

Abstract

In this study, two respective groups of RNA aptamers have been selected against two main classes of glycosaminoglycans (GAGs), heparosan, and chondroitin, as they have proven difficult to specifically detect in biological samples. GAGs are linear, anionic, polydisperse polysaccharides found ubiquitously in nature, yet their detection remains problematic. GAGs comprised repeating disaccharide units, consisting of uronic acid and hexosamine residues that are often also sulfated at various positions. Monoclonal antibodies are frequently used in biology and medicine to recognize various biological analytes with high affinity and specificity. However, GAGs are conserved across the whole animal phylogenic tree and are nonimmunogenic in hosts traditionally used for natural antibody generation. Thus, it has been challenging to obtain high affinity, selective antibodies that recognize various GAGs. In the absence of anti-GAG antibodies, glycobiologists have relied on the use of specific enzymes to convert GAGs to oligosaccharides for analysis by mass spectrometry. Unfortunately, while these methods are sensitive, they can be labor-intensive and cannot be used for in situ detection of intact GAGs in cells and tissues. Aptamers are single-stranded oligonucleotide (DNA or RNA) ligands capable of high selectivity and high affinity detection of biological analytes. Aptamers can be developed in vitro by the systematic evolution of ligands by exponential enrichment (SELEX) to recognize nonimmunogenic targets, including neutral carbohydrates. This study utilizes the SELEX method to generate RNA aptamers, which specifically bind to the unmodified GAGs, heparosan, and chondroitin. Binding confirmation and cross-screening with other GAGs were performed using confocal microscopy to afford three specific GAGs to each target. Affinity constant of each RNA aptamer was obtained by fluorescent output after interaction with the respective GAG target immobilized on plates; the KD values were determined to be 0.71–1.0 μM for all aptamers. Upon the success of chemical modification (to stabilize RNA aptamers in actual biological systems) and fluorescent tagging (to only visualize RNA aptamers) of these aptamers, they would be able to serve as a specific detection reagent of these important GAGs in biological samples.
Introduction
Glycosaminoglycans (GAGs) are linear acidic polysaccharides comprising repeating disaccharide structures of uronic acid and hexosamine residues that are ubiquitously found in all animal tissues.1 These polysaccharides are responsible for critical functions in development, normal physiology, and pathophysiology.2 The study of the GAG structure is essential in developing structure–activity relationships and has been intensively investigated.3,4 Because GAGs are polydispersive and often microheterogenous, structural elucidation is exceedingly difficult and is well behind the structural characterization of other important biopolymers, such as nucleic acids and proteins.5 Structurally simple (unmodified) GAGs, such as heparosan (HE), hyaluronan (HA), and chondroitin (CH), are expressed by human and animal pathogenic and commensal bacteria as capsules and are used as molecular camouflage.6,7 However, animal-derived GAGs are biosynthesized in the golgi by a nontemplate-driven process, making it difficult to rely on deriving structural insights through understanding GAG biosynthesis.1,8 The simple GAG polysaccharide backbone is commonly modified by N- and/or O-sulfation and epimerization in the golgi affording highly complex GAGs. Most of what we know about the GAG structure comes from their controlled chemical or enzymatic depolymerization to disaccharide or oligosaccharide structures that are somewhat easier to characterize than intact GAG polysaccharides.9,10 There have recently been major advances in mass spectrometric methods that have allowed the sequencing of small intact GAGs,5 yet complete structural characterization remains elusive.
Antibodies, particularly monoclonal antibodies prepared using hybridoma technology, have been useful tools for solving the structure of protein biopolymers.11 GAG structures across animal species are so similar that GAGs derived from one animal are generally not immunogenic when administered to another, and the only reported GAG reactivity is in autoimmune disease sera.12 Antibodies generated to GAGs are therefore typically generated against the protein cores of proteoglycans carrying GAGs or unnatural structural features introduced to GAGs through chemical or enzymatic processes.13 This severely limits the repertoire of antibodies available for studying the GAG structure. Proteins obtained using phage display-based directed evolution have been reported to recognize GAG structures,14,15 as these are not dependent on an immune response for their generation. While these specific GAG binding ligands allow in situ assessment of intact GAGs in tissues, they display relatively weak binding affinities.14
A rapidly emerging type of molecular ligand for recognizing targets with high specificity and affinity is the nucleic acid aptamer. Aptamers are single-stranded RNA or DNA of 20–100 nucleotides, which can be generated through the systematic evolution of ligands by the exponential enrichment (SELEX) strategy and can bind to specific molecular targets.16,17 Structural motifs of aptamers, such as stem loops, and ionic interactions play major roles in facilitating specific recognition of a target ligand.18 RNA aptamers having relatively low molecular weights, compared to proteins, are easy to synthesize in large quantities using automated oligonucleotide synthesis19 or in vitro transcription20 and have been applied in biological and clinical applications. Furthermore, RNAs can be modified structurally and chemically to be resistant to nuclease degradation.21
Although RNA aptamers have been used in sandwich assays with lectins to analyze glycoforms,22 there are only a few examples of aptamers that were evolved to recognize carbohydrates.23−26 Basic carbohydrates, such as the amino-substituted trisaccharide tobramycin, strongly interact with an acidic RNA aptamer through ion pairing.23 DNA aptamers designed for neutral carbohydrates, such as cellulose, interact specifically but with lower binding affinities of 3–6 μM.24 Recently, oligonucleotide aptamers for neutral monosaccharides, termed “low-epitope targets”, have been improved through complexation with ternary boronic acids, to become “high-epitope organic receptors” for these targets.26 In addition, RNA aptamers have recently been reported to be capable of recognizing a sialic acid-terminated glycan with relatively high binding affinities of 1–5 nM.25 Nonetheless, no aptamers have been reported to date to interact with polyanionic (highly negatively charged) carbohydrates with high selectivity and good binding affinity.
In the current study, we describe the first preparation and characterization of two groups of RNA aptamers that are evolved using the SELEX strategy and able to interact with two respective polyanionic carbohydrates, HE and CH, with high specificity and submicromolar binding affinity.
Results and Discussion
Aptamer Selection
A directed evolution strategy involving SELEX with a special negative selection procedure was used in this study to obtain RNA aptamers that selectively interact with HE or CH (Figure 1). The RNA aptamer was first chosen over the DNA aptamer because RNA has significantly increased conformational flexibility. Because of the negatively charged nature of both GAG and nucleic acid species, we speculated that the increased conformational flexibility imparted by RNA would fortify the optimal selection of a high binding aptamer to the target GAG. RNAs with certain consensus sequences (mainly G3A clusters) are often predominately enriched during SELEX because of the strong interaction with the solid support (e.g., agarose/magnetic beads and cellulous membrane) used to immobilize the targets of interest, even though such solid support has already been utilized for negative selection.27 The presence of such “background-binding” RNAs makes it much harder and sometimes impossible to enrich desired aptamers that specifically bind to the targets of interest.27 Our initial SELEX trial confirmed this previous observation as the predominant RNA recovered after aptamer selection (against HE immobilized on the magnetic beads) also contained G3A clusters (HE-03 with 20/28 sequence hit, group “A” of Table S1). This was the case even though the blank magnetic beads were used as negative selection in each round of SELEX to remove the “background-binding” RNAs.
Figure 1.
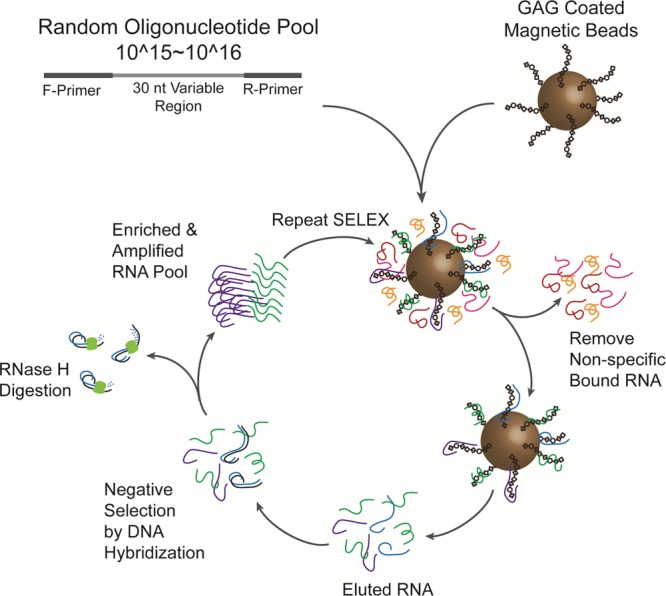
SELEX method for obtaining RNA aptamers to GAGs. A random oligonucleotide library is incubated with GAG-coated magnetic beads. Nonspecifically bound oligonucleotides are washed off, and a negative selection is carried out before reiterating the process for a total of 10 more times.
We used DNA oligonucleotides carrying the sequence complementary to the consensus clusters of “background-binding” RNAs to overcome this problem. After each selection round DNA/RNA duplexes are formed, and the hybridized “background-binding” RNAs are subjected to RNase-H digestion. Therefore, in this modified SELEX process, a group of DNA oligonucleotides (Table S3), including those employed in the study by Shi et al.,27 was used to quench the “background-binding” RNAs in the enriched RNA population. As a result, we obtained two respective groups of candidate RNA aptamers with 12 additional unique sequences against HE (referred as HE-## in group “B” of Table S1) and 39 unique sequences against CH (referred as CH-## in Table S2). More than 85% (73 out of 85) sequenced clones of these newly evolved candidate RNA aptamers did not contain the consensus cluster sequences found in group A, indicating that this sophisticated negative selection strategy was able to effectively suppress the enrichment of “background-binding” RNA in SELEX.
Confirmation of the Interaction of Candidate RNA Aptamers with Respective GAGs
We next used confocal microscopy to directly visualize RNA–GAG interactions suggested by SELEX results to confirm the interaction of evolved candidate RNA aptamers with the respective GAG targets (HE or CH). Each candidate RNA aptamer was first incubated with the same GAG-magnetic bead sample used for its generation. After thorough washing to remove unbound RNA molecules, nucleic acid-specific binding fluorophore, SYBR Green II, was added, and the beads were imaged by confocal microscopy. Additionally, blank magnetic beads were used in the same way to filter out the unique “background-binding” RNAs in the pool of candidate RNA aptamers. As displayed in Figures S4 and S5, 10 HE and 12 CH candidate RNA aptamers (Table 1) were found to show much stronger interaction with the corresponding GAG-bead sample than with blank magnetic beads (Figure 2). On the basis of the sequence alignment (Figure S6), we observed that most of these RNA aptamers do not share a conserved sequence motif. We hypothesize that either some of these aptamers lack binding specificity to the respective GAG targets (discussion below) or they might fold into different tertiary structures that can interact with different regions of the same GAG molecule. The latter hypothesis will require further investigation, including computational simulation approaches.
Table 1. RNA Aptamers that Show Positive Interaction with HE or CHa.
| RNA | sequences |
|---|---|
| HE-01 | GGGAAGAGAAGGACAUAUGAUCCGCAUAAAGUGUAUCCGAUUUGGUUGCUGUUGACUAGUACAUGACCACUUGA |
| HE-04 | GGGAAGAGAAGGACAUAUGAUUCGGAAUGAGCGCAGAAGUAGCGCAUUGGCUUGACUAGUACAUGACCACUUGA |
| HE-06 | GGGAAGAGAAGGACAUAUGAUCCCCAUUGCGGCCAAAAUGACAUAGGCGUGUUGACUAGUACAUGACCACUUGA |
| HE-07 | GGGAAGAGAAGGACAUAUGAUUCGUUCAAGACGGCCUCUGGUUGCGGAUGGCUUGACUAGUACAUGACCACUUGA |
| HE-08 | GGGAAGAGAAGGACAUAUGAUGCGGGAUGUGGUGUACCCGCUAUCCCAGGCUUGACUAGUACAUGACCACUUGAA |
| HE-09 | GGGAAGAGAAGGACAUAUGAUUCGCACUCAAUGAAAGCGGGUCAAUUGCGCGUUGACUAGUACAUGACCACUUGA |
| HE-10 | GGGAAGAGAAGGACAUAUGAUCCAACAGUGGGACAGAGACGACUUAGCGCGUUGACUAGUACAUGACCACUUGA |
| HE-13 | GGGAAGAGAAGGACAUAUGAUCCAACGGUGGGCAGGAUAUCUCAUGCAGUGUUGACUAGUACAUGACCACUUGA |
| HE-14 | GGGAAGAGAAGGACAUAUGAUCUCAAUAAGAGACAGCUUCCGGUGGCUUGGUUGACUAGUACAUGACCACUUGA |
| HE-16 | GGGAAGAGAAGGACAUAUGAUCCCAAACAGGAAAAGGGAUGGGGUCAGCCGUUGACUAGUACAUGACCACUUGA |
| HE-17 | GGGAAGAGAAGGACAUAUGAUCCAAAACAACGUGGUCGAAGCAUGAUCCGCUUGACUAGUACAUGACCACUUGA |
| CH-03 | GGGAAGAGAAGGACAUAUGAUUGCCAAUGGGAUCGCACAAGGAAUGGCCGGUUGACUAGUACAUGACCACUUGA |
| CH-04 | GGGAAGAGAAGGACAUAUGAUCCUGGGGCGGCGAGAGUAUGCGCGCGGCGGUUGACUAGUACAUGACCACUUGA |
| CH-09 | GGGAAGAGAAGGACAUAUGAUCGGAUUGGGCAGAGGCUCGUACGUAUCGGCUUGACUAGUACAUGACCACUUGA |
| CH-17 | GGGAAGAGAAGGACAUAUGAUAGGGAAGGUGCGGGUUGUCCAGUAGUGCAGUUGACUAGUACAUGACCACUUGA |
| CH-20 | GGGAAGAGAAGGACAUAUGAUCUGGAAUCGACGGGCAGGGCUCAGUUGCGCUUGACUAGUACAUGACCACUUGA |
| CH-21 | GGGAAGAGAAGGACAUAUGAUUAGGGCAGGUGUAGGGUUGGUCCUUCGGCGUUGACUAGUACAUGACCACUUGA |
| CH-31 | GGGAAGAGAAGGACAUAUGAUACCGCCGGAGUGGAUAGGCAGGGGUGGUAGUUGACUAGUACAUGACCACUUGA |
| CH-32 | GGGAAGAGAAGGACAUAUGAUCCCGAUGUACGCGGUUUGGGGGCGUCAGGCUUGACUAGUACAUGACCACUUGA |
| CH-34 | GGGAAGAGAAGGACAUAUGAUGAGGUACUGGGGGGGAGGCCGAAUGUGCAUUUGACUAGUACAUGACCACUUGA |
| CH-35 | GGGAAGAGAAGGACAUAUGAUGGAGGGGAUUGGGGGCAUGUUGGGGCGUCCUUGACUAGUACAUGACCACUUGA |
| CH-57 | GGGAAGAGAAGGACAUAUGAUGAGCGCGUCGCCGUGUUACCGCGGGGGGUGUUGACUAGUACAUGACCACUUG |
The constant primer binding regions are indicated by regular font. The sequence of evolved variable region is in bold.
Figure 2.
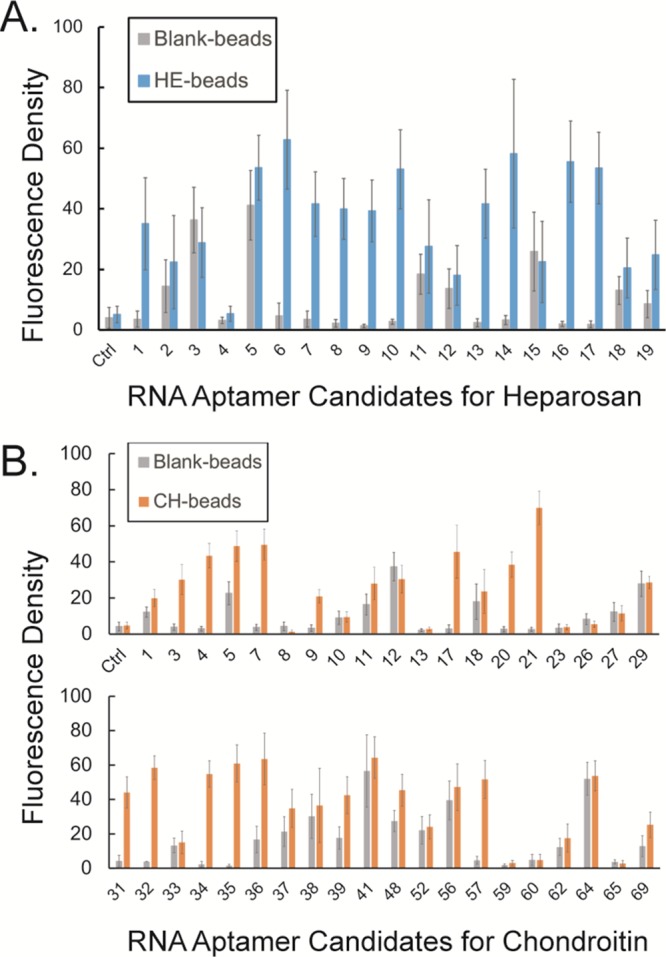
Screening of RNA aptamer candidate interaction with target GAG. The calculated fluorescence density of (A) HE candidate RNA aptamers interacting with blank beads and HE beads, and (B) CH candidate RNA aptamers interacting with blank beads and CH beads.
On the basis of confocal microscopy imaging, we noticed that although the number of HE and HA molecules immobilized to the beads are higher than that of CH, the number of bound aptamers, indicated by the fluorescence density of RNA, is similar for each GAG studied. We speculate that although higher GAG coverage on the beads is supposed to provide more RNA aptamer binding sites, the higher steric hindrance caused by the denser neighboring GAGs and higher molecular repulsions generated between the immobilized GAG and RNA also need to be taken into consideration. In addition, the future study of the minimal aptamer binding domain of the GAG may help to explain such correlation between the fluorescence density and the GAG coverage on the beads.
Aptamer Selectivity Screening
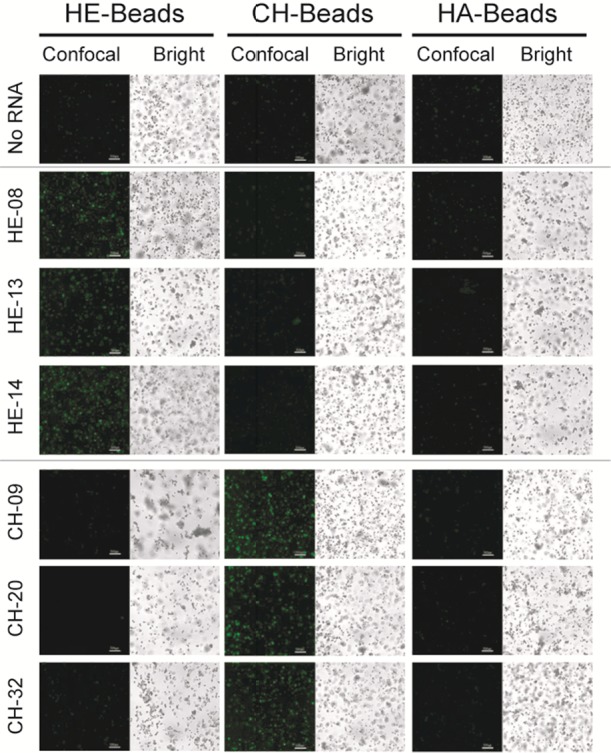
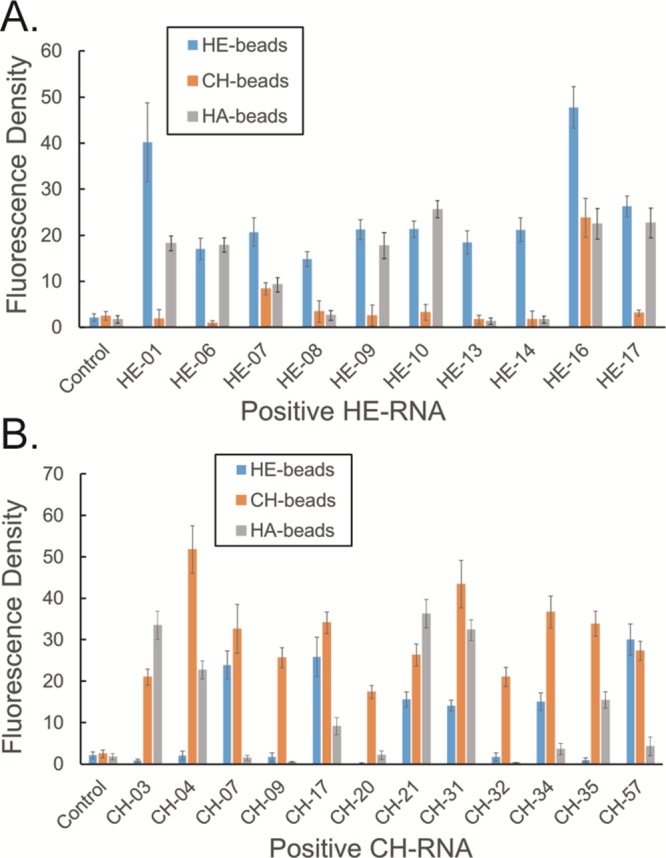
Because different classes of GAG polymers share common sugar backbone structural features, it is highly possible that some of the evolved RNA molecules may cross-react with a closely related GAG class. Therefore, we next examined the binding specificity/selectivity of the evolved RNA aptamers toward their target GAG. For this purpose, we also used the same confocal microscopy-based screening strategy to examine the binding specificity of each RNA aptamer toward the three common unmodified GAG classes, HE, CH, and HA. On the basis of the confocal microscopy imaging data (Figures 3, S7, and S8) and average fluorescence intensity quantification (Figure 4), we identified three HE (HE-08, HE-13 and HE-14) and three CH (CH-09, CH-20 and CH-32) aptamers that were bound specifically and showed no detectable interactions with the other two control GAG molecules. On the basis of the sequence alignment, three HE- or CH-specific binding aptamers share tentative discontinuous sequence motifs (boxed in Figure 5) with at least 3-nt long consensus identity shared by at least two aptamers. In addition to these selective aptamers to HE and CH, we have observed that HE-01, -06, -09, -10, and -17 are also able to recognize HE and HA, whereas CH-03, -04, and -35 recognize CH and HA. On the basis of the targets they have shown to recognize, we speculate that HE-08, -13, and -14 may recognize the GlcNAc residue and α-linkage between GlcNAc and GlcA in HE, whereas HE-01, -06, -09, -10, and -17 can also recognize both GlcA and GlcNAc residues in HE and HA. Similarly, CH-09, -20, and -32 may recognize GalNAc residues and β-linkages in CH, whereas CH-03, -04, and -35 may recognize the β-linkage only in CH and HA. Although we can speculate the interaction was based on the structural components of the GAG targets and what aptamers were able to recognize each GAG, further studies relying on experimental and computational modeling are required to fully elucidate such sequence–function relationship. Nevertheless, our ability to obtain GAG recognizing RNA aptamers with high selectivity sets the stage for future applications of aptamers in polysaccharide recognition in the fields of biology and medicine.
Figure 3.
Cross-screening of RNA aptamers for interaction with other GAGs. The calculated fluorescence density of (A) HE-RNA aptamers on HE, CH, and HA beads and (B) CH-RNA aptamers on HE, CH, and HA beads.
Figure 4.
Cross-screening of RNA–GAG interaction. Confocal microscopy imaging of (A) HE and (B) CH aptamers screened against HE, CH, and HA beads. The “Confocal field” indicates the fluorescent channel image of the confocal microscopy, whereas the “bright field” indicates the bead optical channel image.
Figure 5.

Sequence alignment of HE- and CH-specific binding RNA aptamers. Color-highlighted bases indicate the RNA regions showing no consensus sequence motif among the aptamers within each group. Dash lines indicate the artificial gaps generated by the sequence alignment algorithm to maximize the matched sequence alignment. Boxed sequences indicate the consensus sequence clusters that are longer than 3-nt and shared by at least two of the GAG-specific binding aptamers in each group.
Determination of Aptamer-GAG Binding Affinity
The apparent dissociation constant (KD) was next measured to determine the aptamer–GAG binding affinity. Various concentrations of the RNA aptamer (0–4.5 μM) were first added to a constant amount of its respective GAG immobilized on a 96-well microplate. After washing off unbound RNAs, RiboGreen was added, and the fluorescence intensity was quantified using a plate reader. Saturation curves were obtained by plotting the fluorescence intensity of GAG–RNA binding complex, a function of RNA aptamer concentration.
The saturation curves of RNA aptamers HE-08, HE-13, and HE-14 to HE are shown in Figure 6A. The KD value of each aptamer was extrapolated using nonlinear regression analysis (Table S5). Aptamers HE-08 and HE-14 show very similar KD values for HE, 0.75 and 0.71 μM, respectively, whereas HE-13 shows a slightly larger KD value of 1.0 μM. The saturation curves of aptamers, CH-09, CH-20, and CH-32, binding to CH are shown in Figure 6B. On the basis of the nonlinear regression analyses (Table S5), aptamer CH-20 and CH-32 show higher affinity to CH with KD values of 0.76 and 0.89 μM respectively, whereas CH-09 shows slightly lower affinity with a KD value of 1.0 μM. These aptamers have comparable binding capabilities for their respective target when compared to many GAG-interacting proteins.28 Therefore, we suggest that these aptamers have the potential to serve in practical biological and medical applications.
Figure 6.

Saturation curves for determination of the dissociation constants (KD) of RNA aptamers specific to (A) HE: HE-08, HE-13, and HE-14 and to (B) CH: CH-09, CH-20, and CH-32. A control of immobilized biotin was used in both cases. KD values were calculated by nonlinear regression analysis based on the saturation curves.
Although the HE and CH aptamers have been evolved, screened, and validated to specifically target unsulfated GAG classes, we speculate that they may also interact with their closely-related sulfated GAGs, such as heparan sulfate, a highly sulfated chemical analog of HE,29 heparin that contains some HE sequences,4 and CH sulfate, a modified version of CH. Therefore, in a preliminary test in this report, we have screened the three specific HE aptamers (HE-08, HE-13, and HE-14) against immobilized heparin (Figure S10). Only one aptamer, HE-14, showed binding to heparin with an apparent KD of 0.87 μM (Table S5) but with significantly lower fluorescence intensity compared to interaction with HE. Interactions of HE-08 and HE-13 with heparin were comparable to the control, and affinities were not calculated. Although the disaccharide structural unit of HE is also present in heparin, it contains many sulfated glucosamine, uronic acid residues, and epimerized iduronic acid residues.1 Therefore, although it is possible that the aptamers can interact with these modified versions of their intended target, we speculate that the weaker binding of the HE aptamers to heparin is due to stronger ionic repulsions between the aptamers and this highly sulfated GAG. Because the overall fluorescence intensity of HE-14 binding to HE is much higher than that of HE-14 binding to heparin, we still assert the three HE aptamers are target-specific. SELEX would probably need to be performed starting with the initial RNA library and an immobilized heparin target to obtain an RNA aptamer specific to heparin.
Conclusions
Using SELEX with a sophisticated negative selection strategy, two small libraries of RNA aptamers were generated against the simple (unmodified) GAGs, HE and CH. Positive binding was confirmed for approximately one-third of the members of each RNA aptamer library using a confocal fluorescence assay. When the HE and CH aptamers were cross-screened using the same assay, about one-third of these positive binding aptamers showed specific binding to the simple (unmodified) GAG used in its generation. Ultimately, three positive binding and specific aptamers for each of the two unmodified GAGs were obtained. The binding affinity (KD) of these specific aptamers ranged from high nanomolar to low micromolar and were similar among the HE and CH classes. Typical binding affinities between a protein and its polysaccharide target are often comparable, suggesting that these aptamers have good binding capabilities for their respective targets.28
Though there is much more work that can be done to improve upon these aptamers, the molecular interaction of the RNA aptamers with respective GAGs might be computationally modeled and elucidated in the future, and the suspected RNA bases at the RNA–GAG interface can be further degenerated for another round of SELEX.30 In addition, many modifications are available to tune the binding affinity and stability.21 Aptamer truncation and nucleobase modification can be explored to increase binding affinity,21 whereas incorporation of naturally conserved RNA motifs can be used to provide stability during in vivo applications.31,32
This study also details a robust method for immobilizing GAGs to a solid support that is useful for the selection of aptamers to the target GAGs. Though we immobilized HA onto magnetic beads, we did not select aptamers for this GAG as it had the least potential for therapeutic and diagnostic outcomes. With this experimental setup in place, HA and modified versions of each GAG can undergo selection for RNA aptamers specific to the intended target.
Future Work
In this study, unmodified HE and CH were used for selection and aptamers were only cross-screened for specificity among the unmodified classes (including HA). The HE and CH are modified in vivo, resulting in higher ordered structures of GAGs. These highly modified animal-sourced GAGs contain stretches of unmodified domains identical to the GAGs used to generate the RNA aptamers. Beyond our preliminary test regarding this aspect, it will therefore be important to further screen the selected aptamers against their modified counterparts. In brief, HE aptamers should be tested for interaction with heparan sulfate, and CH aptamers should be tested for interaction with CH sulfate. This could reveal some aptamers that are not wholly specific to their intended target. With their ability to specifically detect the respective GAG targets, these RNA aptamers could serve as novel molecular tags and analytical/molecular biology tools for GAG structural recognition in complex mixtures.
Ultimately, we plan on applying future generations of these RNA aptamers in cell systems. Notable areas of interest include utilizing the aptamers as a means to sense GAGs shed from the glycocalyx under various stressors or disease states, to visualize GAG patterning of the glycocalyx or endothelium, and to detect and regulate the GAG-dependent cell signaling interactions and cell differentiation (Figure 7). For these aptamers to be effectively applicable, they must undergo some modifications such as chemical modifications to the individual bases, RNA truncation for improving RNA stability, and fluorescent labeling for tracking and imaging aptamers.
Figure 7.

Future applications of HE and CH aptamers. (A) RNA aptamers can be modified with molecular beacons for sensing capability, whereby fluorescence occurs after interaction with GAG. (B) Fluorescently labeled aptamers interact with GAGs to visually pattern various mammalian cell types glycocalyx.
These modifications and applications of the aptamers in cell systems are not trivial and will require great effort to carry out systematic studies of the resulting binding interaction before and after modification. As elaborated below, we plan to execute this systematic study to (1) develop a robust modified RNA aptamer capable of sensing and (2) to understand the binding interaction of RNA and GAG and the underlying mechanisms of the aptamer selectivity. (1) “Vanilla” RNA aptamers can be easily degraded in actual biological systems because of the presence of active nucleases. We will need to chemically modify the RNA bases or truncate the RNA aptamer to improve the RNA stability in the presence of nucleases. However, to minimize or avoid the loss of aptamer’s binding specificity and affinity to GAG targets, we need to use both computational modeling and experiments to elucidate the functional core of the RNA aptamers, to find out the bases that are not responsible for actual binding, and to finally test whether the chemical modifications on or deletion of those bases will undermine the binding of those “vanilla” RNA aptamers initially obtained by SELEX. In addition, we need to develop an effective chemistry to covalently label the RNA aptamers with respective fluorophores, which need to be verified as not interfering with the aptamers’ interaction with their respective GAG targets. (2) As the aptamers likely bind to a small portion of the total GAG and that different aptamers act on different domains of the GAG, we plan to test the current RNA aptamers against varying degrees of polymerization of the target GAGs to elucidate the smallest unit required for aptamer–GAG interaction. We would also like to use the binding information on computational studies to model the interaction and to predict the types of sequences that would generate high binding affinity aptamers to various GAGs.
Experimental Procedures
Preparation and Characterization of Unsulfated GAGs
HE33 and CH34 were respectively prepared from E. coli K5 (ATCC #23506) and recombinant E. coli K5 cultured on glucose. HA from Streptococcus zooepidemicus was provided by Professor Toshihiko Toida (Chiba University, Japan). More than 95% purity of each GAG sample is confirmed by nuclear magnetic resonance spectroscopy (Figure S1) and liquid chromatography mass spectrometry (LC–MS, Figures S2 and S3),33,34 which prove the high purity (HE >97%, heparin >99%, CH >95%, and hyaluronic acid >99%) of the GAG samples used in this study. Molecular weight of each GAG sample was determined by high-performance liquid gel permeation chromatography, according to the official monograph for heparin35 with minor modifications.36 Average molecular weight for HE, HA, and CH are 37.7, 125, and 36.4 kDa respectively.
Immobilization of GAGs on the Magnetic Microbeads
Unsulfated GAGs, HE, CH, and HA, were each covalently linked through their reducing ends to amine-functionalized magnetic microparticles (1.0 μm, herein simply referred to as “magnetic beads”) obtained from Chemicell GmbH (Berlin, Germany). More specifically, reductive amination was carried out by mixing 2 mg of GAG (1 mg in the case of CH) and 30 mg of amine-functionalized magnetic beads in 10 mL of dimethylsulfoxide that contained 15% glacial acetic acid (v/v) and reacted in the presence of sodium cyanoborohydride (5 mg) for 120 h at 70 °C. Sodium cyanoborohydride (5 mg) was replenished every 24 h. The reaction was stopped by decreasing the temperature to 25 °C. Then, the beads were washed exhaustively with 1 M NaCl to remove noncovalently associated polysaccharide. Reductive amination afforded immobilized GAGs, in an “end-on” orientation with attachment through their reducing ends, which will allow for larger surface area coverage and greater accessibility of GAG subunits and closely mimic the bacterial capsule and mammalian glycocalyx.
Next, to reduce nonspecific ionic interactions of the nucleic acid backbone with the beads, unreacted primary amines on the beads were blocked by converting to N-acetyl groups with acetic anhydride/water/tetrahydrofuran (v/v/v, 1/4/4). “Blank” magnetic beads utilized in the negative selection or as a negative control in confocal microscopy-based screening were subjected to only the blocking treatment. Amount of each beads-immobilized GAG was quantified (Table S6) by conducting the standard disaccharide analysis following GAG digestion using appropriate polysaccharide lyases.10,37
In Vitro RNA Aptamer Selection
An RNA library that contains 30-nt long degenerated sequences flanked by constant primer binding regions was obtained from TriLink Biotechnologies Inc. (see sequence in Table S4). To select the RNA aptamers targeting HE or CH GAGs, a 5 nmol RNA library (containing ∼3 × 1015 unique RNA molecules) was suspended in the binding buffer (25 mM Tris-HCl, pH 7.6, 100 mM NaCl, 5 mM MgCl2). For the initial round of selection, the library was incubated with 10 μL of GAG-coated magnetic beads for 2 h under shaking (200 rpm) before being thoroughly washed five times to remove the nonspecific-binding RNA. The resulting RNAs bound to the target GAG were recovered by reversing the RNA–GAG interaction by heating at 85 °C for 3 min. The solution containing the eluted RNAs was extracted and concentrated using phenyl/chloroform/isoamyl alcohol (v/v/v, 25/24/1) (PCA) extraction followed by ethanol precipitation. The recovered RNAs from the previous selection round were reverse-transcribed using the SuperScript III First-Strand Synthesis System (ThermoFisher Sci) and PCR-amplified to obtain DNA templates for making the RNA library for the subsequent selection using the MEGAscript T7 Transcription Kit (ThermoFisher Sci). Starting from the second selection round, before mixing the library with the target GAG as described above, the newly transcribed RNA library will be hybridized with the “negative-selection” DNA oligo mixture (Table S3), followed by RNase-H digestion to remove the “background-binding” RNAs, DNase digestion to remove the “negative-selection” DNA, and cleaned by phenol extraction and ethanol precipitation. For each GAG, 10 rounds of selection were conducted before the enriched RNAs were Sanger-sequenced for their identity. It should be noted that to enrich the RNA aptamers for those with high binding affinity, the RNA–GAG incubation conditions varied as the selection progressed: during rounds 1–4, the RNA library was incubated with 20 μL of GAG beads for 2 h with shaking at 200 rpm; for rounds 5–8, the RNA library was incubated with 10 μL of GAG beads for 1 h with shaking at 200 rpm; for rounds 9–10, the RNA library was incubated with 10 μL of GAG beads for only 30 min with shaking on an orbital shaker at 200 rpm.
Sequence Identification and Analysis
After the 10th round of selection, the enriched RNA molecules were RT-PCRed into double-stranded DNAs, which were subsequently cloned into the pCR2.1-TOPO vector using the TOP TA Cloning Kit (Invitrogen Life Technologies) and amplified in E. coli TOP10 cells. Colonies containing cDNAs of 51 HE-targeting and 62 CH-targeting RNAs were Sanger-sequenced by Genscript Biotech. All the sequences were further compiled and analyzed using the alignment algorithm embedded in Geneious (Biomatters Inc). Note that the purified plasmids containing interested cDNA inserts were kept and used in in vitro transcription to produce specific RNA molecules for the downstream binding and selectivity assays.
RNA–GAG Interaction and Selectivity Screening
For each candidate RNA aptamer, 20 μL of 1 μM (quantified by Quant-iT RiboGreen RNA Kit) RNA solution was mixed with 10 μL GAG beads or blank beads in 70 μL binding buffer and incubated in an Eppendrof tube at room temperature for 2 h with shaking at 200 rpm. Excess or unbound RNA molecules were then removed after washing five times using binding buffer. Then, SYBR Green II dye was added according to the manufacture’s protocol, and the interaction of RNA with GAG beads or blank beads was characterized by a Zeiss LSM 510META Spectral Confocal Microscope using 488 nm excitation laser and a 520 nm emission filter. The average fluorescence density of each interaction was quantified in ImageJ (NIH) by normalizing the average fluorescence intensity of the confocal micrographs to their ratio of the fluorescence area to total area. This therefore reflects the amount of GAG-bound RNA molecules. The same procedure was used to screen the binding specificity of the selected candidate HE and CH aptamers to HE, CH, and HA GAGs immobilized on the magnetic beads.
GAG Biotinylation
The carboxyl groups of each GAG were first activated with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysulfosuccinimide (sulfo-NHS) in order to biotinylate the GAGs. GAG (5 mg) was dissolved in 1 mL of 0.1 M 2-[morpholino]ethanesulfonic acid buffer, pH 4.7. EDC (0.5 mg) and sulfo-NHS (1.5 mg) were added and stirred at room temperature for 15 min. Next, concentrated (10×) phosphate buffered saline (PBS) buffer (0.1 M sodium phosphate and 0.15 M NaCl, pH 7.2) was added to adjust the pH to ∼7.0. Finally, amine-PEG3-biotin (2.5 mg) in 1 mL of PBS buffer was added to the activated GAGs and allowed to react for 10 h at room temperature. Postreaction, excess biotin molecules were removed by washing excessively with 1 M NaCl and then water using an Amicon Ultra-0.5 mL centrifugal filter [3 kDa molecular weight cut-off (MWCO)]. Successful GAG biotinylation was confirmed by NMR analysis (Figure S9).
Immobilization of Biotinylated GAG to a Microplate
Black 96-well streptavidin-coated microplate (ThermoFisher Sci) wells were washed three times using 200 μL Trisbuffered saline wash buffer (25 mM Tris, 150 mM NaCl, pH 7.2 containing 0.1% BSA, and 0.05% Tween-20 detergent). Next, a solution containing biotinylated GAG (100 μL of 0.05 mg/mL) was added and incubated at room temperature for 2 h with shaking at 200 rpm. Amine-PEG3-biotin was used as a negative control after being immobilized to a separate microplate. The amount of total GAG immobilized onto the microplate was quantified (Table S6) by disaccharide analysis using LC–MS as described below.
LC–MS-Based Disaccharide Analysis
Standard LC–MS-based disaccharide analysis was carried out to quantify the amount of GAG molecules immobilized to a microplate. For HE, the GAG was hydrolyzed by recombinant Flavobacterial heparin lyases I, II, and III,10 whereas CH and HA were hydrolyzed using CH lyase ABC, all expressed in E. coli.10 The enzymatic digests were then purified by Ultra-0.5 mL Centrifugal Filters (10 kDa MWCO) and lyophilized before the LC–MS analysis (below).
The dried samples were AMAC-labeled by adding 10 μL of 0.1 M AMAC in DMSO/acetic acid (v/v, 17/3) and incubating at room temperature for 10 min, followed by the addition of 10 μL of 1 M aqueous NaBH3CN and incubating for 1 h at 45 °C. The resulting samples were centrifuged at 13 200 rpm for 10 min. Finally, each supernatant was collected and stored in a light-resistant container at room temperature until analyzed by LC–MS.
LC was performed on an Agilent 1200 LC system at 45 °C using an Agilent Poroshell 120 ECC18 (2.7 μm, 3.0 × 50 mm) column with two mobile phases (A: 50 mM ammonium acetate aqueous solution, and B: methanol). The mobile phase passed through the column at a flow rate of 250 μL/min and a gradient of 0–12.5 min, 15–27.5% B; 12.5–13 min, 27.5–100% B; 13–16 min, 100% B; 16–16.2 min, 100–15% B. An Agilent 6300 ion trap equipped with an ESI source was coupled online. The full-scan (300–900Da) MS analysis was performed under the negative ionization mode with a skimmer voltage of −40.0 V, a capillary exit of −40.0 V, and a source temperature of 350 °C. Liquid nitrogen was used as the drying and nebulizing gas at a flow rate of 8 L/min and a pressure of 40 psi, respectively. To quantify the GAGs, unsaturated disaccharide standards of CS (0SCS-0, 4SCS-A, 6SCS-C, 2SCS, 2S4SCS-B, 2S6SCS-D, 4S6SCS-E, TriSCS), HA (0SHA), and HS (0SHS, NSHS, 6SHS, 2SHS, 2SNSHS, NS6SHS, 2S6SHS, TriSHS) purchased from Iduron, were run prior to the samples. Data analysis was performed using Agilent ChemSolution software, and the resulting total sugar for each GAG immobilized is listed in Table S6.
Binding Affinity (KD) Determination of Final RNA Aptamers
The binding affinity (quantified as KD) of the selected aptamers to their targets was determined using a constant amount of GAG over a range of RNA aptamer concentrations (0, 0.25, 0.5, 1, 2, 3, 4, and 4.5 μM). After adding RNA aptamer solution to the microplate wells with immobilized GAG, the mixture was shaken (200 rpm) for 2 h at room temperature. Postincubation, the microplate was washed three times (each shaking for 3 min) with 200 μL binding buffer. Then, 100 μL RiboGreen dye (200-fold dilution) was added to stain any RNA molecule bound to the GAG. The amount of RNA–GAG binding complex was determined by measuring the fluorescence intensity using a microplate reader (excitation: 480 nm; emission: 520 nm). Saturation curves were obtained by fitting the mean fluorescence intensity of aptamer–GAG complex to the range of aptamer concentrations. Nonlinear regression was used to determine KD values.
Acknowledgments
The authors thank RPI’s Center for Biotechnology and Interdisciplinary Studies staff for maintaining core instruments used for this work. The work was supported by Slezak Memorial Fellowship Award to M.K.; RPI faculty startup, CBIS facility award, and HT Materials gift fund to X.W.; National Institutes of Health grant HL125371 to R.J.L.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01853.
Additional information includes NMR spectra of GAGs and biotinylated GAGs, RNA aptamer sequence and alignment, confocal micrographs used for RNA–GAG binding and selectivity screening, and HE aptamer interactions with heparin (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Lindahl U.; Couchman J.; Kimata K.; Esko J. D.. Proteoglycans and Sulfated GLycosaminoglycans. In Essentials of Glycobiology, 3rd ed; Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., Seeberger P. H., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, 2017. [Google Scholar]
- Esko J. D.; Prestegard H.; Linhardt R. J.. Proteins that Bind Sulfated Glycosaminoglycans. In Essentials of Glycobiology, 3rd ed; Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., Seeberger P. H., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, 2017. [Google Scholar]
- Linhardt R. J.; Toida T. Role of Glycosaminoglycans in Cellular Communication. Acc. Chem. Res. 2004, 37, 431–438. 10.1021/ar030138x. [DOI] [PubMed] [Google Scholar]
- Linhardt R. J. 2003 Claude S. Hudson award address in carbohydrate chemistry. Heparin: Structure and activity. J. Med. Chem. 2003, 46, 2551–2564. 10.1021/jm030176m. [DOI] [PubMed] [Google Scholar]
- Ly M.; Leach F. E.; Laremore T. N.; Toida T.; Amster I. J.; Linhardt R. J. The proteoglycan bikunin has a defined sequence. Nat. Chem. Biol. 2011, 7, 827–833. 10.1038/nchembio.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cress B. F.; Englaender J. A.; He W.; Kasper D.; Linhardt R. J.; Koffas M. A. G. Masquerading microbial pathogens: capsular polysaccharides mimic host-tissue molecules. FEMS Microbiol. Rev. 2014, 38, 660–697. 10.1111/1574-6976.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamhi E.; Joo E. J.; Dordick J. S.; Linhardt R. J. Glycosaminoglycans in infectious disease. Biol. Rev. 2013, 88, 928–943. 10.1111/brv.12034. [DOI] [PubMed] [Google Scholar]
- Li J.-P.; Kusche-Gullberg M. Heparan Sulfate: Biosynthesis, Structure, and Function. Int. Rev. Cell Mol. Biol. 2016, 325, 215–273. 10.1016/bs.ircmb.2016.02.009. [DOI] [PubMed] [Google Scholar]
- Conrad H. E.Nitrous Acid Degradation of Glycosaminoglycans. Current Protocols in Molecular Biology; Wiley, 2001; Chapter 17, Unit17.22A. [DOI] [PubMed] [Google Scholar]
- Linhardt R. J.Analysis of Glycosaminoglycans with Polysaccharide Lyases. Current Protocols in Molecular Biology; Wiley, 2001; Chapter 17, Unit 17.13B. [DOI] [PubMed] [Google Scholar]
- Winter G.; Milstein C. Man-made antibodies. Nature 1991, 349, 293–299. 10.1038/349293a0. [DOI] [PubMed] [Google Scholar]
- Pirner K.; Rascu A.; Nürnberg W.; Rubbert A.; Kalden J. R.; Manger B. Evidence for direct anti-heparin-sulphate reactivity in sera of SLE patients. Rheumatol. Int. 1994, 14, 169–174. 10.1007/bf00579703. [DOI] [PubMed] [Google Scholar]
- Hayes A. J.; Hughes C. E.; Caterson B. Antibodies and immunohistochemistry in extracellular matrix research. Methods 2008, 45, 10–21. 10.1016/j.ymeth.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Attreed M.; Desbois M.; van Kuppevelt T. H.; Bülow H. E. Direct visualization of specifically modified extracellular glycans in living animals. Nat. Methods 2012, 9, 477–479. 10.1038/nmeth.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits N. C.; Lensen J. F. M.; Wijnhoven T. J. M.; ten Dam G. B.; Jenniskens G. J.; van Kuppevelt T. H. Phage Display-Derived Human Antibodies Against Specific Glycosaminoglycan Epitopes. Methods Enzymol. 2006, 416, 61–87. 10.1016/s0076-6879(06)16005-x. [DOI] [PubMed] [Google Scholar]
- Ellington A. D.; Szostak J. W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Tuerk C.; Gold L. Systematic Evolution of LIgands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 1990, 249, 505–510. 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Sassanfar M.; Szostak J. W. An RNA motif that binds ATP. Nature 1993, 364, 550–553. 10.1038/364550a0. [DOI] [PubMed] [Google Scholar]
- Wincott F. E.Strategies for oligoribonucleotide synthesis according to the phophoramidite method. Current Protocols in Nucleic Acid Chemistry; Wiley, 2001; Chapter 3, Unit 3.5. [DOI] [PubMed] [Google Scholar]
- Beckert B.; Masqiuda B.. Synthesis of RNA by in Vitro Transcription; Nielsen H., Ed.; Humana Press, 2011; Vol. 703. [DOI] [PubMed] [Google Scholar]
- Gao S.; Zheng X.; Jiao B.; Wang L. Post-SELEX optimization of aptamers. Anal. Bioanal. Chem. 2016, 408, 4567–4573. 10.1007/s00216-016-9556-2. [DOI] [PubMed] [Google Scholar]
- Jolly P.; Damborsky P.; Madaboosi N.; Soares R. R. G.; Chu V.; Conde J. P.; Katrlik J.; Estrela P. DNA aptamer-based sandwich microfluidic assays for dual quantification and multi-glycan profiling of cancer biomarkers. Biosens. Bioelectron. 2016, 79, 313–319. 10.1016/j.bios.2015.12.058. [DOI] [PubMed] [Google Scholar]
- Patel D. J.; Suri A. K.; Jiang F.; Jiang L.; Fan P.; Kumar R. A.; Nonin S. Structure, Recognition and Adaptive Binding in RNA Aptamer Complexes. J. Mol. Biol. 1997, 272, 645–664. 10.1006/jmbi.1997.1281. [DOI] [PubMed] [Google Scholar]
- Yang Q.; Goldstein I. J.; Mei H.-Y.; Engelke D. R. DNA ligands that bind tightly and selectively to cellobiose. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 5462–5467. 10.1073/pnas.95.10.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.; Lee B.-R.; Cho B.-K.; Kim J.-H.; Kim B.-G. In Vitro Selection of Sialic Acid Specific RNA Aptamer and Its Application to the Rapid Sensing of Sialic Acid Modified Sugars. Biotechnol. Bioeng. 2013, 110, 905–913. 10.1002/bit.24737. [DOI] [PubMed] [Google Scholar]
- Yang K.-A.; Barbu M.; Halim M.; Pallavi P.; Kim B.; Kolpashchikov D. M.; Pecic S.; Taylor S.; Worgall T. S.; Stojanovic M. N. Recognition and sensing of low-epitope targets via ternary complexes with oligonucleotides and synthetic receptors. Nat. Chem. 2014, 6, 1003–1008. 10.1038/nchem.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H.; Fan X.; Ni Z.; Lis J. T. Evolutionary dynamics and population control during in vitro selection and amplification with multiple targets. RNA 2002, 8, 1461–1470. 10.1017/s1355838202029941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis R.; Schnaar R.. R-Type Lectins. In Essentials of Glycobiology, 3rd ed; Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., Seeberger P. H., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, 2017. [Google Scholar]
- Wang Z.; Yang B.; Zhang Z.; Ly M.; Takieddin M.; Mousa S.; Liu J.; Dordick J. S.; Linhardt R. J. Control of the heparosan N-deacetylation leads to an improved bioengineered heparin. Appl. Microbiol. Biotechnol. 2011, 91, 91–99. 10.1007/s00253-011-3231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blind M.; Blank M. Aptamer Selection Technology and Recent Advances. Mol. Ther.--Nucleic Acids 2015, 4, e223 10.1038/mtna.2014.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena M. d. l.; Dufour D.; Gallego J. Three-way RNA junctions with remote tertiary contacts: a recurrent and highly versatile fold. RNA 2009, 15, 1949–1964. 10.1261/rna.1889509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter E. B.; Polaski J. T.; Morck M. M.; Batey R. T. Recurrent RNA motifs as scaffolds for genetically encodable small-molecule biosensors. Nat. Chem. Biol. 2017, 13, 295–301. 10.1038/nchembio.2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Zhang Z.; McCallum S. A.; Linhardt R. J. Nuclear magnetic resonance quantification for monitoring heparosan K5 capsular polysaccharide production. Anal. Biochem. 2010, 398, 275–277. 10.1016/j.ab.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W.; Fu L.; Li G.; Andrew Jones J.; Linhardt R. J.; Koffas M. Production of chondroitin in metabolically engineered E. coli. Metab. Eng. 2015, 27, 92–100. 10.1016/j.ymben.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Mulloy B.; Heath A.; Shriver Z.; Jameison F.; Al Hakim A.; Morris T. S.; Szajek A. Y. USP compendial methods for analysis of heparin: chromatographic determination of molecular weight distributions for heparin sodium. Anal. Bioanal. Chem. 2014, 406, 4815–4823. 10.1007/s00216-014-7940-3. [DOI] [PubMed] [Google Scholar]
- Bhaskar U.; Li G.; Fu L.; Onishi A.; Suflita M.; Dordick J. S.; Linhardt R. J. Combinatorial one-pot chemoenzymatic synthesis of heparin. Carbohydr. Polym. 2015, 122, 399–407. 10.1016/j.carbpol.2014.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J. G.; Gupta M.; Xu Y.; Akella S.; Liu J.; Dordick J. S.; Linhardt R. J. Toward an Artificial Golgi: Redesigning the Biological Activities of Heparan Sulfate on a Digital Microfluidic Chip. J. Am. Chem. Soc. 2009, 131, 11041–11048. 10.1021/ja903038d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.