

Abstract
Background
Inflammation signaled by Janus kinases (JAKs) promotes progression of diabetic kidney disease (DKD). Baricitinib is an oral, reversible, selective inhibitor of JAK1 and JAK2. This study tested the efficacy of baricitinib versus placebo on albuminuria in adults with Type 2 diabetes at high risk for progressive DKD.
Methods
In this Phase 2, double-blind, dose-ranging study, participants were randomized 1:1:1:1:1 to receive placebo or baricitinib (0.75 mg daily; 0.75 mg twice daily; 1.5 mg daily; or 4 mg daily), for 24 weeks followed by 4–8 weeks of washout.
Results
Participants (N = 129) were 63±9.1 (mean±standard deviation) years of age, 27.1% (35/129) women and 11.6% (15/129) African-American race. Baseline hemoglobin A1c (HbA1c) was 7.3±1% and estimated glomerular filtration rate was 45.0±12.1 mL/min/1.73 m2 with first morning urine albumin–creatinine ratio (UACR) of 820 (407–1632) (median; interquartile range) mg/g. Baricitinib, 4 mg daily, decreased morning UACR by 41% at Week 24 compared with placebo (ratio to baseline 0.59, 95% confidence interval 0.38–0.93, P = 0.022). UACR was decreased at Weeks 12 and 24 and after 4–8 weeks of washout. Baricitinib 4 mg decreased inflammatory biomarkers over 24 weeks (urine C–X–C motif chemokine 10 and urine C–C motif ligand 2, plasma soluble tumor necrosis factor receptors 1 and 2, intercellular adhesion molecule 1 and serum amyloid A). The only adverse event rate that differed between groups was anemia at 32.0% (8/25) for baricitinib 4 mg daily versus 3.7% (1/27) for placebo.
Conclusions
Baricitinib decreased albuminuria in participants with Type 2 diabetes and DKD. Further research is required to determine if baricitinib reduces DKD progression.
Keywords: albuminuria, biomarkers, diabetic nephropathy, estimated glomerular filtration rate, JAK inhibition
INTRODUCTION
Diabetic kidney disease (DKD) is the most common cause of chronic kidney disease worldwide, and current treatments fail to prevent progression to end-stage renal disease (ESRD) in many cases [1–6]. In the year 2014, it was estimated that over 29 million people, or 9.3% of the US population, had diabetes [7]. Approximately 40% of people with Type 2 diabetes develop DKD, and DKD prevalence has increased due to an increase in the total number of people living with diabetes [8–13]. Overall, diabetes accounts for nearly half of all ESRD cases in the USA and most cases of DKD occur in Type 2 diabetes [8–12, 14]. Renin–angiotensin system inhibition by angiotensin-converting enzyme (ACE) inhibition or angiotensin II receptor blocking (ARB) agents is the prevailing standard of care, but this treatment approach leaves substantial residual risk for progressive DKD [15–18]. Recent clinical cardiovascular outcome trials of newer antihyperglycemic agents, the sodium–glucose cotransporter 2 (SGLT-2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists, have provided evidence that these agents can reduce albuminuria and loss of kidney function. However, kidney outcomes were secondary and remain to be confirmed among patients with DKD at high risk for progression [19–24].
DKD is characterized by sustained inflammation that promotes and directs much of the chronic injury process [25]. One of the major pathways that transduce inflammatory signals in DKD is the Janus kinase-Signal Transducer and Activator of Transcription (JAK-STAT) pathway. The JAK-STAT pathway transmits signals from extracellular ligands, including many cytokines and chemokines as well as growth factors and hormones, directly to the nucleus to induce a variety of cellular responses [26]. While many of these responses are best characterized in lymphoid cells, they have also been reported in intrinsic kidney cells such as podocytes [27], mesangial cells [28] and tubular cells [27]. Gene and protein expression studies of kidney biopsies from people with early- and late-stage DKD have shown increased activation and expression of the JAK-STAT signaling pathway across the spectrum of DKD [27, 29]. In particular, increased expression and activity of JAK1 and JAK2 appear to promote DKD development and progression [30–32]. Moreover, studies have suggested interactions between JAK-STAT and angiotensin signaling, including evidence for activating JAK2 [33].
Baricitinib is an oral, small-molecule inhibitor of the JAK family of protein tyrosine kinases that selectively inhibits JAK1 and JAK2, and has demonstrated clinical efficacy in chronic inflammatory conditions such as rheumatoid arthritis [34–37]. Based on the evidence that JAK-STAT activation is central to DKD pathogenesis, this study was conducted to test the efficacy of baricitinib in participants at high risk for DKD progression defined by persistent macroalbuminuria despite ACE inhibitor or ARB therapy. The aim of the study was to evaluate the effect of baricitinib on albuminuria in participants with Type 2 diabetes and DKD.
MATERIALS AND METHODS
Study participants
Participants were ≥18 years old and had Type 2 diabetes treated with one or more antihyperglycemic agents. Participants also received either an ACE inhibitor or an ARB in a DKD-therapeutic dose for at least 3 months preceding study entry. Doses of antihyperglycemic agents, ACE inhibitors, ARB or other antihypertensive medicines were stable for at least the preceding 4 weeks. Participants had an estimated glomerular filtration rate (eGFR) of 25–70 mL/min/1.73 m2 and documented history of severely increased albuminuria [macroalbuminuria, urine albumin–creatinine ratio (UACR) >300 mg/g and <5000 mg/g]. Main exclusion criteria included systolic blood pressure >150 mmHg or diastolic blood pressure >90 mmHg; hemoglobin <10.0 g/dL; hemoglobin A1c (HbA1c) level >10%; history of diabetic ketoacidosis; previous kidney transplant; non-DKD; major surgical procedure within 8 weeks of study entry; prior treatment with a JAK inhibitor; active or latent infection or immunosuppression.
Study design
This was a randomized, placebo-controlled, double-blind, parallel-group, dose-ranging, Phase 2 trial conducted at 40 sites in Japan, Mexico and the USA, including Puerto Rico (ClinicalTrials.gov identifier NCT01683409). The study design included a 4-week screening period, a 24-week treatment period and a 4- to 8-week washout period (Supplementary data, Figure S3). A sample size of 250 participants (50 per treatment arm) was estimated to provide 82% power for detecting a 35% decrease in UACR compared with placebo, assuming a 5% significance level and one-sided test. At the time of study design, the estimate for the prevalence of macroalbuminuria was >30% for people with Type 2 diabetes and DKD. Due to study recruitment challenges, enrollment was closed at approximately half the planned number. However, blinded reassessment of the primary outcome indicated that a statistically significant between-group difference could still be detected with a smaller sample size.
The study was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonization, and local laws and regulations. The study protocol and informed consent forms were approved by institutional review boards or ethics review boards for each site. All participants provided written informed consent.
Intervention
Eligible participants were randomized 1:1:1:1:1 to placebo or to a baricitinib dose group determined by a computer-generated random sequence using an interactive voice-response system. Participants in each baricitinib dose group were stratified according to their baseline eGFR into high (50–70 mL/min/1.73 m2) and low (25 to <50 mL/min/1.73 m2) eGFR strata [Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation] but were analyzed by assigned treatment arm. For the high eGFR stratum, baricitinib dose groups were 0.75 mg once daily, 0.75 mg twice daily, 1.5 mg once daily or 4 mg once daily. For the low eGFR stratum, baricitinib dose groups were 0.5 mg once daily, 0.5 mg twice daily, 1 mg once daily or 2.75 mg once daily. Dose adjustments for the lower eGFR groups were chosen based on existing pharmacokinetic data held by the study sponsor (Eli Lilly and Company). The study drug was administered as three tablets in the morning and one tablet in the evening for all participants. Study treatment continued for 24 weeks, followed by a 4- to 8-week washout period. Laboratory tests, vital signs and other safety assessments were performed at scheduled visits. As an operational definition for significant anemia, research sites were given specific instructions to interrupt baricitinib dosing if hemoglobin fell below <9 g/dL. The incidence and severity of adverse events were recorded at each visit.
Study outcomes
The primary outcome was the change from baseline to 24 weeks in UACR in the first morning urine sample (any baricitinib dose versus placebo). Secondary outcomes included: 12-week change from baseline in first morning UACR; 24-week change from baseline in 24-h UACR; 24-h total urine albumin excretion; 24-h urine protein and urine protein–creatinine ratio; serum cystatin C-derived eGFR; creatinine clearance and inflammatory biomarkers [urinary C–X–C motif chemokine 10 (CXCL10; also known as interferon gamma-induced protein, IP-10) and chemokine (C–C motif) ligand 2 (CCL2; also known as monocyte chemotactic protein-1, MCP-1); plasma soluble tumor necrosis factor receptors 1 and 2 (sTNFR1 and sTNFR2), serum amyloid A (SAA), vascular cell adhesion marker 1 (VCAM1) and intracellular adhesion marker 1 (ICAM1)]. Serum and urine creatinine levels were measured using the Jaffé method. Results are presented based on eGFR strata, i.e. participants who received an adjusted dose for low eGFR are reported with the higher eGFR group who did not receive an adjusted dose. For example, 2.75 mg daily and 4 mg daily were combined with analogous groupings for other doses.
Inflammatory biomarkers
Plasma and urine biomarkers were measured from stored samples collected at baseline, 2, 4, 12 and 24 weeks following treatment initiation and at 2 and 4 weeks post-treatment. Biomarkers were measured by the following assays: CXCL10 and CCL2 in urine and sTNFR1 and sTNFR2 in plasma were measured in duplicate with commercially available enzyme-linked immunoassays (R+D Systems, Minneapolis, MN, USA); SAA, ICAM1 and VCAM1 in plasma were measured in duplicate with a V-Plex vascular injury 2 assay panel (MesoScale, Gaithersburg, MD, USA). Urine biomarkers were reported as the ratio to creatinine to adjust for differences in urine concentration. Replicate measurements were averaged for the reported values, and mean deviations assessed assay variability.
Statistical analyses
Statistical analyses were performed on the modified intent-to-treat population, defined as all randomized participants who received at least one dose of assigned study drug. The primary analysis of first morning UACR at Week 24 was based on logarithmic values due to the skewness of albuminuria data with a model of mixed-model repeated measures (MMRM) to compare treatment arms. Mean changes from baseline were analyzed using a restricted maximum-likelihood-based repeated measures approach. Analyses included fixed, categorical effects of therapy, stratification variable, visit and treatment-by-visit interaction, as well as continuous, fixed covariates of baseline UACR and baseline UACR-by-visit interaction. An unstructured covariance structure was used to model the within-patient errors. Significance tests were based on least-squares means using a two-sided test with α = 0.1 for the primary analysis. Other tests of treatment effects were conducted with a two-sided alpha level of 0.05. No adjustments were made for multiplicity for this exploratory Phase 2 study. Analyses were implemented using SAS Version 9.2. For the secondary outcome, dichotomous variables were analyzed using Fisher’s exact test. Other continuous outcomes, except for inflammatory biomarker measurements, were analyzed using an MMRM method similar to those for the primary outcome with log transformation applied to urine measurements due to the skewness of the data. Mean changes from baseline for the biomarker outcomes were analyzed using MMRM to compare baricitinib treatment arms to placebo and included fixed, categorical effects of treatment, visit and treatment-by-visit interaction as well as a continuous, fixed covariate of the biomarker level at baseline. Akaike information criterion was used to select the appropriate covariance structure for each biomarker. For outcomes that were log transformed for data analysis, results were back-transformed to original scale for point estimates, confidence intervals and reporting. For the analyses of safety data, discrete outcomes were analyzed using Fisher’s exact test and laboratory measures were analyzed using an analysis of covariance (ANCOVA) model with treatment and baseline value of the test variable as a covariate.
RESULTS
Study participants
Of 376 candidates screened, 130 study participants were randomized. One ineligible participant was randomized but did not receive study drug, and therefore, 129 participants were included in the modified intent-to-treat analyses (Figure 1). Participants (N = 129) were 63±9.1 [mean ± standard deviation (SD)] years of age, 27.1% (35/129) women and 11.6% (15/129) African-American race. Baseline HbA1c was 7.3±1% and eGFR was 45.0±12.1 mL/min/1.73 m2 with first morning UACR of 820 (407–1632) (median; interquartile range) mg/g. Treatment groups were well-balanced with respect to demographic and baseline characteristics (Table 1). For all groups, the median duration of exposure to study drug was 169 days.
FIGURE 1.

Patient disposition through 24 weeks. N = number of participants in each treatment group; n = number of participants in the specified category.
Table 1.
Baseline characteristics and disease activitya
| Placebo (N = 27) | Baricitinib 0.75 mg daily (N = 25) | Baricitinib 0.75 mg twice daily (N = 26) | Baricitinib 1.5 mg daily (N = 26) | Baricitinib 4 mg daily (N = 25) | |
|---|---|---|---|---|---|
| Age (years) | 64 (9.0) | 61 (10.0) | 64 (8.3) | 61 (10.4) | 63 (7.8) |
| Women, n (%) | 7 (25.9) | 8 (32.0) | 5 (19.2) | 5 (19.2) | 10 (40.0) |
| Weight (kg) | 85.9 (26.1) | 87.5 (22.9) | 83.7 (25.5) | 91.5 (24.6) | 86.4 (29.2) |
| Body mass index (kg/m2) | 31.0 (7.3) | 30.4 (6.4) | 30.1 (8.6) | 32.24 (8.6) | 31.4 (8.2) |
| Blood pressure (mmHg) | |||||
| Systolic | 134 (13.7) | 133 (11.3) | 133 (10.6) | 134 (11.1) | 132 (13.5) |
| Diastolic | 75 (10.0) | 76 (9.3) | 77 (9.2) | 77 (12.1) | 74 (10.4) |
| Race, n (%) | |||||
| American Indian or Alaska Native | 2 (7.4) | 2 (8.0) | 2 (7.7) | 2 (7.7) | 1 (4.0) |
| Asian | 14 (51.9) | 12 (48.0) | 12 (46.2) | 11 (42.3) | 11 (44.0) |
| African-American | 2 (7.4) | 7 (28.0) | 3 (11.5) | 0 | 3 (12.0) |
| Native Hawaiian or Other Pacific Islander | 0 | 0 | 0 | 0 | 1 (4.0) |
| White | 8 (29.6) | 4 (16.0) | 9 (34.6) | 13 (50.0) | 9 (36.0) |
| Region, n (%) | |||||
| Japan | 11 (40.7) | 10 (40.0) | 10 (38.5) | 10 (38.5) | 11 (44.0) |
| Mexico | 2 (7.4) | 1 (4.0) | 2 (7.7) | 2 (7.7) | 1 (4.0) |
| USA, including Puerto Rico | 14 (51.9) | 14 (56.0) | 14 (53.8) | 14 (53.8) | 13 (52.0) |
| eGFRb group, n (%) | |||||
| 25–<50 mL/min/1.73 m2 | 18 (66.7) | 16 (64.0) | 18 (69.2) | 18 (69.2) | 17 (68.0) |
| 50–70 mL/min/1.73 m2 | 9 (33.3) | 9 (36.0) | 8 (30.8) | 8 (30.8) | 8 (32.0) |
| eGFR mean (SD) | 44.2 (10.6) | 46.3 (14.3) | 44.8 (13.9) | 44.1 (9.8) | 45.8 (12.3) |
| Creatinine clearance—24-h urine (mL/min), mean (SD) | 63.4 (32.0) | 61.9 (26.8) | 49.0 (20.0) | 54.6 (27.6) | 60.6 (34.1) |
| UACR (FMU) (mg/g) | |||||
| Mean | 1464.6 | 1506.4 | 1040.6 | 1405.3 | 1821.1 |
| Median (IQ range) | 1043.4 | 1204.5 | 833.7 | 1016.9 | 1086.7 |
| (627.5, 2001.0) | (724.8, 1993.9) | (504.5, 1190.3) | (555.8, 1443.4) | (690.3, 2286.8) | |
| HbA1c (%) | 7.2 (1.2) | 7.1 (1.1) | 7.2 (0.9) | 7.4 (1.2) | 7.5 (0.8) |
| MCP-1/creatinine ratio (pg/mg), mean (SD) | 542.1 (484.4) | 516.6 (439.2) | 508.7 (406.7) | 503.4 (405.3) | 841.3 (1303.2) |
FMU, first morning urine; IQ, interquartile (maximum, minimum); N, number of participants in each treatment group; n, number of participants in the specified category.
Data reported as mean (SD) unless otherwise indicated.
eGFR determined by CKD-EPI equation; derived by creatinine.
Compares across treatment arms. Continuous parameters are analyzed using ANCOVA and categorical parameters are analyzed using Chi-square test.
Effect of baricitinib on the primary outcome of albuminuria and secondary outcomes
For the primary outcome of change in first morning UACR from baseline to Week 24, treatment with baricitinib 4 mg daily resulted in a significant decrease of 41% compared with placebo [least squares mean difference (LSMD) 0.59, 95% confidence interval (95% CI) 0.38–0.93, P = 0.022; Figure 2A and B]. Among secondary outcomes, decreases were observed in UACR measured by 24-h urine collection, with significant reductions compared with placebo at Week 12 in the baricitinib 1.5 mg daily (LSMD 0.71, 95% CI 0.51–0.98, P = 0.04) and 4 mg daily (LSMD 0.61, 95% CI 0.44–0.84, P = 0.004) groups and at Week 24 in the baricitinib 1.5 mg daily group (LSMD 0.67, 95% CI 0.47–0.96, P = 0.031; Figure 2C and Supplementary data, Figure S1A). Reductions in 24-h total urinary albumin excretion were observed at Weeks 12 and 24 (Supplementary data, Figure S1B). These treatment-related reductions in 24-h UACR and total urinary albumin excretion were mostly maintained after 4-week washout (Figure 2 and Supplementary data, Figure S1). Measures of kidney function by serum creatinine, 24-h urine creatinine clearance measurements and eGFR (cystatin C-based) did not change in any baricitinib group compared with placebo over the 24-week study (Figure 3).
FIGURE 2.
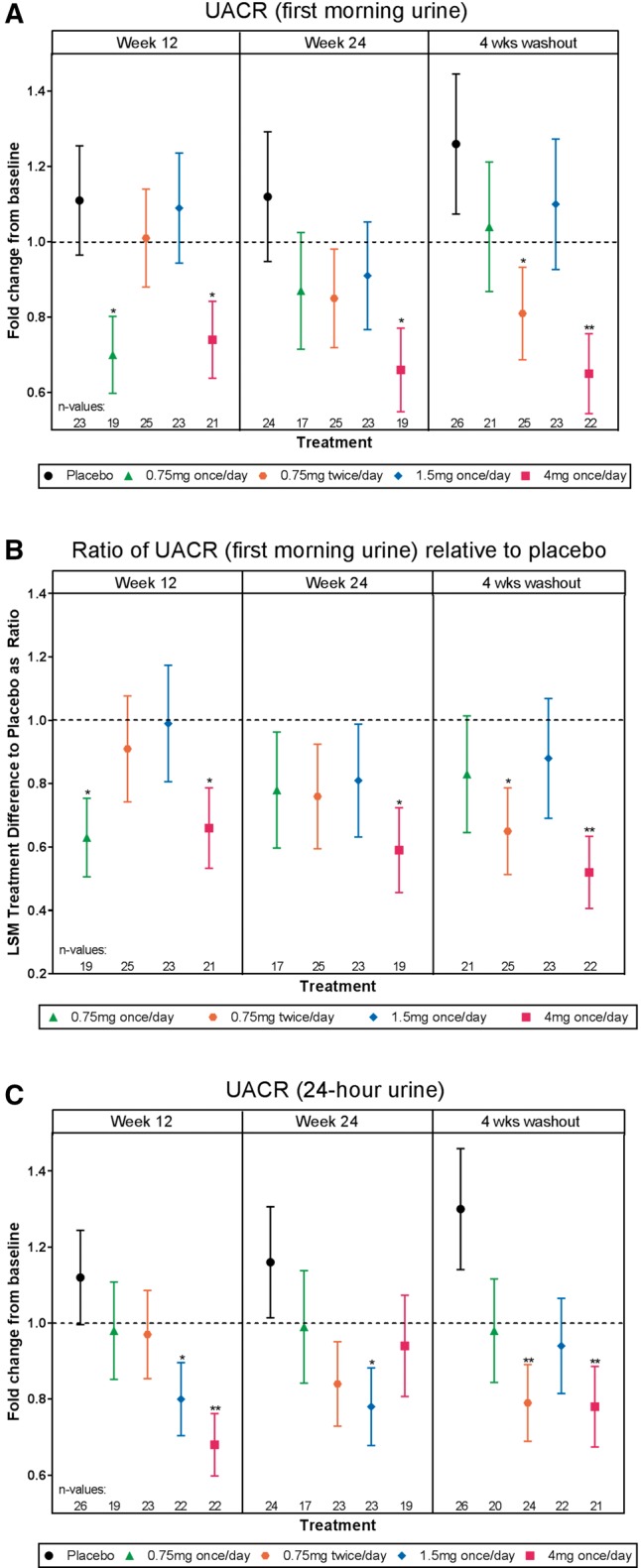
Efficacy analyses. (A) UACR, first morning urine, (B) ratio of UACR (first morning urine) relative to placebo, (C) UACR, 24-h urine. The primary endpoint was change in first morning UACR at Week 24 compared with baseline. The least squares mean (LSM) treatment difference from placebo is displayed as a ratio ± standard error. *P ≤ 0.05, **P ≤ 0.01 versus placebo.
FIGURE 3.

Clinical safety indicators. Observed values (mean ± SD) over time for (A) Hematocrit, (B) Hemoglobin, (C) Plasma creatinine, (D) 24-h urine creatinine clearance, (E) eGFR derived by creatinine, and (F) eGFR derived by cystatin C. For hematocrit and hemoglobin, significance for the difference of treatment comparisons was based on the LSMD and analyzed using an ANCOVA model with treatment and baseline value as covariates. For plasma creatinine, creatinine clearance, and eGFR, significance for the difference of treatment comparisons was based on the LSMD and analyzed using MMRM analyses, which included treatment, eGFR group, visit, treatment-by-visit interaction, baseline, and baseline-by-visit interaction. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 versus placebo. QD, once daily; BID, twice daily.
Other secondary outcomes included effects of baricitinib treatment on pertinent inflammatory biomarkers. Urinary CXCL10, urinary CCL2, plasma sTNFR1 and sTNFR2, SAA [38], ICAM1 and VCAM1 showed dose-dependent decreases from baseline with baricitinib treatment (Figure 4 and Supplementary data, Figure S3). Levels of these inflammatory biomarkers increased above baseline levels during the study drug washout period (Figure 4 and Supplementary data, Figure S2).
FIGURE 4.

Biomarker analyses. The fold change from placebo (±standard error) in (A) urinary CXCL10, (B) urinary CCL2, (C, D) plasma sTNFR1 and sTNFR2 and (E) plasma VCAM1. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 versus placebo.
Effect of baricitinib on safety indicators
There were no deaths during the study. Adverse events led to study drug discontinuation in 11.6% (15/129; Figure 1). Treatment-emergent adverse events (TEAEs) occurred in similar proportions across placebo and baricitinib groups, with the exception of anemia, which was more frequent, 32% (8/25), in the high-dose (4 mg daily) baricitinib group (Table 2). A reduction in hemoglobin of −0.62 mmol/L (95% CI −1.05 to −0.19, P = 0.006) was observed in this group (Figure 3). Of the participants experiencing serious adverse events (SAEs), two were in the placebo group, zero were in the baricitinib 0.75 mg daily group, two were in the baricitinib 0.75 mg twice daily group, two were in the 1.5 mg daily group and six were in the baricitinib 4 mg daily group (Supplementary data, Table S1). SAEs reported in the 4 mg daily group included two incidents of hypoglycemia and one each of dehydration and hyperkalemia not requiring hospitalization. At Week 24, HbA1c decreased from baseline in the baricitinib 4 mg daily group compared with placebo (LSMD −0.5; P = 0.044). There were no significant changes in fructosamine or fasting glucose values for any baricitinib group compared with placebo during the study. A total of 29 participants experienced hypoglycemic episodes without differences in frequency between treatment groups (Supplementary data, Table S2). An increase in weight was observed in some baricitinib groups (Supplementary data, Figure S4). Small increases in alanine aminotransferase and serum creatine phosphokinase were observed with baricitinib treatment (Supplementary data, Table S3). Baricitinib was also associated with modest increases in high-density lipoprotein cholesterol and low-density lipoprotein cholesterol that returned to baseline levels at the 4-week washout visit (Supplementary data, Table S3).
Table 2.
TEAEs reported by more than or equal to five participants in order of frequencya
| Placebo (N = 27) | Baricitinib 0.75 mg daily (N = 25) | Baricitinib 0.75 mg twice daily (N = 26) | Baricitinib 1.5 mg daily (N = 26) | Baricitinib 4 mg daily (N = 25) | |
|---|---|---|---|---|---|
| ≥1 TEAE | 18 (66.7) | 18 (72.0) | 17 (65.4) | 19 (73.1) | 24 (96.0) |
| Nasopharyngitis | 1 (3.7) | 4 (16.0) | 4 (15.4) | 4 (15.4) | 3 (12.0) |
| Anemia | 1 (3.7) | 1 (4.0) | 1 (3.8) | 2 (7.7) | 8 (32.0) |
| Arthralgia | 1 (3.7) | 2 (8.0) | 0 | 2 (7.7) | 2 (8.0) |
| Blood CPK increase | 2 (7.4) | 1 (4.0) | 1 (3.8) | 1 (3.8) | 3 (12.0) |
| Headache | 1 (3.7) | 3 (12.0) | 0 | 1 (3.8) | 2 (8.0) |
| Nausea | 0 | 2 (8.0) | 0 | 1 (3.8) | 3 (12.0) |
| Upper respiratory tract infection | 1 (3.7) | 2 (8.0) | 2 (7.7) | 2 (7.7) | 0 |
| Blood creatinine increase | 0b | 2 (8.0) | 0 | 0 | 3 (12.0) |
| Muscle spasms | 1 (3.7) | 0 | 3 (11.5) | 1 (3.8) | 0 |
| Diarrhea | 2 (7.4) | 1 (4.0) | 1 (3.8) | 0 | 1 (4.0) |
| Dizziness | 2 (7.4) | 2 (8.0) | 0 | 1 (3.8) | 0 |
| Hypoglycemia | 4 (14.8) | 0 | 0 | 0 | 2 (8.0) |
CPK, creatine phosphokinase; N, number of participants in each treatment group; n, number of participants in the specified category.
Data reported as n (%) participants unless otherwise indicated.
Two placebo participants reported renal adverse events: acute kidney injury and renal impairment.
DISCUSSION
This is the first randomized, Phase 2 clinical trial to examine effects of a JAK inhibitor on DKD. The trial met its primary endpoint by demonstrating significant reductions in morning UACR over 24 weeks with baricitinib treatment. Additionally, treatment effects were seen at earlier time points in the high-dose versus lower dose baricitinib groups. Although no unexpected safety signals were detected, the high-dose baricitinib group experienced the adverse event of anemia more frequently with a corresponding decrease in the hemoglobin level.
While specific mechanisms for the protective actions of baricitinib treatment on DKD remain to be fully elucidated, inhibition of inflammatory effects mediated by JAK1 and/or JAK2 is likely a primary target. A great deal of evidence has demonstrated that progression of DKD is promoted by inflammation [25], and the JAK-STAT pathway transduces a number of these signals in both lymphoid cells and resident kidney cells [26–28]. The previous demonstration of increased expression of JAK-STAT isoforms, including JAK1 and JAK2, in kidney glomerular and tubular cells in humans with DKD plus the increased glomerular expression of downstream pro-inflammatory genes in diabetic mice that specifically overexpress JAK2 in podocytes strongly suggest that JAK-STAT activation plays an important role in the pro-inflammatory injury process in the diabetic kidney [27]. The effect of baricitinib in the current study to modulate JAK1- and JAK2-mediated inflammatory processes was supported by dose-related reductions in inflammatory biomarkers including CCL2 (MCP-1), CXCL10 (IP-10), sTNFR1 and sTNFR2, SAA, VCAM1 and ICAM1, all of which have been linked to DKD pathophysiology [38–45]. Although most of these are systemic cytokines, CCL2 (MCP-1) and CXCL10 (IP-10) were measured in urine, and urinary biomarkers appear to more closely track with inflammation in the kidney than systemic inflammation [44]. Indeed, two randomized controlled clinical trials recently have shown reduction in albuminuria among participants with DKD who received inhibitors of CCL2 (MCP-1) signaling [39, 46]. These results support the plausibility that reduction of local inflammation ameliorates DKD and suggest that baricitinib works, at least in part, via local anti-inflammatory effects in the diabetic kidney. In sum, these data support an anti-inflammatory action of JAK1/JAK2 inhibition to reduce albuminuria in high-risk DKD.
A number of clinical trials have aimed to find ways to improve upon ACE inhibition or ARB therapy for DKD, but safer and more effective treatments have been elusive. The combination of ACE and ARB together, as well as other dual renin–angiotensin system blockade therapies, has not been more effective than ACE inhibitor or ARB monotherapy in slowing progression of DKD [47–49]. Notably, increased risks of hyperkalemia and acute kidney injury have been associated with therapies that combine both ACE inhibition and ARB, or which combine other mechanisms of renin–angiotensin system inhibition (e.g. direct renin inhibitor) with an ACE inhibitor or ARB [47–51]. The recent findings that empagliflozin and canagliflozin, inhibitors of SGLT-2, ameliorated DKD progression are encouraging [19, 24]. The GLP-1 receptor agonists, liraglutide and semaglutide, also have been observed to reduce albuminuria and loss of kidney function. However, in none of these studies were kidney outcomes a primary endpoint. Moreover, SGLT-2 inhibitors are not currently approved for use in people with eGFR <45 mL/min/1.73 m2 [19–24]. Since baricitinib reduced albuminuria in a group with eGFRs inclusive of this lower range, JAK1/JAK2 inhibition could be a clinically useful treatment for DKD even at more advanced stages.
Although a safety signal of more frequent anemia and a lower hemoglobin level was observed in the high-dose baricitinib group, these data contrast with studies of baricitinib in rheumatoid arthritis and psoriasis in which the same dose (4 mg daily) was not associated with increased risk of anemia [34–37, 52]. Many people with DKD have impaired erythropoietin production, and erythropoietin signals through a JAK2-dependent pathway [53, 54]. Therefore, JAK2 inhibition could further decrease erythropoietin actions in those who already have low erythropoietin levels. As such, baricitinib may have a relatively narrow therapeutic window. Based on balancing likely benefits and harms, the middle-range dose may be an optimal choice based on the available data. Since both JAK1 and JAK2 isoforms are increased and may contribute to activation of pathogenic signaling pathways in progressive DKD, a JAK1 inhibitor could potentially be effective without a reduction in hemoglobin. However, based on currently published data, it is unclear whether JAK1 or JAK2 has a dominant clinical effect. JAK1-specific inhibitors also reduce pro-inflammatory signaling and have shown efficacy in other inflammatory diseases (e.g. rheumatoid arthritis) [55–57], which are characterized by JAK1/JAK2 activation. In the baricitinib treatment groups, small reductions in creatinine-based eGFR were observed based on slight increases in serum creatinine levels. Notably, there was no directional change in cystatin C-based eGFR, suggesting that increases in serum creatinine levels in the baricitinib groups were related to the drug’s known effect to inhibit tubular creatinine secretion [58].
Limitations include a modest sample size with lower-than-planned power that contributed to variability in UACR and inflammatory biomarker measurements over the relatively short study timeframe. At the time of study design, the estimated prevalence of macroalbuminuria was >30% for patients with Type 2 diabetes and DKD. Lower-than-expected enrollment was consistent with the recent observation that the prevalence of macroalbuminuria in people with DKD has declined and that low eGFR without albuminuria has increased in the USA in concert with better diabetes care and greater use of renin–angiotensin system inhibitors over the past decade [59]. Thus, in future studies of JAK inhibitors it will be critical to enroll participants with normal and lower levels of albuminuria to ensure a representative sample of patients with DKD. With regard to racial distribution, Asian participants were recruited primarily from Japan and comprised about half of the treatment groups. Thus, a relative lack of diversity in the study population introduces a limitation of generalizability for the study findings and is another reason to enroll larger and more varied populations in future studies. Even though small reductions in creatinine-based eGFR occurred in the baricitinib groups, there was no directional change in cystatin C-based eGFR. Therefore, increases in serum creatinine levels in the baricitinib groups were likely related to the drug’s effect to inhibit renal tubular creatinine secretion [58]. Overall, the study still met its primary objective and demonstrated efficacy based on clinically meaningful reductions in albuminuria without change in kidney function.
In conclusion, treatment with baricitinib significantly reduced albuminuria, the most widely accepted clinical biomarker for DKD, in study participants at high risk for disease progression. Therefore, JAK1/JAK2 inhibition shows potential for treating DKD and improving health outcomes in patients who remain at high risk despite standard therapy.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Farsad Afshinnia for assisting with patient screening at the University of Michigan Medical School, Dennis Laska of Eli Lilly and Company for performing the analyses of inflammation biomarkers, and Barbara Coffey and Stephanie Colvin, PhD of Eli Lilly and Company for creating the tables and figures and for assisting with manuscript preparation and process support.
FUNDING
This study was funded by Eli Lilly and Company and Incyte Corporation.
AUTHORS’ CONTRIBUTIONS
All authors participated in the analyses and interpretation of data, provided critical comments and input and reviewed and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The results presented in this manuscript have not been published previously in whole or part, except in abstract form. K.R.T. has received consulting support from Eli Lilly and Company, Boehringer-Ingelheim, Gilead and Astra Zeneca. F.C.B. III, through the University of Michigan, has received grant/research support from Eli Lilly and Company, Bristol-Myers Squibb and Takeda Pharmaceuticals. S.G.A. has received grant/research support and/or consulting fees from Eli Lilly and Company, Baxter, Bayer, Mallinckrodt, Retrophin Pharmaceuticals and Renal Research Institute. M.K., through the University of Michigan, has received grant/research support and/or consulting support from Eli Lilly and Company, Abbvie, Astra-Zeneca, Boehringer-Ingleheim, Novo-Nordisk and Pfizer. R.L.M. has received grant/research support and/or consulting support from Eli Lilly and Company, Abbvie, AM Pharma, Akebia, Ardea, Astute Inc., Baxter, CSL Behring, Ferring Research, Fresenius, Fresenius-Kabi, International Safety Adverse Events Consortium, International Society of Nephrology, Ionis, Relypsa and Regulus. J.A.T. has received grant/research support and/or consulting fees from Eli Lilly and Company, Genkyotec Pharmaceuticals, Gilead Pharmaceuticals, Jansen Pharmaceuticals and Mallinckrodt. Y.T. has received grant/research support and/or consulting fees from AbbVie, Astellas, Asahi-Kasei, Bristol Myers Squibb, Chugai, Daiichi-Sankyo, Eisai, Eli Lilly and Company, Glaxo Smith Kline, Janssen, Kyowa-Kirin, Mitsubishi-Tanabe, MSD, Ono, Pfizer, Sanofi, Takeda, Taisho-Toyama, Teijin, YL Biologics and UCB. M.H. has received grant/research support or consulting support from Eli Lilly and Company. J.L., M.E.S., T.E.C., K.L.D., J.V.H., W.L.M., F.P.N. and J.M.J. are employees of Eli Lilly and Company and may own stock or stock options in Eli Lilly and Company.
REFERENCES
- 1. Saran R,, Li Y, Robinson B. et al. US Renal Data System 2014 annual data report: epidemiology of kidney disease in the United States. Am J Kidney Dis 2015; 66 (Suppl 1): A7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kramer A, Pippias M, Stel VS. et al. Renal replacement therapy in Europe: a summary of the 2013 ERA-EDTA Registry Annual Report with a focus on diabetes mellitus. Clin Kidney J 2016; 9: 457–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tuttle KR, Bakris GL, Bilous RW. et al. Diabetic kidney disease: a report from an ADA Consensus Conference. Diabetes Care 2014; 37: 2864–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Boer IH, Afkarian M, Rue TC. et al. Renal outcomes in patients with type 1 diabetes and macroalbuminuria. J Am Soc Nephrol 2014; 25: 2342–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. ANZDATA Registry. 38th Report: Incidence of End Stage Kidney Disease. Adelaide, Australia: Australia and New Zealand Dialysis and Transplant Registry, 2016 [Google Scholar]
- 6. Afkarian M, Sachs MC, Kestenbaum B. et al. Kidney disease and increased mortality risk in type 2 diabetes. J Am Soc Nephrol 2013; 24: 302–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Centers for Disease Control and Prevention. National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States, 2014. Atlanta, GA: US Department of Health and Human Services, 2014 [Google Scholar]
- 8. Ritz E, Rychlik I, Locatelli F. et al. End-stage renal failure in type 2 diabetes: a medical catastrophe of worldwide dimensions. Am J Kidney Dis 1999; 34: 795–808 [DOI] [PubMed] [Google Scholar]
- 9. Nwankwo E, Bello AK, El Nahas AM.. Chronic kidney disease: stemming the global tide. Am J Kidney Dis 2005; 45: 201–208 [DOI] [PubMed] [Google Scholar]
- 10. Szczech LA, Lazar IL.. Projecting the United States ESRD population: issues regarding treatment of patients with ESRD. Kidney Int Suppl 2004; 90: S3–S7 [DOI] [PubMed] [Google Scholar]
- 11. Atkins RC. The epidemiology of chronic kidney disease. Kidney Int Suppl 2005; 94: S14–S18 [DOI] [PubMed] [Google Scholar]
- 12. McClellan WM. Epidemiology and risk factors for chronic kidney disease. Med Clin North Am 2005; 89: 419–445 [DOI] [PubMed] [Google Scholar]
- 13. de Boer IH. Chronic kidney disease—a challenge for all ages. JAMA 2012; 308: 2401–2402 [DOI] [PubMed] [Google Scholar]
- 14. U.S. Renal Data System. USRDS 2014 Annual Data Report: Atlas of End-Stage Renal Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 2014 [Google Scholar]
- 15. Brenner BM, Cooper ME, de Zeeuw D. et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345: 861–869 [DOI] [PubMed] [Google Scholar]
- 16. Lewis EJ, Hunsicker LG, Clarke WR. et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345: 851–860 [DOI] [PubMed] [Google Scholar]
- 17. Thomas MC, Groop PH.. New approaches to the treatment of nephropathy in diabetes. Expert Opin Investig Drugs 2011; 20: 1057–1071 [DOI] [PubMed] [Google Scholar]
- 18. Abdel-Rahman EM, Saadulla L, Reeves WB. et al. Therapeutic modalities in diabetic nephropathy: standard and emerging approaches. J Gen Intern Med 2012; 27: 458–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Neal B, Perkovic V, Mahaffey KW. et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 2017; 377: 644–657 [DOI] [PubMed] [Google Scholar]
- 20. Marso SP, Daniels GH, Brown-Frandsen K. et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016; 375: 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mann JFE, Orsted DD, Brown-Frandsen K. et al. Liraglutide and renal outcomes in type 2 diabetes. N Engl J Med 2017; 377: 839–848 [DOI] [PubMed] [Google Scholar]
- 22. Marso SP, Bain SC, Consoli A. et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016; 375: 1834–1844 [DOI] [PubMed] [Google Scholar]
- 23. Zinman B, Wanner C, Lachin JM. et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015; 373: 2117–2128 [DOI] [PubMed] [Google Scholar]
- 24. Wanner C, Inzucchi SE, Lachin JM. et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 2016; 375: 323–334 [DOI] [PubMed] [Google Scholar]
- 25. Wada J, Makino H.. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci 2013; 124: 139–152 [DOI] [PubMed] [Google Scholar]
- 26. O’Shea JJ, Plenge R.. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012; 36: 542–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berthier CC, Zhang H, Schin M. et al. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 2009; 58: 469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Choudhury GG, Ghosh-Choudhury N, Abboud HE.. Association and direct activation of signal transducer and activator of transcription1alpha by platelet-derived growth factor receptor. J Clin Invest 1998; 101: 2751–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Woroniecka KI, Park AS, Mohtat D. et al. Transcriptome analysis of human diabetic kidney disease. Diabetes 2011; 60: 2354–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M. et al. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol 2011; 7: 327–340 [DOI] [PubMed] [Google Scholar]
- 31. Brosius FC III, He JC.. JAK inhibition and progressive kidney disease. Curr Opin Nephrol Hypertens 2015; 24: 88–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang H, Nair V, Saha J. et al. Podocyte-specific JAK2 overexpression worsens diabetic kidney disease in mice. Kidney Int 2017; 92: 909–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marrero MB, Banes-Berceli AK, Stern DM. et al. Role of the JAK/STAT signaling pathway in diabetic nephropathy. Am J Physiol Renal Physiol 2006; 290: F762–F768 [DOI] [PubMed] [Google Scholar]
- 34. Genovese MC, Kremer J, Zamani O. et al. Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med 2016; 374: 1243–1252 [DOI] [PubMed] [Google Scholar]
- 35. Fleischmann R, Schiff M, van der Heijde D. et al. Baricitinib, methotrexate, or combination in patients with rheumatoid arthritis and no or limited prior disease-modifying antirheumatic drug treatment. Arthritis Rheumatol 2017; 69: 506–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dougados M, van der Heijde D, Chen YC. et al. Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: results from the RA-BUILD study. Ann Rheum Dis 2017; 76: 88–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taylor PC, Keystone EC, van der Heijde D. et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med 2017; 376: 652–662 [DOI] [PubMed] [Google Scholar]
- 38. Anderberg RJ, Meek RL, Hudkins KL. et al. Serum amyloid A and inflammation in diabetic kidney disease and podocytes. Lab Invest 2015; 95: 250–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. de Zeeuw D, Bekker P, Henkel E. et al. The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. Lancet Diabetes Endocrinol 2015; 3: 687–696 [DOI] [PubMed] [Google Scholar]
- 40. Vaidya VS, Niewczas MA, Ficociello LH. et al. Regression of microalbuminuria in type 1 diabetes is associated with lower levels of urinary tubular injury biomarkers, kidney injury molecule-1, and N-acetyl-beta-D-glucosaminidase. Kidney Int 2011; 79: 464–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Niewczas MA, Gohda T, Skupien J. et al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J Am Soc Nephrol 2012; 23: 507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gohda T, Niewczas MA, Ficociello LH. et al. Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J Am Soc Nephrol 2012; 23: 516–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Titan SM, Vieira JM Jr, Dominguez WV. et al. Urinary MCP-1 and RBP: independent predictors of renal outcome in macroalbuminuric diabetic nephropathy. J Diabetes Complications 2012; 26: 546–553 [DOI] [PubMed] [Google Scholar]
- 44. Wolkow PP, Niewczas MA, Perkins B. et al. Association of urinary inflammatory markers and renal decline in microalbuminuric type 1 diabetics. J Am Soc Nephrol 2008; 19: 789–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pichler R, Afkarian M, Dieter BP. et al. Immunity and inflammation in diabetic kidney disease: translating mechanisms to biomarkers and treatment targets. Am J Physiol Renal Physiol 2017; 312: F716–F731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Menne J, Eulberg D, Beyer D. et al. C–C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transplant 2016; 32: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mann JF, Schmieder RE, McQueen M. et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372: 547–553 [DOI] [PubMed] [Google Scholar]
- 48. Parving HH, Brenner BM, McMurray JJ. et al. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med 2012; 367: 2204–2213 [DOI] [PubMed] [Google Scholar]
- 49. Fried LF, Emanuele N, Zhang JH. et al. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med 2013; 369: 1892–1903 [DOI] [PubMed] [Google Scholar]
- 50. Mann JF, Green D, Jamerson K. et al. Avosentan for overt diabetic nephropathy. J Am Soc Nephrol 2010; 21: 527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Phillips CO, Kashani A, Ko DK. et al. Adverse effects of combination angiotensin II receptor blockers plus angiotensin-converting enzyme inhibitors for left ventricular dysfunction: a quantitative review of data from randomized clinical trials. Arch Intern Med 2007; 167: 1930–1936 [DOI] [PubMed] [Google Scholar]
- 52. Papp KA, Menter MA, Raman M. et al. A randomized phase 2b trial of baricitinib, an oral Janus kinase (JAK) 1/JAK2 inhibitor, in patients with moderate-to-severe psoriasis. Br J Dermatol 2016; 174: 1266–1276 [DOI] [PubMed] [Google Scholar]
- 53. Khoshdel A, Carney S, Gillies A. et al. Potential roles of erythropoietin in the management of anaemia and other complications diabetes. Diabetes Obes Metab 2008; 10: 1–9 [DOI] [PubMed] [Google Scholar]
- 54. Parganas E, Wang D, Stravopodis D. et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell 1998; 93: 385–395 [DOI] [PubMed] [Google Scholar]
- 55. Kremer JM, Emery P, Camp HS. et al. A phase IIb study of ABT-494, a selective JAK-1 inhibitor, in patients with rheumatoid arthritis and an inadequate response to anti-tumor necrosis factor therapy. Arthritis Rheumatol 2016; 68: 2867–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schwartz DM, Kanno Y, Villarino A. et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov 2017; 16: 843–862 [DOI] [PubMed] [Google Scholar]
- 57. Taylor PC, Abdul Azeez M, Kiriakidis S.. Filgotinib for the treatment of rheumatoid arthritis. Expert Opin Investig Drugs 2017; 26: 1181–1187 [DOI] [PubMed] [Google Scholar]
- 58. Payne C, Zhang X, Shahri N. et al. Evaluation of potential drug–drug interactions with baricitinib. Ann Rheum Dis 2015; 74: 1063 [Google Scholar]
- 59. Afkarian M, Zelnick LR, Hall YN. et al. Clinical manifestations of kidney disease among US adults with diabetes, 1988–2014. JAMA 2016; 316: 602–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.