

Abstract
Purpose:
Response to a complex trastuzumab-based regimen is affected by multiple features of the tumor and its microenvironment. Developing a predictive algorithm is key to optimizing HER2-targeting therapy.
Methods:
We analyzed 137 pre-treatment tumors with mRNA-seq and DNA exome sequencing from CALGB 40601, a neoadjuvant phase III trial of paclitaxel plus trastuzumab with or without lapatinib in stage II-III HER2-positive breast cancer. We adopted an Elastic Net regularized regression approach that controls for co-varying features within high-dimensional data. First, we applied 517 known gene expression signatures to develop an Elastic Net model to predict pCR, which we validated on 143 samples from 4 independent trials. Next, we performed integrative analyses incorporating clinicopathologic information with somatic mutation status, DNA copy number alterations (CNAs) and gene signatures.
Results:
The Elastic Net model using only gene signatures predicted pCR in the validation sets (AUC = 0.76). Integrative analyses showed that models containing gene signatures, clinical features, and DNA information were better pCR predictors than models containing a single data type. Frequently selected variables from the multi-platform models included amplifications of chromosome 6p, TP53 mutation, HER2-enriched subtype and immune signatures. Variables predicting resistance included Luminal/ER+ features.
Conclusions:
Models using RNA only, as well as integrated RNA and DNA models, can predict pCR with improved accuracy over clinical variables. Somatic DNA alterations (mutation, CNAs), tumor molecular subtype (HER2E, Luminal), and the microenvironment (immune cells) were independent predictors of response to trastuzumab and paclitaxel-based regimens. This highlights the complexity of predicting response in HER2-positive breast cancer.
Keywords: HER2, breast cancer, trastuzumab, CALGB 40601, Elastic Net
INTRODUCTION
Human epidermal growth factor receptor type 2 (HER2) is overexpressed in ~25% of breast cancers. The anti-HER2 antibody trastuzumab reduces mortality in stage I-III disease by 37% when combined with adjuvant chemotherapy. However, approximately one-fourth of these patients experience recurrence within 10 years and ultimately succumb to their disease(1). Additional HER2-targeting drugs including lapatinib(2), pertuzumab(3), and neratinib(4) have been tested in combination with or following trastuzumab in patients with stage I – III HER2-positive breast cancer, with variable impacts on disease-free survival in terms of statistical significance but all with modest (less than 3%) absolute effects. These results clearly highlight our need to identify those in whom additional therapy is warranted. The MINDACT and similar trials suggest that genomic classifiers may help identify patients with HER2-negative disease who may be treated with less aggressive regimens(5). The identification of a biologic classifier for tailoring therapy in HER2-positive disease would also be very valuable.
It is equally true that HER2-positive breast cancer is highly molecularly heterogeneous. CALGB 40601(6), and the similar trial NeoALTTO(7), have revealed that gene expression signatures of ESR1 and HER2, molecular intrinsic subtype, and immune cell activation are associated with pathological complete response (pCR). Several molecular alterations are thought to contribute to trastuzumab resistance, including PIK3CA mutation(6,8,9), PTEN loss(10,11), and TP53 mutation(12,13), but these possible biomarkers have been inconsistent. In NeoALLTO, mutations in the RhoA pathway were associated with response, which has not yet been further examined(14). In addition to tumor influences, immune cell gene expression has been independently associated with pCR(6,7), and in retrospective/prospective trials, tumor infiltrating lymphocytes (TILs) have been predictive of trastuzumab benefit(15,16). Currently, HER2 overexpression and/or amplification remains the only clinically validated marker to select patients for anti-HER2 therapies.
A number of studies including The Cancer Genome Atlas(17,18) have produced a wealth of genomic data and described disease mechanisms. However, there are still two major challenges when using clinical trial samples: First, most research studies characterize a genomic feature type, such as gene expression, mutation, or copy number, and there are few capable of integrating disparate data types that reflect the continuum of cancer biology and are simultaneously able to address clinical outcomes. Second, because these studies did not utilize samples from prospective clinical trials with prespecified endpoints, they are poorly suited to identify or validate novel predictive biomarkers.
By contrast, in this study we utilized two computational approaches of integrative data analysis, namely Elastic Net and DawnRank, using the samples obtained from Cancer and Leukemia Group B (CALGB) 40601(6), a prospective phase III trial of neoadjuvant chemotherapy with trastuzumab, lapatinib or both. In this analysis, we first developed an Elastic Net model from gene expression data and applied the model onto four different validation datasets. In addition, after combining mutation, DNA copy number alterations, and gene expression data with known clinical features, we developed objective computational models to identify important determinants of response to trastuzumab-based therapy. Our goal was to develop an accurate predictor of response, and at the same time, to learn more about the biology of therapeutic response in HER2-positive breast tumors.
PATIENT AND METHODS
CALGB 40601 Study Design and Patients
The study design and clinical results have been previously published(6); CALGB 40601 is now part of the Alliance for Clinical Trials in Oncology. A total of 305 women with stage II-III HER2-positive disease were randomized to receive paclitaxel (T) at 80 mg/m2 weekly for 16 weeks, with trastuzumab (H, 4 mg/kg loading dose followed by 2 mg/kg), lapatinib (L, 1500 mg/d), or both (L at 1000 mg/d plus the same dose of H) for 16 weeks. The TL arm was closed early based on reports of inferiority and greater toxicity; given that single agent lapatinib is not a clinically relevant treatment and the mechanism of action differs systematically from conventional H or H+L-based therapy, the TL arm was excluded from this analysis. The primary endpoint was pCR, defined as no invasive tumor in the breast, which is a surrogate endpoint of survival in HER2-positive breast cancer(19).
Tumor genomic methods
Participants underwent 4 pretreatment 16-gauge core biopsies: 2 cores were placed into RNA stabilization product (RNALater™Qiagen, Hilden, Germany), and 2 were placed into 10% neutral buffered formalin. CALGB 40601 enrolled 305 patients. Figure 1-A shows the CONSORT diagram for the subset studied here on the genomic level. We eliminated from analysis those patients in whom the RNA or DNA quality was inadequate, those treated on the non-trastuzumab arm (TL), and those with Normal-like intrinsic subtype, which consists mostly of normal tissues. The final training set consisted of 137 patient samples from TH (n= 68) and THL (n= 69) arms; all received trastuzumab-paclitaxel regimens. All 137 patients signed an IRB-approved, protocol-specific informed consent document in accordance with federal and institutional guidelines. This document included consent for the use of RNA and DNA; the consent also covered future biomarker research. DNA exome sequencing was performed at McDonnell Genome Institute (Washington University) and RNA-seq was performed at the UNC High Throughput Sequencing Facility (University of North Carolina). The patient and tumor characteristics of the included samples did not differ significantly from the total dataset (N = 285) including stage, hormonal receptor status, and pCR rates (data not shown).
Figure1. CONSORT diagram of patient selection and characteristics.
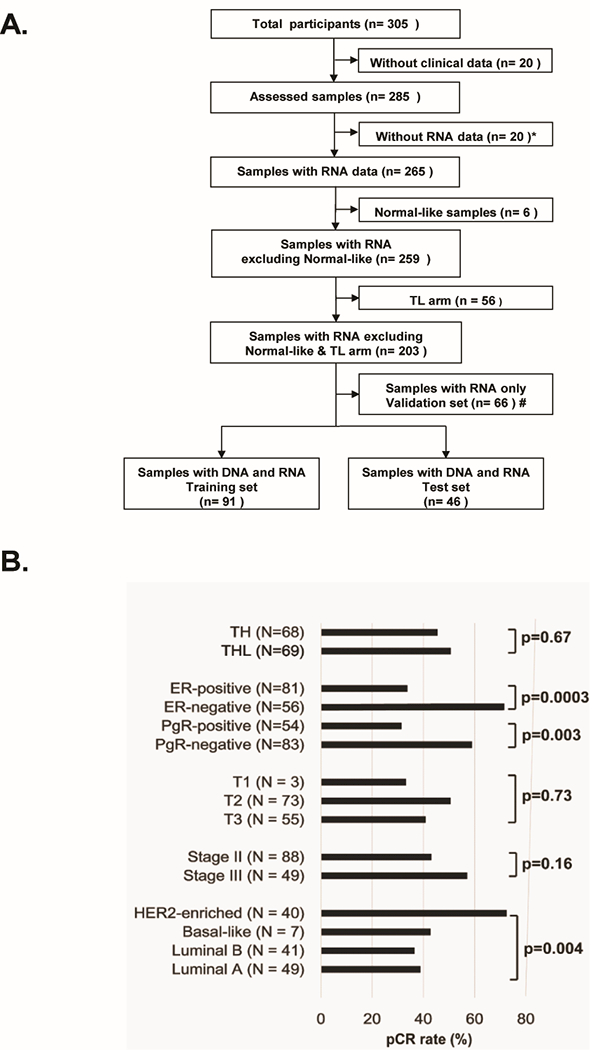
(A) Sample flow chart to show how samples were selected. Starting with 305 patients, specimens were removed for multiple reasons including incomplete clinical data, low RNA yields, a normal-like non-tumor expression profile, being part of the TL= lapatinib and paclitaxel arm, thus leaving 203 patients. Of these, 137 had DNA exomes results, with this final 137 sample set also being split into a training and test set. (B) Clinical and intrinsic expression subtype characteristics with pCR rates using the 137 patient data set. P-values were calculated by Chi-aquare test. TH, trastuzumab and paclitaxel arm; THL, trastuzumab, lapatinib and paclitaxel arm; ER, estrogen-receptor; PgR, progesterone receptor.
Gene expression and signatures
Gene expression profiles were generated by mRNA-sequencing using an Illumina HiSeq 2000 as described in Ciriello et al.(17). Briefly, mRNAseq libraries were made from total RNA using the Illumina TruSeq mRNA sample preparation kit and sequenced on an Illumina HiSeq 2000 using a 2×50bp configuration with an average of 136 million reads per sample. Quality-control-passed reads were aligned to the human reference genome (hg19) using MapSplice(20). The alignment profile was determined by Picard Tools v1.64 (http://broadinstitute.github.io/picard/). Aligned reads were sorted and indexed using SAMtools and translated to transcriptome coordinates then filtered for indels, large inserts, and zero mapping quality using UBU v1.0 (https://github.com/mozack/ubu). Transcript abundance estimates for each sample were performed using RSEM, an expectation-maximization algorithm(21) using the UCSC knownGene transcript and gene definitions. Raw RSEM read counts for all mRNAseq samples were normalized to a fixed upper quartile.
Next, PAM50 subtyping was applied to the gene expression data using a two-step normalization process based on the TCGA(17) cohort as previously described(6). We next applied a collection of 517 gene expression signatures, representing multiple biological pathways and cell types, to all 137 samples. These 517 signatures (all published) were obtained from 73 publications or Gene Set Enrichment Analysis (GSEA)(22) and partially summarized by Fan, et al.(23) (see eTable 1 for the complete list of signatures and their associated references). Using the combined normalized data set with the TCGA data, we applied each signature to the data set in a manner consistent with their derivation. For 478 signatures with homogenous expression across genes within a given set, these represent coordinately regulated sets of either “up” genes, or sets of “down” genes. All the genes were moving in the same direction, therefore we took the median expression value for all genes in a signature. For 39 signatures where gene expression patterns were not homogenous, we calculated correlations to predetermined centroids using previously published training datasets/centroids, or used predetermined special algorithms following their original methods.
Mutation data
We performed hybrid capture exome sequencing on 137 of the tumors (Nimblegen v3.0 SeqCap reagent) and matched peripheral blood mononuclear cells (PBMC) sequenced to average 100x depth coverage using paired-end 2×100bp. Raw sequences were aligned using the BWA-mem algorithm, and refined using our Assembly Based Re-Alignment (ABRA)(24) process to allow for accurate alignment of complex sequence variation. Somatic mutation detection was performed by integrated whole DNA exome and mRNAseq using the UNCeqR analytic tool as previously described(25,26).
Copy number variants
Copy number variation across the genome was determined as follows: The sequence reads were aligned to the genome (hg 19) using the bwa-mem algorithm (https://github.com/lh3/bwa; v0.7.4) with the default parameters. Duplicates were removed using Picard (http://broadinstitute.github.io/picard/). Quality statistics were also generated with Picard including measures of fragment length, sequence content, alignment, capture bias and efficiency, coverage, and variant call metrics. Copy number assessments were performed using SynthEx(27). In brief, counts data for fixed 100kb bins were generated using BEDTools(28). The read ratios were calculated using the “synthetic normal” strategy described in SynthEx. A trending filter procedure was applied to segment the genome. The segment-level copy number values, which are the log2 ratios of normalized signal intensities between tumor and controls, were finally corrected by purity and ploidy estimates from SynthEx, taking whole genome doubling into account for these values. These segment-level values were changed into gene-level values using Switchplus(29), and then we re-calculated the values for 536 predetermined cancer-specific segments (eTable 2) that are frequently altered in multiple types of cancer including breast cancers(30,31); we also calculated chromosome arm based values and included these as features (48 segments). DNA Copy number values derived from exome sequencing were compared with those from SNP6.0 among the TCGA samples(17) with ploidy 1.75 – 2.5, then the thresholds for gain or loss from exome-derived SynthEx values were determined as 0.25 or −0.32, respectively(27); we applied these thresholds to copy number values on the CALGB 40601 samples to call gained and lost segments.
The complete list of DNA mutation somatic variants, DNA copy number segment, and gene expression values from CALGB 40601 samples are provided in Supplementray Data Files 1–3. The accession number for the RNAseq data for CALGB 40601 is GSE116335. Exome data for CALGB 40601 cohort is available via the NCBI dbGAP repository under accession number phs001570.v1.
Statistical analyses
All statistical analyses were performed using R version 3.1.2. All analyses were based on the study database frozen on January 29, 2016.
Elastic Net analysis
For feature selection using a multivariate modeling approach, we used Elastic Net (R package glmnet)(32), which is a regularized regression method that linearly combines the L1 and L2 penalties of the Ridge Regression and Least Absolute Shrinkage and Selection Operator (LASSO)(33). Monte-Carlo cross-validation(34) (R package caret) was conducted using 200 different training sets randomly selected from the 137-sample training set. Models were built to predict pCR in the training set, selecting lambda values over a grid of alpha values from 0.1 to 1 by 0.1 increments via 10-fold cross validation(35) (R package glmnet). Then we calculated accuracy, which equals (sensitivity + specificity) / 2, for each parameter combination. We identified the optimal parameter combination with the highest accuracy during cross-validation, and applied this to the final model using the best parameter combination onto the test set, and then constructed ROC curves and evaluated area under ROC (AUC). The variables with a high or low (negative) coefficient value would be associated with response or resistance to trastuzumab-containing therapy, respectively.
There were two purposes for the Elastic Net analyses, namely model-building and biological discovery through feature selection. First, we wished to evaluate the robustness of the Elastic Net model and use of this approach for pCR prediction. We developed the predictive model using 137 tumors from the CALGB 40601 training set and validated it using gene expression datasets from an independent group of 143 patients who participated in four clinical trials of neoadjuvant chemotherapy plus trastuzumab for patients with HER2-positive breast cancer on whom high-quality gene expression data were available. These included 43 patients in CHERLOB(8) who received anthracycline, taxane, and trastuzumab, with or without lapatinib; 24 patients in XENA(36) who received capecitabine, taxane, and trastuzumab; 10 patients in I-SPY1(37) who received anthracycline, taxane, and trastuzumab; and 66 patients from the CALGB 40601 independent validation set who received taxane and trastuzumab with or without without lapatinib, but who were not included in the training set (because the training set was limited to those with RNA and DNA genomics). All the patients received at least trastuzumab and taxane. Supplementary Figure 1-A diagrams the overall process of the Elastic Net model development and evaluation. In more detail, the gene signature data of all 137 samples from the CALGB 40601 training set were used as the training dataset to construct a model for an expression-only pCR prediction. This model was applied to CHERLOB, XENA, I-SPY, the CALGB 40601 independent validation set and all four test sets combined to construct ROC curves and evaluate AUC.
Second, in order to identify important and novel features contributing to sensitivity and resistance using a multi-dimensional approach, we investigated combining mutation, DNA copy number, and gene expression data with known clinical features, and then used all of these features for Elastic Net model building. Mutation and copy number alterations (CNAs) were used as dichotomous and continuous variables, respectively. By balancing for clinicopathological features, the samples were divided into a training and a test set (R package sampling), then Monte-Carlo cross-validation was conducted using 200 different training sets randomly selected from the larger training set. We developed a set of integrated Elatic Net regression models to predict pCR, varying the features used as input. Because this component of the study was limited to available samples with both RNA and DNA data, which limited sample size, the Elastic Net was performed using 10 rounds of training and testing. The most frequently selected features in the models were identified in order to find reproducible predictive features (Supplementary Figure 1-B).
Survival analysis on METABRIC samples
In order to address the behavior of these models in HER2-positive tumors that did not receive trastuzumab or other HER2-targeted agents, thereby eliminating variables unrelated to HER2-directed therapy, the Elastic Net gene expression-only models from CALGB 40601 were applied onto 216 HER2-positive tumors from the METABRIC(38) dataset from the pre-trastuzumab era. Among the 216 patients studied, 124 did not receive either chemotherapy or HER2-targeting, and 92 received chemotherapy without trastuzumab. The median follow-up period was 7.24 (0.15–26.90) years. The patients were classified into three groups according to the scores derived from the expression only Elastic Net model, and overall survival was assessed by the Kaplan-Meier method.
DawnRank analysis
We used DawnRank(39), a novel computational method that uses within-tumor integrated analyses based upon predetermined protein-protein interactions networks, then populated by patient specific tumor gene expression values, and DNA aberrations, in order to identify those genes with DNA aberrations that have the greatest expression impact on the predefined networks; these Dawnrank scores are calculated on individual patients, then aggregated based upon groups of patients, to find individual genes involved in response to trastuzumab-containing therapies. Using the DawnRank predefined protein-protein interaction networks, we populated this network with mRNA gene expression data for each patient and calculated a score for each gene based upon the expression of the genes directly connected to it in the network. The DawnRank score(s) depend on the three parameters: predetermined protein-protein interaction networks, gene expression values for each patient, and a “damping factor” that represents the extent to which the ranking depends on the structure of the network. These three parameters along with DNA alteration status (i.e. mutation, amplification, or deletion) form the key components to determine “drivers” in individual samples. Log2 transformed normalized mRNAseq gene expression data were median-centered for each gene among 137 CALGB 40601 samples, and further transformed to absolute value scores. Dawnrank was then run for each tumor with a mu = 3, which is the suggested default setting. The genes were then ranked according to the Dawnrank scores.
We then generated a binary matrix of 0 indicating no alteration and 1 indicating any DNA alteration for each gene, and examined somatically altered genes (DNA mutations and/or DNA alterations as described above) by applying DawnRank to the samples with pCR vs those without pCR according to the “percentrank” analysis mode, which aggregates the DawnRank results across a predefined set of samples/patients in order to find drivers based upon groups of patients (i.e. those with a pCR). Briefly, DawnRank applies a modified version of the Condorcet method(40), which is a voting scheme selecting a winning candidate gene by comparing every possible pair of candidate genes. Therefore a pair of candidate genes A and B are compared by the number of alterations in gene A that had higher Dawnrank scores than gene B.
Additional Genomic Analyses
The association between pCR status and the clinicopathological variables, or mutated genes, were investigated using Fisher’s Exact test with Bonferroni correction. Using a two-class unpaired Significance Analysis of Microarrays (SAM)(41), we also conducted a permutation-based, supervised analysis to find features with significant correlation to pCR by comparing pCR samples versus non-pCR samples using gene-level DNA copy number data.
Hierarchical clustering was performed using centroid linkage implemented in software Gene Cluster 3.0(42), and the clustering result was viewed with Java Treeview v1.1.5r2(43) to identify patterns among the features selected in the Elastic Net models. We investigated the significance of gene signatures with unknown roles using the “Investigate Gene Sets” method of Gene Set Enrichment Analysis (GSEA, http://software.broadinstitute.org/gsea/msigdb/annotate.jsp)(22), and also investigated gene signature scores according to PAM50 subtype among 1100 breast cancer patients from TCGA(17). ROC/AUC curves were compared using R package pROC(44). DNA copy number frequency landscapes were generated using Swicthplus(29), which can identify segments with CNAs specific for a user-determined set of tumors, in this case, samples with pCR vs non-pCR. Thus Switchplus provides a supervised method for analysing and visualizing copy number data. Switchplus is provided as a source script in R and available for download at: https://genome.unc.edu/SWITCHplus/.
RESULTS
Cohort characteristics and genomic datasets
On CALGB 40601 specimens, we performed mRNAseq and DNA exomes (N=137, Figure 1-A. We compared non-silent mutation frequencies between HER2-positive breast cancer in TCGA (N=145) and tumors in CALGB 40601 (N=137). Although PIK3CA and CDH1 mutation were more frequent in TCGA HER2+, there were no differences in the list of somatically mutated genes between these two studies after Bonferroni correction (Supplementary Figure 2-A). We also confirmed the similarity between TCGA HER2+ and CALGB 40601 by comparing their DNA copy number frequency landscapes (Supplementary Figure 2-B), and again the results showed very high similarity. Among 10816 non-silent mutations found through exome sequencing of 137 CALGB 40601 tumors, 5106 mutations (47.2%) were detected in the mRNAseq of the corresponding tumors by UNCeqR(26). This frequency is comparable to the 51% frequency seen when performing the same analysis using 871 TCGA lung and breast cancer samples(26).
Among the 137 samples studied on the genomic level in CALGB 40601, clinical estrogen receptor (ER) or progesterone receptor (PgR) status and intrinsic subtype by PAM50 were associated with pCR (Figure 1-B). eTable 3 shows the list of somatically mutated genes with a frequency of >4%, and where only 8 genes occurred in ≥10 patients (≥7 %), and only TP53 mutation status was associated with pCR after Bonferroni correction (eTable 3). Interestingly, there were 4 patients with HER2 somatic mutations, including 2 with a variant allele frequency of greater than 10%; these two included a V777L variant, and a L755S variant. The V777L variant is predicted to be activating and sensitive to lapatinib(45); this patient was in the THL arm and achieved a pCR. The L755S variant is predicted to be transforming, but insensitive to lapatinib; this patient was in the TL arm and had residual disease after therapy. As a resource, multiple individual data type supervised analyses were performed on the 40601 training data set and present as eTable 4; these include results of the supervised analyses using individual gene expression values, gene signatures, DNA segment-level copy number, or gene-level copy number data. Likewise, The supervised results on CALGB 40601 validation set using gene-expression and gene signatures are listed on eTable 5.
Gene expression based prediction of pCR using Elastic Net
We first developed genomic predictors of response to trastuzumab containing regimens using gene expression alone because several validation sets existed that would allow us to develop, then validate, a predictive model based on RNA data. Therefore, we used the Elastic Net method to develop a model for predicting pCR and starting with 517 published expression signatures applied to the 137 patient CALGB 40601 training cohort (Supplementary Figure 1, and Table 1). Included within the positive predictive features were several well-described signatures including “correlation to HER2-Enriched”, two breast cancer recurrence predictors(46,47), and immune signatures. On the other hand, the tumor’s ER status based on clinical assay (cER), the signature of “correlation to luminal A” and a PgR-activity gene signature were negative predictors of pCR. For gene signatures of interest with unknown functions, we used GSEA(22) to gain insight into their function. “HS_Green18” (False discovery rate [FDR] = 3.3e-60) and “HS_Red19” (FDR = 2.6e-52) were correlated with the Luminal B signature and highly expressed in Luminal TCGA tumors(17). “HS_Red12” correlated with the HER2 signature in GSEA (FDR = 3.7e-115) and was highly expressed in TCGA HER2-Enriched tumors (Supplementary Figure 3-A to C). The optimized predictive model using gene expression signatures alone was applied to the CHERLOB(8), XENA(36), I-SPY1(37), and additional CALGB 40601 validation data sets, and all four sets combined (test sets). The AUC values were 0.80 (training), 0.73, 0.71, 0.83, 0.78 and 0.76 (combined); pCR rates in the test sets ranged from 13% in the lowest tertile scores, to 65% in the highest tertile (Figure 2, details in Supplementary Figure 4 and eTable 6).
Table 1.
Elastic Net features selected using the gene signatures only model from CALGB 40601
| Feature | References | Coefficient |
|---|---|---|
| GS_HS_Green18 | PMID.21214954 | −0.07153 |
| GS_HS_Red19 | PMID.21214954 | −0.04255 |
| GS_Duke_Module17_PgR | PMID.20335537 | −0.03614 |
| GS_JANES_Oscillation_JUND_KRT5 | PMID.24658685 | −0.03246 |
| GS_MITO1 | PMID.21214954 | −0.02511 |
| GS_Scorr_LumA_Correlation | PMID.19204204 | −0.01563 |
| GS_MM_Green12 | PMID.21214954 | −0.01218 |
| GS_MUnknown_16 | PMID.21214954 | −0.0108 |
| GS_HS_Green1 | PMID.21214954 | −0.00953 |
| GS_Duke_Module14_p53 | PMID.20335537 | −0.00605 |
| GS_Scorr_P53_Wt_Correlation | PMID.17150101 | −0.00386 |
| GS_Pcorr_NKI70_Good_Correlation | PMID.11823860 | −0.00307 |
| GS_Lim2009_MatureLuminal | PMID.25575446 | −0.0014 |
| GS_Chromogramin | PMID.21214954 | −0.00022 |
| GS_Unknown_12 | PMID.21214954 | 5.94E-05 |
| GS_Duke_Module07_glucosedepletion | PMID.20335537 | 0.002937 |
| GS_GSEA_RB_PATHWAY_BIOCARTA | http.//www.broadinstitute.org/gsea/msigdb/cards | 0.00382 |
| GS_Scorr_P53_Mut_Correlation | PMID.17150101 | 0.004311 |
| GS_HER2_Amplicon | PMID.21214954 | 0.007577 |
| GS_ROR_S_Model | PMID.19204204 | 0.012094 |
| GS_IGG_Cluster | PMID.21214954 | 0.014302 |
| GS_HS_Red12 | PMID.21214954 | 0.018906 |
| GS_GHI_RS_Model | PMID.15591335 | 0.021864 |
| GS_Duke_Module08_her2 | PMID.20335537 | 0.028529 |
| GS_Bcells_Plasmablast | PMID.25800755 | 0.034936 |
| GS_S100A9_A8 | PMID.21214954 | 0.039593 |
| GS_XRTinducedgenes | PMID.24527691 | 0.043993 |
| GS_Scorr_Her2_Correlation | PMID.19204204 | 0.044143 |
Using Elastic Net feature selection, features that made the logistic regression model are shown in this table. All the selected features are weighted and show non-zero coefficient values where positive coefficients predict pCR and negative coefficients predict non-pCR. GS, gene signature; HS, homo sapiens; MM, mammary model; PgR, progesterone receptor; Wt, wild type; Mut, mutation
Figure 2. Performance of the Elastic Net model for pCR prediction using gene signatures on CALGB 40601.
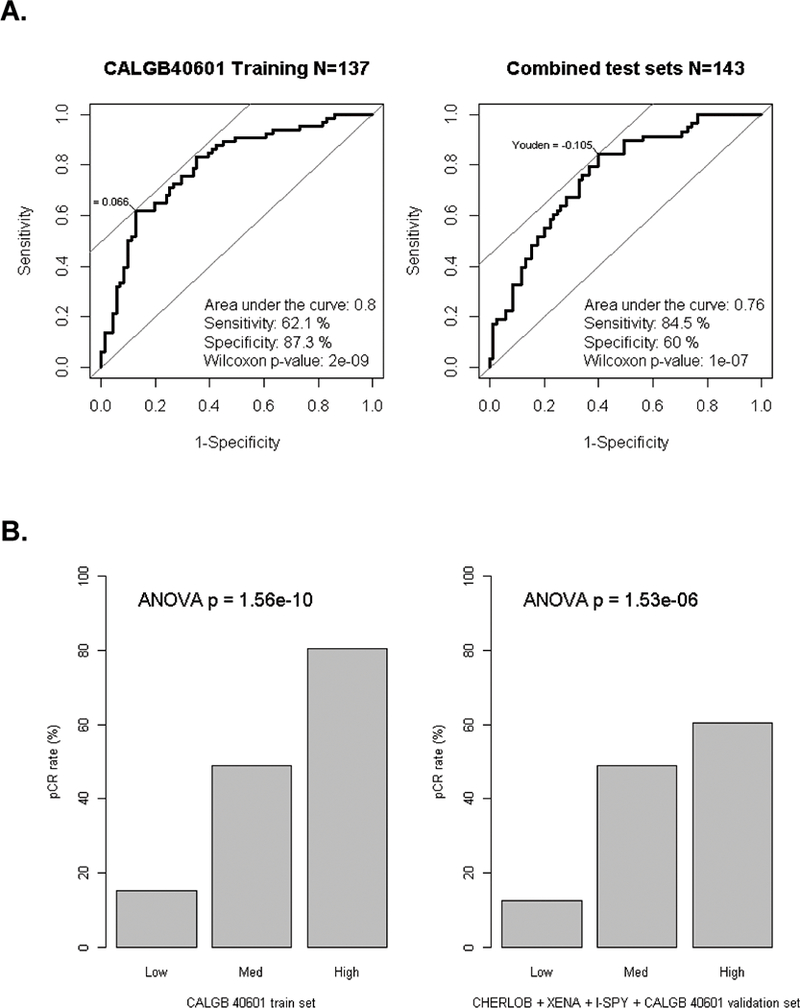
(A) Area under the curve (AUC) from the Receiver operating characteristic curve analysis were estimated for Elastic Net models using gene signatures alone in CALGB 40601. Left, CALGB 40601 as the training set (N = 137), AUC = 0.80; Right, All test sets combined (CHERLOB + XENA + I-SPY + CALGB 40601 validation set, N= 143, AUC = 0.76). Sensitivity and specificity values were selected using Youden’s cutpoint where the sum of sensitivity and specificity is maximal. Mann-Whitney-Wilcoxon test was conduct to calculate p-values. (B) Barplots showing results of the Elastic Net model score split into three rank order groups and then comparing pCR rates for patients in CALGB 40601, or all test sets combined. ANOVA T-test was conducted to calculate p-values by comparing signature scores across all three groups.
Elastic Net analysis using multi-dimensional data
We hypothesized the comprehensive integration of DNA copy number aberrations and mutations, added to gene expression and clinical data, would further improve predictive ability for response to trastuzumab-based therapy. The integrated Elastic Net multi-dimensional modeling assessed 528 DNA copy number segment values using 536 predefined chromosomal segments, 8 genes with somatic mutations in ≥10 patients, and the 517 gene expression signatures. Clinical ER and PgR status were included given consistent associations with pCR in HER2-positive breast cancer trials(6,7). Figure 3-A shows the average AUC values for each unique set of input variables in 10 rounds of repeated Elastic Net analysis (details in eTable 7). The model derived from the combination of gene signatures and CNAs yielded an AUC of 0.76, which was significantly higher than those from each individual data type alone (p <0.05). Gene signatures and CNAs were further combined with either mutation and/or clinical ER/PgR status, but the AUC values did not change significantly. The integrated Elastic Net models largely overlapped the signatures identified in the gene signatures-only model (Table 1). We finally selected the combination of gene signatures, CNAs, mutation and clinical ER/PgR status with the average AUC of 0.75 as the model to most fully explore, because of its objective integration of multiple data types, including TP53 mutation(25), and ER status(7), which have been previously reported as pCR predictors in patients receiving trastuzumab-based regimens. Using serial rounds of training and testing, we identified 33 features that contributed to ≥6 out of the 10 rounds of Elastic Net models using this combination of many distint feature types (Figure 3-B, all features in eTable 8). Included within the most frequently selected positive predictors in the integrated analysis were CNAs at chromosome (Chr.) 6p, TP53 mutation status, the signatures of Correlation to HER2-Enriched, 21-gene Recurrence Score(46), and 2 immune signatures of B-cells. On the other hand, clinical ER status, the signature of Correlation to Luminal A and of PgR-activity remained as negative predictors.
Figure 3. Elastic Net analysis using multi-dimensional data.
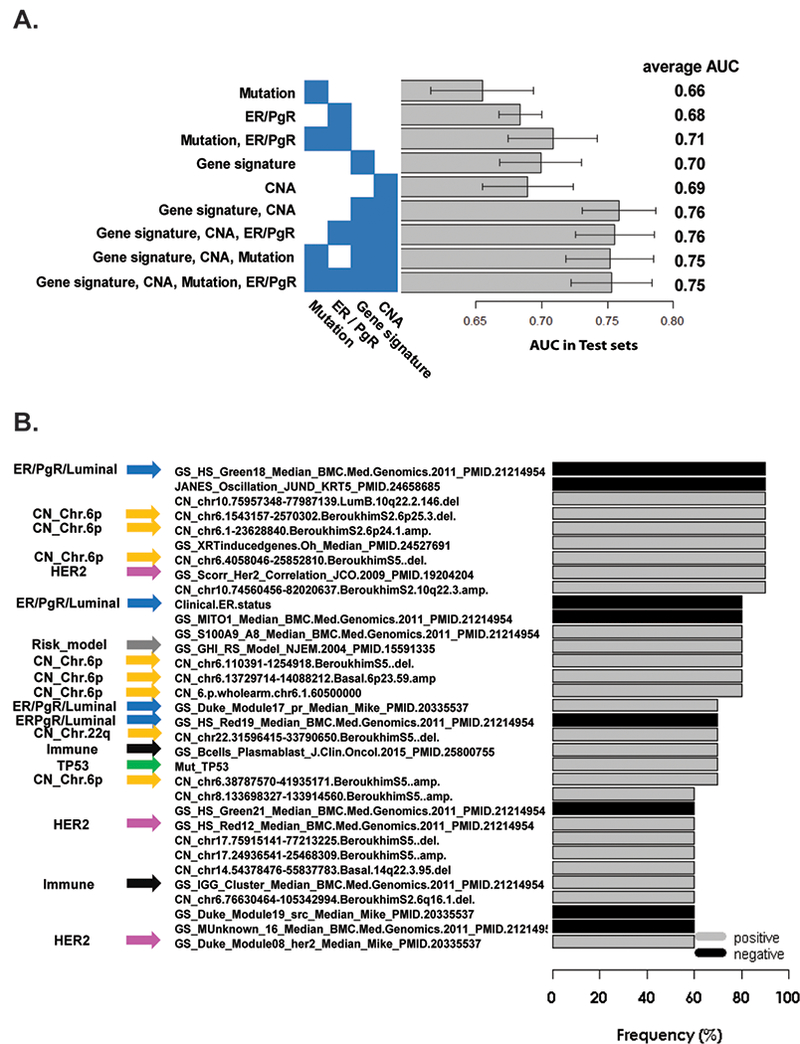
(A) Average AUC scores for various individual data type, or combined data type predictors, using test sets through 10 repeated Elastic Net analyses. Each bar shows the average AUC scores with 95% confidence intervals. (B) Frequently selected Elastic Net features coming from a multi-dimensional predictor. Features contributing to at least 6 out of 10 Elastic Net models using gene signatures, CNAs, mutations, and clinical ER/PgR status. GS, gene signature; CN, copy number; Mut, mutation; Gray and black bars indicate predictors which positively (37) and negatively (53) predict pCR; thus gray predictors are high in pCR samples and black predictors are high in non-pCR samples. Yellow arrows indicate CNAs features at Chromosome 6p; Green arrows indicate TP53 mutation status or signatures; Pink arrows indicate HER2-enriched signatures; A gray arrow indicates 21-gene Recurrence Score; Black arrows indicate immune signatures; Blue arrows indicate Clinical ER status, Luminal signatures and PgR gene signature.
We further conducted supervised clustering of the 33 features among the 137 samples (Figure 4). The features were clustered into two dendrogram nodes according to positive or negative predictors of pCR. The positive features were further aggregated by DNA copy number segments at 6p and 22q, and a grouping of the recurrence predictors, TP53 mutation status, and HER2 signatures. Estrogen-related features as negative predictors including clinical ER status, PgR signature and Correlation to Luminal A, were also clustered together. The samples were next ordered by their average scores derived from the 10 rounds of Elastic Net modeling. The model scores were highly correlated with pCR (logistic regression odds ratio, 1.6; p < 0.001). When samples were trichotomized into top, middle, or lower tertile groups of the model scores, pCR rates were 93.4%, 44.4%, and 6.5%, respectively.
Figure 4. Hierarchical clustering of multi-dimensional features associated with pCR.
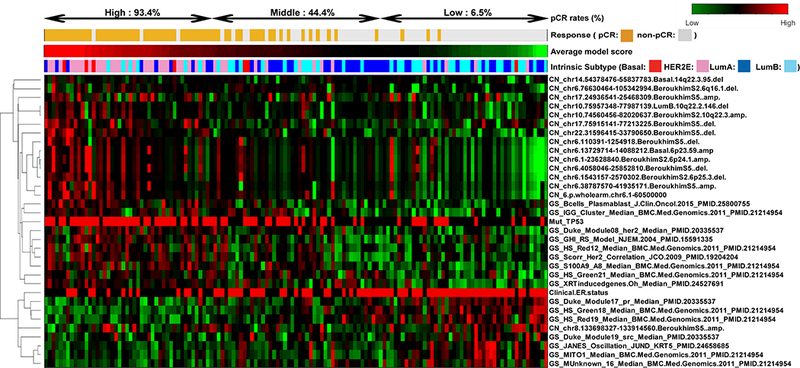
Supervised clustering of the 33 selected features among 137 samples. The features were grouped into two clusters with positive or negative predictors. The samples from left to right were ordered by their average scores derived from the 10 Elastic Net models grouped into high, middle, and low scores.
Survival analysis
The Elastic Net models using gene signatures with/without mutation (i.e. TP53) plus ER/PgR from CALGB 40601 (Table 1 and eTable 9) were applied onto 124 HER2-positive tumors from the METABRIC(38) dataset who did not receive any chemotherapy or trastuzumab, and onto 92 HER2-positive tumors from METABRIC(38) who received chemotherapy but no trastuzumab from the pre-trastuzumab era. Neither of these two models was prognostic (Supplementary Figure 5-A and 5-B) suggesting that these models largely reflect prediction of response to HER2-targeting.
DawnRank analysis
We next ran DawnRank(39), a computational method that uses RNA expression to populate known protein-protein networks, to identify those genetic alterations that alter these networks the most. Using the protein-protein networks comprising 8,248 genes and aggregating the individual patient results based upon those with a pCR and those without a pCR (eTable 10), we sought to identify the genetic drivers of response and resistance. HER2 and TP53, which are regarded as major genetic drivers in breast cancer, were ranked as No.1 or 2 in both pCR and non-pCR samples, supporting the robustness of the analysis, and the importance of TP53 regardless of treatment and response. Next, we extracted the top 1% of the genes from these rankings, then further ranked these genes according to the extent of rank change between pCR and non-pCR samples in order to identify those genes that qualify as drivers differentially present in either responsive (pCR) or resistant (non-pCR) tumors (eTable 11). Among pCR samples, amplified Chr.6p genes were highly ranked compared with non-pCR samples, while deleted Chr.11q genes were highly ranked in non-pCR samples compared with pCR samples.
Identification of gene-level CNAs as candidate biomarkers of trastuzumab resistance/sensitivity
By comparing the DNA copy number landscape plots of pCR vs non-pCR samples (Figure 5-A), we found that gain of Chr.6p12–21 were more frequent in pCR samples, while loss of Chr.22q11–13 were more frequent in non-pCR samples. We next performed computational analysis to find common drivers between 1) copy number altered genes in segments contributing to ≥6 out of the 10 Elastic Net models, 2) the top 1% copy number-altered genes from the DawnRank, and 3) copy number-altered genes with FDR ≤ 1% from SAM analysis (eTable 4). Only MAPK14 and CDKN1A at Chr.6p were identified in all three analyses (Figure 5-B, overlapped genes are listed in eTable 12). This small amount of overlap between the Dawnrank results and other analyses may be because Dawnrank is limited to the 8000 genes in the protein-protein interaction network, or other unknown reasons. Further only MAPK14 had a Pearson correlation ≥0.3 between RNA gene expression and DNA copy number values (MAPK14, 0.38 and CDKN1A, −0.08). Therefore, amplification of wild type MAKP14 (also known as p38), may play a direct role in sensitivity to trastuzumab/paclitaxel-based regimens, but experimental validation is needed.
Figure 5. Identification of DNA copy number alterations as biomarkers of trastuzumab-paclitaxel resistance and sensitivity.
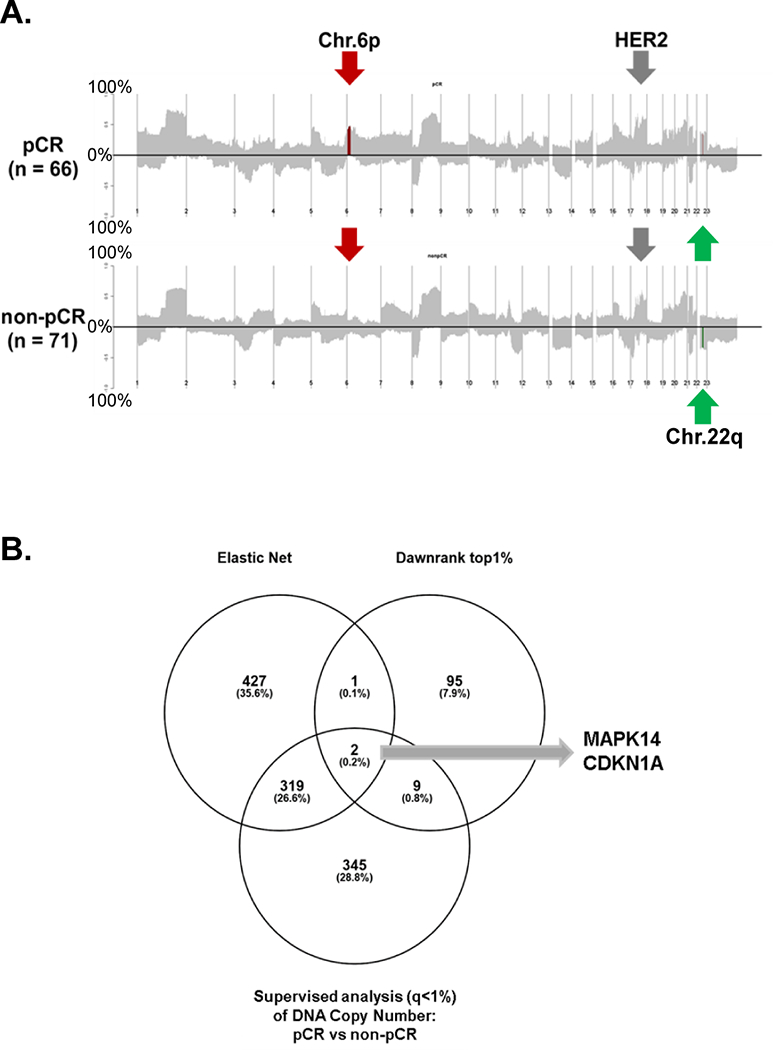
(A) DNA copy number frequency landscape plots for pCR vs non-pCR tumors. The frequency of alterations in each group is indicated on the y-axis from 0 to 100 %. Segments of group-specific copy number gains or loss are plotted above or below the x-axis, respectively. Significantly different regions between pCR vs. non-pCR (t-test p<0.05 after Benjamini and Hochberg correction) are highlighted in red (gain) or in green (loss). (B) A Venn diagram comparing three types of gene-level copy number results. Genes in copy number segments contributing to ≥6 models out of the 10 Elastic Net testing, top 1% copy number genes from the Dawnrank analysis, and copy number genes with false discovery rate ≤ 1% from SAM analysis were plotted and identify MAPK14 and CDKN1A as possible driver genes for trastuzumab-paclitaxel sensitivity.
DISCUSSION
To our knowledge, this study represents one of the first multi-dimensional genomic analyses to integrate DNA mutations, DNA copy number aberrations, and RNA transcriptional expression with clinical variables using prospectively collected frozen tissue samples from a Phase III trial to predict the primary endpoint of the parent trial, pCR. The importance of this approach was suggested at the time we published the primary multivariable analysis of CALGB 40601, in which we found that treatment arm was associated with pCR, but also that gene signatures representing tumor and microenvironmental influences, such as intrinsic subtype and signatures representing activated B-cells, each independently and significantly contributed to pCR, regardless of treatment arm; similar results were found in NeoALTTO, but integrated models were not developed6,7. We found that the most highly correlated negative predictive variables included signatures of the luminal subtypes(7,48), which have been consistently reported as negative pCR predictors and were again in this multi-signature model, while the HER2-enriched subtype(7,48) and activated immune signatures(7) were positive predictors here and in other neoadjuvant trials. These commonalities across studies support the robustness of our results. The predictive model based on gene signatures alone achieved good AUC values of 0.76 (0.71 – 0.83) in the four validation datasets. In actual performance, the low model score tumors had a pCR rate of 13%, and given the association of residual disease with poor outcome, these are tumors that may need additional therapies to achieve higher pCR rates and better outcomes. Conversely, the group with high model scores showed a high response rate of 65%, suggesting that most of these tumors may be receiving adequate treatment with trastuzumab and paclitaxel (Figure 2). Because all 137 patients used for this analysis received at least paclitaxel plus trasuzumab, it is difficult to separate our predictive features for responsiveness to either paclitaxel or trastuzumab. However, this combination is part of standard neoadjuvant chemotherapy regimens for stage II – III HER2-positive breast cancer and is an accepted and low toxicity regimen known to provide distant-disease free survival in excess of 98% for stage I HER2-positive breast cancer(49). It is worth noting that in the one prognostic dataset available, the Elastic Net models were not prognostic in either HER2-positive patients who did not receive chemotherapy nor those who received chemotherapy without trastuzumab. These results suggest that our Elastic Net predictors are not prognostic but truly predictive of drug response. An algorithm integrating relevant genomic predictors, with clinical features, may therefore allow us to safely de-escalate therapy in appropriate patients just as we do with hormone receptor-positive, HER2-negative patients by using commercial genomic assays.
Much of precision medicine is founded upon linking somatic mutations to targeted treatments, however, we found only 8 genes mutated in ≥10/137 patients; we did find two high VAF HER2 mutants, where one achieved a pCR and the other did not, thus foreshadowing the complexities of predicting response in single target based “basket studies”(50). These overall mutation results are comparable to those from a similar neoadjuvant study(14) in which only PIK3CA mutation was associated with lower pCR rates. In our study, only TP53 gene mutation was associated with higher pCR rates with support from two p53 mutation signatures also selected in the Elastic Net approach. Additional encouraging data arose from copy number evaluations; both the Elastic Net and DawnRank analyses made use of the multi-dimensional genomic data and found gain of Chr.6p as a key determinant of sensitivity to trastuzumab-based regimens. Further analysis on the gene-level copy number basis identified amplification of MAPK14 at Chr.6p as being linked to a high likelihood of pCR (Figure 5). The p38 MAPK pathway is activated upon cellular stress and engages pathways that can promote apoptosis(51). Activation of p38 MAPK pathway impaired mammary carcinogenesis in a HER2-positive mouse model(52). Therefore, we hypothesized that trastuzumab-paclitaxel regimens cause stress and that MAPK14 amplification may lead cancer cells to undergo apoptosis and is a potential response biomarker for trastuzumab-paclitaxel containing regimens. We also take note of human leucocyte antigen (HLA) genes because Chr.6p contains all the HLA genes and both the Elastic Net and SAM analysis contained 11 HLA genes (Supplementary eTable 12). Thus, amplification of HLA genes may be involved in the immune response.
The strengths of our analyses were that these studies were performed on prospectively collected frozen tissue samples from a randomized Phase III trial with pCR as the primary endpoint. The Elastic Net gene signature-only model was tested on four different independent validation datasets, and the model predicted pCR with good accuracy on all four. Two types of integrated genomic analyses, Elastic Net and DawnRank, were performed to make use of the multidimensional genomic data, with similar results obtained from each. The weaknesses of our approach were that we lacked an independent validation set for the integrated RNA and DNA Elastic Net predictor, although we did test our methods using 10 rounds of Monte-Carlo training and testing within CALGB 40601 data, and we report these values, that our sample size was relatively small, and that we cannot address the holy grail of anti-HER2 regimens without chemotherapy (although the absence of correlation with outcome in chemotherapy-only treated independent datasets suggests that our findings reflect the HER2-targeted element); we would need to test these models in all-biologic HER2-based regimens.
Collectively, tumor genetics (mutations, CNAs), tumor mRNA subtype (HER2-enriched, Luminal), and the microenvironment (Bcell features) were independently predictive of response to trastuzumab-paclitaxel containing therapies for HER2-overexpressing breast cancer. Elastic Net analysis represents a promising means of developing predictors of pCR for clinical application in part due to its objective ability to select from amongst multiple data types. Additional studies are needed to fully evaluate these multi-platform predictors, but it is clear that integrating all the relevant data types together can improve our predictive abilities and may contribute to rational tailoring approaches for the treatment of HER2-positive breast cancers.
Supplementary Material
Translational relevance.
Response to the increasingly complex trastuzumab-based regimens used in women with HER2-positive breast cancer is affected by multiple clinical and genomic features including immunohistochemical ER positivity, immune cell signatures, and molecular intrinsic subtype. Developing an integrated prediction model of pathologic complete response (pCR) using multi-dimensional genomic data could be key to optimizing HER2-targeting therapy. Here, we applied 517 known gene expression signatures to develop an Elastic Net model with high predictive capability for pCR, which we validated in 4 independent clinical trials. The model included HER2-enriched subtype, immune cell and Luminal/ER features. We further performed integrated analyses incorporating clinicopathologic information with somatic mutation status, DNA copy number alterations (CNAs), and found similar expression features and DNA amplifications of chromosome 6p as a strong predictors of pCR. This highlights the complexity of predicting response and suggests that optimal models to predict response many require multiple data types in addition to the standard clinical features.
ACKNOWLEDGEMENTS
We acknowledge the efforts of the data production and sample intake groups at the McDonnell Genome Institute in producing the exome sequencing data. Analyses were performed using a new Alliance “Data Mart” concept, and the research biopsies were made possible by generous support from The Breast Cancer Research Foundation. We thank our patients, the Alliance Breast Committee, and the Alliance Translational Research Program.
Funding/Support: This work was supported by funds from U10CA180888, U10CA180818 and U10CA180801, the Breast Cancer Research Foundation, the NCI Breast SPORE program (P50-CA58223–09A1), RO1-CA195740, the NIH/NCI Cancer Center Support Grant P30 CA008748-Memorial Sloan Ketterng Cancer, and by the Susan G. Komen Foundation. T. Hyslop and B. Pitcher were supported by the Alliance Statistics and Data Center (U10CA180882–04). M. Tanioka was supported by a Postdoctoral Fellowship Grant from the Susan G. Komen Foundation.
Research support: Lynn N Henry, Abbvie, Pfizer, Innocrin Pharmaceuticals; Sara Tolaney, Genentech, Eisai, Novartis, Pfizer, Merck, BMS, Lilly, Exelixis; Ian E Krop, Genentech/Roche; Chau Dang, Genentech/Roche, PUMA; Lisa A Carey, GlaxoSmithKline, Genentech/Roche,
Footnotes
Trial registrations: clinicaltrials.gov identifier NCT00770809
Conflict of interest
The following authors or their immediate family members indicated a financial interest.
Ownership: Donald A Berry, Berry Consultants LLC; Charles M Perou, Bioclassifier, GeneCentric Diagnostics;
Income: Donald A Berry, Berry Consultants LLC; Charles M Perou, royalties from PAM50 breast cancer gene patent application; Terry Hyslop, Abbie;
Intellectual Property : Charles M Perou and Joel S Parker, PAM50 breast cancer gene patent(s)
The other authors have no conflict of interest.
Presented in part at the 39th San Antonio Breast Cancer Symposium, San Antonio, Dec. 6–10, 2016
REFERENCES
- 1.Perez EA, Romond EH, Suman VJ, Jeong JH, Sledge G, Geyer CE Jr., et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol 2014;32(33):3744–52 doi 10.1200/JCO.2014.55.5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martine J Piccart-Gebhart APH, Jose Baselga, Evandro De Azambuja, Amylou C. Dueck, Giuseppe Viale, et al. First results from the phase III ALTTO trial (BIG 2–06; NCCTG [Alliance] N063D) comparing one year of anti-HER2 therapy with lapatinib alone (L), trastuzumab alone (T), their sequence (T→L), or their combination (T+L) in the adjuvant treatment of HER2-positive early breast cancer (EBC). J Clin Oncol 32:5s, 2014. (suppl; abstr LBA4) 2014. [Google Scholar]
- 3.von Minckwitz G, Procter M, de Azambuja E, Zardavas D, Benyunes M, Viale G, et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N Engl J Med 2017. doi 10.1056/NEJMoa1703643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan A, Delaloge S, Holmes FA, Moy B, Iwata H, Harvey VJ, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2016;17(3):367–77 doi 10.1016/S1470-2045(15)00551-3. [DOI] [PubMed] [Google Scholar]
- 5.Cardoso F, van’t Veer LJ, Bogaerts J, Slaets L, Viale G, Delaloge S, et al. 70-Gene Signature as an Aid to Treatment Decisions in Early-Stage Breast Cancer. N Engl J Med 2016;375(8):717–29 doi 10.1056/NEJMoa1602253. [DOI] [PubMed] [Google Scholar]
- 6.Carey LA BD, Cirrincione CT, Barry WT, Pitcher BN, Harris LN, Ollila DW, Krop IE, Henry NL, Weckstein D, Anders CK, Singh B, Hoadley KA, Iglesia M, Cheang MCU, Mardis E, Perou CM, Winer EP, Hudis CA Molecular heterogeneity and response to neoadjuvant HER2-targeting in CALGB 40601, a randomized phase III trial of paclitaxel plus trastuzumab with or without lapatinib. J Clin Oncol 2016;34(6):542–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fumagalli D, Venet D, Ignatiadis M, Azim HA, Maetens M Jr., Rothe F, et al. RNA Sequencing to Predict Response to Neoadjuvant Anti-HER2 Therapy: A Secondary Analysis of the NeoALTTO Randomized Clinical Trial. JAMA Oncol 2016. doi 10.1001/jamaoncol.2016.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guarneri V, Dieci MV, Frassoldati A, Maiorana A, Ficarra G, Bettelli S, et al. Prospective Biomarker Analysis of the Randomized CHER-LOB Study Evaluating the Dual Anti-HER2 Treatment With Trastuzumab and Lapatinib Plus Chemotherapy as Neoadjuvant Therapy for HER2-Positive Breast Cancer. Oncologist 2015;20(9):1001–10 doi 10.1634/theoncologist.2015-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Majewski IJ, Nuciforo P, Mittempergher L, Bosma AJ, Eidtmann H, Holmes E, et al. PIK3CA mutations are associated with decreased benefit to neoadjuvant human epidermal growth factor receptor 2-targeted therapies in breast cancer. J Clin Oncol 2015;33(12):1334–9 doi 10.1200/JCO.2014.55.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nuciforo PG, Aura C, Holmes E, Prudkin L, Jimenez J, Martinez P, et al. Benefit to neoadjuvant anti-human epidermal growth factor receptor 2 (HER2)-targeted therapies in HER2-positive primary breast cancer is independent of phosphatase and tensin homolog deleted from chromosome 10 (PTEN) status. Ann Oncol 2015;26(7):1494–500 doi 10.1093/annonc/mdv175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stern HM, Gardner H, Burzykowski T, Elatre W, O’Brien C, Lackner MR, et al. PTEN Loss Is Associated with Worse Outcome in HER2-Amplified Breast Cancer Patients but Is Not Associated with Trastuzumab Resistance. Clin Cancer Res 2015;21(9):2065–74 doi 10.1158/1078-0432.CCR-14-2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darb-Esfahani S, Denkert C, Stenzinger A, Salat C, Sinn B, Schem C, et al. Role of TP53 mutations in triple negative and HER2-positive breast cancer treated with neoadjuvant anthracycline/taxane-based chemotherapy. Oncotarget 2016. doi 10.18632/oncotarget.11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fountzilas G, Giannoulatou E, Alexopoulou Z, Zagouri F, Timotheadou E, Papadopoulou K, et al. TP53 mutations and protein immunopositivity may predict for poor outcome but also for trastuzumab benefit in patients with early breast cancer treated in the adjuvant setting. Oncotarget 2016;7(22):32731–53 doi 10.18632/oncotarget.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi W, Jiang T, Nuciforo P, Hatzis C, Holmes E, Harbeck N, et al. Pathway level alterations rather than mutations in single genes predict response to HER2-targeted therapies in the neo-ALTTO trial. Ann Oncol 2017;28(1):128–35 doi 10.1093/annonc/mdw434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salgado R, Denkert C, Campbell C, Savas P, Nucifero P, Aura C, et al. Tumor-Infiltrating Lymphocytes and Associations With Pathological Complete Response and Event-Free Survival in HER2-Positive Early-Stage Breast Cancer Treated With Lapatinib and Trastuzumab: A Secondary Analysis of the NeoALTTO Trial. JAMA Oncol 2015;1(4):448–54 doi 10.1001/jamaoncol.2015.0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol 2015;33(9):983–91 doi 10.1200/JCO.2014.58.1967. [DOI] [PubMed] [Google Scholar]
- 17.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015;163(2):506–19 doi 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511(7511):543–50 doi 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cortazar P, Zhang L, Untch M, Mehta K, Costantino JP, Wolmark N, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 2014;384(9938):164–72 doi 10.1016/S0140-6736(13)62422-8. [DOI] [PubMed] [Google Scholar]
- 20.Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 2010;38(18):e178 doi 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323 doi 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan C, Prat A, Parker JS, Liu Y, Carey LA, Troester MA, et al. Building prognostic models for breast cancer patients using clinical variables and hundreds of gene expression signatures. BMC Med Genomics 2011;4:3 doi 10.1186/1755-8794-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mose LE, Wilkerson MD, Hayes DN, Perou CM, Parker JS. ABRA: improved coding indel detection via assembly-based realignment. Bioinformatics 2014;30(19):2813–5 doi 10.1093/bioinformatics/btu376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katherine A Hoadley WTB, Brandelyn N Pitcher, Joel S Parker, Matthew D Wilkerson, William Irvin Jr., Norah Lynn Henry SMT, Chau Dang, Ian E Krop, Donald A Berry, Elaine R Mardis, Charles M Perou, Eric P Winer,, Clifford A Hudis LAC. Mutational analysis of CALGB 40601 (Alliance), a neoadjuvant phase III trial of weekly paclitaxel (T) and trastuzumab (H) with or without lapatinib (L) for HER2-positive breast cancer. 2014; San Antonio: pS3–08. [Google Scholar]
- 26.Wilkerson MD, Cabanski CR, Sun W, Hoadley KA, Walter V, Mose LE, et al. Integrated RNA and DNA sequencing improves mutation detection in low purity tumors. Nucleic Acids Res 2014;42(13):e107 doi 10.1093/nar/gku489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silva GO, Siegel MB, Mose LE, Parker JS, Sun W, Perou CM, et al. SynthEx: a synthetic-normal-based DNA sequencing tool for copy number alteration detection and tumor heterogeneity profiling. Genome Biol 2017;18(1):66 doi 10.1186/s13059-017-1193-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26(6):841–2 doi 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silva GO, He X, Parker JS, Gatza ML, Carey LA, Hou JP, et al. Cross-species DNA copy number analyses identifies multiple 1q21-q23 subtype-specific driver genes for breast cancer. Breast Cancer Res Treat 2015;152(2):347–56 doi 10.1007/s10549-015-3476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010;463(7283):899–905 doi 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chao HH, He X, Parker JS, Zhao W, Perou CM. Micro-scale genomic DNA copy number aberrations as another means of mutagenesis in breast cancer. PLoS One 2012;7(12):e51719 doi 10.1371/journal.pone.0051719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou H, Hastie T Regularization and variable selection via the Elastic Net Journal of the Royal Statistical Society, Series B 2005:301–20. [Google Scholar]
- 33.Tibshirani R The lasso method for variable selection in the Cox model. Stat Med 1997;16(4):385–95. [DOI] [PubMed] [Google Scholar]
- 34.Qing-Song Xu Y-ZL. Monte Carlo cross validation. Chemometrics and Intelligent Laboratory Systems 2001;56(1):1–11. [Google Scholar]
- 35.Picard RC D Cross-Validation of Regression Models. Journal of the American Statistical Association 1984; 79(387):575–58. [Google Scholar]
- 36.Gluck S, Ross JS, Royce M, McKenna EF, Perou CM Jr., Avisar E, et al. TP53 genomics predict higher clinical and pathologic tumor response in operable early-stage breast cancer treated with docetaxel-capecitabine +/− trastuzumab. Breast Cancer Res Treat 2012;132(3):781–91 doi 10.1007/s10549-011-1412-7. [DOI] [PubMed] [Google Scholar]
- 37.Esserman LJ, Berry DA, Cheang MC, Yau C, Perou CM, Carey L, et al. Chemotherapy response and recurrence-free survival in neoadjuvant breast cancer depends on biomarker profiles: results from the I-SPY 1 TRIAL (CALGB 150007/150012; ACRIN 6657). Breast Cancer Res Treat 2012;132(3):1049–62 doi 10.1007/s10549-011-1895-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486(7403):346–52 doi 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hou JP, Ma J. DawnRank: discovering personalized driver genes in cancer. Genome Med 2014;6(7):56 doi 10.1186/s13073-014-0056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pihur V, Datta S, Datta S. Finding common genes in multiple cancer types through meta-analysis of microarray experiments: a rank aggregation approach. Genomics 2008;92(6):400–3 doi 10.1016/j.ygeno.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 2001;98(9):5116–21 doi 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 1998;95(25):14863–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saldanha AJ. Java Treeview--extensible visualization of microarray data. Bioinformatics 2004;20(17):3246–8 doi 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 44.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011;12:77 doi 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov 2013;3(2):224–37 doi 10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351(27):2817–26 doi 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 47.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27(8):1160–7 doi 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prat A, Bianchini G, Thomas M, Belousov A, Cheang MC, Koehler A, et al. Research-based PAM50 subtype predictor identifies higher responses and improved survival outcomes in HER2-positive breast cancer in the NOAH study. Clin Cancer Res 2014;20(2):511–21 doi 10.1158/1078-0432.CCR-13-0239. [DOI] [PubMed] [Google Scholar]
- 49.Tolaney SM, Barry WT, Dang CT, Yardley DA, Moy B, Marcom PK, et al. Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast cancer. N Engl J Med 2015;372(2):134–41 doi 10.1056/NEJMoa1406281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018;554(7691):189–94 doi 10.1038/nature25475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 2007;11(2):191–205 doi 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 52.Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CW, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet 2004;36(4):343–50 doi 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 53.Pogue-Geile KL, Song N, Jeong JH, Gavin PG, Kim SR, Blackmon NL, et al. Intrinsic subtypes, PIK3CA mutation, and the degree of benefit from adjuvant trastuzumab in the NSABP B-31 trial. J Clin Oncol 2015;33(12):1340–7 doi 10.1200/JCO.2014.56.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.