


Abstract
The spatiotemporal program of metazoan DNA replication is regulated during development and altered in cancers. We have generated novel OK-seq, Repli-seq and RNA-seq data to compare the DNA replication and gene expression programs of twelve cancer and non-cancer human cell types. Changes in replication fork directionality (RFD) determined by OK-seq are widespread but more frequent within GC-poor isochores and largely disconnected from transcription changes. Cancer cell RFD profiles cluster with non-cancer cells of similar developmental origin but not with different cancer types. Importantly, recurrent RFD changes are detected in specific tumour progression pathways. Using a model for establishment and early progression of chronic myeloid leukemia (CML), we identify 1027 replication initiation zones (IZs) that progressively change efficiency during long-term expression of the BCR-ABL1 oncogene, being twice more often downregulated than upregulated. Prolonged expression of BCR-ABL1 results in targeting of new IZs and accentuation of previous efficiency changes. Targeted IZs are predominantly located in GC-poor, late replicating gene deserts and frequently silenced in late CML. Prolonged expression of BCR-ABL1 results in massive deletion of GC-poor, late replicating DNA sequences enriched in origin silencing events. We conclude that BCR-ABL1 expression progressively affects replication and stability of GC-poor, late-replicating regions during CML progression.
INTRODUCTION
Genome duplication is a crucial biological process that ensures accurate transmission of genetic information to daughter cells (1). In eukaryotic cells, multiple functional replication origins are assembled (licensed) during the G1 phase of the cell cycle and are activated (fire) at different times through S phase (2,3). Replication forks emanate from origins and merge wherever they happen to meet rather than at specific sites. Understanding the spatiotemporal program of DNA replication is essential as replication stress (RS), an increased incidence of slowed or stalled replication forks, is today recognized as a major threat to genome stability in stem cells, cancer, development, aging and rare genetic diseases (4–9).
Oncogene expression can induce RS and trigger DNA damage from the earliest tumorigenesis stages (10–14). In precancerous lesions, RS induces a DNA damage response (DDR) that can trigger senescence or apoptosis. Tumorigenesis becomes able to proceed when the DDR is downregulated (e.g. by p53 mutation), favoring cell proliferation with genome instability (10–14). Oncogenes have been proposed to trigger RS by multiple mechanisms: reduced or increased origin firing, exhaustion of limiting nucleotides or replication factors, increased transcription and replication-transcription conflict. For example, in Xenopus egg extracts, in which no transcription takes place, addition of recombinant Myc increases origin firing, fork stalling, and DNA breakage in a manner dependent on Cdc45, a limiting origin firing factor, and these effects are recapitulated by addition of recombinant Cdc45 alone (15,16). In contrast, overexpression of HRASv12 in cultured cells stimulates RNA synthesis and RS in a manner dependent on TBP, a general transcription factor, and these effects are recapitulated by overexpression of TBP alone; increased origin firing seems to be a consequence rather than a cause of RS in this case (17). Recently, a novel nascent DNA mapping assay was used to show that overexpression of Cyclin E1 or MYC, which shortens G1 phase, induces novel intragenic origins, normally erased by transcription during G1, that are particularly prone to fork collapse due to conflict with transcription (18). However, this study only interrogated the earliest-replicating, gene-rich part of the genome, and ectopic origins were only induced in cells with the shortest G1 phase. It remains unclear if oncogene expression can more globally disrupt the spatiotemporal program of DNA replication.
Robust methods to map the mean replication time (MRT) of specific sequences have shown that up to one-half of the genome can switch MRT during development, primarily in units of 400–800 kb (19), to create cell-type specific MRT profiles (20). Deregulation of MRT has been associated with cancer (20,21). A comprehensive study reported that 9–18% of MRT domains from leukemia cells deviated from normal lymphoblastoid cell lines (LCLs), whereas only 2–4% of the MRT domains deviated between LCLs (22). Although leukemic samples were more heterogeneous than LCLs, they shared many replication abnormalities, suggesting early epigenetic alterations of DNA replication in cancer development (22).
Human MRT profiles are not sufficiently resolutive to map individual replication origins (3). However, quantitative analysis of human genome replication was recently achieved by strand-oriented sequencing of purified Okazaki fragments (OK-seq), which reveals the proportions of rightward- (R) or leftward- (L) moving forks along the genome (23). Replication fork directionality (RFD = R – L) profiles disclose replication initiation and termination zones as well as regions of unidirectional fork progression. OK-seq has been used to profile GM06990, an EBV-immortalized lymphoblastoid cell line (LCL) with a near-normal karyotype and HeLa, an epithelial cell line from a cervix adenocarcinoma (23). In both cell lines, replication initiates stochastically within non-transcribed broad (10–100 kb) zones and terminates dispersively between them. One-half of initiation zones (IZs) are circumscribed by active genes, and genes expressed in one cell type are flanked by IZs only in that cell type. Such transcription-associated IZs fire early in S phase. Another half of IZs are not associated with active genes and they fire later in S phase. More IZs were detected in HeLa (9386) than in GM06990 (5684; 4150 shared), but the lack of matched controls precluded attribution of this difference to the cancerous versus non-cancerous origin of the cells. Other properties of IZs were not detectably different between the two cell types.
Here we have generated novel RFD, gene expression and MRT data allowing to compare a total of twelve cell lines, including lymphoid, myeloid and adherent cell types. Lymphoid cell lines, in addition to GM06990, include BL79 and Raji, two independently established Burkitt lymphoma cell lines (BLs), and IARC385, an EBV-immortalized LCL established from the same patient as BL79. Adherent cells, in addition to HeLa, include TLSE19 and IB118, two leiomyosarcoma (LMS) cell lines established from two different patients, and IMR90 primary human fibroblasts. Myeloid cell lines comprise a cellular model for establishment and progression of chronic myeloid leukemia (CML), a malignant disease characterized by the Philadelphia chromosome and the formation of the BCR-ABL1 fusion gene, whose expression is necessary and sufficient for CML formation (24). The tyrosine kinase of ABL1 is constitutively activated by the juxtaposition of BCR. The BCR-ABL1 activity inhibits apoptosis, stimulates proliferation, enhances DNA damage and deregulates DNA repair through complex intracellular signalling (25). To our knowledge, possible effects of BCR-ABL1 on DNA replication have not been reported. We have addressed this question using engineered variants of TF1, a BCR-ABL1 negative erythroleukemia cell line, that express a BCR-ABL-GFP fusion, or GFP as a control, and constitute a validated model for CML establishment and early progression (26–29), and K562, an erythroleukemia cell line derived from a CML patient in blast crisis, which is a late CML model.
The results show that the RFD and RNA-seq profiles of the 12 cell lines cluster in accordance to developmental origin and/or cancerous character, reflecting specific tumour progression pathways. RFD changes between cell lines are widespread through the genome but more frequent in GC-poor regions. In contrast, RNA-seq changes do not vary uniformly with GC content, indicating that replication changes are dissociated from transcription in a cell-type dependent manner. BCR-ABL1 expression in TF1 cells does not trigger large-scale MRT changes comparable to those previously observed between LCLs and leukemia or between different cell types (22). However, many IZs are altered, more often downregulated (∼2/3) than upregulated (∼1/3), predominantly in GC-poor, lowly expressed and late replicating regions, with minimal effects on local MRT. IZ efficiency changes initiated by BCR-ABL1 expression are accentuated during prolonged BCR-ABL1 expression in TF1 and furthermore in the late CML cell line K562. BCR-ABL1 therefore has a long-lasting action on IZ efficiency, in a direction that depends on each IZ but is more often repressive. Strikingly, prolonged BCR-ABL1 expression in TF1 results in massive deletion of GC-poor, late replicating regions enriched in origin silencing events. These results suggest a potential mechanism for generating RS and genome instability independently of transcription by perturbed replication of GC-poor, late-replicating gene deserts.
MATERIALS AND METHODS
Cell lines
GM06990 and HeLa source and culture conditions have been described (23). Raji (Burkitt's lymphoma, BL) and K562 (late stage chronic myeloid leukemia, CML) were purchased from the American Type Culture Collection (ATCC) and cultured as recommended. BL79 and IARC385 were duplicated from the International Agency for Research on Cancer (IARC) library (Lyon, France). BL79 was established from an Epstein-Barr Virus (EBV)-positive BL, while IARC385 was obtained by in vitro EBV immortalization of B lymphocytes from the same patient. BL79 and IARC385 were grown in Roswell Park Memorial Institute (RPMI) 1640 GLUTAMAX medium (Thermofisher), 10% fetal bovine serum (FBS; Thermofisher), 20 mM glucose, 1 mM sodium pyruvate, 1 U ml−1 penicillin, 1 μg ml−1 streptomycin, 2 mM glutamax (Thermofisher). TF1 is a BCR-ABL negative cell line established from a patient with erythroleukemia (ATCC). TF1-GFP and TF1 BCR-ABL were obtained by transduction of green fluorescent protein (GFP) or a BCR-ABL-GFP fusion with a murine stem cell virus (MSCV)-based retroviral vector. EGFP+ cells were sorted using a Becton Dickinson FACSAria and cultured as described previously (27). TF1-BCR-ABL cells were analyzed after culturing for 1 month (TF1-BCRABL-1M) or 6 months (TF1-BCRABL-6M) following transduction. Normal primary fibroblasts (IMR90) were obtained from ATCC. The IMR90-hTERT cell line was obtained by immortalization with h-TERT catalytic subunit at Passage 4. Both IMR90 and IMR90-hTERT were grown in Eagle's Minimum Essential Media (EMEM), 10% FBS, 1 U ml−1 penicillin, 1 μg ml−1 streptomycin. TLSE19 and IB118, two leiomyosarcoma (LMS) cell lines established after surgical resection from a buttock muscle tumor and a cutaneous scalp tumor, respectively, were cultured in RPMI 1640 GLUTAMAX, 10% FBS, 1 U ml−1 penicillin, 1 μg ml−1 streptomycin. All cells were maintained in a humidified atmosphere of 5% CO2 at 37°C.
RFD profiling
RFD profiling of all cell lines was performed by OK-seq as described (23). Briefly, after a 2 min pulse of replicative incorporation of the thymidine analogue 5-ethynyl-2′-deoxyuridine (EdU), DNA was purified, heat-denatured, Okazaki fragments were size-purified on sucrose gradients, biotin-labelled at EdU sites by click-chemistry, captured on streptavidin-coated magnetic beads, amplified by PCR and subjected to Illumina sequencing at the IB2C high-throughput sequencing platform (Gif-sur-Yvette, France). Sequence reads were identified and demultiplexed using the standard Illumina software suite and adaptor sequences were removed by Cutadapt (version 1.2.1 to 1.12). Reads >10 nt were aligned to the human reference genome (hg19) using the BWA (version 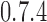 ) software with default parameters. We considered uniquely mapped reads only and counted identical alignments (same site and strand) as one to remove PCR duplicate reads. For GM06990, filtered reads were obtained from the authors of (23). The total number of filtered reads per cell line ranged from 78.4 × 106 (TF1-BCRABL-6M) to 1063.3 × 106 (GM06990). RFD was computed as RFD = (R – F)/(R + F) where R (resp. F) is the number of reads mapped to the reverse (resp. forward) strand of the considered regions.
) software with default parameters. We considered uniquely mapped reads only and counted identical alignments (same site and strand) as one to remove PCR duplicate reads. For GM06990, filtered reads were obtained from the authors of (23). The total number of filtered reads per cell line ranged from 78.4 × 106 (TF1-BCRABL-6M) to 1063.3 × 106 (GM06990). RFD was computed as RFD = (R – F)/(R + F) where R (resp. F) is the number of reads mapped to the reverse (resp. forward) strand of the considered regions.
From 2 (IB118, GM06990) to 6 (BL79) biological replicates were sequenced per cell line. RFD profiles from biological replicates were highly correlated, with Pearson correlation computed in 50 kb non-overlapping windows with >100 mapped reads (R + F) ranging from 0.962 to 0.997. The Pearson correlation between two technical replicates of IMR90 primary cells was 0.996 and their correlations with an RFD profile of the immortalized IMR90-hTERT cell line were 0.989 and 0.992, indicating that immortalization by hTERT did not affect the replication program at 50 kb resolution. Indeed, visual comparison of the RFD profile for primary and immortalized IMR90 cells did not disclose any new or silenced initiation zones. Therefore, the three profiles were pooled together to produce the IMR90 RFD profile.
Copy number of a given region was estimated using OK-seq reads as cRPKG computed as the read coverage expressed in reads per kb per giga (109) filtered reads (RPKG) corrected for the mappability of the region defined as the average 50-mer CRG alignability track (wgEncodeCrgMapabilityAlign50mer.wig) downloaded from UCSC genome browser (http://hgdownload.cse.ucsc.edu/goldenpath/hg19/database/).
MRT profiling
MRT was profiled by Repli-seq (30,31), which consists in sequencing of newly replicated DNA of cells sorted by DNA content into consecutive compartments of S-phase. For GM06990, GM12878, K562, HeLaS3 and IMR90, alignment files of Repli-seq libraries (BAM files) for six S-phase fractions were obtained from the ENCODE project (31,32) at http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeUwRepliSeq/. The number of aligned reads ranged from 8.5 × 106 in K562 to 62.8 × 106 in IMR90 and all reads were used in downstream analyses.
For TF1-GFP and TF1-BCRABL-1M, asynchronously growing cells were incubated with 20 μM EdU for 1 h, pelleted at 200 g for 5 min at room temperature, resuspended to a single-cell suspension (5 × 106 cells/ml) in ice-cold PBS containing 1% FBS, then gently vortexed while 3 vol. of ice-cold ethanol were added dropwise. The fixed cells were stored at –20°C. A total 80 × 106 cells were pelleted at 200 g for 8 min, resuspended in 4 ml of PBS, followed by addition of Triton X-100 and DAPI to final concentrations of 0.1% and 0.02 μg ml−1, respectively, and incubated for 1 h at room temperature. Cells were sorted with MoFlo Astrios (Beckman Coulter) according to DNA content in 6 fractions. An aliquot of unsorted cells was kept as control. Genomic DNA was extracted by a standard proteinase K-phenol-chloroform technique (33), precipitated with isopropanol and resuspended in 10 mM Tris–HCl pH 8.0. For each sample 660 ng of genomic DNA was adjusted to 130 μl and sonicated (Covaris) to ∼300 bp. Biotinylation of nascent DNA was performed by click chemistry for 30 min at room temperature in presence of 2 mM CuSO4, 1.7 mM biotin–TEG–Azide, 10 mM Sodium Ascorbate, followed by purification with Qiagen Min-Elute Clean-Up Kit. End-repair, A-tailing and adapter ligation was performed using Illumina TruSeq DNA LT Sample Prep Kit according to the manufacturer's manual. Biotinylated DNA fragments were captured with 200 μg of Dynabeads® MyOne™ Streptavidin T1 according to the manufacturer's protocol (Thermo). Unligated adapters were removed by washing five times with 5 mM Tris–HCl pH 7.5, 0.5 mM EDTA, 1 M NaCl, 0.05% Tween 20, twice with 10 mM Tris–HCl pH 8.0, 1 mM EDTA, 0.05% Tween 20 and once with water. Libraries were amplified in 12 cycles of PCR with Phusion® polymerase (NEB) using 10 μl of bead suspension as template, purified with Qiagen MinElute PCR purification kit, sequenced on Illumina HiSeq platform and aligned to the human genome (hg19) at the IB2C sequencing platform as for OK-seq libraries. Only uniquely mapping reads (186.0 × 106 for TF1-GFP and 143.3 × 106 for TF1-BCRABL-1M ) were used in downstream analyses.
To compute MRT profiles, tag densities in 100 kb windows were normalized and denoized as in (34,35) using modified thresholds for the two EdU-based datasets (TF1-GFP and TF1-BCRABL-1M). At each locus, the distribution of replication times was obtained by normalizing the denoized tag densities of each S-phase fractions by their sum. MRT values were estimated as the mean of these distributions.
Gene expression profiling
For Raji, GM06990, BL79, or IARC385, total RNA was extracted from 1–10 × 106 exponentially growing cells using Tri reagent (MRC Euromedex) following the manufacturers’ manual and further purified by Turbo DNase (ThermoFisher) treatment, chloroform extraction and precipitation with sodium acetate and ethanol. Library preparation and Illumina sequencing were performed at the Ecole Normale Supérieure Genomic Platform (Paris, France). RNA quality (28S/18S ratio) was checked by Agilent 2100 Bioanalyzer (Agilent). PolyA+ RNA was purified from 1 μg of total RNA using PrepX PolyA mRNA isolation kit (Wafergen). Libraries were prepared using the strand specific RNA-Seq library preparation PrepX RNA-seq kit (Wafergen) and a 41 bp paired-end read sequencing was performed on a NextSeq 500 device (Illumina). Three biological replicates were prepared for each cell line yielding from 75.1 × 106 to 116.1 106 paired-end reads passing Illumina quality filter per replicate.
For TF1-GFP, TF1-BCRABL-1M and TF1-BCRABL-6M, three biological replicates each of total RNA prepared by standard TRIzol (Life Technologies) / chloroform extraction followed by 70% ethanol precipitation were sent for sequencing to ProfileXpert (http://profilexpert.fr), the genomic and microgenomic platform of Université Claude Bernard Lyon 1, France. Quality was checked by Ribogreen/Bioanalyzer. PolyA RNA purification from 2μg samples and indexed sequencing libraries construction were performed using Illumina TrueSeq RNA kit and sequenced on Illumina HiSeq 2500 using 2 flow cell lines (51bp Rapid Single Read run). From 18.2 × 106 to 22.4 × 106 quality-controlled reads were obtained per replicate.
For TLSE19 and IB118, RNA extraction from frozen samples was performed by standard TRIzol (Life Technologies)/chloroform extraction followed by 70% ethanol precipitation. RNA was further purified using the RNeasy Mini Kit (Qiagen) with a DNase treatment (RNase-Free DNase Set, Qiagen). RNAs were quantified using a Nanodrop 1000 spectrophotometer (Thermo Scientific) and qualified with the Agilent 2100 Bioanalyzer (Agilent) using the RNA 6000 Nano Kit according to the manufacturer's instructions. For RNA sequencing an ERCC RNA Spike-In Mix (Life technologies) was added to the RNA as recommended by the manufacturer. Total RNA was ribo-depleted using the Ribo-Zero Gold Kit. RNA profiling was performed using paired-end sequencing (2 × 76 bp), yielding 137.1 × 106 and 104.9 × 106 paired-end RNA-seq reads for TLSE19 and IB118, respectively.
For K562, HeLaS3 and IMR90, we used RNA-seq data from the ENCODE project. We selected two biological replicate paired-end sequence datasets from whole cell PolyA+ RNA per cell line. Read files in fastq format were downloaded from the European Nucleotide Archive https://www.ebi.ac.uk/ena under accession numbers SRR315336 and SRR315337 for K562 and accession numbers SRR315330 and SRR315331 for HeLaS3. For IMR90, fastq files for experiment ‘EncodeCshlLongRnaSeqImr90CellPap’ were downloaded from UCSC http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeCshlLongRnaSeq/; these data correspond to accession numbers SRR534301 and SRR534302.
Gene transcriptional levels for the 12 cell lines were estimated using the same computational pipeline based on the TopHat suite of softwares (36). Tophat (version 2.1.1) and bowtie2 (version 2.2.9) were used to align RNA-seq reads to the human genome (hg19). RNA abundance were computed using Cufflinks (version 2.2.2). We fed the reference transcript annotation provided by Illumina iGenomes (ftp://igenome:G3nom3s4u@ussd-ftp.illumina.com/Homo_sapiens/UCSC/hg19/Homo_sapiens_UCSC_hg19.tar.gz), to tophat (-G option) and cufflinks (-g option) and only considered RNA abundance estimates for genes present in the reference annotation. For each cell line, we thus obtained for the same set of 24 371 genes of size > 300 bp the estimated mRNA level expressed in FPKM (Fragments Per Kilobase of exon model per Million mapped fragments) and a 95% confidence interval on the FPKM ([FPKMlo, FPKMhi]). We considered a gene to be expressed when FPKM ≥ 1 and we filtered out cases where FPKMhi/FPKMlo > 2 (at most 118 genes were filtered out in TF1-BCRABL-6M). The proportion of expressed genes ranged from 43.5% in Raji to 51.3% in TF1-BCRABL-1M. Transcription level of any region 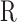 of length lR was estimated based on the FPKM values of all the expressed genes
of length lR was estimated based on the FPKM values of all the expressed genes 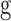 that overlap with the regions as:
that overlap with the regions as: 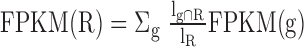 where
where 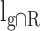 is the overlap length between
is the overlap length between 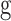 and R.
and R.
GC-content classification of the human genome
GC-content fluctuations recapitulate the non uniform organisation of gene size (37), gene density (38) and replication timing (39) along the human genome. Here, we defined GC-content categories following the five isochores classification of the human genome (40) in light isochores L1 (GC < 37%) and L2 (37% ≤ GC < 41%) and heavy isochores H1 (41% ≤ GC < 46%), H2 (46% ≤ GC < 53%) and H3 (GC ≥ 53%). The genome coverage of L1, L2, H1, H2, H3 was 26.5%, 32%, 24.0%, 13.1% and 4.4%, respectively, after classification based on GC content in non-overlapping 10 kb windows. The proportion of genes in each GC content category was 6.3%, 15.2%, 26.8%, 30.3% and 21.4%, respectively, when attributing the 24,371 genes of size >300 bp to the GC-content category of their promoter region's window.
Computational analyses of RFD, RNA-seq and MRT profiles
All analyses were restricted to the 22 autosomes to avoid artefacts due to the XX or XY karyotypes of the studied cell lines. Computation and figures were implemented in python (version 2.7.13) using numpy (version 1.11.3), scipy (version 0.18.3) and matplotlib (version 2.0.0) scientific computing modules.
RFD profile Pearson correlation CRFD between a pair of cell lines was computed using non-overlapping 10 kb windows with >100 OK-seq reads in both cell lines. RNA-seq correlation CRNA–seq was computed between log10(FPKM) of genes expressed in both cell lines. MRT correlation CMRT was computed between non-overlapping 100 kb windows with a valid MRT estimate. Each set of pair-wise correlation distances (D = 1 – C) was used to hierarchically cluster cell lines using minimum distance as linkage criterion (single linkage clustering). Line and column order of correlation matrices were chosen to be coherent with a dendrogram representation of their hierarchical classification. The results of the correlation analyses were unchanged if calculated using Spearman rank-correlation coefficients instead of Pearson correlations.
When analysing GC-content, FPKM and MRT distributions in 200 kb windows depending on the change of RFD (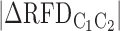 ) between two cell lines (C1, C2), we only considered windows with > 2,000 OK-seq reads in both cell lines to guarantee that the standard deviation, using a Poisson approximation, was <0.023 for both RFD estimates.
) between two cell lines (C1, C2), we only considered windows with > 2,000 OK-seq reads in both cell lines to guarantee that the standard deviation, using a Poisson approximation, was <0.023 for both RFD estimates.
Manual annotation of IZs’ efficiency change in a CML progression model
RFD profiles changes in the first two steps of the CML initiation and progression model (Step 1: TF1-GFP → TF1-BCRABL-1M; Step 2: TF1-BCRABL-1M → TF1-BCR-ABL-6M) were annotated by manually scanning the profiles in 2 Mb windows. An IZ present in the initial state was annotated as Silenced if no IZ was present in the following state or Weakened (resp. Enhanced) when IZ efficiency decreased (resp. increased) between the two consecutive states. IZs inactive in the initial state but active in the final state were annotated New. Moreover, each locus annotated for a Step 1 or Step 2 change (total 1027) was also annotated for its status in Step 3 (TF1-BCRABL-6M → K562). Step 3-specific changes were too numerous to be manually annotated. The 1027 manually annotated loci included 253 IZs which changed efficiency at Step 1 but not Step 2, 551 IZs efficiency changes during Step 2 but not Step 1, and 223 IZs which changed efficiency in both Step 1 and Step 2. In total, this database encompasses 476 and 774 and 716 efficiency changes in Steps 1 and 2 and 3, respectively (Supplementary Table S1).
To minimize human bias inherent to manual annotation, multiple criteria for calling significant differences were elaborated. The vast majority of RFD differences between biological replicates were <0.2 and narrowly localized rather than spread over typical replicon sizes as expected for authentic IZ efficiency changes. Therefore, criteria for calling changes were (i) a change in the amplitude of an ascending RFD segment; (ii) accompanied on one or both sides by extended RFD shift(s) over tens or hundreds of kbs, with (iii) an amplitude >0.2. The latter criterion was modulated according to local noise and was relaxed when long range, correlated RFD shifts on IZ flank(s) were obvious. Annotations were performed by one investigator and curated by a second one.
To further control for false positives and reproducibility, the second investigator blindly annotated multiple RFD profile pairs including Step 1 changes and biological replicates. Zero changes were called between biological replicates, but 791 Step 1 changes were blindly called that included 78% of the 476 changes originally called. The two datasets showed almost identical percentages of new, enhanced, weakened and silenced IZs and similar distributions of MRT, RNA-seq FPKM and GC content, although the second dataset contained a higher proportion of enhanced or weakened IZs close to active genes. Overall, these results indicate very few if any false positive calls and little impact of the investigator on the global properties of the called IZs.
RESULTS
OK-seq RFD profiling of multiple normal and tumor cell lines
We used OK-seq to profile RFD genome-wide in multiple cell lines as previously described for HeLa and GM06990 (23). Four lymphoid (GM06990, Raji, BL79, IARC385), four myeloid (TF1-GFP, TF1-BCRABL-1M, TF1-BCRABL-6M, K562) and four adherent (IMR90, HeLa, TLSE19, IB118) cell types were analyzed.
The 12 RFD profiles of an exemplary 20 Mb segment of chromosome 3 are shown on Figure 1A. RNA-seq and MRT profiles of the same region are shown on Supplementary Figure S1. RFD profiles displayed an alternation of quasi-linear ascending, descending and flat segments (AS, DS, FS) of varying size and slope. RFD often reached values >0.9 or <–0.9, indicating nearly complete purity of Okazaki fragments. A positive (negative) RFD value indicates that forks move predominantly rightward (leftward) in the cell population. AS and DS therefore represent zones of predominant replication initiation (IZ) and termination (TZ), respectively. The amplitude of the RFD shift across each zone reflects its net initiation or termination efficiency. FS of high RFD are unidirectionally replicating regions. FS of null RFD, sometimes found in the middle of a TZ, are replicated equally often in both directions, presumably by random initiation and termination. A few exemplary IZs, TZs and FSs in GM06990 are illustrated at higher resolution for a 6 Mb segment of chromosome 5 on Figure 1B.
Figure 1.

Genome-wide profiling of replication fork directionality. (A) Replication fork directionality (RFD) along a 20 Mb segment of chromosome 3 in 4 lymphoid (top), 4 myeloid (middle) and 4 adherent (bottom) cell lines, as indicated in the top right corners of each panel. (B) RFD profile along a 6 Mb fragment of chromosome 5 where some zones of preferential initiation (IZ, orange), preferential termination (TZ, green) and unidirectional fork progression (thick red (resp. blue) arrows: sense (resp. antisense) fork progression) are marked; the pair of red and blue thin dashed arrows mark a region of null RFD where forks progress in equal proportion in the two directions.
Both shared and cell-type specific RFD patterns were observed. For example, a region replicated differently in GM06990 from other lymphoid cell lines and a region replicated similarly in all lymphoid cell lines are visible at 4.5–5.0 Mb and at 10–12 Mb, respectively, on Figure 1A.
Cell line classification based on DNA replication or transcription profiling
To objectively quantify differences between cell lines, we computed the pairwise correlation coefficients between RFD profiles (averaged from all biological replicates) and ordered them by distance using unsupervised hierarchical clustering (Figure 2A). We similarly analyzed transcription (RNA-seq; Figure 2B) and mean replication time (MRT; Figure 2C) data. All the observed differences between cell lines were larger than the variations between biological replicates (Pearson correlations 0.961–0.996 for RFD computed at 50 kb; 0.954–0.995 for RNA-seq; >0.99 for MRT; Supplementary Figure S2). Lymphoid, myeloid and adherent cells formed three separate RFD and MRT clusters. A similar classification was observed by RNA-seq except that HeLa clustered with myeloid instead of adherent cells. In contrast to RFD profiles, RNA-seq and MRT profiles were generated by different labs using different methods. We cannot exclude that HeLa clustered with myeloid cells by RNA-seq because HeLa, IMR90 and K562 RNA-seq profiles were from Encode whereas other myeloid and adherent cell profiles were generated differently. Despite the higher homogeneity of RFD methods, the correlation coefficients were generally smaller by RFD than by RNA-seq or MRT. Within-group correlation distances were similar by RFD so that the three groups were recovered by cutting the dendrogram at level 0.3. The situation was more heterogeneous by RNA-seq where within-group correlation distances increased from lymphoid to myeloid to adherent cells and the three groups could not be recovered by cutting the dendrogram at a constant level.
Figure 2.

Cell line classification based on correlations between replication and gene expression profiles. (A–C) Correlation matrices between RFD profiles (CRFD ; A), RNA-seq (CRNA–seq ; B) and MRT profiles (CMRT ; C); Pearson correlation coefficient values are color-coded from blue (0.4) to red (1.0) using the colour bar on the right (Materials and Methods). (Top) A corresponding dendrogram representation of the hierarchical classification of cell lines is shown on top of each correlation matrix; ordinate is the correlation distance (Materials and Methods). (D) Cumulative distributions of the absolute MRT changes (|ΔMRT|) between cell lines. Each curve is color-coded according to the pair of cell lines indicated in the insert. The considered threshold of significance (45) (|ΔMRT| >0.2) is indicated by a vertical dotted line.
Within the myeloid group, RFD profiles clustered in accordance to CML progression (Figure 2A). Profile differences accumulated with BCR-ABL1 expression time in TF1, which increased resemblance to K562, a late CML. A similar, albeit weaker, progression from early to late CML was also observed by RNA-seq (Figure 2B). In contrast, MRT profiles did not reflect this progression, since K562 appeared more correlated to TF1-GFP than to TF1-BCRABL-1M (Figure 2C). This cannot be explained by variations in MRT methodology since both TF1 derivatives were profiled by the same method. In summay, RFD and RNA-seq profiles suggested the existence of CML-specific replication and transcription changes not necessarily reflected in MRT profiles. To further investigate this, we compared the cumulative distributions of MRT changes between TF1-GFP, TF1-BCRABL-1M and K562 to those observed between distinct LCLs (GM06990 versus GM12878) and distinct cell types (K562, GM06990, IMR90) (Figure 2D). The proportion of MRT changes with |ΔMRT| > 0.2 after one month of BCR-ABL1 expression in TF1 cells (1.13%) was slightly higher than between LCLs (0.3%), but much lower than between distinct cell types (K562 versus IMR90: 22.9%; K562 versus GM06990: 12.8%; IMR90 versus GM06990: 21.8%). In contrast, the proportion of |ΔMRT| > 0.2 changes between TF1-GFP and K562 (9.45%) was more similar to that previously observed between LCLs and leukemia . We conclude that RFD changes induced in our model for CML establishment and early progression are not accompanied by large-scale shifts in MRT, but that such large-scale shifts may appear during progression to late CML (K562).
Within the lymphoid cell group, a similar classification of cell lines was again obtained by RNA-seq and RFD (Figure 2A, B). The two BLs (Raji, BL79) were more correlated to each other than to either LCL (GM06990; IARC385), suggesting the existence of BL-specific replication and transcription patterns. BL79 was more correlated to IARC385 than to GM06990. This was expected since IARC385 was established from the same patient as BL79, as confirmed by HLA typing. Intriguingly, Raji and IARC385 were more correlated to each other than to GM06990. This suggested that IARC385 may share some replication and transcription signatures of BLs, and may not be as ‘normal' as GM06990. Indeed, the two LCLs IARC385 and GM06990 were the least correlated cell lines within this group. A previous analysis reported that MRT profiles of nonleukemic cells are much less variant than those from leukemias . In agreement, we observed a strong correlation of the MRT profiles from GM06990 and GM12878, two LCLs with a near-normal karyotype (Figure 2C). All types of BLs are characterized by dysregulation of the C-MYC gene, located on 8q24, by one of three possible chromosomal translocations. Karyotype analysis showed that BL79 carries a t(8;14)(q24;q32) translocation confirming its identification as a BL cell line. In contrast, a large fraction of IARC385 cells contained a t(4;11) translocation, which is not typical of BLs, but none of the three possible diagnostic translocation of BLs. Genome-wide CGH array analysis revealed few copy number variations (CNVs) in GM06990 and BL79, more in Raji and even more in IARC385 (Supplementary Figure S3). These results suggest either that IARC385 was established from an abnormal, but non-BL blood cell from the same patient as BL79, which seems unlikely, or that IARC385 was destabilized during immortalization, which is more plausible. Either scenario may explain the observed genomic instability associated with replication and transcription changes reminiscent of BLs. EBV transformation occurred in vivo for the BLs but in vitro for the LCLs. It is possible that differential expression of viral latency proteins in BLs and LCLs explains why the RFD profile of BL79 is more related to Raji than to its sister cell line IARC385. Alternatively, this correlation may result from other oncogenic events common to BLs.
Within the adherent cells, different classifications were obtained by RFD and RNA-seq (Figure 2A, B). By RNA-seq, the two LMS IB118 and TLSE19 were more correlated to each other than to IMR90 and less correlated to HeLa, which in fact clustered with myeloid cells. By RFD, however, the strongest resemblance was observed between TLSE19 and IMR90. TLSE19 was only slightly more correlated to IB118 than to HeLa. IB118 was only slightly more correlated to TLSE19 than to IMR90, but was more distant to HeLa. The cell of origin and driver mutations of LMSs are currently unclear. These results may help to distinguish different types of LMS and suggest a possible differentiation of TLSE19 and IB118, which were derived from a buttock muscle tumor and a scalp tumor, respectively. The strong correlation of the RNA-seq profiles of the two LMSs may be due to the use of the different RNA-seq methodologies for LMSs and for other cell types. Alternatively, the shared expression of cancer-specific genes, in two LMSs of different cellular origins, may have resulted in strong convergence of RNA-seq but not RFD profiles. It is notable the RNA-seq and RFD data better matched each other for IB118 than for TLSE19. This suggests that replication is more dissociated from transcription in TLSE19 than in IB118.
We investigated whether CNVs may affect the classification of cell lines. Filtering out aneuploid regions detected by CGH array analysis in the lymphoid cell group (Supplementary Figure S3) did not affect the classification (Supplementary Figure S4A). We observed a good correlation between CGH array signal and OK-seq coverage corrected for mappability (Supplementary Figure S4B). OK-seq coverage for the TF1 cell lines formed distinct clusters allowing us to further select shared diploid regions between lymphoid and TF1 cells covering 12% of the sequenced genome. The cell line classification obtained from these regions alone was again unchanged (Supplementary Figure S4C). Thus, the RFD correlation analysis was robust to CNVs determined by either CGH arrays or OK-seq coverage.
In summary, the global correlation analysis clustered the RFD profiles of the 12 cell lines in accordance to their developmental origin and/or cancerous character, reflecting progression along specific tumour progression pathways. Globally similar results were obtained by RNA-seq, but divergences between RNA-seq and RFD classifications were also observed. These results suggest that recurrent replication changes occur in specific tumour types but that the tightness of their connection with transcription may depend on cell type.
Replication and transcription changes are widespread along the genome
To investigate whether RFD, RNA-seq and MRT differences between cell lines were concentrated in particular regions of the genome, we repeated the analyses shown in Figure 2ABC for each chromosome separately (Supplementary Figure S5). The results obtained for the entire genome were recapitulated for each chromosome, with minor exceptions detailed in the legend to Supplementary Figure S5. The correlation coefficients were again smaller by RFD than by RNA-seq or MRT for each chromosome, but this was more pronounced for GC-poor chromosomes. Indeed, we demonstrate below that RFD changes, although widespread, are more frequent in GC-poor compartments of the genome.
The correlation between the global RFD correlation matrix and each chromosome's RFD correlation matrix was high (>0.893), albeit sensitive to chromosome size as expected (Figure 3A). Correlations between individual chromosomes were also high (>0.79) but again sensitive to chromosome size (Figure 3A). To more precisely assess how widespread RFD changes are, we generated a large number of random probes, 50 kb to 50 Mb in size, consisting each of 5–5000 randomly located 10 kb windows. For each probe, we computed its RFD correlation matrix and the correlation thereof with the global genome correlation matrix. We then determined the statistical fluctuation of the latter correlation and the probability of observing the same classification of cell lines as with the global genome (Figure 3B). We found that a random probe size ≥5 Mb was sufficient to reach a >90% correlation with the global genome matrix, and to observe with a probability >0.95, the same three-group classification of cell lines. This demonstrates that cell-line specific RFD changes are widely distributed over the entire genome. The RFD-based cell line classification therefore does not reflect outlier data points but is representative of the global genome.
Figure 3.

Replication changes between cell lines are widespread through the genome. (A) Correlation coefficients between the global genome correlation matrix between RFD profiles shown in Figure 2A and the individual chromosomes' correlation matrices shown in Supplementary Figure S5. Pearson correlation coefficient values are colour-coded from blue (0.4) to red (1.0) using the colour bar on the right. (B) Random 5 Mb probes are sufficient to recover the cell line classification. Blue, boxplot of the distribution of the correlation coefficient between the global matrix shown in Figure 2A and random probes of indicated scale (Mb) consisting of 5–5000 randomly located 10 kb windows. Orange, probability of observing the same three-group classification of cell lines as in Figure 2A.
Replication but not transcription changes concentrate in GC-poor regions
We repeated the above correlation analyses separately for five increasing GC-content classes reflecting the GC content of the five isochores (L1, L2, H1, H2, H3) of the human genome (Figure 4A–E). GC content has been correlated genome-wide with gene density (38), gene expression and MRT (39). A similar hierarchical clustering of cell lines to that obtained with genome-wide correlation analysis was recovered in each case (Supplementary Figure S6), suggesting that RFD changes between cell lines are widespread through the five isochores. Nevertheless, the RFD correlation coefficients increased with GC content most of the time (Figure 4A–E). When the correlation coefficient differences between each GC-content class and the entire genome were computed (Figure 4F–J), most differences were negative in L1, null in L2 and increasingly positive in H1 to H3. In other words, the RFD profiles were less, equally, or more similar to each other in the L1, L2 or H1-3 fractions, respectively, than in the total genome. Therefore, RFD changes were more frequent in the GC-poor fractions of the genome. These observations were not due to a higher technical noise in GC-poor regions. If due to noise differences, correlations differences should vanish when the scale of analysis is increased. However, the cell classification (Figure 2A) and the GC dependence of correlation differences (Figure 4) were conserved or even enhanced when the scale of analysis was increased from 10 kb to 100 kb, 200 kb and 1Mb (Supplementary Figures S7 and S8).
Figure 4.

RFD profiles are more conserved in high GC-content regions. (A–E) Correlation matrix of RFD profiles depending on the GC content; 10 kb windows were grouped in GC-content categories following the 5 isochores classification of the human genome in light isochores L1 (GC < 37% ; 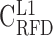 ; A) and L2 (37% ≤ GC < 41% ;
; A) and L2 (37% ≤ GC < 41% ; 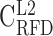 ; B), and heavy isochores H1 (41% ≤ GC < 46% ;
; B), and heavy isochores H1 (41% ≤ GC < 46% ; 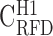 ; C), H2 (46% ≤ GC < 53% ;
; C), H2 (46% ≤ GC < 53% ; 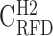 ; D) and H3 (GC ≥ 53% ;
; D) and H3 (GC ≥ 53% ; 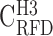 ; E); coverage (Cov) of the sequenced human genome by each isochore family is provided in the header of each column; Pearson correlation coefficient values are colour-coded from blue (0.4) to red (1.0) using the color bar on the right (Materials and Methods). (F-J) Matrices of correlation differences
; E); coverage (Cov) of the sequenced human genome by each isochore family is provided in the header of each column; Pearson correlation coefficient values are colour-coded from blue (0.4) to red (1.0) using the color bar on the right (Materials and Methods). (F-J) Matrices of correlation differences 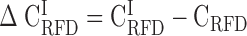 where I can be L1, L2, H1, H2 or H3; correlation difference values are colour coded blue (resp. red) for negative (resp. positive) differences using the color bar on the right; a blue (resp. red) colour indicates that RFD profiles are less (resp. more) conserved in the considered isochore than in the 22 autosomes. Matrices row and column order is the same as in Figure 2A.
where I can be L1, L2, H1, H2 or H3; correlation difference values are colour coded blue (resp. red) for negative (resp. positive) differences using the color bar on the right; a blue (resp. red) colour indicates that RFD profiles are less (resp. more) conserved in the considered isochore than in the 22 autosomes. Matrices row and column order is the same as in Figure 2A.
There were a few exceptions to the general trend of increasing RFD correlation in the L1 < L2 < H1 < H2 < H3 order (Figure 4). The correlations between HeLa and GM06990, and between IB118 and Raji, decreased rather than increased with GC content, in the order H2 < H3 < H1 < L1 < L2. In addition, the order was L1 < L2 < H3 < H1 = H2 for HeLa versus IB118, H1 = L1 < H2 = L2 < H3 for HeLa vs. IMR90 and L1 < H3 < L2 < H1 < H2 for IARC385 versus Raji. We previously observed that in HeLa and GM06990, many IZs border active genes, and isolated genes expressed in only one cell type are flanked by IZs only in that cell type (23). Given that genes are enriched in GC-rich isochores (38), such transcription-dependent changes in IZ activity are expected to decrease the RFD profile correlation more strongly in GC-rich than in GC-poor regions. The comparison of IZs in GM06990 and HeLa also revealed that in addition to these gene-bordering IZs, many cell-type specific IZs are found away from active genes, in late-replicating (23) GC-poor regions. Since GC-poor isochores form a much larger fraction of the genome than GC-rich isochores, the net density of RFD changes between HeLa and GM06990 is higher in GC-rich than in GC-poor regions, due to a predominant contribution of transcription-associated changes in GC-rich regions. Inversely, the net density of RFD changes between other cell lines is most often higher in GC-poor regions, suggesting a predominance of transcription-independent RFD changes in GC-poor regions.
A similar GC-content analysis was performed with the RNA-seq data (Supplementary Figure S9). GC content-dependent changes in correlation coefficients were less marked than for RFD (Supplementary Figure S9A–E). Unlike RFD, correlation difference matrices of RNA-seq data showed no general tendency to follow GC content (Supplementary Figure S9F–J). Caution is required, as RNA-seq profiles, unlike RFD profiles, were obtained by four different methodologies for (i) the lymphoid cells; (ii) the TF1 group; (iii) the LMSs; (iv) the ENCODE cell lines HeLa, K562 and IMR190. Inside the lymphoid group, correlations increased with GC-content. The only exception was the IARC385 versus Raji comparison, with increasing RNA-seq correlations in the order L1 < L2 < H3 < H1 < H2, which in fact closely followed the atypical RFD correlation order L1 < H3 < L2 < H1 < H2 noted above for this pair of cell lines. Thus, in the lymphoid group, the GC-dependence of RFD and RNA-seq correlations closely matched each other. This was also the case for the TF1 group and the LMS group. A more complex situation was observed for the ENCODE cell lines. The order for adherent cells HeLa versus IMR90 was H1 = L1 < H2 = L2 < H3 by RFD but H3 < L1 < L2 < H2 < H1 by RNA-seq, thus deviating from GC-content in a different manner for RNA-seq and RFD. Within the ENCODE group, the myeloid versus adherent comparisons revealed a more subtle deviation of replication and transcription changes. For K562 versus HeLa, the order was L1 < L2 < H1 < H2 = H3 by RFD but L2 < L1 < H3 < H2 < H1 by RNA-seq. For K562 versus IMR90, the order was L1 < L2 < H1 < H2 < H3 by RFD but L1 < L2 < H3 < H2 = H1 by RNA-seq. Other inter-group comparisons also showed a different ordering of RNA-seq and RFD correlations, but in these cases we cannot exclude an effect of the different RNA-seq methodologies. To summarize, the RNA-seq profiles did not reveal an as consistent tendency for transcription changes to increase or decrease with GC content as RFD changes. This suggests that replication changes were at least partly dissociated from transcription changes, to an extent that depended on the cellular context.
BCR-ABL1 expression continuously induces replication changes in gene deserts
We focussed further analysis on the early CML progression model, where changes in RFD can be directly attributed to changes in expression of the BCR-ABL1 oncogene in TF1 cells. We studied the distribution of GC-content, transcription level and MRT over the entire genome or over regions of RFD change between cell lines. The largest RFD changes observed after 1 month (Step 1; Figure 5A–C), or between 1 month and 6 months (Step 2; Supplementary Figure S10) of BCR-ABL1 expression, were observed in GC-poor, lowly expressed and late replicating regions. GC content and MRT are strongly correlated genome-wide (Figure 5D) and even more at the largest RFD changes (Figure 5E). In order to deconvolute the effects of these two parameters on RFD changes, we computed the fold-enrichment of the 5% largest RFD changes after stratifying the data according to both factors. The enrichment kept increasing from high to low GC after controlling MRT, and from early to late MRT after controlling for GC content (Figure 5F). Therefore, neither factor was the sole driver of RFD changes; both showed an impact on RFD changes beyond their correlation.
Figure 5.

The largest RFD changes induced by 1 month of BCR-ABL1 expression are observed in GC-poor, lowly-expressed and late replicating regions. Cumulative distribution functions (cdf) of GC content (A), transcription in TF1-BrcAbl-1M (FPKM1M) (B) and MRT in TF1-BrcAbl-1M (C) computed in non-overlapping 200 kb windows of the 22 autosomes (Materials and Methods). Cdfs were determined for all windows (all, black) or selected windows with the 20% (blue), 10% (green), 5% (yellow) and 1% (red) largest RFD changes between TF1-GFP and TF1-BrcAbl-1M (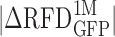 ). Similar results are observed after 6 months of induced BCR-ABL1 expression (Supplementary Figure S10). (D,E) 2D-histograms of GC content vs. MRT (computed in 200 kb windows) of the whole genome (D) or the 5% largest RFD changes following 1 month of BCR-ABL expression. (E) Fold-enrichment of the 5% largest RFD changes after subdividing the genomic bins in four MRT classes (MRT ≤ 0.67; 0.67 < MRT ≤0.718; 0.718 < MRT ≤ 0.75; and MRT > 0.75) and four GC content classes (GC ≤ 0.35; 0.35 < GC ≤ 0.37; 0.37 < GC ≤ 0.4; and GC > 0.4). These subdivisions correspond to quartiles of each parameter distribution for the windows of largest RFD changes, which ensures that each of the 16 bins contains sufficient data for statistically meaningful comparison.
). Similar results are observed after 6 months of induced BCR-ABL1 expression (Supplementary Figure S10). (D,E) 2D-histograms of GC content vs. MRT (computed in 200 kb windows) of the whole genome (D) or the 5% largest RFD changes following 1 month of BCR-ABL expression. (E) Fold-enrichment of the 5% largest RFD changes after subdividing the genomic bins in four MRT classes (MRT ≤ 0.67; 0.67 < MRT ≤0.718; 0.718 < MRT ≤ 0.75; and MRT > 0.75) and four GC content classes (GC ≤ 0.35; 0.35 < GC ≤ 0.37; 0.37 < GC ≤ 0.4; and GC > 0.4). These subdivisions correspond to quartiles of each parameter distribution for the windows of largest RFD changes, which ensures that each of the 16 bins contains sufficient data for statistically meaningful comparison.
Consistent with the high global correlation coefficients (>0.95) of the RFD profiles of the three TF1 cell lines, visual inspection verified a striking identity of the three profiles over most of the genome, as exemplified in Figure 6A. This facilitated detection and manual annotation of IZ efficiency changes, scored as new, enhanced, weakened or silenced IZs (see Materials and Methods), at each step of CML progression. Exemplary annotated 2Mb segments are shown on Figure 6. RNA-seq and MRT profiles of the same regions are shown on Supplementary Figure S11. In total, 476 changes during Step 1 and 774 changes during Step 2 were observed (Supplementary Table S1). The distributions of IZ efficiency changes were strikingly similar at Step 1 and Step 2 (Figure 7A). Weakened IZs were by far the most frequent at each step (55% and 66%, respectively) and enhanced IZs the second most frequent. We confirmed that Step 1 changes preferentially occurred in GC-poor, lowly expressed and late replicating regions (Figure 8). Interestingly, this tendency was more pronounced for new or silenced IZs than for weakened or enhanced IZs, in other words for more extreme changes. Similar results were obtained whether RNA-seq and MRT data from TF1-BCRABL-1M (Figure 8) or from TF1-GFP (Supplementary Figure S12) were used.
Figure 6.

Manual annotation of RFD profile changes during the first two steps of the CML progression model. (A–D) RFD profiles computed in 10 kb non-overlapping windows for TF1-GFP (black), TF1-BrcAbl-1M (yellow) and TF1-BrcAbl-6M (blue) are visualised in 2 Mb regions along the 22 human autosomes. (A) Region along chromosome 12 where RFD profiles in the 3 cell lines do not present any significant difference. (B) Region along chromosome 2 illustrating IZs whose efficiency is repeatedly enhanced (green) or weakened (purple) in Steps 1 and 2 of the model CML progression. (C) Region along chromosome 18 having two IZ efficiency changes in Step 1 (enhanced, leftmost green; weakened, purple) that are confirmed in Step 2. The rightmost green line marks an IZ which is repeatedly enhanced at Steps 1 and 2 (as in B). (D) Region along chromosome 5 illustrating an IZ activated during Step 1 (red line) and silenced during Step 2.
Figure 7.

Persistence of replication initiation zones efficiency changes in the CML progression model. (A) Distribution of IZ efficiency changes (New, Enhanced, Weakened and Silenced; Materials and Methods) in Step 1 (black; n = 476) and Step 2 (yellow; n = 774) of CML progression). (B) Evolution of Step 1 changes during Step 2, reported separately for each type of change. (C) Evolution of Step 2 changes in K562, reported as in (B). Error bars represent a 95% confidence interval assuming data counts are from a Poisson distribution.
Figure 8.

Initiation zones efficiency changes in response to 1 month of BCR-ABL1 expression are observed in GC-poor, lowly-transcribed and late replicating regions. Cumulative distribution functions (cdf) of GC content (A), transcription in TF1-BCRABL-1M (FPKM1M) (B) and MRT in TF1-BCRABL-1M (C) computed in non-overlapping 200 kb windows of the 22 autosomes (Materials and Methods). Cdfs were determined for all windows (all, black) or limited to windows with Silenced (blue), Weakened (green), Enhanced (violet) and New (red) IZ. Similar results are obtained using transcription and MRT data of TF1-GFP (Supplementary Figure S12).
A similar situation was observed for Step 2 changes except that weakened IZs were more often found in GC-rich, highly expressed, early replicating DNA (Supplementary Figure S13). To address whether these weakened IZs were associated with nearby gene transcription changes, we plotted the RNA expression (by 200 kb windows) ratio in TF1-BCRABL-6M over TF1-BCRABL-1M, as a function of their mean expression level (Supplementary Figure S14A) and computed the cumulative distribution of expression changes (Supplementary Figure S14B), for the whole genome or for windows containing at least one such IZ. The results indicate that weakened IZ at Step 2 were significantly, but weakly, associated with nearby transcription repression.
We observed that correlation coefficients between cell lines were generally smaller by RFD than by RNA-seq (Figure 2). We investigated in the CML system whether, when focussing on early replicating regions, changes in RNA predicted changes in RFD and IZs. The distribution of RNA expression changes in early replicating windows with the largest RFD changes at Step 2, was shifted towards RNA repression when compared to all early windows (Supplementary Figure S15A, B), consistent with the association of weakened IZs with transcription repression (Supplementary Figure S14). However, we found no dependence of the distribution of Step 2 RFD changes on RNA expression changes in early replicating regions (Supplementary Figure S15C). Therefore, although the largest RFD changes in early replicating regions were detectably associated with transcription repression, changes in RNA expression did not reciprocally predict RFD changes.
We then analyzed the evolution of the 476 Step 1 changes during Step 2 (Figure 7B). The behavior of the Step 1 changes during Step 2 significantly depended on the type of change at Step 1 (P < 0.001 using a χ2 test of independence). Step 1 changes, whatever their direction, were most frequently (253/476) confirmed during Step 2. New or enhanced IZs at Step 1 that changed again at Step 2 (n = 88) were most frequently enhanced (n = 50). In contrast, weakened IZs at Step 1 that changed again at Step 2 (n = 131) were most frequently further weakened (n = 89) or silenced (n = 33). Therefore, there was a significant tendency for Step 2 changes to follow the same direction as Step 1. These results demonstrate that long-term BCR-ABL1 expression gradually altered the efficiency of specific IZs in TF1 cells thus destabilizing their replication program.
We then analyzed how IZs that changed during Step 2 behaved in K562 cells, a model for advanced CML (Figure 7C). This behavior significantly depended of the type of Step 2 change (P < 0.001, χ2 test). Among the 85 silenced IZs, 75 (88%) remained silent in K562. Among the 514 weakened IZs, a majority (65%) were further weakened (n = 165) or silenced (n = 168). In contrast, a majority (58%) of the 127 enhanced IZs were further enhanced (n = 53) or confirmed (n = 21). Among the 47 new IZs, 43% were confirmed (n = 6) or enhanced (n = 14) but 57% were weakened (n = 3) or silenced (n = 24) in K562. In short, these 773 IZs tended to further change activity in K562 in the same direction as at previous steps, but silencing was stronger (Supplementary Table S1).
In summary, BCR-ABL1 expression during early CML progression changed replication predominantly in GC-poor, lowly expressed and late replicating regions. The targeted IZs were more frequently weakened than enhanced. Targeted IZs in early CML tended to further change activity in the same direction at later tumor progression stages. Therefore, BCR-ABL1 had a long-lasting action on IZs but the direction of the change depended on the targeted region.
BCR-ABL1 induces massive deletions in GC-poor, late-replicating regions
To examine potential links between replication changes and genome instability in the CML model, we used OK-seq coverage to estimate copy number in TF1 cells, as justified above (Supplementary Figure S4). Two-dimensional analysis of OK-seq coverage in TF1-GFP and TF1-BCRABL-1M revealed two clearly demarcated populations consistent with diploid (2N) and triploid (3N) regions and no detectable ploidy changes between the two cell lines (Figure 9A; Supplementary Figure S16A, B). No difference in the distribution of Step 1 RFD changes was observed between 2N and 3N regions (Figure 9C). In contrast, the comparison of TF1-BCRABL-1M and TF1-BCRABL-6M revealed frequent copy number losses, affecting 35% of 2N regions and 34% of 3N regions, and a slightly broader distribution of Step 2 RFD changes in unstable (2Nto1N and 3Nto2N) than in stable (2Nto2N and 3Nto3N) sequences (Figure 9B, D; Supplementary Figure S16B, C). Note that the tilting of the two coma-shaped patterns on Figure 9B indicates that unstable regions were already slightly less abundant than stable regions at Step 1. This suggests that each deletion affected a minor fraction of the cells at Step 1, but was present in a much larger fraction of the cells at Step 2. Importantly, GC-poor and late-replicating regions, where replication changes concentrate (Figure 5), were over-represented in deletion events in either 2N or 3N regions (Figure 10).
Figure 9.

BCR-ABL1 induces genomic deletions associated with replication changes. (A, B) 2D histograms of OK-seq coverage (log2(cRPKG)) in TF1-BCRABL-1M versus TF1-GFP (A) and TF1-BCRABL-6M vs. TF1-BCRABL-1M (B). Stable and unstable diploid and triploid regions are outlined. Normalized histogram values are colour-coded from blue (0.0) to red (3.2) using the colour bar on the right. (C, D) Cdfs of Step 1 (C) and Step 2 (D) RFD changes in indicated regions of panels A and B. Regions where cdfs separate in panel D are enlarged (insets).
Figure 10.

Genomic instability induced by prolonged BCR-ABL1 expression affects GC-poor, late-replicating sequences irrespective of initial ploidy in TF1. Cdfs of GC content (A, B) and MRT in TF1 GFP (C, D) according to ploidy status at Step 1 (A, C) and evolution at Step 2 (B, D) of CML progression.
We computed the density of the different types of IZ efficiency changes at Step 1 and Step 2 in stable and unstable regions (Figure 11). The density of IZ efficiency changes at Step 1 or Step 2 was not significantly different in 2N and 3N regions (Figure 11A, B). However, the density of silenced origins at either Step 1 or Step 2 was from 2- to 3-fold higher in unstable than in stable regions (Figure 11C, D), irrespective of ploidy status (Supplementary Figure S17). In contrast, moderate changes in origin efficiency (enhanced and weakened origins) were depleted in unstable regions at Step 2 (Figure 11D). It is remarkable that even though genomic destabilization only became obvious at Step 2, origin silencing was already enriched in unstable regions, and depleted in stable regions, as strongly at Step 1 than at Step 2 (Figure 11C,D). Thus, replication plasticity, but not copy number, was correlated with genome instability. Although we cannot exclude that both perturbations were independent consequences of BCR-ABL1 expression, origin silencing clearly preceded, and therefore may have triggered, destabilization of GC-poor, late-replicating regions.
Figure 11.

Origin silencing events are enriched in genomic regions destabilized by BCR-ABL1 expression, independently of initial ploidy and before obvious genome instability. Relative densities of IZ efficiency changes (New, Enhanced, Weakened and Silenced) at Step 1 (black; n = 476) or Step 2 (yellow; n = 774) of CML progression according to ploidy status at Step 1 (A, B) and ploidy evolution at Step 2 (C, D). Absolute densities of each type of IZ efficiency change in indicated regions (2N, 3N, Stable, Unstable) were normalized by their average over the whole genome (dashed lines). Error bars represent a 95% confidence interval assuming data counts are from a Poisson distribution.
DISCUSSION
We have generated novel RFD, gene expression and MRT datasets and have explored several approaches to compare the replication and transcription programs of twelve cancer and non-cancer cell types.
A global, unbiased correlation approach revealed that the RFD and MRT profiles clustered in three separate groups corresponding to lymphoid, myeloid and adherent cells. Similar results were obtained by RNA-seq, except that HeLa clustered with myeloid instead of adherent cells. Therefore, cancer-associated changes in replication do not blur their developmental origin signature, although changes in gene expression may sometimes do so. It is notable that the two LMSs were more correlated to each other by RNA-seq than by RFD, which suggests that their cell of origin may be different and that the selection for a tumour phenotype may have resulted in stronger convergence of their transcription than their replication program. We did not detect any convergence of the replication or transcription programs of cancer cells from different developmental origins (e.g. LMS versus BLs). In contrast, within lymphoid cells, we found evidence for BL-specific replication and transcription patterns. Furthermore, within myeloid cells, we found that expression of the BCR-ABL1 oncogene in TF1 cells, which models the establishment and early progression of CML, altered RFD and transcription of TF1 cells in a manner that increased their resemblance to K562, a late CML cell line. Overall, our global correlation analyses provide evidence for recurrent replication and transcription changes along specific tumour progression pathways. Interestingly, the RFD changes induced by BCR-ABL1 expression in early CML were not associated with large-scale MRT switches comparable to those observed between early and late CML or previously reported between leukemias and control LCLs (22). Local RFD changes do predict MRT changes, but of lower amplitude than current MRT profiles can detect. The global correlation analyses further revealed that RFD changes are widespread through the genome but more frequent in GC-poor regions. In contrast, RNA-seq changes do not vary uniformly with GC content. These results strengthen that replication changes are dissociated from transcription changes, to an extent that depends on the compared cell types.
More detailed investigations of the CML model strengthened these conclusions. The largest RFD changes induced by 1 month of BCR-ABL1 expression in TF1 cells were concentrated in GC-poor, lowly-expressed and late replicating regions. A similar, albeit less pronounced, tendency was observed after 6 months. For example, 98% and 92% of the 1% largest RFD changes after 1 month and 6 months, respectively, of BCR-ABL1 expression, occurred in the latest half of S phase (MRT > 0.5). Visual examination of the TF1 RFD profiles after 0, 1 or 6 months of BCR-ABL1 expression showed strikingly identical profiles over most of the genome, consistent with their high global correlation coefficients (>0.95). This conservation facilitated the manual annotation of BCR-ABL1-induced changes of IZ efficiency at 1027 loci and their fate during early and late CML progression. Control analyses indicated that manual annotation was free of false positive calls and that the GC-content and MRT distributions of called IZs were not significantly impacted by investigator bias, although the detection of enhanced or weakened IZ near active genes showed a small investigator bias (see Materials and Methods). State-of-the-art, automatic detection of IZs resulted in as much as 15% of divergent IZs between biological replicates (23). Further progress is required before automatic measurement of IZ efficiency changes can outperform manual annotation. That said, our automatic, reproducible analyses of RFD correlation coefficients (Figures 2 and 4) and largest RFD changes (Figures 5 and 9, Supplementary Figure S10) were fully consistent with manual annotation (Figures 6–8, 11, Supplementary Figures S12, S13, S16).
The BCR-ABL1-targeted IZs were more often downregulated (∼2/3) than upregulated (∼1/3), and these changes were more often enhanced than reverted over months of BCR-ABL1 expression in TF1 and in the late CML K562. Other RFD changes were restricted to the vicinity of affected IZs, consistent with the predicted redistribution of termination events. Large (domain-scale) MRT shifts, which are believed to reflect the altered firing time of multiple coordinated origins, were not observed. Local MRT changes predicted at individual IZs were below current resolution of MRT profiles. Similarly to global RFD changes, IZ efficiency changes were enriched in GC-poor, lowly-expressed and late-replicating regions and this tendency was less pronounced after 6 months than after 1 month of BCR-ABL1 expression. More specifically, we observed that IZ weakening events became more frequent in the GC-rich, early replicating, highly expressed portion of the genome after 6 months of BCR-ABL1 expression, and such changes were associated with transcription repression. In summary, BCR-ABL1 has a long-lasting action on IZ efficiency, in a direction that depends on each IZ but is more often repressive, with an initially strong preference for late-replicating gene deserts and a progressive shift to other genome compartments during prolonged expression.
Our previous study of HeLa and GM06990 RFD profiles revealed the existence of three types of IZs (23). Type 1 and type 2 IZs are circumscribed on one or both sides by active genes, and fire early in S phase. Type 3 IZs, on the other hand, are not associated with active genes and fire predominantly late in S phase. The mechanisms that delimit type 3 IZs remain unclear, but the findings reported here suggest that BCR-ABL1 specifically affects non-transcriptional mechanisms that set the boundaries and/or regulate the activity of type 3 IZs, although BCR-ABL1 can also weaken some type 1/2 IZs, in association with transcription repression, after 6 months of expression.
Changes in IZ efficiencies induced by BCR-ABL1 mostly occur in GC-poor, lowly expressed, late-replicating DNA regions and are predominantly repressive. This is in apparent contrast to other oncogenes such as MYC or Cyclin E which, by shortening G1 phase, induce early firing of intragenic origins normally erased by transcription during G1 phase (18). However, late-replicating, gene-poor regions of the genome were not interrogated in the latter study.
The BCR-ABL1 kinase affects a wide range of intracellular signalling pathways (25) which might directly or indirectly modulate origin firing. The origin licensing factor Cdc6 is upregulated in a BCR-ABL1-dependent manner in primary CML and K562 cells (41), which may explain the increased activity of some origins. The DNA damage response (DDR) pathway is activated in CML (42), and DDR activation is known to repress late-firing origins. IZ changes were predominantly repressive, although activation events were also observed, and the direction of most changes was conserved during CML progression, suggesting that specific classes of late IZs may have opposite reponses to BCR-ABL1. The downregulation of some late IZs by BCR-ABL1 may also indirectly stimulate other late IZs, due to increased availability of limiting origin firing factors and/or decreased inactivation by passive replication. Finally, RFD changes between cell lines, cancerous or not, are more frequent in GC-poor than GC-rich regions, suggesting that type 3 IZs are generally more plastic than type 1 and 2 IZs. Signalling by BCR-ABL1 and other oncogenes may reactivate this plasticity during transformation.
These results suggest that BCR-ABL1 expression may generate RS independently of transcription in GC-poor, late-replicating gene deserts. Indeed, prolonged expression of BCR-ABL1 resulted in a striking, massive deletion of GC-poor, late-replicating sequences covering one-third of the genome. The density of silenced origins was three-fold higher in unstable than in stable sequences, even before their effective deletion. Origin silencing in late-replicating DNA may significantly increase the probability of incomplete replication before entry into mitosis, resulting in a specific chromosomal fragility landscape. On the other hand, origin silencing was not frequent enough to directly perturb replication of all unstable sequences. We have previously argued that replication of the human genome involves a superposition of efficient initiation at ‘master' IZs detected in RFD profiles followed by more random, cryptic initiation between them (23,34,43,44). While we only detect silencing of master IZs, it is possible that BCR-ABL1 has a much broader effect on dispersed initiation. Alternatively, we cannot exclude that genomic instability and origin silencing are two independent effects of BCR-ABL1 expression that both affect late-replicating, GC-poor regions. Finally, it remains possible that early DNA damage induced by BCR-ABL1 expression activates a DDR that strongly represses late origin firing even before deletion events become obvious.
DATA AVAILABILITY
The new sequence data generated in this study have been deposited at the European Nucleotide Archive (ENA, www.ebi.ac.uk/ena) under primary accession number PRJEB25180.
Supplementary Material
ACKNOWLEDGEMENTS
This work has benefited from facilities and expertise of the High-throughput Sequencing Platform of IB2C, the Genomic platform of IBENS (http://genomique.biologie.ens.fr) and the genomic and microgenomic platform of Université Claude Bernard Lyon 1, France (ProfileXpert, http://profilexpert.fr).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ligue Nationale Contre le Cancer (Comité de Paris); Agence Nationale de la Recherche [ANR 2010 BLAN 161501]; Association pour la Recherche sur le Cancer; the Fondation pour la Recherche Médicale [FRM DEI201512344404]; Cancéropôle Ile-de-France and the INCa [ERABL, 2009-2-INV-03 and PL-BIO]; programme ‘Investissements d’Avenir' launched by the French Government and implemented by the ANR [ANR-10-IDEX-0001-02 PSL*Research University]; France Génomique national infrastructure, funded as part of the ‘Investissements d’Avenir' program managed by the ANR [ANR-10-INBS-09]; X.W. was supported by a Ph-D fellowship from the Chinese Research Council; B.A. acknowledges support from Science and Technology Commission of Shanghai Municipality [15520711500]; Joint Research Institute for Science and Society (JoRISS). Funding for open access charge: Fondation pour la Recherche Médicale (FRM DEI201512344404).
Conflict of interest statement. None declared.
REFERENCES
- 1. DePamphilis M.L., Bell S.D.. Genome Duplication. 2011; Garland Science. [Google Scholar]
- 2. Hyrien O. Peaks cloaked in the mist: the landscape of mammalian replication origins. J. Cell Biol. 2015; 208:147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hyrien O. Kaplan D. The Initiation of DNA Replication in Eukaryotes. 2016; Springer. [Google Scholar]
- 4. Hills S.A., Diffley J.F.. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014; 24:R435–R444. [DOI] [PubMed] [Google Scholar]
- 5. Zeman M.K., Cimprich K.A.. Causes and consequences of replication stress. Nat. Cell Biol. 2014; 16:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Munoz S., Mendez J.. DNA replication stress: from molecular mechanisms to human disease. Chromosoma. 2016; 26:1–15. [DOI] [PubMed] [Google Scholar]
- 7. Ahuja A.K., Jodkowska K., Teloni F., Bizard A.H., Zellweger R., Herrador R., Ortega S., Hickson I.D., Altmeyer M., Mendez J. et al. A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nat. Commun. 2016; 7:10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alvarez S., Diaz M., Flach J., Rodriguez-Acebes S., Lopez-Contreras A.J., Martinez D., Canamero M., Fernandez-Capetillo O., Isern J., Passegue E. et al. Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat. Commun. 2015; 6:8548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Flach J., Bakker S.T., Mohrin M., Conroy P.C., Pietras E.M., Reynaud D., Alvarez S., Diolaiti M.E., Ugarte F., Forsberg E.C. et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014; 512:198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bartkova J., Horejsi Z., Koed K., Kramer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J.M., Lukas C. et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005; 434:864–870. [DOI] [PubMed] [Google Scholar]
- 11. Bartkova J., Rezaei N., Liontos M., Karakaidos P., Kletsas D., Issaeva N., Vassiliou L.V., Kolettas E., Niforou K., Zoumpourlis V.C. et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006; 444:633–637. [DOI] [PubMed] [Google Scholar]
- 12. Gorgoulis V.G., Vassiliou L.V., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T., Venere M., Ditullio R.A. Jr, Kastrinakis N.G., Levy B. et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005; 434:907–913. [DOI] [PubMed] [Google Scholar]
- 13. Halazonetis T.D., Gorgoulis V.G., Bartek J.. An oncogene-induced DNA damage model for cancer development. Science. 2008; 319:1352–1355. [DOI] [PubMed] [Google Scholar]
- 14. Gaillard H., Garcia-Muse T., Aguilera A.. Replication stress and cancer. Nat. Rev. Cancer. 2015; 15:276–289. [DOI] [PubMed] [Google Scholar]
- 15. Dominguez-Sola D., Ying C.Y., Grandori C., Ruggiero L., Chen B., Li M., Galloway D.A., Gu W., Gautier J., Dalla-Favera R.. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007; 448:445–451. [DOI] [PubMed] [Google Scholar]
- 16. Srinivasan S.V., Dominguez-Sola D., Wang L.C., Hyrien O., Gautier J.. Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep. 2013; 3:1629–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kotsantis P., Silva L.M., Irmscher S., Jones R.M., Folkes L., Gromak N., Petermann E.. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016; 7:13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Macheret M., Halazonetis T.D.. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature. 2018; 555:112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pope B.D., Ryba T., Dileep V., Yue F., Wu W., Denas O., Vera D.L., Wang Y., Hansen R.S., Canfield T.K. et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014; 515:402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hiratani I., Gilbert D.M.. Replication timing as an epigenetic mark. Epigenetics. 2009; 4:93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Donley N., Thayer M.J.. DNA replication timing, genome stability and cancer: late and/or delayed DNA replication timing is associated with increased genomic instability. Semin. Cancer Biol. 2013; 23:80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ryba T., Battaglia D., Chang B.H., Shirley J.W., Buckley Q., Pope B.D., Devidas M., Druker B.J., Gilbert D.M.. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 2012; 22:1833–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Petryk N., Kahli M., d’Aubenton-Carafa Y., Jaszczyszyn Y., Shen Y., Silvain M., Thermes C., Chen C.L., Hyrien O.. Replication landscape of the human genome. Nat. Commun. 2016; 7:10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daley G.Q., Van Etten R.A., Baltimore D.. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990; 247:824–830. [DOI] [PubMed] [Google Scholar]
- 25. Casolari D.A., Melo J.V.. Rowley DJ. Chromosomal Translocations and Genome Rearrangements in Cancer. 2015; Switzerland: Springer International Publishing; 107–138. [Google Scholar]
- 26. Zhao R.C., Jiang Y., Verfaillie C.M.. A model of human p210(bcr/ABL)-mediated chronic myelogenous leukemia by transduction of primary normal human CD34(+) cells with a BCR/ABL-containing retroviral vector. Blood. 2001; 97:2406–2412. [DOI] [PubMed] [Google Scholar]
- 27. Laperrousaz B., Jeanpierre S., Sagorny K., Voeltzel T., Ramas S., Kaniewski B., Ffrench M., Salesse S., Nicolini F.E., Maguer-Satta V.. Primitive CML cell expansion relies on abnormal levels of BMPs provided by the niche and on BMPRIb overexpression. Blood. 2013; 122:3767–3777. [DOI] [PubMed] [Google Scholar]
- 28. Asmussen J., Lasater E.A., Tajon C., Oses-Prieto J., Jun Y.W., Taylor B.S., Burlingame A., Craik C.S., Shah N.P.. MEK-dependent negative feedback underlies BCR-ABL-mediated oncogene addiction. Cancer Discov. 2014; 4:200–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grockowiak E., Laperrousaz B., Jeanpierre S., Voeltzel T., Guyot B., Gobert S., Nicolini F.E., Maguer-Satta V.. Immature CML cells implement a BMP autocrine loop to escape TKI treatment. Blood. 2017; 130:2860–2871. [DOI] [PubMed] [Google Scholar]
- 30. Chen C.L., Rappailles A., Duquenne L., Huvet M., Guilbaud G., Farinelli L., Audit B., d’Aubenton-Carafa Y., Arneodo A., Hyrien O. et al. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 2010; 20:447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hansen R.S., Thomas S., Sandstrom R., Canfield T.K., Thurman R.E., Weaver M., Dorschner M.O., Gartler S.M., Stamatoyannopoulos J.A.. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thurman R.E., Day N., Noble W.S., Stamatoyannopoulos J.A.. Identification of higher-order functional domains in the human ENCODE regions. Genome Res. 2007; 17:917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Green M.R., Sambrook J.. Molecular Cloning: A Laboratory Manual. 2012; NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 34. Baker A., Audit B., Chen C.-L., Moindrot B., Leleu A., Guilbaud G., Rappailles A.e., Vaillant C.e., Goldar A., Mongelard F. et al. Replication fork polarity gradients revealed by megabase-sized U-shaped replication timing domains in human cell lines. PLoS Comput. Biol. 2012; 8:e1002443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Audit B., Baker A., Chen C.L., Rappailles A., Guilbaud G., Julienne H., Goldar A., d’Aubenton-Carafa Y., Hyrien O., Thermes C. et al. Multiscale analysis of genome-wide replication timing profiles using a wavelet-based signal-processing algorithm. Nat. Protoc. 2013; 8:98–110. [DOI] [PubMed] [Google Scholar]
- 36. Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L.. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012; 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Duret L., Mouchiroud D., Gautier C.. Statistical analysis of vertebrate sequences reveals that long genes are scarce in GC-rich isochores. J. Mol. Evol. 1995; 40:308–317. [DOI] [PubMed] [Google Scholar]
- 38. Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W. et al. Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921. [DOI] [PubMed] [Google Scholar]
- 39. Woodfine K., Fiegler H., Beare D.M., Collins J.E., McCann O.T., Young B.D., Debernardi S., Mott R., Dunham I., Carter N.P.. Replication timing of the human genome. Hum. Mol. Genet. 2004; 13:191–202. [DOI] [PubMed] [Google Scholar]
- 40. Bernardi G. Misunderstandings about isochores. Part 1. Gene. 2001; 276:3–13. [DOI] [PubMed] [Google Scholar]
- 41. Zhang J.H., He Y.L., Zhu R., Du W., Xiao J.H.. Deregulated expression of Cdc6 as BCR/ABL-dependent survival factor in chronic myeloid leukemia cells. Tumour Biol. 2017; 39:1010428317713394. [DOI] [PubMed] [Google Scholar]
- 42. Takagi M., Sato M., Piao J., Miyamoto S., Isoda T., Kitagawa M., Honda H., Mizutani S.. ATM-dependent DNA damage-response pathway as a determinant in chronic myelogenous leukemia. DNA Repair (Amst.). 2013; 12:500–507. [DOI] [PubMed] [Google Scholar]
- 43. Guilbaud G., Rappailles A., Baker A., Chen C.L., Arneodo A., Goldar A., d’Aubenton-Carafa Y., Thermes C., Audit B., Hyrien O.. Evidence for sequential and Increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput. Biol. 2011; 7:e1002322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hyrien O., Rappailles A., Guilbaud G., Baker A., Chen C.L., Goldar A., Petryk N., Kahli M., Ma E., d’Aubenton-Carafa Y. et al. From simple bacterial and archaeal replicons to replication N/U-domains. J. Mol. Biol. 2013; 425:4673–4689. [DOI] [PubMed] [Google Scholar]
- 45. Julienne H., Audit B., Arneodo A.. Embryonic stem cell specific ‘master’ replication origins at the heart of the loss of pluripotency. PLoS Comput. Biol. 2015; 11:e1003969. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The new sequence data generated in this study have been deposited at the European Nucleotide Archive (ENA, www.ebi.ac.uk/ena) under primary accession number PRJEB25180.