

Abstract
Gain-of-function mutations in fibroblast growth factor receptors (FGFRs) cause congenital skeletal anomalies, including craniosynostosis (CS), which is characterized by the premature closure of craniofacial sutures. Apert syndrome (AS) is one of the severest forms of CS, and the only treatment is surgical expansion of prematurely fused sutures in infants. Previously, we demonstrated that the prolyl isomerase peptidyl-prolyl cis–trans isomerase interacting 1 (PIN1) plays a critical role in mediating FGFR signaling and that Pin1+/− mice exhibit delayed closure of cranial sutures. In this study, using both genetic and pharmacological approaches, we tested whether PIN1 modulation could be used as a therapeutic regimen against AS. In the genetic approach, we crossbred Fgfr2S252W/+, a mouse model of AS, and Pin1+/− mice. Downregulation of Pin1 gene dosage attenuated premature cranial suture closure and other phenotypes of AS in Fgfr2S252W/+ mutant mice. In the pharmacological approach, we intraperitoneally administered juglone, a PIN1 enzyme inhibitor, to pregnant Fgfr2S252W/+ mutant mice and found that this treatment successfully interrupted fetal development of AS phenotypes. Primary cultured osteoblasts from Fgfr2S252W/+ mutant mice expressed high levels of FGFR2 downstream target genes, but this phenotype was attenuated by PIN1 inhibition. Post-translational stabilization and activation of Runt-related transcription factor 2 (RUNX2) in Fgfr2S252W/+ osteoblasts were also attenuated by PIN1 inhibition. Based on these observations, we conclude that PIN1 enzyme activity is important for FGFR2-induced RUNX2 activation and craniofacial suture morphogenesis. Moreover, these findings highlight that juglone or other PIN1 inhibitors represent viable alternatives to surgical intervention for treatment of CS and other hyperostotic diseases.
Introduction
Craniosynostosis (CS), defined as premature fusion of one or more cranial sutures, occurs in approximately 1 in 2500 live births (1). Normally, the mammalian cranial vault consists of five major flat bones joined by structures known as cranial sutures (2), which play a key role in cranial growth during development (3). To permit brain growth in the early stage of life, the sutures should remain patent (1). However, in CS, premature fusion prevents skull expansion to accommodate brain growth, potentially resulting in elevated intracranial pressure and multiple sequelae, including brain dysfunction, hydrocephalus, reduced intelligence and visual impairment due to pressure on the optic nerve (1,4). Currently, the only treatments for these disorders involve post-natal surgery.
In a prospective analysis of a cohort presenting with CS, the genes most frequently mutated were FGFR2 (32%), FGFR3 (25%), TWIST1 (19%) and EFNB1 (7%) (1). Heterozygous mutations of FGFR2 cause three classical CS syndromes: Apert, Crouzon and Pfeiffer (5). Apert syndrome (AS), one of the severest forms of CS (6,7), is accompanied by facial abnormalities such as bicoronal synostosis, hypertelorism, mid-face hypoplasia, narrow arched palate and mitten-like syndactyly of fingers and toes (8). Over 98% of cases are caused by specific missense mutations of FGFR2, either Ser252Trp (66%) or Pro253Arg (32%), in the linker between the immunoglobulin-like domain II (IgII) and IgIII domains (1,9). These substitutions increase the affinity and broaden the specificity of fibroblast growth factor (FGF)-ligand binding, explaining the cellular consequences of mutation, including accelerated proliferation and differentiation of osteoblasts in the cranial suture; premature differentiation is probably the most important factor leading to CS (9,10). In this study, we focused on early coronal suture closure using a mouse model of AS that harbors a targeted mutation (S252W) in Fgfr2.
Peptidyl-prolyl cis–trans isomerase NIMA-interacting 1 (PIN1) catalyzes post-phosphorylation conformational regulation. It binds only to specific pSer/Thr-Pro motifs, causing a cis–trans conformational change of the substrate (11). In a previous study, we demonstrated that RUNX2 expression is regulated by the FGF/fibroblast growth factor receptor (FGFR) signaling pathway (12) and that prolyl isomerization of Runt-related transcription factor 2 (RUNX2) mediated by PIN1 is crucial for acetylation and stabilization of RUNX2 in FGF/FGFR2 signaling (13,14). This molecular modification is essential for proper bone development (13–17). Excessive FGFR2 downstream signaling, including elevated RUNX2 activity and stability due to FGFR2 mutations, could be a crucial driver of CS phenotypes.
Juglone (5-hydroxy-1,4-naphthoquinone) is a brown dye isolated from walnut shells and leaves of walnut trees (genus Juglans) (18). Numerous in vitro and in vivo studies have shown that juglone effectively inhibits PIN1 activity (18–21). We hypothesized that suppression of PIN1 would normalize hyperactivated FGFR2 signaling, thereby rescuing the premature obliteration of coronal sutures that is the hallmark symptom of CS. To test our hypothesis, we pursued two independent approaches in Fgfr2S252W/+ mice, a model for human AS: a genetic approach targeting one allele of PIN1 and a pharmacological approach in which PIN1 activity was suppressed by administration of juglone during fetal development. Both approaches successfully attenuated CS phenotypes in the disease model mice, suggesting that PIN1 represents a promising molecular target for overcoming CS without surgery.
Results
Inactivation of PIN1 prevents premature fusion of coronal suture in AS model mice
PIN1 is crucial for osteoblast differentiation mediated by FGF/FGFR2 signaling (13,14). To further investigate the relationship between PIN1 and skeletal abnormalities in our AS mouse model, we reduced the dose of PIN1 by crossing Pin1+/−Fgfr2Neo-S252W/+ with Pin1+/−EIIA-Cre+/− (Pin1+/−;Fgfr2S252W/+) to generate compound mutant mice. These genetic manipulations resulted in significantly different overall phenotypes (Supplementary Material Fig. S1). Micro-computed tomography (CT) scans, performed to observe mineralized tissues, revealed that newborn Fgfr2S252W/+ mice had abnormalities, including a dome-shaped skull and a shortened skull length (Fig. 1A and B), compared with wild-type (WT) mice, as described previously (22). In the case of a normal suture in WT newborn mice, the suture is not completely fused and mineralized. So it appears as empty on micro-CT scan images of the skull as previously described (23–27). However, the CSs of coronal sutures of Fgfr2S252W/+ mice were completely fused, which were rescued in Pin1+/−;Fgfr2S252W/+ mice (Fig. 1A and B, yellow arrows). As a reference, a schematic diagram (24) is shown in Figure 1C and D. Pin1+/−;Fgfr2S252W/+ mice had longer skull (Fig. 1E) and nasal region (Fig. 1F) and smaller skull height (Fig. 1G) and intercanthal distance than Fgfr2S252W/+ mice (Fig. 1H). To observe histological changes, we prepared sagittal sections of the skull. As reported previously, Fgfr2S252W/+ coronal sutures were completely fused (Fig. 1I). In contrast, Pin1+/−;Fgfr2S252W/+ coronal sutures remained in the differentiated state and were not yet completely fused (Fig. 1I). The incidence of coronal and frontal–nasal suture fusion depended on the genotype (Table 1). Reduced body length in Fgfr2S252W/+ mice was also restored by genetic deletion of one allele of Pin1; this effect was statistically significant (Supplementary Material Fig. S1). These observations suggest that inactivation of PIN1 prevents CS of coronal sutures in Fgfr2S252W/+ mice.
Figure 1.
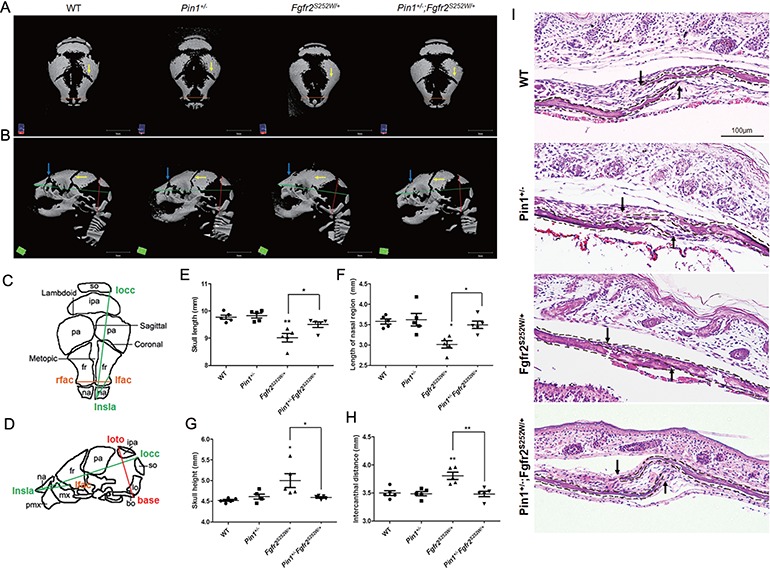
Rescue of premature fusion of coronal suture in Fgfr2S252W/+ mice by removal of one allele of Pin1. (A, B) Representative micro-CTs of skulls from WT, Pin1+/−, Fgfr2S252W/+ and Pin1+/−;Fgfr2S252W/+ mice (n = 5). Superior views (A) and lateral views (B) of skull calvaria. Removal of one allele of Pin1 rescued premature fusion of coronal suture (yellow arrows), a CS calvarial phenotype, and the frontal–nasal suture (blue arrows). (C, D) Schematic diagram and landmarks of mouse skull vault. The mouse skull consists of paired frontal bones (fr), paired parietal bones (pa) and the interparietal bone (ipa), intervened by the coronal, sagittal, metopic and lambdoid sutures. The 3D coordinates of specific craniofacial landmarks, shown in (C) and (D), were used for morphometric analyses of the skulls. Green lines represent linear distances corresponding to the length of the nasal region (lnsla–lflac) and the skull length (rnsla–rocc). Red line indicates skull height (lpto–bas). Orange lines indicating intercanthal distance (lflac–rflac) show linear distances corresponding to facial width. (E–H) Representative micro-CT results of skulls, shown as bar graphs (n = 5 in each group; *P < 0.05; **P < 0.005; ***P < 0.001). (I) Histological sections of coronal sutures in newborn mice. Arrows indicate growing fronts of frontal and parietal bones. The calvarial bone is highlighted with dotted lines.
Table 1.
The incidence of coronal and frontal–nasal suture fusion according to genotype
| Genotype | Coronal suture fusion (between frontal and parietal bone) | Ratio (%) | Frontal–nasal suture fusion (between nasal and frontal bone) | Ratio (%) |
|---|---|---|---|---|
| WT (n = 5) | 0/5 | 0 | 0/5 | 0 |
| Pin1+/− (n = 5) | 0/5 | 0 | 0/5 | 0 |
| Fgfr2S252W/+ (n = 5) | 5/5 | 100 | 5/5 | 100 |
| Pin1+/−Fgfr2S252W/+ (n = 5) | 0/5 | 0 | 2/5 | 40 |
Restoration of craniofacial skeletal abnormalities in juglone-treated Fgfr2S252W/+ mice
To assess the relevance of these findings to patients, we treated our model mice with juglone, a pharmacological inhibitor of PIN1 (18). Prior to these experiments, we optimized the dose of juglone based on toxicity tests (Supplementary Material Fig. S2). For this purpose, pregnant mice were injected once daily with juglone (0, 0.01, 0.1, 1 or 2.5 mg/kg) from E14.5 to E18.5; dynamic skeletogenesis of the calvaria occurs around E12.5 (28). Newborn mice were sacrificed and examined to determine the highest concentration that did not decrease the number of littermates (Supplementary Material Fig. S2A) but significantly affected the skeletal phenotype (Supplementary Material Fig. S2B); based on the results of this analysis, we set the concentration of juglone to 1 mg/kg. When pregnant mice were given daily injection of 1 mg/kg juglone, littermates which comprised WT and Fgfr2S252W/+ mice were birthed as an equal proportion in control and juglone-injected groups (Supplementary Material Fig. S2C).
The level of skull development was evaluated by embryonic skeletal staining and histological analysis (Fig. 2A–C). As in the experiments described earlier, Fgfr2S252W/+ mice exhibited multiple abnormalities, including a dome-shaped skull (Fig. 2A), a shortened skull length (Fig. 2A and B), widely spaced eyes (Fig. 2B, yellow line), premature closure of the coronal suture (Fig. 2A, white arrowhead; Fig. 2B, arrow) and nasal suture (Fig. 2A, black arrowhead), whereas juglone-treated Fgfr2S252W/+ mice showed recovery from premature coronal suture fusion (5/5, 100%) and frontal–nasal suture fusion (2/5, 40%) (Table 2). Next, we made histological observations of the coronal sections in sagittal sections of calvaria (Fig. 2C). Fgfr2S252W/+ mice exhibited premature closure of coronal sutures, whereas closure occurred normally in juglone-treated Fgfr2S252W/+ mice despite expression of the mutant Fgfr2S252W allele (Fig. 2C). To quantify restoration of CS calvaria phenotypes by juglone treatment, we obtained micro-CT scans of skulls from each group and littermate controls at birth (Fig. 2D and E). Images from the top (Fig. 2D) and the side (Fig. 2E) of Fgfr2S252W/+ mice calvaria showed that coronal (yellow arrow) and nasal (blue arrow) sutures were completely overlapping and calcified. In contrast, juglone-treated Fgfr2S252W/+ mice had calvarial phenotypes similar to those of WT mice. The incidence of coronal and frontal–nasal suture fusion depended on the experimental group (Table 2). We then looked for morphological differences in specific craniofacial features between groups, in comparison with WT littermate mice (Fig. 2D and E), using Figure 1C and D and reference landmarks (24,29). The lengths of the skull and nasal region were significantly shorter in Fgfr2S252W/+ mice than in WT mice (Fig. 2F and G). In addition, cranial height and interocular distance were larger in Fgfr2S252W/+ mice than in WT mice, likely due to brain development under size limitations (Fig. 2H and I); this difference was statistically significant. The reduced body length of Fgfr2S252W/+ mice was rescued by juglone treatment (Supplementary Material Fig. S3), and the overall phenotypes of juglone-treated Fgfr2S252W/+ mice were similar to those of WT mice. Together, these results indicate that juglone attenuates abnormal calvaria bone formation in Fgfr2S252W/+ mice.
Figure 2.
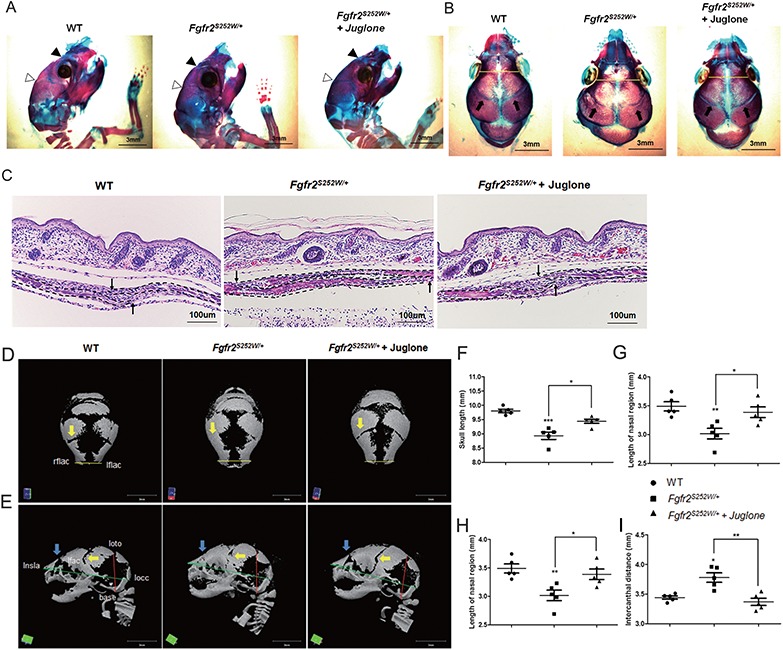
Craniosynostosis phenotypes in AS model mice are rescued by treatment with the PIN1 inhibitor juglone. Pregnant mice were intraperitoneally injected with 1 mg/kg juglone (Fgfr2S252W/+ + juglone) or vehicle (Fgfr2S252W/+) once a day from E14.5 to E18.5. WT and Fgfr2S252W/+ mice were sacrificed at birth. (A, B) Mouse calvaria were examined after Alizarin Red S and Alcian Blue staining. Black arrowheads, frontal–nasal suture; white arrowheads, coronal suture (A). Both sutures were already closed in Fgfr2S252W/+ mice, whereas those of WT and Fgfr2S252W/+ + juglone mice remained open. Coronal views of the heads of the same animals are shown in (B). Intercanthal distance (yellow lines) was significantly wider in Fgfr2S252W/+ mice than in control mice. Arrows indicate a coronal suture that is almost overlapping in Fgfr2S252W/+, whereas sutures in the other group remain open. (C) Histological sections of coronal sutures in newborn mice. Arrows indicate growing fronts of frontal and parietal bones. Note that suture overlap is much more pronounced in Fgfr2S252W/+ mice than in the other groups. The calvarial bone is highlighted with dotted lines. (D, E) Representative micro-CTs of skulls from WT, Fgfr2S252W/+ and juglone-treated Fgfr2S252W/+ mice (n = 5). Superior views (D) and lateral views (F) of skull calvaria. Yellow arrows indicate rescue of early fusion of coronal suture (5/5, 100%), and blue arrows indicate rescue of the distorted frontal–nasal suture (2/5, 40%) of mutant mice by juglone. Green lines represent linear distances corresponding to the lengths of the nasal region and the skull. The red line indicates skull height, whereas the yellow lines, such as the neurocranial width at intercanthal distance (lflac–rflac), indicate linear distances corresponding to facial width. (F–I) Representative micro-CT results of skulls are shown in bar graphs (n = 5 in each group; *P < 0.05; **P < 0.005; ***P < 0.001). Statistically significant differences indicate rescue of the CS calvarial phenotype.
Table 2.
The incidence of suture fusion according to experimental group
| Genotype | Coronal suture fusion (between frontal and parietal bone) | Ratio (%) | Frontal–nasal suture fusion (between nasal and frontal bone) | Ratio (%) |
|---|---|---|---|---|
| WT (n = 5) | 0/5 | 0 | 0/5 | 0 |
| Fgfr2S252W/+ (n = 5) | 5/5 | 100 | 5/5 | 100 |
| Fgfr2S252W/+ + juglone (n = 5) | 1/5 | 20 | 3/5 | 60 |
Juglone affects expression of genes downstream of FGF/FGFR2
Previously, we found links between FGF/FGFR2 signaling and PIN1 in cell culture (14,30). Therefore, we examined whether rescue by juglone, which has an inhibitory concentration 50 (IC50) value of 13.55 μm in primary osteoblasts (Supplementary Material Fig. S4), affected expression modulators and downstream genes of FGFR signaling, including Dusp6 and two genes in the Sprouty family, Spry2 and Spry3 (22). Extracellular signal-regulated kinase 1/2 (ERK1/2) activation triggers the transcription of Dusp6 mRNA, and the Dusp6 protein inactivates ERK1/2, resulting in a negative feedback loop activated by FGF during somite development (31). Similarly, members of the Sprouty family antagonize FGF and FGFR signaling (32). To assess the effect of juglone on FGF/FGFR2 signaling, we measured mRNA expression of Dusp6, Spry2 and Spry3 by reverse transcription polymerase chain reaction (RT-PCR). Expression of all three genes was elevated in primary mouse calvarial cells from Fgfr2S252W/+ mice (Fig. 3A–C) and MC3T3-E1 cells overexpressing Fgfr2S252W from a plasmid (Fig. 3D–F), in which FGF/FGFR2 signaling was hyperactivated in comparison with WT primary mouse calvarial cells and Fgfr2WT-overexpressing MC3T3-E1 cells. After juglone treatment, expression of these genes decreased in a dose-dependent manner (Fig. 3A–E). We measured the expression of 18s rRNA as a negative control. As expected, expression of 18s rRNA gene was not affected by mutation of Fgfr2 in primary mouse calvarial cells (Supplementary Material Fig. S5A). We also confirmed that the increased expression of target genes in Fgfr2S252W/+ primary mouse calvarial cells was suppressed by U0126 treatment, as reported previously (Supplementary Material Fig. S5B–D) (22). These observations indicate that juglone inhibits the downstream genes of FGF/FGFR2 signaling.
Figure 3.
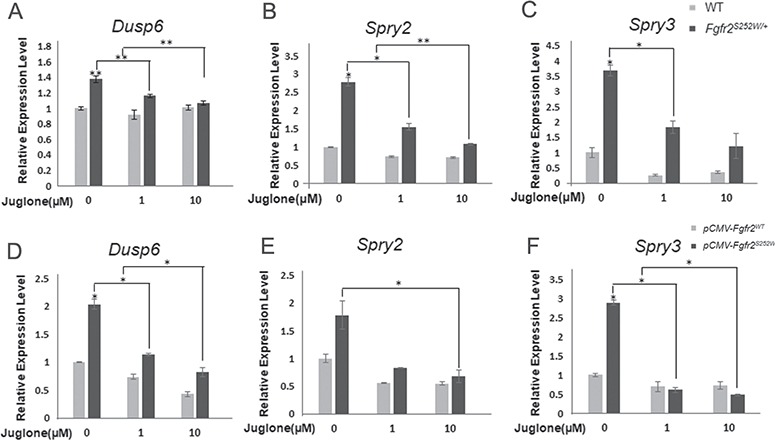
Expression of target genes changes upon treatment with juglone. (A, B) Relative expression levels of FGFR2 signaling modulators and downstream genes, based on qPCR analysis of WT and Fgfr2S252W/+ calvaria cells (n = 3) following treatment with different dosages of juglone for 1 day. (C–E) 70% confluent MC3T3-E1 cells were transfected with Fgfr2WT or Fgfr2S252W mutant expression plasmids. After 24 h, cells were treated with 0, 1 and 10 μm of juglone for 1 day. mRNA levels of FGFR signaling modulators and downstream genes, such as Dusp6 (C) and the Sprouty family genes (Spry2, 3) (D, E), were analyzed quantitatively (n = 3 in each group; *P < 0.01; **P < 0.005; ***P < 0.001). The level of each mRNA was normalized to that of Gapdh in the same sample.
Juglone restores RUNX2 hyperstability in Fgfr2S252W/+ mouse calvarial cells by decreasing acetylation of RUNX2
RUNX2, a key transcription factor in osteoblast differentiation, triggers mesenchymal stem cells to differentiate into osteoblasts (33). The importance of RUNX2 in mammalian skeletal development has been clearly demonstrated by the observation that RUNX2 knockout mice lack both mature osteoblasts and a mineralized skeleton, including the calvaria (34). In addition, RUNX2 expression is localized to the critical area of cranial suture closure (35). To determine the mechanism underlying restoration of the calvarial phenotype of Fgfr2S252W/+ mice by juglone, we assessed endogenous RUNX2 expression in WT and Fgfr2S252W/+ primary mouse calvarial cells by immunofluorescence analysis (Fig. 4A). In WT cells, we observed weak staining for endogenous RUNX2 in the nucleus (Fig. 4A, first row). After treatment of WT cells with juglone, the level of endogenous RUNX2 was slightly reduced (Fig. 4A, second row). Fgfr2S252W/+ cells had more endogenous RUNX2 in the nucleus than WT cells (Fig. 4A, third row), but juglone treatment significantly reduced the level of RUNX2 in these Fgfr2S252W/+ primary mouse calvarial cells (Fig. 4A, fourth row), which was quantified by measuring the intensity of the nuclear staining (green) area (Fig. 4B).
Figure 4.
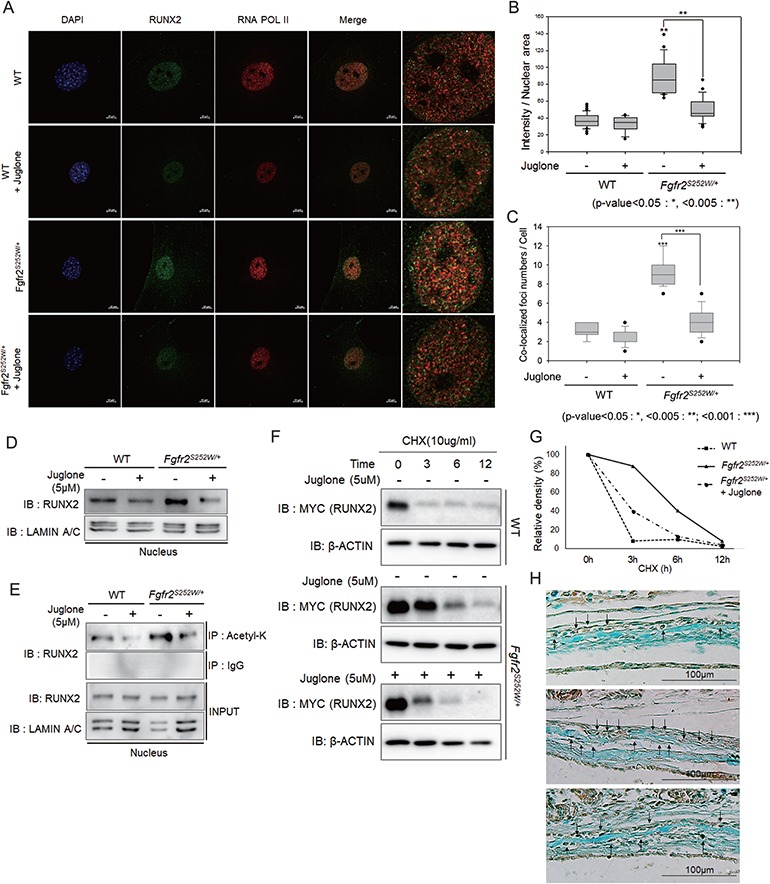
RUNX2 is overactivated in Fgfr2S252W/+ mice, and juglone destabilizes RUNX2 by decreasing acetylation. (A) Endogenous RUNX2 was detected by immunofluorescence and immunocytochemistry. Primary mouse calvarial cells from WT and Fgfr2S252W/+ mice were treated with 5 μm juglone for 24 h. Cells were fixed, and endogenous proteins were immunostained with anti-RUNX2 (green) and anti-RNA POL II (red) antibodies, as well as 4′,6-diamidino-2-phenylindole (blue). In the merged image, RUNX2 and RNA POL II colocalization (yellow) was observed as foci, particularly in Fgfr2S252W mutant-expressing cells. (B) Endogenous intranuclear RUNX2 was quantified, and results are shown as a sigma plot (n = 20 in each group; *P < 0.01; **P < 0.005; ***P < 0.001). (C) The numbers of RUNX2/RNA POL II-colocalized foci were quantified (n = 10 in each group; *P < 0.01; **P < 0.005; ***P < 0.001). (D) Abundance of endogenous RUNX2 in dissociated nuclear extracts, as determined by immunoblot assay. (E) Degree of acetylation of RUNX2, as determined by IP with anti-acetyl-lysine antibody from dissociated nuclear extracts. (F, G) RUNX2 stability analysis. Myc-tagged Runx2 plasmid was transfected into primary mouse calvaria cells. After 24 h, cells were pre-treated with or without 5 μm juglone for 1 h and then treated with 10 μg/ml CHX for the indicated times. Cells were harvested and followed by immunoblotting with the indicated antibodies. β-Actin was used as a loading control. (G) Band intensities of RUNX2 in F were quantitated and plotted against time. (H) RUNX2 in calvaria tissue from coronal sutures of newborn mice was detected by IHC using Alcian Blue counterstaining.
Increased nuclear localization of RUNX2 in Fgfr2S252W/+ cells was not enough to conclude that its activity as a transcription factor in these cells was increased. Many previous papers have reported that active transcription factors appear in the nucleus as foci with RNA polymerase (POL) II (14,36–39). Thus, to determine whether its increased localization to the nucleus coincides with an increase in its activity, we quantified the number of RUNX2/RNA POL II-colocalized foci. Indeed, the number of these foci was higher in Fgfr2S252W/+ cells than in WT cells and decreased after juglone treatment (Fig. 4C).
We conducted nuclear fractionation and western blot analysis; we found that the increased levels of nuclear endogenous RUNX2 compared with that in WT cells were reduced by juglone treatment (Fig. 4D). We showed previously that post-translational modification (PTM) is important for RUNX2 stabilization (13,14,30). In particular, PIN1-mediated structural modification of RUNX2 is an indispensable step connecting phosphorylation, acetylation and transcriptional activation of RUNX2 by FGF signaling (14). To determine the mechanism by which juglone restored nuclear RUNX2 to WT level, we examined the acetylation of RUNX2. Immunoprecipitation (IP) assays revealed that acetylation of RUNX2 was higher in Fgfr2S252W/+ mouse calvarial cells than in WT cells (Fig. 4E). In contrast, the acetylated RUNX2 level returned to normal in juglone-treated Fgfr2S252W/+ mouse calvarial cells (Fig. 4E). Next, we investigated whether juglone affects the stability of the RUNX2. FGF signaling promotes Runx2 expression in osteoblasts (12); however, we wanted to test the effects of juglone on PTM of RUNX2, as this affects the stability of RUNX2. To this end, we transfected Myc-tagged Runx2 plasmid into WT and Fgfr2S252W/+ mouse calvarial cells and monitored the stability of exogenous RUNX2 (Fig. 4F). In WT mouse calvarial cells, treatment with cycloheximide (CHX), an inhibitor of protein synthesis, almost decreased the level of exogenous RUNX2 within 3 h, whereas in Fgfr2S252W/+ cells, its level remained stable in for over 9 h (Fig. 4F). However, when Fgfr2S252W/+ mouse calvarial cells were treated with juglone and CHX, the level of RUNX2 was reduced appreciably (Fig. 4F). We quantified the changes in RUNX2 expression over time (Fig. 4G). These results strongly indicate that isomerization of RUNX2 by PIN1, which is critical for RUNX2 stabilization through acetylation, is inhibited by juglone, thereby reversing the effects of the Fgfr2S252W mutation and the associated stabilization of RUNX2. Using immunohistochemistry (IHC), we confirmed that elevated RUNX2 levels in Fgfr2S252W/+ mouse coronal sutures were attenuated by juglone treatment (Fig. 4H). Cell morphology was also different because cuboidal cells, such as WT and juglone-treated Fgfr2S252W/+, were undergoing differentiation (40). However, the bone lining cells in Fgfr2S252W/+ are already differentiated (40); RUNX2-stained cells had a different histological appearance. Together, these results suggest that the Fgfr2 mutation in osteoblasts increase the amount of RUNX2 and juglone restored RUNX2 to near its normal WT level.
Juglone attenuates Fgfr2 mutation-induced osteoblast proliferation and differentiation in CS
As shown in Figure 4, juglone prevented excessive RUNX2 acetylation and stabilization. Because acetylated and stabilized RUNX2 has transactivation activity and regulates cell proliferation and differentiation (41,42), we examined the in vitro effects of juglone on osteoblast differentiation. RUNX2 activity was assessed in a reporter assay using 6XOSE2, a construct that contains six tandem repeats of osteoblast-specific cis-element 2 (12). Primary mouse calvarial cells of Fgfr2S252W/+ mice exhibited approximately 2.5-fold higher levels of RUNX2 activity than those of WT mice (Fig. 5A). However, the elevated RUNX2 activity caused by Fgfr2S252W mutation decreased in a dose-dependent manner following juglone treatment (Fig. 5A); this observation has important implications for cell proliferation and differentiation, which are regulated by RUNX2. The FGF/FGFR2 signaling pathway is critical for proliferation in skeletogenesis (43). To determine the effects of juglone on osteoblast proliferation and differentiation, we performed a proliferation assay and assessed the mRNA levels of marker genes in WT and Fgfr2S252W/+ primary mouse calvarial cells. Cell growth (Fig. 5B) and expression of proliferation marker genes such as CyclinD1, Cdk2, Cdk4 and PCNA were higher in Fgfr2S252W/+ calvarial cells than in WT cells (Fig. 5C–F). Administration of juglone decreased cell growth (Fig. 5B), as well as the expression of proliferation marker genes (Fig. 5C–F), in a dose-dependent manner. Transcript levels of bone marker genes followed a similar pattern (Fig. 5G and H). Suppression of PIN1 by juglone almost completely abrogated the higher expression of specific genes induced by Fgfr2S252W mutation. During osteoblast maturation, Fgfr2S252W/+ calvarial cells exhibited enhanced production of mineralized bone matrix compared with WT cells, as detected by alizarin red staining (Fig. 5I); juglone rescued this phenotype in a concentration-dependent manner (Fig. 5I). Together, these data suggest that juglone reduces the transcriptional activity of RUNX2 and abrogates the effects of Fgfr2 mutation, namely, excessive osteoblast differentiation resulting in CS.
Figure 5.
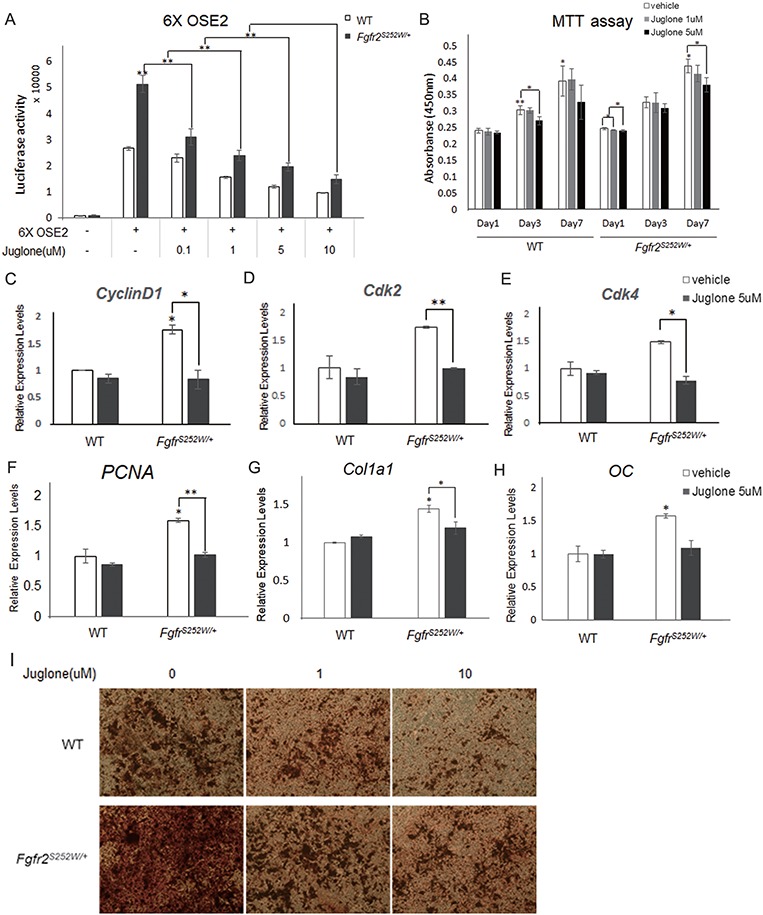
Effects of juglone treatment on osteoblast proliferation and differentiation. (A) Relative RUNX2 transacting activity was assessed using a 6XOSE2 luciferase reporter construct. Following transfection with the 6XOSE2 reporter gene, cells were treated with juglone at various concentrations for 24 h. Luciferase assays were performed on both WT and Fgfr2S252W/+ mouse calvarial cells. (B) Proliferation of WT and Fgfr2S252W/+ mouse calvarial cells following treatment with various doses of juglone for 1, 3 and 7 days. Cell proliferation was assessed by water-soluble tetrazolium assay. (C–F) Relative expression of proliferation-related genes in WT and Fgfr2S252W/+ mouse calvarial cells, as determined by qPCR. (G–H) Relative expression of osteoblast differentiation markers in both genotypes of primary mouse calvarial cells, as determined by qPCR. Cells were treated with vehicle or 5 μm juglone for 24 h and were more cultured for 2 days in osteogenic media. Expression of each marker gene was normalized against that of Gapdh in the same sample. (I) Late-stage primary osteoblast differentiation was confirmed by Alizarin Red S staining. Both genotypes of primary calvarial cells were cultured for 3 weeks in osteogenic media after 48 h culture with the indicated concentrations of juglone (n = 3 in each group; *P < 0.01; **P < 0.005; ***P < 0.001).
Discussion
The majority of severe CS cases are due to de novo mutations and are therefore usually unanticipated (1). Currently, the only way to treat CS patients is surgical intervention, involving creation of an expanded but rigid skull vault to accommodate further increases in brain growth, with some additional remodeling of the inner surface of the calvaria (1). However, recent developments in prenatal diagnostic testing for mutations in amniotic fluid, especially FGFR2 mutations, raise the possibility of early diagnosis (44,45). Detection of CS during pregnancy could allow premature closure of the cranial suture to be prevented by inhibiting excessive growth at the developmental stage of the fetus using a drug. This approach is promising in that it not only alleviates the burden of surgery in infants but could also prevent mental retardation caused by delayed surgery or surgery sequela arising during brain growth.
In this study, we generated Pin1;Fgfr2 compound mutant mice and investigated the effects of juglone, an inhibitor of PIN1, on the recovery of CS phenotypes such as early suture closure in Fgfr2S252W/+ mice (Figs. 1 and 2 and Supplementary Material Figs. S1 and S3). Many studies have been conducted to elucidate the factors that cause CS or identify therapeutic targets for CS. Wang et al. demonstrated the importance of the p38 mitogen-activated protein kinase (MAPK) pathway in AS, which is caused by mutations in FGFR2 (24). Tanimoto et al. showed that the CS phenotype of mice homozygous for the Gli3 deletion can be rescued through the removal of one allele of Runx2 (46). In addition to this genetic approach, Shukla et al. reported that U0126, which suppresses ERK/MAPK signaling, prevents premature cranial suture closure in Fgfr2S252W/+ mice (22). Usually, the ERK/MAPK enzyme action products are shared with a substrate for the PIN1 enzyme, because PIN1 binds only to the pSer/Thr-Pro motif (14,47). Therefore, inhibition of PIN1 disrupts signaling downstream of the ERK/MAP kinase pathway, and it is possible that it would result in fewer non-specific effects than the inhibition of ERK-MAP kinase. In addition, in our genetic approach, we demonstrated that targeting the Pin1 gene in Fgfr2S252W/+ mice leads to AS phenotype recovery (Fig. 1), supporting the conclusion drawn from our pharmacological approach that juglone-mediated rescue is indeed due to inhibition of PIN1 (Fig. 2). Accordingly, we propose that PIN1-targeted therapy would be valuable for treatment of CS.
This approach could also be applied to alleviating other phenotypes associated with FGFR-related CS. These phenotypes differ slightly different depending on the type of FGFR mutation (8,48). The FGFR S252W mutation, as in AS, causes premature fusion of the coronal suture with syndactyly of the hands or feet, mid-face hypoplasia and dwarfism (1,49,50). As described previously (22,24,50), we also observed abnormalities in endochondral bone formation, as evidenced by shorter body length (Supplementary Material Figs. S1 and S3) and shortening of the skull (Figs. 1 and 2) in Fgfr2S252W mutant mice. In addition to fusion of the calvarial sutures, mid-face growth is also an important factor affecting skull length (51), and these phenotypes are caused by increased endochondral ossification due to gain-of-function mutation in FGFRs (52). We found that inactivation of PIN1 by genetic manipulation or drug treatment restored body size (Supplementary Material Figs. S1 and S3) and skull length (Figs. 1 and 2). These effects of PIN1 targeting might also be applied to endochondral ossification processes. Furthermore, these bone phenotypes are similar to those in patients with AS and achondroplasia, the most common genetic dwarfism in humans, which is caused by mutation in FGFR3 (53). CS can be classified as syndromic based on FGFR gene mutations and associated phenotypes. Although we used only the Fgfr2S252W/+ CS mouse model in this study, CS is also caused by mutations in other FGFRs, including FGFR1, FGFR2 or FGFR3 (1,8). Some of these mutations, such as FGFR2S252W, increase the affinity of the receptor for ligand, whereas others cause excessive signaling via ligand-independent receptor activation (8,48). However, we found that early fused cranial sutures are rescued by targeting PIN1, which suppresses hyperactivated downstream signaling of the FGF/FGFR signaling, a common feature of FGFR mutants. Therefore, we propose that this approach might also be applicable for the therapy of other CS syndromes, such as those caused by gain-of-function mutations in other FGFRs.
Recovery rates from nasal–frontal suture fusion by genetic deletion or inhibition of PIN1 were different from observed recovery rates from coronal suture fusion (Tables 1 and 2). We proposed two reasons for these. First, it seems that suture fusion patterns are a suture-specific consequence of a complex combination of biogenesis or signaling in the sutures and variation in individual timing specific to each suture (54). Therefore, the degree or timing of the FGFR2 signaling effect differs depending on the type of suture (55–57). Second, areas of the skull derived from the lineage in the coronal suture are developmentally distinct from areas in nasal–frontal suture, because of the mixed developmental origin of the mammalian skull vault (54,58,59): the frontal and sagittal suture are neural crest derived, whereas the parietal bone and coronal suture are of mesodermal origin (54). The main symptom of CS caused by the Fgfr2S252W mutation is the coronal suture early fusion, so our study has focused on the effect of recovery of coronal suture fusion.
Juglone is one of the best-known PIN1 inhibitors, and its IC50 values have been reported to vary with cell type (60–62). In primary osteoblasts, we found that the IC50 value was 13.55 μm (Supplementary Material Fig. S4). In addition, juglone passes the placenta barrier, as demonstrated by the observation that high concentrations of juglone could induce abortion (Supplementary Material Fig. S2). Juglone undergoes biotransformation in the liver to yield various metabolites, which are detected primarily in urine (63), indicating that the compound has access to liver cells (63,64) These observations imply that juglone has the potential to regulate liver function and that it could also be used for medicinal purposes in other parts of the body (18,63,64). We established the appropriate concentration of juglone through toxicity testing in pregnant mice and determined a dose that did not affect the number of littermates or overall skull phenotypes (Supplementary Material Fig. S2). We confirmed that development of the abnormal phenotype of Fgfr2S252W/+ mice (small, shortened skull and premature fusion of the coronal suture) could be prevented by juglone treatment (Fig. 2). Also, although many previous studies have reported that the survival rate of AS is low (50,65–67), optimized concentrations of juglone increased the number of littermates in comparison with the control group (average control litter: 5.25 pups; average litter of juglone-injected mother: 7.11 pups). This observation suggests that juglone improves the survival of newborn mice with the Fgfr2S252W/+ mutation.
Together, these genetic and pharmacological approaches using juglone indicate that PIN1 is the target for juglone-mediated attenuation of hyperactivated FGF/FGFR signaling in Fgfr2S252W/+ mice. However, pure juglone is used only for research purposes and is categorized as an ‘Unapproved Herb’ in the German Commission E monograph owing to its probably mutagenic and carcinogenic properties (64). In addition, it has been designated as a potentially toxic natural product by the National Toxicology Program in the USA (63,64). Therefore, for clinical use, it would be important to develop PIN1 inhibitors that are both effective and safe.
It is widely accepted that PIN1 is a molecular switch that determines the fate of numerous osteogenic transcription factors, especially those related to stabilization of substrate proteins (13–15,17,68). In addition, direct delivery of PIN1 protein into mesenchymal cells stimulates their osteogenic differentiation (69). We previously suggested that FGF2 induces PTMs of RUNX2. In particular, we showed that RUNX2 is acetylated by p300 (70) and that FGF2-induced phosphorylation (12,71) and subsequent acetylation (72) of RUNX2 stimulates its in vitro transactivation activity by stabilizing the protein. Also, we reported that prolyl isomerization of RUNX2 alters the conformation to an acetylation-friendly structure, resulting in stabilization and activation RUNX2 (14). In this study, we found that Fgfr2S252W mutation increased the RUNX2 level by promoting these PTMs, which in turn increased the nuclear RUNX2 level in Fgfr2S252W/+ primary calvarial cells or tissues (Fig. 4). This increase in RUNX2 level, in addition to elevation of general FGFR signaling, resulted in excessive osteoblast proliferation and differentiation (Fig. 5), which may be the molecular mechanism underlying premature closure of the coronal sutures in CS. Moreover, we showed that the PIN1 inhibitor juglone decreased excessive RUNX2 stabilization by inhibiting its isomerization and consequent acetylation (Figs. 4 and 5), confirming the importance of PIN1 in the RUNX2 PTM process in vivo. However, although we showed that premature coronal suture fusion rescued by controlling PIN1 was only due to decrease of RUNX2 levels, many previous studies using AS mouse models reported that phosphorylation levels of ERK1/2, AKT and p38 are elevated in the calvaria by gain-of-function mutations of FGFR2 such as S252W and P253R (22,24,73,74). In addition, inhibition of ERK, an important mediator of FGF signaling, restores the phenotype of AS mouse (22). In a previous study, we showed that PIN1 is crucial for regulation of the FGF/FGFR signaling pathway (14,68) and that FGF target genes are affected by juglone (Fig. 3). Therefore, we suspect that restoration of CS phenotypes via alteration of RUNX2 PTMs represents only one of many potential mediators downstream of the FGF signaling pathway.
In summary, our results indicate that PIN1 is a new therapeutic target for CS. Rescue of CS by juglone was associated with alterations in PIN1 activity and RUNX2 stability (Fig. 6). Administration of the PIN1 inhibitor juglone during pregnancy significantly suppressed CS phenotypes, especially premature fusion of the coronal suture, in an Fgfr2S252W mutant mouse model. These data clearly establish the regulatory role of PIN1 in abnormalities associated with FGF/FGFR2 hyperactivation and highlight the feasibility of developing specific targeted prevention and therapeutic strategies for CS.
Figure 6.
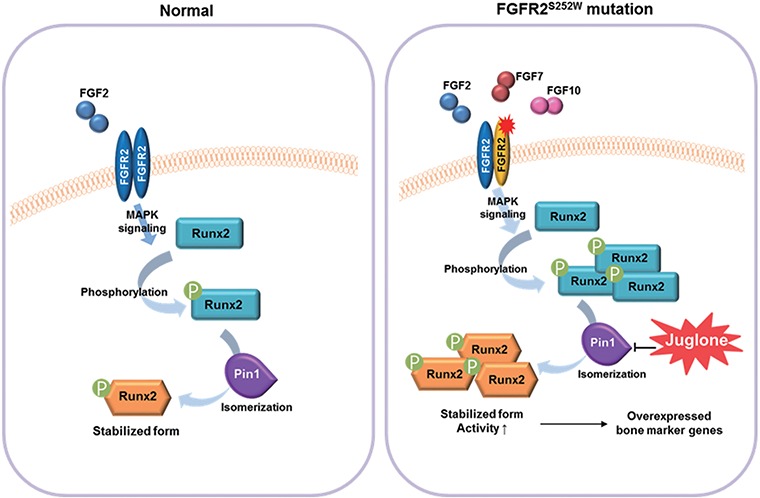
Mechanisms underlying restoration of the normal phenotype by juglone in calvaria with gain-of-function mutations in FGFR2. Juglone treatment lowers abnormally elevated RUNX2 levels in Fgfr2S252W/+ mice by inhibiting RUNX2 isomerization, thereby restoring proper downstream transcriptional activity (9,83). The S252W mutation of FGFR2 increases ligand affinity and alters ligand specificity, resulting in hyperactive FGF signaling. In this genetic background, RUNX2, one of the targets of FGF signaling, undergoes excessive PTM, such as isomerization and acetylation. Repression of RUNX2 isomerization by juglone, a PIN1 inhibitor, decreases the RUNX2 level and normalizes the expression of RUNX2 target genes, thereby rescuing the abnormal calvaria phenotypes of Fgfr2S252W/+ mice.
Materials and Methods
Animal experiments
Fgfr2S252W/+ and Pin1-deficient mice (22,75) were maintained under specific pathogen-free conditions. The loxP-neo-loxP cassette, a blocker sequence, can be deleted to activate the Fgfr2S252W allele upon crossing with mice carrying a ubiquitous Cre transgene (EIIA-Cre, the Jackson Laboratory, Bar Harbor, Maine, USA) (22,76). Mice carrying the Fgfr2S252W and EIIA-Cre alleles were mated, and Pin1;Fgfr2 compound mutant mice were generated by mating Pin1+/−Fgfr2Neo-S252W/+ mice with Pin1+/−EIIa-Cre/+ mice. The genetic background of Fgfr2Neo-S252W/+ mice is mouse strain (FVB) (22), and the EIIa-Cre transgene (B6.FVB-TgN(EIIa-cre)C5379Lm, No. 003724) was from the Jackson Laboratory. The genetic background of the Pin1+/− mice is C57BL/6 (34). Pregnant mice were injected intraperitoneally with juglone (Calbiochem, Darmstadt, Germany). Skeletal staining and micro-CT analysis were performed as described previously (77). The numbers of animals in experimental groups are indicated in the figure legends. Animals were housed in cages following Institutional Animal Care and Use Committee (IACUC) policies. All animal studies were reviewed and approved by the Special Committee on Animal Welfare, Seoul National University, Seoul, Republic of Korea.
Cell culture, cell proliferation assay and luciferase reporter assay
WT and Fgfr2S252W/+ mice calvarial cells were isolated from newborn mice. Primary calvarial cells and pre-osteoblast MC3T3-E1 cells were cultured in α-minimum essential medium (α-MEM) with 10% fetal bovine serum containing 1% penicillin/streptomycin. For osteogenic differentiation, the medium was supplemented with 5 mm β-glycerophosphate and 50 μg/ml ascorbic acid. Cell proliferation was assayed as previously described (77), and luciferase activity was detected using the Luciferase Assay Kit (Promega, Madison, WI, USA) (17).
Histology analysis
For hematoxylin and eosin (H&E) staining and IHC of tissue sections, newborn mice were fixed in 4% paraformaldehyde for 24 h, dehydrated and embedded in paraffin using standard procedures. Serial paraffin sections (5 μm thickness) were subjected to H&E staining as described previously (13) or IHC using the EnVision™ G|2 Doublestain System (Dako, Glostrup, Denmark) (77). Stained tissues were imaged on a conventional microscope equipped with a DP72 digital camera (Olympus, Tokyo, Japan). Antibodies used for these experiments are listed in Supplementary Material Table S1.
Antibodies and reagents
Antibodies and reagents are described in detail in Supplementary Material Tables S1 and S2.
Micro-CT analysis
Newborn mice were sacrificed and fixed with 4% paraformaldehyde overnight at 4°C. Micro-CT was performed on an inspeXio SMX-100CT system, and data were analyzed using the TriBON software (RATOC, Tokyo, Japan). The use of these methods for bone analyses was described previously (78–80).
Extraction of total RNAs, RT-PCR and quantitative real-time PCR
Total RNA extraction from cultured cells, RT-PCR and real-time PCR (qPCR) were performed as described previously (22,77,80). The primer sets used for qPCR are listed in Supplementary Material Table S3 and in (22,77,80). mRNA expression was determined by qPCR, and the results were normalized to the Gapdh expression level.
DNA construction and transfection
Construction of Fgfr2WT and Fgfr2S252W mutant expression vectors was described previously (81). Transactivation activity of RUNX2 was evaluated using the 6XOSE2-Luc reporter vector (12). The Neon transfection system (Invitrogen, San Diego, CA, USA) was used for DNA transfection of primary mouse calvarial cells or MC3T3-E1 cells.
Nuclear–cytoplasmic fractionation and immunoassays
Nuclear–cytoplasmic fractionation was conducted as previously described (17). Detection of cellular proteins by IP and immunoblot assays and detection of RUNX2 by immunofluorescence were carried out as described previously (14).
Alizarin Red S staining
Detection of mineralized bone matrix production was performed as described previously (82).
Statistics
All quantitative data are presented as means ± SD (in vitro data) or means ± SEM (in vivo data). Each experiment was performed at least three times, and results from representative individual experiments are shown in the figures. Statistical analysis was performed by either unpaired two-tailed Student’s t-test or one-way analysis of variance followed by Bonferroni’s test using the Prism 5.0 software (GraphPad Software, San Diego, CA, USA). P < 0.05 was considered significant.
Study approval
The Seoul National University IACUCs approved the animal study (Approval No. SNU-160121-1).
Supplementary Material
Acknowledgements
The authors thank C.X. Deng (University of Macau, China) for providing the Fgfr2S252W knock-in mouse and S.C. Bae (Chungbuk National University, Cheongju, Korea) for providing valuable advice on pharmacokinetics.
Conflict of Interest statement. None declared.
Funding
General Researcher Program of the National Research Foundation of Korea (NRF-2014R1A2A2A01004865, NRF-2017R1A2B3011778).
References
- 1. Johnson D. and Wilkie A.O. (2011) Craniosynostosis. Eur. J. Hum. Genet., 19, 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clendenning D.E. and Mortlock D.P. (2012) The BMP ligand Gdf6 prevents differentiation of coronal suture mesenchyme in early cranial development. PloS One, 7, e36789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jin S.W., Sim K.B. and Kim S.D. (2016) Development and growth of the normal cranial vault: an embryologic review. J. Korean Neurosurg. Soc., 59, 192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Derderian C. and Seaward J. (2012) Syndromic craniosynostosis. Semin. Plast. Surg., 26, 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bagheri-Fam S., Ono M., Li L., Zhao L., Ryan J., Lai R., Katsura Y., Rossello F.J., Koopman P., Scherer G. et al. (2015) FGFR2 mutation in 46,XY sex reversal with craniosynostosis. Hum. Mol. Genet., 24, 6699–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Du X., Weng T., Sun Q., Su N., Chen Z., Qi H., Jin M., Yin L., He Q., and Chen L. (2010) Dynamic morphological changes in the skulls of mice mimicking human Apert syndrome resulting from gain-of-function mutation of FGFR2 (P253R). J. Anat., 217, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koca T.T. (2016) Apert syndrome: a case report and review of the literature. North. Clin. Istanb., 3, 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Robin N.H., Falk M.J. and Haldeman-Englert C.R. (1993) Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., Ledbetter N., Mefford H.C., Smith R.J.H. and Stephens K. (eds.), In GeneReviews(R), Seattle (WA), in press. [Google Scholar]
- 9. Ibrahimi O.A., Zhang F., Eliseenkova A.V., Itoh N., Linhardt R.J. and Mohammadi M. (2004) Biochemical analysis of pathogenic ligand-dependent FGFR2 mutations suggests distinct pathophysiological mechanisms for craniofacial and limb abnormalities. Hum. Mol. Genet., 13, 2313–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anderson J., Burns H.D., Enriquez-Harris P., Wilkie A.O. and Heath J.K. (1998) Apert syndrome mutations in fibroblast growth factor receptor 2 exhibit increased affinity for FGF ligand. Hum. Mol. Genet., 7, 1475–1483. [DOI] [PubMed] [Google Scholar]
- 11. Nakamura K., Kosugi I., Lee D.Y., Hafner A., Sinclair D.A., Ryo A. and Lu K.P. (2012) Prolyl isomerase Pin1 regulates neuronal differentiation via beta-catenin. Mol. Cell. Biol., 32, 2966–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim H.J., Kim J.H., Bae S.C., Choi J.Y., Kim H.J. and Ryoo H.M. (2003) The protein kinase C pathway plays a central role in the fibroblast growth factor-stimulated expression and transactivation activity of Runx2. J. Biol. Chem., 278, 319–326. [DOI] [PubMed] [Google Scholar]
- 13. Yoon W.J., Islam R., Cho Y.D., Woo K.M., Baek J.H., Uchida T., Komori T., Wijnen A., Stein J.L., Lian J.B. et al. (2013) Pin1-mediated Runx2 modification is critical for skeletal development. J. Cell. Physiol., 228, 2377-2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoon W.J., Cho Y.D., Kim W.J., Bae H.S., Islam R., Woo K.M., Baek J.H., Bae S.C. and Ryoo H.M. (2014) Prolyl isomerase Pin1-mediated conformational change and subnuclear focal accumulation of Runx2 are crucial for fibroblast growth factor 2 (FGF2)-induced osteoblast differentiation. J. Biol. Chem., 289, 8828–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoon W.J., Islam R., Cho Y.D., Ryu K.M., Shin H.R., Woo K.M., Baek J.H. and Ryoo H.M. (2015) Pin1 plays a critical role as a molecular switch in canonical BMP signaling. J. Cell. Physiol., 230, 640–647. [DOI] [PubMed] [Google Scholar]
- 16. Kim W.J., Islam R., Kim B.S., Cho Y.D., Yoon W.J., Baek J.H., Woo K.M. and Ryoo H.M. (2016) Direct delivery of recombinant Pin1 protein rescued osteoblast differentiation of Pin1 deficient cells. J. Cell. Physiol., in press. [DOI] [PubMed] [Google Scholar]
- 17. Shin H.R., Islam R., Yoon W.J., Lee T., Cho Y.D., Bae H.S., Kim B.S., Woo K.M., Baek J.H. and Ryoo H.M. (2016) Pin1-mediated modification prolongs the nuclear retention of beta-catenin in Wnt3a-induced osteoblast differentiation. J. Biol. Chem., 291, 5555–5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hennig L., Christner C., Kipping M., Schelbert B., Rucknagel K.P., Grabley S., Kullertz G. and Fischer G. (1998) Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry, 37, 5953–5960. [DOI] [PubMed] [Google Scholar]
- 19. Reese S., Vidyasagar A., Jacobson L., Acun Z., Esnault S., Hullett D., Malter J.S. and Djamali A. (2010) The Pin 1 inhibitor juglone attenuates kidney fibrogenesis via Pin 1-independent mechanisms in the unilateral ureteral occlusion model. Fibrogenesis Tissue Repair, 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jeong H.G., Pokharel Y.R., Lim S.C., Hwang Y.P., Han E.H., Yoon J.H., Ahn S.G., Lee K.Y. and Kang K.W. (2009) Novel role of Pin1 induction in type II collagen-mediated rheumatoid arthritis. J. Immunol., 183, 6689–6697. [DOI] [PubMed] [Google Scholar]
- 21. Aithal K.B., Kumar S., Rao B.N., Udupa N. and Rao S.B. (2012) Tumor growth inhibitory effect of juglone and its radiation sensitizing potential: in vivo and in vitro studies. Integr. Cancer. Ther., 11, 68–80. [DOI] [PubMed] [Google Scholar]
- 22. Shukla V., Coumoul X., Wang R.H., Kim H.S. and Deng C.X. (2007) RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat. Genet., 39, 1145–1150. [DOI] [PubMed] [Google Scholar]
- 23. Miura T., Perlyn C.A., Kinboshi M., Ogihara N., Kobayashi-Miura M., Morriss-Kay G.M. and Shiota K. (2009) Mechanism of skull suture maintenance and interdigitation. J. Anat., 215, 642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y., Sun M., Uhlhorn V.L., Zhou X., Peter I., Martinez-Abadias N., Hill C.A., Percival C.J., Richtsmeier J.T., Huso D.L. et al. (2010) Activation of p38 MAPK pathway in the skull abnormalities of Apert syndrome Fgfr2(+P253R) mice. BMC Dev. Biol., 10, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deckelbaum R.A., Majithia A., Booker T., Henderson J.E. and Loomis C.A. (2006) The homeoprotein engrailed 1 has pleiotropic functions in calvarial intramembranous bone formation and remodeling. Development, 133, 63–74. [DOI] [PubMed] [Google Scholar]
- 26. Motch Perrine S.M., Stecko T., Neuberger T., Jabs E.W., Ryan T.M. and Richtsmeier J.T. (2017) Integration of brain and skull in prenatal mouse models of Apert and Crouzon syndromes. Front. Hum. Neurosci., 11, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Purushothaman R., Cox T.C., Maga A.M. and Cunningham M.L. (2011) Facial suture synostosis of newborn Fgfr1(P250R/+) and Fgfr2(S252W/+) mouse models of Pfeiffer and Apert syndromes. Birth Defects Res. A Clin. Mol. Teratol., 91, 603–609. [DOI] [PubMed] [Google Scholar]
- 28. Rice D.P. (2008) Craniofacial sutures. Development, disease and treatment. Preface Front. Oral Biol., 12 xi. [PubMed]
- 29. Maga A.M., Navarro N., Cunningham M.L. and Cox T.C. (2015) Quantitative trait loci affecting the 3D skull shape and size in mouse and prioritization of candidate genes in-silico. Front. Physiol., 6, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Islam R., Yoon W.J., and Ryoo H.M. (2016) Pin1, the master orchestrator of bone cell differentiation. J. Cell. Physiol., in press. [DOI] [PubMed] [Google Scholar]
- 31. Smith T.G., Sweetman D., Patterson M., Keyse S.M. and Munsterberg A. (2005) Feedback interactions between MKP3 and ERK MAP kinase control scleraxis expression and the specification of rib progenitors in the developing chick somite. Development, 132, 1305–1314. [DOI] [PubMed] [Google Scholar]
- 32. Faedo A., Borello U. and Rubenstein J.L. (2010) Repression of Fgf signaling by sprouty1-2 regulates cortical patterning in two distinct regions and times. J. Neurosci., 30, 4015–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu J., Li Z., Hou Y. and Fang W. (2015) Potential mechanisms underlying the Runx2 induced osteogenesis of bone marrow mesenchymal stem cells. Am. J. Transl. Res., 7, 2527–2535. [PMC free article] [PubMed] [Google Scholar]
- 34. Komori T., Yagi H., Nomura S., Yamaguchi A., Sasaki K., Deguchi K., Shimizu Y., Bronson R.T., Gao Y.H., Inada M. et al. (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell, 89, 755–764. [DOI] [PubMed] [Google Scholar]
- 35. Park M.H., Shin H.I., Choi J.Y., Nam S.H., Kim Y.J., Kim H.J. and Ryoo H.M. (2001) Differential expression patterns of Runx2 isoforms in cranial suture morphogenesis. J. Bone Miner. Res. , 16, 885–892. [DOI] [PubMed] [Google Scholar]
- 36. Misteli T. and Spector D.L. (1998) The cellular organization of gene expression. Curr. Opin. Cell. Biol., 10, 323–331. [DOI] [PubMed] [Google Scholar]
- 37. Iborra F.J., Pombo A., McManus J., Jackson D.A. and Cook P.R. (1996) The topology of transcription by immobilized polymerases. Exp. Cell. Res., 229, 167–173. [DOI] [PubMed] [Google Scholar]
- 38. Grande M.A., Kraan I., Jong L. and Driel R. (1997) Nuclear distribution of transcription factors in relation to sites of transcription and RNA polymerase II. J. Cell. Sci., 110 ( Pt 15), 1781-1791. [DOI] [PubMed] [Google Scholar]
- 39. Szentirmay M.N. and Sawadogo M. (2000) Spatial organization of RNA polymerase II transcription in the nucleus. Nucleic Acids Res., 28, 2019–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Florencio-Silva R., Sasso G.R., Sasso-Cerri E., Simoes M.J., and Cerri P.S. (2015) Biology of bone tissue: structure, function, and factors that influence bone cells. Biomed. Res. Int., 2015, 421746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Galindo M., Pratap J., Young D.W., Hovhannisyan H., Im H.J., Choi J.Y., Lian J.B., Stein J.L., Stein G.S. and Wijnen A.J. (2005) The bone-specific expression of Runx2 oscillates during the cell cycle to support a G1-related antiproliferative function in osteoblasts. J. Biol. Chem., 280, 20274-20285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Komori T. (2010) Regulation of osteoblast differentiation by Runx2. Adv. Exp. Med. Biol., 658, 43–49. [DOI] [PubMed] [Google Scholar]
- 43. Su N., Jin M. and Chen L. (2014) Role of FGF/FGFR signaling in skeletal development and homeostasis: learning from mouse models. Bone Res., 2, 14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Twigg S.R. and Wilkie A.O. (2015) A genetic-pathphysiological framework for craniosynostosis. Am. J. Hum. Genet., 97, 359–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Phupong V., Srichomthong C. and Shotelersuk V. (2004) Prenatal exclusion of Crouzon syndrome by mutation analysis of FGFR2. Southeast Asian J. Trop. Med. Public Health, 35, 977–979. [PubMed] [Google Scholar]
- 46. Tanimoto Y., Veistinen L., Alakurtti K., Takatalo M. and Rice D.P. (2012) Prevention of premature fusion of calvarial suture in GLI-Kruppel family member 3 (Gli3)-deficient mice by removing one allele of Runt-related transcription factor 2 (Runx2). J. Biol. Chem., 287, 21429–21438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hsu T., McRackan D., Vincent T.S. and Gert de Couet H. (2001) DrosophilaPin1 prolyl isomerase Dodo is a MAP kinase signal responder during oogenesis. Nat Cell Biol, 3, 538–543. [DOI] [PubMed] [Google Scholar]
- 48. Teven C.M., Farina E.M., Rivas J. and Reid R.R. (2014) Fibroblast growth factor (FGF) signaling in development and skeletal diseases. Genes Dis., 1, 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen L., Li D., Li C., Engel A. and Deng C.X. (2003) A Ser252Trp [corrected] substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone, 33, 169–178. [DOI] [PubMed] [Google Scholar]
- 50. Wang Y., Xiao R., Yang F., Karim B.O., Iacovelli A.J., Cai J., Lerner C.P., Richtsmeier J.T., Leszl J.M., Hill C.A. et al. (2005) Abnormalities in cartilage and bone development in the Apert syndrome FGFR2(+/S252W) mouse. Development, 132, 3537–3548. [DOI] [PubMed] [Google Scholar]
- 51. Morriss-Kay G.M. and Wilkie A.O. (2005) Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J. Anat., 207, 637–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen P., Zhang L., Weng T., Zhang S., Sun S., Chang M., Li Y., Zhang B. and Zhang L. (2014) A Ser252Trp mutation in fibroblast growth factor receptor 2 (FGFR2) mimicking human Apert syndrome reveals an essential role for FGF signaling in the regulation of endochondral bone formation. PLoS One, 9, e87311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee Y.C., Song I.W., Pai Y.J., Chen S.D. and Chen Y.T. (2017) Knock-in human FGFR3 achondroplasia mutation as a mouse model for human skeletal dysplasia. Sci Rep, 7, 43220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Heuze Y., Singh N., Basilico C., Jabs E.W., Holmes G. and Richtsmeier J.T. (2014) Morphological comparison of the craniofacial phenotypes of mouse models expressing the Apert FGFR2 S252W mutation in neural crest- or mesoderm-derived tissues. Bone, 63, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cowan C.M., Quarto N., Warren S.M., Salim A. and Longaker M.T. (2003) Age-related changes in the biomolecular mechanisms of calvarial osteoblast biology affect fibroblast growth factor-2 signaling and osteogenesis. J. Biol. Chem., 278, 32005–32013. [DOI] [PubMed] [Google Scholar]
- 56. Fakhry A., Ratisoontorn C., Vedhachalam C., Salhab I., Koyama E., Leboy P., Pacifici M., Kirschner R.E. and Nah H.D. (2005) Effects of FGF-2/-9 in calvarial bone cell cultures: differentiation stage-dependent mitogenic effect, inverse regulation of BMP-2 and noggin, and enhancement of osteogenic potential. Bone, 36, 254–266. [DOI] [PubMed] [Google Scholar]
- 57. Quarto N. and Longaker M.T. (2006) FGF-2 inhibits osteogenesis in mouse adipose tissue-derived stromal cells and sustains their proliferative and osteogenic potential state. Tissue Eng., 12, 1405–1418. [DOI] [PubMed] [Google Scholar]
- 58. Quarto N., Behr B., Li S. and Longaker M.T. (2009) Differential FGF ligands and FGF receptors expression pattern in frontal and parietal calvarial bones. Cells Tissues Organs, 190, 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jiang X., Iseki S., Maxson R.E., Sucov H.M., and Morriss-Kay G.M. (2002) Tissue origins and interactions in the mammalian skull vault. Dev. Biol., 241, 106–116. [DOI] [PubMed] [Google Scholar]
- 60. Avci E., Arikoglu H. and Erkoc Kaya D. (2016) Investigation of juglone effects on metastasis and angiogenesis in pancreatic cancer cells. Gene, 588, 74–78. [DOI] [PubMed] [Google Scholar]
- 61. Zhang W., Li Y., Luo J., Lu X., Chen M., Zhu W. and Jiang Y. (2015) Juglone inhibits proliferation and induces apoptosis of human cervical squamous cancer SiHa cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi, 31, 186–189. [PubMed] [Google Scholar]
- 62. Chao S.H., Greenleaf A.L. and Price D.H. (2001) Juglone, an inhibitor of the peptidyl-prolyl isomerase Pin1, also directly blocks transcription. Nucleic Acids Res., 29, 767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Saling S.C., Comar J.F., Mito M.S., Peralta R.M. and Bracht A. (2011) Actions of juglone on energy metabolism in the rat liver. Toxicol. Appl. Pharmacol., 257, 319–327. [DOI] [PubMed] [Google Scholar]
- 64. Chen L.J., Lebetkin E.H. and Burka L.T. (2005) Metabolism and disposition of juglone in male F344 rats. Xenobiotica, 35, 1019–1034. [DOI] [PubMed] [Google Scholar]
- 65. Yang F., Wang Y., Zhang Z., Hsu B., Jabs E.W. and Elisseeff J.H. (2008) The study of abnormal bone development in the Apert syndrome Fgfr2+/S252W mouse using a 3D hydrogel culture model. Bone, 43, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Park W.J., Theda C., Maestri N.E., Meyers G.A., Fryburg J.S., Dufresne C., Cohen M.M. Jr. and Jabs E.W. (1995) Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am. J. Hum. Genet., 57, 321–328. [PMC free article] [PubMed] [Google Scholar]
- 67. Slaney S.F., Oldridge M., Hurst J.A., Moriss-Kay G.M., Hall C.M., Poole M.D. and Wilkie A.O. (1996) Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am. J. Hum. Genet., 58, 923–932. [PMC free article] [PubMed] [Google Scholar]
- 68. Islam R., Yoon W.J. and Ryoo H.M. (2017) Pin1, the master orchestrator of bone cell differentiation. J. Cell. Physiol., 232, 2339–2347. [DOI] [PubMed] [Google Scholar]
- 69. Kim W.J., Islam R., Kim B.S., Cho Y.D., Yoon W.J., Baek J.H., Woo K.M. and Ryoo H.M. (2017) Direct delivery of recombinant Pin1 protein rescued osteoblast differentiation of Pin1-deficient cells. J. Cell. Physiol., 232, 2798–2805. [DOI] [PubMed] [Google Scholar]
- 70. Jeon E.J., Lee K.Y., Choi N.S., Lee M.H., Kim H.N., Jin Y.H., Ryoo H.M., Choi J.Y., Yoshida M., Nishino N. et al. (2006) Bone morphogenetic protein-2 stimulates Runx2 acetylation. J. Biol. Chem., 281, 16502–16511. [DOI] [PubMed] [Google Scholar]
- 71. Kim H.J., Park H.D., Kim J.H., Cho J.Y., Choi J.Y., Kim J.K., Kim H.J., Shin H.I. and Ryoo H.M. (2004) Establishment and characterization of a stable cell line to evaluate cellular Runx2 activity. J. Cell. Biochem., 91, 1239–1247. [DOI] [PubMed] [Google Scholar]
- 72. Park O.J., Kim H.J., Woo K.M., Baek J.H. and Ryoo H.M. (2010) FGF2-activated ERK mitogen-activated protein kinase enhances Runx2 acetylation and stabilization. J. Biol. Chem., 285, 3568–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yin L., Du X., Li C., Xu X., Chen Z., Su N., Zhao L., Qi H., Li F., Xue J. et al. (2008) A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone, 42, 631–643. [DOI] [PubMed] [Google Scholar]
- 74. Holmes G., Rothschild G., Roy U.B., Deng C.X., Mansukhani A. and Basilico C. (2009) Early onset of craniosynostosis in an Apert mouse model reveals critical features of this pathology. Dev. Biol., 328, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fujimori F., Takahashi K., Uchida C. and Uchida T. (1999) Mice lacking Pin1 develop normally, but are defective in entering cell cycle from G(0) arrest. Biochem. Biophys. Res. Commun., 265, 658–663. [DOI] [PubMed] [Google Scholar]
- 76. Lakso M., Pichel J.G., Gorman J.R., Sauer B., Okamoto Y., Lee E., Alt F.W. and Westphal H. (1996) Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. USA, 93, 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bae H.S., Yoon W.J., Cho Y.D., Islam R., Shin H.R., Kim B.S., Lim J.M., Seo M.S., Cho S.A., Choi K.Y. et al. (2017) An HDAC inhibitor, Entinostat/MS-275, partially prevents delayed cranial suture closure in heterozygous Runx2 null mice. J. Bone. Miner. Res., in press. [DOI] [PubMed] [Google Scholar]
- 78. Ruan M.Z., Dawson B., Jiang M.M., Gannon F., Heggeness M. and Lee B.H. (2013) Quantitative imaging of murine osteoarthritic cartilage by phase-contrast micro-computed tomography. Arthritis Rheum., 65, 388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ruan M.Z., Cerullo V., Cela R., Clarke C., Lundgren-Akerlund E., Barry M.A. and Lee B.H. (2016) Treatment of osteoarthritis using a helper-dependent adenoviral vector retargeted to chondrocytes. Mol. Ther. Methods Clin. Dev., 3, 16008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cho Y.D., Bae H.S., Lee D.S., Yoon W.J., Woo K.M., Baek J.H., Lee G., Park J.C., Ku Y. and Ryoo H.M. (2016) Epigenetic priming confers direct cell trans-differentiation from adipocyte to osteoblast in a transgene-free state. J. Cell. Physiol., 231, 1484–1494. [DOI] [PubMed] [Google Scholar]
- 81. Tanimoto Y., Yokozeki M., Hiura K., Matsumoto K., Nakanishi H., Matsumoto T., Marie P.J. and Moriyama K. (2004) A soluble form of fibroblast growth factor receptor 2 (FGFR2) with S252W mutation acts as an efficient inhibitor for the enhanced osteoblastic differentiation caused by FGFR2 activation in Apert syndrome. J. Biol. Chem., 279, 45926–45934. [DOI] [PubMed] [Google Scholar]
- 82. Cho Y., Hong J., Ryoo H., Kim D., Park J. and Han J. (2015) Osteogenic responses to zirconia with hydroxyapatite coating by aerosol deposition. J. Dent. Res., 94, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yu K., Herr A.B., Waksman G. and Ornitz D.M. (2000) Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc. Natl. Acad. Sci. USA, 97, 14536–14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.