

Summary
The role of maternal and embryonic leucine zipper kinase (MELK) in cancer cell proliferation has been contentious, with recent studies arriving at disparate conclusions. We investigated the in vitro dependency of cancer cells on MELK under a range of assay conditions. Abrogation of MELK expression has little effect under common culture conditions, in which cells are seeded at high densities and reach confluence in 3–5 days. However, MELK dependency becomes clearly apparent in clonogenic growth assays using either RNAi or CRISPR technologies to modulate MELK expression. This dependency is in sharp contrast to that of essential genes, such as those encoding classic mitotic kinases, but is similar to that of other oncogenes including MYC and KRAS. Our study provides an example demonstrating some of the challenges encountered in cancer target validation, and reveals how subtle, but important, technical variations can ultimately lead to divergent outcomes and conclusions.
Subject Areas: Techniques in Genetics, Technical Aspects of Cell Biology, Cancer
Graphical Abstract
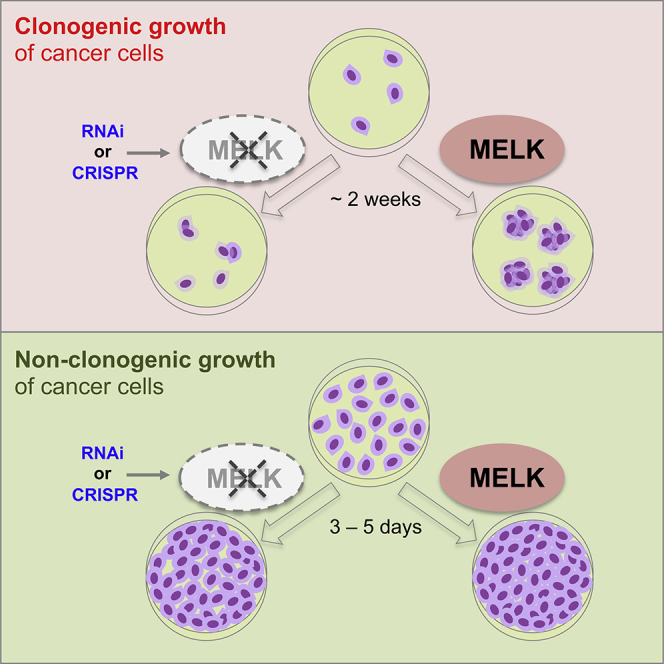
Highlights
-
•
Inhibiting MELK expression compromises clonogenic growth of cancer cells
-
•
MELK depletion minimally affects non-clonogenic cell growth
-
•
MELK depletion by RNAi or CRISPR has similar effects on cell growth
-
•
Cancer cell dependency on MELK is similar to that on classic oncogenes
Techniques in Genetics; Technical Aspects of Cell Biology; Cancer
Introduction
Maternal and embryonic leucine zipper kinase (MELK) is overexpressed in multiple cancers, including breast cancer (Gray et al., 2005, Wang et al., 2014), and is frequently included in gene sets used to predict the risk of breast tumor recurrence (Parker et al., 2009, Van De Vijver et al., 2002, Van't Veer et al., 2002). Notably, in breast tumors, MELK is most highly overexpressed in triple-negative breast cancer (TNBC) (Wang et al., 2014), a subgroup that lacks expression of hormone receptors or human epidermal growth factor receptor 2 (HER2) and is extremely aggressive with few effective therapies.
To investigate the functional role of MELK, we and others previously used an inducible short hairpin gene knockdown approach and found that MELK knockdown suppresses TNBC cell growth (Wang et al., 2014, Touré et al., 2016). In addition, hormone receptor-positive breast cancer cells and untransformed cells are insensitive to MELK depletion, indicating a selective dependency of TNBC cells on MELK (Wang et al., 2014, Touré et al., 2016). Moreover, recent studies using RNA interference (RNAi) techniques have found MELK to be a promising target in other cancer types, including melanoma (Janostiak et al., 2017), prostate cancer (Jurmeister et al., 2018), high-risk neuroblastoma (Guan et al., 2018), adrenocortical carcinoma (Kiseljak-Vassiliades et al., 2018), chronic lymphocytic leukemia (Zhang et al., 2018), and diffuse intrinsic pontine glioma (Meel et al., 2018).
Despite these studies suggesting an important role for MELK in cancer, the validity of MELK as a cancer target was recently challenged. Gene editing of MELK, mediated by transducing Cas9-expressing cells with guide RNA-encoding lentivirus, was found to have little effect on cancer cell proliferation (Lin et al., 2017). Specifically, lentiviral transduction of MELK-targeting guides abrogated MELK protein expression in TNBC lines (CAL-51, MDA-MB-231) and a melanoma cell line (A375), but did not alter cancer cell growth in any of these cases (Lin et al., 2017). In addition, the authors noted that multiple high-throughput genetic screens had not identified MELK as a potential cancer target (Hart et al., 2015, Marcotte et al., 2012, Marcotte et al., 2016). The authors further derived MELK knockout clones of cancer cells and demonstrated that these clonal cells grew normally both in cell culture and in xenograft mouse models (Giuliano et al., 2018, Lin et al., 2017).
Consistent with these findings, our laboratories and those of our collaborators also generated clones featuring CRISPR-mediated MELK knockout in MDA-MB-468 cells, a TNBC line that is highly sensitive to inducible short hairpin RNA (shRNA)-mediated knockdown of MELK (Touré et al., 2016, Wang et al., 2014). Intriguingly, the MELK CRISPR knockout clones have similar response as the parental cells to five MELK-targeting shRNAs (Huang et al., 2017), raising a number of possibilities including (1) growth inhibition caused by multiple independent shMELK is due to off-target effects or (2) certain functional, shMELK-sensitive MELK isoforms may be present in the knockout cells but remain unaffected by CRISPR reagents. Notably, the MDA-MB-468 cell line harbors gene amplification of MELK and a splice mutation in the MELK gene (Figure S1; Barretina et al., 2012), bringing further challenges in data interpretation.
These conflicting observations not only initiated a debate into whether MELK is indeed a cancer target, but also more generally contributed to broader questions on how best to identify and validate cancer targets. In this instance, one possibility is that these particular disparities stem from differences in RNAi and CRISPR-Cas9 technologies. However, these studies utilized shRNA and single guide RNA (sgRNA) of multiple sequences, and the results have been independently reproduced. Moreover, a comparison between RNAi and CRISPR screens in cancer cell lines reveals consistency regarding cancer gene dependencies (McDonald et al., 2017). Therefore, we hypothesize that these disparities may originate from fundamental differences in the target validation assays, rather than the choice of genetic tools.
We examined in depth the experimental procedures of the studies concerned (Wang et al., 2014, Touré et al., 2016, Hart et al., 2015: Lin et al., 2017, Marcotte et al., 2012, Marcotte et al., 2016) and noted that among other technical variations there are considerable differences in assay formats between studies that identified MELK as a target and those that showed the contrary or did not find MELK as potential target. Specifically, the former studies (Wang et al., 2014, Touré et al., 2016) tested MELK dependency using clonogenic growth assays, which examined the ability of single cells to proliferate into colonies (Franken et al., 2006, Puck and Marcus, 1956), whereas the latter studies (Hart et al., 2015, Lin et al., 2017, Marcotte et al., 2012, Marcotte et al., 2016) primarily employed common cell growth assays, wherein cells are seeded at relatively high density to allow them to reach confluence within 3–5 days for analysis or are sub-passaged if necessary. In this study, we demonstrate that this subtle technical variation leads to dramatic differences in experimental outcomes—that is, MELK is required for clonogenic growth of TNBC cells regardless of how its expression is abrogated, but the effects of its abrogation are largely negligible under the common “short-term, high-density” culture conditions.
Results
RNAi Study Reveals MELK Dependence for Clonogenic Cell Growth
We evaluated whether the outcome of proliferation assays testing MELK dependence is influenced by different assay formats, using inducible RNAi technology to modulate MELK expression. We confirmed that efficient MELK knockdown occurred as early as 1 day after doxycycline induction (Figure 1A), supporting the suitability of this approach for studying functional effects in short-term assays. In addition, MELK knockdown efficiency is comparable between triple-negative (BT549) and estrogen receptor-positive (ER+, MCF7) cells (Figure 1B). In both cell lines, MELK knockdown did not cause any significant effects on cell growth when cells were seeded at a density such that they reached confluence in 3–5 days (Figure 1C). In the TNBC cell line, however, a growth inhibitory effect of shMELK appeared with decreasing plating density and became clearly apparent under long-term (11-day) clonogenic conditions (Figure 1C, left column). In contrast, ER+ breast cancer cells were substantially more resistant to MELK knockdown, even under long-term clonogenic conditions (Figure 1C, right column). These findings demonstrate that determination of MELK requirement for proliferation is strongly dependent on the assay conditions.
Figure 1.

Doxycycline-Inducible Knockdown of MELK Impairs the Clonogenic Growth of TNBC Cells
(A) Fluorescent western blot analysis of MELK in BT549 cells that were stably transduced with tet-on-shMELK1 (Wang et al., 2014). Cells were either left untreated or treated with doxycycline (100 ng/mL) for the indicated time periods. The images were acquired via Odyssey CLx Infrared Imaging System (LI-COR Biosciences). Note that the first lane (M) was loaded with PageRuler Plus Prestained Protein Ladder (Thermo Fisher Scientific, cat# 26619), which have near-infrared fluorescence (molecular weight of the markers indicated).
(B) Fluorescent western blot analysis of MELK in BT549 and MCF7 cells that were stably transduced with tet-on-shMELK1. Cells were either left untreated or treated with doxycycline (100 ng/mL) for 2 days before lysate preparation and immunoblotting.
(C) Cells were seeded in 12-well plates at the indicated densities and were either left untreated or treated with doxycycline (100 ng/mL). Upon harvest, cells were fixed and stained with crystal violet. The staining was then extracted for quantification of cell growth (mean ± SD; n = 3).
Identifying MELK Guide Sequences for Efficient Gene Editing
We next attempted to confirm these RNAi-based findings using CRISPR/Cas9-mediated gene editing approaches, to rule out any potential artifacts conferred by RNAi technology. A lack of understanding of the biology of MELK isoforms (UniProt Consortium, 2016; Figure 2A) brings challenges to the rational design of guide sequences. However, the kinase domain of MELK constitutes the prioritized target, given its location at the amino terminus as well as its essential role for MELK functions. We evaluated the seven sgRNA sequences used in the previous study by Lin et al., five of which target early exons encoding MELK kinase domain and two of which target the kinase-associated domain 1 (KA1) residing in the carboxyl terminus (Lin et al., 2017).
Figure 2.

Identifying Guide Sequences for Efficient Gene Editing of MELK
(A) Schematic diagram of human MELK transcripts. The longest full-length one is isoform 1, shown at the bottom. The target locations of seven guide sequences (Lin et al., 2017), as well as the exon number of the MELK gene, are indicated.
(B) Fluorescent western blot analysis of MELK in MDA-MB-231 cells infected with control or MELK-targeting lentiCRISPR. Cells were harvested 7 days after infection. Total cell lysates were resolved on freshly cast 8% SDS-PAGE, transferred onto nitrocellulose membranes. Membranes were incubated with the indicated primary antibodies against MELK. The images were acquired using the Odyssey CLx Infrared Imaging System. Note that among the total seven guide sequences, sg_6 caused the most efficient reduction of MELK protein abundance.
We cloned these guide sequences into an all-in-one lentiCRISPR vector (Sanjana et al., 2014, Shalem et al., 2014) and generated lentiviral particles. Target cells were transduced twice with freshly prepared virus, and 7 days after initial transduction, they were tested for MELK expression by immunoblotting.
Notably, we found that different target sequences had distinctly varied effects on the protein abundance of MELK (Figure 2B), assayed by two antibodies raised against epitopes in the amino or carboxyl terminus of MELK. We also noted the same effects of guide sequences in another cancer cell line (Figure S2A). Among the seven guide sequences targeting MELK, sg_6 caused the greatest reduction in MELK expression (Figures 2B and S2A). Using sg_6, we further demonstrated that its effect on MELK protein abundance lasted until at least 11 days after transduction (Figure S2B), indicating the suitability of this tool for testing clonogenic cell growth.
CRISPR/Cas9-Mediated Gene Editing Reveals a Role of MELK for the Clonogenic Growth of Cancer Cells
After identifying a guide sequence (sgMELK_6) that can efficiently reduce MELK protein abundance, we proceeded with subsequent experiments to examine whether MELK depletion elicits any functional consequences.
We infected cells with virus encoding all-in-one lentiCRISPR (Figure 3A), confirmed efficient genomic editing (Figure 3B), and observed an apparent decrease of MELK protein level via the use of multiple independent anti-MELK antibodies (Figure 3C). Concurrently, cells were seeded at a relatively high density (50,000 cells per well, 12-well plates) for short-term cell proliferation assays and at a low density (500 cells per well) for clonogenic growth assays. Consistent with our RNAi-based findings, little growth reduction was observed in 3-day assays, whereas approximately 80% growth inhibition was observed under clonogenic growth conditions in 10-day colony formation assays (Figure 3D). Consistent with this observation, we also used the dual-vector lentiCRISPR platform (Lin et al., 2017) and found a similar inhibition on clonogenic cell growth by MELK gene editing (Wang et al., 2018).
Figure 3.

CRISPR/Cas9-Mediated Gene Editing of MELK in TNBC Cells
(A) Workflow of lentiCRISPR-mediated gene editing in cancer cells. Asterisk denotes that following the treatment of puromycin (1.5 μg/mL; 48 hr), all uninfected cells died, whereas the viability or proliferation status of infected cells were unaffected by puromycin selection.
(B) Sanger sequence traces of PCR products that amplify exon 5 of MELK. The predicted cutting site (red triangle) and protospacer adjacent motif (PAM) are indicated. The target site of sgMELK_6 is highlighted yellow.
(C) Fluorescent western blot analysis of MELK in MDA-MB-231 cells infected with control or MELK-targeting lentiCRISPR (harvested 11 days after infection). Information about MELK antibodies (vendor, catalogue number, and position of the used peptide immunogen) is indicated.
(D) lentiCRISPR-mediated gene editing of MELK suppresses the clonogenic growth of MDA-MB-231 cells. Four days after initial virus infection, cells were harvested and seeded in 12-well plates. Cell proliferation was measured by Celigo Imaging Cytometer (Nexcelom Bioscience) on indicated days post plating (mean ± SD; n = 3). The whole-well images filled in with a green color indicate cell confluence.
Based on these findings, we reasoned that cells introduced with MELK guide would likely “drop out” during culture under the same conditions. The lentiGuide vector (LRG2.1) encodes GFP (Lin et al., 2017, Tarumoto et al., 2018), providing an excellent tool for measuring the depletion or enrichment of sub-population of cells. Despite a lack of antibiotic-resistant gene on LRG2.1 for selection, we were able to achieve an efficient decrease of MELK protein abundance in cells infected with MELK guide, as evidenced by fluorescent immunoblotting (Figure 4B). After Cas9-expressing MDA-MB-231 cells were introduced with lentiGuide, the cells were mixed with parental cells (GFP negative) and seeded for clonogenic assays (Figure 4A). Using this protocol, we found that cells expressing MELK guide were depleted 3-fold more than cells transduced with lentivirus encoding control sgRNA (Figure 4C), indicating a compromised fitness of MELK-edited cells under the clonogenic culture conditions.
Figure 4.

Depletion of Cancer Cells following CRIPSR/Cas9-Mediated Gene Editing of MELK
(A) Workflow of the experimental procedures. Note that the cells (MD-MB-231) express Cas9 and that the lentiGuide vector encodes GFP.
(B) Fluorescent western blot analysis of MELK in Cas9-expressing MDA-MB-231 cells infected with control or MELK-targeting lentiGuide. Cells were harvested 4 days after the initial virus infection. Information of MELK antibodies is indicated.
(C) Cas9-expressing MDA-MB-231 cells were infected with GFP-encoding lentiGUIDE that targets ROSA or MELK. Four days after the first (of total two) infection, cells were harvested and mixed at 1:1 ratio with uninfected cells (MDA-MB-231-Cas9). Part of the cells was fixed for flow cytometric analysis (top panel). The remaining cells were seeded in 6-well plates (1,000 cells per well) and harvested in 11 days for measuring the percentage of GFP+ cells (bottom panel). The quantification of fold depletion from six duplicates is shown in the histogram (right).
CRISPR/Cas9-Mediated Editing of Essential Genes and Oncogenes
We next compared the effects of MELK gene editing with those of essential genes, particularly classic mitotic kinases that more often scored in genome-wide functional screens (Hart et al., 2015, Marcotte et al., 2012, Marcotte et al., 2016). CRISPR-Cas9-mediated editing of PLK1 or AURKB yielded modest loss of protein expression (Figures 5A, S3A, and S3B) but had dramatic suppression of cell growth under both regular and clonogenic culture conditions (Figures 5B and S3C). These observations suggest that reliance of cells on expression of essential genes is evident independent of assay conditions.
Figure 5.

CRISPR/Cas9-Mediated Gene Editing of Potential Cancer Targets in TNBC Cells
(A) Fluorescent western blot analysis of PLK1 in MDA-MB-231 cells infected with control or all-in-one lentiCRISPR targeting PLK1. Note that cells were harvested 5 days after the initial infection.
(B) lentiCRISPR-mediated gene editing of PLK1 suppresses the growth of cancer cells, largely independent of assay formats. Four days after initial virus infection, cells were harvested and seeded in 24-well plates. Cell proliferation was measured by Celigo Imaging Cytometer (Nexcelom Bioscience) on indicated days post plating (mean ± SD; n = 4). The whole-well images filled in with a green color indicate cell confluence.
(C) Fluorescent western blot analysis of lysates that were harvested from MDA-MB-231 cells infected with all-in-one lentiCRISPR vectors targeting control sequence (sgCon) or those of MELK, KRAS, or MYC. Cells were lysed 4 days after initial infection.
(D) Cells were harvested 3 days after initial all-in-one lentiCRISPR infection and seeded in 24-well plates at the indicated densities. Cell proliferation was measured by cell confluence.
We further asked whether the requirement of MELK for clonogenic growth is a unique feature of this gene, or whether it is a characteristic shared with other (proto-)oncogenes. To this end, we performed a side-by-side comparison of MELK gene editing with that of KRAS and MYC in MDA-MB-231 cells, a cell line that harbors KRAS-activating mutation G13D and is sensitive to the depletion of KRAS (Kopp et al., 2014; approximately 30% inhibition of cell growth 7 days after transfection of KRAS-targeting small interfering RNA) or MYC (Kessler et al., 2012; approximately 80% inhibition of clonogenicity following viral transduction of shMYC). Similar to MELK, KRAS or MYC gene editing also caused cell growth inhibition that is largely dependent on the initial plating density (Figure 5C). Consistent with these genetic studies, a recent study found that spheroid assays have been found to be required for the majority of KRAS G12C cancer cell lines to show sensitivity to a covalent inhibitor of KRAS G12C, whereas under regular short-term monolayer culture conditions only a minority of G12C mutant lines are sensitive to the treatment (Janes et al., 2018).
Correlation between Gene Dependency and Self-Expression
Having made consistent observations between RNAi- and CRISPR/Cas9-mediated perturbation of MELK, we next analyzed existing large-scale datasets to identify any potential pattern of MELK dependency. We analyzed the gene dependency scores from Project Achilles and gene expression values from the Cancer Cell Line Encyclopedia among the hundreds of cancer cell lines that were scored in both datasets (Figure 6A) (Broad Institute; Barretina et al., 2012, Tsherniak et al., 2017). In Project Achilles, gene dependency scores were generated from a genome-scale RNAi screen that was performed across 501 cancer cell lines, using a computation model to distinguish between on- and off-target effects of RNAi (Tsherniak et al., 2017). Despite the practice of cell culture at a high seeding density, the long-term cell culture (at least 28 days or 16 population doublings) combined with the quantitative readout of the screen enables the comparison of gene dependence among different cancer cell lines.
Figure 6.

Correlation between MELK Expression and Cancer Cell Dependence on MELK
(A) Workflow of data analysis. Genome-wide RNAi screen was performed in 501 cancer cell lines, followed by DEMETER computational analysis for exclusion of off-target effects of RNAi (Tsherniak et al., 2017). The dependency score values were then subject to correlation analysis with gene expression, a database generated by Cancer Cell Line Encyclopedia (Barretina et al., 2012).
(B and C) Analysis of indicated genes for the correlation between cancer cell dependency scores and gene expression. The left plots demonstrate exmples where expression of a given gene is reversely (B) or positively (C) correlated with the scores of cancer cell dependence on the gene (each dot represents one cancer cell line). The right tables include correlation values for the indicated genes. Pearson correlation coefficient (r) is shown, with p values denoting the statistical significance test for Pearson correlation (GraphPad Prism 7). Note that greater depletion of cells expressing shRNAs targeting specific genes causes more negative dependency scores, indicating greater dependency of cancer cells on these genes.
Using the Cancer Cell Line Encyclopedia, we focused on its gene expression data since genetic mutations and gene amplifications of MELK are quite rare in cancer. Interestingly, by analyzing the data from the two data sets, we found a moderate, but statistically significant, negative correlation between MELK dependency scores and its gene expression (Figure 6B). This is indicative of cells with higher expression of MELK having greater dependence (and more negative dependency scores). We further used the same approach and analyzed well-established (proto-)oncogenes, including MYC, KRAS, ERBB2, EGFR, MCL, and β-catenin. Similar to MELK, the dependency scores of these oncogenic drivers were all negatively correlated with their expression levels (Figure 6B).
In contrast, the expression levels of genes encoding classic mitotic kinase (e.g., AURKB, CDK1, MPS1, BUB1, BUB1B) or components of essential cellular machinery (e.g. EIF3B), were positively correlated with dependency scores (Figure 6C). This is consistent with the notion of these serving as essential genes, whereby cell lines with low expression of such genes—due to either genomic or epigenetic alterations—are likely more sensitive to gene knockdown. Indeed, a previous study documented the CYCLOPS (copy number alterations yielding cancer liabilities owing to partial loss) phenomenon, and found that CYCLOPS genes are enriched for components of fundamental cellular processes such as RNA splicing and proteasome-mediated protein degradation (Nijhawan et al., 2012). Together, these analyses suggest that MELK behaves in a more similar manner to an oncogenic factor than an essential gene.
Discussion
Driven by the recently arising controversy on the validity of MELK as a cancer target, we utilized both inducible RNAi and lentiCRISPR tools for modulating the expression of MELK, and investigated the impact of gene expression perturbation under a range of assay conditions. Our study demonstrates that MELK is required for clonogenic proliferation of TNBC cells but that its abrogation has negligible effects under regular culture conditions that allow cells to reach confluency after short-term culture. This is in contrast to essential genes, such as classic mitotic kinases, whose necessity for cell proliferation is observable independent of such assay conditions. Notably, such a role of MELK for cancer cell growth was also observed for oncogenes including MYC and mutant KRAS. Furthermore, analysis of gene dependency across hundreds of cancer cell lines revealed that MELK dependency has a drastically different pattern from that of essential genes, but is similar to that of established oncogenic drivers.
Is MELK a Viable Cancer Target?
The functional dependence on MELK has been reported in models representing an array of tumor types (reviewed by Settleman et al., 2018), and more recently in a few others including prostate cancer (Jurmeister et al., 2018), adrenocortical carcinoma (Kiseljak-Vassiliades et al., 2018), chronic lymphocytic leukemia (Zhang et al., 2018), and diffuse intrinsic pontine glioma (Meel et al., 2018). These studies, including our earlier report of MELK as a target for TNBC (Wang et al., 2014), all used RNAi as the genetic approach. The conclusions of these studies were recently questioned because of the potential off-target effects of RNAi and upon the observations that, in other studies, CRISPR/Cas9-mediated gene editing had no appreciable impacts on tumor cell growth (Giuliano et al., 2018, Lin et al., 2017). We believe that our current findings underline the importance of assay design and technical considerations in cancer target validation efforts and begin to explain and reconcile some of the disparities in the current literature concerning the role of MELK in cancer cell proliferation.
Our study lends confidence toward continued investigation of MELK modulation as a potential approach for cancer therapy. The role of MELK for clonogenic growth of TNBC cells was evidenced by the use of three genetic tools: doxycycline-inducible shRNA, all-in-one lentiCRISPR, and dual-vector lentiCRISPR. We also found that both the functional reliance on MELK by our experimental cell models and MELK dependency across hundreds of cancer cell lines (Tsherniak et al., 2017) is surprisingly similar to established oncogenes, and in contrast to essential genes. These data support the notion of MELK dependence by specific types of tumor cells.
Nevertheless, a number of critical issues need to be addressed before anti-MELK strategies are considered for clinical investigation. For MELK-targeting approaches, their magnitude of efficacy in vivo remains a key question. Will perturbing MELK activity or expression effectively decrease tumor burden or improve response to existing therapies? An inherent demand of these studies is the availability of MELK-targeting methods with sufficient potency and selectivity. Directions for future investigation may include the construction of cell models with inducible gene editing of MELK and development of MELK inhibitors with desired potency and pharmacokinetic features.
Given the widespread utility of small molecules in cancer research and treatment, we summarize MELK-targeting compounds that were recently developed or identified from compound library screens (Table 1). Among these studies, one interesting strategy is to find MELK as an off-target of drugs that are either approved or in clinical development, and to leverage the information on scaffold and chemical groups for further design and optimization (Edupuganti et al., 2017, Klaeger et al., 2017).
Table 1.
MELK Inhibitors
| Compound | Biochemical IC50 (nM)a | Reference | Description |
|---|---|---|---|
| OTSSP167 | 0.41 | Chung et al., 2012 | Highly potent but unselective |
| 0.5 | Huang et al., 2017 | ||
| Klaeger et al., 2017 | |||
| NVS-MELK8a | 2 | Touré et al., 2016 | Highly selective; inhibiting TNBC cell growth |
| 11.9 | Huang et al., 2017 | ||
| 17 | 3 ± 0.8 | Edupuganti et al., 2017 | Inhibiting TNBC cell growth |
| HTH-01-091 | 10.5 | Huang et al., 2017 | Low potency in TNBC cells |
| PF-3758309 | ∼30 | Klaeger et al., 2017 | An inhibitor of PAK4 |
| Nintedanib | 43 | Klaeger et al., 2017 | A multi-kinase inhibitor approved for idiopathic pulmonary fibrosis |
| ∼100 | Edupuganti et al., 2017 | ||
| BI-847325 | ∼100 | Klaeger et al., 2017 | An MEK and aurora kinase inhibitor |
The biochemical assays vary in the use of different forms of MELK recombinant protein (such as full-length versus kinase domain only), substrates, and readouts.
RNAi versus CRISPR: Which Is the Right Choice?
Our study uses both RNAi and CRISPR approaches in examining MELK dependency. From this direct comparison, we hope to provide some insights into the choice of genetic tools for perturbing gene expression in cancer biology studies.
With regard to the efficiency of targeting gene expression, it is tempting to term RNAi as a “knockdown” and CRISPR as a “knockout” technique. Our study, however, fails to tell which tool excels, but does indicate that CRISPR is not equal to gene knockout, at least in the context of using non-clonally-derived, pooled populations of cells generated from lentiviral transduction of a single guide sequence and antibiotic selection. This is consistent with the occurrence of in-frame mutations during CRISPR/Cas9-mediated gene editing (Koike-Yusa et al., 2014). Another feature of CRISPR, similar to RNAi, is the unpredictability on gene editing effect. It is common to observe that some guides are completely ineffective in altering target protein abundance (Figures 2 and S3B). The observation might be explained by the possibility that certain loci remain inaccessible to the gene editing machinery. As such, our studies indicate that neither tool is able to entirely overcome the deficiencies of the other, but that the two tools—CRISPR and RNAi—are likely to be complementary, especially in the settings of studying gene function in pooled population of cells.
In summary, we provide evidence—based on both RNAi and CRISPR tools—that MELK is required for clonogenic cell growth. This feature, together with the observed pattern of MELK dependency among hundreds of cancer cell lines, points toward MELK as an oncogenic kinase. We expect the current study to contribute to a valuable, and necessary, discussion about how best to design target validation assays and evaluate the fitness of such assays for their designed purposes.
Limitations of the Study
The current study focuses on MELK in MDA-MB-231, a cell line that was used in both our previous RNAi-based study (Wang et al., 2014) and two recent ones that leveraged the tool of CRISPR/Cas9-mediated gene editing (Giuliano et al., 2018, Lin et al., 2017). Although we believe that the current study solves some of the discrepancies among these different observations, it does not explain how MELK knockdown still compromises cell growth in clonal MELK-null MDA-MB-468 cells (Huang et al., 2017). Although the phenotype was considered to evidence off-target effects of a total of five independent shMELKs, data interpretation can be challenged by the MELK gene amplification status in this cell line, a situation that tends to introduce difficulties in creating homozygous MELK-null clonal cells by CRIPSR technique. Nevertheless, we expect that if given sufficient time and selection pressure, MELK-resistant clones could be generated from parental cancer cells that have MELK dependence, similar to previous observations for Kras (Mou et al., 2017). It would be interesting to study factor(s) substituting for or forming synthetic lethal interactions with MELK.
Another limitation of the current study concerns the genetic tool used for MELK knockdown. The constitutive expression of both Cas9 and guide RNA in cells transduced with all-in-one lentiCRISPR limits the ability to examine MELK dependency in established tumors in vivo. Further study of MELK as a cancer target would necessitate the development of models whereby efficient MELK knockdown can be triggered by inducible expression of Cas9 and guide sequences.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Drs. Dirk Heckl (Hannover Medical School) and Haoquan Wu (Texas Tech University Health Sciences) for their advices on the use of CRISPR/Cas9-mediated gene editing technology. We appreciate the efforts from the groups of Drs. Feng Zhang (Broad Institute) and Chris Vakoc (Cold Spring Harbor Laboratory) in developing the tools of lentiCRISPR. We thank Dr. Jason Sheltzer (Cold Spring Harbor Laboratory) for the generous gifts of Cas9-expressing cancer cells and lentiGuide constructs and his efforts in initiating the series of scientific discussion on MELK. We are grateful to Drs. Nathanael Gray, Timothy Mitchison, Rosalind Segal, and Charles Stiles for constructive discussions. This study was supported by the Breast Cancer Research Foundation Award BCRF-17-176 (J.J.Z.), NIH grants R35 CA210057 (J.J.Z.), and R01 CA187918 (T.M.R. and J.J.Z.).
Author Contributions
Y.W., B.B.L., T.M.R., and J.J.Z. designed the experiments. Y.W. and J.L. conducted the experiments. Y.W., B.B.L., T.M.R., and J.J.Z. wrote the paper.
Declaration of Interests
Y.W. and J.J.Z. are inventors on patent applications WO2014110163, WO2015073509, WO2016141296, and WO2016141279. The other authors declare no completing interests.
Published: November 30, 2018
Footnotes
Supplemental Information includes Transparent Methods and three figures and can be found with this article online at https://doi.org/10.1016/j.isci.2018.10.015.
Contributor Information
Yubao Wang, Email: yubao_wang@dfci.harvard.edu.
Jean J. Zhao, Email: jean_zhao@dfci.harvard.edu.
Supplemental Information
References
- Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D., Reddy A. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S., Suzuki H., Miyamoto T., Takamatsu N., Tatsuguchi A., Ueda K., Kijima K., Nakamura Y., Matsuo Y. Development of an orally-administrative MELK-targeting inhibitor that suppresses the growth of various types of human cancer. Oncotarget. 2012;3:1629–1640. doi: 10.18632/oncotarget.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edupuganti R., Taliaferro J.M., Wang Q., Xie X., Cho E.J., Vidhu F., Ren P., Anslyn E.V., Bartholomeusz C., Dalby K.N. Discovery of a potent inhibitor of MELK that inhibits expression of the anti-apoptotic protein Mcl-1 and TNBC cell growth. Bioorg. Med. Chem. 2017;25:2609–2616. doi: 10.1016/j.bmc.2017.03.018. [DOI] [PubMed] [Google Scholar]
- Franken N.A., Rodermond H.M., Stap J., Haveman J., Van Bree C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- Giuliano C.J., Lin A., Smith J.C., Palladino A.C., Sheltzer J.M. MELK expression correlates with tumor mitotic activity but is not required for cancer growth. Elife. 2018;7:e32838. doi: 10.7554/eLife.32838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray D., Jubb A.M., Hogue D., Dowd P., Kljavin N., Yi S., Bai W., Frantz G., Zhang Z., Koeppen H., de Sauvage F.J. Maternal embryonic leucine zipper kinase/murine protein serine-threonine kinase 38 is a promising therapeutic target for multiple cancers. Cancer Res. 2005;65:9751–9761. doi: 10.1158/0008-5472.CAN-04-4531. [DOI] [PubMed] [Google Scholar]
- Guan S., Lu J., Zhao Y., Yu Y., Li H., Chen Z., Shi Z., Liang H., Wang M., Guo K., Chen X. MELK is a novel therapeutic target in high-risk neuroblastoma. Oncotarget. 2018;9:2591–2602. doi: 10.18632/oncotarget.23515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K.R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S., Mero P. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015;163:1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Huang H.T., Seo H.S., Zhang T., Wang Y., Jiang B., Li Q., Buckley D.L., Nabet B., Roberts J.M., Paulk J., Dastjerdi S. MELK is not necessary for the proliferation of basal-like breast cancer cells. Elife. 2017;6:e26693. doi: 10.7554/eLife.26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes M.R., Zhang J., Li L.S., Hansen R., Peters U., Guo X., Chen Y., Babbar A., Firdaus S.J., Darjania L., Feng J. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell. 2018;172:578–589. doi: 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Janostiak R., Rauniyar N., Lam T.T., Ou J., Zhu L.J., Green M.R., Wajapeyee N. MELK promotes melanoma growth by stimulating the NF-κB pathway. Cell Rep. 2017;21:2829–2841. doi: 10.1016/j.celrep.2017.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurmeister S., Ramos-Montoya A., Sandi C., Pértega-Gomes N., Wadhwa K., Lamb A.D., Dunning M.J., Attig J., Carroll J.S., Fryer L.G., Felisbino S.L. Identification of potential therapeutic targets in prostate cancer through a cross-species approach. EMBO Mol. Med. 2018;10:e8274. doi: 10.15252/emmm.201708274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler J.D., Kahle K.T., Sun T., Meerbrey K.L., Schlabach M.R., Schmitt E.M., Skinner S.O., Xu Q., Li M.Z., Hartman Z.C., Rao M. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012;335:348–353. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiseljak-Vassiliades K., Zhang Y., Kar A., Razzaghi R., Xu M., Gowan K., Raeburn C.D., Albuja-Cruz M., Jones K.L., Somerset H. Elucidating the role of the maternal embryonic leucine zipper kinase (MELK) in adrenocortical carcinoma. Endocrinology. 2018;159:2532–2544. doi: 10.1210/en.2018-00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaeger S., Heinzlmeir S., Wilhelm M., Polzer H., Vick B., Koenig P.A., Reinecke M., Ruprecht B., Petzoldt S., Meng C., Zecha J. The target landscape of clinical kinase drugs. Science. 2017;358:eaan4368. doi: 10.1126/science.aan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike-Yusa H., Li Y., Tan E.P., Velasco-Herrera M.D.C., Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014;32:267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- Kopp F., Wagner E., Roidl A. The proto-oncogene KRAS is targeted by miR-200c. Oncotarget. 2014;5:185–195. doi: 10.18632/oncotarget.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A., Giuliano C.J., Sayles N.M., Sheltzer J.M. CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials. Elife. 2017;6:e24179. doi: 10.7554/eLife.24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R., Brown K.R., Suarez F., Sayad A., Karamboulas K., Krzyzanowski P.M., Sircoulomb F., Medrano M., Fedyshyn Y., Koh J.L., van Dyk D. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012;2:172–189. doi: 10.1158/2159-8290.CD-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R., Sayad A., Brown K.R., Sanchez-Garcia F., Reimand J., Haider M., Virtanen C., Bradner J.E., Bader G.D., Mills G.B., Pe’er D. Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell. 2016;164:293–309. doi: 10.1016/j.cell.2015.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald E.R., de Weck A., Schlabach M.R., Billy E., Mavrakis K.J., Hoffman G.R., Belur D., Castelletti D., Frias E., Gampa K., Golji J. Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell. 2017;170:577–592.e10. doi: 10.1016/j.cell.2017.07.005. [DOI] [PubMed] [Google Scholar]
- Meel M.H., de Gooijer M.C., Navarro M.G., Waranecki P., Breur M., Buil L., Wedekind L.E., Twisk J.W., Koster J., Hashizume R., Raabe E.H. MELK inhibition in diffuse intrinsic pontine glioma. Clin. Cancer Res. 2018 doi: 10.1158/1078-0432.CCR-18-0924. [DOI] [PubMed] [Google Scholar]
- Mou H., Moore J., Malonia S.K., Li Y., Ozata D.M., Hough S., Song C.Q., Smith J.L., Fischer A., Weng Z., Green M.R. Genetic disruption of oncogenic Kras sensitizes lung cancer cells to Fas receptor-mediated apoptosis. Proc. Natl. Acad. Sci. U S A. 2017;114:3648–3653. doi: 10.1073/pnas.1620861114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhawan D., Zack T.I., Ren Y., Strickland M.R., Lamothe R., Schumacher S.E., Tsherniak A., Besche H.C., Rosenbluh J., Shehata S., Cowley G.S. Cancer vulnerabilities unveiled by genomic loss. Cell. 2012;150:842–854. doi: 10.1016/j.cell.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J.S., Mullins M., Cheang M.C., Leung S., Voduc D., Vickery T., Davies S., Fauron C., He X., Hu Z., Quackenbush J.F. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puck T.T., Marcus P.I. Action of x-rays on mammalian cells. J. Exp. Med. 1956;103:653–666. doi: 10.1084/jem.103.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settleman J., Sawyers C.L., Hunter T. Science Forum: challenges in validating candidate therapeutic targets in cancer. Elife. 2018;7:e32402. doi: 10.7554/eLife.32402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelsen T.S., Heckl D., Ebert B.L., Root D.E., Doench J.G., Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarumoto Y., Lu B., Somerville T.D., Huang Y.H., Milazzo J.P., Wu X.S., Klingbeil O., El Demerdash O., Shi J., Vakoc C.R. LKB1, salt-inducible kinases, and MEF2C are linked dependencies in acute myeloid leukemia. Mol. Cell. 2018;69:1017–1027. doi: 10.1016/j.molcel.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touré B.B., Giraldes J., Smith T., Sprague E.R., Wang Y., Mathieu S., Chen Z., Mishina Y., Feng Y., Yan-Neale Y., Shakya S. Toward the validation of maternal embryonic leucine zipper kinase: discovery, optimization of highly potent and selective inhibitors, and preliminary biology insight. J. Med. Chem. 2016;59:4711–4723. doi: 10.1021/acs.jmedchem.6b00052. [DOI] [PubMed] [Google Scholar]
- Tsherniak A., Vazquez F., Montgomery P.G., Weir B.A., Kryukov G., Cowley G.S., Gill S., Harrington W.F., Pantel S., Krill-Burger J.M., Meyers R.M. Defining a cancer dependency map. Cell. 2017;170:564–576. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2016;45:D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van De Vijver M.J., He Y.D., Van't Veer L.J., Dai H., Hart A.A., Voskuil D.W., Schreiber G.J., Peterse J.L., Roberts C., Marton M.J., Parrish M. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Van't Veer L.J., Dai H., Van De Vijver M.J., He Y.D., Hart A.A., Mao M., Peterse H.L., van der Kooy K., Marton M.J., Witteveen A.T., Schreiber G.J. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Wang Y., Lee Y.M., Baitsch L., Huang A., Xiang Y., Tong H., Lako A., Von T., Choi C., Lim E., Min J. MELK is an oncogenic kinase essential for mitotic progression in basal-like breast cancer cells. Elife. 2014;3:e01763. doi: 10.7554/eLife.01763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Li Y.M., Baitsch L., Huang A., Xiang Y., Tong H., Lako A., Von T., Choi C., Lim E., Min J. Correction: MELK is an oncogenic kinase essential for mitotic progression in basal-like breast cancer cells. Elife. 2018;7:e36414. doi: 10.7554/eLife.36414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Zhou X., Li Y., Xu Y., Lu K., Li P., Wang X. Inhibition of maternal embryonic leucine zipper kinase with OTSSP167 displays potent anti-leukemic effects in chronic lymphocytic leukemia. Oncogene. 2018 doi: 10.1038/s41388-018-0333-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.