

Summary
α-Synuclein (αS) regulates vesicle exocytosis but forms insoluble deposits in Parkinson’s disease. Developing disease-modifying therapies requires animal models that reproduce cardinal features of PD. We recently described a previously unrecognized physiological form of αS, α-helical tetramers, and showed that fPD-causing missense mutations shift tetramers to aggregation-prone monomers. Here, we generated mice expressing the fPD E46K mutation plus 2 homologous E→K mutations in adjacent KTKEGV motifs. This tetramer-abrogating mutant causes phenotypes similar to PD. αS monomers accumulate at membranes and form vesicle-rich inclusions. αS becomes insoluble, proteinase K-resistant, Ser129-phosphorylated and C-terminally truncated, as in PD. These changes affect regions controlling motor behavior, including decrease in nigrostriatal dopaminergic neurons. The outcome is a progressive motor syndrome including tremor and gait and limb deficits responsive to L-DOPA. This fully-penetrant phenotype indicates that tetramers are required for normal αS homeostasis and chronically shifting tetramers to monomers may result in PD, with attendant therapeutic implications.
ETOC
Nuber et al. demonstrate that tetramers are required for the physiological state of α-synuclein in vivo by developing a novel mouse model of Parkinson’s disease based on destabilizing tetramers. This produces neuropathology and a progressive motor syndrome resembling PD.
Introduction
Parkinson’s disease (PD), dementia with Lewy bodies (DLB) and related neurodegenerative disorders involve the age-dependent accumulation of α-synuclein (αS) in cytoplasmic inclusions termed Lewy bodies and neurites. In PD, the inclusions occur in midbrain dopaminergic and diverse cortical neurons, dysfunction and degeneration of which underlie the motor and cognitive impairments (Surmeier et al, 2017). Point mutations and duplications in the SNCA gene cause familial forms of PD and DLB with extensive brainstem, striatal and cortical αS cytopathology and neuronal loss (Fujishiro et al, 2013).
Studies of the normal cellular function of αS suggest that it transiently interacts with vesicle membranes and regulates the trafficking and exocytosis of synaptic and other vesicles (Wang et al, 2014, Logan et al, 2017, Burre et al, 2010). On the other hand, in vitro experiments indicate that abnormal αS oligomers can bind to and perturb membrane function and contribute to cell death (Fusco et al, 2017, Sharon et al, 2003, Winner et al, 2011). However, some fPD subjects carry αS missense mutations (e.g., A30P, G51D) that have less membrane affinity than the wild-type (wt) protein and may therefore induce cytotoxicity via aqueously soluble oligomers (Ysselstein et al, 2015), arguing for a pathogenic role of aggregation-prone monomers independent of their abnormal association with membranes. Understanding the conversion of physiological αS to pathogenic oligomers requires knowledge of the normal forms of the protein in healthy neurons. Beginning in 2011, our laboratory described and characterized a previously unrecognized native form of cellular αS: metastable, α-helically folded tetramers of ~58 kDa (4 × 14,502 monomers) (Bartels et al, 2011, Dettmer et al, 2013, Dettmer et al, 2015a, Dettmer et al, 2015b). Heretofore, cellular αS had been described as ‘natively unfolded’ monomers that could aggregat into β-sheet-rich pathological oligomers. Although the initial description of cellular α-helical tetramers and related multimers proved controversial (Fauvet et al, 2012, Burre et al, 2013), other laboratories also obtained evidence for the existence of apparently physiological tetramers and related conformers (Wang et al, 2011, Westphal and Chandra, 2013, Burre et al, 2014).
We hypothesize that αS normally occurs in a dynamic equilibrium of unfolded ~14 kDa monomers with α-helically-folded ~60 kDa tetramers. We reported that αS purified under non-denaturing conditions from human erythrocytes and neuroblastoma cells occurs in part as tetramers that resist aggregation in vitro (Bartels et al, 2011). Intact-cell crosslinking showed that not only αS but also the non-pathogenic βS and γS homologues occur as both tetramers and monomers (Dettmer et al., 2013). In accord, the insertion of tetramer-abrogating mutations into the 6 imperfectly repeated KTKEGV motifs slowed the kinetics of vesicle exocytosis conferred by wt αS in living neurons, suggesting that tetramers participate in a normal function of αS in vesicle trafficking (Wang et al, 2014). Regarding the relevance of tetramers to disease, we found that all fPD-causing αS missense mutations decrease the physiological tetramer:monomer (T:M) ratio in neurons, thereby increasing the levels of aggregation-prone monomers (Dettmer et al, 2015a). One of these fPD-causing mutations (E46K) occurs within a KTKEGV motif that is imperfectly repeated 6–9 times and helps confer helicity to αS in vitro (Davidson et al, 1998).
Based on this information, we examined the effect of amplifying the tetramer → monomer shift induced by E46K by adding one or two homologous E-to-K mutations into the adjacent KTKEGV repeats (i.e., at residues 35 and/or 61). This resulted in an E-to-K dose-dependent loss of tetramers and gain of monomers in cells (Dettmer et al, 2015a). The resultant K-mutant monomers displayed increased interaction with vesicular membranes, leading to round cytoplasmic inclusions of clustered vesicles associated with neurotoxicity (Dettmer et al, 2015a).
The distinct properties of αS tetramers vs. monomers as regards transient lipid binding (Bartels et al, 2011) were underscored by the finding that unfolded αS monomers but not purified cellular tetramers could bend and stabilize vesicle membranes in vitro (Westphal and Chandra, 2013), which may in turn inhibit membrane fusion events during exocytosis (Wang et al, 2014). In accord, another report provided evidence for a physiological function of αS multimers (tetramers, octamers) in facilitating SNARE complex formation and vesicle fusion (Burre et al, 2014). The relevance of the tetramers for neuronal physiology is further supported by the recent discovery that human neurons bearing loss-of-function mutations in glucocerebrosidase (GBA1) that cause Gaucher’s disease (Brady et al, 1965) and PD [e.g.(Aharon-Peretz et al, 2004)] have decreased T:M ratios of their wt αS protein, and genetic or pharmacological restoration of normal glucosylceramide levels corrects this decrease (Kim et al, 2018).
These steadily accruing data notwithstanding, the biological importance of the αS T:M equilibrium remains controversial. To establish its role in vivo, we correlated tetramer-abrogating mutants of αS with cytotoxicity in neuronal culture and identified a triple mutant (E35K+E46K+E61K) that more robustly decreases tetramers and induces cytotoxicity than E46K alone. Based on this culture finding, we generated transgenic mouse lines that express this tetramer-lowering “3K” αS mutation. When compared to WT and fPD E46K (“1K”) mutant mice, the 3K mutant more strongly shifts tetramers to excess monomers, leading to striking neuropathological, biochemical and behavioral features resembling PD, including degeneration of DAergic striatal and cortical fibers and neurons, reduced DA levels, and an age-dependent, progressive motor phenotype of tremor, decreased limb function and abnormal gait that is transiently improved by L-DOPA. The tetramer-incompetent monomers become insoluble, abnormally phosphorylated and C-terminally truncated (as in PD) and accumulate at vesicles in association with lipid-rich autofluorescent deposits (lipofuscin). This unique PD model shows that αS tetramerization is a required physiological process and its chronic impairment results in key features of PD, thus both validating and enabling the novel therapeutic approach of stabilizing tetramers to prevent PD, DLB and other synucleinopathies.
Results
Amplifying the E46K fPD mutation in vivo shifts αS tetramers toward excess full-length and truncated monomers and enhances αS membrane association
Our previous studies showed that all PD-causing αS missense mutations decrease the physiological T:M ratio in neurons (Dettmer et al, 2015a) and that inserting tetramer-lowering E → K mutations into the KTKEGV motifs induces cytotoxicity and inclusions in cell culture (Dettmer et al, 2015a, Dettmer et al, 2015b) (Table S1). To enhance PD-like phenotypes in αS tg mice, which usually display a rather late onset of PD-relevant motor symptoms and little or no L-DOPA responsiveness (Table S2), we first confirmed the correlation between tetramer abrogation and neurotoxicity by expressing each single E→ K mutant and their amplification (3K) vs. WT αS in hu M17D neuroblastoma cells. Each of the 3 E→ K mutants alone significantly reduced the WT T:M ratio in cells by ~40%, but the 3K mutant markedly exacerbated this to ~90% (Fig. 1a,b). Associated cytotoxicity was then quantified in 3 distinct assays: PARP-cleavage (Fig. 1c,d), cell confluence (Fig. 1e, upper panel) and LDH release (Fig. 1e, lower panel). The single K mutations increased toxicity significantly in the first and third assays but did not change confluence, whereas 3K further enhanced cytotoxicity in all 3 assays. Analysis of LDH release vs. tetramer-abrogation showed a significant correlation dependent on the number of E→ K mutations (Fig. 1f). Thus, inserting tetramer-lowering E→ K mutations in 3 adjacent KTKEGV motifs led to much more T:M ratio lowering and attendant cytotoxicity than any single mutation.
Figure 1.
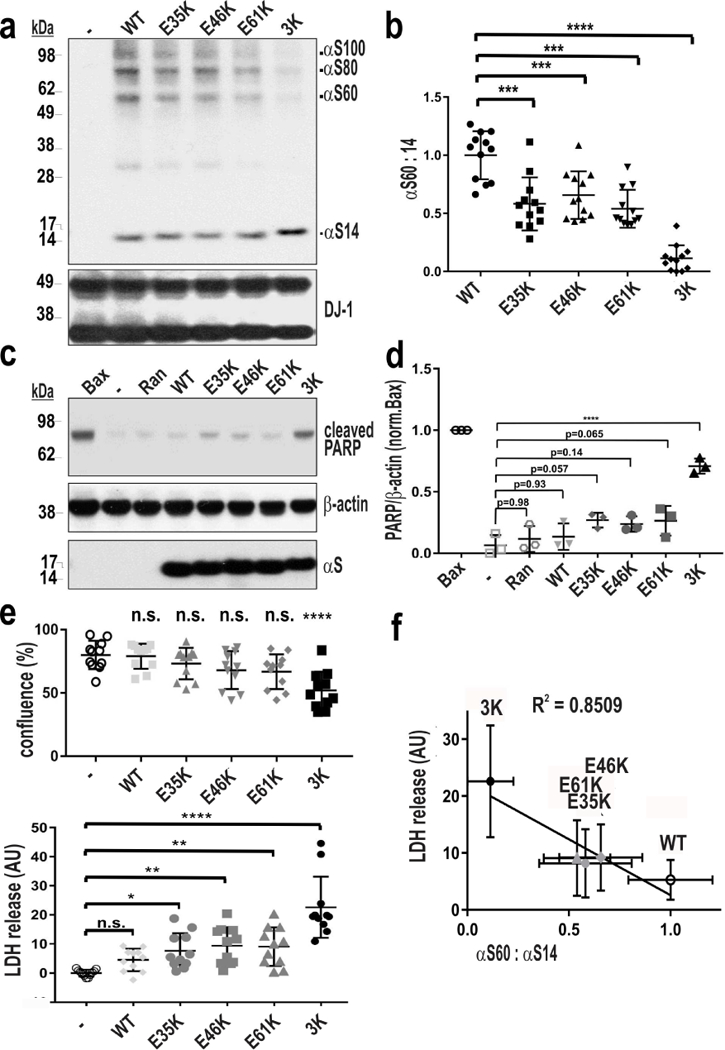
Biochemical characterization of αS multimers and cytotoxicity by individual E→K mutants. (a) Intact-cell crosslinking of αS using the cell-penetrant crosslinker DSG on M17D cells transiently expressing the indicated αS variants were lysed in PBS/1% Triton-X100; (b) αS60:14 ratios of all 5 genotypes by WB (Syn1) in n=7 ind. exps. (c-f): M17D cytotoxicity assays: (c) WB for cleaved PARP relative to the positive control (Bax); Ran is a negative control protein. n=3 ind. exps., each transfected as indicated, or mock (−); relative cleaved PARP is graphed in (d). (e) IncuCyte cell confluency assay and LDH-release cytotoxicity assay, n=11 ind. exps. (f) Correlation between decrease in T:M ratio and increase in LDH release. Means ± SD; n.s. non-significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, 1way-ANOVA, Dunnett’s multiple comparisons test.
To examine in vivo the amplifying nature of the 3K mutation and its relevance to human (hu) PD, we performed a comparative analysis of our novel 3K tg mice to a genomically humanized fPD E46K mouse (see Methods) and to new hu WT αS mice (Fig. 2a). We had previously described a method to trap the cell-lysis-sensitive tetramers by intact-cell crosslinking of fresh, minced brain tissue bits with the cell-penetrant crosslinker DSG (Dettmer et al, 2015a, Dettmer et al, 2013). Similar to our cell culture studies (Fig. 1), the E46K (1K) mouse brain had a significant decrease in the T:M ratio vs. WT (p<0.05), and it was further lowered in the 3K brain (p<0.01, one-way ANOVA, post Tukey) (Fig. 2b,c). Sequential extractions of cerebral cortex (without crosslinking) revealed a shift from buffer-soluble (cytosolic) αS to more TX-solubilized αS (membrane fraction) in 1K vs. WT cortex, and this shift was increased further in 3K cortex (Fig. S1a,b). These changes in tetramerization and solubility between WT, 1K and 3K were observed despite similar total hu αS brain levels, using a hu αS-specific ELISA on the 3 genotypes (Fig. 2d) and by immunostaining brains with the hu αS-specific 15G7 (Fig. 2e). The stepwise K-mutant decreases in T:M ratio were confirmed in an additional lower-expressing 3K line (line #3798) we bred to homozygosity (designated 3KL+/+; Fig. S1c,e,f). Because our present and prior studies in cultured neurons identified the 3K mutant to strongly decrease tetramer levels and induce more cytotoxic αS-positive inclusions (Dettmer et al, 2015a), we focused our analyses on the higher expressing 3K mouse (line #3817), since it can be compared to the expression-matched WT line (line #3877). By using abs against hu and hu+mouse αS, we quantified a ~2-fold overexpression of hu 3K αS compared to endogenous αS (Fig. S1g,h), and this achieves a total αS level similar to that of hu brain (Paleologou et al, 2009). We used intact-cell crosslinking (see above) to evaluate whether the 3K variant caused a major decrease in αS tetramers (designated αS60) and related multimers (αS80 and αS100) and increase in monomers (αS14) in brain regions relevant for motor function. Comparing the 3K and WT hu αS mice, we found a large and significant decline of the T:M ratio in all brain regions analyzed (p<0.01, n=5; Student’s t-test) (Fig. 2f,g). Densitometry of 3K αS60 showed either a trend (striatum, p=0.2) or a significant reduction (substantia nigra (SN), p=0.039; cortex, p=0.013) in T:M compared to the WT brain levels. In accord, 3K αS brains showed a clear-cut excess of monomers: 58% in striatum (p<0.01), 51% in SN (p<0.01), and 41% in cortex (p<0.05) vs. the respective monomer levels in the expression-matched WT hu αS mice (Fig. 2g).
Figure 2.
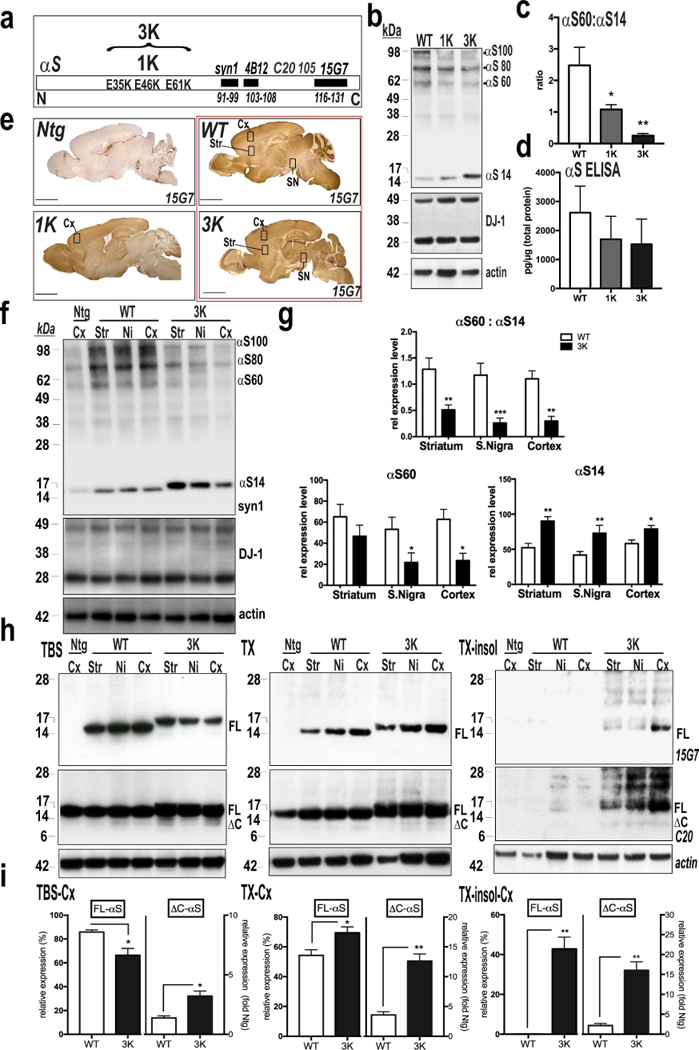
Immunohistochemical (IHC) and biochemical characterization of WT, 1K and 3K αS tg mice. (a) Schematic of the amplification of the E46K fPD mutation by adding two E→ K mutations (“3K”) in the adjacent KTKEGV motifs. Also shown: epitope loci of the principal abs. (b) Intact-cell crosslinking of αS in cortical brain bits from WT, 1K and 3K tg mice. Syn1 detects monomeric (αS14) and tetrameric (αS60) αS and probable conformers of the tetramer (αS80, αS100) (Dettmer et al, 2013). DJ-1 monomers/dimers serve as control for equal crosslinking and loading. Note the decrease in tetramers associates with a stepwise increase in monomers in 1K and 3K vs. WT. (c) Quant. of WBs like those in (b) reveals significant decreases in T:M ratio in 1K vs. WT and 3K vs. WT (n=3 mice per genotype, 3 ind. exps). (d) hu-specific αS ELISA show equal αS levels in cortical brain homogenates of 6 mo old WT, 1K and 3K mice (n=3). (e) IHC (hu αS 15G7) of sagittal brain sections from WT, 1K and 3K tg mice and a non-tg (Ntg) littermate control. Boxes indicate regions dissected for WBs shown below. (f) Intact-cell crosslinking of αS from the indicated regions of WT vs. 3K mice. (g) Quant. WBs of αS monomers (~14 kDa) and tetramers (~60 kDa) reveals a significant decrease in T:M ratio in 3K vs. WT (n=5 mice per genotype run in 5 ind. exps.). (h) Representative WBs (non-crosslinked) of sequential extractions of TBS-soluble (cytosolic), TX-soluble (membrane) and TX-insoluble brain homogenates (15G7: hu αS; C20: hu+mouse αS). (i) Densitometry (Mean ± SEM) of WBs of full-length (FL) αS (15G7) and ΔC αS (C20) in indicated brain regions (n=3 mice per genotype run in 3 ind. exps.). *p<0.05, **p<0.01, ***p<0.001; unpaired 2-tailed Student’s t test.
Next, we sequentially extracted homogenates of key brain regions in buffers of increasing solubilization strength, thereby separating the TBS-soluble fraction (cytosolic) from the Triton X-100-solublized membrane fraction; the TX-insoluble membrane pellets were then solubilized in RIPA. Whether blotting with abs specific for the C-terminal region of hu αS (15G7; aa 116–131) or detecting both rodent and human αS (C20), the 3K mutation led to significantly less αS in cytosol and more in the TX-soluble and TX-insoluble fractions vs. the corresponding levels in the WT and 1K brains (Figs. 2h,i; S1a,b). Thus, the 3K mutation markedly reduced the cytosolic T:M ratio (due to both decreased tetramers and increased monomers) (Fig. 2f,g), with the excess monomers having decreased solubility in physiological buffer and enhanced association with membranes (Fig. 2h,i). Moreover, a substantial amount of αS accumulated in the TX-insoluble fraction (Fig. 2h, right panel), with a clear difference in the pattern of these SDS-stable oligomers (Fig. S1d) from that of the DSG-captured native αS multimers (Fig. S1c). In addition, we observed minor αS species in 3K brains migrating just below the major full-length monomers that were immunoreactive with C20 (to a large C-terminal region) but not with 15G7 (to the extreme C-terminus) (Fig. 2h,i). We designate this C-terminally truncated hu species ΔC-αS (confirmed by MS below). Small amounts of ΔC-αS were detectable in cortex, striatum and nigra, were relatively higher in the Tx-insoluble than TX-soluble and cytosol fractions, and were consistently greater (~15% increase) in 3K than WT brains (Fig. 2i). ΔC-αS species occur in PD and/or αS tg rodents [eg.(Nuber et al, 2014, Nuber et al, 2013, Mishizen-Eberz et al, 2005)] and can arise in part from calpain-1 cleavage. Activating calpain-1 could have at least two adverse effects: cleaving αS and thereby enhancing its aggregation and neurotoxicity, and inhibiting autophagy and thus the proteolytic degradation of αS (Nuber and Selkoe, 2016). To assess whether calpain-1 might contribute to the ΔC-αS species, hu WT and 3K brains were analyzed by mass spectrometry (MS). MS detected hu αS tryptic fragments spanning aa 13–119 in the 3K brain (and containing the 3 E→ K mutations); this fragment matched a calpain-1 generated fragment of recombinant, N-acetylated and fibrillized hu αS (Fig. S1i) that served as a positive control. Only shorter (aa 35–96) hu ΔC-αS fragments were seen in the hu WT mouse brain (Fig S1j). ΔC-αS aa 1–119 has been identified in PD brain (Liu et al, 2005)].
The tetramer-monomer shift caused by the 3K mutation is associated with a progressive motor syndrome with multiple features of PD
Our initial breeding of the 3K tg line soon revealed spontaneously developing behavioral changes that included abnormal gait and gradually worsening head and body tremor. At weaning (~1 mo), 3K male and female mice showed no apparent motor phenotype, but in the next 2–4 mo, all 3K mice developed an increasingly apparent body tremor (Movie S1 for 6 mo males; Movie S2 for 6 mo females; Movie S3 for 10 mo males; Movie S4 for 12 mo females). In general, the motor syndrome appeared earlier and increased more robustly in the male 3K mice. Indeed, the body tremor of 3K males interfered with breeding after the age of ~2 mo. Thus, 3K females were used to maintain the nascent heterozygous colony, precluding their inclusion in our initial motor behavior longitudinal analyses. Tremor was the most striking feature of the age-dependent motor phenotype. To quantify its degree, we made 8–10 sec videos of 6 mo old 3K vs. WT mice and stacked (superimposed) ~60 frames, which were converted to a stack mode ‘mean’ to sum the extent of body movement. This method approximates the distance of movement between the captured profiles of the animal’s back and also their sharpness (Fig. 3a,d). By age 6 mo, 3K mice had a stiff, uncoordinated gait, which was preceded by hind limb clasping that began at ~3–4 mo (Fig. 3b,d). At age 3 mo, 3K mice trended toward less spontaneous movement during open field (OF) testing, and this became significantly reduced at 6 mo vs. the age- and αS level-matched hu WT mice (Fig. 3e). The lack of coordinated limb movement was substantiated by a markedly impaired ability of 3K mice to maintain balance while walking on a slowly accelerating rotarod (Fig. 3e) both at 3 and 6 mo. In a pole climbing task, 3 and 6 mo 3K mice had an increased number of falls after turning downward on the pole (3K vs. WT 6 mo: p<0.001) that progressed with age (3K 3 mo vs. 6 mo, p=0.01), and a rise in the time to climb down the pole (Fig. 3c) (Movie S5 for a 3K male at 6 mo; Movie S6 for WT male). We detected none of these motor deficits in the expression-matched WT hu αS mice at all time points (Fig. 3a-e).
Figure 3.
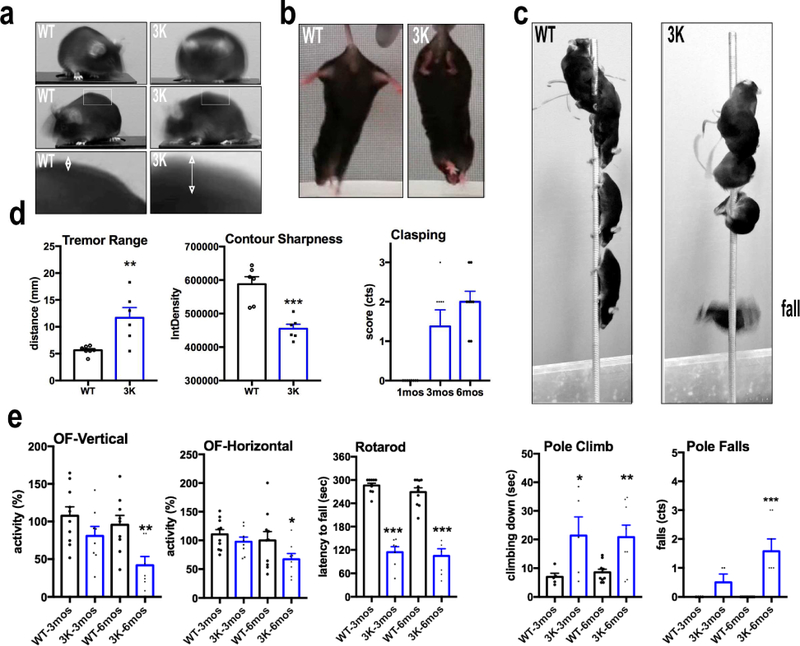
Progressive motor deficits in tetramer-deficient 3K αS tg mice. (a) Stacked images to assess spontaneous head and body tremor in 3K mice at 6 mo by videography and ImageJ software (representative images of n=2 3K and WT). (b) Limb clasping in 3K (n=8) vs. WT (n=10) αS tg mice at 1–6 mo. (c) Quant. of the tests in a and b (Methods). (d) Stacked images of a single WT and a single 3K mouse during the pole test show typical abnormal climbing and subsequent fall of a 3K mouse after turning around to climb down. (e) Graphs quantify time to descend the pole and number of falls in 3 mo and 6 mo mice. Left graphs quantify spontaneous exploration in an open field (OF) chamber and fine motor and balancing skills in a 4–40 rpm accelerating rotarod test. Means ± SEM. *p<0.05, **p<0.01; ***p<0.001. (c) (tremor range and contour sharpness): unpaired 2-tailed Student’s t test, or else one-way ANOVA, post Tukey.
Excess αS monomers associate with vesicles and are truncated, phosphorylated and PK-resistant in 3K mouse brain
To assess neuropathological changes associated with the shift of tetramers to excess, buffer-insoluble and partially truncated monomers (Fig. 2) and the accompanying progressive motor deficits (Fig. 3), we searched for proteinase K (PK) resistant αS in cryostat sections of striatum, substantia nigra (SN)/midbrain and motor cortex (mCx). In 6 mo 3K mice, PK-resistant αS granules/dot-like structures were detected in the neuropil of striatum and cerebral cortex, whereas in age- and hu αS level-matched WT controls, the much lower amounts of αS accumulation displayed less or no PK resistance (Fig. 4a,b). In the 3K midbrain and mCx, dense round perikaryal inclusions were observed, often situated near the nucleus, and some of these were partially PK resistant whereas only background staining was observed in the simultaneously processed WT hu αS brain sections (Fig. 4a,b).
Figure 4.
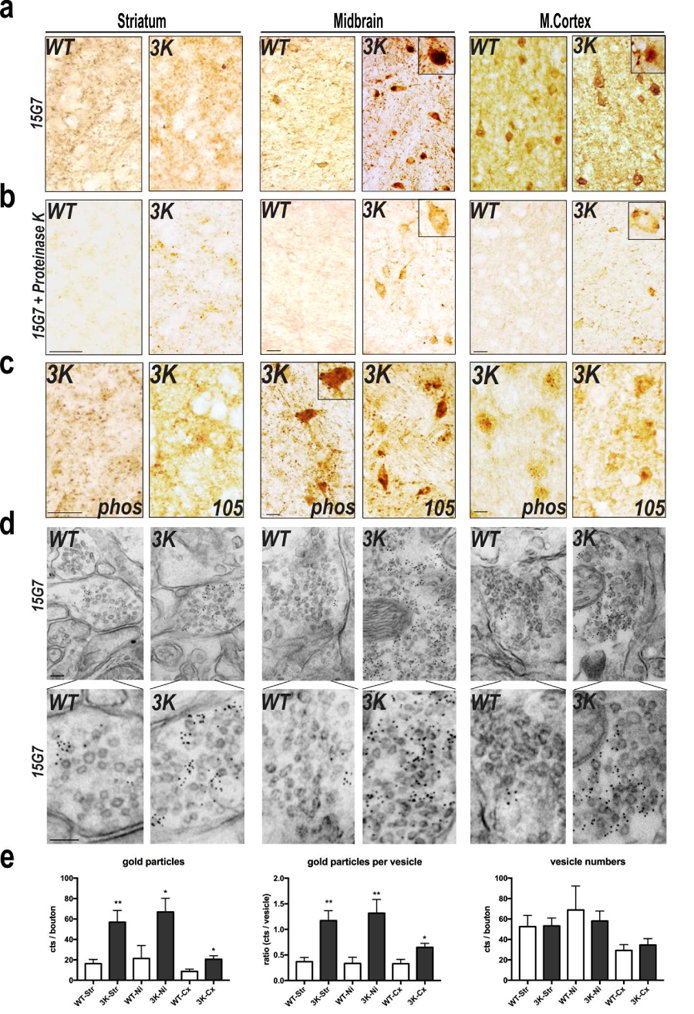
3K αS accumulates at vesicles and forms proteinase K resistant, phosphorylated, and truncated αS deposits mouse brain. (a, b) Hu (15G7) αS IHC (age 6 mo) without (a) and with (b) proteinase K pre-treatment. (c) IHC with abs to ser129-phosphorylated (phos) and ΔC-truncated (105) αS. (d) Immunogold EM analysis of 6 mo brains with 15G7 (bottom row: high power). (e) Quant. of αS immunogold particles at synaptic vesicles. 10 fields each from n=2 brains per genotype run in 2 ind. exps. (blinded counting). Means ± SEM. *p<0.05, **p<0.01, unpaired two-tailed Student’s t test. Scale bars (a-c) 20 µm, (d) 60 nm.
To confirm that the abundant αS monomers in 3K brain (Fig. 2g) form PD-relevant pathological aggregates, we applied a mAb (51253, Abcam) specific for phosphorylated (pSer129) αS or a mAb (105, gift of W. Zago, Prothena Biosciences) favoring the detection of C-terminally truncated (ΔC) αS. Both modifications are commonly detected in hu Lewy pathology and mouse models thereof [e.g., (Nuber and Selkoe, 2016)]. We observed dot-like patterns in cortical neuropil with both mAbs, staining of round and granular perikaryal deposits in midbrain neurons, and dots and varicosities in the striatal neuropil (Fig. 4c).
To address the ultrastructural correlates of the abnormal αS-immunoreactive profiles in the neuropil and neuronal somata, we prepared ultrathin sections for immunogold labeling. Consistent with the many, small dot-like inclusions detected by light microscopy in 3K mouse neuropil, we readily identified a co-localization of the mutant αS with membrane vesicles in synaptic boutons, while far fewer gold particles were seen in the expression-matched WT hu αS brains (Fig. 4d, higher mag in lower panels). We quantified these data: there were no differences in vesicle numbers per se; however, significantly more total gold particles and particles per vesicle occurred in the 3K synaptic boutons than in the expression-matched hu WT brains (Fig. 4e).
To further assess whether the rising level of αS cytopathology correlated with the progressive motor abnormalities and was a feature of both 3K and fPD E46K, we compared these changes in the 3K, 1K and hu WT αS tg mice at 3 vs. 6 mo (Fig. 5). Notably, we found a significant multi-fold rise in pSer+ labeling as early as 3 mo in 3K vs. 1K and WT mice (Fig. 5a,b,d). By 6 mo, levels in 3K mice had risen to very high levels, and 1K mice now also showed a small (~2-fold) but significant rise vs. 3 mo (Fig. 5a, higher mag in 5b; quant. in 5d). Interestingly, by age 6 mo, somewhat less pSer129+ neuropil fibers but more perikaryal inclusions with these features were observed in cortical neurons of the 3K mice (less noticeably in the 1K mice) (Fig. 5b). To substantiate this apparent neurite loss, we impregnated cortical tissue blocks with Golgi-Cox solution, providing a 3-dimensional view of some neurons and their processes. We observed shortened neurites and less dense dendritic arbors by Golgi-Cox in the pyramidal neuropil of 3K cortex (Fig. 5c), a reduced neurite pattern also reported from loss of DAergic cortical input in a 6-OH DA rat model (Solis et al, 2007). Quantifying the % area of neuropil covered by Golgi-Cox-impregnated neurons revealed a significant decrease in 6 mo 3K mice (Fig. 5d). The time-dependent pSer129 accumulation and neurite loss was associated with decreased performance in pole climbing at ~3 and 6 mo (Fig. S2a). For example, the smaller increase in pSer129+ aggregates in 1K mice at 6 mo was associated with an intermediate pole performance (not reaching significance vs. WT or 3K) (Figs. 5d, S2a). Compared to these data on the male 3K mice, female 3K mice developed both motoric phenotypes vs. gender-matched hu WT mice by age 6 mo (exemplified by the challenging rotarod test; Fig. 2b) and neurochemical changes (αS pSer129+ aggregates; Fig. S3a), but at a slower rate. Similarly, a lower expressing 3K line (3KL+/+) displayed an intermediate phenotype (Fig. S2c; see below).
Figure 5.
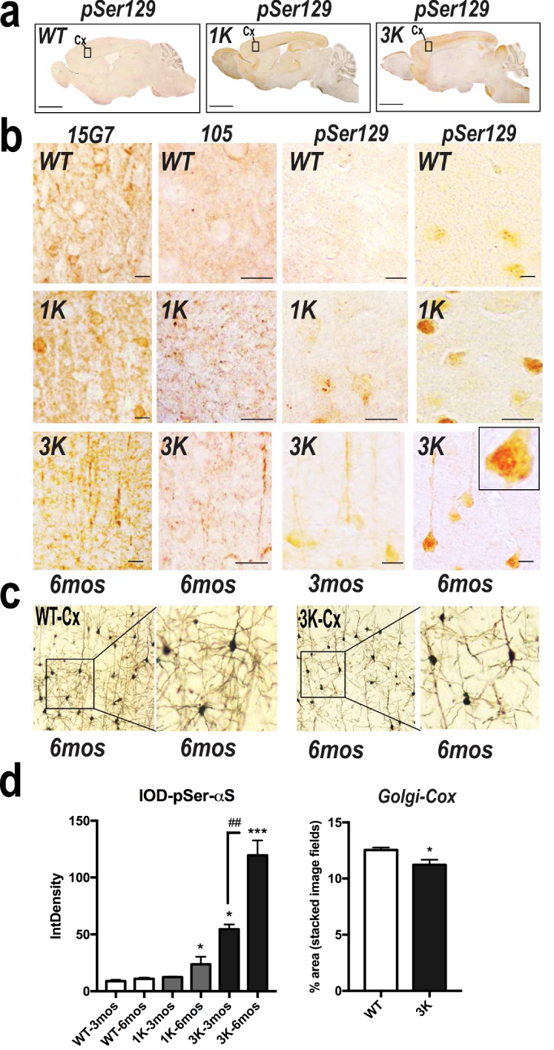
IHC characterization of WT, 1K and 3K αS aggregate pathology in cerebral cortex. (a) Representative IHC photomicrographs to ser129-phosphorylated αS of sagittal brain sections from WT, 1K and 3K mice. Boxes indicate regions shown in panels in (b). (b) Cortical sections (age 3 or 6 mo) stained with 15G7 or else fortruncated (105 ab) or pSer129 in WT, 1K and 3K mice. Note the relative decrease in pSer129+ neurites in the 6 mo 3K cortex (also seen by Golgi Cox in (c)). (c) Golgi-Cox impregnated cortical sections of WT, 3K tg mice (n=6 each of brains also subjected to 15G7 and pSer129 staining in (b). (d) Quant. of relative pSer129 optical density and % area covered by Golgi Cox impregnates profiles (n=10 fields each of cortical sections from n=3 mice per age and genotype). Means ± SEM. *,#p<0.05; **,##p<0.01;***p<0.001 in 1K and 3K vs.WT; left graph: one-way ANOVA, post Tukey; Golgi Cox: unpaired two-tailed Student’s t test.
By quantitative immunoblotting, we observed a robust ~3-fold rise of pSer129+ αS in 3K over hu WT in whole brain extracts at 6 mo (Fig. S3a). We further evaluated pSer129+ accumulation in cerebellar and cortical sections, since increasing evidence suggests their involvement in PD-related tremor (Caligiore et al, 2016). We found more abundant and age-dependent accumulation of pSer129+ αS in 3K cortical regions, whereas relatively minor pSer129+ αS immunoreactivity occurred in 3K cerebellar cortex (Fig. S3b). pSer129+ inclusions were also detected in some DAergic and/or noradrenergic TH-positive perikarya, including in SN and locus coeruleus and occasionally in the VTA (Fig. S3c), but we detected no significant loss of TH+ VTA cells in 3K mice at 6 mo (Fig. S3d), a neuronal selectivity that again resembles PD.
Homozygous 3K mice have severe motor deficits that preclude survival after weaning, while a second, lower-expressing 3K line shows deficits intermediate between 1K and 3K lines
To assess whether high levels of toxic αS 3K monomers in the brain that derived from two copies of the tetramer-abrogating 3K construct further increases pathology, we attempted breedings between 3K heterozygous mice. One 3K male was able to fertilize a 3K female, producing 4 homozygous (3K+/+) pups. Their severe motor phenotype was apparent shortly after birth and led to their being unable to reach food and water independently after weaning, thus requiring euthanasia by age ~4 wk) (Fig. S3e, Movie S7). By immunoblotting brain extracts, we observed a doubling of monomer levels vs. 3K+/− mice (data not shown). A striking finding of these 3K+/+ mice was the severe pSer129+ aggregation pathology already at 4 wk, twice the level of the 3K+/− line at the same age (Fig. S3f,g). Their hu αS aggregates co-localized with LAMP-1 (not shown), suggesting they accumulated in lysosomes. Further phenotyping was precluded by the small number of pups born and their very brief survival.
To validate the striking PD-like phenotypes described so far in our 3K+/− line, we characterized mice from a second and lower-expressing 3K line that had been generated simultaneously from the same 3K transgene. This low-expressing line is designated 3KL, and the males bred normally, in contrast to the breeding deficit of the high-expressing 3K males. Accordingly, the 3KL line could be bred to homozygosity, yielding 3KL+/+ mice, and we have performed limited phenotyping of these. By both neuropathological and motor assessments, the 3KL+/+ mice displayed phenotypes that were intermediate between the 1K and 3K mice: brain extracts showed a significant decrease in T:M ratio (Figs. S1c,e,f). Brain sections analyzed simultaneously from our four mouse lines showed a stepwise accumulation of pSer129+ deposits in neuronal perikarya and neuropil (WT<1K<3KL+/+<3K) (Fig. S3h,i). The challenging rotarod test revealed that 3KL+/+ mice began to show a small but significant age-dependent motor impairment at 8 mo vs. age-matched Ntg controls, but this was far less than in the 3K line at 6 mo (Fig. S2c). The generation of homozygous 3K pups, as well as the independent tetramer-deficient 3KL+/+ line from the same transgene and its intermediate phenotype, rule out insertional mutagenesis as an artifactual explanation for the phenotype observed in our principal 3K line.
αS 3K monomers accumulate at vesicles in synapses and cell bodies that also display lipid-rich deposits of endo-lysosomal origin
EM analyses of the 3K mice revealed αS gold immunoreactivity closely adjacent to both small synaptic vesicles (~60 nm) and larger, perinuclear, vesicles (~300 nm). To explore the ultrastructure of the intraneuronal deposits further, we performed conventional and immunogold EM in mCx and SN of 3K and hu WT αS mice at 3 mo. 3K mice had medium- and large-sized lipofuscin-like deposits that were less abundant in WT neurons, which exhibited more finely granular deposits (Fig.6a). The 3K deposits often contained lipid droplets (Fig.6a, white arrowheads). The deposits were often single-membrane bound and closely associated with late endo-lysosomal structures such as multivesicular bodies (Fig.6a, white asterisks). Some deposits showed αS immunoreactivity associated with the vesicular membrane (Fig.6a, black arrowheads) and contained lamellar membrane whorls typical of late endosomes/lysosomes (Fig 6a, upper right panel, black asterisk), accumulating in close proximity to the nucleus (Fig. 6a, lower right panel). Abnormally sized endo-lysosomes have been found to accumulate LAMP-1 foci embedded within pSer129+ inclusions in αS culture models of PD (Volpicelli-Daley et al, 2014) and are a common feature of fPD and sPD (Kett and Dauer, 2016). Also, previous neuropathological analyses described accumulations of such vesicles close to (Watanabe et al, 1977) or as part of (Forno, 1996) human LBs, and their sizes have been postulated to increase in early Lewy-type pathology (Kuusisto et al, 2003). Hypothesizing an early Lewy-like pathology from our tetramer-lowering αS mutations, we quantified the lipofuscin-like deposits by double-staining our sections with the neuronal cytoskeleton marker βIII-tubulin and the late endo-lysosomal membrane marker LAMP-1 (Fig. 6b) and analyzed both LAMP-1 accumulation and autofluorescence. A significant (>2-fold) accumulation of LAMP-1 was observed in 6 mo old 3K mutant mice (for LAMP-1 single staining, see Fig. 6c; graph in d), and the neurons often contained an increase in autofluorescent aggregates typical of lipofuscin deposits (Fig. 6c,d). We next immunostained sections for synaptotagmins, which are widely used markers for vesicular membranes and are implicated in calcium-dependent exocytosis (Cheng et al, 2015). While synaptotagmin-1 (syt-1) is localized to small synaptic vesicles, synaptotagmin-7 (syt-7) localizes to the neuronal plasma membrane and lysosomes (Zhang et al, 2011); thus, these two markers can help differentiate between vesicular pools. As expected from our ultrastructural findings of αS-rich vesicular accumulations (Fig. 4d,e), 3K αS monomers strongly co-localized with syt-1 at nerve terminals and with syt-7 in neuronal somata, while much less co-localization was detected in the expression-matched hu WT αS brain (Fig. S4a,b). To validate the finding, we conducted co-staining for syt-7 and LAMP-1. We found these markers co-localized in the αS-immunoreactive neuronal inclusions and in some enlarged processes in the 3K mice, but these were less apparent in WT and 1K (fPD) αS mice (Fig. S4c). The finding of an age- and K-dependent progressive increase of vesicle-rich αS+ lesions going from WT to 1K to 3K brains was further supported by quantifying the large foci of co-reactivity of either pSer129 and LAMP1 (Figure 6e,f) or syt-7 and total (15G7+) hu αS (1K-3mo vs. 1K-6mo: p<0.05; 3K-3mo vs. 3K-6mo: p<0.0.001; one-way ANOVA, post Tukey) (Fig. 6f).
Figure 6.
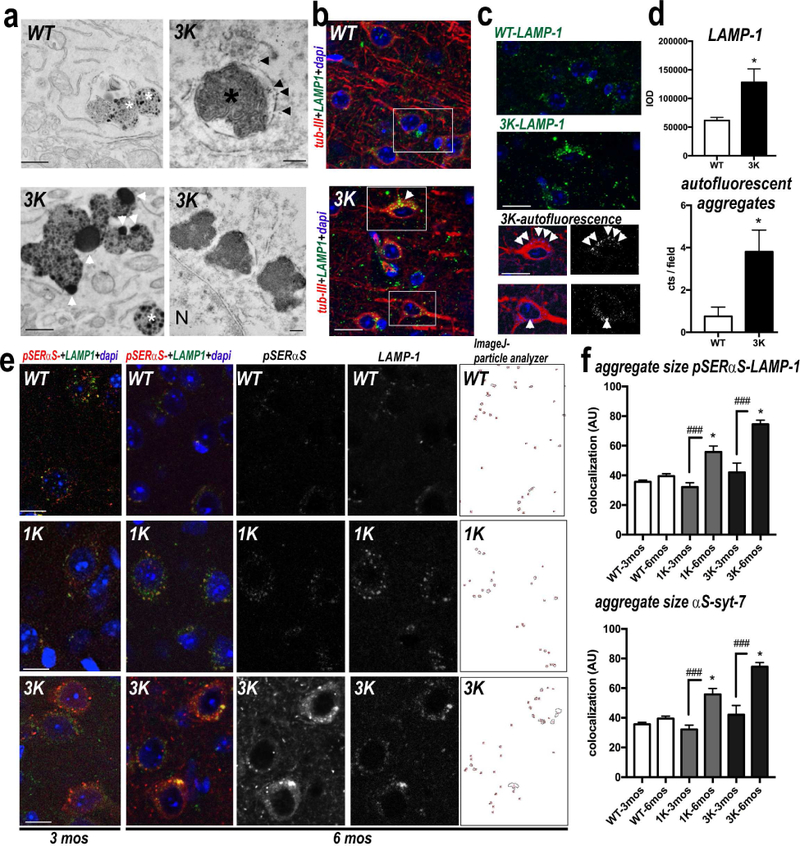
Abnormal αS accumulation at large perikaryal vesicles in motor cortex of 3K mice. (a) EM reveals large lipid-rich lipofuscin-like deposits (white arrowheads) in neurons of 6 mo old 3K (but not WT) brain. The deposits tended to occur near multivesicular bodies (white asterisks), were often single membrane bound (black arrowheads) and occasionally contained lamellar membrane whorls typical of late endosomes/lysosomes (black asterisk) and close to the nucleus (N). (b) Confocal microscopy (merged images) of brain sections triple-labeled for β-III tubulin (red), LAMP-1 (green) and nuclei (DAPI, blue). (c) Spectrally unmixed images of boxes from the merged images are enlarged to show overlap of LAMP-1 and autofluorescence in β-III positive neurons. (d): Quant. LAMP-1 reactivity and autofluorescent deposits in cortical neurons of 6 mo 3K vs. WT mice (n=10 fields each of brain sections of n=3 mice per genotype). (e) Confocal microscopy of cortical brain sections labeled for pSer129-αS (red) and LAMP-1 (green) as indicated. Far right: colocalized points were analyzed using Image J colocalization highlighter and particle analyzer plugin and (f) quant. (n= 3 mice per genotype at 3 and 6 mo). Adjacent brain sections labeled for hu (15G7) αS and syt-7 (Fig. S4) were quantified for co-localized aggregate sizes in n=10 fields each of sections from each genotype. Note significant age-dependent increases in 1K and 3K but not in the age-and (αS)-matched WT mice. Means ± SEM. *p<0.05, **p<0.01, (d: unpaired 2-tailed Student’s t test; f: one-way ANOVA post Tukey. Scale bars: (a) 1 µm, (b) 25 µm, (c, e) 20 µm.
Vesicle- and lipid-rich granules such as we observe in the 3K brain are reported to accumulate in midbrain and cortical neurons in PD and DLB and are hypothesized to contribute to the formation of mature Lewy bodies over time (Meredith et al, 2004). Thus, we searched for Lewy-type inclusions in the oldest 3K male mouse available to date (16 mo). We found infrequent large (~8 µm diameter) spherical inclusions close to the nucleus in neuronal sonata of cortex and substantia nigra (Fig. S5a). These Lewy body-like inclusions had a lipofuscin-rich center surrounded by densely packed filamentous material that was immunoreactive with the 15G7 hu αS (Fig. S5b). We also observed scattered dark, degenerated neurons and synapses in 3K cortical and midbrain sections and degenerated synapses and neurons that contained large intraneuronal lipid deposits (Fig. S5c).
3K mice have dopaminergic neurodegeneration, and the associated motor deficits are partly responsive to L-DOPA
To assess whether the pathological accumulation of 3K αS monomers at synaptic vesicles and endo-lysosomes and the associated motor phenotype correlated with a decrease in DAergic integrity, we quantified tyrosine hydroxylase (TH-) immunopositive axons and their terminals. We conducted the studies in male mice, since subtle differences in sex hormones can contribute to variances between male and female mice in neurochemistry [e.g., estrogen facilitates dopamine neurotransmission (Barth et al, 2015)]. We analyzed the dorsal striatum (caudate/putamen, CPu), which is rich in projections from the DAergic neurons of the substantia nigra pars compacta (SNpc). We measured a ~30% reduction in the density of TH+ fibers in in 6 mo old 3K mice compared to age- and expression-matched WT hu αS mice, which were in turn indistinguishable from Ntg mice (Fig. 7a,b). Since nerve fiber degeneration may precede DAergic cell loss in PD (Cheng et al, 2010), we used unbiased stereological cell counting of TH-immunopositive neurons in the SNpc of 6 mo 3K vs. hu WT tg mice and Ntg littermates. In addition to the significant decrease of TH-positive striatal fibers, we found a substantial reduction of TH-positive SNpc neurons (~27%; p<0.01) in 3K compared to Ntg mice (Fig. 7b). In addition to the loss of TH-immunopositive striatal terminals and nigral cell bodies, 3K mice displayed a ~39% decrease of striatal DA measured by HPLC (p<0.01 vs. Ntg) (Fig. 7b). We also observed a trend toward reduced striatal DA in the WT hu αS tg mice by HPLC measurements (~20%, p=0.09 vs. Ntg). The WT mice had no significant reduction in TH fibers, and their decrease in SNpc somata missed reaching significance (Fig. 7b). Notably, we found a discrete punctate pattern and numerous larger hu αS-immunopositive aggregates throughout the striatum and in the vicinity of SN DAergic neurons in 3K vs. WT mice (Fig. 7c, with graphs), consistent with our EM finding of a significant increase in hu αS accumulation on synaptic vesicles (Fig. 4d,e). In light of the reduction in TH fibers in 3K striatum, we asked whether other dopaminergic synaptic markers were decreased. In addition to TH, striatal protein levels of the vesicular marker VMAT-2 showed significant reductions (p<0.05) (Fig. 7d), further substantiating degeneration of the dopaminergic neuropil.
Figure 7.
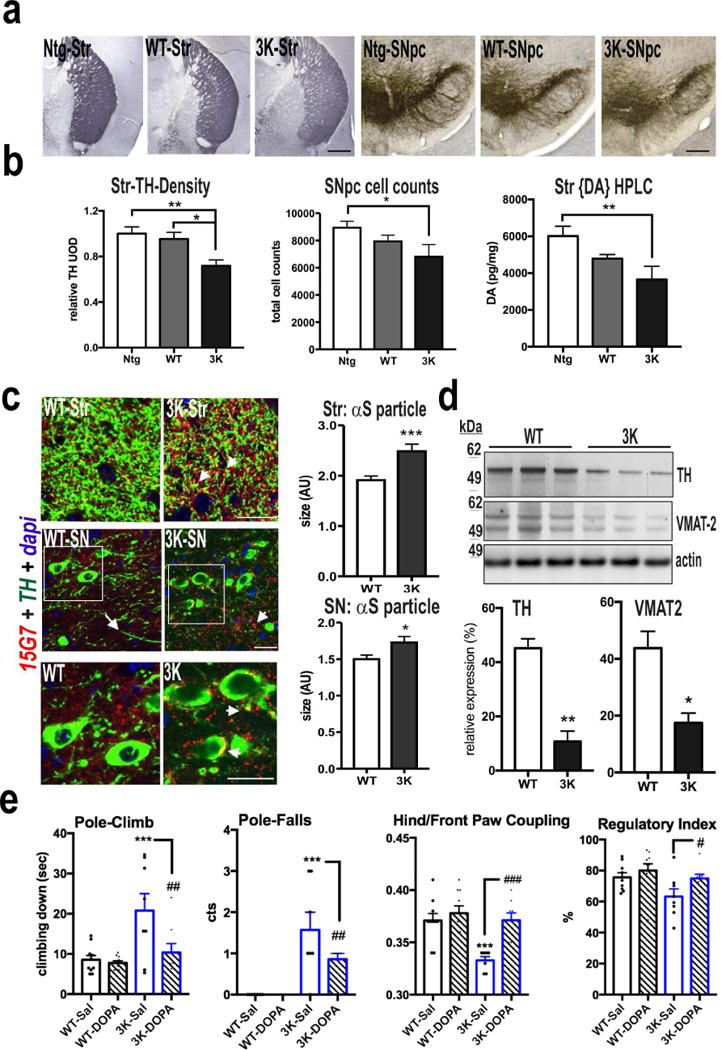
Dopaminergic neurodegeneration in 3K mice. (a) Representative images of tyrosine hydroxylase (TH) positive nerve terminals (left panel) and SNpc neurons (right panel) of Ntg, WT and 3K mice. (b) Relative TH optical density (total of 18 sections each group) was analyzed in the CPu and unbiased stereological counting determined the number of TH-positive neurons in the SNpc of 6 mo mice (Ntg n=4, WT n=5, 3K n=3); and HPLC assay of striatal DA in 6 mo male mice (Ntg n=5, WT n=7, 3K n=5). (c) IHC analysis of αS (15G7) co-localization in DAergic neurons of 3K mice shows small dot-like hu αS accumulations (arrowhead) and a decrease in TH-immunoreactive neuropil in TH-positive nigral neurons (arrow) in 3K brain sections and quantification of the hu αS-positive particle size in striatum and SNpc. (d) WBs of DAergic proteins in striatum of 6 mo old 3K and WT mice. (e) Pharmacological rescue of motor deficits of 3K mice by L-DOPA: increase in pole climbing performance and improved hind and front paw coupling and regulatory step index on a treadmill. Mean ± SEM. Ntg vs. WT vs. 3K, One-way ANOVA, post LSD, *,#p < 0.05; **,##p < 0.01, ***,###p<0.001; *WT-Sal vs. 3K Sal: #3K-Sal vs. 3K-DOPA: p<0.05, p<0.01, unpaired 2-tailed Student’s t test. Scale bars (a) 0.5 mm, (b) 25 µm.
Given the numerous phenotypic parallels of the 3K mice to PD, we analyzed the effects of L-DOPA treatment on motor coordination in the pole-climbing test and in the regularity of the step pattern on a treadmill (quantified by gait analysis software), using a blinded, randomized crossover trial. We used a relatively low dose of L-DOPA previously published to ameliorate a moderate DAergic phenotype and avoid induction of stereotypic behaviors in mice [e.g.(Chartoff et al, 2001)]. A single L-DOPA injection significantly improved pole test performance of the 3K mice, as measured by a decrease in falls while turning on the pole as well as a shorter time to climb down the pole (Fig. 7e: 3K-Sal vs. 3K-DOPA, p<0.01). L-DOPA treatment also reduced clasping and improved performance on a 4-limb wire hanging test, suggesting that it improved muscle tone in 3K mice (Fig. S6a). However, L-DOPA did not improve the highly abnormal 3K performance on the rotarod (Fig. S6a) and did not noticeably alter the tremor (as can be the case in L-DOPA-treated PD), suggesting a contribution from the clear-cut cortical pathology to these motor phenotypes. The deficits in wire hanging, in addition to changes gait patterns and pole climbing (Movies S1–S6) suggest abnormalities in the functional coupling of the limbs in 3K mice. We therefore photographically analyzed the fine paw patterns of mice running on a motorized belt. Saline-treated 3K mice displayed a significant abnormality of hind and front paw coupling and a decrease in the regularity of steps (Fig. 7e: WT-Sal vs. 3K-Sal, p<0.001 for hind/front paw coupling and p<0.05 for the overall step regularity index). 3K mice also showed less normal diagonal body support (2-paw body support), while the three-paw support was significantly increased, suggesting an insecure gait (Fig. S6b). Importantly, L-DOPA treatment normalized the impaired paw coupling (Fig. 7e: 3K-Sal vs. 3K-DOPA p<0.001) and the regularity of steps (Fig. 7e: 3K-Sal vs. 3K-DOPA p<0.05) Moreover, L-DOPA-treated 3K mice significantly increased their diagonal body support (Fig. S6b), and the stance graphs of the same 3K mouse treated with saline or L-DOPA revealed a marked improvement in gait after L-DOPA, with no such difference in the normal WT αS tg mice (Fig. S6c).
Discussion
Creating faithful animal models of human neurodegenerative disease is critical for therapeutic development but has proved to be a major challenge. For PD, DLB and related synucleinopathies, we describe a new mouse line that has an excess of membrane-bound pathogenic monomers derived from the unique approach of lowering a physiological form of αS, the α-helical tetramer. This results in multiple features of PD at the neuropathological, biochemical and behavioral levels.
Chronically shifting normal tetramers to excess monomers in vivo is associated with a PD-like state
Consistent with studies from our and other labs (Table S1), we detect physiological tetramers and related multimers in healthy, intact brain tissues of non-tg and WT hu αS tg mice (Fig. 2b-g), and these animals do not develop features of PD. Our intact-cell crosslinking approach confirms a significant decrease of intracellular tetramers and increase in monomers in mice with the fPD E46K (1K) mutation, and a much greater lowering of the T:M ratio in mice with an amplification of this fPD mutant (3K), in accord with previous findings (Dettmer et al, 2015a) and new data (Fig. 1) in cultured neural cells. The phenotypes of the 1K and 3K lines are not due to differences in αS expression, as αS levels are similar and both were even slightly lower than in the hu WT tg brains (Fig. 2d). The only fPD E46K (1K) line available for this study expressed a BAC construct that includes the entire hu αS gene, in contrast to the transgene encoding just the hu αS protein in our 3K line. A disadvantage of this comparison is that the 3K line lacks the hu regulatory elements that the 1K line has. On the other hand, the two lines have closely matching overexpression of hu αS, and the fact that there is a graded effect of hu WT vs. 1K vs. 3K across the 3 lines means that distinct expression constructs (BAC vs. transgene) can each produce the biochemical phenotype. We observe all of our key PD-like changes in the 1K vs. WT line, and these changes are more robust in the 3K mice, with all 3 lines having similar αS expression. The changes documented in the 1K mice include a significant shift in the T:M ratio (Figs. 2,S1), Ser129 phosphorylation (Figs. 5,S3), age-dependent inclusion formation (Figs. 5,6) and motor signs (Fig. S2). The lesser amount of pathogenic 1K αS monomers is a result of the smaller decrease in T:M ratio, as expected from just one E→ K substitution in the αS amphipathic helix, and as we established in our earlier culture work on 1K vs. 2K vs. 3K (Dettmer et al, 2015a) and confirmed with our new data in Fig. 1. While only the 1K is driven by the hu αS regulatory sequences (BAC construct), one can view this as a molecular biological plus, in that we observe the PD-type changes of pathologically excessive αS monomers PD-type changes from the loss of aS tetramers in an entirely independent line.
The shift from cytosolic to membrane-associated αS by the 3K mutation is observed both biochemically (Figs. 2,S1) and ultrastructurally (Fig. 4d,e). The robust PD-like posttranslational modifications of αS in 3K mice notwithstanding, the decrease in the brain T:M ratio was the largest biochemical change we observed in this mouse (~70% decrease vs. WT hu αS) (Figs. 2b-g) and was already present by age 3 mo (Fig. 2b). However, this may not be the common pathogenic mechanism of all fPD-causing missense mutations, as the documented T:M decreases of A30P and G51D αS (Dettmer et al., 2015a) involve amino acid changes which result in less, not more, membrane association and thus act independently of an increase in membrane affinity. Interestingly, CD spectroscopy of wt and fPD recombinant αS showed decreasing α-helical content in the order wt>A30P>A53T>E46K (Wang et al, 2011), closely resembling the relative decreases in cellular T:M ratios of our previous neuronal culture studies (Dettmer et al, 2015a). The reported ages of onset of clinical symptoms in carriers of the five fPD missense mutations (Kruger et al, 1998, Appel-Cresswell et al, 2013, Proukakis et al., 2013 (Neurology 80, 1062–1064; DOI: 10.1212/WNL.0b013e31828727ba) may also agree in general with the relative decrease in T:M ratio in our culture studies. Therefore, creating new mouse models with an amplified effect of G51D on the T:M ratio could help clarify the PD-biochemical mechanisms apparently occurring in the absence of the enhanced membrane binding seen with E46K.
The central finding of this study is that an excess of monomers resulting from amplifying the known tetramer-lowering effect (Dettmer et al, 2015a) of the E46K mutation by inserting analogous mutations into two adjacent KTKEGV motifs can induce vesicle-rich, αS-containing aggregates, and this leads to age-dependent αS neuropathology associated with a progressive motor phenotype including prominent resting tremor and deficits in limb function and gait that respond in part to L-DOPA. DAergic SNpc neuron loss (vs. VTA neurons) coupled with decreases in striatal TH fibers and DA levels as well as prominent cortical αS aggregates and degeneration of the cortical neuropil underlie the tremor and limb and gait deficits that resemble cardinal features of human PD. Intriguingly, although 3K male and female mice have similar αS expression levels (Fig. S1), 3K male mice have more prominent motor changes than 3K females (Fig. S2), reminiscent of what occurs in hu PD. Several studies have reported sex differences in PD clinical profiles that manifest in women with delayed average onset (by ~2 years), slower rate of motor decline and more tremor, whereas rigidity is more prominent in men (Gillies et al, 2014) and female sex hormones are reported to facilitate striatal dopamine neurotransmission (Barth et al, 2015). αS is normally involved in membrane vesicle trafficking, and altered αS-membrane interaction is emerging as a key factor in PD (Logan et al, 2017). Thus, sex hormones likely contribute to the male/female differences in severity of our 3K mice, an important topic to address further using this line.
Decreases in the αS tetramer:monomer ratio may underlie certain genetic forms of PD
Before 2011, αS was assumed to exist as a natively unfolded monomer in cells, although it was long known that recombinant monomers could become α-helically folded in vitro upon contact with small, highly curved lipid vesicles (Davidson et al, 1998). We and others (Table S1) reported evidence that a portion of cellular αS existed in apparent α-helical tetramers if isolated under non-denaturing conditions or crosslinked in vivo with cell-penetrant crosslinkers. We found that the tetramers were highly metastable, as cell lysis disassembled them unless performed at high protein concentrations (‘molecular crowding’) (Dettmer et al, 2013). This suggests the presence of a “limiting factor”, perhaps a small lipid, that stabilizes the tetramers in vivo. Tetramers are soluble and physiological (β- and γ-synuclein also exist as tetramers), and neurons require a normal T:M equilibrium to avoid excess monomers misfolding into abnormal oligomers, which can be associated with clustering of cytoplasmic vesicles into round inclusions associated with neurotoxicity (Dettmer et al., 2015a, Dettmer et al., 2015b; Dettmer et al., 2017).
αS missense mutations that cause aggressive, early-onset forms of fPD have all been shown to decrease the T:M ratio in neurons, including in iPSC-derived human neurons at endogenous expression levels and in A53T transgenic mouse brain (Dettmer et al, 2015a). Recently, hu neurons bearing Gaucher-causing mutations in GBA1 were found to also shift endogenous WT αS tetramers to excess monomers, and the decreased T:M ratio could be corrected by genetic or pharmacological lowering of the accumulated glucosylceramide (Kim et al, 2018). This striking finding [replicated in our group (Dettmer et al., unpubl. data)] strongly supports the relevance of the T:M equilibrium to ‘sporadic’ PD, since GBA1 loss-of-function mutations are a major genetic cause of PD, and such patients were previously considered to be part of idiopathic PD. Importantly, αS is wild-type in Gaucher’s carriers and patients, so pathogenic shifts in the T:M ratio do not require mutating the protein. These hu genetic findings, coupled with the striking PD-like phenotypes of our tetramer-abrogating mice, support a new paradigm for PD pathogenesis in which a disturbed T:M equilibrium can lead to progressive synucleinopathy.
Here, we correlated a shift of αS T:M ratio with the development of PD-relevant neurotoxicity in vivo. The significant decreases in both TH protein and DA neurotransmission (Fig. 7b) as well as the abnormal Golgi-Cox stained neuropil in the 3K cortex (Fig. 5c) suggest that cell and fiber loss from abrogating tetramers is likely mediated by the pathological vesicle association of the excess monomers.
C-terminal αS truncation and pSer129 phosphorylation are consequences of a pathogenic T:M shift
C-terminally truncated αS species are reported to increase significantly in fPD (Li et al, 2005), sPD and DLB hu brains (Anderson et al, 2006, Nuber et al, 2014, Nuber et al, 2013), and they may promote aggregation of αS (Mishizen-Eberz et al, 2005) and induce DAergic neuronal dysfunction in models (Tofaris et al, 2006). Our findings that a ΔC-αS truncation site in 3K brains matches a calpain-1 cleavage site (aa 119) of pure, fibrillized αS in vitro and that this ΔC truncation is prevented by a calpain-specific inhibitor in neural cells expressing the tetramer-lowering 3K or 3R mutants (Nuber et al, unpubl. data) suggest that calpain-1 activity contributes to pathological C-terminal truncation in our PD models.
The predominant post-translational modification of 3K αS, however, is extensive Ser129 phosphorylation (Figs. 4–6, S3). This modification is prominent in fPD (Lesage et al, 2013) and DLB (Obi et al, 2008) as well as in various tg models, including mice overexpressing fPD A53T or A30P αS (Wakamatsu et al, 2007, Schell et al, 2009). The increases in pSer reactivity and inclusion sizes seen in 1K mice are markedly augmented by the excess monomers in heterozygous 3K+/− mice and even more so in homozygous 3K+/+ mice, and the hyperphosphorylation is associated with phenotypic progression, consistent with a positive correlation between accumulation of the pSer129 modification and the severity of PD symptoms in humans (Walker et al, 2013).
An early, functionally relevant dopaminergic deficit in the tetramer-abrogating 3K mice
To our knowledge, the early DAergic deficiency, resting tremor, and progressive limb and gait impairments partially responsive to L-DOPA are unique for a PD mouse model (Table S1), thus representing a useful model for mechanistic studies and therapeutic screening. In cultured neurons, modestly increased expression of WT αS not exceeding the levels in human brain allows retention of the physiological T:M ratio and preserves neuronal integrity (Dettmer et al, 2015a). Consistent with this concept, expression of WT αS (Richfield et al, 2002) or expressing the single E46K (1K) fPD mutant by conventional transgenesis (1–2 fold above endogenous αS) caused only subtle neurological deficits in aged (~20 mo) homozygous mice or else required an additional insult with PD-related neurotoxins (Emmer et al, 2011, Cannon et al, 2013), whereas double-mutant (A30P+A53T) hu αS promotes frank cytopathology in mouse brain (Table S2). In contrast to single-mutant (1K) tg rodents in prior reports (Emmer et al, 2011, Cannon et al, 2013) and the present study, 3K synucleinopathy is readily apparent beginning at 3 mo, and the resultant decreases in nigrostriatal TH+ somata and fibers and increases in less soluble and more phosphorylated αS coincide with an early-onset, progressive motor syndrome.
Membrane alterations and αS filamentous inclusions resembling Lewy bodies occur in 3K mice.
Our data demonstrate that 3K monomers interact with vesicular membranes more than WT αS, and this includes synaptic vesicles and endo-lysosomal structures that co-label for LAMP-1 and syt-7. Synaptotagmins (syt) are calcium-sensitive proteins capable of aggregating membranes and thereby stimulating fusion if SNARE complexes are in place (Hui et al, 2011). Since αS monomers can stabilize some membranes against fusion (Westphal and Chandra, 2013), monomer accumulation could perturb neurotransmission and alter endo-lysosomal protein degradation, particularly in neurons with high calcium flux (Burbulla et al, 2017). The increase in LAMP-1 puncta that co-localize with pSer+ aggregates and associate with endo-lysosomal changes in our 3K mice (Figs. 6,S4) may serve as an early feature of eventual Lewy-type pathology (Volpicelli-Daley et al, 2014). In hu EM studies, such lysosomal/vesicle accumulation was observed in fPD and sPD and discussed as a potential step toward formation of so-called pale bodies and perhaps later Lewy bodies (Kuusisto et al, 2003). Our new model did not display clear-cut accumulation of filamentous αS lesions at age 6 months, but a few appeared in the areas of αS-rich vesicle clusters at a much later age (16 mo).
Thus, the presence of vesicle accumulations and elevated full-length, truncated and phosphorylated αS monomer levels in PD patient brains support the PD relevance of the T:M shift and the ensuing round, phosphorylated cytoplasmic inclusions that are robust features of our middle-aged mice and may be a prerequisite to developing mature Lewy-type inclusions at an older age.
Implications of the new mouse model for mechanistic and therapeutic studies of PD and DLB
Mechanistically, we hypothesize that the reason this new mouse model closely mirrors key features of human PD (Table S2) is that the underlying event – chronically altering the neuronal T:M equilibrium – is likely to occur in PD, DLB and other human synucleinopathies. Genetic evidence for this concept comes from human fPD αS missense mutations (Dettmer et al, 2015a) and now from GBA deficiency (Kim et al, 2018). A recent analysis of the molecular mechanism underlying 3K and other tetramer-reducing mutations suggests an alteration of proper transient formation of the amphipathic helix that is induced in WT αS by membrane binding (Dettmer et al, 2017). Previous experimental studies reported that E46K (1K) αS has a higher affinity for binding to negatively charged vesicles than does WT αS (Choi et al, 2004), likely via abnormally increased, membrane-induced amphipathic helix formation (Wise-Scira et al, 2013), and may thereby lead to αS oligomer cytotoxicity (Winner et al, 2011); 3K may enhance this abnormality. Thus, reversible αS tetramerization and amphipathic helix formation appear to regulate a normal, transient vesicle-binding function of αS (Wang et al, 2014), and abrogating normal tetramers interferes with this function and has pathogenic consequences, as we now show in vivo.
Finally, the therapeutic implications of the recapitulation of numerous PD phenotypes in the 3K mice include screening for small molecules that can stabilize physiological tetramers or decrease the levels of excess full-length and truncated monomers and their associated vesicle aggregates. The reproducibility of inclusion formation and neuronal dysfunction in both cultured cells and living mice undergoing tetramer-to-monomer disequilibrium provides a combined in vitro and in vivo platform to discover and validate such compounds. This approach could be analogous to the successful biochemical stabilization of physiological tetramers of transthyretin by the approved drug, tafamadis, which has clinical benefits for patients with that progressive amyloidosis (Coelho et al, 2013). In addition, our mouse models can be used to identify and characterize small molecules that enhance the clearance of αS inclusions and otherwise normalize the several PD-like posttranslational modifications of αS by restoring the physiological T:M ratio.
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dennis J. Selkoe (dselkoe@bwh.harvard.edu).
Experimental Model and Subject Details
Generation of E→ K mutant mice and their treatment.
Transgene expression in pThy1 is controlled by the Thy1.2 neuronal regulator. To generate Thy1-WT, Thy1–2K and Thy1–3K αS tg mice, the full-length human WT αS cDNA was ligated into pTS (2) vector (Zhang et al, 2014), and mutagenesis was used to generate the 2K and 3K variants, as previously reported (Dettmer et al, 2015a). Briefly, pThy1-WT-αS, pThy1–2K-αS and pThy1–3K-αS plasmids were generated by XhoI restriction enzyme (New England Biolabs, Ipswich, MA) and digestion of pThy1/Flpbow3 (Addgene plasmid #45181, gift of Dr. Joshua Sanes) (Cai et al, 2013) was followed by insertion of the respective SNCA gene variant by In-Fusion cloning (Clontech, Mountain View, CA). The WT or K-mutant cDNAs were micro-injected into C57BL/6J one-cell embryos. All animal procedures were approved by the Institutional Animal Care and Use Committee at BWH (IACUC protocol #05022). Founder animals were identified by PCR of DNA from ear biopsies using primers specific for the Thy1 promoter and the transgene construct (Thy1-F: 5´- tctgagtggcaaaggaccttagg-3´, Syn-R: 5´-gtggggctccttcttcatt-3´). Out of 5–6 potential founders per line, we established two lines (2K; #3520; 3K: #3798) with low overexpression (similar level to mouse endogenous αS); one 2K line (#3515) with abundant overexpression (~4 fold); one 3K line (#3817) with moderately high overexpression (~2–3 fold); and one WT line (#3877) with a moderately high overexpression (2–3 fold). The 3KL+/+ total hu αS expression level is ~20% compared to either WT, 1K or 3K αS. We chose to focus our characterization on 3K (#3817), which has moderate overexpression at a similar level to our WT line to examine the effects of strong tetramer abrogation. We plan to make this unique new 3K model available to the research community (through a central mouse laboratory) to pursue PD/DLB mechanisms and drug discovery.
All mice were bred to the C57BL/6J background, were viable and displayed similar expression levels in males and females. Mice of all lines bred successfully, except 3K-3817 males due to their severe motor phenotype. Therefore, the 3K-3817 colony was maintained by breeding 3K-3817 female mice to non-tg C57BL/6J males. We characterized WT-3877 (designated “WT”) vs. 3K-3817 (designated “3K”) in the present study; mouse numbers of the other lines were not yet sufficient to conduct the extensive characterization at the biochemical, structural and longitudinal behavior levels we report here for line 3K-3817 vs. the expression-matched WT-3877 line. B6N.Cg-Tg(SNCA*E46K)3Elan/J mice (designated 1K) were purchased by Jackson Laboratories. An initial characterization of the latter mice that includes DAergic integrity is found online (https://www.michaeljfox.org/files/MJFF_SfN_aSyn_Poster.pdf). We used 3–6 mos 3K (#line3817) as well as 6–8 mos 3KL (#line 3798) and 3–6 mos 1K (B6N.Cg-tg(SNCA*E46K) male and female mice to analyze details in hu αS (Fig. 2 and related S1), and pSer aggregation patterns (Fig. 5 and related Fig. S3), and motor phenotypes (Movie S1–4, and related Fig. S2) vs. age-and gender-matched WT ctl mice (#3817) or Ntg littermates (indicated in each figure). We used 3–16 mos 3K (#line 3817) male mice for longitudinal behaviour phenotypes (Fig.3 and related Movie S5–7), evaluation of DAergic integrity (Fig.7, related Fig. S6) and ultrastructural analyses (Fig. 4–6, related Fig. S5) vs. age-matched WT and Ntg littermates (indicated in each figure). All mice were kept in normal 12 h light/12 h dark cycles and had free access to food and water. For L-DOPA treatment: mice received benserazide (Sigma, 12.5 mg/kg, IP) 20 min prior to L-DOPA (Sigma, 12.5 mg/kg, IP) and the L-DOPA 10 min prior to the start of behavioral testing in a randomized, blinded crossover design. Behavioral studies were conducted at the NeuroBehavior Laboratory of the Harvard NeuroDiscovery Center (HNDC).
Cell culture and transfection
BE(2)-M17 is a clone of the SK-N-BE(2) neuroblastoma cell line that was established in November of 1972 from a bone marrow biopsy taken from a 2 year old male with disseminated neuroblastoma.
Human neuroblastoma cells (BE(2)-M17D ATCC number CRL-2267) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 50 units/ml penicillin, 50 µg/ml streptomycin, and 2 mM L-glutamine. M17D cells were transfected at 60% confluency using Lipofectamine 2000 according to the manufacturer’s protocol. The single (E35K, E46K, E61K) and 3K constructs were synthesized as a GeneArt String DNA fragment (Gene Art/Life Technologies) and inserted into pcDNA4 with the In-Fusion HD Cloning Kit (Clontech) as previously described(Dettmer et al, 2015a, Dettmer et al, 2017). Cells were harvested 48 h after transfection. DSG crosslinking and western blotting was performed as previously published(Dettmer et al, 2015a, Dettmer et al, 2017). The cytotoxic effect of the single K-mutants was measured 24h after transfection using the LDH based CytoTox 96®Non-Radioactive Cytotoxicity Assay according to the manufacturer’s instructions (Promega). Cells were also measured by percentage confluency at the same time point using the proprietary IncuCyte program.
Method Details
Intact-cell crosslinking of brain tissue
Dissected brain regions were gently minced into small bits with a razor blade, and the brain bits were washed free of released cytosol and re-suspended in PBS with EDTA-free Complete protease inhibitors (Roche Applied Science). Intact-cell crosslinking was then conducted on the washed brain bits as previously described(Dettmer et al, 2013) with minor modifications. Briefly, the cell-permeable crosslinker DSG was prepared at 1 mM final concentration in DMSO immediately before use. Samples were incubated with crosslinker for 40 min at 37°C with rotation. The r eaction was quenched by adding Tris, pH 7.6, at 100 mM final concentration and incubated for 10 min at RT. After quenching and aspiration of the supernatant, proteins in intact tissue were extracted directly in TBS/1% Triton X-100 (below).
Sequential tissue extractions
The regional expression pattern of αS was initially examined at age 6 mo. Mice were anesthetized, decapitated and the brains dissected on a chilled stage. Sequential extractions were performed as described(Nuber et al, 2013). Briefly, tissues were homogenized in 2.5 volumes of TBS+ [50 mM Tris-HCl, pH 7.4, 175 mM NaCl; 5 mM EDTA; protease inhibitor cocktail (Calbiochem, CA)] and spun for 30 min at 120,000g. The pellet was subsequently extracted in TBS+ containing 1% Triton X-100, then in TBS+ containing 1 M sucrose, The TX-insoluble pellet was then extracted in RIPA buffer (TBS+, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate), with each extraction step followed by ultracentrifugation for 30 min at 120,000g.
Western blot analyses
For western blotting, 10–25 µg total protein of sequential extracts of dissected mouse brain regions or 0.5 µg N-acetylated recombinant fibrillized αS that had been cleaved by calpain were each run on 4–12% Bis-Tris gels (Invitrogen, Carlsbad, CA) and electroblotted onto nitrocellulose membranes (Millipore, Bedford, MA). For improved immunodetection of αS (monomers of which are prone to washing off filters(Newman et al, 2013, Lee and Kamitani, 2011)), the membranes were fixed in 0.4% paraformaldehyde (PFA) for 20 min. After washing in phosphate-buffered saline (PBS), membranes were blocked for 1 h at RT in PBST (phosphate-buffered saline with 0.2% Tween-20) containing 5% bovine serum albumin (BSA). Blots were then incubated with human-specific αS antibody (ab) (15G7, Enzo; 1:500), or abs that recognize both rodent and human αS (syn1, clone 42, BD Bioscience; or C20, Santa Cruz) (each 1:2000) or ab against phosphorylated (ser129) αS (51253; Abcam; 1:5000) in PBST containing 5% BSA overnight. After washing with PBST, membranes were probed with appropriate secondary antibodies (1:5000, American Qualex, CA), visualized with enhanced chemiluminescence (ECL, PerkinElmer, Boston, MA), and analyzed with the VersaDoc gel imaging system. Proteins were normalized to □-actin (A5441, Sigma; 1:3000) used as a loading control. Quantification of signal intensities was performed as described (Nuber et al, 2008).
Synthetic αS fibril assembly and calpain digestion
Purified recombinant αS was fibrillized as reported (Herrera et al, 2008). Calpain digestion of the recombinant αS was performed as described(Nuber et al, 2013). Briefly, 15 µg of recombinant, fibrillized αS was incubated at 37°C with calpain I (2 U) in buffer containing 40 mM HEPES and 1 mM calcium. To identify fragments specific to calpain cleavage, 4 mM calpeptin was added to calpain buffer and co-incubated for 60 min. To stop proteolysis, LDS sample buffer with 100 mM DTT (Invitrogen) was added and samples either heated at 90 °C for 5 min and used f or Western blot analyses or stored at −20°C.
High pressure liquid chromatography
HPLC was conducted as previously described (Nuber et al, 2013). To estimate striatal monoamine levels at age 6 mo in WT (n=7), 2K or 3K αS tg (n=7) and non-tg (Ntg) control mice (n=7), the mice were deeply anesthetized by CO2, quickly decapitated, and the striata dissected on ice, homogenized in 0.5 M perchloric acid, centrifuged, filtered, and stored at −80°C un til analysis for monoamine content. Samples containing 500 pg dihydroxybenzylamine as an internal standard were analyzed by HPLC with electrochemical detection. The column was an ET 125/2, Nucleosil 120–5, C-18 reversed phase column (Macherey & Nagel, Düren, GER). The mobile phase consisted of 75 mM Na H2PO4, 4 mM KCl, 20 µM EDTA, 1.5 mM SDS, 100 µl/l diethylamine, 12% methanol and 12% acetonitrile adjusted to pH 6.0 using phosphoric acid. The electrochemical detector (Intro, Antec, Leyden, NL) was set at 500 mV vs. an ISAAC reference electrode (Antec) at 30°C. Data are reported as means ± SEMs. Statistical analysis used Student’s t test for pairwise comparisons; a value of p < 0.05 was considered significant.
Immunohistochemistry
Mice were anesthetized with an intraperitoneal injection of a mixture of xylazine (10mg/kg)/ ketamine (100mg/kg) followed by intracardiac perfusion with PBS and ice-cold 4% (w/v) PFA in PBS (pH 7.4). The brain was dissected from the skull and post-fixed in 4% PFA for another 48 h at 4°C. Brains were cut i nto 25 µm cryotome sections and immunostained as described(Nuber et al, 2008). Briefly, after treatment with H202 (0.3% in PBS, 20 min) and blocking (10% normal goat serum, 0.1% Triton X-100, 1 h), sections were incubated for 12 h at 4°C with anti-human αS (15G7; 1:50, Enzo), anti phosphorylated (pSER129) αS (51253; 1:50.000; Abcam) or 105 (gift of Dr. Zago, Prothena Biosciences, SF, CA; 1:1000) in 10% normal goat serum. After washing with PBS, sections were incubated with the respective biotinylated secondary antibodies (1:200 in PBS; Vector; and subsequently transferred into ABC solution (1:500 in PBS; Vectastain Elite Kit, Vector Laboratories) for 1 h and visualized with 3,3’-diaminobenzidin (DAB). Double-labeling was performed as described(Nuber et al, 2013). Briefly, sections were blocked in 10% normal donkey serum and incubated overnight at 4°C with antibodies to hu αS (15G7, 1:100; Enzo), anti phosphorylated (pSER 129) αS (51253; 1:2000; Abcam), synaptotagmin-1 (syt-1) (MAB 4364, 1:5000; RD), synaptotagmin-7 (syt-7) (SAB 4200160, 1:2500; Sigma), tyrosine hydroxylase (TH) (AB 152, 1:500; Millipore), mouse-III tubulin (78078, 1:5000; Abcam), or LAMP-1 (25245, 1:1000; Abcam). This was followed by incubation with FITC-conjugated secondary antibodies (1:1000 in PBS) for 4 hours at RT. Confocal microscopy was conducted with an Axiovert 35 microscope (Zeiss) mounted on a MRC1024 laser scanning confocal microscope (Bio-Rad). For confocal images of either syt-1, syt-7 or TH with 15G7-reactive human-αS, each image was color-balanced. An Image J plug-in called “Colocalization highlighter” created a mask of either syt-7, LAMP-1 or TH pixels that overlapped with hu-αS pixels. The sizes of the co-localized pixels on the resultant 8-bit images were quantified using the analyze particle function plugin of ImageJ. For Golgi-Cox stainings, brains of n=6 mice per group were dissected from the skull and incubated in rapid Golgi-Cox solutions for 14 days, following manufacturer’s instructions (FD Rapid GolgiStain™ Kit, FD Neurotechnologies).
Electron microscopy
Mice underwent transcardiac perfusion with 4% PFA and the dissected brains were fixed in 4% PFA for another 48 h, subsequently in 2% PFA for 24 h, and then transferred into 0.5% PFA. Dissected brain regions were quenched with 0.2 M glycine in PBS for 10–15 min at RT and then permeabilized by washing for several times in PBS containing 0.1% Triton X-100 (PBST), then blocked for 30 min in 1% (w/v) bovine serum albumin in PBST at RT. A rat anti-human αS antibody (15G7,1:50, Enzo) in PBST containing 1% BSA was added to sections and incubated overnight at 4°C. Sections were washed five times for 10 min i n PBS followed by incubation with rabbit anti-rat bridging antibody (1:100 Abcam ab6703) in PBS containing 1% BSA for 1h at room temperature, then washed five times in PBS, followed by incubation with 15 nm Protein A-gold particles (1:50, Utrecht University Med Ctr) in PBS containing 1% BSA for an hour at room temperature. Sections were subsequently washed in PBS and then fixed in 1% (v/v) glutaraldehyde in PBS for 30 min. For Epon embedding, slices were incubated in 0.5% (w/v) osmium in ddH2O for 30 min, washed three times in ddH2O then stepwise dehydrated (each step for 10 min) in 70% (v/v) ethanol, 95% (v/v) ethanol and twice in 100% (v/v) ethanol. The slices were incubated in propyleneoxide, infiltrated in 1:1 propylenoxide/TAAB Epon, embedded in fresh TAAB Epon (Marivac Canada Inc) and polymerized at 60 °C for 48 h. Each block was cut into 60 nm ultrathin sections using a Reichert Ultracut-S microtome. Sections were placed onto copper grids and stained with uranyl acetate and lead citrate. The sections were examined using a JEOL 1200EX transmission electron microscope. Images were recorded with an AMT 2k CCD camera at 20,000–30,000× magnification.
Assessment of striatal DA fiber integrity and stereological counting
40-µm-free floating sections were rinsed in TBS (0.15M NaCl, 0.1M Tris-HCl, pH 7.5) and endogenous peroxidase activity quenched with 0.6% H2O2 in TBS for 30 min at RT. Unspecific protein binding was blocked with 3% normal donkey serum in TBS containing 0.3% Triton X-100. Sections were incubated with rabbit TH (AB152, 1:500, Millipore) over night at 4°C. Following 3 rinses with TBS, sections were incubated with secondary anti-rabbit (1:1000, Dianova 711–065-152) in TBS containing 3% normal donkey serum, washed, and subsequently transferred into ABC solution (1:500 in PBS; Vectastain Elite Kit, Vector Laboratories) for 1 h and visualized with 3,3’-diaminobenzidin (DAB). Brain sections across a +4.8 to +3.5 interaural range were chosen, referring to the Paxinos and Franklin mouse brain atlas(Paxinos and Franklin, 2001). Sections from all genotypes were simultaneously stained and digitized using constant imaging settings (Stereoinvestigator, MicroBrightField, Colchester, VT), and subsequent analyses were performed on a blinded basis. Images were converted to gray scale, and the mean gray value intensity was measured in the caudate/putamen (CPu) and in the adjacent corpus callosum (cc) to correct for signal background. Mean gray values were converted to uncalibrated optical density (UOD) using ImageJ 1.46r software (NIH). The UOD of TH signal in the CPu was calculated by the formula CPufinal(UOD) = CPu(UOD) – cc(UOD), similar to a previously published study (Schlachetzki et al, 2014).
For stereological counting, every fourth free-floating section of 40 µm thickness, encompassing the full rostro-caudal extent of the substantia nigra (SN) or ventral tegmental area (VTA), was stained for TH (for 36 as described above. This approach resulted in 9 to 10 sections in which the SN or VTA was identified. The number of TH positive neurons in the SN pars compacta (SNpc) or VTA of one hemisphere was stereologically assessed using the optical fractionator principle and using the Stereo Investigator software (MicroBrightField, Colchester, VT). The region of interest was outlined using a 2.5× objective and sampled with a 40× objective using a square optical frame ( SNpc and VTA: 50 × 50 µm) and sampling grid (SNpc: 60 × 60 µm and VTA: 75 × 75 µm), with 2 µm guard zones above and below the counting. The estimated coefficient of error (Gundersen, m=1) ranged from 0.05 to 0.07, and the estimated section thickness was ~19 µm. The reported “total” number of TH positive neurons is the number estimated by number-weighted section thickness multiplied by a factor of 2, representing both hemispheres.
Behavioral testing
Tremor assessment
Tremor assessment was conducted by an investigator blinded to the genotype. To measure the depth of tremor in 6 mo old 3K and WT mice (n=6 per group), we used short-term (8–10 sec) video recording of mice standing on a small platform (5×3cm) (thus excluding gross posture changes) and subsequent stacking of ~60 frames. The layers were converted to a smart object and then to the stack mode ‘mean’ in order tosum the body movements in a single frame image using Adobe Photoshop software. This method allowed evaluation of the distance between the captured tremulous spine images, which were inverted for clarity. The contrast of the tremulous spine images after inversion was used to evaluate their ‘sharpness’ by mageJI pixel intensity measurement.
Clasping
For clasping scores, mice were held by their tails and analyzed for their behavior for 10 sec. Animals would receive a score if a dystonic clasping event occurred for longer than 2 sec. One point was scored for dystonic movements of one hind-limb, 2 points for both hind-limbs and 3 points if mice displayed a combination of hind- and fore-limb and trunk clasping.
Open field
For assessment of spontaneous exploratory behaviour, mice were randomly placed in 27 × 27 cm Plexiglas boxes (20.3 cm high) and activity was recorded for 30 min with the Activity Monitor, Med Associates (St. Albans, VT) (Version 5.9). Activity is expressed as sum of beam breaks (measured per 1 min interval) and as relative % to WT activity. Locomotor activity was studied in 3 and 6 mo old animals (see Results).
Pole test
Mice were placed on top of a 50 cm vertical pole (all-thread metal rod) with a diameter of 1 cm and tested for their ability to descend from a round (‘assistant’) platform (2.5 cm diameter; head-down guidance) and/or mice were placed head-up on the metal rod and tested for their ability to turn around and descend the pole (snout first). Scoring began when all four paws reached the pole and/or the animal initiated the turning movement. The time to make a complete 180° turn and latency to reach the cage floor were recorded. Maximal duration time was set to 60 sec to avoid exhaustion. If the mouse made a turn and fell, the maximum duration time was assigned. The test consisted of 5 consecutive trials with the platform (climbing down) followed by an inter-trial pause of 30 min and then 5 consecutive trials without platform (turn). Average times to ‘climb down’ and to ‘turn’ were calculated from the best 4 out of 5 consecutive trials, and the sums of falls were calculated for each mouse.
Gait scan
To assess motor function and coordination in walking mice, automated gait analysis was performed using Treadscan (Cleversys Inc, Reston, VA). Gait patterns of 3–6 mo mice were measured for 25 sec at a speed of 13 cm/sec on a transparent running belt illuminated by a LED light and reflecting footprints captured by a video camera positioned underneath the walkway.
Rotarod
Motor coordination and motor skill learning were evaluated using an accelerating rotarod (Ugo Basile), and time spent on the rod was recorded. The first two days consisted of a habituation trial at constant speed (4 rpm for 5 min), followed by two trials of 4–40 rpm progressive acceleration within 5 min for 5 consecutive days, including 2 trials each day. On the next two days, the mice were tested only on the accelerating trials (4–40 rpm, 5 min). An inter-trial pause of at least 1 h was applied to avoid fatigue and stress, and a maximum cutoff of 5 min was used. Motor coordination was evaluated by comparing the mean latency to fall over the 4 consecutive days between groups.
Quantification and Statistical Analysis
Details regarding each statistical test, biological sample size (n) and p value can be found in the corresponding figure legends. All data are represented as mean ± SEM. SEM represents variance within a group. In all experiments, the genotypes can be found in the corresponding legends. Data were collected and processed side by side in randomized order for all experiments; most analyses were routinely performed blind to the conditions of the experiments. Unpaired, two-tailed t tests were used for comparison between two groups, with p < 0.05 considered significant. For all comparisons involving multiple variables, one-way or two-way ANOVA was performed followed by Tukey’s or Fisher’s LSD post hoc test for multiple comparison using p < 0.05 for significance. For all experiments, between 3–6 (biochemistry, histology) and 8–12 (behavior) animals per experiment were used, with the number per group stated in each figure legend. Additional bar graphs have been included in the supplemental figures to show the appropriate statistical information (mean ± SEM). All statistical analyses were preformed using GraphPad Prism.
Supplementary Material
Highlights.
Abrogating αS tetramers by PD-type E → K mutations leads to excess monomers in vivo
Excess αS monomers are post-translationally modified and aggregated similar to PD
Excess monomers cluster in nigral & cortical neurons and induce fiber & dopamine loss
Tetramer:monomer dyshomeostasis induces L-DOPA responsive, PD-like motor deficits
Acknowledgments:
We thank A. Newman for cloning the initial DNA construct and A. Sharpe (HMS-Transgenic Mouse Core, Boston, MA) for injection services, U. Neumann (FAU Erlangen) and A. Nam for assistance with brain sectioning and for neuronal counts, J. Sanderson (T. Bartels lab, BWH) for help with rec. αS and ELISA, G. Majid for MS analyses (Biomolecular/Proteomics Mass Spectrometry Facility, UCSD, CA), P. Lorello (HNDC Neurobehavior Laboratory, Boston MA) for assistance with behavior, and the members of the Selkoe, Dettmer and Bartels labs for helpful discussons. Supported by NIH grants NS083845 (to D.J.S.), NS099328 (to U.D.), NS103123 (to S.N.), and a research grant from the HNDC (to S.N.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: DJS is a director of and consultant to Prothena Biosciences. Other authors declare no conflict of interest.
References
- Aharon-Peretz J, Rosenbaum H & Gershoni-Baruch R (2004). Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med, 351, 1972–7. [DOI] [PubMed] [Google Scholar]
- Anderson JP, Walker DE, Goldstein JM, et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. The Journal of biological chemistry, 281, 29739–52. [DOI] [PubMed] [Google Scholar]
- Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, et al. (2013). Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord, 28, 811–3. [DOI] [PubMed] [Google Scholar]
- Bartels T, Choi JG & Selkoe DJ (2011). alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature, 477, 107–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth C, Villringer A & Sacher J (2015). Sex hormones affect neurotransmitters and shape the adult female brain during hormonal transition periods. Front Neurosci, 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady RO, Kanfer JN & Shapiro D (1965). Metabolism of Glucocerebrosides. Ii. Evidence of an Enzymatic Deficiency in Gaucher’s Disease. Biochem Biophys Res Commun, 18, 221–5. [DOI] [PubMed] [Google Scholar]
- Burbulla LF, Song P, Mazzulli JR, et al. (2017). Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science, 357, 1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J, Sharma M & Sudhof TC (2014). alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A, 111, E4274–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J, Sharma M, Tsetsenis T, et al. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science, 329, 1663–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J, Vivona S, Diao J, et al. (2013). Properties of native brain alpha-synuclein. Nature, 498, E4–6; discussion E6–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Cohen KB, Luo T, et al. (2013). Improved tools for the Brainbow toolbox. Nat Methods, 10, 540–7. [PubMed] [Google Scholar]
- Caligiore D, Helmich RC, Hallett M, et al. (2016). Parkinson’s disease as a system-level disorder. NPJ Parkinsons Dis, 2, 16025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon JR, Geghman KD, Tapias V, et al. (2013). Expression of human E46K-mutated alpha-synuclein in BAC-transgenic rats replicates early-stage Parkinson’s disease features and enhances vulnerability to mitochondrial impairment. Exp Neurol, 240, 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartoff EH, Marck BT, Matsumoto AM, et al. (2001). Induction of stereotypy in dopamine-deficient mice requires striatal D1 receptor activation. Proc Natl Acad Sci U S A, 98, 10451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HC, Ulane CM & Burke RE (2010). Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol, 67, 715–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Zhang X, Yu L, et al. (2015). Calcium signaling in membrane repair. Semin Cell Dev Biol, 45, 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W, Zibaee S, Jakes R, et al. (2004). Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS letters, 576, 363–8. [DOI] [PubMed] [Google Scholar]
- Coelho T, Maia LF, Da Silva AM, et al. (2013). Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol, 260, 2802–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, et al. (1998). Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem, 273, 9443–9. [DOI] [PubMed] [Google Scholar]
- Dettmer U, Newman AJ, Luth ES, et al. (2013). In vivo cross-linking reveals principally oligomeric forms of alpha-synuclein and beta-synuclein in neurons and non-neural cells. J Biol Chem, 288, 6371–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U, Newman AJ, Soldner F, et al. (2015a). Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun, 6, 7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U, Newman AJ, Von Saucken VE, et al. (2015b). KTKEGV repeat motifs are key mediators of normal alpha-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity. Proc Natl Acad Sci U S A, 112, 9596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U, Ramalingam N, Von Saucken VE, et al. (2017). Loss of native alpha-synuclein multimerization by strategically mutating its amphipathic helix causes abnormal vesicle interactions in neuronal cells. Hum Mol Genet, 26, 3466–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmer KL, Waxman EA, Covy JP, et al. (2011). E46K human alpha-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. J Biol Chem, 286, 35104–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvet B, Mbefo MK, Fares MB, et al. (2012). alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem, 287, 15345–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forno LS (1996). Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol, 55, 259–72. [DOI] [PubMed] [Google Scholar]
- Fujishiro H, Imamura AY, Lin WL, et al. (2013). Diversity of pathological features other than Lewy bodies in familial Parkinson’s disease due to SNCA mutations. Am J Neurodegener Dis, 2, 266–75. [PMC free article] [PubMed] [Google Scholar]
- Fusco G, Chen SW, Williamson PTF, et al. (2017). Structural basis of membrane disruption and cellular toxicity by alpha-synuclein oligomers. Science, 358, 1440–1443. [DOI] [PubMed] [Google Scholar]
- Gillies GE, Pienaar IS, Vohra S, et al. (2014). Sex differences in Parkinson’s disease. Front Neuroendocrinol, 35, 370–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera FE, Chesi A, Paleologou KE, et al. (2008). Inhibition of alpha-synuclein fibrillization by dopamine is mediated by interactions with five C-terminal residues and with E83 in the NAC region. PLoS ONE, 3, e3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Gaffaney JD, Wang Z, et al. (2011). Mechanism and function of synaptotagmin-mediated membrane apposition. Nat Struct Mol Biol, 18, 813–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kett LR & Dauer WT (2016). Endolysosomal dysfunction in Parkinson’s disease: Recent developments and future challenges. Mov Disord, 31, 1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Yun SP, Lee S, et al. (2018). GBA1 deficiency negatively affects physiological alpha-synuclein tetramers and related multimers. Proc Natl Acad Sci U S A, 115, 798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, et al. (1998). Ala30Pro mutation in the gene encoding a-synuclein in Parkinsons’s disease. Nat. Genet, 18, 106–108. [DOI] [PubMed] [Google Scholar]
- Kuusisto E, Parkkinen L & Alafuzoff I (2003). Morphogenesis of Lewy bodies: dissimilar incorporation of alpha-synuclein, ubiquitin, and p62. J Neuropathol Exp Neurol, 62, 1241–53. [DOI] [PubMed] [Google Scholar]
- Lee BR & Kamitani T (2011). Improved immunodetection of endogenous alpha-synuclein. PLoS One, 6, e23939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Anheim M, Letournel F, et al. (2013). G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol, 73, 459–71. [DOI] [PubMed] [Google Scholar]
- Li W, West N, Colla E, et al. (2005). Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci U S A, 102, 2162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CW, Giasson BI, Lewis KA, et al. (2005). A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson disease. J Biol Chem, 280, 22670–8. [DOI] [PubMed] [Google Scholar]
- Logan T, Bendor J, Toupin C, et al. (2017). alpha-Synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci, 20, 681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith GE, Halliday GM & Totterdell S (2004). A critical review of the development and importance of proteinaceous aggregates in animal models of Parkinson’s disease: new insights into Lewy body formation. Parkinsonism Relat Disord, 10, 191–202. [DOI] [PubMed] [Google Scholar]
- Mishizen-Eberz AJ, Norris EH, Giasson BI, et al. (2005). Cleavage of alpha-synuclein by calpain: potential role in degradation of fibrillized and nitrated species of alpha-synuclein. Biochemistry, 44, 7818–29. [DOI] [PubMed] [Google Scholar]
- Newman AJ, Selkoe D & Dettmer U (2013). A new method for quantitative immunoblotting of endogenous alpha-synuclein. PLoS One, 8, e81314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuber S, Harmuth F, Kohl Z, et al. (2013). A progressive dopaminergic phenotype associated with neurotoxic conversion of alpha-synuclein in BAC-transgenic rats. Brain, 136, 412–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuber S, Petrasch-Parwez E, Winner B, et al. (2008). Neurodegeneration and motor dysfunction in a conditional model of Parkinson’s disease. J Neurosci, 28, 2471–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuber S & Selkoe DJ (2016). Caspase-1 clipping causes complications for alpha-synuclein. Proc Natl Acad Sci U S A [DOI] [PMC free article] [PubMed]
- Nuber S, Tadros D, Fields J, et al. (2014). Environmental neurotoxic challenge of conditional alpha-synuclein transgenic mice predicts a dopaminergic olfactory-striatal interplay in early PD. Acta Neuropathol, 127, 477–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obi K, Akiyama H, Kondo H, et al. (2008). Relationship of phosphorylated alpha-synuclein and tau accumulation to Abeta deposition in the cerebral cortex of dementia with Lewy bodies. Exp Neurol, 210, 409–20. [DOI] [PubMed] [Google Scholar]
- Paleologou KE, Kragh CL, Mann DM, et al. (2009). Detection of elevated levels of soluble alpha-synuclein oligomers in post-mortem brain extracts from patients with dementia with Lewy bodies. Brain, 132, 1093–101. [DOI] [PubMed] [Google Scholar]
- Paxinos G & Franklin KBJ (2001). The mouse brain in stereotaxic coordinates, San Diego, Academic Press. [Google Scholar]
- Proukakis C, Moore D, Labrum R, et al. (2011). Detection of novel mutations and review of published data suggests that hereditary spastic paraplegia caused by spastin (SPAST) mutations is found more often in males. J Neurol Sci, 306, 62–5. [DOI] [PubMed] [Google Scholar]
- Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, et al. (2002). Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp Neurol, 175, 35–48. [DOI] [PubMed] [Google Scholar]
- Schell H, Hasegawa T, Neumann M, et al. (2009). Nuclear and neuritic distribution of serine-129 phosphorylated alpha-synuclein in transgenic mice. Neuroscience, 160, 796–804. [DOI] [PubMed] [Google Scholar]
- Schlachetzki JC, Marxreiter F, Regensburger M, et al. (2014). Increased tyrosine hydroxylase expression accompanied by glial changes within the non-lesioned hemisphere in the 6-hydroxydopamine model of Parkinson’s disease. Restor Neurol Neurosci, 32, 447–62. [DOI] [PubMed] [Google Scholar]
- Sharon R, Bar-Joseph I, Frosch MP, et al. (2003). The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron, 37, 583–595. [DOI] [PubMed] [Google Scholar]
- Solis O, Limon DI, Flores-Hernandez J, et al. (2007). Alterations in dendritic morphology of the prefrontal cortical and striatum neurons in the unilateral 6-OHDA-rat model of Parkinson’s disease. Synapse, 61, 450–8. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Obeso JA & Halliday GM (2017). Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci, 18, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Garcia Reitbock P, Humby T, et al. (2006). Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1–120): implications for Lewy body disorders. J Neurosci, 26, 3942–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Gamble KL, Schultheiss CE, et al. (2014). Formation of alpha-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell, 25, 4010–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamatsu M, Ishii A, Ukai Y, et al. (2007). Accumulation of phosphorylated alpha-synuclein in dopaminergic neurons of transgenic mice that express human alpha-synuclein. J Neurosci Res, 85, 1819–25. [DOI] [PubMed] [Google Scholar]
- Walker DG, Lue LF, Adler CH, et al. (2013). Changes in properties of serine 129 phosphorylated alpha-synuclein with progression of Lewy-type histopathology in human brains. Exp Neurol, 240, 190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Das U, Scott DA, et al. (2014). alpha-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr Biol, 24, 2319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Perovic I, Chittuluru J, et al. (2011). A soluble alpha-synuclein construct forms a dynamic tetramer. Proceedings of the National Academy of Sciences of the United States of America, 108, 17797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe I, Vachal E & Tomita T (1977). Dense core vesicles around the Lewy body in incidental Parkinson’s disease: an electron microscopic study. Acta Neuropathol, 39, 173–5. [DOI] [PubMed] [Google Scholar]
- Westphal CH & Chandra SS (2013). Monomeric synucleins generate membrane curvature. J Biol Chem, 288, 1829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B, Jappelli R, Maji SK, et al. (2011). In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A, 108, 4194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise-Scira O, Dunn A, Aloglu AK, et al. (2013). Structures of the E46K mutant-type alpha-synuclein protein and impact of E46K mutation on the structures of the wild-type alpha-synuclein protein. ACS Chem Neurosci, 4, 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ysselstein D, Joshi M, Mishra V, et al. (2015). Effects of impaired membrane interactions on alpha-synuclein aggregation and neurotoxicity. Neurobiol Dis, 79, 150–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Du WX, Fregevu C, et al. (2014). Expression, purification, and characterization of almond (Prunus dulcis) allergen Pru du 4. J Agric Food Chem, 62, 12695–700. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wu Y, Wang Z, et al. (2011). Release mode of large and small dense-core vesicles specified by different synaptotagmin isoforms in PC12 cells. Mol Biol Cell, 22, 2324–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.