responsible for many diseases with huge global significance in the medical and veterinary field. Plasmodium spp is an apicomplexa parasitic alveolate and is the cause of malaria in different parts of the world. Malaria afflicts millions of individuals in the tropical zones, causing serious morbidity and mortality. Toxoplasma gondii is the etiological agent of toxoplasmosis. This opportunistic pathogen has worldwide distribution, may cause cerebral pathology in immunocompromised individuals, and can produce several congenital anomalies including induction of miscarriage and neurological disorders [1,2]. T. gondii is one of the leading causes of focal central nervous system (CNS) disease in AIDS patients. Other apicomplexan organisms such as Cryptosporidium parvum and Cyclospora spp. also cause opportunistic infections in humans and have relevance in developing countries by causing diarrhoea outbreaks [3,4].
Other members of this phylum including Theileria, Sarcocystis and Babesia spp. are important pathogens of animals. The limited arsenal of drugs available against these organisms is insufficient due to the rapid emergence of resistance, as is the case for Plasmodium or incomplete activity, as the case for chronic Toxoplasmosis. Thus, there is a continuous need develop new chemotherapies in order to combat these diseases in a unique mode of action [5,6].
Significant efforts have been undertaken in the last decades to eradicate malaria [7]. Between 2000 and 2015 there was a registered reduction in malaria total estimated cases of more than 14% in 43 out of the 99 countries with transmission. Due to the development of resistance, antimalarial drugs which have been used to date since the advent of chloroquine, have lost effectiveness around the World [8]. Currently, artemisinin-based combination therapy (ACT) is the mainstay of malaria treatment. Unfortunately, there are an increasing number of countries (mostly in East Asia) in which resistance to artemisinin has been reported [9,10]. Additionally, there is an alarming emergence of multiresistant strains in distinct parts of the globe. Furthermore, the resistance in the mosquito vector to pyrethroids has also been reported [11]. This scenario has led governmental and non-governmental organizations to focus their efforts to finding new compounds that serve to combat the disease [12].
Polyamines are essential metabolites that are implicated in many physiological functions including DNA replication and repair, being essential for normal cell growth and viability [13,14]. The apparent universal distribution of polyamines, and the complexity of compensatory mechanisms that are involved to maintain polyamine homeostasis, suggest that these molecules are critical for cell survival.
The biosynthesis of polyamines typically starts with the conversion of arginine to putrescine which occurs through one of two distinct pathways: arginase coupled with ornithine decarboxylase (ODC), or arginine decarboxylase (ADC) coupled with agmatine iminohydrolase. Putrescine can be further converted into spermi-dine and spermine by spermidine and spermine synthases, respectively. When polyamines are available, spermine or spermi-dine can be retro-converted into spermidine and putrescine by the consecutive action of spermine/spermidine N1−acetyltransferase (SSAT) and polyamine oxidase (PAO) [15–17].
The phylum Apicomplexa has a polyamine metabolism that involves a combination of several sensitive feedback systems regulating their synthesis, degradation, and transport [18,19]. Regulation of polyamine biosynthesis is complex and the key biosynthetic enzyme, ODC is one of the most highly regulated enzymes. It is present in P. falciparum, but extracts of T. gondii had no detectable ODC and ADC activity. Thus, T. gondii lacks a forward-directed polyamine biosynthetic pathway and elucidate the need to discover an alternate mechanism to tackle the pathogenicity of this organism [19].
Previously reported studies from Bitonti et al. shown a series that bis-(benzyl)-polyamines analogs with antimalarial activity against both, a chloroquine-sensitive and -resistant strains of P. falciparum. [20] A selected analog of the series, MDL-27695, (Fig. 1) was markedly active and presented a synergistic effect with D,L-alpha-difluoromethylornithine (DFMO), being able to cure P. berghei infected mice. Initially, MDL-27695 was proposed to target the polyamines biosynthesis. Nevertheless, the mechanism of action of these compounds looks likely to be more complex. On one hand, Bitonti et al. observed that DNA/RNA synthesis was depleted faster with MDL-27695 than upon DFMO treatment, on the other polyamines biosynthesis was possibly repressed but might not be the only cause of the cytotoxic event [21].
Fig. 1.
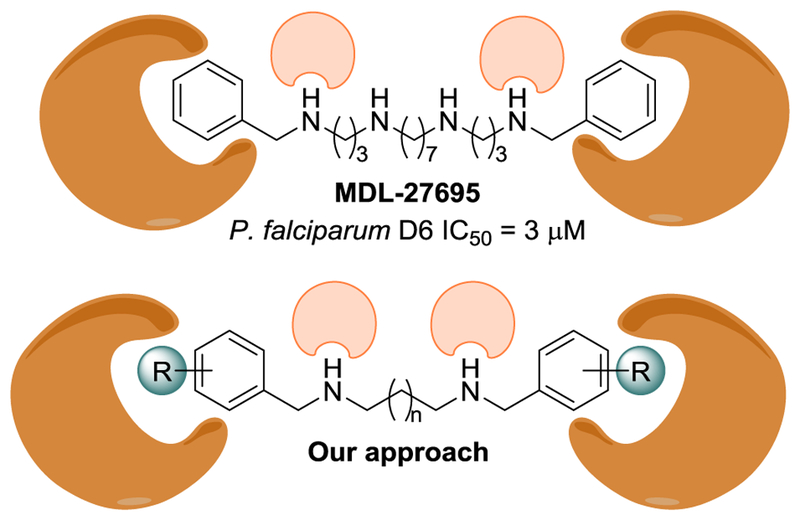
Structure of MDL-27695 and our strategy.
In previous work, we have synthesized N,N′-disubstituted aliphatic diamines and determined their activity against parasites of the kinestoplastida group [22]. Many analogs were active against T. cruzi, T. brucei and L. donovani showing low cytotoxicity. Here, we wanted to expand the analysis of the initial collection to determine activity against Toxoplasma and Plasmodium, looking to link the activity with the pathophysiological differences between these two parasites. Moreover, we intended to understand their mechanism of action linking their phenotypical differences with their metabolic differences. Finally, we were eager to determine the relationship between the physicochemical properties and activity of the relevant compounds.
2. Results and discussion
2.1. Chemistry
The molecular scaffold of our library was based on the assumption that MDL-27695 interacts with a molecular target anchored by two nitrogens requiring a bulky hydrophobic substituent in both terminal nitrogens (Fig. 1). Following that hypothesis, a minimal version of that scaffold should be a bis-substituted benzyl N,N′-disubstituted diamine. That analog should fulfil the steric and polar requirements and at the same time be considerably simpler. The library has two sources of diversity: 1) the carbon size chain, which brings flexibility and more grades of freedom and 2) the substituent on the aromatic ring. The library was prepared in a single step by a double reductive amination between the diamines and the benzaldehydes using the procedure by Phanstiel and co-workers [23] with minimal modifications (Scheme 1). The library was built with all the possible combinations of commercial aliphatic diamines with a 3, 4, 6, 8, 10 and 12 carbon chain length and five aromatic aldehydes (benzaldehyde, 4-methoxy-benzaldehyde, 4-benzyloxy benzaldehyde, 3-hydroxy-4-benzyloxy benzaldehyde and 3-methoxy-4-benzyloxy benzalde-hyde). The library of thirty N,N′ disubstituted diamines was prepared in solution, including the dodecyldiamine analogs that were not previously reported [22].
Scheme 1.

Synthesis of N,N′−derivatives.
The synthesis produced all the desired products with good yield (Table 1, average 77%, ranging 30–99%). The yield was similar across each series (around 80%).
Table 1.
Anti-apicomplexan activity, cytotoxicity and cluster family of N, N′-substituted diamines.
| Compound | n | R | Yield*(%) | P. falciparum D6 IC50 (μM) | SIa | P. falciparum W2 IC50 (μM) | SIa | T. gondii Growth inhibition at 1 μMb | CytotoxicityVero cells IC50 (μM)c | Cluster |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | H | 79 | 2.35 | >7.9 | 2.63 | >7.1 | + | NC | A |
| 2 | 4 | H | 73 | 2.98 | >5.9 | 2.49 | >7.1 | + | NC | A |
| 3 | 6 | H | 46 | 2.09 | >7.7 | 2.09 | >7.7 | + | NC | B |
| 4 | 8 | H | 94 | 0.37 | >39.7 | 0.37 | >39.7 | + | NC | B |
| 5 | 10 | H | 89 | 0.48 | >28.0 | 0.28 | >47.6 | +++ | NC | B |
| 6 | 12 | H | 79 | 0.97 | >12.9 | 1.02 | >12.2 | + | NC | B |
| 7 | 3 | 4-OMe | 78 | 3.18 | >4.8 | 5.41 | >2.8 | ++ | NC | C |
| 8 | 4 | 4-OMe | 77 | 4.57 | >3.2 | 6.69 | >2.2 | + | NC | D |
| 9 | 6 | 4-OMe | 30 | 3.08 | 2.4 | 3.36 | 2.5 | + | 8.32 | E |
| 10 | 8 | 4-OMe | 59 | 1.48 | >8.4 | 1.69 | >7.3 | + | NC | F |
| + | ||||||||||
| 11 | 10 | 4-OMe | 66 | 0.60 | >19.0 | 0.67 | >17.0 | ++ | NC | F |
| 12 | 12 | 4-OMe | 99 | 1.58 | >6.8 | 1.86 | >5.8 | +++ | NC | F |
| 13 | 3 | 4-OBn | 67 | 1.22 | 4.9 | 1.43 | 4.2 | +++ | 6.00 | G |
| 14 | 4 | 4-OBn | 77 | 2.08 | 2.8 | 3.12 | 1.9 | ++ | 5.83 | H |
| 15 | 6 | 4-OBn | 86 | 0.39 | >24.0 | 0.29 | >32.2 | + | NC | I |
| 16 | 8 | 4-OBn | 99 | 3.72 | >2.4 | 4.09 | >2.2 | + | NC | J |
| 17 | 10 | 4-OBn | 60 | 2.47 | >3.4 | 3.50 | >2.4 | +++ | NC | J |
| 18 | 12 | 4-OBn | 99 | 8.00 | >1.2 | 8.00 | >1.2 | ND | NC | J |
| 19 | 3 | 3-OH, 4-OMe | 96 | >8.00 | - | >8.00 | - | ND | NC | C |
| 20 | 4 | 3-OH, 4-OMe | 80 | >8.00 | - | >8.00 | - | +++ | NC | D |
| 3-OH, 4-OMe | ||||||||||
| 21 | 6 | 3-OH, 4-OMe | 82 | >8.00 | - | >8.00 | - | ++ | NC | E |
| 22 | 8 | 3-OH, 4-OMe | 78 | 4.80 | >2.38 | 5.52 | >2.1 | + | NC | F |
| 23 | 10 | 3-OH, 4-OMe | 72 | 1.97 | >5.41 | 1.88 | >5.7 | + | NC | F |
| 24 | 12 | 3-OH, 4-OMe | 46 | 1.97 | >5.10 | 2.88 | >3.5 | + | NC | F |
| 25 | 3 | 3-OMe, 4-OBn | 50 | 0.42 | 15.9 | 0.47 | 14.1 | ++++ | 6.65 | G |
| 3-OMe, 4-OBn | ||||||||||
| 26 | 4 | 3-OMe, 4-OBn | 84 | 0.33 | >27.4 | 0.49 | >18.4 | ++ | NC | H |
| 27 | 6 | 3-OMe, 4-OBn | 81 | 0.26 | >31.7 | 0.44 | >19.0 | + | NC | I |
| 28 | 8 | 3-OMe, 4-OBn | 88 | 0.31 | 14.6 | 0.43 | 10.5 | - | 4.52 | J |
| 29 | 10 | 3-OMe, 4-OBn | 89 | 1.12 | >6.8 | 1.05 | >7.2 | + | NC | J |
| 3-OMe, 4-OBn | ||||||||||
| 30 | 12 | 3-OMe, 4-OBn | 99 | 1.28 | >5.67 | 1.37 | >5.3 | + | NC | J |
| 31 | 3 | H tetrasubstituted | 17 | >8.00 | - | >8.00 | - | ND | NC | K |
| 32 | 4 | H tetrasubstituted | 26 | >8.00 | - | >8.00 | - | ND | NC | K |
| 33 | 6 | H tetrasubstituted | 17 | >8.00 | - | >8.00 | - | ND | NC | L |
| 34 | 8 | H tetrasubstituted | 27 | >8.00 | - | >8.00 | - | ND | NC | L |
| 35 | 10 | H tetrasubstituted | 28 | >8.00 | - | >8.00 | - | ND | NC | L |
| 36 | 3 | 4-OMe Tetrasubt. | 30 | >8.00 | - | >8.00 | - | ND | NC | K |
| 37 | 4 | 4-OMe Tetrasubt. | 25 | >8.00 | - | >8.00 | - | ND | NC | K |
| 38 | 6 | 4-OMe Tetrasubt. | 10 | >8.00 | - | >8.00 | - | ND | NC | L |
| 39 | 8 | 4-OMe Tetrasubt. | 16 | >8.00 | - | >8.00 | - | ND | NC | L |
| 40 | 10 | 4-OMe Tetrasubt. | 28 | >8.00 | - | >8.00 | - | ND | NC | L |
| CQ | 0.083 | 0.422 | ||||||||
| Art | 0.094 | 0.094 | ||||||||
| Pyr | - |
NC = not cytotoxic at the maximum concentration tested (4.46 μg/mL) CQ= Chloroquine Art = Arteminsin Pyr = pyremethamine.
SI (Selectivity Index: IC50 Vero Cells/IC50 Parasites).
Scale: ++++ total growth inhibition of parasite with fibroblast integrity, +++ Some tachizoite growth but approximately at 75% of control with fibroblast integrity, ++ correspond at 50% of parasite growth, + at 25% and - no growth inhibition.
Tested at a maximum concentration of 8 μM.
2.2. Biological evaluation
The polyamines pathway in Plasmosdium spp. has been studied during the last 25 years with different grades of success. Target-oriented drug design directed to enzymes in the polyamine pathway provided promising hits, without showing considerable progress towards becoming lead compounds. Therefore, having in mind all the successful examples via phenotypic screening, and looking to a wide spectrum candidate, we decided to assay our collection not only against P. falciparum strains but also against T. gondii. Also, cytotoxicity was evaluated in two different cell lines, African green monkey kidney epithelial (VERO) cells and human fibroblasts.
2.2.1. Antiplasmodium activity
All the analogs were assayed against the chloroquine sensitive Sierra Leona clone (D6) and the chloroquine resistant, Indochina clone (W2) strains of P. falciparum. To our satisfaction, most of the compounds were active against both the strains. (Table 1). Ninety percent of the collection has an IC50 below 5 μM, with 73% of the library being more potent than MDL-27695. In general, there are no substantial variations in the potency between the sensitive and resistant strain, suggesting that the mechanism that confer chloroquine resistance to the W2 strain does not affect sensitivity to this family of compounds.
When comparing activities based on the benzyl substituent some clear tendencies emerge. First, there is a narrow difference between the more and the less potent member of each series, which ranges from 7.61 for R = 4-BnO to 1.02 for R = 3-MeO,4-BnO (Fig. 2).
Fig. 2.

Antimalarial activity (D6 Clone) by substituent. The average value of each subset is shown.
These slight differences in antiplasmodium efficacy of the analogs indicate that the diamine chain length does not have a strong effect the activity. At the same time, comparing the activity by the same diamine linker it seems that it does not considerably affect the potency.
The analysis of the unsubstituted benzyl derivatives (1 to 6) reveal that compounds with large chains (compounds 4, 5 and 6) are very potent (below 1 μM against both strains). A comparison with our template compound MDL-27695 reveal that these analogs are 3–8 times more active being shorter in length and having only two nitrogen atoms. The R = 4-MeO series exhibited a similar activity profile, but in this case, compound 11 with a C10 diamine was the most active with IC50 of 0.60 μM. A different activity profile was noticed in case of 4-benzyloxy analogs (13 to 18). The tendency changes, with 15 was the most active (n = 8, IC50 = 0.39 for D6 and 0.29 μM for W2) and 18 was the least active (n = 12, IC50 = 8.00 μM for both D6 and W2 strains).
Isovanillin analogs 19, 20 and 21 did not show activity up to the maximum concentration tested (8.00 μM). That anomaly could be related to the presence of a 3-OH group, which by substantially increasing hydrophilicity would alter membrane permeability. Compound 27 was the most active analog of the whole library with an IC50 of 260 nM against the P. falciparum D6 strain and SI > 31.7, only 3-fold less active than chloroquine and artemisinin and 12-fold more active than MDL-27695. However, compound 4 showed an IC50 of 370 nM but presents an even higher SI (>39.7).
As for the resistant strain analog 5, which is 1.5-fold more active than chloroquine, combines the best activity and selectivity, with an IC50 280 nM. Finally, the 3-methoxy-4-benzyloxy benzyl analogs are the most active series, with an average of 0.62 μM, with compound 27 (n = 6, IC50 = 0.26 μM) the most active and 30 the less active (n 12, IC50 = 1.28 μM). Interestingly, there is slight differ-activity among the compounds in this series, suggesting that increasing the chain has minimal effect on activity.
Having completed the analysis of the chain length and the aromatic ring substitution influence on the antiplamodial activity, it was important to establish the impact of the nitrogen substitution. There are many literature reports of monosubstituted diamines, but surprisingly, no precedents of bioactive tetrasubstituted aliphatic diamine analogs as antiparasitic agents [24]. With that idea in mind, ten tetrasubstituted analogs were prepared. Only the simplest aromatic substituents (R = H and 4-OMe) and five diamines (n = 3,4,6,8 and 10) were selected, looking to comply with Lipinsky’s rules (MW < 500 Da and logP<5), additionally dodecylamine analogs were not considered because they were, on average, less active. In this opportunity, a different methodology for the synthesis was required. A one-pot procedure was selected, where two consecutive reductive aminations occurred using sodium triacetoxy borohydride, acetic acid in DCM art room temperature [25,26]. Ten new products were prepared (Scheme 2) with 22% global yield (Table 1). The main drawback of the reaction was the aldehyde reduction that forced using at least 4.5 equivalents of that component. Unfortunately, when more equivalents were used, the amount of the benzyl alcohol increased, impeding a clean separation of the product by column chromatography.
Scheme 2.

Synthesis of N,N,N′,N′−derivatives.
The new analogs were evaluated against the D6 and W2 strains of P. falciparum and did not show activity at the maximum concentration tested (8 μM). A more detailed analysis revealed that the most active member of each series of the disubstituted analogs were at least 40 times (Compound 4, R = H, IC50 P.f. D6 = 0.37 μM) and 19 times (compound 11, R = 4-MeO, IC50 P.f. D6 = 0.60 μM) more active than the tetrasubstituted analogs 34 and 40 (both P.f. D6 > 8.0 μM), respectively.
The antimalarial activity of N,N−disubstituted putrescine and trimethylenediamine had been previously reported [27]. Comparing the activity of the reported N,N−disubstituted with the corresponding N,N′-disubstituted analogs has shown that the last ones were always more active (between 1.5 and 7 times more potent). These results clearly demonstrate that the secondary amine is an important structural feature required for the antimalarial activity, being the N,N′-disubstitution the pattern that provided the best results.
2.2.2. Anti-Toxoplasma gondii activity
To test activity of our library of diamines against T. gondii we cultured parasites on human foreskin fibroblasts in the presence of drug at 1 μM or of DMSO as solvent control for 5 days, at which time point cells were fixed and stained to monitor parasite growth. The results, presented in Table 1, show that in general the molecules in our library are not as effective in inhibiting T. gondii growth as they are with P. falciparum. Compounds 5, 12, 13, 17 and 25 (20% of the collection) were the most effective against T. gondii and exhibited at least 75% of inhibition compared with the untreated parasites. Interestingly, compound 5 was also the most active member of the benzyl series against P. falciparum (IC50 = 0.48 μM). Compound 12 is the most active on T. gondii of the 4-methoxy benzyl series, and is just two carbons larger than the analog of that family with best antiplasmodial activity. The 4-benzyloxy benzyl containing compounds 13 and 17, which have 3 and 10 carbon chain respectively, show 75% of T. gondii growth inhibition. Compound 15, which did not affect T. gondii growth was the member of that series (4-benzyloxy benzyl) with the best antiplasmodial activity. The 3-methoxy-4-hydroxy benzyl and the 3-methoxy-4-benzyloxy benzyl series contains a second oxygen on the benzyl ring. For the isovanillin analogs, short chains derivatives (20, 21) were the most active, behaving differently on P. falciparum were the longer analogs were the most potent.
Finally, the IC50 of most active analogs was determined, revealing that compounds 13 and 25 has an IC50 = 2.97 μM and 25, the most active member of the series has an IC50 =1.53 μM. Interestingly, none of these compounds displayed cytotoxicity on fibroblast at 10 μM, providing an SI as anti-T. gondii of 3.37 and 6.54 μM for 13 and 25, respectively. In summary, the overall library behavior was different between the two parasite species, showing that tachyzoites were not affected in the same way that Plasmodium erythrocytic stage (trophozoites) suggesting a different action mechanism in both parasites.
2.2.3. Cytotoxicity
Vero cells are an excellent model for test cytotoxicity in vitro because is an aneuploid and a continuous cell linage. The library was tested for their effect on Vero cells to a maximum concentration of 8 μM. Only five compounds, (9, 13, 14, 25 and 28), which corresponds to 16% of our library, were cytotoxic with a range of IC50 between 4 and 8 μM. These five diamines analogs have a short carbon chain length, and all, with the exemption of compound 9, have benzyloxy substituents on position 4 of the aromatic ring. Nonetheless, these five compounds, particularly 25 and 28 exhibit excellent activities against apicomplexa parasites. Therefore, even being cytotoxic at low micromolar they are 10-fold higher as anti-malarial providing good SI (15.9 and 14.6, respectively).
2.3. Cheminformatics and clusters analysis
The rationale used to design this targeted library of symmetrical N,N′-disubstituted diamines allowed only two entries for scaffold modifications providing a collection that has low chemical diversity. However, using the ChemMineTools online platform [28], it was possible to cluster the collection based on their structural matrixes. This information was useful to establish a structural-activity relationship correlating the activity and topological parameters. (Fig. 3). The outcome of this calculation revealed the precedence of three main clusters (marked in green, red and blue in Fig. 3) and ten secondary clusters (labelled from A to J in Table 1).
Fig. 3.

Cluster and dendrogram analysis of N,N0-di substituted diamine library.
The main cluster (green) contains the most active analogs in both parasites. This cluster has two sets of subclusters: one is represented for compounds 13, 14, 25 and 26 (clusters G and H) and the second is represented by 15, 16, 17, 18, 27, 28, 29 and 30 (clusters I and J). When activities are considered, the first subcluster includes analogs with the highest potency towards both parasites. The second subcluster is more active against P. falciparum. The percentage of growth inhibition of Toxoplasma is low with the exception of compound 17.
2.3.1. Physicochemical characteristics and ADME-Tox
Addressing pharmacokinetic properties at the early stages of drug development reduces the chances of failure on clinical trials. To characterize the profile of our analogs, we performed computational studies of all compounds to predict their adsorption, distribution, metabolism and excretion (ADME) properties, Lipinski’s rule of five, toxicity liabilities and drug likeness. The in silico toxicology calculated by the OSIRIS property explorer platform [29] indicates that none of the products prepared are potentially mutagenic, irritant, teratogenic, or toxic for sexual reproduction. The only exception is the isovanillin subfamily derivatives, which are potentially mutagenic. Likewise, this subset demonstrated to possess the lowest levels of biological activities, therefore they are not good candidates for future optimizing steps.
On the other hand, the most relevant parameters (molecular weight, polar surface area, volume, logBB and logP) of the collection allow us to predict the potential of the compounds as leaders, possible administration routes and to enrich SAR. To that purpose the Molinspiration [30] and ChemAxon [31] platforms were used to calculate these physicochemical parameters including the predominant molecular form at physiological pH (Table 2).
Table 2.
Physicochemical properties of N,N′−substituted diamines.
| Compound | n | R | MW | LogP | LogD | LogBB | TPSA | Volume |
|---|---|---|---|---|---|---|---|---|
| 1 | 3 | H | 254.377 | 2.538 | 0,46 | 0.373 | 24.06 | 264.27 |
| 2 | 4 | H | 268.404 | 2.994 | −0,88 | 0.436 | 24.06 | 281.07 |
| 3 | 6 | H | 296.458 | 3.906 | −0,01 | 0.584 | 24.06 | 314.67 |
| 4 | 8 | H | 324.512 | 4.818 | 0,88 | 0.735 | 24.06 | 348.27 |
| 5 | 10 | H | 352.566 | 5.730 | 1,77 | 0.851 | 24.06 | 381.88 |
| 6 | 12 | H | 380.620 | 6.642 | 2,66 | 1.005 | 24.06 | 415.48 |
| 7 | 3 | 4-OMe | 314.429 | 2.807 | 0,26 | 0.119 | 42.52 | 315.36 |
| 8 | 4 | 4-OMe | 328.456 | 3.261 | −1,09 | 0.193 | 42.52 | 332.16 |
| 9 | 6 | 4-OMe | 356.510 | 4.095 | −0,16 | 0.670 | 42.52 | 365.76 |
| 10 | 8 | 4-OMe | 384.564 | 4.930 | 0,73 | 0.747 | 42.52 | 399.37 |
| 11 | 10 | 4-OMe | 412.618 | 5.764 | 1,62 | 0.929 | 42.52 | 432.97 |
| 12 | 12 | 4-OMe | 440.672 | 6.599 | 2,51 | 1.082 | 42.52 | 466.57 |
| 13 | 3 | 4-OBn | 466.625 | 6.272 | 3,64 | 0.300 | 42.52 | 458.66 |
| 14 | 4 | 4-OBn | 480.652 | 6.726 | 2,35 | 0.370 | 42.52 | 475.46 |
| 15 | 6 | 4-OBn | 508.706 | 7.560 | 3,23 | 0.812 | 42.52 | 509.06 |
| 16 | 8 | 4-OBn | 536.760 | 8.395 | 4,12 | 0.944 | 42.52 | 542.66 |
| 17 | 10 | 4-OBn | 564.814 | 9.230 | 5,01 | 1.098 | 42.52 | 576.27 |
| 18 | 12 | 4-OBn | 592.868 | 10.064 | 5,89 | 1.252 | 42.52 | 609.87 |
| 19 | 3 | 3-OH, 4-OMe | 346.427 | 2.028 | 0,23 | −0.308 | 82.98 | 331.39 |
| 20 | 4 | 3-OH, 4-OMe | 360.454 | 2.482 | −0,83 | −0.294 | 82.98 | 348.19 |
| 3-OH, 4-OMe | ||||||||
| 21 | 6 | 3-OH, 4-OMe | 388.508 | 3.316 | 0,05 | 0.142 | 82.98 | 381.80 |
| 22 | 8 | 3-OH, 4-OMe | 416.562 | 4.151 | 0,94 | 0.297 | 82.98 | 415.40 |
| 23 | 10 | 3-OH, 4-OMe | 444.616 | 4.985 | 1,83 | 0.448 | 82.98 | 449.01 |
| 24 | 12 | 3-OH, 4-OMe | 472.670 | 5.820 | 2,71 | 0.604 | 82.98 | 482.61 |
| 25 | 3 | 3-OMe, 4-OBn | 526.680 | 6.030 | 3,48 | 0.116 | 60.98 | 509.75 |
| 3-OMe, 4-OBn | ||||||||
| 26 | 4 | 3-OMe, 4-OBn | 540.704 | 6.473 | 2,26 | 0.130 | 60.98 | 526.55 |
| 27 | 6 | 3-OMe, 4-OBn | 568.758 | 7.308 | 3,12 | 0.593 | 60.98 | 560.15 |
| 28 | 8 | 3-OMe, 4-OBn | 596.812 | 8.142 | 4,02 | 0.776 | 60.98 | 593.76 |
| 29 | 10 | 3-OMe, 4-OBn | 624.866 | 8.977 | 4,95 | 0.942 | 60.98 | 627.36 |
| 3-OMe, 4-OBn | ||||||||
| 30 | 12 | 3-OMe, 4-OBn | 652.920 | 8.977 | 5,92 | 1.057 | 60.98 | 660.96 |
| 31 | 3 | H TetraSubst. | 434.63 | 6.24 | 4.84 | NC | 6.48 | 441.45 |
| 32 | 4 | H TetraSubst. | 448.65 | 6.51 | 3.32 | NC | 6.48 | 458.25 |
| 33 | 6 | H TetraSubst. | 476.71 | 7.52 | 4.20 | NC | 6.48 | 491.86 |
| 34 | 8 | H TetraSubst. | 504.76 | 8.42 | 5.09 | NC | 6.48 | 525.46 |
| 35 | 10 | H TetraSubst. | 532.82 | 8.93 | 5.97 | NC | 6.48 | 559.06 |
| 36 | 3 | 4-OMe TetrSubst. | 498.62 | 4.32 | 3.95 | NC | 87.39 | 473.52 |
| 37 | 4 | 4-OMe TetrSubst. | 512.65 | 4.59 | 2.61 | NC | 87.39 | 490.32 |
| 38 | 6 | 4-OMe TetrSubst. | 540.70 | 5.60 | 3.49 | NC | 87.39 | 523.93 |
| 39 | 8 | 4-OMe TetrSubst. | 568.76 | 6.61 | 4.38 | NC | 87.39 | 557.53 |
| 40 | 12 | 4-OMe TetrSubst. | 596.81 | 7.62 | 5.27 | NC | 87.39 | 591.13 |
NC = not calculated.
According to Lipinski’s rule of five, a potential drug candidate should be orally active if the molecular weight is ≤ 500 Da, logP 5, the number of hydrogen bond acceptors ≤10 and the number of hydrogen bond donors ≤ 5. The analysis of the collection revealed that 66% of the collection has MW < 500 (256–654 Da) and 83% has logP < 5 (−1.14 to 7.79) (Fig. 4).
Fig. 4.

Distribution of compounds of synthetic library vs. physicochemical properties.
According to Hammett and Hansch’s principle, a relationship between lipophilicity descriptors like logP and biological activity most commonly produce a parabolic correlation. In the case of our collection, the logP values of the 30 compounds fluctuate between −1.14 and 7.79. A correlation is clearly observed for P. falciparum (Fig. 4), where most active analogs have logP between 2.47 and 4.95. By contrast, the correlation for T. gondii is not clear (Fig. 5). However, we could constrain the logP range values between 2.66 and 3.75 for the highest values of activity against T. gondii.
Fig. 5.

Parasitic activities (P. falciparum and T. gondii) vs. logP.
However, our library contains ionizable groups that are charged at physiological pH, which make logD a better descriptor of the lipophilicity of these molecules. The calculated logD at physiological pH (7.4) of the collection is presented in Table 2, with 90% of the collection being ≤ 5 (−1.09 to 5.92). Given the chemical nature of the amine groups in the designed structures, the lipophilicity decreases markedly as the pH drops. This fact would suggest that the molecule could have a better interaction in the acidic environment of some organelles, in the same way as ethambutol does within lysosomes and autophagosomes [32]. In summary, this analysis reveals that most of the compounds has good potential to be orally active drug candidates.
The distribution of the compounds between blood and brain is a very important consideration for new candidate drug molecules. In case T. gondii, which effectively infects neurons and causes disease in the brain, good drug candidates must be able to cross the blood brain barrier (BBB). To predict that properly we calculated the logBB using COSMOquick [33]. This calculation shows that 75% of our compounds have logBB > 0.3. It has been shown that molecules with logBB > 0.3 cross the BBB readily while molecules with logBB < −1 are poorly distributed to the brain [34,35], thus our library includes numerous compounds that would be expected to enter the brain.
2.4. Analysis of the molecular electrostatic potential of the aromatic portion
Molecular electrostatic potentials (MEPs) were used for analyzing drug-receptor and enzyme-substrate interaction and other recognition processes, based on the assumption that those potentials described how different molecular regions will interact with other approaching chemical species.
In order to determine how the electronic potential of the aromatic portion of the analogs affects the activity, a tridimensional molecular electrostatic potential map of the aromatic portion of the prepared N,N′-dibenzyl aliphatic diamines derivatives were performed at the van der Waals contact surface. To simplify the calculations, only substituted N-methyl benzyl were used (Fig. 6a. R = H; R 4-OMe; R = 4-OBn; R = .3-OH,4-OMe; R = 3-MeO, 4-BnO) The geometries of the groups were optimized using B3LYP functional along with 6−311++G(d,p) (Fig. 6b). The electrostatic potentials, superimposed onto a surface of constant electron charge density were calculated (0.001 e/au3, Fig. 6c).
Fig. 6.

N-Methyl benzylamine portion of the N,N′−dibenzyldiamines prepared. A) Structure of the five benzylamines, B) Minimum energy conformation for each compound, C) Molecular electronic potential maps onto a van der Waals surface (isodensity 0.001 e/au3) for the N−Methyl benzylamines. The color-coded is shown at the left (in the range of 0.1408 au (deepest red) to +0.1152 au (deepest blue)). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
It is well known that electrostatic potential has been defined as the energy of interaction of a positively charge point with the nuclei and the electrons of a molecule. On Fig. 6C the coloring area represents the electrostatic potential, V(r), providing a measurement of the molecular charge distribution. The calculated MEP maps for the five analogs that go from −0.1408 au in deepest red for the strongest attraction, to +0.1152 au in deepest blue for the strongest repulsion. Negative regions V(r) are usually associated with a lone pair of electronegative atoms and the π electrons of the unsatu-rated hydrocarbons. In this case, the MEPs are different for each of the five analogs studied and, as expected, is affected by the aromatic ring subtituents. The negative potentials are localized at the oxygens of the phenols and methoxy groups. On the other side, the positive potential is localized on the nitrogen amine and is significantly affected by the substituent on the aromatic ring.
When the activity of each series was correlated with the electrotactic potential, two clear tendencies emerged. On one side, the activity of the analogs with benzyloxy group seems governed by steric effects. On the other side, the activity of the series is clearly associated to the negative electrostatic potential, being detrimental to the antiplasmodial activity.
3. Conclusions
Enzymes are usually arranged into symmetrical homodimers or tetramers, with the consequent active site organization in a highly symmetrical fashion. Thus, symmetrical inhibitors or biological actuators will correspond generally to their binding site [36]. Numerous examples of symmetrical active compounds have been reported including anticancer [37,38], antiparasitic [39,40], anti-tubercular [41] and enzyme inhibitors [42,43], between others. The compounds designed and prepared on this work were inspired on the symmetrical compound MDL-27695. We were looking to simplify that compound’s structure, removing nitrogens from the carbon chain and inquiring the effect of oxygenated substituents of the aromatic ring on the activity. Therefore, we searched for a fast and easy way to explore the chemical space of the proposed disubstituted N,N′- aliphatic diamines. A major advantage of the proposed scaffold lies in the fact that the synthesis of symmetrical derivatives is extremely straightforward and only required commercially available starting materials. The synthesis did not need additional protection/deprotection steps, minimizing the synthesis to a single-step, being a clear example of atom economy and green chemistry [44,45].
It has been suggested that symmetry and planarity disruption is a valuable strategy to improve aqueous solubility [46]. The synthesized compounds are very flexible preventing a good packing that facilitates the dissolution process. Also, the two nitrogen atoms on the aliphatic chain provide the required polarity to improve the water solubility and bioavailability. Therefore, from the solubility point of view, the symmetrical scaffold prepared did not represent a disadvantage.
The one-pot synthetic procedure provided 30 symmetric derivatives in good yields. In vitro studies have demonstrated that most of the analogs (26 of 30 compounds) are active against P. falciparum at micromolar level or below (9 compounds at submicromolar level). On the other hand, the related parasite T. gondii does not seem as sensitive to these type of compounds (6 of 30 compounds shows at least 75% of activity at 1 μM). The differences on the sensitivity between T. gondii and P. falciparum could be attributed to a favoured incorporation to the red bloods cells or the interaction with different molecular targets due to their metabolic divergences.
A series of tetrasubstituted N,N′−aliphatic diamines were prepared being considerable less active against both P. falciparum strains than the bis-substituted counterpart demonstrating the importance of the secondary amine on the activity.
A summary of the structure-activity analysis is detailed on Fig. 7. As can be seen, the antimalarial activity of the compounds is fairly responsive to the structural variations in most of the series, with certain substituents increasing activity, like R = H, or R = 3-MeO,4-BnO. MEPs results clearly shown that the aromatic subtituents affect the electrostatic potential of the nitrogen and the aromatic ring and that, together with the steric factors, can be correlated with the activity. Chain length appears to slightly influence potency, being the electronic and the steric the most critical factors modulating the activity.
Fig. 7.

Structure-activity relationship of our library against apicomplexan and selected analogs.
The selection of the best drug candidate as antimalarial and anti-T. gondii should combine a low IC50 values with Lipinski’s rules compliance, low cytotoxicity (high SI) and ideal range of logBB (only for T. gondii). Following that premise, analogs 4, 5, 15, 26 and 27 are the most promising antimalarial candidates, having submicromolar IC50 and SI > 20. Filtering out the selection by the ADME parameters points to compound 4 as the best candidate. In the case of T. gondii, analogs 13 and 25 based on the activity are the most promising, but of these only 13 is predicted to cross the BBB, which would make it the best candidate for future studies (Fig. 5). It is likely that the mechanism of action of these compounds differs between these two parasites. Additionally, the targets and mechanisms of action in each parasite might be different across the different series. Future studies with the best candidates will focus on elucidating mode of action. The fact that similar compounds act by different mechanisms in trypanosomatids [22,47] and the pathogenic fungus Pneumocystis carinii [48] support this hypothesis. Polyamine oxidase have been studied as a possible target of MDL-27695 on P. falciparum [49]. The oxidation of that compound by purified rat liver oxidase release benzaldehyde and the N−monosubstituted analog, then goes to another oxidation cycle releasing the free polyamine. Also, polyamine concentration increases inside P. falciparum infected red blood cells by newly recruited transporters.
Similar degradation products have been studied on Plasmodium falciparum [50], displaying similar potency to the bis-benzyl disubstituted analogs but holding a bulky anthracene ring. Having in mind that monosubstituted diamines have been described as polyamine transport inhibitors [51–53], a possible action mechanism could involve oxidation to activate the compound that then inhibit the polyamine transport.
In conclusion, the structural simplification of MDL-27695 provided potent antimalarial analogs validating our initial hypothesis. The fact that compounds 4 and 5, which has strong similarities to MDL-27695 but only have 2 nitrogens, supports the idea that the bis-N−benzyl moiety could be acting as the pharmacophore within those structures. Lastly, the N,N′−disubstituted aliphatic diamine analogs provides unique tools to develop new antiparasitic agents and to study biological processes related to polyamines metabolism. A possible multitarget action mechanism should be validated by non-directed metabolomics and proteomics study.
4. Material and methods
4.1. General information
1H and 13C NMR spectra were acquired on a Bruker Avance II 300 MHz (75.13 MHz) using CDCl3 as solvent. Chemical shifts (d) were reported in ppm downfield from tetramethylsilane (TMS) at 0 ppm as internal standard and coupling constants (J) are in hertz (Hz). Chemical shifts for carbon nuclear magnetic resonance (13C NMR) spectra are reported in parts per million relative to the center line of the CDCl3 triplet at 76.9 ppm. The following abbreviations are used to indicate NMR signal multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, p = pentet br = broad signal. High-resolution mass spectra (HRMS) were recorded on a Bruker MicrOTOf-QII with lock spray source. IR spectra were obtained using an FTIR Shimadzu spectrometer and only partial spectral data are listed. Chemical reagents were purchased from commercial suppliers and used without further puri-fication, unless otherwise noted. Solvents were analytical grade or were purified by standard procedures prior to use. Yields were calculated for material judged homogeneous by thin layer chromatography (TLC) and nuclear magnetic resonance (1H NMR). All reactions were monitored by thin layer chromatography performed on silica gel 60 F254 pre-coated aluminium sheets, visualized by a 254 nm UV lamp, and stained with an ethanolic solution of 4-anisaldehyde. Column flash chromatography was performed using silica gel 60 (230–400 mesh).
4.2. In vitro activity against P. falciparum (IC50)
Antimalarial activity was determined in vitro on the chloroquine sensitive (D6, Sierra Leone) and resistant (W2, Indo China) strains of P. falciparum. The 96-well microplate assay was based on the evaluation of the effect of compounds on the growth of asynchronous cultures of P. falciparum, determined by the assay of parasite lactate dehydrogenase (pLDH) activity. The appropriate dilutions of the compounds were prepared in DMSO or RPMI-1640 medium and added to the cultures of P. falciparum (2% hematocrit, 2% parasitemia) setup in clear flat bottomed 96-well plates. The plates were placed into the humidified chamber and flushed with a gas mixture of 90% N2, 5% CO2 and 5% O2. The cultures were incubated at 37 °C for 48 h. Growth of the parasite in each well was determined by pLDH assay using Malstat reagent. The medium and red blood cells (RBC) controls were also setup in each plate. The standard antimalarial agent chloroquine and artemisinin were used as the positive controls.
4.3. In vitro activity against T. gondii
Toxoplasma parasites (RH strain parasites expressing the green fluorescent protein, GFP) were maintained in human foreskin fibroblast (HFF) monolayers in Dulbecco’s medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, and 100 g/mL streptomycin. Uninfected and infected HFFs cells were maintained in a humidified incubator at 37 °C with 5% CO2. For primary screen, 103 parasites were allowed to infect a confluent HFF monolayer. At 8 h post-infection, the medium was replaced with medium containing 1 or 10 μM of each compound. Three days later cultures were monitored by fluorescence microscopy to monitor parasite growth. To determine IC50 HFFs were infected with 80 parasites and after four hours extracellular parasites were washed off and media with drug at a range of concentrations was added. At 5 days post-infection, cultures were fixed with methanol for 2 min and stained with crystal violet to visualized plaques which were enumerated. Percentage survival was calculated by dividing the number of plaques in drug over that in equivalent amount of DMSO. Experiments were performed in three experimental and biological replicates.
4.4. Cytotoxicity assay on Vero cells
The in vitro cytotoxicity was determined against mammalian kidney fibroblasts (VERO). The assay was performed in 96-well tissue culture-treated plates as described earlier. Briefly, cells were seeded to the wells of the plate (25,000 cells/well) and incubated for 24 h. Samples were added and plates were again incubated for 48 h. The number of viable cells was counted and the plot for EC50 performed.
4.5. ADME/toxicity predictions
Computational modeling to estimate the bioavailability, aqueous solubility, blood brain barrier potential, human intestinal absorption, mutagenicity, and toxicity for the compounds was performed using the ChemMineTools and Osiris Property.
4.6. General procedure for N,N-diamines disubstituted synthesis
Aldehyde (3 eq dissolved in the same mixture of solvents) was added to a solution of diamine (1 eq) in an anhydrous mixture of DCM:methanol (3:1). The mixture was refluxed overnight, the solvent was evaporated and the crude di-imine was dissolved in an anhydrous mixture of DCM:methanol (1:1). The solution was cooled at 0 °C, and sodium borohydride (3 eq) was added portionwise. The reaction was allowed to warm to room temperature and stirred for additional 12 h. Excess of sodium borohydride was quenched acidifying with 10% HCl, with subsequent addition of ammonium hydroxide to reach a basic pH. Phases were separated and the aqueous phase extracted with DCM (3 × 20 mL). Combined organic extracts were dried with sodium sulphate and evaporated. Products were purified by flash chromatography.
4.6.1. Synthesis of N1,N3-dibenzylpropane-1,3-diamine (1)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 97 mg of white solid (isolated yield: 79%). 1H NMR (CDCl3) δ 7.31 (m, 10H, aromatics); 3.78 (s, 4H, Ar-CH2); 2.72 (t, 4H, J = 6.6 Hz, N-CH2); 1.74 (m, 2H, C2-H). 13C NMR (CDCl3) δ 140.3 (C); 128.4 (CH); 128.2 (CH); 126.9 (CH); 54.1 (CH2); 48.0 (CH2); 30.0 (CH2). IR υ 3296 (R-NH-R1); 3026 (Ar-H); 1119 (C-N-C); 697 (monosubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C17H23N2 (M + H+) 255.1861; found 255.1853.
4.6.2. Synthesis of N1,N4-dibenzylbutane-1,4-diamine (2)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 88 mg of white solid (isolated yield: 73%). 1H NMR (CDCl3) δ 7.28 (m, 10H, aromatics); 3.78 (s, 4H, Ar-CH2); 2.64 (t, 4H, J = 6.6 Hz, N-CH2); 1.57 (m, 4H, C2-H and C3-H). 13C NMR (CDCl3) δ 139.6 (C); 128.4 (CH); 128.2 (CH); 127.1 (CH); 53.6 (CH2); 48.9 (CH2); 27.6 (CH2). IR υ 3311 (R-NH-R1); 1120 (C-N-C); 697 (mono-substituted aromatic) cm−1. ESI-HRMS: calculated mass for C18H25N2 (M + H+) 269.2018; found 269.2011.
4.6.3. Synthesis of N1,N6-dibenzylhexane-1,6-diamine (3)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 47 mg of white solid (isolated yield: 46%). 1H NMR (CDCl3) δ 7.28 (m, 10H, aromatics); 3.78 (s, 4H, Ar-CH2); 2.61 (t, 4H, J = 7.2 Hz, N-CH2); 1.49 (m, 4H, C2-H and C5-H); 1.34 (m, 4H, C3-H and C4-H). 13C NMR (CDCl3) δ 140.3 (C); 128.4 (CH); 128.2 (CH); 126.9 (CH); 54.0 (CH2); 49.3 (CH2); 29.9 (CH2); 27.3 (CH2). IR υ 3312 (R-NH-R1); 3026 (Ar-H); 1119 (C-N-C); 697 (monosubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C20H29N2 (M + H+) 297.2331; found 297.2328.
4.6.4. Synthesis of N1,N8-dibenzyloctane-1,8-diamine (4)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 85 mg of white solid (isolated yield: 94%). 1H NMR (CDCl3) δ 7.30 (m, 10H, aromatics); 3.82 (s, 4H, Ar-CH2); 2.67 (t, 4H, J = 7.2 Hz, NCH2); 1.52 (m, 4H, C2-H and C7-H); 1.30 (s, 8H, C3-H to C6-H). 13C NMR (CDCl3) δ 140.2 (C); 128.4 (CH); 128.2 (CH); 126.9 (CH); 53.9 (CH2); 49.3 (CH2); 29.9 (CH2); 29.5 (CH2); 27.5 (CH2). IR υ 3307 (R-NH-R1); 3026 (Ar-H); 1121 (C-N-C); 697 (monosubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C22H33N2 (M + H+) 325.2644; found 325.2660.
4.6.5. Synthesis of N1,N10-dibenzyldecane-1,10-diamine (5)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 73 mg of white solid (isolated yield: 89%). 1H NMR (CDCl3) δ 7.31 (m, 10H, Ar-H); 3.84 (s, 4H, Ar-CH2); 2.65 (t, 4H, J = 7.20 Hz, N CH2); 1.55 (m, 4H, C2-H and C9-H); 1.25 (m, 12H, C3-H=to C8-H). 13C NMR (CDCl3) δ 136.7 (C); 128.9 (CH); 128.6 (CH); 127.7 (CH); 52.4 (CH2); 47.9 (CH2); 29.3 (CH2); 29.2 (CH2); 28.2 (CH2); 27.0 (CH2). IR υ 3307 (R-NH-R1); 3026 (Ar-H); 1121 (C-N-C); 697 (monosubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C24H37N2 (M + H+) 353.2957; found 353.2966.
4.6.6. Synthesis of N1,N12-dibenzyldodecane-1,12-diamine (6)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 97 mg of white solid (isolated yield: 79%). 1H NMR (CDCl3) δ 7.31 (m, 10H, Ar-H); 3.84 (s, 4H, Ar-CH2); 2.65 (t, 4H, J = 7.20 Hz, NCH2); 1.55 (m, 4H, C2-H and C11-H); 1.25 (m, 16H, C3-H = to C10-H). 13C NMR (CDCl3) δ 136.7 (C); 128.9 (CH); 128.6 (CH); 127.7 (CH); 52.4 (CH2); 47.9 (CH2); 29.3 (CH2); 29.2 (CH2); 28.2 (CH2); 27.0 (CH2). IR υ 3307 (R-NH-R1); 3026 (Ar-H); 1121 (C-N-C); 697 (monosubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C26H41N2 (M + H+) 381.3255; found 381.3264.
4.6.7. Synthesis of N1,N3-bis(4-methoxybenzyl)propane-1,3-diamine (7)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 132 mg of colourless oil (isolated yield: 78%). 1H NMR (CDCl3) δ 7.21 (d, 4H, J = 8.7 Hz, C2′−H); 6.85 (d, 4H, J = 8.7 Hz, C3′−H); 3.79 (s, 6H, O-CH3); 3.71 (s, 4H, Ar-CH2); 2.68 (t, 4H, J = 6.6 Hz, N-CH2); 1.71 (m, 2H, C2-H). 13C NMR (CDCl3) δ 158.6 (C);=132.7 (C); 129.3 (CH); 113.7 (CH); 55.2 (CH3); 53.5(CH2); 47.9 (CH2); 30.2 (CH2). IR υ 3301 (R-NH-R1); 3062 (Ar-H); 1176 (C-N-C); 1243 and 1036 (Ar-O-C); 818 (disubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C19H27N2O2 (M + H+) 315.2073; found 315.2087.
4.6.8. Synthesis of N1,N4-bis(4-methoxybenzyl)butane-1,4-diamine (8)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 115 mg of white solid (isolated yield: 77%). 1H NMR (CDCl3) δ 7.19 (d, 4H, J = 8.7 Hz, C2′−H); 6.83 (d, 4H, J = 8.7 Hz, C3′−H); 3.75 (s, 6H, O-CH3); 3.68 (s, 4H, Ar-CH2); 2.58 (t, 4H, J = 6.6 Hz, N-CH2); 1.51 (m, 4H, C2-H and C3-H). 13C NMR (CDCl = 3) δ 158.6 (C); 132.4 (C); 129.3 (CH); 113.8 (CH); 55.2 (CH3); 53.3 (CH2); 49.1 (CH2); 27.8 (CH2). IR υ 3314(R-NH-R1); 2997 (Ar-H); 1246 and 1036 (Ar-O-C); 1174 (C-N-C); 818 (disubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C20H29N2O2 (M + H+) 329.229; found 329.2233.
4.6.9. Synthesis of N1,N6-bis(4-methoxybenzyl)hexane-1,6-diamine (9)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 37 mg of white solid (isolated yield: 30%). 1H NMR (CDCl3) δ 7.23 (d, 4H, J = 8.7 Hz, C2′−H); 6.86 (d, 4H, J 8.7 Hz, C3′−H); 3.79 (s, 6H, -CH3); 3.71 (s, 4H, Ar-CH2); 2.59 (t, 4H, J = 6.9 Hz, N-CH2); 1.48 (m, 4H, C2-H and C5-H); 1.33 (m, 4H, C3-H and = C4-H). 13C NMR (CDCl3) δ 158.6 (C); 132.5 (C); 129.3 (CH); 113.8 (CH); 55.2 (CH3); 53.4(CH2); 49.2 (CH2); 29.9 (CH2); 27.3 (CH2). IR υ 3296(R-NH-R1); 2957 (Ar-H); 1252 and 1033 (Ar-O-C); 1103 (C-N-C); 818 (disubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C22H33N2O2 (M + H+) 357.2542; found 357.2531.
4.6.10. Synthesis of N1,N8-bis(4-methoxybenzyl)octane-1,8-diamine (10)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 95 mg of white solid (isolated yield: 59%). 1H NMR (CDCl3) δ 7.23 (d, 4H, J = 8.7 Hz, C2′−H); 6.85 (d, 4H, J = 8.7 Hz, C3′−H); 3.78 (s, 6H, O-CH3); 3.71 (s, 4H, Ar-CH2); 2.59 (t, 4H, J = 6.9 Hz, N-CH2); 1.48 (m, 4H, C2-H and C7-H); 1.28 (m, 8H, C3-H to = C6-H). 13C NMR (CDCl3) δ 158.6 (C); 132.4 (C); 129.3 (CH); 113.8 (CH); 55.2 (CH3); 53.4 (CH2); 49.3 (CH2); 29.9 (CH2); 29.5 (CH2); 27.3 (CH2). IR υ 3307 (R-NH-R1); 3026 (Ar-H); 1121 (C-N-C); 697 (monosubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C24H37N2O2 (M +H+) 385.2855; found: 385.2857.
4.6.11. Synthesis of N1,N10-bis(4-methoxybenzyl)decane-1,10-diamine (11)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 63 mg of white solid (isolated yield: 66%). 1H NMR (CDCl3) δ 7.26 (d, 4H, J = 8.7 Hz, C2′−H); 6.86 (d, 4H, J 8.7 Hz, C3′−H); 3.79 (s, 6H, O-CH3); 3.75 (s, 4H, Ar-CH2); 2.62 (t, 4H, J = 7.2 Hz, NH-CH2); 1.52 (m, 4H, C2-H and C9-H); 1.26 (m, 12H, C3-H=to C8-H). 13C NMR (CDCl3) δ 158.9 (C); 130.2 (C); 129.8 (CH); 113.9 (CH); 55.2 (CH3); 52.4 (CH2); 48.9 (CH2); 29.4 (CH2); 29.3 (CH2); 28.9 (CH2); 27.2 (CH2). IR υ 3392 (R-NH-R1); 1253 and 1033 (Ar-O-C); 1177 (C-N-C); 818 (disubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C26H41N2O2 (M + H+) 413.3168. found 413.3172.
4.6.12. Synthesis of N1,N12-bis(4-methoxybenzyl)dodecane-1,12-diamine (12)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 109 mg of white solid (isolated yield: 99%). 1H NMR (CDCl3) δ 7.26 (d, 4H, J = 8.7 Hz, C2′−H); 6.86 (d, 4H, J = 8.7 Hz, C3′−H); 3.79 (s, 6H, O-CH3); 3.75 (s, 4H, Ar-CH2); 2.62 (t, 4H, J = 7.2 Hz, N-CH2); 1.52 (m, 4H, C2-H and C11-H); 1.26 (m, 16H, C3-H= to C10-H).13C NMR (CDCl3) δ 158.9 (C); 130.2 (C); 129.8 (CH); 113.9 (CH); 55.2 (CH3); 52.4 (CH2); 48.9 (CH2); 29.4 (CH2); 29.3 (CH2); 28.9 (CH2); 27.2 (CH2). IR υ 3392 (R-NH-R1); 1253 and 1033 (Ar-O-C); 1177 (CN-C); 818 (disubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C28H45N2O2 (M + H+) 441.3475; found 441.3462.
4.6.13. Synthesis of N1,N3-bis(4-(benzyloxy)benzyl)propane-1,3-diamine (13)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 170 mg of white solid (isolated yield: 67%). 1H NMR (CDCl3) δ7.41 (m, 10H, aromatics 4′−benzyloxy) 7.21 (d, 4H, J = 8.7 Hz, C2′- H); 6.93 (d, 4H, J = 8.7 Hz, C3′−H); 5.05 (s, 4H, Ar-CH = 2-O); 3.71 (s, 4H, Ar-CH2−N); 2.69 (t, 4H, J = 6.9 Hz, N-CH); 1.72 2H, 13 2 (m, C2). C NMR (CDCl3) δ 157.9 (C); 137.1 (C); 132.7 (C); 129.4 (CH); 128.6 (CH); 128.0 (CH); 127.5 (CH); 114.8 (CH); 70.1 (CH2); 53.4 (CH2); 47.9 (CH2); 29.9 (CH2). IR υ 3289 (R-NH-R1); 3032 (Ar-H); 1106 (C-N-C); 1239 and 1026 (Ar-O-C) cm−1. ESI-HRMS: calculated mass for C31H35N2O2 (M + H+) 467.2699; found 467.2678.
4.6.14. Synthesis of N1,N4-bis(4-(benzyloxy)benzyl)butane-1,4-diamine (14)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 168 mg of white solid (isolated yield: 77%). 1H NMR (CDCl3) δ7.42 (m, 10H, aromatics 4′−benzyloxy); 7.22 (d, 4H, J = 8.7 Hz, C2′- H); 6.93 (d, 4H, J = 8.7 Hz, C3′−H); 5.05 (s, 4H, Ar-CH = 2-O); 3.71 (s, 4H, Ar-CH2−N); 2.62 (t, 4H, J = 6.6 Hz, N-CH2); 1.53 (m, 4H, C2-H and C4-H). 13C NMR (CDCl3) δ 157.8 (C); 137.1 (C); 132.9 (C); 129.3 (CH); 128.6 (CH); 127.9 (CH); 127.5 (CH); 114.8 (CH); 70.1 (CH2); 53.4 (CH2); 49.2 (CH2); 27.9 (CH2). IR υ 3294 (R-NH-R1); 3035 (Ar-H); 1167 (C-N-C); 1247 and 1015 (Ar-O-C); 814 (disubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C32H37N2O2 (M + H+)
4.6.15. Synthesis of N1,N6-bis(4-(benzyloxy)benzyl)hexane-1,6-diamine (15)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 155 mg of white solid (isolated yield: 86%). 1H NMR (CDCl3) δ7.38 (m, 10H, aromatics 4′−benzyloxy); 7.23 (d, 4H, J = 8.7 Hz, C2′- H); 6.93 (d, 4H, J = 8.7 Hz, C3′−H); 5.05 (s, 4H, Ar-CH = 2-O); 3.72 (s, 4H, Ar-CH2−N); 2.61 (t, 4H, J = 6.9 Hz, N-CH2); 1.51 (m, 4H, C2-H and C5-H); 1.32 (m, 4H, C3-H and C4-H). 13C NMR (CDCl3) δ 157.9 (C); 137.1 (C); 132.3 (C); 129.5 (CH); 128.6 (CH); 127.9 (CH); 127.5 (CH); 114.8 (CH); 70.0 (CH2); 53.2 (CH2); 49.1 (CH2); 29.7 (CH2); 27.2 (CH2). IR υ 3293 (R-NH-R1); 3048 (Ar-H); 1104 (C-N-C); 1251 and 1016 (Ar-O-C); 815 (disubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C34H41N2O2 (M + H+) 509.3168; found 509.3146.
4.6.16. Synthesis of N1,N8-bis(4-(benzyloxy)benzyl)octane-1,8-diamine (16)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 147 mg of white solid (isolated yield: 99%). 1H NMR (CDCl3) δ7.42 (m, 10H, aromatics 4′−benzyloxy); 7.23 (d, 4H, J = 8.7 Hz, C2′- H); 6.93 (d, 4H, J = 8.7 Hz, C3′−H); 5.05 (s, 4H, Ar-CH = 2-O); 3.72 (s, 4H, Ar-CH2−N); 2.60 (t, 4H, J = 7.2 Hz, N-CH2); 1.49 (m, 4H, C2-H and C7-H); 1.29 (m, 8H, CH2. C3-H to C6-H). 13C NMR (CDCl3) δ 157.8 (C); 137.1 (C); 133.0 (C); 129.3 (CH), 128.6 (CH), 127.9 (CH), 127.5 (CH); 114.8 (CH); 70.0 (CH2); 53.5 (CH2); 49.4 (CH2); 30.1 (CH2); 29.5 (CH2); 27.3 (CH2). IR υ 3246 (R-NH-R1); 3035 (Ar-H); 1104 (C-N-C); 1249 and 1015 (Ar-O-C); 813 (disubstituted aromatic) cm−1. ESI HRMS: calculated mass for C36H45N2O2 (M + H+) 537.3481; found 537.3477.
4.6.17. Synthesis of N1,N10-bis(4-(benzyloxy)benzyl)decane-1,10-diamine (17)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 79 mg of white solid (isolated yield: 60%). 1H NMR (CDCl3) d7.42 (m, 10H, aromatics 4′−benzyloxy); 7.24 (d, 4H, J = 8.7 Hz,C2′- H); 6.93 (d, 4H, J = 8.7 Hz, C3′−H); 5.05 (s, 4H, Ar-CH = 2-O); 3.72 (s, 4H, Ar-CH2−N); 2.61 (t, 4H, J = 6.9 Hz, N-CH2); 1.49 (m, 4H, C2-H and C9-H); 1.27 (m, 12H, C3-H to C8-H). 13C NMR (CDCl3) δ 157.9 (C); 137.1 (C); 132.7 (C); 129.4 (CH), 128.6 (CH), 128.0 (CH), 127.5 (CH); 114.8 (CH); 70.0 (CH2); 53.4 (CH2); 49.4 (CH2); 30.0 (CH2); 29.5 (CH2); 27.6 (CH2). IR υ 3311 (R-NH-R1); 3049 (Ar-H); 1105 (C-N-C); 1250 and 1016 (Ar-O-C); 814 (disubstituted aromatic) cm−1. ESI HRMS: calculated mass for C38H49N2O2 (M + H+) 565.3794; found 565.3802.
4.6.18. Synthesis of N1,N12-bis(4-(benzyloxy)benzyl)dodecane-1,12-diamine (18)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 143 mg of white solid (isolated yield: 99%). 1H NMR (CDCl3) δ7.42 (m, 10H, aromatics 4′−benzyloxy); 7.24 (d, 4H, J = 8.7 Hz, C2′- H); 6.93 (d, 4H, J = 8.7 Hz, C3′−H); 5.05 (s, 4H, Ar-CH = 2-O); 3.72 (s, 4H, Ar-CH2−N); 2.61 (t, 4H, J = 6.9 Hz, N-CH2); 1.49 (m, 4H, C2-H and C11-H); 1.27 (m, 16H, C3-H to C10-H). 13C NMR (CDCl3) δ 157.9 (C); 137.1 (C); 132.7 (C); 129.4 (CH); 128.6 (CH); 128.0 (CH); 127.5 (CH); 114.8 (CH); 70.0 (CH2); 53.4 (CH2); 49.4 (CH2); 30.0 (CH2); 29.5 (CH2); 27.6 (CH2). IR υ 3311 (R-NH-R1); 3049 (Ar-H); 1105 (C-N-C); 1250 and 1016 (Ar-O-C); 814 (disubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C38H49N2O2 (M + H+) 565.3794; found 565.3802.
4.6.19. Synthesis of 5,5’-((propane-1,3-diylbis(azanediyl)) bis(methylene))bis(2-methoxyphenol) (19)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 180 mg of white solid (isolated yield: 96%). 1H NMR (CDCl3) δ 6.79 (m, 6H, aromatics); 3.87 (s, 6H, O-CH3); 3.69 (s, 4H, Ar-CH2); 2.73 (t, 4H, J = 6.6 Hz, N-CH); 1.74 (m, 2H, C2-H). 13 2 C NMR (CDCl3) δ 146.8 (C); 146.7 (C); 134.1 (C); 118.9 (CH); 115.9 (CH); 112.4 (CH); 56.1 (CH3); 53.1 (CH2); 47.5 (CH2); 30.0 (CH2). IR (KBr) υ 3449 (RNH-R1); 3029 (Ar-H); 2464 (Ar-O-H); 1280 and 1028 (Ar-O-C); 1132 (C-N-C) cm−1. ESI-HRMS: calculated mass for C19H27N2O4 (M + H+) 347.1971; found 347.1969.
4.6.20. Synthesis of 5,5’-((butane-1,4-diylbis(azanediyl)) bis(methylene))bis(2-methoxyphenol) (20)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 131 mg of white solid (isolated yield: 80%). 1H NMR (CDCl3) δ 6.77 (m, 6H, aromatics); 3.87 (s, 6H, O-CH3); 3.69 (s, 4H, Ar-CH2); 2.62 (t, 4H, J = 6.6 Hz, N-CH2); 1.56 (m, 4H, C2-H and C3-H). 13C NMR (CDCl3) δ 146.7 (C); 146.7 (C); 134.1 (C); 118.9 (CH); 115.8 (CH); 112.4 (CH); 56.1 (CH3); 53.0 (CH2); 49.0 (CH2); 27.8 (CH2). IR (KBr) υ 3441(R-NH-R1); 3040 (Ar-H); 2319 (Ar-O-H); 1255 and 1026 (Ar-O C); 1138 (C-N-C); 866 cm−1. ESI-HRMS: calculated mass for C20H29N2O4 (M + H+) 361.2127; found 361.2121.
4.6.21. Synthesis of 5,5’-((hexane-1,6-diylbis(azanediyl)) bis(methylene))bis(2-methoxyphenol) (21)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 110 mg of white solid (isolated yield: 82%). 1H NMR (CDCl3) δ 6.80 (m, 6H, aromatics); 3.86 (s, 6H, O-CH3); 3.68 (s, 4H, Ar-CH2); 2.59 (t, 4H, J = 7.2 Hz, N-CH2); 1.47 (m, 4H, C2-H and C5-H); 1.30 (m, 4H, C3-H and C4-H). 13C NMR (CDCl3) δ 146.7 (C); 146.7 (C); 134.1 (C); 118.9 (CH); 115.8 (CH); 112.4 (CH); 56.1 (CH3); 53.1 (CH2); 49.0 (CH2); 29.9 (CH2); 27.4 (CH2). IR (KBr) υ 3465(R-NH-R1); 3031 (Ar-H); 2441 (Ar-O-H); 1286 and 1035 (Ar-O-C); 1131 (C-N-C) cm−1. ESI HRMS: calculated mass for C22H33N2O4 (M + H+) 389.2440; found 389.2442.
4.6.22. Synthesis of 5,5’-((octane-1,8-diylbis(azanediyl)) bis(methylene))bis(2-methoxyphenol) (22)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 135 mg of white solid (isolated yield: 72%). 1H NMR (CDCl3) δ 6.80 (m, 6H, aromatics); 3.86 (s, 6H, O-CH3); 3.69 (s, 4H, Ar-CH2); 2.58 (t, 4H, J = 7.2 Hz, N-CH2); 1.47 (m, 4H, C2-H and C7-H); 1.25 (m, 8H, C3-H to C6-H). 13C NMR (CDCl3) δ 146.8 (C); 146.7 (C); 134.0 (C); 119.0 (CH); 115.9 (CH); 112.4 (CH); 56.1 (CH3); 53.1 (CH2); 49.0 (CH2); 29.9 (CH2); 29.5 (CH2); 27.3 (CH2). IR (KBr) υ 3447(R-NH-R1); 2991 (Ar-H); 2445 (Ar-O-H); 1282 and 1033 (Ar-O-C); 1131 (C-N-C); 855 (trisubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C24H37N2O4 (M + H+) 417.2753; found 417.2740.
4.6.23. Synthesis of 5,5’-((decane-1,10-diylbis(azanediyl)) bis(methylene)) bis(2-methoxyphenol) (23)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 74 mg of white solid (isolated yield: 72%). 1H NMR (CDCl3) δ 6.82 (m, 6H, aromatics); 3.86 (s, 6H, O-CH3); 3.70 (s, 4H, Ar-CH2); 2.60 (t, 4H, J = 7.2 Hz, N-CH2); 1.48 (m, 4H, C2-H and C9-H); 1.25 (m, 12H, CH2 C3-H to C8-H). 13C NMR (CDCl3) δ 146.7 (C); 146.7 (C); 134.1 (C); 118.9 (CH); 115.8 (CH); 112.4 (CH); 56.1 (CH3); 53.1 (CH2); 49.0 (CH2); 29.9 (CH2); 29.5 (CH2); 27.6 (CH2). IR (KBr) υ 3418 (RNH-R1); 3033 (Ar-H); 2444 (Ar-O-H); 1282 and 1033 (Ar-O-C); 1131 (C-N-C); 873 (trisubstituted aromatic) cm−1. ESI-HRMS: calculated mass for C24H41N2O4 (M + H+) 445.3066; found 445.3050.
4.6.24. Synthesis of 5,5’-((dodecane-1,12-diylbis(azanediyl)) bis(methylene))bis(2-methoxyphenol) (24)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 86 mg of white solid (isolated yield: 46%). 1H NMR (CDCl3) δ 6.82 (m, 6H, aromatics); 3.86 (s, 6H, O-CH3); 3.70 (s, 4H, Ar-CH2); 2.60 (t, 4H, J = 7.2 Hz, N-CH2); 1.48 (m, 4H, C2-H and C11-H); 1.25 (m, 16H, CH2 C3-H to C12-H). 13C NMR (CDCl3) δ 146.7 (C); 146.7 (C); 134.1 (C); 118.9 (CH); 115.8 (CH); 112.4 (CH); 56.1 (CH3); 53.1 (CH2); 49.0 (CH2); 29.9 (CH2); 29.5 (CH2); 27.6 (CH2). IR (KBr) υ 3418(R-NH-R1); 3033 (Ar-H); 2444 (Ar-O-H); 1282 and 1033 (Ar-O C); 1131 (C-N-C); 873 (trisubstituted aromatic) cm−1.
4.6.25. Synthesis of N1,N3-bis(4-(benzyloxy)-3-methoxybenzyl) propane-1,3-diamine (25)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 133 mg of white solid (isolated yield: 50%). 1H NMR (CDCl3) δ7.37 (m, 10H, aromatics 4′-benzyloxy); 6.82 (m, 6H, aromatics); 5.13 (s, 4H, Ar-CH2−O); 3.87 (s, 6H, O-CH3); 3.70 (s, 4H, Ar-CH2−N); 2.69 (t, 4H, J = 6.6 Hz, N-CH2−); 1.72 (m, 2H, C2-H). 13C NMR (CDCl3) δ 149.7 (C); 147.2 (C); 137.3 (C); 133.8 (C); 128.5 (CH); 127.8 (CH); 127.3 (CH); 120.2 (CH); 114.1 (CH); 112.0 (CH); 71.1 (CH2); 56.0 (CH3); 53.9 (CH2); 48.0 (CH2); 30.2 (CH2). IR υ 3302 (R-NH-R1); 3063 (Ar-H); 1264 and 1026 (Ar-O-C); 1139 (C-N-C) cm−1. ESI HRMS: calculated mass for C33H39N2O4 (M + H+) 527.2910; found 527.2886.
4.6.26. Synthesis of N1,N4-bis(4-(benzyloxy)-3-methoxybenzyl) butane-1,4-diamine (26)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 205 mg of white solid (isolated yield: 84%). 1H NMR (CDCl3) δ7.39 (m, 10H, aromatics 4′-benzyloxy); 6.82 (m, 6H, aromatics); 5.13 (s, 4H, Ar-CH2−O); 3.89 (s, 6H, O-CH3); 3.70 (s, 4H, Ar-CH2−N); 2.64 (t, 4H, J = 6.6 Hz, N-CH2); 1.56 (m, 4H, C2-H and C3-H). 13C NMR (CDCl3) δ 149.7 (C); 147.2 (C); 137.3 (C); 133.2 (C); 128.5 (CH), 127.8 (CH),127.3 (CH); 120.3 (CH); 114.0 (CH); 112.0 (CH); 71.1 (CH2); 56.0 (CH3); 53.6 (CH2); 49.1 (CH2); 27.7 (CH2). IR υ 3299 (R NH-R1); 3066 (Ar-H); 1261 and 1032 (Ar-O-C); 1131 (C-N-C) cm−1. ESI-HRMS: calculated mass for C34H41N2O2 (M + H+) 541.3066; found 541.3045.
4.6.27. Synthesis of N1,N6-bis(4-(benzyloxy)-3-methoxybenzyl) hexane-1,6-diamine (27)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 159 mg of white solid (isolated yield: 81%). 1H NMR (CDCl3) δ7.40 (m, 10H, aromatics 4′-benzyloxy); 6.84 (m, 6H, aromatics); 5.13 (s, 4H, Ar-CH2−O); 3.89 (s, 6H, O-CH3); 3.71 (s, 4H, Ar-CH2−N); 2.61 (t, 4H, J = 7.2 Hz, N-CH2); 1.51 (m, 4H, C2-H and C5-H); 1.33 (m, 4H, C3-H and C4-H). 13C NMR (CDCl3) δ 149.7 (C); 147.6 (C); 137.3 (C); 133.4 (C); 128.5 (CH); 127.8 (CH); 127.3 (CH); 120.3 (CH); 114.0 (CH); 112.0 (CH); 71.1 (CH2); 56.0 (CH3); 53.7 (CH2); 49.3 (CH2); 29.8 (CH2); 27.3 (CH2). IR υ 3409 (R-NH-R1); 1264 and 1004 (Ar-O-C); 1145 (C-N-C) cm−1. ESI-HRMS: calculated mass for C36H45N2O4 (M + H+) 569.3379; found 569.3380.
4.6.28. Synthesis of N1,N8-bis(4-(benzyloxy)-3-methoxybenzyl) octane-1,8-diamine (28)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 155 mg of white solid (isolated yield: 88%). 1H NMR (CDCl3) δ7.39 (m, 10H, aromatics 4′-benzyloxy); 6.84 (m, 6H, aromatics); 5.14 (s, 4H, Ar-CH2−O); 3.89 (s, 6H, O-CH3); 3.71 (s, 4H, Ar-CH2−N); 2.61 (t, 4H, J = 7.2 Hz, N-CH2); 1.46 (m, 4H, C2-H and C7-H); 1.29 (m, 8H, C3-H to C6-H). 13C NMR (CDCl3) δ 149.7 (C); 147.2 (C); 137.3 (C); 133.2 (C); 128.5 (CH); 127.8 (CH); 127.3 (CH); 120.3 (CH); 114.0 (CH); 112.0 (CH); 71.1 (CH2); 56.0 (CH3); 53.7 (CH2); 49.3 (CH2); 29.8 (CH2); 29.4 (CH2); 27.2 (CH2). IR υ 3410 (R-NH-R1); 3006 (Ar-H); 1259 and 1029 (Ar-O-C); 1162 (C-N-C) cm−1. ESI-HRMS: calculated mass for C38H49N2O4 (M + H+) 597.3692; found 597.3717.
4.6.29. Synthesis of N1,N10-bis(4-(benzyloxy)-3-methoxybenzyl) decane-1,10-diamine (29)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 130 mg of white solid (isolated yield: 89%). 1H NMR (CDCl3) δ7.40 (m, 10H, aromatics 4′−benzyloxy); 6.81 (m, 6H, aromatics); 5.14 (s, 4H, Ar-CH2−O); 3.89 (s, 6H, O-CH3); 3.71 (s, 4H, Ar-CH2−N); 2.61 (t, 4H, N-CH2); 1.51 (m, 4H, C2-H and C9-H); 1.27 (m, 12H, C3-H to C8-H). 13C NMR (CDCl3) δ 149.7 (C); 147.2 (C); 137.3 (C); 133.6(C); 128.5 (CH); 127.8 (CH); 127.3 (CH); 120.3 (CH); 114.0 (CH); 112.0 (CH); 71.1 (CH2); 56.0 (CH3); 53.8 (CH2); 49.4 (CH2); 30.0 (CH2); 29.5 (CH2); 27.4 (CH2). IR υ 3422 (R-NH-R1); 1263 and 1029 (Ar-O-C); 1140 (C-N-C) cm−1. ESI-HRMS: calculated mass for C40H53N2O4 (M + H+) 625.4005; found 625.4030.
4.6.30. Synthesis of N1,N12-bis(4-(benzyloxy)-3-methoxybenzyl) dodecane-1,12-diamine (30)
Following the general reaction conditions for the disubstituted diamines synthesis, the reaction was worked up and purified to afford 135 mg of white solid (isolated yield: 90%). 1H NMR (CDCl3) δ7.40 (m, 10H, aromatics 4′−benzyloxy); 6.81 (m, 6H, aromatics); 5.14 (s, 4H, Ar-CH2−O); 3.89 (s, 6H, O-CH3); 3.71 (s, 4H, Ar-CH2−N); 2.61 (t, 4H, N-CH2); 1.51 (m, 4H, C2-H and C11-H); 1.27 (m, 16H, C3-H to C10-H). 13C NMR (CDCl3) δ 149.7 (C); 147.2 (C); 137.3 (C); 133.6 (C); 128.5 (CH); 127.8 (CH); 127.3 (CH); 120.3 (CH); 114.0 (CH); 112.0 (CH); 71.1 (CH2); 56.0 (CH3); 53.8 (CH2); 49.4 (CH2); 30.0 (CH2); 29.5 (CH2); 27.4 (CH2). IR υ 3422 (R-NH-R1); 1263 and 1029 (Ar-O-C); 1140 (C-N-C) cm−1. ESI-HRMS: calculated mass for C42H57N2O4 (M + H+) 653.4313; found 653.4313.
4.7. General procedure for N,N,N′,N′−diamines tetrasubstituted synthesis
To a solution of aldehyde (4.5 eq.) and diamine (1 eq) in anhydrous DCM, NaBH(AcO)3 (7.2 eq.) and acetic acid (6.4 eq.) were added at room temperature. The reaction was stirred under inert atmosphere for 24 h and then was quenched with a 10% NaHCO3. Solution. Phases were separated and the aqueous phase extracted with DCM (3 × 20 mL). Combined organic extracts were dried with sodium sulphate and evaporated. Products were purified by flash chromatography.
4.7.1. Synthesis of N1,N1,N3,N3-tetrabenzyl-propane-1,3-diamine (31)
Following the general reaction conditions for the tetrasubstituted diamines synthesis, the reaction was worked up and purified to afford 29 mg of white solid (isolated yield: 17%). 1H NMR (CDCl3) δ 7.28 (m, 20H, aromatics); 3.51 (s, 8H, Ar-CH2); 2.42 (t, 4H, J = 6.9 Hz, N-CH; 1.73 (m, 2H, C2-H). 13 2 C NMR (CDCl3) δ 139.7 (C); 128.9 (CH); 128.2 (CH); 126.8 (CH); 58.4 (CH2); 51.6 (CH2); 24.5 (CH2). IR υ 3026 (Ar-H); 1127 (C-N); 697 (monosubstituted aromatic) cm 1. ESI-HRMS: calculated mass for C31H35N2 (M + H+) 435.2800; found 435.2781.
4.7.2. Synthesis of N1,N1,N4,N4-tetrabenzylbutane-1,4-diamine (32)
Following the general reaction conditions for the tetrasubstituted diamines synthesis, the reaction was worked up and purified to afford 42 mg of white solid (isolated yield: 26%). 1H NMR (CDCl3) δ 7.31 (m, 20H, aromatics); 3.50 (s, 8H, Ar-CH2); 2.35 (t, 4H, J = 4.8 Hz, N-CH2); 1.49 (m, 4H, C2-H and C3-H). 13C NMR (CDCl3) δ 140.0 (C); 128.8 (CH); 128.2 (CH); 126.07 (CH); 58.3 (CH2); 53.4 (CH2); 24.8 (CH2). IR υ 3028 (Ar-H); 1123 (C-N); 693 (mono-substituted aromatic) cm−1. ESI-HRMS: mass calculated for C32H37N2 (M + H+) 449.2957; found 449.2964.
4.7.3. Synthesis of N1,N1,N6,N6-tetrabenzylhexane-1,6-diamine (33)
Following the general reaction conditions for the tetrasubstituted diamines synthesis, the reaction was worked up and purified to afford 27 mg of white solid (isolated yield: 17%). 1H NMR (CDCl3) δ 7.31 (m, 20H, aromatics); 3.53 (s, 8H, Ar-CH2); 2.36 (t, 4H, J = 6.9 Hz, N-CH2); 1.45 (m, 4H, C2-H and C5-H); 1.17 (m, 4H, C3-H and C4-H). 13C NMR (CDCl3) δ 140.1 (C); 128.8 (CH); 128.1 (CH); 126.7 (CH); 58.3 (CH2); 53.4 (CH2); 27.1 (CH2); 27.0 (CH2). IR υ 3026 (Ar-H); 1120 (C-N); 698 (monosubstituted aromatic) cm−1. ESI HRMS: mass calculated for C34H41N2 (M + H+) 477.3270; found 477.3273.
4.7.4. Synthesis of N1,N1,N8,N8-tetrabenzyloctane-1,8-diamine (34)
Following the general reaction conditions for the tetrasubstituted diamines synthesis, the reaction was worked up and purified to afford 28 mg of white solid (isolated yield: 27%). 1H NMR (CDCl3) δ 7.32 (m, 20H, aromatics); 3.54 (s, 8H, Ar-CH2); 2.39 (t, 4H, J = 6.9, N-CH2); 1.47 (m, 4H, C2-H and C7-H); 1.22 (m, 8H, C3-H to C6-H). 13C NMR (CDCl3) δ140.1 (C); 128.8 (CH); 128.1 (CH); 126.7 (CH); 58.3 (CH2); 53.4 (CH2); 29.5 (CH2); 27.3 (CH2); 27.0 (CH2). IR υ 3026 (Ar-H); 1126 (C-N); 697 (monosubstituted aromatic) cm−1.
4.7.5. Synthesis of N1,N1,N10,N10-tetrabenzyldecane-1,10-diamine (35)
Following the general reaction conditions for the tetrasubstituted diamines synthesis, the reaction was worked up and purified to afford 26 mg of white solid (isolated yield: 28%). 1H NMR (CDCl3) δ 7.32 (m, 20H, aromatics); 3.54 (s, 8H, Ar-CH2); 2.39 (t, 4H, J = 7.2 Hz, N-CH2); 1.48 (m, 4H, C2-H and C9-H); 1.17 (s, 12H, C3-H to C8-H). 13C NMR (CDCl3) δ 140.1 (C); 128.8 (CH); 128.1 (CH); 126.7 (CH); 58.3 (CH2); 53.4 (CH2); 29.6 (CH2); 29.5 (CH2); 27.3 (CH2); 27.0 (CH2). ESI-HRMS: calculated mass for C38H49N2 (M + H+) 533.3892, found 533.3896.
4.7.6. Synthesis of N1,N1,N3,N3-tetrakis(4-methoxybenzyl)propane portion wise-1,3-diamine (36)
Following the general reaction conditions for the tetrasubstituted diamines synthesis, the reaction was worked up and purified to afford 194 mg of yellowish oil (isolated yield: 30%). 1H NMR (CDCl3) δ 7.16 (d, 8H, J = 8.7 Hz, aromatics); 6.81 (d, 8H, J = 8.7 Hz, aromatics); 3.79 (s, 12H, O-CH3); 3.40 (s, 8H, Ar-CH = 2); 2.36 (t, 4H, J = 6,9 Hz, N-CH2); 1.67 (m, 2H, C2). 13C NMR (CDCl3) δ 158.5 (C); 132.0 (C); 130.0 (CH); 113.5 (CH); 57.5 (CH2); 55.2 (CH3); 51.3 (CH2); 24.5 (CH2). IR υ 2997 (Ar-H); 1247 and 1034 (Ar-O-C); 1179 (C-N) cm−1. ESI-HRMS: calculated mass for C35H43N2O4 (M + H+) 555.3223; found 555.3235.
4.7.7. Synthesis of N1,N1,N4,N4-tetrakis(4-methoxybenzyl)butane-1,4-diamine (37)
Following the general reaction conditions for the tetrasubstituted diamines synthesis, the reaction was worked up and purified to afford 45 mg of white solid (isolated yield: 25%). 1H NMR (CDCl3) δ 7.22 (d, 8H, J = 8.7 Hz, aromatics); 6.83 (d, 8H, J = 8.7 Hz, aromatics); 3.79 (s, 12H, -O-CH3); 3.42 (s, 8H, Ar-CH = 2); 2.31 (t, 4H, N-CH2); 1.44 (m, 4H, C2-H and C3-H). 13C NMR (CDCl3) δ158.5 (C); 132.1 (C); 129.9 (CH); 113.5 (CH); 57.4 (CH2); 55.3 (CH3); 51.3 (CH2); 24.8 (CH2). IR υ 2998 (Ar-H); 1247 and 1037 (Ar-O-C); 1178 (C-N) cm−1. ESI-HRMS: mass calculated for C36H45N2O4 (M + H+) 569.3379; found 569.3385.
4.7.8. Synthesis of N1,N1,N6,N6-tetrakis(4-methoxybenzyl)hexane-1,6-diamine (38)
Following the general reaction conditions for the tetrasubstituted diamines synthesis, the reaction was worked up and purified to afford 21 mg of white solid (isolated yield: 10%). 1H NMR (CDCl3) δ 7.24 (d, 8H, J = 8.7 Hz, aromatics); 6.83 (d, 8H, J = 8.7 Hz, aromatics); 3.79 (s, 12H, O-CH3); 3.45 (s, 8H, Ar-CH = 2); 2.33 (t, 4H, J = 7.2 Hz, N-CH2); 1.46 (m, 4H, C2-H and C5-H); 1.16 (m, 4H, C3-H and C4-H). 13C NMR (CDCl3) δ 158.4 (C); 132.0 (C); 129.8 (CH); 113.5 (CH); 57.3 (CH2); 55.2 (CH3); 51.1 (CH2); 27.2 (CH2); 27.0 (CH2). IR υ 3032 (Ar-H); 1247 and 1036 (Ar-O-C); 1170 (C-N) cm−1. ESI-HRMS: mass calculated for C38H49N2O4 (M + H+) 597.3692; found 597.3718.
4.7.9. Synthesis of N1,N1,N8,N8-tetrakis(4-methoxybenzyl)octane-1,8-diamine (39)
Following the general reaction conditions for the tetrasubstitutes diamines synthesis, the reaction was worked up and puri-fied to afford 20 mg of white solid (isolated yield: 16%). 1H NMR (CDCl3) δ 7.25 (d, 8H, J = 8.7 Hz, aromatics); 6.84 (d, 8H, J = 8.7 Hz, aromatics); 3.79 (s, 12H, O-CH3); 3.46 (s, 8H, Ar-CH = 2); 2.37 (t, 4H, J = 7.2 Hz, N-CH2); 1.46 (m, 4H, C2-H and C7-H); 1.16 (s, 8H, C3-H to C6-H). 13C NMR (CDCl3) δ 158.4 (C); 132.1 (C); 129.9 (CH); 113.5 (CH); 57.4 (CH2); 55.2 (CH3); 53.1 (CH2); 29.6 (CH2); 27.3 (CH2); 27.3 (CH2). ESI-HRMS: mass calculated for C40H53N2O4 (M + H+) 625.4005; found 625.4036.
4.7.10. Synthesis of N1,N1,N10,N10-tetrakis(4-methoxybenzyl) decane-1,10 diamine (40)
Following the general reaction conditions for the tetrasubstitutes diamines synthesis, the reaction was worked up and puri-fied to afford 21 mg of white solid (isolated yield: 28%). 1H NMR (CDCl3) δ7.25 (d, 8H, J = 8.7 Hz, aromatics); 6.84 (d, 8H, J = 8.7 Hz, aromatics); 3.79 (s, 12H, O-CH3); 3.46 (s, 8H, Ar-CH = 2); 2.36 (t, 4H, J = 7.5 Hz, N-CH2); 1.47 (m, 4H, C2-H and C9-H); 1.18 (s, 12H, CH2, C3-H to C8-H). 13C NMR (CDCl3) δ 158.4 (C); 132.0 (C); 129.9 (CH); 113.5 (CH); 57.4 (CH2); 55.2 (CH3); 53.0 (CH2); 29.8 (CH2); 29.5 (CH2); 27.3 (CH2); 26.9 (CH2). ESI-HRMS: mass calculated for C42H57N2O4 (M + H+) 653.4318; found 653.4340.
4.8. Computational methods
The geometries of the products were optimized using B3LYP functional [54–56] together with 6–311 ++G(d, p) basis set. The B3LYP has shown to be appropriate to obtain reliable geometries and for describing electronic density surfaces. All calculations were performed with the Gaussian 09 package [57].
Acknowledgments
The authors wish to express their gratitude to Andrea P. Caminos for the help on the initial steps of the project. UNR (Universidad Nacional de Rosario) and Fundación Josefina Prats have contributed to support this work. Financial support through CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas, PIP 2009–11/0796 and PIP 2012–14/0448), Agencia Nacional de Promocion (ANPCyT - PICT 2011/0589) and Fundacion Científica y Tecnologica Bunge y Born (FBB 31/10) are gratefully acknowledged. This investigation also received financial support from the UNICEF/UNDP/WORLD BANK/WHO Special Programme for Research and Training in Tropical Diseases (TDR, A50987) to GRL. GRL is member of the Research Career of CONICET. E.A.P.Z., L.F. and E.O.J.P. thanks CONICET for the award of a Fellowship.
Footnotes
Competing interests
The authors declare that they have no competing interest.
Appendix A. Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.ejmech.2017.11.069.
References
- [1].Pappas G, Roussos N, Falagas ME, Int. J. Parasitol 39 (2009) 1385. [DOI] [PubMed] [Google Scholar]
- [2].Fereig RM, Nishikawa Y, in: Thomas S (Ed.), Vaccine Design: Methods and Protocols, Volume 2: Vaccines for Veterinary Diseases, Springer New York, New York, NY, 2016, p. 153. [Google Scholar]
- [3].McLeod R, Berry PF, Marshall WH, Hunt SA, Ryning FW, Remington JS, Am. J. Med 67 (1979) 711. [DOI] [PubMed] [Google Scholar]
- [4].Zhang M, Joyce BR, Sullivan WJ Jr., V. Nussenzweig. Eukaryot. Cell 12 (2013) 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Talevich E, Mirza A, Kannan N, BMC Evol. Biol 11 (2011) 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Terkawi MA, Igarashi I, Apicomplexan Parasites, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, p. 453. [Google Scholar]
- [7].(a) Hay SI, Guerra CA, Gething PW, Patil AP, Tatem AJ, Noor AM, Kabaria CW, Manh BH, Elyazar IRF, Brooker S, Smith DL, Moyeed RA, Snow RW, PLoS Med 6 (2009); [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) World Malaria Report of World Health Organization, 2016. [Google Scholar]
- [8].Mehlotra RK, Fujioka H, Roepe PD, Janneh O, Ursos LM, Jacobs-Lorena V, McNamara DT, Bockarie MJ, Kazura JW, Kyle DE, Fidock DA, Zimmerman PA, Proc. Natl. Acad. Sci. U. S. A 98 (2001) 12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kamal-Yanni MM, Potet J, Saunders PM, Malar. J 11 (2012) 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Meshnick SR, Int. J. Parasitol 32 (2002) 1655. [DOI] [PubMed] [Google Scholar]
- [11].Kumar S, Kumari R, Pandey P, Protoplasma 252 (2015) 717. [DOI] [PubMed] [Google Scholar]
- [12].Global Plan to Combat Neglected Tropical Diseases 2008e2015, World Health Organization, 2007. [Google Scholar]
- [13].Das B, Gupta R, Madhubala R, Pharmacol. Res 31 (1995) 189. [DOI] [PubMed] [Google Scholar]
- [14].Murray-Stewart TR, Woster PM, Casero RA, Biochem. J 473 (2016) 2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pegg AE, Poulin R, Coward JK, Int. J. Biochem. Cell Biol 27 (1995) 425. [DOI] [PubMed] [Google Scholar]
- [16].Sprenger J, Carey J, Svensson B, Wengel V, Persson L, PLoS One 11 (2016) e0163442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Casero RA, Pegg AE, FASEB J 7 (1993) 653. [PubMed] [Google Scholar]
- [18].Muller IB, Das Gupta R, Luersen K, Wrenger C, Walter RD, Mol. Biochem. Parasitol 160 (2008) 1. [DOI] [PubMed] [Google Scholar]
- [19].Cook T, Roos D, Morada M, Zhu G, Keithly JS, Feagin JE, Wu G, Yarlett N, Microbiology 153 (2007) 1123. [DOI] [PubMed] [Google Scholar]
- [20].Bitonti AJ, Dumont JA, Bush TL, Edwards ML, Stemerick DM, McCann PP, Sjoerdsma A, Proc. Natl. Acad. Sci. U.S.A 86 (1989) 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bitonti AJ, McCann PP, Sjoerdsma A, Adv. Exp. Med. Biol 250 (1988) 717. [DOI] [PubMed] [Google Scholar]
- [22].Caminos AP, Panozzo-Zenere EA, Wilkinson SR, Tekwani BL, Labadie GR, Bioorg. Med. Chem. Lett 22 (2012) 1712. [DOI] [PubMed] [Google Scholar]
- [23].(a) Gardner RA, Belting M, Svensson K, Phanstiel O. t., J. Med. Chem 50 (2007) 308; [DOI] [PubMed] [Google Scholar]; (b) Kaur N, Delcros J-G, Imran J, Khaled A, Chehtane M, Tschammer N, Martin B, Phanstiel O. t., J. Med. Chem 51 (2008) 1393. [DOI] [PubMed] [Google Scholar]
- [24].Jagu E, Pomel S, Pethe S, Loiseau PM, Labruére R, Eur. J. Med. Chem 139 (2017) 982. [DOI] [PubMed] [Google Scholar]
- [25].Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD, J. Org. Chem 61 (1996) 3849. [DOI] [PubMed] [Google Scholar]
- [26].Porta EO, Carvalho PB, Avery MA, Tekwani BL, Labadie GR, Steroids 79 (2014) 28. [DOI] [PubMed] [Google Scholar]
- [27].Labadie GR, Choi S-R, Avery MA, Bioorg. Med. Chem. Lett 14 (2004) 615. [DOI] [PubMed] [Google Scholar]
- [28].Backman TWH, Cao Y, Girke T. Nucleic Acids Res 39 (2011) W486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].http://www.organic-chemistry.org/prog/peo/26/03/2014: version 2 (Accessed 20 October 2015).
- [30].http://www.molinspiration.com.v2014.11. (Accessed 6 March 2017).
- [31]. Marvin 17.6.0, ChemAxon. http://www.chemaxon.com.
- [32].Yamada D, Saiki S, Furuya N, Ishikawa KI, Imamichi Y, Kambe T, Fujimura T, Ueno T, Koike M, Sumiyoshi K, Hattori N, Biochem. Biophys. Res. Commun 471 (2016) 109. [DOI] [PubMed] [Google Scholar]
- [33].Loschen C, Klamt A, Ind. Eng. Chem. Res 51 (2012) 14303. [Google Scholar]
- [34].Clark DE, J. Pharm. Sci 88 (1999) 815. [DOI] [PubMed] [Google Scholar]
- [35].Abraham MH, Takács-Nov ak K, Mitchell RC, J. Pharm. Sci 86 (1997) 310. [DOI] [PubMed] [Google Scholar]
- [36].Wermuth CG, The Practice of Medicinal Chemistry, Academic Press, 2011. [Google Scholar]
- [37].Sánchez-Martín R, Campos JM, Conejo-García A, Cruz-López O, Bánez-Coronel M, Rodríguez-Gonzalez A, Gallo MA, Lacal JC, Espinosa A, J. Med. Chem 48 (2005) 3354. [DOI] [PubMed] [Google Scholar]
- [38].Bai R, Liang Z, Yoon Y, Liu S, Gaines T, Oum Y, Shi Q, Mooring SR, Shim H, Eur. J. Med. Chem 118 (2016) 340e350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Thelemann J, Illarionov B, Barylyuk K, Geist J, Kirchmair J, Schneider P, Anthore L, Root K, Trapp N, Bacher A, Witschel M, Zenobi R, Fischer M, Schneider G, Diederich F, ChemMedChem 10 (2015) 2090. [DOI] [PubMed] [Google Scholar]
- [40].Ersmark K, Feierberg I, Bjelic S, Hamelink E, Hackett F, Blackman MJ, Hulten J, Samuelsson B, Åqvist J, Hallberg A, J. Med. Chem 47 (2004) 110. [DOI] [PubMed] [Google Scholar]
- [41].Pathak AK, Pathak V, Riordan JR, Suling WJ, Gurcha SS, Besra GS, Reynolds RC, Bioorg. Med. Chem. Lett 17 (2007) 4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bhattacharya S, Chaudhuri P, Jain AK, Paul A, Bioconjugate Chem 21 (2010) 1148. [DOI] [PubMed] [Google Scholar]
- [43].Cumpstey I, Sundin A, Leffler H, Nilsson UJ, Angew. Chem. Int. Ed 44 (2005) 5110. [DOI] [PubMed] [Google Scholar]
- [44].Alfonsi K, Colberg J, Dunn PJ, Fevig T, Jennings S, Johnson TA, Kleine HP, Knight C, Nagy MA, Perry DA, Stefaniak M, Green Chem 10 (2008) 31. [Google Scholar]
- [45].Wender PA, Verma VA, Paxton TJ, Pillow TH, Acc. Chem. Res 41 (2008) 40. [DOI] [PubMed] [Google Scholar]
- [46].Ishikawa M, Hashimoto Y, J. Med. Chem 54 (2011) 1539. [DOI] [PubMed] [Google Scholar]
- [47].Majumder S, Kierszenbaum F, Antimicrob. Agents Chemother 37 (1993) 2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Merali S, Chin K, Clarkson AB, Antimicrob. Agents Chemother 44 (2000) 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bitonti AJ, Dumont JA, Bush TL, Stemerick DM, Edwards ML, McCann PPJ, J. Biol. Chem 265 (1990) 382. [PubMed] [Google Scholar]
- [50].Niemand J, Burger P, Verlinden BK, Reader J, Joubert AM, Kaiser A, Louw AI, Kirk K, Phanstiel O, Birkholtz LM, Antimicrob. Agents Chemother 57 (2013) 2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Muth A, Kaur N, Shicora AC, Ayene IS, Gilmour SK, Phanstiel O, J. Med. Chem 56 (2013) 5819. [DOI] [PubMed] [Google Scholar]
- [52].Muth A, Madan M, Archer JJ, Ocampo N, Rodriguez L, Phanstiel O, Med. Chem 57 (2014) 348. [DOI] [PubMed] [Google Scholar]
- [53].Wang C, Delcros JG, Cannon L, Konate F, Carias H, Biggerstaff J, Gardner RA, Phanstiel O, J. Med. Chem 46 (2003) 5129. [DOI] [PubMed] [Google Scholar]
- [54].Becke ADJ, Chem. Phys 98 (1993) 5648. [Google Scholar]
- [55].Lee C, Yang W, Parr RG, Phys. Rev. B 37 (1988) 785. [DOI] [PubMed] [Google Scholar]
- [56].Hehre WJ, Acc. Chem. Res 9 (1976) 399. [Google Scholar]
- [57].Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich A, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016. [Google Scholar]