

Abstract
Changes in cis-regulatory regions are thought to play a major role in the genetic basis of adaptation. However, few studies have linked cis-regulatory variation with adaptation in natural populations. Here, using a combination of exome and RNA-seq data, we performed expression quantitative trait locus (eQTL) mapping and allele-specific expression analyses to study the genetic architecture of regulatory variation in wild house mice (Mus musculus domesticus) using individuals from five populations collected along a latitudinal cline in eastern North America. Mice in this transect showed clinal patterns of variation in several traits, including body mass. Mice were larger in more northern latitudes, in accordance with Bergmann's rule. We identified 17 genes where cis-eQTLs were clinal outliers and for which expression level was correlated with latitude. Among these clinal outliers, we identified two genes (Adam17 and Bcat2) with cis-eQTLs that were associated with adaptive body mass variation and for which expression is correlated with body mass both within and between populations. Finally, we performed a weighted gene co-expression network analysis (WGCNA) to identify expression modules associated with measures of body size variation in these mice. These findings demonstrate the power of combining gene expression data with scans for selection to identify genes involved in adaptive phenotypic evolution, and also provide strong evidence for cis-regulatory elements as essential loci of environmental adaptation in natural populations.
Understanding the genetic basis of adaptation is a major goal in evolutionary biology. Cis-regulatory mutations, which can change the expression of proximal genes, have long been predicted to be important targets for adaptive phenotypic evolution (King and Wilson 1975; Wray 2007; Stern and Orgogozo 2008; Wittkopp and Kalay 2012). One reason for this is that cis-regulatory mutations may have fewer deleterious pleiotropic effects than protein-coding changes. While protein-coding mutations may affect protein products across tissues and developmental stages, cis-regulatory mutations can affect the expression of genes in spatially and temporally specific ways. In apparent support of this idea, several studies have identified positive selection on noncoding regions (e.g., Jenkins et al. 1995; Crawford et al. 1999; Kohn et al. 2004; Andolfatto 2005; MacDonald and Long 2005; Holloway et al. 2007; Jeong et al. 2008; Torgerson et al. 2009) and an important role for noncoding variation in local adaptation (e.g., Jones et al. 2012; Fraser 2013).
Despite the accumulating evidence that regulatory loci play an important role in adaptive evolution, there are still only a handful of cases where cis-regulatory mutations have been linked to ecologically important traits. Among the best examples are adaptive coat color differences in deer mice (Linnen et al. 2013), the ability to digest lactose in humans (Tishkoff et al. 2007), and pelvic reduction in sticklebacks (Chan et al. 2010). Most examples of adaptive gene expression have been identified through candidate gene approaches, which typically favor traits for which components of a pathway are already known and the genetic basis of the trait is relatively simple. However, most traits are influenced by many loci of small to modest effect. Thus, identifying genetic variants associated with adaptation at complex traits is key to understanding the genetic basis of adaptation.
One avenue for linking adaptive non-coding variation to either molecular or organismal phenotypes is through gene expression. In expression quantitative trait loci (eQTLs) mapping, gene expression levels are tested for associations with genetic markers to identify variants that contribute to expression phenotypes. Expression quantitative trait mapping is an effective method for identifying regulatory variants because gene expression is frequently influenced by nearby cis-eQTLs (Nica and Dermitzakis 2013). Cis-eQTLs have been successfully detected with small sample sizes (Montgomery and Dermitzakis 2011; Tung et al. 2015) and in wild individuals from natural populations (Tung et al. 2015). Combining eQTL mapping with genomic scans for selection can be a powerful method for identifying the gene targets of adaptive genetic variation (Fraser 2013; Ye et al. 2013) and potentially linking this variation to adaptive organismal phenotypes.
House mice (Mus musculus domesticus) provide a useful model for studying the genetic basis of adaptation. House mice are an important biomedical model and have a distribution that mirrors that of human populations (Phifer-Rixey and Nachman 2015). In the eastern United States, house mice show latitudinal variation consistent with local adaptation. Mice collected at northern latitudes are heavier than mice at southern latitudes, and their progeny also show differences in a common laboratory environment, indicating that this difference is genetic (Lynch 1992; Phifer-Rixey et al. 2018). This observation conforms to the classic ecogeographic observation known as Bergmann's rule that animals in colder climates have larger mass to reduce heat loss (Bergmann 1847). While Bergmann's rule has been observed in many groups, including humans (Ashton et al. 2000; Ruff 2002; Foster and Collard 2013), no study so far has linked this pattern to variation at specific genes. Consistent with energetic adaptation of mice from eastern North America, laboratory strains founded from northern and southern locations also show differences in aspects of blood chemistry, including leptin, glucose, and triglyceride levels (Phifer-Rixey et al. 2018).
Recent work with these populations identified hundreds of genes with environmental associations in North American (Phifer-Rixey et al. 2018). Here, we combine a genomic scan for selection with expression quantitative trait mapping to identify regulatory variants that contribute to gene expression differences and show signals of selection in these populations, identifying two strong candidate genes for adaptive phenotypic variation. To our knowledge, this study represents the first case where genomic scans have been combined with eQTL mapping to identify regulatory variants in natural populations that underlie an adaptive organismal phenotype.
Results
Cis-regulatory variation in wild house mice
To characterize regulatory variation in wild mice, we sequenced liver transcriptomes from 50 mice collected from five populations along a latitudinal transect on the east coast of North America (Fig. 1; Supplemental Table S1; Supplemental File S1). Mice were collected from 29°N to 44°N latitude. Liver was collected in RNAlater, and body mass and length were recorded for each individual. From these individuals, we produced a total of ∼1.2 billion RNA-seq reads with an average of 15,473,949 uniquely mapped exonic reads per sample, which were used to quantify gene-wise mRNA abundance (hereafter, gene expression). We also analyzed DNA sequence data generated from exome-capture of the same individuals (Phifer-Rixey et al. 2018). Exome and RNA-seq data were used to identify variants segregating in M. m. domesticus (see Methods).
Figure 1.
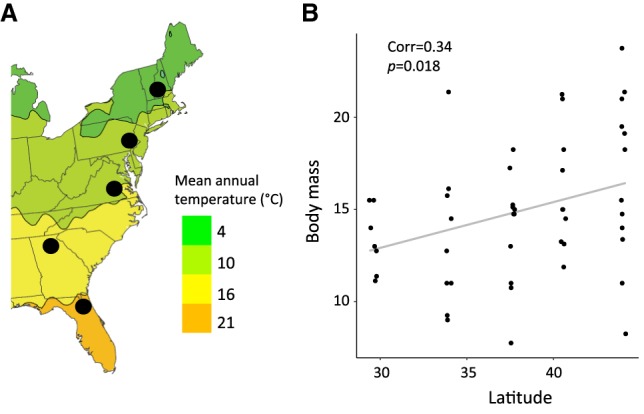
(A) Sampling locations along the east coast of North America (climate map obtained from NOAA, National Weather Service). (B) Consistent with Bergmann's rule, body mass in mice increases with increasing latitude (Pearson's correlation = 0.34, P = 0.018) (see Supplemental Table S9; Supplemental Methods).
We identified cis-regulatory variation using two complementary approaches, expression quantitative trait loci mapping and allele-specific expression (ASE). To identify cis-eQTLs, we tested for associations between variants within 200 kb of a gene and expression level using a linear mixed model. Variants near a gene are more likely to act in cis to affect gene expression. Cis-eQTLs typically have larger effect sizes than trans-eQTLs, making them easier to detect in small sample sizes (Montgomery and Dermitzakis 2011). After filtering, a total of 406,999 variants were identified using exome data and tested for associations with expression at 13,080 genes. We identified cis-eQTLs for 849 of these genes (6.5% of genes surveyed). Reflecting the probe set, the majority of cis-eQTLs were identified in gene bodies (57%) and introns (18%) (Supplemental Fig. S1).
Allele-specific expression (i.e., differences in expression between parental alleles) can also be used to infer epigenetic or genetic variation acting in cis (Cowles et al. 2002). As the two parental alleles are exposed to the same trans-acting environment within an individual, differences in expression at heterozygous sites can be used to infer cis-regulatory variation. A total of 28,234 exonic heterozygous sites, corresponding to 6738 genes, could be tested for ASE. Across all individuals, we found evidence for ASE for 442 genes at a false discovery rate of 5% (6.7% of genes surveyed) (Supplemental Table S2).
In investigating the power to detect cis-regulatory variation, we found that cis-eQTLs were more likely to be detected when SNP density is higher near and within the gene of interest (Mann-Whitney U test, P < 2.2 × 10−16) (Supplemental Fig. S2). We were more likely to detect ASE for genes with higher expression and higher SNP density (Mann-Whitney U test, P = 3.1 × 10−11 and P < 2.2 × 10−16, respectively) (Supplemental Figs. S2, S3). While differences in the power to detect ASE and cis-eQTLs can lead to the identification of different gene sets, we found significant overlap between the gene sets identified with these analyses (hypergeometric test, P = 5 × 10−6) (Supplemental Table S2).
Evidence for adaptive regulatory variation
To assess whether the regulatory variation documented above underlies adaptive differences among populations, we studied sequence and gene expression variation along a latitudinal cline (Fig. 1A). Clinal patterns of variation can reflect local adaptation as a response to spatially varying selection (Endler 1977). Regulatory variants with clinal frequencies that mediate clinal patterns of gene expression would be strong candidates for adaptive regulatory evolution. To identify such variants, we searched for cases where (1) gene expression is clinal, (2) gene expression is associated with a cis-eQTL, and (3) allele frequencies of the cis-eQTL vary clinally (Fig. 2). While geographic clines may alternatively be explained by isolation by distance, there is no evidence for isolation by distance for these populations (see Supplemental Methods).
Figure 2.

Overlap between genomic scans identifies regulatory variants that are candidates for clinal adaptation. (A) The LFMM |z-scores| for each SNP vs. chromosome position. SNPs with |z-scores| > 2 were considered clinal outliers. (B) Manhattan plot of cis-eQTL. Shown in red are significant SNPs. (C) Manhattan plot of gene starting position versus the correlation between gene expression and latitude. Points labeled in orange are genes for which expression is significantly correlated with latitude (P < 0.05). On the outside are ideograms with the location of genes for which these three signals (A–C) overlap. Figure created with Circos (Krzywinski et al. 2009).
To identify clinal patterns of gene expression, we tested for correlations between latitude and expression levels in the liver transcriptomes of the 50 wild individuals. We identified 1488 genes for which expression was significantly correlated with latitude (P < 0.05), 132 of which were associated with a cis-eQTL (Fig. 2). We also tested for differential expression between the most northern population (New Hampshire/Vermont) and the most southern population (Florida) and identified 458 genes with differential expression between the ends of the cline (Supplemental Fig. S4), 48 of which were associated with a cis-eQTL (at q < 0.1) (Supplemental Table S3).
To connect these patterns to clinal sequence variation, a genome scan using the program Latent Factor Mixed Models (LFMM) was performed to test for correlations between latitude and genetic variation while accounting for population structure (see Methods; Frichot et al. 2013). For this study, LFMM has an advantage over other methods because it does not assume a specific demographic model but still accounts for demographic history by estimating genome-wide covariance among allele frequencies. We focused on SNPs in the 5% tail of the distribution and considered these clinal outliers (|z-scores| > 2) (Fig. 2A). Blocks of linkage disequilibrium (LD) (Supplemental Fig. S5; Gabriel et al. 2002) were then inferred to identify colocalization between outlier SNPs and cis-eQTLs. Of cis-eQTLs that fell within the same LD block as an outlier, 17 were associated with genes that also show significant clinal patterns of gene expression (Table 1; Fig. 2; Supplemental Table S4). When comparing the latitudinal extremes, average estimates of Fst for these candidate loci were significantly higher than that of the full list of loci (full list average Fst = 0.10, candidate average Fst = 0.34; permutation test, P = 0.0014). Eight of these genes were also significantly differentially expressed between the ends of the cline (Supplemental Table S5). These 17 genes represent cases where cis-eQTLs contribute to expression differences between populations and show signals of local adaptation, making them strong candidates for adaptive regulatory variation.
Table 1.
cis-eQTLs that colocalize or are within the same LD block as a clinal outlier that also show expression changes correlated with latitude

Linking adaptive regulatory variation to specific traits
The liver plays a central role in metabolic processes in the body, and regulatory changes in this tissue may contribute to latitudinal variation in traits related to metabolism. Body mass varies clinally (Fig. 1A), and lab-born progeny from populations at the ends of the transect also show differences in blood glucose, triglyceride, adiponectin, and leptin levels (Lynch 1992; Phifer-Rixey et al. 2018). Four of the 17 candidate genes identified as strong candidates also have mutant phenotypes related to body weight and metabolism. Laboratory mutants for Cox7c and Hmgb1 are associated with changes in glucose levels (Blake et al. 2017), and mutants for Adam17 and Bcat2 are also associated with changes in body mass (Wu et al. 2004; She et al. 2007; Gelling et al. 2008; Blake et al. 2017), glucose (Serino et al. 2007; She et al. 2007; Blake et al. 2017), leptin (She et al. 2007; Gelling et al. 2008), and adiponectin levels (Serino et al. 2007; Blake et al. 2017). Another gene identified in this analysis, Iah1, transcriptionally regulates genes with important roles in lipid metabolism and triglyceride synthesis and falls under a QTL for fatty liver in mice (Kobayashi et al. 2016; Suzuki et al. 2016).
Adam17 and Bcat2 are candidates for adaptive differences in body mass
While knockout models can provide a link between genotypes and putative phenotypes, these models may not reflect the phenotypic consequences of mutations found in natural populations (Palopoli and Patel 1996). Changes in body weight are also among the most common effects of gene knockouts in mice and may often reflect downstream consequences of other phenotypic changes (Reed et al. 2008; White et al. 2013). While identifying the genetic basis of complex adaptive traits is challenging, gene expression provides an intermediate phenotype that may link sequence variants to organismal traits. To connect adaptive variation in body mass in these populations to genetic variation, we asked whether body mass differences were associated with gene expression differences in the set of candidate genes (Table 1). Since latitude and body mass covary in this sample (Fig. 1B), we controlled for latitude by regressing it out as a variable. We identified two genes, a disintegrin and metallopeptidase domain 17 (Adam17) (Fig. 3A–F) and branched chain amino acid transaminase 2 (Bcat2), for which expression was significantly correlated with body mass, after accounting for latitude as a covariable (Pearson's correlation, Adam17: P = 4.6 × 10−4, R2 = 0.22; Bcat2: P = 4.5 × 10−3, R2 = 0.17) (see also Supplemental Table S6; Supplemental Fig. S6). To further account for the possible confounding effects of population structure, we also looked at the correlation between expression level and body mass within each of the five populations. Replicating the pattern seen across populations, Adam17 expression was negatively associated with body mass in four of the five populations, and Bcat2 expression was positively associated with body mass in four of the five populations (Supplemental Figs. S7, S8). Despite a lack of power for within-population comparisons, the association between Adam17 expression and body mass was significant in New Hampshire/Vermont (Pearson's correlation, P = 3.5 × 10−3) and the association between Bcat2 expression and body mass was significant in Pennsylvania (Pearson's correlation, P = 0.03) and Georgia (Pearson's correlation, P = 1.8 × 10−3).
Figure 3.

Adam17 is a candidate for adaptive differences in body mass among mice in eastern North America. (A) Expression of Adam17 is correlated with latitude (P = 0.032, Pearson's correlation = −0.30). Sex was not a significant predictor of Adam17 expression. (B) A SNP at Chr 12: 21,332,631 was identified as a cis-eQTL for Adam17. (C) Allele frequencies of Chr 12: 21,332,631 in five populations. (D) The LFMM |z-scores| for sites on Chromosome 12 versus position. Points above the red line were considered clinal outliers in this study. The red box represents the peak in which Chr 12: 21,332,631 is found. (E) Nearby outlier SNPs in LD with Chr 12: 21,332,631. Correlations (r2, %) are given in each block. The z-scores for each site's association with latitude are given in parentheses. (F) Adam17 expression is significantly associated with body mass when controlling for latitude (Pearson's correlation, P = 4.6 × 10−4, R2 = 0.22). (G) Genotype at Chr 12: 21,332,631, the cis-eQTL for Adam17, significantly trends with body size when latitude is controlled for (Cochran-Armitage trend test, P = 0.034).
The cis-eQTLs for Adam17 and Bcat2 explain 34% and 29.7% of the variance in expression for these genes, respectively. Genotypes at these sites were also associated with differences in body mass (Mann-Whitney U test, Bcat2: TT > CC, P = 0.024; Adam17: CC > TT, P = 0.036) (Supplemental Fig. S9). Again, covariation between latitude and body mass can confound relationships between body mass and candidate genes. After regressing latitude from body mass to control for covariation between these variables, the Adam17 cis-eQTL was significantly associated with body mass (Cochran-Armitage trend test, P = 0.034) (Fig. 3G), although the Bcat2 cis-eQTL was not (Cochran-Armitage trend test, P = 0.14). The Adam17 and Bcat2 cis-eQTLs explain an estimated 8.35% and 1.51% of the variation in body mass, respectively. These estimates should be treated as approximations since they may be influenced by (1) unmeasured environmental differences between populations, (2) population structure (even when population structure is accounted for using principle components, as was done here) (see Browning and Browning 2011; Dandine-Roulland et al. 2016), (3) imperfect linkage disequilibrium between the surveyed SNPs and causal variants (Wray et al. 2013), and small sample size (Xu 2003). Nonetheless, it is likely that the effect size for the Adam17 cis-eQTL is large compared to what is seen in most human GWAS for complex traits (Stranger et al. 2011). Large-effect mutations may be favored in situations where populations are initially far from an optimum (Orr 1998; Dittmar et al. 2016). For example, variation at one gene accounts for a >2-kg weight difference between Europeans and Inuits (Fumagalli et al. 2015), and a single IGF1 allele in dogs accounts for 15% of variance in dog skeletal size (Sutter et al. 2007). House mice in this transect descended from mice in western Europe adapted to a Mediterranean climate and thus likely experienced strong selection pressures in a novel environment, potentially favoring some mutations of large effect.
To investigate regulatory variation at Adam17 in western Europe, we retrieved available liver RNA-seq and genomic data from European mice (Harr et al. 2016). We found that the Adam17 cis-eQTL is segregating within European populations (Supplemental Fig. S10A) and is significantly associated with liver expression in European individuals (P = 3.2 × 10−6) (Supplemental Fig. S10B; see Supplemental Methods). This suggests that adaptation by the large-effect regulatory variation at Adam17 in the United States is a product of selection on standing genetic variation.
Notably, Adam17 and Bcat2 are the two candidate genes from Table 1 with known lab mouse mutants that affect body mass (Wu et al. 2004; She et al. 2007; Gelling et al. 2008; Blake et al. 2017). Bcat2 encodes a protein that catalyzes the first step of branched-chain amino acid (BCAA) metabolism, which affects metabolism and body mass in humans and rodents (Newgard et al. 2009). Adam17 encodes a protein that regulates several signaling pathways. Adult Adam17 heterozygous and null mutants show differences in metabolic phenotypes including body mass, susceptibility to diet-induced obesity, and energy homeostasis (Serino et al. 2007; Gelling et al. 2008). ADAM17 and its physiological inhibitor, TIMP3, have also been reported to be involved in glucose homeostasis and adipose, hepatic, and vascular inflammation in both genetic and nutritional models of obesity in mice (Fiorentino et al. 2010; Menghini et al. 2012; Matsui et al. 2014). In addition to its association with body mass and metabolism in mice, in humans variation at ADAM17 has been linked to differences in body weight, BMI, waist circumference, and obesity risk (Junyent et al. 2010) and shows signatures of selection (Pickrell et al. 2009; Parnell et al. 2010; Fumagalli et al. 2011).
One target of ADAM17 activity is the epidermal growth factor receptor (EGFR) signaling pathway (Lee et al. 2003). Phenotypes observed in mice with mutant EGF receptors (including changes in body weight [Blake et al. 2017]) suggest that changes in EGFR signaling as a consequence of deficient ADAM17 activity may contribute to the metabolic phenotypes seen in Adam17 mutants (Gelling et al. 2008). We tested for an overrepresentation of genes in the EGFR signaling pathway in the set of genes with clinal expression by annotating genes to pathways using the PANTHER database (Thomas et al. 2003). We saw a 1.57-fold enrichment of genes in this pathway compared to a background set of genes expressed in the liver (hypergeometric test, P = 0.018). We also find that the gene that encodes the only known physiological inhibitor of Adam17, Timp3 (Le Gall et al. 2010), is differentially expressed between the northern and southern populations (q = 0.09) (Supplemental Fig. S11) and has expression that is correlated with that of Adam17 (Pearson's correlation, P = 0.02, R2 = 0.09) (Supplemental Fig. S11). Unlike Adam17, Timp3 expression is not associated with body mass (Pearson's correlation, P = 0.054), although our sample size may not be sufficient to detect an association.
The data above clearly suggest that regulatory variation at Adam17 and Bcat2 underlies adaptive differences in body mass, but they do not identify the specific causal mutations. To identify candidate casual mutations, we used annotations from the mouse ENCODE project (Mouse ENCODE Consortium et al. 2012) to search for putative regulatory elements near the Adam17 and Bcat2 cis-eQTLs. The Adam17 cis-eQTL is in LD with SNPs through a proximal enhancer and in the Adam17 promoter, both of which are active in the livers of adult mice. Low-coverage whole-genome data show that there are variants segregating within this enhancer in these populations (Supplemental Figs. S12, S13; whole genome data from Phifer-Rixey et al. 2018). Two of the Adam17 promoter variants are also clinal outliers (Supplemental Fig. S14). The Bcat2 cis-eQTL is within an intronic region and is not in LD with annotated regulatory elements that are active in liver tissue.
Expression modules are correlated with body size variation in natural populations of house mice
Finally, we used a gene co-expression network approach to identify biologically related gene sets associated with phenotypic variation in these populations. Weighted Gene Co-expression Network Analysis (WGCNA) was used to identify groups of genes with highly correlated expression, called co-expression modules (see Methods; Langfelder and Horvath 2008). Expression modules were assigned for male and female mice separately, and then male-female consensus modules were created to identify co-expression patterns shared across sexes.
Co-expression modules were then tested for correlations with measures of body size (Supplemental Figs. S15–S17). Five expression modules in males and five expression modules in females were correlated with trait variation (Supplemental Fig. S18). Trait-associated modules were enriched for a number of Gene Ontology (GO) categories compared to the background set of genes expressed in the liver, including growth factor binding (q = 5.3 × 10−8) and lipid metabolic process (q = 1.2 × 10−2). None of the male-female consensus modules were significantly correlated with organismal traits, indicating that associations between co-expression modules and traits are sex-specific (Supplemental Fig. S17).
Focusing on the modules with the highest trait correlations (royalblue module in females, corr = 0.92, P = 2 × 10−8 and black module in males, corr = 0.8, P = 5 × 10−8, for body mass index), we annotated genes with mutant phenotypes collected from Mouse Genome Informatics (MGI) (Blake et al. 2017). Supporting the association between these expression modules and phenotypic variation, we found that many of the genes with high connectivity in these modules have mutant phenotypes related to body size or metabolism (Fig. 4). For example, the most connected gene in the female royalblue module is Nr2c2. Mutant phenotypes for Nr2c2 include changes in eating behavior, energy homeostasis, body mass, size, and blood chemistry. Similarly, highly connected genes in the male black module (e.g., Col3a1, Col1a1, Col1a2, Col5a2, Sparc, Bcam, Fstl1, Igfbp5, Cpe, Cav1, Lamc1, Ltbp3, Krt7) show mutant phenotypes related to body mass and body size. Four of these genes (Adamts2, Col1a1, Col1a2, Sparc) were also identified as hub genes in the module most highly correlated with mouse body weight in another study utilizing an F2 laboratory cross (Ghazalpour et al. 2006).
Figure 4.

Visualization of the most connected genes in the female “royalblue” (A) and the male “black” (B) co-expression modules with VisANT (Hu et al. 2008). The royalblue module is associated with BMI (P = 2 × 10−8) and body length variation (P = 6 × 10−6). The black module is associated with BMI (P = 5 × 10−8), body mass (P = 0.001), and body length variation (P = 3 × 10−10). Blue circles represent genes for which we identified a cis-eQTL that explains a component of expression variation. Circles with black borders are genes with mutant phenotypes related to body size or metabolism. Phenotype information was collected from MGI (Blake et al. 2017).
Finally, we used the co-expression data set to identify regulatory variation within modules associated with body size. Within the body size-associated modules (Supplemental Fig. S18), we associated 189 genes with a cis-eQTL, including several highly connected genes in the sex-specific modules with the highest trait correlations (Fig. 4). As in the previous analysis, we then searched for genes with a cis-eQTL that colocalized with a clinal sequence variant. We identified 15 genes with clinally varying cis-eQTLs in the body size-associated modules (Supplemental Table S7). We found that gene expression for four of these 15 genes was significantly correlated with BMI in one sex (females: Ube2q2, P = 0.0002; 3110082I17Rik, P = 0.0027; Cep85, P = 0.017; males: Pygb P = 0.035). Cis-eQTLs associated with these genes were not significantly associated with BMI; however, our study is also underpowered for identifying sex-specific associations. The correlation between gene expression and BMI and the presence of clinal cis-eQTLs make these genes of interest for future study.
Discussion
Identifying loci and genes that underlie adaptive variation within and between populations is a major goal in evolutionary biology. One method used to identify such variants is genomic scans for selection. While many genomic scans attempt to link sequence variants to phenotypes through gene annotations and knockout models, most fail to connect genotypes to phenotypes in natural populations. Here, we used expression data from natural populations of house mice collected along an environmental gradient to link regulatory variation at two genes (Adam17 and Bcat2) with body mass variation. We have linked these genes to body mass variation by (1) associating cis-eQTLs with the expression of Adam17 and Bcat2, (2) associating the Adam17 and Bcat2 cis-eQTLs with body mass variation, and (3) associating the expression of these two genes with body mass variation. Supporting the association we see between these genes and body mass, mutant alleles for Adam17 and Bcat2 in laboratory mice are associated with changes in body mass and metabolism (Wu et al. 2004; Serino et al. 2007; She et al. 2007; Gelling et al. 2008; Blake et al. 2017). These two genes account for a substantial proportion of phenotypic variation in body mass among the mice studied here, with large effect sizes compared to those measured in GWAS for most complex traits. For traits under stabilizing selection within populations (as in virtually all human GWAS), effect sizes are expected to be much smaller than in comparisons between populations experiencing strong divergent selection, as is the case here. The effect size of mutations underlying traits under stabilizing selection within populations is expected to be smaller than the effect sizes of mutations in the early stages of an adaptive walk (Orr 1998; Remington 2015).
In addition to identifying regulatory variation at specific genes associated with body mass, we also used a systems biology approach to identify co-expression patterns associated with body size variation in wild mice. Gene co-expression networks capture biologically relevant relationships between genes that can be useful for understanding gene functions and interactions. Here, we have used this information to characterize co-expression modules that were associated with body size and identified regulatory variation within these co-expressed gene sets that may play a role in body size variation.
The tendency for body size to increase with latitude (i.e., Bergmann's rule) has been documented in many species, including humans (Ashton et al. 2000; Ruff 2002; Foster and Collard 2013), and reflects an evolved response to differences in temperature (Bergmann 1847). In humans, many candidate genes for metabolic disorders, such as obesity, also show evidence of climatic adaptation (Hancock et al. 2008). In humans, both ADAM17 and BCAT2 have been implicated in metabolic disease (Arribas and Esselens 2009; Newgard et al. 2009; Junyent et al. 2010; Menghini et al. 2013), and variation at ADAM17 has been identified in genome scans for selection (Pickrell et al. 2009; Parnell et al. 2010; Fumagalli et al. 2011) in addition to its association with body weight and obesity risk (Junyent et al. 2010).
Finally, this study provides evidence for the role of cis-regulatory variation in environmental adaptation in natural populations. While cis-regulatory variation has long been hypothesized to play a major role in adaptive phenotypic evolution, connecting regulatory variation with adaptive organismal phenotypes remains tricky. Combining eQTL mapping with genomic scans, as was done here, may be a fruitful approach for identifying adaptive regulatory variation in other natural systems.
Methods
Sampling
Mice used in this study were collected from five sampling locations (Supplemental Table S1; Supplemental File S1) along a latitudinal gradient in the eastern United States. Mice were sacrificed in the field and measurements (body weight, total body length, tail length) were taken at time of collection. Body mass index (BMI) was calculated as body weight/length2 (g/mm2). Liver tissue was collected in RNAlater and stored at 4°C overnight and then frozen to −80°C until RNA extraction with the Qiagen's RNeasy Mini kit.
mRNA-sequencing and mapping
For each sample, 100-base pair paired-end reads were sequenced on the Illumina HiSeq 4000 platform. RNA-seq reads were mapped with TopHat2 (Kim et al. 2013) to personal reference genomes, created by inserting variants into the mouse reference (GRCm38) and masking indels (see Supplemental Methods). We removed genes with fewer than 500 reads across samples (i.e., an average of 10 reads per sample). Gene expression was then quantile normalized and corrected for hidden factors and known covariates (individual sex and the first six principle components from genotype data to account for population structure) using a Bayesian approach (Supplemental Figs. S19, S20; Stegle et al. 2010, 2012).
Exome capture sequencing and identification of clinal outliers
The exome-sequence data was used to identify clinal outliers (Phifer-Rixey et al. 2018; see also Supplemental Methods). Libraries were enriched for exonic target regions and subsequently 100-bp paired-end reads were sequenced on the Illumina HiSeq 2000 platform, resulting in 2 GB of raw sequence data per individual. Forty-one of the 50 individuals for which there is exome-sequence data have matched RNA-seq libraries (see Supplemental Table S1). Reads were mapped with Bowtie 2 (Langmead and Salzberg 2012), and allele frequencies were estimated with ANGSD (Korneliussen et al. 2014). LFMM (Frichot et al. 2013) was used to identify covariance between environmental and genetic variation (see Supplemental Methods).
Cis-eQTL discovery
We performed cis-eQTL mapping using variant calls from RNA-seq and exome data (see Supplemental Methods). One limitation of this method is that the genotyping data set is limited to sites represented by these data (i.e., variant calls are largely limited to exomic regions of the genome). Consequently, many causal sites may not be typed and variants associated with expression may be tagging causal sites in LD. For the exome data set, depth per site of the targeted exome was ∼15×. For genes represented in the analysis, on average per individual we had sufficient coverage for ∼32% of bases within gene boundaries and ∼15% of bases in the 200-kb boundary used as the cut-off for cis-eQTL mapping (Supplemental Table S8).
To identify cis-acting eQTLs, we used a linear mixed model applied in the program GEMMA (Zhou and Stephens 2012) on expression residuals to associate expression with sequence variants (see Supplemental Methods). A relatedness matrix was computed and included as a covariate. We retained the variant with the lowest p-value for each gene and then performed a Bonferroni's correction. Variants with Bonferroni-corrected P-values of <0.05 were considered significant.
Weighted gene co-expression network analysis
We carried out a weighted gene co-expression network analysis on expression residuals following WGCNA protocols (Langfelder and Horvath 2008) to create expression modules. Each module is summarized by a representative eigengene, the first principle component of a given module. Each gene's expression was correlated with the module eigengene as a measure of the gene's centrality to the module, called module membership.
Data access
Illumina sequencing data from this study have been submitted to the NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA407812. Museum accession numbers for samples used in this study are available in Supplemental File S1.
Supplementary Material
Acknowledgments
We thank members of the Nachman lab and three anonymous reviewers for their thoughtful comments and suggestions. This work was supported by a National Institutes of Health grant (R01 GM074245) to M.W.N. This work was facilitated by an Extreme Science and Engineering Discovery Environment (XSEDE) allocation to M.W.N. and M.P.-R. XSEDE is supported by National Science Foundation (NSF) grant number ACI-1548562. K.L.M. was supported by an NSF Doctoral Dissertation Improvement Grant (DEB 1601699). M.A.B. was supported by an NSF Graduate Research Fellowship (DGE 1106400).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.238998.118.
References
- Andolfatto P. 2005. Adaptive evolution of non-coding DNA in Drosophila. Nature 437: 1149–1152. [DOI] [PubMed] [Google Scholar]
- Arribas J, Esselens C. 2009. ADAM17 as a therapeutic target in multiple diseases. Curr Pharm Des 15: 2319–2335. [DOI] [PubMed] [Google Scholar]
- Ashton KG, Tracy MC, Queiroz AD. 2000. Is Bergmann's rule valid for mammals? Am Nat 156: 390–415. [DOI] [PubMed] [Google Scholar]
- Bergmann C. 1847. Ueber die Verhaltnisse der warmeokonomie der thiere zu ihrer grosse. Gottinger Studien 3: 595–708. [Google Scholar]
- Blake JA, Eppig JT, Kadin JA, Richardson JE, Smith CL, Bult CJ; the Mouse Genome Database Group. 2017. Mouse Genome Database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucl Acids Res 45: D723–D729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Browning BL. 2011. Population structure can inflate SNP-based heritability estimates. Am J Hum Genet 89: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YF, Marks ME, Jones FC, Villarreal G, Shapiro MD, Brady SD, Southwick AM, Absher DM, Grimwood J, Schmutz J, et al. 2010. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327: 302–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowles CR, Hirschhorn JN, Altshuler D, Lander ES. 2002. Detection of regulatory variation in mouse genes. Nat Genet 32: 432–437. [DOI] [PubMed] [Google Scholar]
- Crawford DL, Segal JA, Barnett JL. 1999. Evolutionary analysis of TATA-less proximal promoter function. Mol Biol Evol 16: 194–207. [DOI] [PubMed] [Google Scholar]
- Dandine-Roulland C, Bellenguez C, Debette S, Amouyel P, Génin E, Perdry H. 2016. Accuracy of heritability estimations in presence of hidden population stratification. Sci Rep 6: 26471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar EL, Oakley CG, Conner JK, Gould BA, Schemske DW. 2016. Factors influencing the effect size distribution of adaptive substitutions. Proc R Soc B 283: 20153065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endler JA. 1977. Geographic variation, speciation, and clines (MPB-10). Princeton University Press, Princeton, NJ. [PubMed] [Google Scholar]
- Fiorentino L, Vivanti A, Cavalera M, Marzano V, Ronci M, Fabrizi M, Menini S, Pugliese G, Menghini R, Khokha R, et al. 2010. Increased tumor necrosis factor α–converting enzyme activity induces insulin resistance and hepatosteatosis in mice. Hepatology 51: 103–110. [DOI] [PubMed] [Google Scholar]
- Foster F, Collard M. 2013. A reassessment of Bergmann's rule in modern humans. PLoS One 8: e72269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB. 2013. Gene expression drives local adaptation in humans. Genome Res 23: 1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frichot E, Schoville SD, Bouchard G, François O. 2013. Testing for associations between loci and environmental gradients using latent factor mixed models. Mol Biol Evol 30: 1687–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Sironi M, Pozzoli U, Ferrer-Admettla A, Pattini L, Nielsen R. 2011. Signatures of environmental genetic adaptation pinpoint pathogens as the main selective pressure through human evolution. PLoS Genet 7: e1002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Moltke I, Grarup N, Racimo F, Bjerregaard P, Jørgensen ME, Korneliussen TS, Gerbault P, Skotte L, Linneberg A, et al. 2015. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science 349: 1343–1347. [DOI] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, et al. 2002. The structure of haplotype blocks in the human genome. Science 296: 2225–2229. [DOI] [PubMed] [Google Scholar]
- Gelling RW, Yan W, Al-Noori S, Pardini A, Morton GJ, Ogimoto K, Schwartz MW, Dempsey PJ. 2008. Deficiency of TNFα converting enzyme (TACE/ADAM17) causes a lean, hypermetabolic phenotype in mice. Endocrinology 149: 6053–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazalpour A, Doss S, Zhang B, Wang S, Plaisier C, Castellanos R, Brozell A, Schadt EE, Drake TA, Lusis AJ, et al. 2006. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet 2: e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock AM, Witonsky DB, Gordon AS, Eshel G, Pritchard JK, Coop G, Di Rienzo A. 2008. Adaptations to climate in candidate genes for common metabolic disorders. PLoS Genet 4: e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr B, Karakoc E, Neme R, Teschke M, Pfeifle C, Pezer Ž, Babiker H, Linnenbrink M, Montero I, Scavetta R, et al. 2016. Genomic resources for wild populations of the house mouse, Mus musculus and its close relative Mus spretus. Sci Data 3: 160075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway AK, Lawniczak MKN, Mezey JG, Begun DJ, Jones CD. 2007. Adaptive gene expression divergence inferred from population genomics. PLoS Genet 3: e187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Snitkin ES, DeLisi C. 2008. VisANT: an integrative framework for networks in systems biology. Brief Bioinform 9: 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins DL, Ortori CA, Brookfield JFY. 1995. A test for adaptive change in DNA sequences controlling transcription. Proc Biol Sci 261: 203–207. [DOI] [PubMed] [Google Scholar]
- Jeong S, Rebeiz M, Andolfatto P, Werner T, True J, Carroll SB. 2008. The evolution of gene regulation underlies a morphological difference between two Drosophila sister species. Cell 132: 783–793. [DOI] [PubMed] [Google Scholar]
- Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, Swofford R, Pirun M, Zody MC, White S, et al. 2012. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484: 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junyent M, Parnell LD, Lai CQ, Arnett DK, Tsai MY, Kabagambe EK, Straka RJ, Province M, An P, Smith E, et al. 2010. ADAM17_i33708A > G polymorphism interacts with dietary n-6 polyunsaturated fatty acids to modulate obesity risk in the Genetics of Lipid Lowering Drugs and Diet Network study. Nutr Metab Cardiovasc Dis 20: 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, Wilson AC. 1975. Evolution at two levels in humans and chimpanzees. Science 108: 107–116. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Suzuki M, Ohno T, Tsuzuki K, Taguchi C, Tateishi S, Kawada T, Kim YI, Murai A, Horio F. 2016. Detection of differentially expressed candidate genes for a fatty liver QTL on mouse chromosome 12. BMC Genet 17: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn MH, Fang S, Wu C-I. 2004. Inference of positive and negative selection on the 5′ regulatory regions of Drosophila genes. Mol Biol Evol 21: 374–383. [DOI] [PubMed] [Google Scholar]
- Korneliussen TS, Albrechtsen A, Nielsen R. 2014. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15: 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski MI, Schein JE, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gall SM, Maretzky T, Issuree PD, Niu XD, Reiss K, Saftig P, Khokha R, Lundell D, Blobel CP. 2010. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J Cell Sci 123: 3913–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DC, Sunnarborg SW, Hinkle CL, Myers TJ, Stevenson M, Russell W, Castner BJ, Gerhart MJ, Paxton RJ, Black RA, et al. 2003. TACE/ADAM17 processing of EGFR ligands indicates a role as a physiological convertase. Ann NY Acad Sci 995: 22–38. [DOI] [PubMed] [Google Scholar]
- Linnen CR, Poh YP, Peterson BK, Barrett RD, Larson JG, Jensen JD, Hoekstra HE. 2013. Adaptive evolution of multiple traits through multiple mutations at a single gene. Science 339: 1312–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch CB. 1992. Clinal variation in cold adaptation in Mus domesticus: verification of predictions from laboratory populations. Am Nat 139: 1219–1236. [Google Scholar]
- MacDonald SJ, Long AD. 2005. Prospects for identifying functional variation across the genome. Proc Natl Acad Sci 102: 6614–6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui Y, Tomaru U, Miyoshi A, Ito T, Fukaya S, Miyoshi H, Atsumi T, Ishizu A. 2014. Overexpression of TNF-α converting enzyme promotes adipose tissue inflammation and fibrosis induced by high fat diet. Exp Mol Pathol 97: 354–358. [DOI] [PubMed] [Google Scholar]
- Menghini R, Casagrande V, Menini S, Marino A, Marzano V, Hribal ML, Gentileschi P, Lauro D, Schillaci O, Pugliese G, et al. 2012. TIMP3 overexpression in macrophages protects from insulin resistance, adipose inflammation, and nonalcoholic fatty liver disease in mice. Diabetes 61: 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menghini R, Fiorentino L, Casagrande V, Lauro R, Federici M. 2013. The role of ADAM17 in metabolic inflammation. Atherosclerosis 228: 12–17. [DOI] [PubMed] [Google Scholar]
- Montgomery SB, Dermitzakis ET. 2011. From expression QTLs to personalized transcriptomics. Nat Rev Genet 12: 277–282. [DOI] [PubMed] [Google Scholar]
- Mouse ENCODE Consortium, Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, Gilbert DM, Groudine M, Bender M, Kaul R, et al. 2012. An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol 13: 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. 2009. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9: 311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nica AC, Dermitzakis ET. 2013. Expression quantitative trait loci: present and future. Philos Trans R Soc Lond B Biol Sci 368: 20120362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA. 1998. The population genetics of adaptation: the distribution of factors fixed during adaptive evolution. Evolution 52: 935–949. [DOI] [PubMed] [Google Scholar]
- Palopoli MF, Patel NH. 1996. Neo-Darwinian developmental evolution: Can we bridge the gap between pattern and process? Curr Opin Genet Dev 6: 502–508. [DOI] [PubMed] [Google Scholar]
- Parnell LD, Lee YC, Lai CQ. 2010. Adaptive genetic variation and heart disease risk. Curr Opin Lipidol 21: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phifer-Rixey M, Nachman MW. 2015. The natural history of model organisms: insights into mammalian biology from the wild house mouse Mus musculus. eLife 4: e05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phifer-Rixey M, Bi K, Ferris KG, Sheehan MJ, Lin D, Mack KL, Keeble SM, Suzuki TA, Good JM, Nachman MW. 2018. The genomic basis of environmental adaptation in house mice. PLoS Genet 14: e1007672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, Absher D, Srinivasan BS, Barsh GS, Myers RM, Feldman MW, et al. 2009. Signals of recent positive selection in a worldwide sample of human populations. Genome Res 19: 826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed DR, Lawler MP, Tordoff MG. 2008. Reduced body weight is a common effect of gene knockout in mice. BMC Genet 9: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remington DL. 2015. Alleles versus mutations: Understanding the evolution of genetic architecture requires a molecular perspective on allelic origins. Evolution 69: 3025–3038. [DOI] [PubMed] [Google Scholar]
- Ruff C. 2002. Variation in human body size and shape. Annu Rev Anthropol 31: 211–232. [Google Scholar]
- Serino M, Menghini R, Fiorentino L, Amoruso R, Mauriello A, Lauro D, Sbraccia P, Hribal ML, Lauro R, Federici M. 2007. Mice heterozygous for tumor necrosis factor-α converting enzyme are protected from obesity-induced insulin resistance and diabetes. Diabetes 56: 2541–2546. [DOI] [PubMed] [Google Scholar]
- She P, Reid TM, Bronson SK, Vary TC, Hajnal A, Lynch CJ, Hutson M. 2007. Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab 6: 181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegle O, Parts L, Durbin R, Winn J. 2010. A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput Biol 6: e1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegle O, Parts L, Piipari M, Winn J, Durbin R. 2012. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc 7: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DL, Orgogozo V. 2008. The loci of evolution: How predictable is genetic evolution? Evolution 62: 2155–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Stahl EA, Raj T. 2011. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics 187: 367–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter NB, Bustamante CD, Chase K, Gray MM, Zhao K, Zhu L, Padhukasahasram B, Karlins E, Davis S, Jones P, et al. 2007. A single IGF1 allele is a major determinant of small size in dogs. Science 316: 112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Kobayashi M, Ohno T, Kanamori S, Tateishi S, Murai A, Horio F. 2016. Genetic dissection of the fatty liver QTL Fl1sa by using congenic mice and identification of candidate genes in the liver and epididymal fat. BMC Genet 17: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. 2003. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13: 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Reed FA, Ranciaro A, Voight BF, Babbitt CC, Silverman JS, Powell K, Mortensen HM, Hirbo JB, Osman M, et al. 2007. Convergent adaptation of human lactase persistence in Africa and Europe. Nat Genet 39: 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torgerson DG, Boyko AR, Hernandez RD, Indap A, Hu X, White TJ, Sninsky JJ, Cargill M, Adams MD, Bustamante CD, et al. 2009. Evolutionary processes acting on candidate cis-regulatory regions in humans inferred from patterns of polymorphism and divergence. PLoS Genet 5: e1000592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung J, Zhou X, Alberts SC, Stephens M, Gilad Y. 2015. The genetic architecture of gene expression levels in wild baboons. eLife 4: e04729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JK, Gerdin AK, Karp NA, Ryder E, Buljan M, Bussell JN, Salisbury J, Clare S, Ingham NJ, Podrini C, et al. 2013. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell 154: 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ, Kalay G. 2012. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat Rev Genet 13: 59–69. [DOI] [PubMed] [Google Scholar]
- Wray GA. 2007. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet 8: 206. [DOI] [PubMed] [Google Scholar]
- Wray NR, Yang J, Hayes BJ, Price AL, Goddard ME, Visscher PM. 2013. Pitfalls of predicting complex traits from SNPs. Nat Rev Genet 14: 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY, Kao HJ, Li SC, Stevens R, Hillman S, Millington D, Chen YT. 2004. ENU mutagenesis identifies mice with mitochondrial branched-chain aminotransferase deficiency resembling human maple syrup urine disease. J Clin Invest 113: 434–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S. 2003. Theoretical basis of the Beavis effect. Genetics 165: 2259–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Lu J, Raj SM, Gu Z. 2013. Human expression QTLs are enriched in signals of environmental adaptation. Genome Biol Evol 5: 1689–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Stephens M. 2012. Genome-wide efficient mixed-model analysis for association studies. Nat Genet 44: 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.