

Highly aligned GO gel fibers are fabricated under high extensional flow for efficient nanofluidic transport of ion species.
Abstract
Colloidal dispersions with liquid crystallinity hold great promise for fabricating their superstructures. As an example, when graphene oxide (GO) sheets are assembled in the liquid crystalline state, they can turn into ordered macroscopic forms of GO such as fibers via the wet spinning process. Here, we report that by reinforcing intersheet interactions, GO liquid crystals (LCs) turn into mechanically robust hydrogels that can be readily drawn into highly aligned fibrillar structures. GO hydrogel fibers with highly aligned sheets (orientation factor, f = 0.71) exhibit more than twice the ionic conductivity compared to those with partially aligned structures (f = 0.01). The hierarchically interconnected two-dimensional nanochannels within these neatly aligned GOLC hydrogel fibers may facilitate controlled transport of charge carriers and could be potentially explored as cables for interconnecting biosystems and/or human-made devices.
INTRODUCTION
Transporting ionic species selectively and rapidly is critical not only for operating electrochemical devices but also for life-sustaining chemical transformations in all living creatures (1, 2). Controllable transport of ions confined in nanopores and nanochannels is of substantial interest due to their similarity to ionic systems found in nature, such as transmembrane proteins and vascular plants (3). Unlike microscale channels, nanosized channels support selective transport of counterions and exhibit exceptional ionic conductivity when the Debye length of ion species matches the channel dimensions (4, 5). Recently, notable advances have been reported in ion transport within nanocapillaries, using hydrated surfaces of well-packed colloids (6), especially those made of two-dimensional (2D) sheets (7–11). These bottom-up assembled material systems can also take unusual form factors, such as macroscopic fibrillar structure, which can potentially act as nanofluidic ionic cables (NICs) to conduct ionic signals and power (12, 13). However, controlling the internal microstructure and surface chemistry of the nanofluidic channels for these ionic cables remains challenging.
Colloidal liquid crystals (LCs) exhibit anisotropic and dynamic alignment of the building blocks in response to various free-energy gradients (14–16). Using colloidal LCs for macroscopic assembly is beneficial in that they flow like a liquid, which enables directed self-assembly under a flow field with controlled shear and vorticity. Preserving the spontaneous conformation of the colloids throughout the assembly process holds great promise for rational engineering of assembled macrostructures with novel anisotropic properties. In particular, charged colloids in a fluid can be readily oriented by an extensional flow-field gradient and simultaneously coagulated into a fibrillar form with hierarchical microstructure (17). To achieve high anisotropy of the assembled material properties as well as the structures, a steep flow velocity gradient is necessary (12, 18). However, colloidal gel networks are easily ruptured by mechanical perturbation due to the absence of strong interactions such as chemical bonding and the relatively short length of constituents (19). Therefore, strengthening the gel networks and cross-linking the colloids, by controlling the channel size, should be considered from the assembly step for the facile design of nanofluidic channels.
Anisotropically ordered structures are required for highly conductive NICs to shorten the ionic pathway (i.e., interconnected channel length) and decrease the channel height, which, in turn, increases the surface charge density. 2D giant colloids such as graphene oxide (GO) could be a prospective constituent building block of NICs because its high specific surface area and specific charge density guarantee a high volume fraction and surface charge density of the interconnected 2D nanochannels (7, 8). Here, we describe flow-driven dynamic self-assembly of highly aligned GO gel fibers and their application as ionic cables. We developed a straightforward method to reinforce the GO gel network, which made it sufficiently stable to withstand mechanical perturbations. In situ observation of the assembly process showed that GO gel fibers coagulated in the CaCl2 coagulation solution ruptured under a low extensional flow velocity gradient before the LC domains were aligned. However, after adding NH4OH to the coagulation solution, application of a higher flow velocity gradient was possible. We investigated the effects of the NH4OH-containing coagulation solution on the mechanical and viscoelastic properties of the GO gels. In addition, the origin of the enhanced rigidity of the GO gel, arising from fundamental changes in its intermolecular interaction, is discussed. The fundamental understanding of kinetics of fiber solidification allowed the tailoring of the macrostructure of GO gel fibers to be highly aligned on a meter-long scale during the dynamic self-assembly. We demonstrate the efficacy of highly aligned GO gel fibers as NICs by comparing the ion transport behavior with that of partially aligned fibers in various salt concentrations.
RESULTS
Assembly of GOLC into GO hydrogel fibers
Figure 1A shows a schematic of the apparatus used for in situ observation of the dynamic assembly of GOLC dispersion to make gel fibers. A coagulation bath, a syringe mounted on a syringe pump, and a roller for collecting and drawing the fiber were installed on the stage of a polarized optical microscope (POM). GO dispersion [0.5 weight % (wt %)] with GO sheets with an average lateral size of 1.7 μm (fig. S1) was loaded in the syringe and ejected through a glass nozzle (inner diameter, 400 μm) into a 5 wt % CaCl2 coagulation solution under varying flow rates. The initial rate of ejected fluid was referred to as v1, indicating freely ejected fluid. The assembly process was recorded using a high-speed camera focused on the nozzle end with increasing extensional flow rate (i.e., linear velocity of the take-up roller, v2) until the gel fiber fractured (Fig. 1, A and B). In Fig. 1A, a photo image shows a meter-long GO gel fiber collected on a stainless steel bobbin.
Fig. 1. In situ observation of dynamic self-assembly of GO gel fibers.
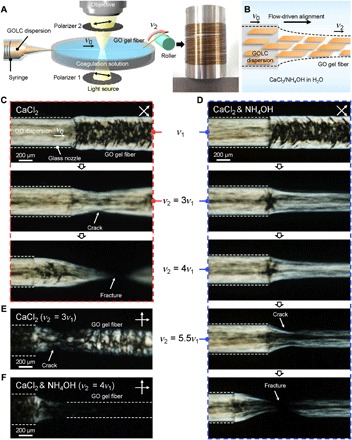
(A) Schematic of the experimental set-up: The assembly apparatus was installed on the stage of a POM, and the assembly process was recorded with a high-speed camera focused on the glass nozzle tip. The final GO hydrogel fibers can be reeled on a bobbin as shown in the photo. (B) Schematic illustration showing flow-driven alignment of GO sheets and formation of hydrogel fiber. (C and D) POM snapshots capturing different assembly behaviors of GO gel fibers (C) without and (D) with the addition of NH4OH in the CaCl2 coagulation solution. Snapshots were taken at various rates of the take-up roller (v2). Note that the crossed polarizers were rotated 45° from the fiber axis. (E and F) POM images showing degree of alignment in GO gel fibers obtained at (E) 3v1 in the CaCl2-only coagulation solution and (F) 4v1 in the NH4OH and CaCl2 coagulation solution.
Using our experimental setup, we directly observed dynamic changes in LC alignment in response to v2 during coagulation, as shown in the series of POM images in Fig. 1C. The topmost image displays a GO gel fiber assembled at the initial state (i.e., v2 = v1). Birefringent optical texture reflects the local orientation of the LC domains (14). GOLC fluid flowing inside the glass nozzle (white dashed lines) was subjected to a laminar flow [mean velocity (v0) of 21 mm s−1] with a parabolic velocity profile due to the drag force of the nozzle wall acting on the surface of the GOLC (fig. S2B), which affects the rotational dynamics of GOLC domains (20). The alignment of the GOLC is given by the local shear; thus, GO is aligned closer to the nozzle walls and more isotropic at the core. The GO sheets will not respond independently to shear. Because the fluid has an LC behavior, the effect on their orientation and alignment relies on their domains. When the fluid was ejected, the mean velocity of fluid (v1) was slightly decreased to 17 mm s−1 due to the relaxation of the laminar velocity profile (fig. S2C). This velocity-profile relaxation significantly affects the preferential GO arrangement inside the LC fluid (fig. S2 and movie S1). The birefringence of the ejected fluid indicates highly aligned LC domains along the sheath of fluid but more rotated ones in its core region. The disturbed core fluid became gradually coagulated with the distance from the nozzle due to the diffusion of ionic cross-linkers (Ca2+) from the coagulation solution (fig. S3) (21). The most straightforward and well-supported explanation to this behavior is that the core fluid is subjected to a retardation, which causes the switch from alignment in the flow direction to alignment in the cross direction (22). This can actually be seen directly at the exit from the nozzle, where there is a retardation, which is competing with the extensional flow. The resulting nonhomogeneous structural arrangement hinders facile ion transport and mechanical robustness along the fiber axis. Therefore, the alignment of GOLC should be tailored along the fiber axis not only to promote guided transport of ions but also to increase load transfer along the longitudinal direction. As a consequence, to design the preferential structure, extensional flow provides a mechanism that may induce homogeneous alignment throughout the LC fluid, which can be obtained using the drawing effect utilizing the mechanism of solidification. However, this GO gel fiber showed low drawability because of its poor mechanical properties. When v2 is increased to 3v1 (i.e., 51 mm s−1), the gel fiber tended to crack easily and fracture at the velocity difference of 3v1 − v0 (second and third images in Fig. 1C). Figure 1E shows the nematic Schlieren textures around the cracking point of the GO gel fibers.
We modified the coagulation solution by addition of NH4OH (Fig. 1D) to improve the drawability of GO gel fibers. The assembly behavior of GOLC in the NH4OH-containing solution at v1 was similar to that shown in the original coagulation solution (first images in Fig. 1, C and D). However, when v2 becomes 3v1, perpendicular LC alignment in the core became parallel (second image in Fig. 1D). Movie S2 shows the in situ observation recorded at a constant v2 of 3v1. The LC domains in the ejected core fluid were dynamically rearranged in parallel to the axis of fiber by the high extensional flow field. In addition, the higher velocity difference (4v1 − v0) led to alignment of the LC domains to the fiber axis (third image in Fig. 1D) and the high anisotropy of the aligned gel fiber was certainly observed in Fig. 1F. Last, a crack was generated at the sheath layer when v2 reached 5.5v1 (fourth image in Fig. 1D). The crack propagated rapidly through the opposite side and ultimately fractured the GO gel fiber (fifth image in Fig. 1D). The applied mechanical drawing acts as a tensile stress and increases proportionally to the axial velocity gradient [i.e., dv1(x)/dx, extension rate] (23, 24). We derived the distance dependence of extension rate from the fourth image in Fig. 1D as the following equation (more details are available in the supplementary materials)
| (1) |
where D1(x) is the diameter of the GO gel fiber at distance x (e.g., x = 0 at the nozzle end) and Q0 is the volumetric flow velocity (2.7 μl s−1) of the GOLC fluid. As shown in fig. S4, the extension rate was highest near the nozzle end and gradually decreased to zero with the distance as the diameter of ejected fluid became constant to 230 μm. The ultimate extension rate at the fractured position (x = ~200 μm) was 88 s−1. Under modified coagulation conditions, the gel fiber sustained a 2.6-fold higher ultimate extension rate than the pristine GO gel fiber (34 s−1). In addition, the normalized birefringence intensity of the pristine gel fiber at v2 = 3v1 was 1.7-fold lower than that of the modified GO gel fiber at v2 = 4v1, indicating that the pristine gel fiber had a relatively low degree of orientation (fig. S5D) (16). Together, these results indicated that highly oriented gel fibers could not be obtained using the CaCl2-only coagulation solution because the pristine gel was sensitive to high-speed mechanical drawing, which is essential to obtain highly aligned LC domains.
Enhancement of molecular interactions between GO sheets during coagulation
We next investigated the effect of the NH4OH-containing coagulation solution on the mechanical and viscoelastic properties of GOLC gels. The 1D characteristics and fast drying rate (less than 10 min) of the gel fibers made them unsuitable for in-depth investigations of intact gel properties. Consequently, we prepared freestanding GO gels with a cylindrical shape to study its coagulation behavior (Fig. 2A and fig. S6A). Two types of GO gels were formed in an aqueous bath containing CaCl2 only and both CaCl2 and NH4OH, which are termed as “original GO gel” and “modified GO gel,” respectively (Fig. 2, A and B). Figure 2C shows typical stress-strain curves of GO gels under uniaxial compression. Neither of the two gel types was brittle enough to exhibit a discernible failure point, so we compared their stresses at the same strain (60%). The original GO gel had a stress of 3.1 ± 0.3 kPa, while the modified GO gel sustained an almost sixfold higher stress (18.1 ± 3.1 kPa). The high mechanical strength of the GO gel prepared using the NH4OH-containing CaCl2 coagulation solution explains why the modified GO gel fiber could sustain a higher extension rate (88 s−1) than the original gel fiber during self-assembly.
Fig. 2. Enhanced molecular interactions of GO.
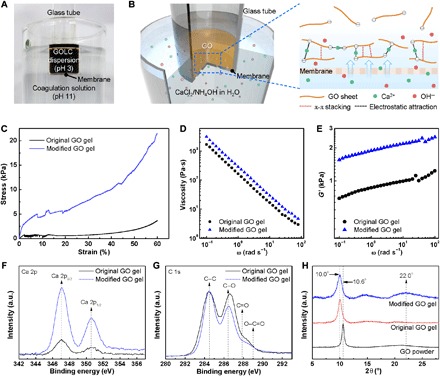
Photo (A) and schematic illustrations (B) showing experimental setup to observe the formation of GO hydrogel by diffusion of coagulants. The coagulant (Ca2+) diffuses into GOLC dispersion through the pores in membrane and induces gelation. GO sheets also undergo partial deoxygenation after the addition of NH4OH, which increases their π-π interaction. (C) Stress-strain curves of both the original and modified GO gels under uniaxial compression. (D and E) Frequency dependence of (D) viscosity and (E) storage modulus (G′) of both types of GO gels. (F and G) XPS spectra of the samples showing (F) Ca 2p and (G) C 1s bands. a.u., arbitrary units. (H) XRD patterns of GO powder and freeze-dried GO gels.
We next examined the viscoelastic properties and dynamic rheological responses of the gels. As shown in Fig. 2D, the viscosity of both gels rapidly decreased as the frequency increased, indicating that intermolecular interactions are composed primarily of noncovalent interactions (e.g., hydrogen bond, van der Waals, and electrostatic bond) (25). The viscosity of the modified GO gel was 1.7-fold higher than that of the original GO gel over the entire frequency range (0.1 to 100 rad s−1), indicating that the modified GO gel had a strengthened physical network relative to the original GO gel. This increase in strength was further supported by the results for storage modulus (G′), a measure of the field strength of an internal structure based on reversible deformation of a gel under shear stress (26). The G′ value of the modified GO gel was 1.4-fold higher than that of the original GO gel (Fig. 2E). The strength of both gels was quantitatively analyzed by yield stress (τy), which is the minimum stress required to break a physical structure (27). The τy values, derived from Casson plots in fig. S6B, are listed in table S1. The modified GO gel had a τy of 308 Pa, an almost 1.7-fold greater value than that of the original GO gel (181 Pa). Moreover, the τy value of the modified GO gel increased in proportion to the concentration of added NH4OH to the coagulation solution (table S1). This relationship implies that a high pH coagulation solution can modify the microstructure of modified GO gels to make them more rigid, allowing GO gel fibers to resist a high extension rate.
The origin of the enhanced rigidity of the GO gels made using the NH4OH-containing coagulation solution was investigated using x-ray photoelectron spectroscopy (XPS), and the results are shown in Fig. 2 (F and G). High-resolution Ca 2p curves in XPS indicated that the modified GO gel [2.3 atomic % (at %)] had a higher calcium content than the original GO gel (1.1 at %). Ca2+ plays a key role in sustaining gel networks by cross-linking negatively charged adjacent GO sheets through electrostatic attraction (Fig. 2B). Our results suggest that a larger number of electrostatic cross-links exist in the modified GO gel network than the original gel network. We attributed this to the increased negative surface charge of the GO colloids due to base-promoted ionization of the oxygen functional groups on the surface of the GO sheets (28). The pH values of the CaCl2-only and modified coagulation solution were 9.5 and 11, respectively. The corresponding zeta potential (~40 mV) of GO dispersion at pH 11 was higher than that (~36 mV) of GO dispersion at pH 9.5 (fig. S7). High-resolution C 1s curves revealed that the modified GO gel had a lower amount of oxygenated carbons (O/C ratio, 0.43) than the original gel (0.50) because of partial deoxygenation of GO sheets in the high pH coagulation solution (table S2). Alkaline conditions can induce mild but rapid recovery of graphitic domains on GO sheets even at room temperature, although whether this is due to removal of oxygen functionalities or stripping of oxidative debris from GO is still under debate (29, 30). In addition, the similarity in XPS curves of original and modified GO gel fibers demonstrates that there is not much difference in deoxygenation level and calcium content from the different fabrication time of cylindrical gels and gel fibers (fig. S8 and table S2). Figure 2H shows x-ray diffraction (XRD) patterns of GO powder and freeze-dried gels. A 2θ peak at 10.6° corresponding to interlayer spacing (d-spacing, 8.3 Å) between GO sheets was observed for the powder sample. However, the d-spacing slightly increased to 8.8 Å (2θ = 10.0°) in both gel types because of the presence of Ca2+ (ionic radius, 1.1 Å) that functioned as intercalated ionic cross-linkers between the GO sheets (31). The 2θ peak at 22.0° (4.0 Å), which we assigned to d-spacing between graphitic crystallites (~26.5°) on the GO sheets, appeared only in the modified GO gel. This indicates that additional π-π stacking interactions between locally recovered graphitic domains exist in the GO gel network. The 2θ peak position (22.0°) was slightly lower than previously reported positions of graphene chemically converted with a strong reductant (24° to 25°), indicating incomplete reduction of GO sheets under the mild reduction conditions, consistent with the C 1s XPS analysis (32, 33). Raman analysis also proves the partial restoration of graphitic domains (fig. S9). The higher ID/IG ratio of modified GO gels implies that the average size of the graphitic domains was decreased because of the occurrence of numerous small-sized graphitic domains (32). Therefore, the remarkable rigidity of the GO gel prepared in the NH4OH-containing coagulation solution was due not only to abundant electrostatic cross-links but also to additional π-π stacking interactions.
Interconnected 2D nanochannels and ionic conductivities of GO gel fibers
The macrostructures of the GO gel fibers could be tailored by v2 on the meter scale because of the strength of the gel network. Two types of GO gel fibers were assembled in the NH4OH-containing coagulation solution at v2 = 4v1 and v1, which we refer to as 4v1-GO and v1-GO gel fibers, respectively. Many light brown lines parallel to the fiber axis are visible in the POM images of the 4v1-GO gel fiber shown in Fig. 3A. The bright light almost disappeared when the crossed polarizers were arranged parallel to the fiber axis (Fig. 3C). In contrast, LC textures of the v1-GO gel fiber were parabolic with respect to the fiber axis (Fig. 3, B and D). The inner structures of both GO gel fibers were investigated with scanning electron microscopy (SEM) by peeling the sheath layer of freeze-dried gel fibers (Fig. 3, E and F). Parallel and parabolic arrangements of interconnected GO sheets were observed, consistent with the corresponding LC textures observed by POM. Differences in the orientation of both GO gel fibers were verified by small-angle x-ray scattering (SAXS) experiments (Fig. 3, G and H). The SAXS pattern of 4v1-GO gel fibers had a fan-like shape that extended along the horizontal direction, reflecting the preferred orientation of the LC domains to the fiber axis. In contrast, the round-square shape of the SAXS pattern of the v1-GO gel fibers indicated nonpreferential but symmetric alignment of the LC domains. To quantitatively verify differences in orientation, we derived Herman’s orientation function (f) from azimuthal scans of the SAXS patterns (fig. S10). The value of f approaches 1 when GO sheets are perfectly aligned with the fiber axis or 0 when the sheets are completely randomly oriented (34). Calculated values of f were 0.71 and 0.01 for 4v1-GO and v1-GO gel fibers, respectively. Controlled alignment of the GO gel fiber is expected to strongly affect the mechanical properties of dried GO fibers. Figure S11 shows typical stress-strain curves of 4v1-GO and v1-GO fibers. 4v1-GO fibers exhibited 1.4-fold higher Young’s modulus (38.3 GPa) than did v1-GO fibers (27.6 GPa) (table S3). This can be attributed to the higher value of f (0.93) of 4v1-GO fibers compared to v1-GO fibers (0.82; fig. S12). The tensile strength (301.4 MPa) of 4v1-GO fibers was almost two times higher than that (152.3 MPa) of v1-GO fibers, indicating the defectless compact structure of 4v1-GO fibers (21). The improved compactness of 4v1-GO fibers was further confirmed with apparent density and porosity of both GO fibers, as summarized in table S4.
Fig. 3. Tailoring the microstructure of GO gel fibers.
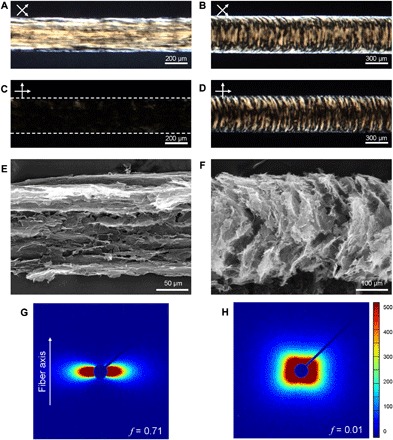
(A to D) POM images of (A and C) 4v1-GO gel fibers and (B and D) v1-GO gel fibers taken between (top) 45°-rotated and (bottom) parallel crossed polarizers with respect to the gel fiber axis. (E and F) SEM images of (E) 4v1-GO gel fibers and (F) v1-GO gel fibers show different sheet alignment. (G and H) SAXS patterns of (G) 4v1-GO gel fibers and (H) v1-GO gel fibers. f denotes Herman’s orientation function.
Along with the well-controlled macrostructure, interconnecting 2D nanochannels between coaligned GO sheets makes GO gel fibers promising materials for NICs, which can carry ionic current through nanochannels. We assessed the efficacy of anisotropically ordered structures in NICs by directly measuring ionic conductivity (Fig. 4A). First, preexisting ions such as Ca2+ and NH4+ in dried gel fibers were removed and exchanged for K+ and Cl− ions by immersing the fibers in target KCl solution at a certain concentration (1 μM to 1 M) for at least 24 hours. Then, a GO gel fiber was directly bridged between drain and source reservoirs containing the predetermined salt solution. Ag/AgCl electrodes were connected to the reservoirs to measure the ionic current along the GO gel fiber. The ionic conductivity of gel fibers at various salt concentrations was measured with linear sweep voltammetry (LSV; Fig. 4C).
Fig. 4. Effect of ordered NIC structures on ionic conductivity.
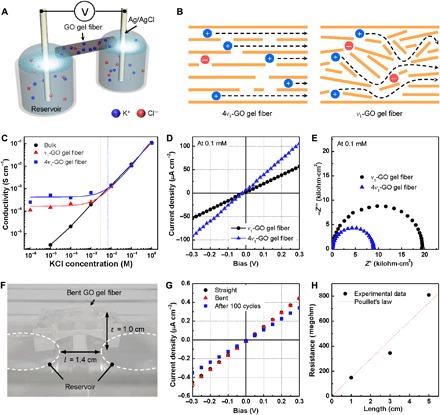
(A) Schematic illustration showing the experimental setup for measuring the ionic conductivity of NICs. (B) Schematic illustrating the different ion transport pathways in 4v1- and v1-GO gel fibers. (C) Ionic conductivity of the NICs at various salt (KCl) concentrations from 1 μM to 1 M. (D) Typical I-V curves and (E) Nyquist plots measured in 0.1 mM KCl solution. (F) Digital photograph of an arched GO gel fiber bridging the two reservoirs. The bending ratio is represented as arch height (t) divided by length (l). (G) I-V curves of NIC at bent and straight states and after 100 cycles of bending. (H) Ionic resistance depending on the length of NICs derived from fig. S13.
At a high salt concentration, the ionic conductivity of 4v1-GO and v1-GO gel fibers was similar to that of the bulk electrolyte solution, which was proportional to the salt concentration. However, at a low salt concentration, the ionic conductivity of 4v1-GO gel fibers began to deviate from that of the bulk solution at ~7 mM (blue dotted line), while this deviation occurred at 3 mM for v1-GO gel fibers (red dotted line). Below the critical concentration, the conductivity of both GO gel fibers was almost invariant with respect to the salt concentration, because the concentration inside the nanochannels was maintained with counterions (K+) by electroosmosis. This is a characteristic feature of nanofluidic ion transport in a surface charge–governed regime (4). 4v1-GO gel fibers consistently maintained a 2.4-fold higher conductivity than v1-GO gel fibers (1.7 × 10−4 S cm−1) in this regime (Fig. 4D). As shown in Fig. 4B, well-oriented 2D nanochannels along the 4v1-GO gel fibers shortened the ion pathways by guiding counterions along the longitudinal direction of the fibers. In contrast, cations in the v1-GO gel fibers struggled to move along the relatively disordered 2D nanochannels. In addition, the average surface charge density of the nanochannels inside the gel fibers was calculated to be 0.23 and 0.13 mC cm−2 for 4v1-GO and v1-GO gel fibers, respectively (see details in the supplementary materials). This observation indicates that 4v1-GO gel fibers with compact, parallel 2D nanochannels are better suited for preferential transport of counterions and exclusion of co-ions (Cl−) than v1-GO gel fibers. Structure-related facilitation of efficient ion transport was further confirmed by electrochemical impedance spectroscopy (EIS). Figure 4E shows Nyquist plots with semicircular curves measured in 0.1 mM KCl solution over the frequency range of 1 MHz to 100 mHz. The diameters of the semicircles represent the charge-transfer resistance (Rct) of the gel fibers. Rct of the v1-GO gel fiber (19.6 kilohm·cm2) was 2.5-fold higher than that of the 4v1-GO gel fiber (6.8 kilohm·cm2). The ionic conductivity of the 4v1-GO gel fiber in the surface charge–governed regime was comparable and even higher than those of previously reported nanosheet films, such as boron nitride (BN) nanosheets (table S5).
The flexibility and length dependence of NICs were further investigated using 4v1-GO gel fibers to demonstrate their potential as novel ionic cables. A bent GO gel fiber was bridged between two reservoirs, where the ratio of arch height (t) to length (l) was 0.71 (Fig. 4F). The I-V curves of the GO gel fiber for bending and straight states were similar, indicating that nanochannels in the GO gel fiber can stably transport ions under mechanical deformation (Fig. 4G). In addition, the GO gel fiber was mechanically flexible and robust enough to retain its ionic conductivity after 100 bending-unbending cycles. Last, the length dependence was explored by measuring the resistance of GO gel fibers with different lengths (fig. S13). Ionic resistance increased proportionally to length, satisfying Pouillet’s law for electrical conductors (Fig. 4H).
DISCUSSION
We demonstrated that in flow-driven fiber formation from GOLC, strengthening the GO gel network is an essential prerequisite as it allows fiber pulling under a high velocity gradient (~88 s−1), leading to highly aligned GO fibers. Adding NH4OH to the CaCl2 coagulation bath significantly strengthens GO gels, leading to markedly increased stiffness, allowing them to sustain higher extensional flow. The effects of NH4OH on GO gels can be summarized by the following: Initially, GO sheets become more negatively charged under the high pH condition and thereby provide more electrostatic cross-linking points for the cationic coagulant (Ca2+). Next, partial deoxygenation of GO sheets by NH4OH induces additional attractive π-π interactions. Therefore, by adjusting the flow gradient, GO gel fibers with largely random and highly oriented microstructures can be obtained. The resulting GO gel fibers were tested as NICs. Highly aligned GO gel fibers with an f value of 0.71 exhibited 2.4-fold higher ionic conductivity (4.2 × 10−4 S cm−1) than partially aligned GO gel fibers with an f value of 0.01, indicating that the degree of sheet alignment (f) within the fiber is a critical determinant of the performance. These ionic cables could be used for a wide range of system operated in ionic media for the transmission of ionic signals or power. The control of coagulation kinetics is not limited to fiber assembly and could be potentially applied to the dynamic assembly of other types of hierarchical structures of GO.
MATERIALS AND METHODS
Synthesis and characterization of GOLC dispersion
GO was chemically exfoliated from graphite powder (grade 2012, Asbury Carbons) by a modified Hummers method reported elsewhere (35, 36). GO powders were dispersed in acetone (99.5%; Daejung Chemicals and Metals Co. Ltd.) and washed with excess acetone to remove the residual acids and metal impurities, using a vacuum-assisted filtration kit (35, 36). The dried GO powder was redispersed in deionized water (DIW; Direct Q3, Millipore) at a concentration of 0.2 wt % and sonicated for 30 min. GO dispersion was centrifuged at 4000 rpm for 1 hour, and the precipitate was discarded. Then, after centrifugation at 8000 rpm for 1 hour, extremely small debris was also removed by dispensing the supernatant. The typical concentration of the final stock GO dispersion was approximately 1.0 wt %. The nematic liquid crystalline nature of GO dispersion was examined using a POM, as reported elsewhere (fig. S1) (14, 21).
Wet spinning of GO gel fibers
A 0.5 wt % GO dispersion was loaded into a syringe with a single nozzle (inner diameter, 400 μm). In a typical spinning process, the GOLC dispersion was ejected into a coagulation bath containing 5 wt % CaCl2 solution with 0.25 M NH4OH (30% NH3 solution, Sigma-Aldrich) at a constant rate (v1) of 17 mm s−1. The coagulated gel fiber was drawn at a constant rate (v2) by controlling the revolutions per minute of a take-up roller. v2 was set to 68 mm s−1 for obtaining 4v1-GO gel fibers. v1-GO gel fibers mean that the fibers are assembled by only the injection force of GO dispersion without the aid of the roller. For this, we set v2 (i.e., linear velocity of roller) to v1. After washing in DIW bath, the gel fibers were collected on a bobbin. For SEM, gel fibers were freeze-dried using a lyophilizer (FDB-5503, Operon) after quickly quenching them with liquid nitrogen.
Preparation of bulk freestanding GO gels
We designed a diffusion-induced gelation process for GO colloids in a static state to prepare bulk freestanding GO gels as reflecting coagulation behavior (Fig. 2, A and B). Open-ended glass tubes (inner diameter, 14 mm) were used as a mold. In a typical process, one side of the tube was closed with a dialysis membrane (molecular weight cutoff, 6000 to 8000; Spectra Dialysis Membrane), and 2 ml of the GO dispersion (0.5 wt %) was injected into the tube through the other side. Then, the tube was submerged in a beaker containing 50 ml of the coagulation solution, and the level of the GO dispersion was adjusted to be equivalent to that of the coagulation solution. The membrane was semipermeable with an average pore size small enough to prevent infiltration of GO sheets (average diameter, 1.7 μm) and therefore mediated ion diffusion at the interface only toward the GO dispersion (Fig. 2B). Once the tube was submerged, the sol-gel transition occurred, and the GO dispersion was fully converted to a gel after 12 hours (fig. S6A). The freestanding GO hydrogel was separated from the tube and washed by soaking in 50 ml of DIW. The DIW was replaced more than five times at fixed time intervals of 6 hours.
Ionic conductivity measurement of NICs
A measurement cell, fabricated using a polydimethylsiloxane elastomer (Sylgard 184, Dow Corning), consisted of source and drain reservoirs (15 ml) that were 30 mm in diameter with a 10-mm-length gap between them (fig. S14A). The two reservoirs were filled with KCl solution (99.5%, Sigma-Aldrich) at the same concentration (1 M to 1 μM). As shown in fig. S14B, a GO gel fiber was bridged between the two reservoirs after removing the preexisting ions (e.g., Ca2+ and NH4+) and then exchanging them for K+ ions in the following three steps: GO gel fibers were washed with excess DIW, dried in an 80°C oven for 30 min, and rehydrated for at least 24 hours in the target KCl solution. The removal and exchange of ions in the GO gel fiber were confirmed by XPS analysis (fig. S15). Last, the two reservoirs were connected to a potentiostat (SP-200, Bio-Logic) using two Ag/AgCl electrodes (fig. S14, C and D). LSV in the potential range of −0.4 to 0.4 V and EIS in the frequency range of 0.1 Hz to 7 MHz were collected using the potentiostat in a custom-made faraday cage. The ionic conductivity of the bulk electrolyte solution was measured using a glass tube with a diameter of 475 μm and a length of 10 mm.
Characterization
In situ observation of the dynamic self-assembly process was carried out using a BX-51 polarizing microscope (Olympus) under identical light intensity. The process was simultaneously recorded using a high-speed camera (M320S, Phantom Miro) at a frame rate of 3000 frames s−1 with an exposure time of 5.5 μs. The birefringence intensity in the POM images in fig. S5 was quantified using the ImageJ freeware. The birefringence intensity profiles of the two images along the lines inside the glass nozzle were similar (fig. S5C). Compression tests were carried out with an Instron 5966 tester (Instron Engineering Corporation) with a load cell of 10 N. A cylindrical GO hydrogel (diameter, 14 mm; height, 13 mm) was placed between two parallel plates and compressed at a strain rate of 10% min−1. Tensile tests of single dried GO fibers were also carried out at a strain rate of 10% min−1. The GO fibers were fixed onto a rectangle frame with 25 mm of gauge length using epoxy glue to prevent the slip of fibers. The mechanical strengths of fibers were calculated by the force divided by the cross-sectional area. The mechanical properties of GO fibers were averaged over at least 10 samples. The linear densities of dried GO fibers were measured with a FAVIMAT sing fiber tester (Textechno H. Stein) using a vibroscope method. XRD patterns of GO powder and freeze-dried bulk gels were obtained with an x-ray diffractometer (Rigaku, SmartLab) using a Cu Kα radiation source (λ = 1.5418 Å). XPS analysis was carried out with an XPS spectrometer (Theta Probe, Thermo Fisher Scientific) using monochromatic Al Kα radiation. The zeta potential of aqueous GO dispersion with a concentration of 0.05 mg ml−1 was measured using a Zetasizer Nano ZS90 (Malvern Instruments). The pH of GO dispersion was adjusted with dilute NH4OH solution. Raman analysis of GO gels was performed on a NRS-3100 (JASCO Inc.) with 532-nm laser excitation. Dynamic rheological properties of GO hydrogels were measured with the Advanced Rheometric Expansion System (AR50, TA Instruments). A parallel-plate geometry with a gap of 8 mm was adopted. Dynamic frequency sweep experiments were carried out over the frequency sweep range of 0.1 to 100 rad s−1 at a fixed oscillatory strain of 1%. Synchrotron SAXS measurements were carried out at the 4C SAXS II beamline of the Pohang Accelerator Laboratory (Pohang, Republic of Korea) with an x-ray beam wavelength of 0.675 Å at a sample-to-detector distance of 4 m. A single GO gel fiber was loaded on an open-circle holder, and both sides of the holder were sealed with polyimide tape to prevent drying of the gel fiber during SAXS measurements for an exposure time of 30 s. SEM images of the freeze-dried gel fibers and dried fibers were obtained using a Hitachi S-4800 microscope (Hitachi High-Technologies Co.). The topography of the single-layered GO sheet was characterized using an atomic force microscope (XE-70, Park Systems).
Calculation of flow velocity and extension rate
The mean flow velocity of GOLC fluid (v0) flowing inside the glass nozzle was calculated as follows
| (2) |
where Q0, A0, and D0 are the volumetric flow velocity (2.7 μl s−1) of the GOLC fluid and cross-sectional area and diameter (400 μm) of the glass nozzle, respectively. In addition, the flow velocities of v0 and v1 (flow velocity of ejected fluid) are related as follows
| (3) |
However, as high extensional flow is applied, the flow velocity and diameter of the ejected fluid are dependent on the distance from the nozzle end as follows
| (4) |
where D1(x) is the diameter of the GO gel fiber at x and was obtained by analyzing the in situ observation images (Fig. 1, C and D). Hence, the extension rate [dv1(x)/dx] is defined as follows
| (5) |
Determination of the degree of orientation in fibers
Herman’s orientation function (f) is defined as follows (37)
| (6) |
where 〈cos2θ〉 is the average value of the square of the cosine of the angle θ. Assuming rotational symmetry of the fiber axis, the following equation is obtained
| (7) |
where I(θ) is the intensity at θ in the azimuthal scan, collected from the SAXS data (figs. S10 and S12).
Calculation of the surface charge density of the 2D nanochannels
The ionic conductance (G) of a single nanochannel filled with KCl solution is approximately equal to the sum of the bulk conductance, q(μK + μCl)CBNAwh0/L, and the surface charge–governed conductance, 2μKσSw/L, where q is the elementary charge; μK and μCl are the mobility of K+ and Cl−, respectively; CB is the salt concentration; NA is Avogadro’s number; w, h0, and L are the width, height, and length of the single channel, respectively; and σS is the surface charge on the nanochannel walls (7). Bulk conductance dominates at high concentrations, whereas at low concentrations, surface charge–governed conductance dominates. In the case of the GO gel fiber, conductance was measured from the collective contributions of numerous single-channel arrays in the GO gel fiber (fig. S16, A and B). Therefore, G of a single nanochannel should be multiplied by the number of channels (n). We hypothesized that curved nanochannels confined in a cylindrical fiber with radius (r) could be regarded as rectangular parallelepipeds, like GO films (fig. S16D). When the length (L) of the rectangular parallelepiped is equal to that of the cylinder (~10 mm), the width (W) and height (H) of the rectangular parallelepiped can be denoted as 2πr and r/2, respectively. Hence, the number of the existing channels can be estimated by dividing H by the interlayer spacing (h). The measured values of r were 28 and 70 μm for the 4v1-GO and v1-GO gel fibers, respectively. The XRD pattern in fig. S16C indicates that the average interlayer spacing (h) of the GO gel fibers was 1.38 nm. Considering the thickness of the graphene basal plane (i.e., the extension of electronic clouds, 0.35 nm) (38), the height available for the electrolyte solution in the single nanochannel (h0) was 1.03 nm. By fitting the conductance data in the high concentration regions, we derived the value of W to be 179 and 396 μm for 4v1-GO and v1-GO gel fibers, respectively. These values agree well with the corresponding theoretical values (i.e., 2πr, 176 and 440 μm, respectively), indicating that our hypothesis is reasonable. Similarly, by fitting the conductance data in the low concentration regions, we derived the σS values.
Supplementary Material
Acknowledgments
We are grateful to H. S. Leese for helpful discussion. Funding: This research was supported by the Basic Science Research Program (2017R1A2B4010771), the program for fostering next-generation researchers in engineering (2017H1D8A2032495), and the Nano·Material Technology Development Program (2016M3A7B4905609) of the National Research Foundation of Korea funded by the Ministry of Science, ICT and Future Planning. J.H. acknowledges the support from the U.S. Office of Naval Research (ONR-N000141612838). Author contributions: Conceptualization: T.H.H. and J.H.; methodology and investigation: H.P., K.H.L., Y.B.K., S.B.A., S.H.N., W.E., J.Y.H., T.H.H., J.H., and W.J.L.; supervision: T.H.H., J.H., and W.J.L. All authors contributed to writing and reviewing the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/11/eaau2104/DC1
Fig. S1. Characterization of GOLC dispersion.
Fig. S2. Analysis of velocity profile.
Fig. S3. Estimation of time scale for Ca2+ diffusion.
Fig. S4. Diameter and extension rate of ejected fluid at v2 = 5.5v1 as a function of distance.
Fig. S5. POM images and birefringence intensity profiles of gel fibers.
Fig. S6. Sol-gel transition and gel behavior of GO solution.
Fig. S7. Zeta potential of GO as a function of pH in aqueous dispersion at a concentration of 0.05 mg ml−1.
Fig. S8. XPS spectra of GO gels.
Fig. S9. Raman spectra of GO gels.
Fig. S10. Determination of the degree of orientation in gel fibers.
Fig. S11. Stress-strain curves of dried GO fibers under 10% min−1 of tensile strain.
Fig. S12. Determination of the degree of orientation in dried fibers.
Fig. S13. I-V curves of NICs of different lengths.
Fig. S14. Experimental setup for measuring the ionic conductivity of NICs.
Fig. S15. Removal of Ca2+ by ion exchange in GO gel fibers.
Fig. S16. Modeling of GO gel fiber nanochannels using the configuration of GO films.
Table S1. Yield stress values of GO gels prepared with different amounts of NH4OH.
Table S2. Quantitative XPS analyses of cylindrical gels and gel fibers.
Table S3. Comparison of mechanical properties of our GO fibers with previously reported GO fibers and other nanocarbon-based fibers.
Table S4. Structural parameters of dried GO fibers.
Table S5. Comparison of ionic conductivity of GO gel fibers with various nanosheet films.
Movie S1. In situ observation of dynamic assembly of GO gel fiber at v2 = v1.
Movie S2. In situ observation of dynamic assembly of GO gel fiber at v2 = 3v1.
REFERENCES AND NOTES
- 1.Gouaux E., MacKinnon R., Principles of selective ion transport in channels and pumps. Science 310, 1461–1465 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Wang C., Fu K. K., Dai J., Lacey S. D., Yao Y., Pastel G., Xu L., Zhang J., Hu L., Inverted battery design as ion generator for interfacing with biosystems. Nat. Commun. 8, 15609 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hou X., Jiang L., Learning from nature: Building bio-inspired smart nanochannels. ACS Nano 3, 3339–3342 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Duan C., Majumdar A., Anomalous ion transport in 2-nm hydrophilic nanochannels. Nat. Nanotechnol. 5, 848–852 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Tagliazucchi M., Szleifer I., Transport mechanisms in nanopores and nanochannels: Can we mimic nature? Mater. Today 18, 131–142 (2015). [Google Scholar]
- 6.Choi E., Wang C., Chang G. T., Park J., High current ionic diode using homogeneously charged asymmetric nanochannel network membrane. Nano Lett. 16, 2189–2197 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Raidongia K., Huang J., Nanofluidic ion transport through reconstructed layered materials. J. Am. Chem. Soc. 134, 16528–16531 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Koltonow A. R., Huang J., Two-dimensional nanofluidics. Science 351, 1395–1396 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Cheng H., Zhou Y., Feng Y., Geng W., Liu Q., Guo W., Jiang L., Electrokinetic energy conversion in self-assembled 2D nanofluidic channels with Janus nanobuilding blocks. Adv. Mater. 29, 1700177 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Fan R., Huh S., Yan R., Arnold J., Yang P. D., Gated proton transport in aligned mesoporous silica films. Nat. Mater. 7, 303–307 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Gao J., Koltonow A. R., Raidongia K., Beckerman B., Boon N., Luijten E., Olvera de la Cruz M., Hunag J., Kirigami nanofluidics. Mater. Chem. Front. 2, 475–482 (2018). [Google Scholar]
- 12.Kiriya D., Kawano R., Onoe H., Takeuchi S., Microfluidic control of the internal morphology in nanofiber-based macroscopic cables. Angew. Chem. Int. Ed. Engl. 51, 7942–7947 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Grena B., Alayrac J.-B., Levy E., Stolyarov A. M., Joannopoulos J. D., Fink Y., Thermally-drawn fibers with spatially-selective porous domains. Nat. Commun. 8, 364 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J. E., Han T. H., Lee S. H., Kim J. Y., Ahn C. W., Yun J. M., Kim S. O., Graphene oxide liquid crystals. Angew. Chem. Int. Ed. Engl. 50, 3043–3047 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Kim J., Michelin S., Hilbers M., Martinelli L., Chaudan E., Amselem G., Fradet E., Boilot J.-P., Brouwer A. M., Baroud C. N., Peretti J., Gacoin T., Monitoring the orientation of rare-earth-doped nanorods for flow shear tomography. Nat. Nanotechnol. 12, 914–919 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Nakayama M., Kajiyama S., Kumamoto A., Nishimura T., Ikuhara Y., Yamato M., Kato T., Stimuli-responsive hydroxyapatite liquid crystal with macroscopically controllable ordering and magneto-optical functions. Nat. Commun. 9, 568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang K.-S., Lin C.-A., Lin C.-H., Preparation of high-strength and high-modulus poly(vinyl alcohol) fibers by crosslinking wet spinning multistep drawing method. J. Appl. Polym. Sci. 52, 1181–1189 (1994). [Google Scholar]
- 18.Håkansson K. M. O., Fall A. B., Lundell F., Yu S., Krywka C., Roth S. V., Santoro G., Kvick M., Wittberg L. P., Wågberg L., Söderberg L. D., Hydrodynamic alignment and assembly of nanofibrils resulting in strong cellulose filaments. Nat. Commun. 5, 4018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D. J. Shaw, Introduction to Colloid and Surface Chemistry (Butterworth-Heinemann, ed. 4, 1999). [Google Scholar]
- 20.Tao Y.-G., den Otter W. K., Briels W. J., Kayaking and wagging of rods in shear flow. Phys. Rev. Lett. 95, 237802 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Eom W., Park H., Noh S. H., Koh K. H., Lee K., Lee W. J., Han T. H., Strengthening and stiffening graphene oxide fiber with trivalent metal ion binders. Part. Part. Syst. Charact. 34, 1600401 (2017). [Google Scholar]
- 22.Trebbin M., Steinhauser D., Perlich J., Buffet A., Roth S. V., Zimmermann W., Thiele J., Förster S., Anisotropic particles align perpendicular to the flow direction in narrow microchannels. Proc. Natl. Acad. Sci. U.S.A. 110, 6706–6711 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han C. D., Segal L., Study of fiber extrusion in wet spinning. I. Experimental determination of elongational viscosity. J. Appl. Polym. Sci. 14, 2973–2998 (1970). [Google Scholar]
- 24.Han C. D., Segal L., Study of fiber extrusion in wet spinning. II. Effects of spinning conditions on fiber formation. J. Appl. Polym. Sci. 14, 2999–3019 (1970). [Google Scholar]
- 25.Xu Y., Sheng K., Li C., Shi G., Self-assembled graphene hydrogel via a one-step hydrothermal process. ACS Nano 4, 4324–4330 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Hu B., Fuchs A., Huseyin S., Gordaninejad F., Evrensel C., Supramolecular magnetorheological polymer gels. J. Appl. Polym. Sci. 100, 2464–2479 (2006). [Google Scholar]
- 27.Mitra S. B., Wang B., Rozzi S. M., Clark T. R., Polyacid-modified composite (compomer) systems containing polymers of N-vinylpyrrolidone. Polym. Adv. Technol. 12, 380–386 (2001). [Google Scholar]
- 28.Li D., Müller M. B., Gilje S., Kaner R. B., Wallace G. G., Processable aqueous dispersions of graphene nanosheets. Nat. Nanotechnol. 3, 101–105 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Rourke J. P., Pandey P. A., Moore J. J., Bates M., Kinloch I. A., Young R. J., Wilson N. R., The real graphene oxide revealed: Stripping the oxidative debris from the graphene-like sheets. Angew. Chem. Int. Ed. Engl. 50, 3173–3177 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Fan X., Peng W., Li Y., Li X., Wang S., Zhang G., Zhang F., Deoxygenation of exfoliated graphite oxide under alkaline conditions: A green route to graphene preparation. Adv. Mater. 20, 4490–4493 (2008). [Google Scholar]
- 31.Park S., Lee K.-S., Bozoklu G., Cai W., Nguyen S. T., Ruoff R. S., Graphene oxide papers modified by divalent ions—Enhancing mechanical properties via chemical cross-linking. ACS Nano 2, 572–578 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Feng H., Cheng R., Zhao X., Duan X., Li J., A low-temperature method to produce highly reduced graphene oxide. Nat. Commun. 4, 1539 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Pei S., Zhao J., Du J., Ren W., Cheng H.-M., Direct reduction of graphene oxide films into highly conductive and flexible graphene films by hydrohalic acids. Carbon 48, 4466–4474 (2010). [Google Scholar]
- 34.Kongkhlang T., Tashiro K., Kotaki M., Chirachanchai S., Electrospinning as a new technique to control the crystal morphology and molecular orientation of polyoxymethylene nanofibers. J. Am. Chem. Soc. 130, 15460–15466 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Kim F., Luo J., Cruz-Silva R., Cote L. J., Sohn K., Huang J., Self-propagating domino-like reactions in oxidized graphite. Adv. Funct. Mater. 20, 2867–2873 (2010). [Google Scholar]
- 36.Han T. H., Huang Y.-K., Tan A. T. L., Dravid V. P., Huang J., Steam etched porous graphene oxide network for chemical sensing. J. Am. Chem. Soc. 133, 15264–15267 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Um I. C., Ki C. S., Kweon H., Lee K. G., Ihm D. W., Park Y. H., Wet spinning of silk polymer. II. Effect of drawing on the structural characteristics and properties of filament. Int. J. Biol. Macromol. 34, 107–119 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Nair R. R., Wu H. A., Jayaram P. N., Grigorieva I. V., Geim A. K., Unimpeded permeation of water through helium-leak–tight graphene-based membranes. Science 335, 442–444 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Guo F., Kim F., Han T. H., Shenoy V. B., Huang J., Hurt R. H., Hydration-responsive folding and unfolding in graphene oxide liquid crystal phases. ACS Nano 5, 8019–8025 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bai H., Li C., Wang X., Shi G., On the gelation of graphene oxide. J. Phys. Chem. C 115, 5545–5551 (2011). [Google Scholar]
- 41.Dikin D. A., Stankovich S., Zimney E. J., Piner R. D., Dommett G. H. B., Evmenenko G., Nguyen S. T., Ruoff R. S., Preparation and characterization of graphene oxide paper. Nature 448, 457–460 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Xu Z., Sun H., Zhao X., Gao C., Ultrastrong fibers assembled from giant graphene oxide sheets. Adv. Mater. 25, 188–193 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Yoon S. S., Lee K. E., Cha H.-J., Seong D. G., Um M.-K., Byun J.-H., Oh Y., Oh J. H., Lee W., Lee J. U., Highly conductive graphene/Ag hybrid fibers for flexible fiber-type transistors. Sci. Rep. 5, 16366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seyedin S., Romano M. S., Minett A. I., Razal J. M., Towards the knittability of graphene oxide fibres. Sci. Rep. 5, 14946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ci L., Punbusayakul N., Wei J., Vajtai R., Talapatra S., Ajayan P. M., Multifunctional macroarchitectures of double-walled carbon nanotube fibers. Adv. Mater. 19, 1719–1723 (2007). [Google Scholar]
- 46.Lundahl M. J., Cunha A. G., Rojo E., Papageorgiou A. C., Rautkari L., Arboleda J. C., Rojas O. J., Strength and water interactions of cellulose I filaments wet-spun from cellulose nanofibril hydrogels. Sci. Rep. 6, 30695 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mittal N., Ansari F., Gowda.V K., Brouzet C., Chen P., Larsson P. T., Roth S. V., Lundell F., Wågberg L., Kotov N. A., Söderberg L. D., Multiscale control of nanocellulose assembly: Transferring remarkable nanoscale fibril mechanics to macroscale fibers. ACS Nano 12, 6378–6388 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Xiang C., Behabtu N., Liu Y., Chae H. G., Young C. C., Genorio B., Tsentalovich D. E., Zhang C., Kosynkin D. V., Lomeda J. R., Hwang C.-C., Kumar S., Pasquali M., Tour J. M., Graphene nanoribbons as an advanced precursor for making carbon fiber. ACS Nano 7, 1628–1637 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Qin S., Liu D., Wang G., Portehault D., Garvey C. J., Gogotsi Y., Lei W., Chen Y., High and stable ionic conductivity in 2D nanofluidic ion channels between boron nitride layers. J. Am. Chem. Soc. 139, 6314–6320 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Shao J.-J., Raidongia K., Koltonow A. R., Huang J., Self-assembled two-dimensional nanofluidic proton channels with high thermal stability. Nat. Commun. 6, 7602 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/11/eaau2104/DC1
Fig. S1. Characterization of GOLC dispersion.
Fig. S2. Analysis of velocity profile.
Fig. S3. Estimation of time scale for Ca2+ diffusion.
Fig. S4. Diameter and extension rate of ejected fluid at v2 = 5.5v1 as a function of distance.
Fig. S5. POM images and birefringence intensity profiles of gel fibers.
Fig. S6. Sol-gel transition and gel behavior of GO solution.
Fig. S7. Zeta potential of GO as a function of pH in aqueous dispersion at a concentration of 0.05 mg ml−1.
Fig. S8. XPS spectra of GO gels.
Fig. S9. Raman spectra of GO gels.
Fig. S10. Determination of the degree of orientation in gel fibers.
Fig. S11. Stress-strain curves of dried GO fibers under 10% min−1 of tensile strain.
Fig. S12. Determination of the degree of orientation in dried fibers.
Fig. S13. I-V curves of NICs of different lengths.
Fig. S14. Experimental setup for measuring the ionic conductivity of NICs.
Fig. S15. Removal of Ca2+ by ion exchange in GO gel fibers.
Fig. S16. Modeling of GO gel fiber nanochannels using the configuration of GO films.
Table S1. Yield stress values of GO gels prepared with different amounts of NH4OH.
Table S2. Quantitative XPS analyses of cylindrical gels and gel fibers.
Table S3. Comparison of mechanical properties of our GO fibers with previously reported GO fibers and other nanocarbon-based fibers.
Table S4. Structural parameters of dried GO fibers.
Table S5. Comparison of ionic conductivity of GO gel fibers with various nanosheet films.
Movie S1. In situ observation of dynamic assembly of GO gel fiber at v2 = v1.
Movie S2. In situ observation of dynamic assembly of GO gel fiber at v2 = 3v1.