

Abstract
The 1555 A to G substitution in mitochondrial 12S A‐site rRNA is associated with maternally transmitted deafness of variable penetrance in the absence of otherwise overt disease. Here, we recapitulate the suggested A1555G‐mediated pathomechanism in an experimental model of mitoribosomal mistranslation by directed mutagenesis of mitoribosomal protein MRPS5. We first establish that the ratio of cysteine/methionine incorporation and read‐through of mtDNA‐encoded MT‐CO1 protein constitute reliable measures of mitoribosomal misreading. Next, we demonstrate that human HEK293 cells expressing mutant V336Y MRPS5 show increased mitoribosomal mistranslation. As for immortalized lymphocytes of individuals with the pathogenic A1555G mutation, we find little changes in the transcriptome of mutant V336Y MRPS5 HEK cells, except for a coordinated upregulation of transcripts for cytoplasmic ribosomal proteins. Homozygous knock‐in mutant Mrps5 V338Y mice show impaired mitochondrial function and a phenotype composed of enhanced susceptibility to noise‐induced hearing damage and anxiety‐related behavioral alterations. The experimental data in V338Y mutant mice point to a key role of mitochondrial translation and function in stress‐related behavioral and physiological adaptations.
Keywords: aging, disease, misreading, mitochondria, protein synthesis
Subject Categories: Molecular Biology of Disease, Protein Biosynthesis & Quality Control
Introduction
Mitochondria are multi‐functional organelles involved in oxidative metabolism, cellular homeostasis, and signal transduction. They are the primary energy‐providing system in eukaryotic cells by accommodating the enzymatic machinery of oxidative phosphorylation for ATP production. The mitochondrial oxidative phosphorylation system is made up of more than 100 different proteins, 13 of which are encoded by mitochondrial DNA and all the others by nuclear genes 1. Mitochondrial DNA is maternally inherited, and the human mtDNA encodes in addition to the 13 proteins of the respiratory electron transport chain (ETC) for 22 tRNA and two ribosomal RNA genes required for their translation 2, 3.
Mitochondria are of endosymbiotic origin 4, and the mitochondrial ribosomes are more closely related to bacterial ribosomes than to eukaryotic cytosolic ribosomes 5. Mitochondrial protein synthesis allows for the translation of the 13 respiratory subunits encoded by the mitochondrial genome 6. All of the more than 1,000 other proteins of the mammalian mitochondrial proteome are synthesized by the cytosolic ribosome 7, 8, 9, 10.
Classical mitochondrial diseases result from mutations in mtDNA or nuclear genes that disrupt mitochondrial function 11, 12. Back in 1993, a pivotal study established that the nucleotide 1555 A to G substitution in the mitochondrial 12S rRNA gene predisposes to maternally transmitted, non‐syndromic sensorineural deafness 13. The A1555G mutation produces a clinical phenotype that may range from normal hearing to progressive hearing loss or severe congenital deafness 14. Early biochemical investigations of the A1555G mutation using transmitochondrial cell lines pointed to a role for mitochondrial protein synthesis 15, but the molecular mechanisms involved remained obscure 16, 17, 18. In the absence of experimental models of mtDNA manipulation, we have previously used bacterial genetics to reconstruct the corresponding rRNA substitution in bacterial ribosomes. These studies suggested that the pathogenic A1555G mutation affects ribosomal accuracy 19. The postulated mechanism, however, was difficult to reconcile with the significant and diverse pathologies associated with mutations in nuclear‐encoded mitochondrial aminoacyl‐tRNA synthetases 20, 21, 22 and the apparent absence of A1555G‐associated disease in non‐cochlear tissues. Here, to address this question, we mutated the nuclear‐encoded mitochondrial ribosomal protein MRPS5 to result in an experimental model of mitoribosomal mistranslation.
Results
Identification of a potentially misreading mutation in mitoribosomal protein MRPS5 and in homologous bacterial ribosomal protein RpsE
The amino acid replacement S200Y in yeast cytosolic ribosomal protein Rps2 (uS5) is a ribosomal ambiguity mutation (ram) that confers misreading in yeast cytosolic ribosomes 23. We modeled the corresponding amino acid replacement on available crystal structures of the homologous mitochondrial protein MRPS5 (uS5m) and homologous bacterial protein RpsE (uS5) 24, 25. Modeling indicates that replacement of the corresponding native amino acid with the large aromatic tyrosine residue in both mitochondrial MRPS5 (human MRPS5 V336Y) and bacterial RpsE (Escherichia coli rpsE A127Y) would result in steric hindrance within the protein C‐terminal domain, similar to what is observed for S200Y in the yeast Rps2 protein (Appendix Fig S1).
RpsE A127Y is a misreading mutation in bacterial ribosomes
To study whether the mutation rpsE A127Y is a ram mutation in bacterial ribosomes, we generated bacterial mutants by introducing the corresponding mutation into E. coli (rpsE A127Y) and Mycobacterium smegmatis (rpsE S152Y) (Appendix Fig S1). We determined ribosomal misreading and read‐through in cell‐free translation assays using dual luciferase gain‐of‐function reporters. In addition, we included bacterial mitohybrid ribosomes with and without the pathogenic A1555G mutation in the analysis. Reconstructions of mitochondrial 12S rRNA in bacterial ribosomes, with the bacterial decoding A‐site in rRNA helix 44 replaced by the corresponding mitochondrial sequence, have established that the A1555G mutation affects translation accuracy 19. E. coli rpsE A127Y, M. smegmatis rpsE S152Y, and bacterial mitohybrid ribosomes with the pathogenic A1555G mutation all showed significantly increased levels of misreading of near‐cognate codons and read‐through in comparison with their respective controls (Fig 1A and B).
Figure 1. Determination of translational accuracy in cell‐free translation assays.
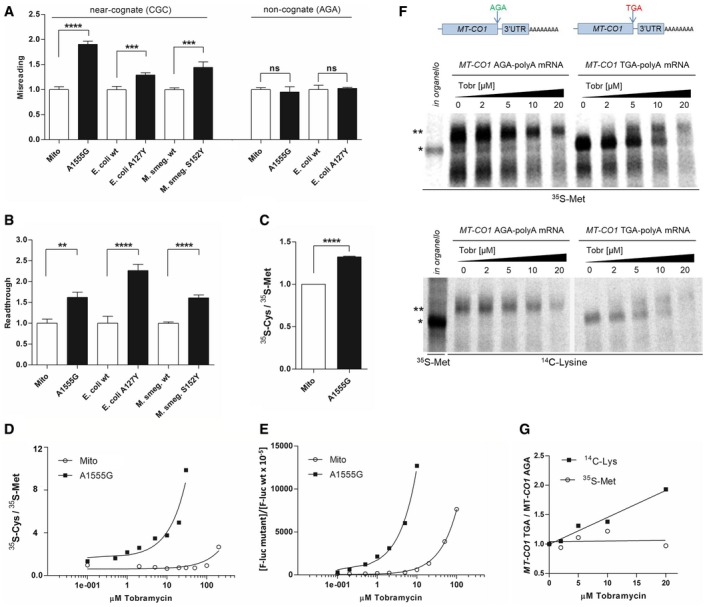
-
A, BMisreading (A) and read‐through (B) were measured by dual luciferase gain‐of‐function assays using wild‐type mitohybrid ribosomes (n = 8 misreading, n = 4 read‐through), mutant A1555G mitohybrid ribosomes (n = 8 misreading, n = 4 read‐through), E. coli wild‐type ribosomes (n = 9), E. coli mutant A127Y ribosomes (n = 9), M. smegmatis merodiploid wild‐type ribosomes (n = 9), and M. smegmatis merodiploid mutant S152Y ribosomes (n = 9). Mistranslation was determined by assessing the misreading at His245 (CAC codon) of the F‐luc gene that was replaced by the near‐cognate codon CGC or the non‐cognate codon AGA, both encoding for Arg. To determine read‐through, the Asp357 (GAC codon) of the F‐luc gene was replaced by the nonsense TGA stop codon. Results are derived by calculating mutant hFluc/hRluc activity related to wild‐type hFluc/hRluc activity. Wild‐type samples were set as 1, error bars indicate SEM; **P < 0.01, ***P < 0.005, ****P < 0.001, ns, not significant (Student's t‐test).
-
CRatio of 35S‐cysteine/35S‐methionine incorporation by in vitro translation using wild‐type mitohybrid ribosomes, mutant A1555G mitohybrid ribosomes, and MT‐CO1 mRNA. Following immunoprecipitation, the MT‐CO1 band was quantified by autoradiography, and wild‐type mitohybrid ribosomes were set as 1 (n = 3), error bars indicate SEM; ****P < 0.001 (Student's t‐test). Products of immunoprecipitation as resolved by SDS–PAGE are shown exemplarily in Appendix Fig S2E.
-
DRatio of 35S‐cysteine/35S‐methionine incorporation by in vitro translation in the presence of tobramycin using wild‐type mitohybrid ribosomes, mutant A1555G mitohybrid ribosomes, and MT‐CO1 mRNA; the MT‐CO1 band was quantified by autoradiography.
-
EMisreading of wild‐type mitohybrid ribosomes and mutant A1555G mitohybrid ribosomes in the presence of tobramycin. Misreading was assessed by dual luciferase assay, and results derived by calculating mutant hFluc/hRluc activity are related to wild‐type hFluc/hRluc activity.
-
FIn vitro translation of MT‐CO1‐TGA mRNA and MT‐CO1‐AGA mRNA using mutant A1555G mitohybrid ribosomes in the presence of tobramycin (0–20 μM). Autoradiography of immunoprecipitated MT‐CO1 proteins; top: 35S‐Met labeling, bottom: 14C‐Lys labeling. In organello‐translated 35S‐Met‐labeled MT‐CO1 was used as a marker. * MT‐CO1, ** MT‐CO1 extended by read‐through; MT‐CO1 AGA‐polyA and MT‐CO1 TGA‐polyA constructs used for in vitro translation are schematized.
-
GRatio of 14C‐Lys/35S‐Met‐labeled immunoprecipitated MT‐CO1 proteins. In vitro translation using MT‐CO1‐TGA mRNA, MT‐CO1‐AGA mRNA, and mutant A1555G mitohybrid ribosomes in the presence of tobramycin (0–20 μM). The corresponding MT‐CO1 band was quantified by autoradiography.
Ratio of cysteine/methionine incorporation and increased read‐through upon translation of MT‐CO1 mRNA are markers for ribosomal misreading
Mitochondrial ribosomes are neither suitable for cell‐free translation assays, nor is it technically feasible to introduce reporter genes to probe mitoribosomal accuracy, given the current limitations in genetic manipulation of mitochondria of higher eukaryotes 26.
Amino acid sequence analysis of the 13 mtDNA‐encoded human proteins (NCBI, JO1415.2) revealed that the mitochondrial cytochrome oxidase subunit I MT‐CO1 (COX1) protein contains a single cysteine and 32 methionines. Additionally, the nucleic acid sequence encoding MT‐CO1 shows 96 near‐cognate codons for cysteine and 48 near‐cognate codons for methionine. This yields a ratio of near‐cognate/cognate codons for cysteine versus near‐cognate/cognate codons for methionine of 64.0 [(96/1)/(48/32); for calculation of cognate and near‐cognate frequencies, see Appendix Table S1]. Based on these calculations, we hypothesized that misreading would increase the ratio of cysteine/methionine incorporation and decided to assess ribosomal accuracy by determining the amount of cysteine versus methionine incorporation in MT‐CO1 protein.
We used bacterial A1555G mutant and wild‐type mitohybrid ribosomes to demonstrate that experimental determination of cysteine/methionine incorporation in MT‐CO1 can be used to assess misreading. The MT‐CO1 sequence adapted to bacterial codon usage resulted in a change in the near‐cognate/cognate codon ratio for cysteine versus methionine to 86.7 [(130/1)/(48/32); the ratio is different from the mitochondrial ratio because bacteria have fewer near‐cognate methionine codons]. The ratio of cysteine/methionine incorporated upon in vitro translation of MT‐CO1 mRNA was significantly increased in the mutant A1555G mitohybrid ribosomes as compared to wild‐type mitohybrid ribosomes (Fig 1C). Further support for this observation was provided by the finding that treatment with the aminoglycoside tobramycin increased misreading in a similar manner when assessing MT‐CO1 cysteine/methionine incorporation or when using a luciferase gain‐of‐function assay, for both A1555G mitohybrid ribosomes and wild‐type mitohybrid ribosomes (Fig 1D and E; note that the pathogenic A1555G mutation confers hypersusceptibility to tobramycin‐induced misreading 19).
The hungry AGA codon at the 3′ end of the MT‐CO1 protein results in a −1 frameshift on the ribosome, allowing a UAG stop codon to be recognized by mitochondrial release factor mtRF1a 27. We hypothesized that misreading mitochondrial ribosomes should bypass the MT‐CO1 stop signal by read‐through, resulting in an extended protein with a C‐terminal poly‐lysine stretch (no stop codon is present in the MT‐CO1 3′ UTR in‐frame or in −1 frame, facilitating translation of the polyA tail). We first validated this assay using bacterial A1555G mitohybrid ribosomes because of their hypersusceptibility to aminoglycosides 19. Using the MT‐CO1 sequence adapted to bacterial codon usage, two synthetic mRNAs were generated—one MT‐CO1 mRNA with the native AGA codon (MT‐CO1 AGA, AGA encoding arginine in bacterial translation) and a second MT‐CO1 mRNA with the AGA codon replaced by a UGA stop codon (MT‐CO1 TGA). Both constructs carry the 3′ UTR of MT‐CO1 (69 nt) followed by a polyA tail of 51 nucleotides. In the absence of aminoglycosides, A1555G hybrid ribosomes produced a 57.1 kD‐sized protein corresponding to the native MT‐CO1 protein when translating MT‐CO1 UGA mRNA and an extended MT‐CO1 62.1 kD‐sized protein when translating MT‐CO1 AGA mRNA (Fig 1F). Two effects were observed in the presence of increasing amounts of aminoglycoside. First, protein synthesis became inhibited due to the inhibitory effect of tobramycin on ribosomal translocation. Second, increasing amounts of the extended 62.1 kD MT‐CO1 protein were produced for the MT‐CO1 UGA mRNA due to tobramycin‐induced UGA stop codon read‐through, 3′ UTR translation, and synthesis of a poly‐lysine stretch (Fig 1F). In further support of our conclusion, in vitro translation in the presence of 14C‐lysine showed that the ratio of 14C‐lysine/35S‐methionine incorporation increased in the presence of tobramycin (Fig 1G), as expected when aminoglycoside‐induced read‐through results in the synthesis of a poly‐lysine stretch. No extended MT‐CO1 protein was synthesized in the presence of tobramycin upon translation of MT‐CO1 mRNA with a TAA stop codon lacking 3′ UTR and polyA tail (Appendix Fig S2A).
Collectively, these data demonstrate that both the ratio of cysteine/methionine incorporation and the increased read‐through upon translation of MT‐CO1 mRNA constitute valid markers for ribosomal misreading.
Mutation V336Y in MRPS5 confers mitoribosomal misreading in in organello translation
We next generated stably transfected human HEK cells of mutant V336Y MRPS5 and wild‐type MRPS5 controls. We found evidence that the transgene‐encoded MRPS5 protein localizes to the mitochondrial fraction using myc‐tagged MRPS5 (Appendix Fig S2B). A GFP reporter was added downstream of MRPS5 via an internal ribosomal entry site (IRES) to facilitate monitoring of transgene expression. V336Y mutants and wild‐type control transfectants showed similar GFP fluorescence (Appendix Fig S2C). We quantified MRPS5 mRNA expression by RT–qPCR to demonstrate that transfected V336Y mutant MRPS5 and transfected wild‐type MRPS5 produce similar levels of MRPS5 mRNA (Appendix Fig S2D).
To investigate the accuracy of mitochondrial translation, we used in organello translation and determined the ratio of cysteine/methionine incorporation in MT‐CO1 protein. In organello translation in the presence of the misreading agent tobramycin resulted in a significantly increased ratio of cysteine/methionine incorporation in MT‐CO1 protein (Fig 2A). Compared to MT‐CO1 with a ratio of 64.0 for near‐cognate/cognate codons for cysteine versus methionine, this ratio is only 3.8 for the mtDNA‐encoded cytochrome oxidase subunit 2 MT‐CO2 (near‐cognate/cognate codons for cysteine versus near‐cognate/cognate codons for methionine: 29/3 versus 26/10, see Appendix Table S1). We thus predicted that compared to MT‐CO1, in organello translation in the presence of the misreading agent tobramycin should not grossly affect the ratio of cysteine/methionine incorporation in MT‐CO2. As shown in Fig 2B, treatment with tobramycin resulted in a dose‐dependent increase in the ratio of cysteine/methionine incorporation in MT‐CO1 protein, but did not measurably affect this ratio in MT‐CO2 protein. We next studied the ratio of cysteine/methionine incorporation in MT‐CO1 protein in mutant V336Y MRPS5 and wild‐type control transfectants. In comparison with the wild‐type control mutant, V336Y MRPS5 transfectants showed a significantly increased ratio of cysteine/methionine incorporation in MT‐CO1 protein (Fig 2C).
Figure 2. Assessment of mitoribosomal misreading in in organello translation.
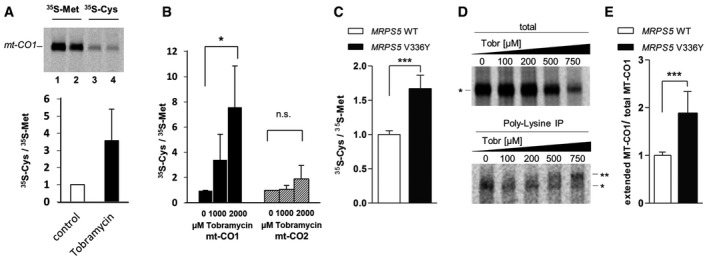
- Autoradiography of in organello mitochondrial translation products derived from HEK293 wild‐type cells in the presence of tobramycin, incorporation of 35S‐Met and 35S‐Cys in MT‐CO1 protein. Following in organello translation proteins were immunoprecipitated and analyzed by autoradiography. Lane 1, 3: controls; lanes 2, 4: in organello translation in the presence of 1,000 μM tobramycin. MT‐CO1 bands were quantified and ratio of 35S‐Cys/35S‐Met calculated, and the 35S‐Cys/35S‐Met ratio in the absence of tobramycin was set as 1 (n = 3), error bars indicate SEM.
- Ratio of 35S‐Cys/35S‐Met‐labeled MT‐CO1 and MT‐CO2 proteins synthesized in in organello translation and effect of tobramycin. Following in organello translation in the presence of tobramycin (1,000 μM, 2,000 μM), proteins were immunoprecipitated and analyzed by autoradiography. MT‐CO1 and MT‐CO2 bands were quantified, and ratio of 35S‐Cys/35S‐Met incorporation was calculated (n = 3), error bars indicate SEM; *P < 0.05 (Student's t‐test), ns, not significant.
- Ratio of 35S‐Cys/35S‐Met‐labeled MT‐CO1 protein synthesized in in organello translation using mitochondria from HEK293 MRPS5 wild‐type cells and HEK MRPS5 mutant V336Y cells. Following in organello translation, proteins were immunoprecipitated and analyzed by autoradiography. MT‐CO1 bands quantified and ratios calculated. The ratio of the MRPS5 wild‐type was set as 1 (n = 8 clones), error bars indicate SD; ***P < 0.0001 (Student's t‐test).
- Autoradiography of in organello mitochondrial translation products derived from HEK293 MRPS5 wild‐type cells in the presence of tobramycin (0–750 μM). Proteins were 35S‐Met‐labeled and loaded either directly on SDS–PAGE (total, top) or following poly‐lysine immunoprecipitation (bottom). * MT‐CO1 and ** MT‐CO1 with poly‐lysine extension.
- Quantification of MT‐CO1 proteins in HEK293 MRPS5 wild‐type cells and HEK293 MRPS5 mutant V336Y cells. In organello mitochondrial translation in the presence of 35S‐Met and 750 μM tobramycin. Proteins were loaded either directly on SDS–PAGE (total) or following poly‐lysine immunoprecipitation and analyzed by autoradiography. For total protein samples, the MT‐CO1 band was quantified, for poly‐lysine immunoprecipitated samples, the extended MT‐CO1 band was quantified and the ratio of extended MT‐CO1/total MT‐CO1 was calculated. The ratio of MRPS5 wild‐type was set as 1 (MRPS5 wild‐type, n = 5 clones, MRPS5 V336Y, n = 7 clones), error bars indicate SD; ***P < 0.0001 (Student's t‐test).
Immunoprecipitation with anti‐lysine antibodies was used to study read‐through of MT‐CO1 in mitochondrial in organello translation. In the presence of increasing tobramycin concentrations, increasing amounts of extended MT‐CO1 protein were produced (Fig 2D). Compared to wild‐type MRPS5 transfected cells, mitochondrial extracts from mutant V336Y cells showed significantly increased read‐through of the MT‐CO1 protein (Fig 2E).
Together, and consistent with our modeling predictions, these findings demonstrate that the MRPS5 V336Y mutation confers mitoribosomal misreading.
Effects of MRPS5 V336Y‐mediated mitoribosomal misreading in HEK293 cells
Measurements of ATP levels, mitochondrial mass, mtDNA/nuclear DNA ratio, and oxygen consumption showed no significant differences between wild‐type MRPS5‐transfected and mutant V336Y MRPS5‐transfected cells (Appendix Fig S3A–D). Measurements of generation time and quantitative (35S‐Met incorporation) and qualitative (SDS–PAGE of 35S‐Met‐labeled proteins) assessments of mitochondrial translation (Fig 3A–C) also showed no differences. We corroborated the finding that generation time and protein synthesis efficacy were unaffected by using the corresponding mutation in the bacterial system (E. coli—rpsE A127Y, M. smegmatis—rspE S152Y; see Appendix Fig S3E and F). These data indicate that, despite conferring misreading, the homologous mutations in ribosomal proteins MRPS5 and rpsE do not grossly affect efficiency of protein synthesis in phylogenetically related translation systems. Together, these data demonstrate that the mitoribosomal misreading mutation MRPS5 V336Y little affects HEK293 cells.
Figure 3. Analysis of HEK293 mutant MRPS5 V336Y and HEK293 MRPS5 wild‐type cells.
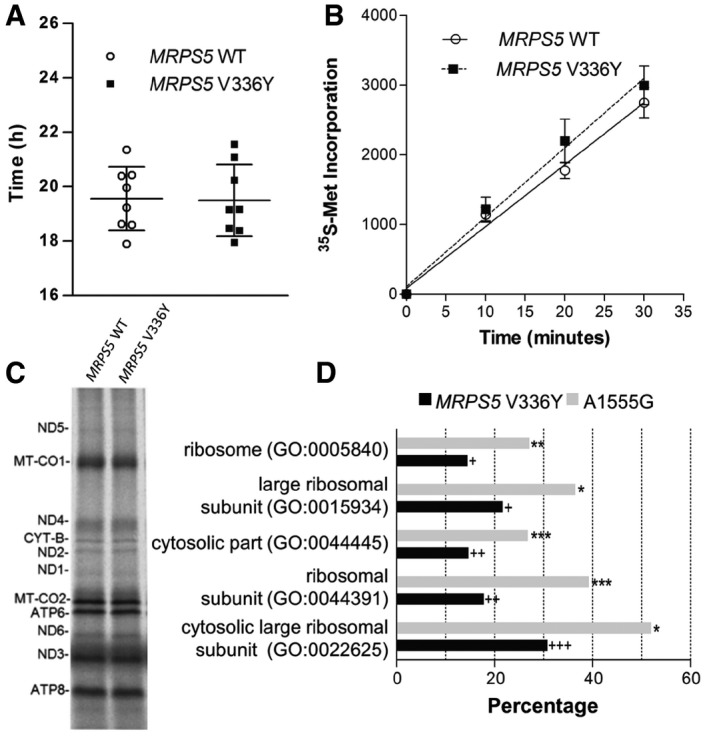
- Generation time (n = 8 clones, ± SD).
- Whole cell in vivo mitochondrial translation as determined by 35S‐Met incorporation (n = 4 clones, ± SEM).
- Autoradiography of in organello translation using 35S‐Met labeling.
- Top five GO terms (cellular component) associated with genes upregulated in HEK293 mutant MRPS5 V336Y compared to HEK293 wild‐type MRPS5. Corresponding GO terms associated with genes upregulated in mutant MT‐RNR1 A1555G lymphocytes (Hollis et al, 2015) were plotted for comparison. The bars represent the percentage of genes that map to a GO term and which are upregulated. Fisher's exact t‐test, + P < 10−6, ++ P < 10−7, +++ P < 10−8 (MRPS5 V336Y n = 4, MRPS5 wild‐type n = 6); *P < 10−30, **P < 10−40, ***P < 10−50 (A1555G n = 28, wild‐type n = 15).
In comparison with wild‐type MRPS5‐transfected cells, transcriptomic analysis of the V336Y mutants by RNA sequencing revealed little differences, except for an overexpression of cytoplasmic ribosomal proteins. The top five affected GO terms were all cytoplasmic ribosome‐related (GO term ribosome, adjusted P‐value 2.46 × 10−4; see Fig 3D).
Mutation V338Y in MRPS5 affects ETC function in brain cells in vivo
Next, we constructed homozygous knock‐in mutant Mrps5 mice (Mrps5 V338Y/V338Y; in mice, the homologous position in Mrps5 is V338Y, see Appendix Fig S1) using a FLEx strategy; for further details, see Materials and Methods and Appendix Fig S4. Mutant Mrps5 V338Y mice are viable, born at the expected frequencies, have normal physical appearance, and show no overt signs of disease.
We evaluated the capacity of the oxidative phosphorylation system in brain cortical mitochondria of both mature (9‐month‐old) and aged (19‐month‐old) Mrps5 V338Y/V338Y and wild‐type control mice using the Seahorse XF24 flux analyzer system (Fig 4A–D). Total oxygen consumption rate (OCR) was significantly reduced in the mitochondria of 9‐ and 19‐month‐old Mrps5 V338Y mice compared to mitochondria of age‐matched wild‐type mice. Specifically, basal activity (state 2) and maximal respiration (state 3, which measures the capacity of mitochondria to metabolize oxygen in the presence of the ATP synthase substrate ADP) showed an age‐ and mutation‐related decline (Fig 4B and C). In addition, respiration in the absence of a proton gradient (uncoupled state 3u following injection of FCCP) was significantly impaired in the mutant mice (Fig 4D).
Figure 4. Mutation‐related mitochondrial function in brain.
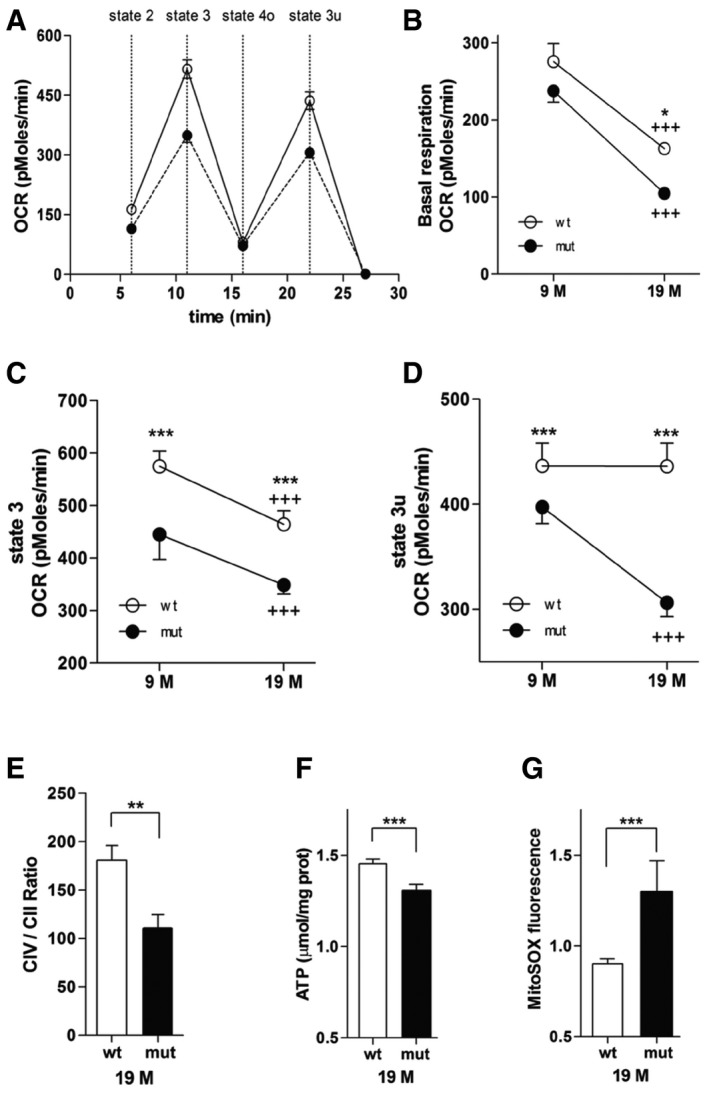
-
A–DOxygen consumption rate (OCR) was measured in freshly isolated mitochondria from wild‐type and mutant mouse cortex. (A) OCR in brain mitochondria from 19‐month‐old mutant mice (black circle) compared with OCR from wild‐type mice (white circle). The sequential injection of mitochondrial substrates and inhibitors is indicated by dotted lines (see details in the Materials and Methods section). Values corresponding to the different respiratory states are shown. (B) Baseline OCR, effect of age (P < 0.0001), and genotype (P < 0.05). (C) State 3 OCR (after addition of ADP), effect of age (P < 0.001), and genotype (P < 0.0001). (D) State 3 uncoupled OCR (state 3u after addition of FCCP), effect of genotype (P < 0.0001), and age–genotype interaction (P < 0.0001).
-
ERatio of mitochondrial complex IV/complex II activities.
-
FATP levels.
-
GSuperoxide anion radicals.
Deterioration of mitochondrial function is often related to an age‐dependent decrease of ATP and increased levels of ROS as a result of ETC leakage, prompting us to examine ATP and ROS levels in the mitochondria of 19‐month‐old animals, in addition to measurements of complex II and complex IV activities. In contrast to complex II which is exclusively encoded by nuclear genes, complex IV is encoded by both nuclear and mitochondrial genes. Thus, comparing complex IV activity to that of complex II (complex IV/complex II ratio) highlights the impact of the Mrps5 V338Y mutation. Compared to wild‐type control mice, the complex IV/complex II ratio was significantly reduced in cortical mitochondria from mutant mice (Fig 4E). Consistent with the finding of a mutation‐related decline in coupling efficiency of ETC, we observed reduced ATP levels and increased amounts of mitochondrial reactive oxygen species (mtROS) in mitochondria of Mrps5 V338Y/V338Y mice (Fig 4F and G).
Mutant MRPS5 mice show age‐related and stress‐induced behavioral alterations
Mutant Mrps5 V338Y/V338Y mice and littermates did not present any overt difference in spontaneous behavior. To assess a large spectrum of more subtle behavioral phenotypes, we subjected Mrps5 V338Y/V338Y mice and their littermates to a comprehensive battery of tests for changes in learning and memory, species‐typical behavior, anxiety, exploration, and sensorimotor function. We studied mice at the age of 9 and 19 months to detect early‐onset and age‐related phenotypes; selected tests were also recorded at 14 months of age.
Locomotor activities in the home cage, in the large open field, during light–dark transition, and in the elevated O‐maze were affected neither by genotype nor by age (Appendix Fig S5A–D). Swim speed in the water‐maze, performance on the rotarod, grip test, hot plate test, and acoustic startle (Appendix Fig S5E–H) did not show any mutational effect on sensorimotor function. Responses in the acoustic startle were reduced in 19‐month‐old mice, most likely due to the age‐related hearing loss typically seen in C57BL/6 mice 28.
To evaluate anxiety‐related and exploratory behavior, we tested mice in the large open field, light–dark transition, elevated O‐maze, and IntelliCage. In the large open field, littermate controls showed an age‐dependent increase in center avoidance and a decrease in rearing activity from 9 to 19 months of age. In contrast, Mrps5 V338Y/V338Y animals showed pronounced center avoidance and low rearing activity at all ages, with 9‐month‐old Mrps5 V338Y/V338Y mice already resembling aged animals (Fig 5A and B). Area explored in the large open field (Appendix Fig S5I) and avoidance of the brightly illuminated chamber in the light–dark transition test (Fig 5C) showed similar age dependency, with Mrps5 V338Y/V338Y being less explorative than controls at the age of 9 months but not at the age of 19 months. On the elevated O‐maze, responses of Mrps5 V338Y/V338Y animals were statistically indistinguishable from those of controls even though on average they spent slightly less time in the open arms (Appendix Fig S5J). Explorative analysis of individual cohorts during the free adaptation stage in IntelliCage revealed decreased activity for Mrps5 V338Y/V338Y mice at 9 months of age (Fig 5D).
Figure 5. Exploration and anxiety, home cage learning, and place learning in the water‐maze.
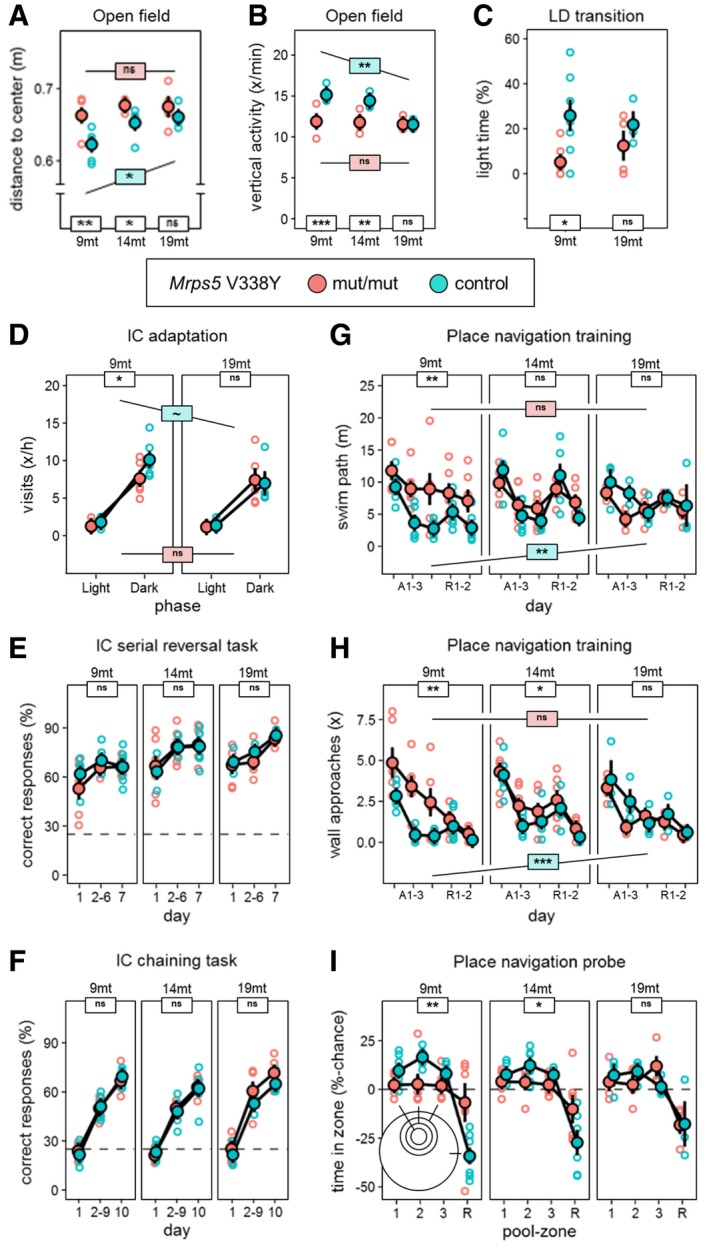
- Average distance to center as measure of anxiety in the open field (ANOVA: genotype F 1,24 = 16.24, P = 0.0005, age F 2,24 = 5.217, P = 0.0131). Mutants show increased anxiety levels at 9 and 14 months.
- Estimated frequency of rearing activity as measure of exploratory drive in the open field (ANOVA: genotype F 1,24 = 39.40, P < 0.0001, age × genotype F 2,26 = 8.204, P = 0.0017). Mutants showed little exploratory activity at 9 and 14 months.
- Light–dark transition test, percentage of time spent in the bright compartment (ANOVA: genotype F 1,16 = 7.393, P = 0.0152). Reduced scores at 9‐month‐old animals indicate increased anxiety.
- Exploratory corner visits in dark and light phase during adaptation to the IntelliCage (ANOVA: phase F 1,18 = 617.2, P < 0.0001). Corner visits during the dark phase as a correlate of exploratory activity showed premature reduction in 9‐month‐old animals.
- Learning in the hippocampus‐dependent IntelliCage serial reversal task (ANOVA: day F 2,56 = 26.71, P < 0.0001). Scores provided no evidence for a mutation effect on learning rate or performance.
- Learning in the hippocampus‐dependent IntelliCage chaining task (ANOVA: day F 2,54 = 450.1, P < 0.0001). Scores provided no evidence for a mutation effect on learning rate or performance.
- Water‐maze place navigation, swim path during acquisition, and reversal (ANOVA: day F 4,104 = 21.14, P < 0.0001, genotype F 1,26 = 13.11, P = 0.0013, age × genotype F 2,26 = 8.204, P = 0.0017). All groups learned: Controls showed reduced performance with increasing age, and mutants showed poor performance already at 9 and 14 months.
- Number of wall approaches as indicator of poor coping with the test and non‐spatial strategy choice (ANOVA: genotype F 1,26 = 18.31, P = 0.0002, age F 2,26 = 4.486, P = 0.0212, age × genotype F 2,26 = 7.068, P = 0.0035). Number of wall approaches increased with age in controls and was prematurely increased in mutants at 9 and 14 months.
- Water‐maze probe trial, spatial retention score for target zones 1–3 comprising 5, 10, and 10% of the pool surface versus remaining 75% of surface (ANOVA: zone F 3,78 = 36.04, P < 0.0001, genotype × zone F 3,78 = 7.807, P = 0.0001). Spatial selectivity declined with age in controls and was prematurely reduced in mutants at 9 and 14 months.
Learning and memory were evaluated in the T‐maze spontaneous alternation test and the IntelliCage and compared to performance in the more adverse environment of the water‐maze place and cue navigation tasks. Performance in the T‐maze spontaneous alternation test (Appendix Fig S5K), and learning in the serial reversal (Fig 5E) and chaining (Fig 5F) tasks in IntelliCage were not affected by genotype. In the water‐maze place navigation task, the performance during acquisition and reversal training worsened significantly in the controls with aging. In contrast, Mrps5 V338Y/V338Y mice performed poorly already at young age and were significantly impaired compared to the controls at 9 months of age (Fig 5G). The poor escape of the Mrps5 mutants in the place navigation task was accompanied by an increased number of wall approaches, again with 9‐month‐old mutants earning scores similar to those of aged animals (Fig 5H). In the probe trial (Fig 5I), Mrps5 mutants showed overall significantly reduced preference for the trained target zone. Consistent with an age‐dependent phenotype of the mutation, a significant zone × genotype interaction was observed for the 9‐ and 14‐month‐old cohorts, but not for the 19‐month‐old animals. The cue navigation protocol in the water‐maze showed a small overall deficit in Mrps5 mutant mice (Appendix Fig S5L).
From the above results, we conclude that mutation effects on learning task performance were strongly context‐dependent. While we observed no adverse mutation effect or age‐related performance declines in T‐maze alteration and IntelliCage learning, performance in the water‐maze place navigation task was strongly affected by the mutation in an age‐dependent manner. As the performance of the mutants was only affected in the water‐maze and not in other learning tasks, it is unlikely that this deficit is reflective of a genuine cognitive impairment, also supported by the finding that hippocampus‐dependent burrowing and nesting were unaffected by the mutation (Appendix Fig S5M and N). Rather, as also supported by the results of our tests on anxiety and exploration, water‐maze performance was probably disrupted by an excessive sensitivity of Mrps5 mutants to the aversive stress conditions associated with this test. In less aversive environments such as the T‐maze or IntelliCage, Mrps5 mutants were able to learn normally.
Comparative brain histopathological analyses of 9‐month‐old Mrps5 V338Y/V338Y mice and age‐matched wild‐type controls did not reveal any difference. In particular, no signs of neurodegeneration were found in the mutant mice.
Mutation V338Y heightens susceptibility to noise‐related auditory damage
We selected auditory function in vivo as a sensory modality susceptible to environmental stress on mitochondria 29. The non‐invasive procedure of auditory brain stem responses (ABR) records neuronal activity along the auditory pathways and provides information on the functional integrity of the cochlea. Mrps5 V338Y/V338Y and control mice had comparable baseline thresholds of around 20 dB sound pressure level (SPL), indicating that the mutation per se did not affect fundamental auditory function (Fig 6). Following an exposure to noise (2–10 kHz broadband at intensities of 108 and 110 dB), ABR thresholds were determined 3 weeks later when damage to the auditory system was stabilized. Exposure to noise resulted in increases in ABR thresholds at 12, 24 and 32 kHz. The noise‐induced auditory damage was significantly more pronounced in Mrps5 V338Y/V338Y mice than in the control littermates as evidenced by consistently larger threshold shifts at all three frequencies. Furthermore, while control mice showed a graded response to noise with increasing thresholds from 108 to 110 dB, V338Y mutants demonstrated a ceiling effect already at 108 dB corroborating their heightened susceptibility to noise trauma (Fig 6).
Figure 6. Noise‐induced hearing damage.
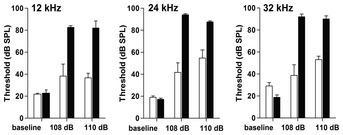
Noise exposure and ABR measurements. Exposure of mice to broadband noise (2–10 kHz) of either 108 or 110 dB SPL resulted in elevations of auditory thresholds. Thresholds in Mrps5 V338Y/V338Y mutant mice (black bars) were significantly higher after exposure than thresholds in littermates (open bars); P < 0.0001 (two‐way ANOVA for combined 108 and 110 dB data). Frequencies of 12, 24, and 32 kHz were selected for measurement to cover the mid‐frequency to high‐frequency regions of the cochlea. n = 6 for baseline littermates; n = 6 for baseline mutants; n = 3 for all noise exposures. Graphs show mean and SEM.
Discussion
By genetic manipulation of the nuclear‐encoded mitochondrial ribosomal protein MRPS5, we have established the first‐ever reported mammalian model of a mitochondrial ram mutant. Following the in silico identification of a putative ram mutation in mammalian MRPS5 (human V336Y, mouse V338Y), phylogenetic reconstructions provided evidence that the corresponding mutation A127Y in homologous bacterial ribosomal protein rpsE (E. coli) results in mistranslation as evidenced by increased incorporation of near‐cognate aa‐tRNA species and stop codon read‐through. In the absence of established means to introduce reporter genes into mitochondria of higher eukaryotes, we identified the mtDNA‐encoded MT‐CO1 protein as an endogenous reporter of mitoribosomal misreading. Using bacterial mitohybrid ribosomes with and without the pathogenic A1555G mutation and translating MT‐CO1 mRNA in the absence and presence of the misreading agent tobramycin, we established that both the ratio of cysteine/methionine incorporation and the read‐through in MT‐CO1 protein translation constitute markers of ribosomal misreading. Subsequently, we directly demonstrated by mitochondrial in organello translation that the MRPS5 mutation V336Y increases both the ratio of cysteine/methionine incorporation and read‐through of endogenous MT‐CO1, as does treatment with tobramycin.
We found that mutant MRPS5‐mediated mitochondrial mistranslation in homozygous knock‐in Mrps5 V338Y/V338Y mice is associated with impaired mitochondrial function in post‐mitotic cells in vivo. Brain cortical mitochondria showed a significant reduction in basal respiration and maximal respiratory capacity, decreased ATP levels, and increased formation of ROS. The cognitive performance of mutant Mrps5 mice was apparently normal under non‐aversive conditions. However, we observed age‐ and stress‐related alterations in three behavioral traits in Mrps5 V338Y mutant mice: first, increased anxiety responses as evidenced by less time spent in the center of the open field and more time spent in the dark chamber of the light–dark transition test; second, decreased explorative drive as revealed by diminished vertical activity and a smaller area explored in the open field; and third, impaired learning and memory under stressful conditions as shown by poor training and probe trial scores in the water‐maze place navigation tests. The same combination of behavioral differences characterized the transition from adulthood to old age in the wild‐type control mice, reflecting normal behavioral aging. The triad of symptoms observed in Mrps5 mutant mice—increased vigilance to threat, reduced exploratory drive, and impaired coping with stress—is typical for physiological behavioral aging in humans and rodents 30, 31, 32, 33. Thus, impaired mitochondrial respiration in part phenocopied non‐cognitive age‐associated alterations in brain function. The pathological phenotype of neurological stress intolerance and anxiety‐related behavioral alterations in Mrps5 V338Y/V338Y mice points to a key role for mitochondrial function in stress‐associated behavioral adaptations 34, 35, 36.
Auditory studies revealed another compelling example of a mitochondria‐related pathology in the mutants. Auditory thresholds, reflecting the integrity of sensory hair cells and ascending auditory pathways, are normal in V338Y mutant animals compared to littermate controls. This mirrors the normal hearing phenotype of most A1555G patients. The A1555G mutation shows a variable phenotype from lifelong normal hearing to severe hearing loss, pointing to a role for additional genetic and environmental stress factors necessary for expression and exacerbation of a pathological phenotype 37, 38. Notable stressors for the A1555G mutation are aminoglycoside antibiotics whose administration can lead to sudden profound hearing loss 13. The hypersusceptibility to aminoglycoside ototoxicity in A1555G patients is at least in part conferred by increased binding of the drug to its target—rRNA residue 1555 is part of the drug binding pocket in the ribosomal A‐site 19. In the absence of increased binding of aminoglycosides to mutant V338Y ribosomes—the ribosomal protein MRPS5 is not part of the drug binding pocket—we decided to use noise trauma as a particular pertinent stressor because of its traumatic mechanisms impinge on sensory hair cell mitochondria 29. Compared to wild‐type control mice, mutant V338Y mice showed significantly heightened susceptibility to noise trauma. The increased loss of auditory function in Mrps5 V338Y/V338Y mutant mice observed upon exposure to noise apparently results from noise‐induced enhanced ROS formation, ATP depletion, and mitochondrial cell death signaling 39, 40, superimposed on an already mutation‐mediated compromised mitochondrial function.
In contrast to several human disease‐causing mutations in mitochondrial tRNAs and nuclear genes encoding mitochondrial aminoacyl‐tRNA synthetases 11, 22, the mutant MRPS5 mice display a remarkable absence of overt pathology in vivo. While this surprising observation may reflect a threshold effect of the mutation studied, our findings are supported by the only discrete alterations observed in humans with the mechanistically similar pathogenic A1555G mutation 37. Our results from whole genome transcriptome analyses of the V336Y mutant HEK cells are in agreement with microarray‐based studies in immortalized lymphocytes of individuals with the mitochondrial A1555G mutation which revealed little changes, except for a coordinated upregulation of cytosolic ribosomal proteins, an effect that was suggested as a compensatory mechanism for the mitochondrial dysfunction associated with the A1555G mutation 41. In addition to a possible threshold effect, the most probable explanation for the lack of overt pathology in A1555G patients and MRPS5 V338Y mutant mice may reside in the nature of ribosomal misreading. Disease‐causing mutations in mitochondrial tRNA or aminoacyl‐tRNA synthetases affect a specific tRNA and every molecule thereof with detrimental consequences for translation 22, 42, 43. In contrast, misreading by ram mutations affects translational accuracy in a random and stochastic manner, which apparently can be in part tolerated. In addition, while ram mutations, such as A1555G or MRPS5 V338Y, may allow for increased rates of near‐cognate misreading, the incorporation of non‐cognate amino acids during translation is prevented by the difference in the free binding energy between cognate and non‐cognate aa‐tRNAs 44.
In conclusion, by mutagenesis of nuclear‐encoded mitochondrial protein MRPS5 we have established a unique model of mitoribosomal misreading that recapitulates the suggested A1555G pathomechanism 15, 16, 17, 18, 19. Evidence is provided that increased mitochondrial misreading is indeed the cause of hearing loss in the A1555G patients. The model reproduces the hearing‐related deficit as well as the absence of overt non‐cochlear pathology that distinguishes A1555G from classical mitochondrial diseases 11, 22. Further, the neurological alterations observed under adverse conditions and the noise‐induced auditory trauma found in MPRS5 mutant mice point to a key role for mitochondrial function in stress‐related behavioral and physiological adaptations.
Materials and Methods
Bacterial strains
For generation of merodiploid M. smegmatis rpsE S152Y, genomic DNA from M. smegmatis served as template for rpsE gene amplification. Fusion PCR via complementary overhanging sequences was used for gene construction. The first PCR used 5′ primer fwd1 (5′‐ATA CAA AAC CAG GTC AGG AGA TCG AAT GGC CGA GCA GGC TGG CGC‐3′, native leader sequence of the rpsE operon at the 5′ end) and 3′ primer rev1 (5′‐ACT ACT AGT TTA TGC CGA TCC CTC ACG CG‐3′, SpeI ACTAGT recognition site at the 3′ end) to amplify the rpsE coding DNA sequence (CDS). The second PCR used plasmid pMIH 45 as template and 5′ primer fwd2 (5′‐ACT AAG CTT CGG TGA CCA CAA CGC GCC CG‐3′) and 3′ primer rev2 (5′‐TCG ATC TCC TGA CCT GGT TTT GTA TCG CAA TTG TCT TGG CCA TTG‐3′) to amplify the hsp60 promotor with a HindIII AAGCTT recognition site at the 5′ end. The two amplicons overlap at the leader sequence and were fused using pfu polymerase (Promega), subcloned into the pGEM®‐T Easy vector (Promega), and controlled by DNA sequencing. PCR‐mediated site‐directed mutagenesis was used to introduce the S152Y mutation. The constructs were cloned into integrative vector pMIH using restriction enzymes HindIII and SpeI (Thermo Fisher) resulting in vectors pMIH‐rpsE‐wt and pMIH‐rpsE‐S152Y, respectively. Vectors pMIH‐rpsE‐wt and pMIH‐rpsE‐S152Y were electroporated into M. smegmatis mc2155 ΔrrnB 46. Single colonies were picked, propagated for further analysis, and checked by PCR and DNA sequence analysis.
For the generation of E. coli rpsE A127Y, Oligo λ Red‐mediated recombination was used as described previously 47. In brief, two steps of recombineering were conducted, using E. coli MG1655 and oligonucleotides that comprised the A127Y mutation or the wild‐type sequence, respectively. Positive clones were screened by PCR and confirmed by DNA sequencing. The recombinant E. coli strains were a kind gift of Diarmaid Hughes and Douglas Huseby, Uppsala University, Sweden.
For growth experiments, bacterial strains were streaked on agar plates and grown at 37°C until single colonies were visible. Single colonies were used to inoculate liquid LB medium. Growth experiments were started at an initial OD600 of 0.002 and further incubated at 37°C. OD600 was measured every 30 min for E. coli and every 3 h for M. smegmatis. Signal intensities at time point 0 h were set as one, and growth curves were plotted. Doubling time was calculated as t D = ln(2)/slope.
Cell‐free luciferase translation assays
Purified bacterial 70S ribosomes were used in dual luciferase translation assays as previously described 48, 49. A typical translation reaction (30 μl) contained 0.25 μM 70S ribosomes, 4 μg firefly (F‐luc) and 0.4 μg Renilla (R‐luc) mRNA, 40% (vol/vol) of M. smegmatis S100 extract, 200 μM amino acid mixture, 24 units of RiboLock (Thermo Fisher), and 0.4 mg/ml of tRNAs—energy was supplied by the addition of 12 μl of commercial S30 Premix without amino acids (Promega). The reaction mixture was incubated at 37°C for 35 min, stopped on ice, and assayed for F‐luc and R‐luc luciferase activities according to supplier's protocol (Promega). Luminescence was measured using the FLx800 luminometer (BioTek Instruments).
Mistranslation was determined as described previously 50, 51. In brief, to assess misreading His245 (CAC codon) of the F‐luc gene was replaced by the near‐cognate codon CGC or the non‐cognate codon AGA, both encoding for Arg. To determine read‐through, the Asp357 (GAC codon) of the F‐luc gene was replaced by the nonsense TGA stop codon. Arg245F‐luc mRNA, X357TGA F‐luc mRNA, and WT F‐luc mRNA were used in in vitro translation reactions, and R‐luc mRNA was used as internal control. Mistranslation was calculated by the ratio of mutant firefly/Renilla luciferase activity to wild‐type firefly/Renilla luciferase activity.
Cell culture and transfection of HEK293 cells
Human embryonic kidney cells (HEK293, Innoprot) were maintained in complete Dulbecco's modified Eagle's medium (DMEM) (Life Technologies) containing 10% fetal bovine serum (FBS) (Life Technologies), at 37°C in 5% CO2.
Plasmid pmouseMrps5‐WT containing the mouse Mrps5 coding region under the control of the chicken β‐actin promotor and the CMV enhancer (CAGGS promotor) was constructed by Genoway (Lyon, France). In brief, the mouse Mrps5 wild‐type CDS was synthesized, and restriction sites BamHI and AvrII were introduced at the 5′ and the 3′ ends. Using the BamHI and AvrII restriction sites, the 1,318‐nt‐sized fragment was ligated into an eukaryotic expression vector, which in addition to the CAGGS promotor carries the human growth hormone polyadenylation signal, resulting in pmouseMrps5‐WT.
The plasmid expressing the human MRPS5 gene was constructed as follows. A cDNA fragment containing human MRPS5 wild‐type CDS was generated by PCR, using cDNA reverse‐transcribed from total RNA extracted from HEK293 cells. Total RNA was prepared using TRIzol (Life Technologies) and DNase‐treated (DNase I, Life Technologies) by incubation for 15 min at 37°C; 1 μl of 25 mM EDTA was added, and samples were incubated for 10 min at 65°C and stored at −80°C. RNA was reverse‐transcribed using a High Capacity RNA‐to‐cDNA Kit (Life Technologies) according to the manufacturer's instructions. V336Y (GTC→TAC) point mutation was introduced by PCR‐mediated site‐directed mutagenesis, and restriction sites for BamHI and XhoI were introduced at the 5′ and 3′ ends, respectively. The mouse Mrps5 wild‐type CDS in the pmouseMrps5‐WT vector was replaced with human MRPS5 wild‐type or human MRPS5 V336Y using BamHI/XhoI restriction sites, resulting in vectors pMRPS5WT and pMRPS5V336Y. IRES‐eGFP was amplified from plasmid pLZRSpBMN‐IRES‐eGFP (a kind courtesy of Saule Zhanybekova, University of Basel, Switzerland) and inserted downstream of the MRPS5 CDS using restriction sites for BstBI and ClaI, resulting in vectors pMRPS5WT‐IRES‐eGFP and pMRPS5V336Y‐IRES‐eGFP. The hygromycin B resistance cassette was amplified by PCR from vector pGL4.14 (Promega), subcloned into pGEM®‐T Easy and inserted into pMRPS5WT‐IRES‐eGFP and pMRPS5V336Y‐IRES‐eGFP using MluI‐specific restriction sites.
For generation of stable transfectants, HEK293 cells were transfected with vectors pMRPS5 V336Y‐IRES‐eGFP and pMRPS5WT‐IRES‐eGFP using TurboFect (Thermo Fisher) according to the manufacturer's instructions. Cells were cultured in DMEM supplemented with 10% FBS and propagated under hygromycin B (100 μg/ml) selection for 5–7 weeks. GFP‐expressing colonies were picked for further characterization. GFP fluorescence was analyzed by flow cytometry using the BD FACSCanto II (BD Biosciences) and the FlowJo data analysis software (Tree Star Inc.).
Determination of MRPS5 mRNA expression
Quantitative PCR was used to determine MRPS5 transgene mRNA expression relative to endogenous MRPS5 mRNA levels. Total RNA was prepared using TRIzol extraction and reverse‐transcribed using the High Capacity RNA‐to‐cDNA Kit (Life Technologies) according to the manufacturer's instructions. A 221‐nt fragment from MRPS5 cDNA was amplified by using 5′ primers binding specifically either to the endogenous or the transgene MRPS5 5′‐UTR (5′‐TGC CCT GGG CGG AGG CCG AGG CGC GGC TC‐3′—forward endogenous gene‐specific; 5′‐GAT CAG AAG CTT CGT TAA CTG AGC TCA GG‐3′—forward transgene‐specific) and a 3′ primer binding to both endogenous and transgene MRPS5 (5′‐GGC GTA GGG ATG GGT GTC TCT GGT TCC CAG‐3′—reverse). For each sample, SYBR Green qPCR was conducted in triplicate using EvaGreen qPCR Mix (Bio&Sell) and ABI 7500 Fast Real‐Time PCR System (Life Technologies). Amplification consisted of 40 cycles (95°C for 20 s and 60°C for 45 s), and specificity of amplification was confirmed by melting curve analysis. Expression levels of MRPS5 were quantified as previously described 52.
The ratio of MRPS5 transgene mRNA to endogenous MRPS5 mRNA for cell lines expressing mutant V336Y MRPS5 was determined using MRPS5 forward 5′‐CTG CCA CAG GGC CAT CAT CAC CAT CTG C‐3′ and MRPS5 reverse 5′‐CGG AAG AGG CCC TGG GTG AGG CTG AGC‐3′ primers flanking the site of mutation. These primers amplify a 108‐nt PCR fragment of both endogenous and transgene MRPS5 mRNA. Discrimination was achieved by using TaqMan probes that recognize specifically human wild‐type endogenous MRPS5 (5′‐CAT GTA TGC CAA GGT C‐3′, conjugated to NED) or human mutant transgene MRPS5 V336Y (5′‐TAT GCC AAG TAC TCT G‐3′, conjugated to FAM). For each sample, TaqMan qPCR was conducted in triplicate using the TaqMan Kit (Life Technologies) and the ABI 7500 Fast Real‐Time PCR System (Life Technologies). Amplification consisted of 40 cycles (95°C for 20 s and 60°C for 45 s), and the ratio of transgene versus endogenous MRPS5 was calculated as described previously 52.
Cellular localization of the transfected MRPS5 protein
For cellular localization of the MRPS5 protein, we introduced a 33‐nt fragment GAG CAA AAG CTC ATT TCT GAA GAG GAC TTG AAT coding for myc tag peptide EQKLISEEDLN at the C‐termini of both MRPS5 WT and MRPS5 V338Y by PCR, resulting in vectors pMRPS5WTmyc‐IRES‐eGFP and pMRPS5V336Ymyc‐IRES‐eGFP. HEK293 cells were transiently transfected using TurboFect and cultured in DMEM supplemented with 10% FBS for 72 h. Cellular localization of MRPS5 protein was assessed by Western blot of mitochondrial and cytosolic fractions using anti‐myc antibody (ab9106, Abcam).
Determination of ratio of mitochondrial DNA (mtDNA)/nuclear DNA (nDNA), mitochondrial mass, ATP content, oxygen consumption, and generation time in HEK293 cells
The mtDNA:nDNA ratio was determined as previously described 53. In brief, total DNA was extracted from cultured cells with QIAamp DNA Mini Kit (Qiagen). For mtDNA, a fragment of MT‐TL1 gene (tRNA leucine) was amplified with forward primer (5′‐CAC CCA AGA ACA GGG TTT GT‐3′) and reverse primer (5′‐TGG CCA TGG GTA TGT TGT TA‐3′), and for nDNA, a fragment of B2M gene (beta‐2‐microglobulin) was amplified with forward primer (5′‐TGC TGT CTC CAT GTT TGA TGT ATC T‐3′) and reverse primer (5′‐TCT CTG CTC CCC ACC TCT AAG T‐3′). Amplification was performed in an ABI 7500 real‐time PCR system (Life Technologies) and consisted of 40 cycles (95°C for 20 s, 62°C for 30 s), and specificity of amplification was confirmed by melting curve analysis. Each sample was measured in triplicate.
Mitochondrial mass was measured using MitoTracker Deep Red FM and FACSCanto II (BD Biosciences) equipped with helium–neon laser according to the manufacturer's recommendations. HEK293 cells were seeded in a MULTIWELL™ 12‐well plate (BD Falcon) at a density of 5 × 104 cells per well and cultured overnight in DMEM supplemented with 10% FBS. Cell culture medium was replaced with DMEM containing 200 nM MitoTracker Deep Red FM, and cells were incubated for 30 min at 37°C and 5% CO2. Cells were washed twice with PBS and detached from surface with Accutase (Thermo Fisher) and analyzed by FACS.
Total ATP content was quantified using the bioluminescence‐based assay ViaLight Plus Kit (Lonza). 104 cells were seeded in triplicate in a black 96‐well plate (BD Falcon) and incubated for 48 h in 100 μl DMEM supplemented with 10% FBS. 5 μl Alamar Blue (Life Technologies) were added to each well and incubated for 3 h. Alamar Blue was measured as an internal standard, followed by measurement of ATP content according to the manufacturer's protocol.
Mitochondrial respiration was measured using the Seahorse Bioscience XF24 analyzer. XF24 cell culture microplates (Seahorse Bioscience) were coated using 50 mg/ml poly‐D‐lysine, and cells were seeded at a density of 105 cells/well in 100 μl DMEM supplemented with 10% FBS. Cells were incubated for 3 h at 37°C and 5% CO2 for attachment, medium was aspirated, replaced by DMEM without FBS, and cells were incubated overnight. Prior to the experiment, the plate was incubated 30 min at 37°C in a CO2‐free incubator. Cells were subsequently washed with Mitochondrial Assay Solution (MAS; 70 mM sucrose, 220 mM mannitol, 10 mM KH2PO, 4.5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, 0.2% (w/v) fatty acid‐free BSA, pH 7.2 at 37°C) according to the manufacturer's protocol (Seahorse Bioscience). Measurements were performed in MAS, supplemented with 10 mM pyruvate, 10 mM succinate, 2 mM malate, and 0.2 nM Plasma Membrane Permeabilizer (PMP) reagent (Seahorse Bioscience). The experiment started in a coupled state in the presence of succinate, pyruvate, and malate (state 2, basal respiration). State 3 was initiated following the injection of 55 μl of 40 mM ADP (4 mM final). State 4o was induced by addition of 62 μl of 25 μg/ml oligomycin (3 μg/ml final). State 3 uncoupled (3u) was assessed after injection of 68 μl 40 μM FCCP (4 μM final) before shutting down mitochondrial respiration by injecting 76 μl of 40 μM and 20 μM (4 μM and 2 μM final) of antimycin A and rotenone, respectively. Drugs were purchased from Seahorse Bioscience. Data were extracted from the Seahorse XF‐24 software, and bioenergetic parameters were calculated according to the manufacturer's guidelines.
Cellular growth was monitored using Alamar Blue (Life Technologies). HEK293 cells were seeded on 24‐well plates (BD Falcon) at low density and incubated in DMEM with 10% FBS at 37°C. 10% Alamar Blue was added to the cells (v/v) at time points 0, 24, 48 and 72 h, and fluorescence was measured (excitation: 530 nm, emission: 590 nm) after 3 h of incubation. Signal intensities at time point 0 h were set as one, and growth curves were plotted. Doubling time was calculated as t D = ln(2)/slope.
Cell‐free MT‐CO1 translation assays
Purified 70S M. smegmatis hybrid ribosomes were used in cell‐free translation assays. The mitochondrial COX1 gene (MT‐CO1) was redesigned for efficient bacterial translation: (i) The codon usage was optimized for bacterial translation, (ii) an additional alanine GCG codon was introduced after the ATG start codon to create Kozak context, and (iii) the TAA stop codon was introduced instead of AGA (hungry codon). To measure read‐through, constructs MT‐CO1‐AGA‐polyA and MT‐CO1‐TGA‐polyA were designed. MT‐CO1‐AGA‐polyA has the 69‐nt‐long native 3′ UTR of human MT‐CO1 mRNA followed by a 51‐nt‐long polyA sequence, MT‐CO1‐TGA‐polyA has a TGA stop codon instead of AGA. The constructs were ordered from GenScript USA Inc. and cloned into the pUC57 vector (Thermo Fisher). A typical translation reaction with the total volume of 30 μl contained 0.25 μM 70S ribosomes, 4 μg of in vitro‐transcribed mRNA, 40% (vol/vol) of M. smegmatis S100 extract, 200 μM amino acid mixture without methionine, cysteine or lysine, 24 units of RiboLock (Thermo Fisher), and 0.4 mg/ml of tRNAs. Energy was supplied by the addition of 12 μl of commercial S30 Premix without amino acids (Promega). Depending on the labeled amino acid, 35S‐methionine (KSM‐01; Hartmann Analytic), 35S‐cysteine (ARS 0101; Hartmann Analytic), or 14C‐lysine (ARS‐0673; Hartmann Analytic) were added; cold methionine, cysteine, or lysine was subsequently added to a final concentration of 0.2 mM. The reaction mixture was incubated at 37°C for 1 h and stopped on ice. To reduce background and for quantification of radioactively labeled bands, the products of the in vitro translation reaction were immunoprecipitated. The purified proteins were analyzed on 15% SDS–PAGE. As a control for MT‐CO1, we used in organello translation products labeled with 35S‐Methionine. The gel was fixed, dried, and exposed on phosphor‐imager screen and scanned with a FLA‐5100 laser scanner (Fujifilm). Intensity of MT‐CO1 bands was quantified using AIDA (version 3.52.046, raytest Isotopenmessgeräte GmbH).
Mitochondrial in organello translation
Mitochondria were isolated from HEK293 cells as described 54. In brief, HEK293 cells were collected from two 90% confluent petri dishes, washed 3 times with PBS, and resuspended in 1 ml of extraction buffer (20 mM HEPES‐KOH (pH 7.5), 0.25 M sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT). Cells were broken by passing ten times through a syringe needle (0.45 × 12 mm), centrifuged 5 min at 800 g, and supernatant was transferred to a new tube. The pellet was resuspended in 1 ml of extraction buffer, passed five times through a syringe needle (0.45 × 12 mm), and centrifuged for 5 min at 800 g. The supernatant was collected, combined with the previous supernatant, and centrifuged 5 min at 800 g. The supernatant was collected and centrifuged for 15 min at 10,000 g, and the resulting pellet was used as the mitochondria‐enriched fraction.
Mitochondrial in organello translation was done as described, with slight modifications 55, 56, 57. In brief, the mitochondria‐enriched pellet was resuspended in 1 ml of mitochondria reaction buffer (MR‐buffer) containing 20 mM Tris–HCl (pH 7.2), 90 mM KCl, 4 mM MgSO4, 1.5 mM KH2PO4, 20 mM glutamate, 0.5 mM malate, 14 mM sucrose, 44 mM sorbitol, 4 mM ADP, 0.1 mM amino acids (without methionine and cysteine), 1 mg/ml BSA (fatty acids free), and 0.1 mg/ml cycloheximide. Depending on the labeled amino acid, 15 μl of 35S‐methionine or 35S‐cysteine was added. Cold methionine or cysteine was subsequently added to a final concentration of 0.1 mM. The resulting master mix was split into 100 μl per translation reaction. In organello translation was conducted by incubation at 30°C for 2 h on a shaker (800 rpm). After incubation, the reaction mixtures were centrifuged for 5 min at 15,000 g, and the supernatants were discarded. The pellets were washed with cold washing buffer containing 10 mM Tris–HCl (pH 7.4), 320 mM sucrose, 1 mM EDTA, centrifuged for 5 min at 15,000 g, and resuspended in 20 μl of 1× SDS loading buffer or in 30 μl H2O for subsequent immunoprecipitation. The samples were analyzed on 15% SDS–PAGE. The gel was fixed, dried, and exposed on phosphor‐imager screen and scanned with laser scanner FLA‐5100. Intensity of bands was quantified using AIDA.
Immunoprecipitation
In brief, for one reaction 50 μl of Dynabeads M‐280 sheep anti‐mouse IgG (11202D, Thermo Fisher) or sheep anti‐rabbit IgG (11204D, Thermo Fisher) was washed 3 times with 1 ml of blocking solution (PBS with 0.5% BSA). For immobilization of antibodies, Dynabeads were incubated with 5 μg of monoclonal MT‐CO1 antibody (ab14705, Abcam), monoclonal MT‐CO2 antibody (ab110258, Abcam), or polyclonal antibody against Lys14 (customized antibody, GenScript USA Inc.) in 1 ml of blocking solution overnight at 4°C, subsequently washed three times with blocking solution, and resuspended in 50 μl of blocking solution.
To 30 μl of bacterial or in organello translation reaction, 20 μl of 20× IP buffer (0.4 M Tris–HCl, pH 7.5, 1.5 M NaCl, 0.5% SDS, 10% Triton X‐100, 10 mM EDTA) was added, incubated for 30 min at room temperature, and H2O was added to a final volume of 350 μl. 50 μl of Dynabeads with the immobilized antibodies was added to the translation mixture and incubated overnight at 4°C while shaking. The Dynabeads were washed with blocking solution, pelleted, and resuspended in 20 μl of H2O. 6 μl of 4× SDS loading buffer was added and vortexed. The samples were heated at 95°C for 5 min and resolved on 15% SDS–PAGE. The gel was fixed, dried, and exposed on phosphor‐imager screen and scanned with laser scanner FLA‐5100. Intensity of MT‐CO1 and MT‐CO2 bands was quantified using AIDA. For validation of antibodies used, see Fig 2F and G.
MT‐CO1 translation in rabbit reticulocyte lysate
MT‐CO1‐AGA‐polyA mRNA and MT‐CO1‐TGA‐polyA mRNA were used for translation in rabbit reticulocyte lysates. A typical translation reaction with a total volume of 30 μl contained 20 μl of rabbit reticulocyte lysate (Promega), 4 μg of in vitro‐transcribed mRNA, 200 μM amino acid mixture without methionine, 24 units of RiboLock (Thermo Fisher) and 1 μl of 35S‐methionine (KSM‐01; Hartmann Analytic). The reaction mixture was incubated at 37°C for 1 h and stopped on ice. The products of the in vitro translation reaction were immunoprecipitated using MT‐CO1 or poly‐lysine antibodies. The immunoprecipitated proteins were analyzed on 15% SDS–PAGE. As a control for MT‐CO1, we used in organello translation products labeled with 35S‐methionine. The gel was fixed, dried, and exposed on a phosphor‐imager screen and scanned with a FLA‐5100 laser scanner (Fujifilm).
Mitochondrial translation
HEK293 cells were grown in 10‐cm petri dishes (BD Falcon) in DMEM with 10% FBS to 80% confluence, washed, and incubated for 30 min in DMEM without methionine. Media were exchanged to DMEM without methionine supplemented with 100 μM emetine (Sigma‐Aldrich) and incubated for 15 min. The medium was replaced with 500 μl methionine‐free DMEM supplemented with 2 μl of 35S‐methionine and 100 μM emetine, and cells were incubated for 2 h at 37°C. The supernatant was carefully aspirated, and cells were detached by pipetting with 1 ml of PBS. The cells were washed 2 times with PBS and resuspended in 200 μl of 1× SDS loading buffer. The samples were analyzed on a 15% SDS–PAGE. The gel was fixed, dried, and exposed on a phosphor‐imager screen and scanned with the FLA‐5100 laser scanner. A pilot experiment was performed to control that emetine effectively inhibited cytosolic protein synthesis and that the residual protein synthesis reflects mitochondrial translation as shown by susceptibility to linezolid (Pfizer) (Appendix Fig S3G).
Western Blot
Cells were grown to 70% confluence in DMEM with 10% FBS. Cells were lysed with 1× Passive Lysis Buffer (Promega) and ultrasonicated. Lysates were centrifuged (15,000 g, 10 min), and protein concentration in the supernatant was measured by the Micro BCA Protein Assay Kit (Thermo Fisher). Ten micrograms of total protein was resolved on 10% SDS–PAGE and blotted on nitrocellulose membranes, which were probed with specific antibodies. Amersham ECL Prime Western blotting detection reagent (RPN2232; GE Healthcare) was used as substrate for horseradish peroxidase (HRP). The specific antibodies used in this study were as follows: anti‐myc tag (ab9106, Abcam); anti HSP60 (ab46798, Abcam), anti‐ATP6 (MS508, MitoScience), and anti‐β‐tubulin (ab15568, Abcam); HRP‐conjugated goat anti‐rabbit (G‐21234, Thermo Fisher); and goat anti‐mouse antibodies (A10551, Thermo Fisher).
Generation of Knock‐In Mrps5 transgenic mice
Mrps5 V338Y mice were generated using a FLEx approach 58. This strategy is based on the duplication of the exon to be mutated and its insertion in the antisense orientation. The duplicated regions are flanked with loxP and lox511 cloned in opposite directions (Mrps5 FLEx allele). Upon Cre induction, the mutated exon will be flipped resulting in the deletion of the wild‐type exon and expression of the mutant protein (Mrps5 V338Y allele).
The gene‐targeting vectors were constructed from genomic 129Sv mouse‐strain DNA. For Mrps5 V338Y, exon 10 was duplicated and the mutant exon inserted in antisense within intron 10. The insertion points were selected in order not to disrupt any potential regulatory region identified by consensus sequences. The duplicated exons were flanked by loxP and mutated lox511 sites for deletion of the WT exon and switch of the mutant exon upon Cre‐mediated action. A Flippase Recognition Target (FRT)‐flanked neomycin cassette was cloned into the targeting vector. Linearized targeting vectors were transfected into 129Sv ES cells, and positive selection was started 48 h after electroporation by addition of G418. Resistant clones were isolated, amplified, duplicated, and genotyped by both PCR and Southern blot analysis. The following primer pairs were designed to specifically amplify the targeted locus—34690sa: 5′‐GGG AAC TTC CTG ACT AGG GGA GGA GTA G‐3′, 34691sa: 5′‐GCA AAG ACT CAG ACA AAC AAC CGA CG‐3′. Targeted locus was confirmed by Southern blot analysis using internal and external probes on both 5′ and 3′ ends. PCR and Southern blot genotyping led to the identification of two targeted clones for Mrps5 V338Y.
Recombined ES cell clones were microinjected into C57BL/6 blastocysts and gave rise to male chimeras. Breeding with C57BL/6 Flp deleter mice (CAG‐Flp) was performed to produce the Mrps5 heterozygous inducible line devoid of neomycin cassette: Mrps5 FLEx/WT. Inducible mice were backcrossed to C57BL/6 for four generations. The inducible Mrps5 V338Y line was crossed to C57BL/6 CRE deleter mice (CMV‐Cre) to produce the Mrps5 V338Y heterozygous induced mutant line. Cre‐recombination resulted in flipping of the duplicated inverted exon bearing the mutation of interest (FLEx strategy) and expression of the mutant allele, resulting in heterozygous mutant line Mrps5 V338Y/WT referred to as induced mice. For each line, heterozygous mice were genotyped by PCR, Southern blot, and sequencing (Appendix Fig S4).
Heterozygous inducible mice (Mrps5 FLEx/WT) were first identified by PCR using primer 017 (5′‐GCC AAA GAG ACA TGC AGT GAG AAG AGT ACC‐3′) and primer 018 (5′‐CCA CCA TGA GTC CAA TGA TTG CAC C‐3′). The primers result in amplification of a specific 1.0 kb fragment for the Mrps5 FLEx allele, while wild‐type Mrps5 does not result in an amplicon. PCR‐positive mice (Mrps5 FLEx/WT ) were confirmed by Southern blot analysis using a 3′ external probe (amplified using the following primers: 5′‐ACT TTA CAG CCT AGC TCG TCA GCA CAG C‐3′, 5′‐CCC AAG CTA AGA CCC CTT TCA ACA GC‐3′). Upon digestion with NheI, a 5.1 kb fragment is expected for the Mrps5 WT allele, and a 5.5 kb fragment is expected for the Mrps5 FLEx allele. Heterozygous induced mice (Mrps5 V338Y/WT) were genotyped by PCR using primers 34712hom (5′‐GCT TCT GTT TGT GGC TTG TGT TGC C‐3′) and 34713hom (5′‐TTG TTG GAC TGG TGA AAC ACC T CGG‐3′). The primers result in a 0.6 kb gene fragment for the Mrps5 WT allele, and a 0.8 kb gene fragment for the Mrps5 V338Y allele. Heterozygous induced mice (Mrps5 V338Y/WT) were confirmed by Southern blot analysis using a 5′ external probe (amplified using the following primers: 5′‐GGT GGC TTT TGC AGT AAT TTT ATT TACT TGC‐3′, 5′‐AAA CTC CCT AAG CTT CTC CCC ACA CTC‐3′). Following digestion with AvrII/HpaI, the following fragment sizes are expected for each allele: 9.6 kb for Mrps5 WT, 7.9 kb for Mrps5 FLEx, and 7.2 kb for Mrps5 V338Y. The presence of the mutation in the induced mice (Mrps5 V338Y/WT) was controlled by sequencing of a 2.8 kb product specific for the induced allele using primers hybridizing to the loxP site (remaining upstream of exon 10 upon induction) and downstream of exon 11. The primers used (primer 017: 5′‐GCC AAA GAG ACA TGC AGT GAG AAG AGT ACC‐3′, primer 34709cof: 5′‐ATG AGG GGA GGG GCA GGA ACC TAA‐3′) will not amplify the wild‐type allele.
Mice tissue sections were frozen until RNA extraction was performed. RNA was extracted using TRIzol as described above. cDNA synthesis was performed using SuperScript II cDNA Synthesis Kit (Life Technologies) using random hexamers and 40 ng RNA in 20 μl reagent according to the manufacturer's protocol. Expression of the mutant gene was assessed from brain, inner ear, and retina recovered from 6‐ to 7‐week‐old animals. Heterozygous Mrsp5‐induced mice (Mrps5 V338Y/WT) were compared to wild‐type mice for expression of the Mrps5 gene. RT–qPCR analysis showed no significant difference in Mrps5 total mRNA between heterozygous induced mutant mice and controls. Expression of the V338Y mutant allele was demonstrated by sequencing of the RT–PCR product. Primers used for RT–PCR and sequencing were EBO4‐SeqF1 (5′‐GCT ATT GGG AAA GCT GCT GA‐3′) and EBO4‐SeqR2 (5′‐TCA CGT CCT GCC AGT CCA GC‐3′). Primers used for RT–qPCR analysis were EBO4‐TOT‐F1 (5′‐CCA TGA ACA TGC TCA ACC TC‐3′) and EBO4‐TOT‐R2 (5′‐ATA GGC AGA GGC CCA CAT T‐3′).
Heterozygous Mrps5 V338Y/WT mice are viable, are fertile, and do not show any gross phenotype. Heterozygous mutant mice bred within the expected Mendelian ratio, suggesting that the mutation has no significant effect on the viability of the line. Interbreeding Mrps5 V338Y/WT resulted in homozygous Mrps5 V338Y/V338Y mice. Analysis of Mrps5 gene expression in the homozygous mutant mice revealed Mrps5 mutant mRNA only in all tissues tested (Appendix Fig S4). The overall expression level of mutant Mrps5 mRNA was comparable to wild‐type mice, indicating that the mutation did not lead to deregulation of Mrps5 gene expression. Homozygous mutant mice were able to breed and did not show any gross phenotype.
RNA sequencing and data analysis
RNA sequencing (RNA‐seq) was performed at the UZH/ETH Functional Genomics Center Zurich (FGCZ) according to the Illumina RNA sequencing protocol. For transcriptome analysis, four clones of Mrps5 V336Y and six clones of Mrps5 wild‐type transfected cells were compared. RNA was extracted using TRIzol. The quality of the isolated RNA was determined using a Qubit® (1.0) Fluorometer (Life Technologies) and a Bioanalyzer 2100 (Agilent). Only those samples with a 260 nm/280 nm ratio between 1.8 and 2.1 and a 28S/18S ratio within 1.5‐2 were further processed. The TruSeq Stranded mRNA Sample Prep Kit (Illumina) was used in the succeeding steps. Briefly, total RNA samples (100–1,000 ng) were ribosome‐depleted and then reverse‐transcribed into double‐stranded cDNA with actinomycin added during first‐strand synthesis. The cDNA samples were fragmented, end‐repaired, and polyadenylated. TruSeq adapters containing the index for multiplexing were ligated to the fragmented DNA samples. Fragments containing TruSeq adapters on both ends were selectively enriched with PCR. The quality and quantity of the enriched libraries were validated using the Qubit® (1.0) Fluorometer and LabChip® GX (Caliper Life Sciences). The product is a smear with an average fragment size of approximately 360 bp. The libraries were normalized to 10 nM in Tris‐Cl 10 mM, pH 8.5 with 0.1% Tween‐20. The TruSeq SR Cluster Kit v4‐cBot‐HS (Illumina) was used for cluster generation using 8 pM of pooled normalized libraries on the cBOT. Sequencing was performed on the Illumina HiSeq 2500 single end 126 bp using the TruSeq SBS Kit v4‐HS (Illumina). The quality of the reads was assessed using FastQC (Babraham Bioinformatics), and potential contaminations were evaluated with FastQ Screen (Babraham Bioinformatics) using bowtie2 v. 2.1.0 default parameters 59. Quantification of gene expression was performed using the RSEM package (version 1.2.18) 60 mapping against the ensembl 75 annotations derived from the human genome assembly GRCh37 and mouse genome assembly GRCm37. Genes not present (< 10 counts per gene) in at least 50% of samples from one condition were discarded from further analyses. Differential gene expression analysis between sample groups of interest was performed using the R/bioconductor package edgeR 61. To evaluate functional activities, differentially expressed genes were mapped to known biological ontologies based on the GO project using gene annotation tool Enrichr 62, 63.
Transcriptome data are available in Gene Expression Omnibus (GEO), accession no. GSE101503.
Histopathological analyses
Mutant Mrps5 V338Y/V338Y mice of 9 months of age were histologically analyzed in comparison with age‐matched wild‐type littermates (three animals per group). 4‐μm sections of formalin‐fixed, paraffin‐embedded (FFPE) brain and cerebellum were subjected to hematoxylin–eosin (H&E) staining. In addition, FFPE brain sections were Holmes–Luxol‐stained (to assess axon integrity and myelination status) and processed for immunohistochemistry (IHC) using anti‐GFAP from DAKO (1:500) and anti‐Iba1 from Wako (1:3,000); secondary antibody was anti‐rabbit IgG (Vector; ImmPRESS Reagent Kit).
Cortical brain homogenate preparation
Isolated cortical hemispheres were dissected on ice, washed in ice‐cold buffer 1 (138 mM NaCl, 5.4 mM KCl, 0.17 mM Na2HPO4, 0.22 mM K2HPO4, 5.5 mM glucose, 58.4 mM sucrose, pH 7.35), and homogenized with a glass homogenizer (10–15 strokes, 400 rpm) in 2 ml of ice‐cold buffer 2 (210 mM mannitol, 70 mM sucrose, 10 mM HEPES, 1 mM EDTA, 0.45% BSA, 0.5 mM DTT, 5× Complete Protease Inhibitor (Roche Diagnostics).
Preparation of isolated mitochondria
Cortical brain homogenates were centrifuged at 1,450 g for 7 min at 4°C to remove nuclei and tissue particles; centrifugation was repeated with the supernatant fraction for 3 min 64. The resulting supernatant fraction was centrifuged at 10,000 g for 5 min at 4°C to pellet mitochondria. The resulting pellet was resuspended in 1 ml of ice‐cold buffer 2 and centrifuged at 1,450 g for 3 min at 4°C to remove debris. The mitochondria‐enriched supernatant was centrifuged at 10,000 g for 5 min at 4°C to obtain the mitochondrial fraction. This fraction was resuspended in 300 μl of PBS and stored at 4°C until use, followed by determination of protein content.
Oxygen consumption and ATP measurements in isolated brain mitochondria
Rates of oxygen consumption were measured in isolated cortical mitochondria using a Seahorse Bioscience XF24 Analyzer, following the manufacturer's protocol and as previously described 64. Briefly, mitochondria were diluted 1:10 in cold 1× MAS containing 10 mM succinate, 2 mM malate, and 10 mM pyruvate. 50 μl of mitochondrial suspension (5 μg mitochondrial protein/well) was delivered to each well of a XF Cell Culture microplate (except for background correction wells) and centrifuged at 2,000 g for 20 min at 4°C to let mitochondria adhere to the wells. After centrifugation, 450 μl of pre‐warmed (37°C) 1× MAS plus substrates was added to each well and the plate was incubated 5 min at 37°C in a CO2‐free incubator prior to the experiment. The plate was placed in a XF24 Analyzer, and oxygen consumption rates were assessed under different respiratory states as described above for HEK293 cells, except that oligomycin was added to a final concentration of 2.5 μg/ml.
ATP content was determined using ViaLight Plus Kit (Lonza) following the manufacturer's instructions and normalization per protein content.
Complex IV and complex II measurements in isolated brain mitochondria
Mitochondrial complex IV activity was determined in intact isolated mitochondria (1 μg) using the Cytochrome c Oxidase Assay Kit (Sigma) as previously described 65. The colorimetric assay is based on the observation that a decrease in absorbance at 550 nm of ferrocytochrome c is caused by its oxidation to ferricytochrome c by cytochrome c oxidase. Briefly, the assay was performed in a 96‐well plate. Isolated mitochondria were diluted in enzyme dilution buffer (10 mM Tris–HCl, 250 mM sucrose, pH 7) to a final concentration of 0.2 μg/μl. 5 μl of the mitochondrial solution was added per well. Subsequently, 195 μl of assay buffer (10 mM Tris–HCl, 120 mM KCl, pH 7) was added, then 10 μl of ferrocytochrome c was injected in each well, and the absorbance at 550 nm was measured for 60 s using a Victor X5 multi‐wavelength reader (PerkinElmer).
Mitochondrial complex II activity was determined in intact isolated mitochondria using complex II‐mediated reduction of 2,6‐dichlorophenolindophenol (DCIP). Isolated mitochondria were placed in 1 ml of medium containing 60 mM KH2PO4 (pH 7.4), 3 mM KCN, 20 μg/ml rotenone, 20 mM succinate, and 5 μg mitochondrial protein. The reaction was initiated by the addition of 1.3 mM phenazine methosulfate (PMS) and 0.18 mM DCIP. Reduction of DCIP was followed by measuring the decreasing absorbance at 600 nm. The extinction coefficient used for DCIP was 21 mM/cm. Complex IV and II activities were normalized as per mg protein content.
Determination of superoxide anion radicals
MitoSOX™ Red reagent (Thermo Fisher) is a fluorogenic dye specifically targeted to mitochondria in live cells. Oxidation of MitoSOX™ Red reagent by superoxide produces red fluorescence. Cortical brain homogenate samples were adjusted to 1 mg protein/ml in HBSS. 150 μl of a 5 μM MitoSOX™ reagent working solution (prepared according to the manufacturer's protocol) was added to 250 μl sample, followed by incubation at 37°C for 10 min, protected from light. Then, samples were centrifuged for 3 min at 500 g. After discarding the supernatant, the pellets were washed three times with 250 μl HBSS (3 min at 500 g). Finally, the samples were transferred into a 96‐well plate (final volume 100 μl per well) and fluorescence was detected using the Victor X5 multiplate reader at 510 nm (excitation) and 580 nm (emission). The intensity of fluorescence is proportional to superoxide anion radicals in mitochondria.
Noise exposure and auditory brain stem response
Male C57BL/6 mice were first exposed for 2 h to broadband noise (2–10 kHz) at intensities of 104, 106, 108, and 110 dB. ABR thresholds were determined 3 weeks later when damage to the auditory system has stabilized and is no longer confounded by transient impairment of sound processing. The frequencies of 12, 24, and 32 kHz were selected for measurement to cover the mid‐frequency to high‐frequency regions of the cochlea. Exposures at noise intensities of 108 and 110 dB resulted in robust elevations of ABR thresholds and were chosen as treatment for mutant Mrps5 V338Y/V338Y mice and littermate controls. Sham‐exposed controls (2 h in the exposure chamber without sound) showed no effect on ABR.
Animals for behavioral analysis
Mice were housed under a 12/12‐h light–dark cycle (lights on at 20:00) in groups of 2–5, unless individual housing was required by experimental protocols. Mice were tested during the dark phase of the cycle under indirect dim light (ca. 12 lux) unless specified otherwise. Before behavioral testing, each mouse was tagged by subcutaneous injection of a RFID microchip (Planet ID GmbH) under brief isoflurane inhalation anesthesia. Three cohorts comprising a total of 34 female mice were examined in the same sequence of behavioral tests (in the order listed below) starting at the age of approximately 9 and 19 months, and selected tests were also recorded at 14 months.
IntelliCage system
The IntelliCage apparatus (TSE Systems) was placed in a polycarbonate cage (20.5 cm high, 58 × 40 cm top, 55 × 37.5 cm bottom, Techniplast, 2000P) and accommodated up to 16 mice 66. A food rack provided standard mouse chow ad libitum. Woodchip bedding as well as four standard red shelters were provided. Four triangular conditioning chambers (15 × 15 × 21 cm) were fitted in the cage corners. Two drinking bottles were accessible in each of them behind motorized doors. Visits, nosepokes, and licks were recorded and assigned to individual mice through a RFID antenna at the chamber entrance. A PC running IntelliCage Plus software (TSE Systems) collected the data and controlled the experiments. Testing began by free adaptation (7 days) providing access to all bottles with doors open at all times. During subsequent nosepoke adaptation (3–7 days), doors were closed but could be opened at any time by a nosepoke. Finally, drinking session adaptation (4–7 days) shaped mice to drink only during 4 1‐h drinking sessions each day. The following learning tasks were based on the same drinking session schedule 67. In all of them, percentage of rewarded visits with nosepokes served as measure of performance. In the corner‐preference task, each mouse could obtain water only in one of the four corners during 4 days. In the corner‐preference serial reversal task (7 days), the mice had to learn a new corner during every drinking session. During the final chaining task (10 days), the mice had to visit corners in a clock or anti‐clockwise sequence to get water.
Individual home cage activity
Diurnal home cage activity of individually housed mice was recorded during 7 days under the standard light cycle using a rack of type II long mouse cages (365 mm long × 207 mm wide × 140 mm high) equipped with one passive IR sensor per mouse (ActiviScope, New Behavior Inc) 68.
Video tracking
During open field and water‐maze tests, on the elevated O‐maze, and during the light–dark transition test, the mice were video‐tracked using a Noldus EthoVision 3.00 system (Noldus Information Technology). The system recorded position, object area, and the status of defined event recorder keys on the keyboard. Raw data were then transferred to public domain software Wintrack 2.4 (http://www.dpwolfer.ch/wintrack) for further analysis.
Open field
The round arena had a diameter of 150 and 35 cm high sidewalls made of white polypropylene 68. Each subject was released near the wall and observed for 10 min on two subsequent days. Vertical activity was estimated by counting reductions in object size in the absence of locomotion.
Elevated O‐maze
A 5.5‐cm‐wide annular runway with an outer diameter of 46 cm was positioned 40 cm above the floor 68. Two opposing 90° closed sectors were protected by inner and outer walls (height 16 cm). The remaining two open sectors had no walls. Animals were released near a closed sector and observed for 10 min. Head dips were recorded using the keyboard event recorder. Head dips that occurred while the animal kept its body between the protection walls were classified as protected head dips.
Grip test
The grip meter consisted of a mechanical newton meter (max. force: 300 g) that was positioned horizontally 9 cm above a wooden plate and attached to a metallic T‐bar of 5 cm width and 3 mm thickness. Mice were held by the tail and allowed to grasp the bar with both forepaws. They were then gently pulled away until they released the bar. Five consecutive measurements were obtained each during two sessions with an interval of approximately 1 h 69.
Light–dark transition test
The apparatus consisted of a transparent chamber (20 × 30 cm, 20 cm high) exposed to direct room light (ca. 450 lux) and connected to an enclosed dark box (20 × 15 cm, 20 cm high) via a small opening 68. Mice were released in the bright compartment and observed for 5 min.
Water‐maze place navigation
The round white polypropylene pool had a diameter of 150 cm with 68 cm high walls. It was filled with water (24–26°C, depth 15 cm), which was rendered opaque by addition of 1 l of milk (UHT whole milk 3.5% fat, Coop). The white quadratic goal platform (14 × 14 cm) was made of metallic wire mesh and painted white. It was hidden 0.5 cm below the water surface in the center of one of the four quadrants, approximately 30 cm from the side wall. Salient extra‐maze cues were placed on the walls of the testing room. Animals performed 30 training trials (max. duration 120 s), 6 per day with intertrial intervals of 30–60 min and varying starting positions. During the first 18 trials, the hidden platform was held in the same position (acquisition phase) and then moved to the opposite quadrant for the remaining 12 trials (reversal phase). The first 60 s of the first reversal trial served as probe trial to test for spatial retention.
Nesting test
Mice were provided with 3 g of compressed cotton (Plexx) in a single cage and given 24 h to build a nest. Nests were assessed on a rating scale of 1–5 70.
Burrowing test
A gray plastic tube (inner diameter 6.3 cm, length 18.2 cm) with one open end was filled with 350 g of standard diet food pellets and placed at a slight angle into a large standard transparent type III cage. Each mouse was given 24 h to remove pellets from the tube 71.
T‐maze spontaneous alternation
Arms of the T‐shaped maze measured 30 × 10 cm with 20 cm high side walls. Goal and start arms could be blocked with manually operated doors. Three trials each was run on two subsequent days with an interval of about 60 min 72. During the sample phase, the animal was allowed to choose a goal arm and was confined to the chosen goal arm for 30 s. After this, the animal was transferred back to the start arm and again allowed to choose one of the goal arms (choice phase).
Acoustic startle response
The SR‐LAB™ SDI startle response system (San Diego Instruments) was controlled by proprietary SR‐LAB software. The cylindrical animal enclosure (28 × 89 mm) was made of clear Perspex, rested on a plate with a motion sensor and was enclosed in a sound‐attenuated ventilated cabinet. The loudspeaker was located above the animal and produced white noise pulses. To record a startle response profile, 66 trials were presented in total after a habituation time of 300 s. Nine different sound levels (dB) used were as follows: 64, 68, 72, 76, 80, 90, 100, 110, and 120. Each stimulus was 40 ms and presented 6 times in pseudorandom order such that each sound level occurred once within a block of nine trials. The series began and ended with six presentations each of the 120 dB stimulus. The average intertrial interval was 15 s (ranging from 10 to 20 s). The startle response was recorded for 250 ms (measuring the response every 1 ms) starting with the onset of the startle stimulus. Background noise was set at 64 dB during the entire session.
Hotplate test
To assess acute thermal nociception, the incremental hot/cold plate analgesia meter (model PE34, IITC Life Science) was set to 55°C. The animal was observed until it licked a hindpaw, jumped, or until the cutoff time (60 s) was reached.
Water‐maze cue navigation
Testing apparatus and procedures were the same as used for the place navigation task, except that the platform was marked with a salient local cue and moved to a new position for every trial. Each mouse was trained for 2 days with six trials each.
Statistical analysis
Data from biochemical assays were analyzed with Prism 5.0 software using unpaired Student's t‐test to determine the significance of difference unless otherwise specified.
Data from behavioral studies were analyzed using an ANOVA model with genotype (Mrps5 V338Y/V338Y, Mrps5 WT/WT) and age cohort (9 months, 19 months) as between‐subject factors. Within‐subject factors were added as needed to explore the dependence of genotype effects on place, time, or stimulus. Interactions were further explored by pairwise t‐tests or by splitting the ANOVA model, as appropriate. Variables with strongly skewed distributions or strong correlations between variances and group means were subjected to Box‐Cox transformation before statistical analysis. The false discovery rate (FDR) control procedure of Hochberg was applied to groups of conceptually related variables within single tests to correct significance thresholds for multiple comparisons. Similarly, FDR correction was applied to t‐tests during post hoc testing. Statistical analyses and graphs were produced using R version 3.2.3, complemented with the packages ggplot2, psych, and moments. All tests were two‐sided with significance set at P < 0.05.
Ethics statement
Animal experiments were approved by the Veterinary Office of the Canton of Zurich (licenses 29/2012 and 44/2015). For ABR measurements, animal care was supervised by the University of Michigan's Unit for Laboratory Animal Medicine and experimental protocols were approved by the University of Michigan Committee on Use and Care of Animals (protocol no. PRO00006852).
Data availability
RNA‐seq data are available under Gene Expression Omnibus (GEO) GSE101503.
Author contributions
Concept and design of study, coordination, and supervision of experiments: ECB; MT‐CO1 and MT‐CO2 assays: RA and PF; modeling, generation of stable transfectants, luciferase misreading, and read‐through assays: DS and SD; analysis of stable transfectants: DS, SD, RA, RJ, YT, and HB; behavioral studies: A‐KF and DW; construction and generation of transgenic mice: PI‐P and KT; analysis of brain cortical mitochondria: KS and AE; ABR studies: NO and JS; transcriptome studies: RJ and HR; histopathology studies: BO and SF; writing of the manuscript with input from all authors: ECB, EW, SD, JS, and DW. All authors analyzed and discussed the data.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
David P. Wolfer is a member of the Neuroscience Center Zurich (ZNZ) and the Zurich Center for Integrative Human Physiology (ZIHP). We are indebted to Diarmaid Hughes and Douglas Huseby, Uppsala University (Sweden), for providing recombinant E. coli strains. We thank the Institute of Medical Microbiology and the University of Zurich for continuous support.
EMBO Reports (2018) 19: e46193
References
- 1. Scheffler IE (2001) A century of mitochondrial research: achievements and perspectives. Mitochondrion 1: 3–31 [DOI] [PubMed] [Google Scholar]
- 2. Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA (1981) Sequence and gene organization of mouse mitochondrial DNA. Cell 26: 167–180 [DOI] [PubMed] [Google Scholar]
- 3. Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F et al (1981) Sequence and organization of the human mitochondrial genome. Nature 290: 457–465 [DOI] [PubMed] [Google Scholar]
- 4. Sagan L (1967) On the origin of mitosing cells. J Theor Biol 14: 255–274 [DOI] [PubMed] [Google Scholar]
- 5. Greber BJ, Ban N (2016) Structure and function of the mitochondrial ribosome. Annu Rev Biochem 85: 103–132 [DOI] [PubMed] [Google Scholar]
- 6. Christian BE, Spremulli LL (2012) Mechanism of protein biosynthesis in mammalian mitochondria. Biochim Biophys Acta 1819: 1035–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, Agrawal RK (2003) Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell 115: 97–108 [DOI] [PubMed] [Google Scholar]
- 8. Neupert W, Herrmann JM (2007) Translocation of proteins into mitochondria. Annu Rev Biochem 76: 723–749 [DOI] [PubMed] [Google Scholar]
- 9. Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N (2009) Importing mitochondrial proteins: machineries and mechanisms. Cell 138: 628–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szklarczyk R, Huynen MA (2010) Mosaic origin of the mitochondrial proteome. Proteomics 10: 4012–4024 [DOI] [PubMed] [Google Scholar]
- 11. Chinnery PF (2015) Mitochondrial disease in adults: what's old and what's new? EMBO Mol Med 7: 1503–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW (2017) The genetics and pathology of mitochondrial disease. J Pathol 241: 236–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI et al (1993) Mitochondrial ribosomal RNA mutation associated with both antibiotic‐induced and non‐syndromic deafness. Nat Genet 4: 289–294 [DOI] [PubMed] [Google Scholar]
- 14. Fischel‐Ghodsian N (1999) Mitochondrial deafness mutations reviewed. Hum Mutat 13: 261–270 [DOI] [PubMed] [Google Scholar]
- 15. Guan MX, Fischel‐Ghodsian N, Attardi G (2001) Nuclear background determines biochemical phenotype in the deafness‐associated mitochondrial 12S rRNA mutation. Hum Mol Genet 10: 573–580 [DOI] [PubMed] [Google Scholar]
- 16. Raimundo N, Song L, Shutt TE, McKay SE, Cotney J, Guan MX, Gilliland TC, Hohuan D, Santos‐Sacchi J, Shadel GS (2012) Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell 148: 716–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O'Sullivan M, Rutland P, Lucas D, Ashton E, Hendricks S, Rahman S, Bitner‐Glindzicz M (2015) Mitochondrial m.1584A 12S m62A rRNA methylation in families with m.1555A>G associated hearing loss. Hum Mol Genet 24: 1036–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee S, Rose S, Metodiev MD, Becker L, Vernaleken A, Klopstock T, Gailus‐Durner V, Fuchs H, Hrabě De Angelis M, Douthwaite S et al (2015) Overexpression of the mitochondrial methyltransferase TFB1M in the mouse does not impact mitoribosomal methylation status or hearing. Hum Mol Genet 24: 7286–7294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hobbie SN, Bruell CM, Akshay S, Kalapala SK, Shcherbakov D, Böttger EC (2008) Mitochondrial deafness alleles confer misreading of the genetic code. Proc Natl Acad Sci USA 105: 3244–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scheper GC, van der Klok T, van Andel RJ, van Berkel CG, Sissler M, Smet J, Muravina TI, Serkov SV, Uziel G, Bugiani M et al (2007) Mitochondrial aspartyl‐tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet 39: 534–539 [DOI] [PubMed] [Google Scholar]
- 21. Konovalova S, Tyynismaa H (2013) Mitochondrial aminoacyl‐tRNA synthetases in human disease. Mol Genet Metab 108: 206–211 [DOI] [PubMed] [Google Scholar]
- 22. Sissler M, González‐Serrano LE, Westhof E (2017) Recent advances in mitochondrial aminoacyl‐tRNA synthetases and disease. Trends Mol Med 23: 693–708 [DOI] [PubMed] [Google Scholar]
- 23. Synetos D, Frantziou CP, Alksne LE (1996) Mutations in yeast ribosomal proteins S28 and S4 affect the accuracy of translation and alter the sensitivity of the ribosomes to paromomycin. Biochem Biophys Acta 1309: 156–166 [DOI] [PubMed] [Google Scholar]
- 24. Greber BJ, Bieri P, Leibundgut M, Leitner A, Aebersold R, Boehringer D, Ban N (2015) Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome. Science 348: 303–308 [DOI] [PubMed] [Google Scholar]
- 25. Amunts A, Brown A, Toots J, Scheres SH, Ramakrishnan V (2015) Ribosome. The structure of the human mitochondrial ribosome. Science 348: 95–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Patananan AN, Wu TH, Chiou PY, Teitell MA (2016) Modifying the mitochondrial genome. Cell Metab 23: 785–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Temperley R, Richter R, Dennerlein S, Lightowlers RN, Chrzanowska‐Lightowlers ZM (2010) Hungry codons promote frameshifting in human mitochondrial ribosomes. Science 327: 301 [DOI] [PubMed] [Google Scholar]
- 28. Zheng QY, Johnson KR, Erway LC (1999) Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res 130: 94–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Böttger EC, Schacht J (2013) The mitochondrion: a perpetrator of acquired hearing loss. Hear Res 303: 12–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prenderville JA, Kennedy PJ, Dinan TG, Cryan JF (2015) Adding fuel to the fire: the impact of stress on the ageing brain. Trends Neurosci 38: 13–25 [DOI] [PubMed] [Google Scholar]
- 31. Mata R, Wilke A, Czienskowski U (2013) Foraging across the life span: is there a reduction in exploration with aging? Front Neurosci 7: 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Perna G, Iannone G, Alciati A, Caldirola D (2016) Are anxiety disorders associated with accelerated aging? A focus on neuroprogression. Neural Plast 2016: 8457612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beaudreau SA, O'Hara R (2008) Late‐life anxiety and cognitive impairment: a review. Am J Geriatr Psychiatry 16: 790–803 [DOI] [PubMed] [Google Scholar]
- 34. Hovatta I, Tennant RS, Helton R, Marr RA, Singer O, Redwine JM, Ellison JA, Schadt EE, Verma IM, Lockhart DJ et al (2005) Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature 438: 662–666 [DOI] [PubMed] [Google Scholar]
- 35. Einat H, Yuan P, Manji HK (2005) Increased anxiety‐like behaviors and mitochondrial dysfunction in mice with targeted mutation of the Bcl‐2 gene: further support for the involvement of mitochondrial function in anxiety disorders. Behav Brain Res 165: 172–180 [DOI] [PubMed] [Google Scholar]
- 36. Hollis F, van der Kooij MA, Zanoletti O, Lozano L, Canto C, Sandi C (2015) Mitochondrial function in the brain links anxiety with social subordination. Proc Natl Acad Sci USA 112: 15486–15491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bitner‐Glindzicz M, Pembrey M, Duncan A, Heron J, Ring SM, Hall A, Rahman S (2009) Prevalence of mitochondrial 1555A–>G mutation in European children. N Engl J Med 360: 640–642 [DOI] [PubMed] [Google Scholar]
- 38. Bravo O, Ballana E, Estivill X (2006) Cochlear alterations in deaf and unaffected subjects carrying the deafness‐associated A1555G mutation in the mitochondrial 12S rRNA gene. Biochem Biophys Res Commun 344: 511–516 [DOI] [PubMed] [Google Scholar]
- 39. Vicente‐Torres MA, Schacht J (2006) A BAD link to mitochondrial cell death in the cochlea of mice with noise‐induced hearing loss. J Neurosci Res 83: 1564–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen FQ, Zheng HW, Hill K, Sha SH (2012) Traumatic noise activates Rho‐family GTPases through transient cellular energy depletion. J Neurosci 32: 12421–12430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bykhovskaya Y, Mengesha E, Fischel‐Ghodsian N (2009) Phenotypic expression of maternally inherited deafness is affected by RNA modification and cytoplasmic ribosomal proteins. Mol Genet Metab 97: 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goto Y, Nonaka I, Horai S (1990) A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348: 651–653 [DOI] [PubMed] [Google Scholar]
- 43. Ruiz‐Pesini E, Lott MT, Procaccio V, Poole JC, Brandon MC, Mishmar D, Yi C, Kreuziger J, Baldi P, Wallace DC (2007) An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res 35: D823–D828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ogle JM, Ramakrishnan V (2005) Structural insights into translational fidelity. Annu Rev Biochem 74: 129–177 [DOI] [PubMed] [Google Scholar]
- 45. Hobbie SN, Kalapala SK, Akshay S, Bruell C, Schmidt S, Dabow S, Vasella A, Sander P, Böttger EC (2007) Engineering the rRNA decoding site of eukaryotic cytosolic ribosomes in bacteria. Nucleic Acids Res 35: 6086–6093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hobbie SN, Pfister P, Brull C, Westhof E, Böttger EC (2005) Analysis of the contribution of individual substituents in 4,6‐aminoglycoside‐ribosome interaction. Antimicrob Agents Chemother 49: 5112–5118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sawitzke JA, Costantino N, Li XT, Thomason LC, Bubunenko M, Court C, Court DL (2011) Probing cellular processes with oligo‐mediated recombination and using the knowledge gained to optimize recombineering. J Mol Biol 407: 45–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bruell CM, Eichholz C, Kubarenko A, Post V, Katunin VI, Hobbie SN, Rodnina MV, Böttger EC (2008) Conservation of bacterial protein synthesis machinery: initiation and elongation in Mycobacterium smegmatis . Biochemistry 47: 8828–8839 [DOI] [PubMed] [Google Scholar]
- 49. Hobbie SN, Akshay S, Kalapala SK, Bruell CM, Shcherbakov D, Böttger EC (2008) Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc Natl Acad Sci USA 105: 20888–20893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matt T, Ng CL, Lang K, Sha SH, Akbergenov R, Shcherbakov D, Meyer M, Duscha S, Xie J, Dubbaka SR et al (2012) Dissociation of antibacterial activity and aminoglycoside ototoxicity in the 4‐monosubstituted 2‐deoxystreptamine apramycin. Proc Natl Acad Sci USA 109: 10984–10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Duscha S, Boukari H, Shcherbakov D, Salian S, Silva S, Kendall A, Kato T, Akbergenov R, Perez‐Fernandez D, Bernet B et al (2014) Identification and evaluation of improved 4′‐O‐(alkyl) 4,5‐disubstituted 2‐deoxystreptamines as next‐generation aminoglycoside antibiotics. MBio 5: e01827–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- 53. Venegas V, Halberg MC (2012) Measurement of mitochondrial DNA copy number. Methods Mol Biol 837: 327–335 [DOI] [PubMed] [Google Scholar]
- 54. Pallotti F, Lenaz G (2001) Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods Cell Biol 65: 1–35 [DOI] [PubMed] [Google Scholar]
- 55. McKee EE, Grier BL, Thompson GS, McCourt JD (1990) Isolation and incubation conditions to study heart mitochondrial protein synthesis. Am J Physiol 258: E492–E502 [DOI] [PubMed] [Google Scholar]
- 56. Fernández‐Silva P, Acín‐Pérez R, Fernández‐Vizarra E, Pérez‐Martos A, Enriquez JA (2007) In vivo and in organello analyses of mitochondrial translation. Methods Cell Biol 80: 571–588 [DOI] [PubMed] [Google Scholar]
- 57. McKee EE, Ferguson M, Bentley AT, Marks TA (2006) Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrob Agents Chemother 50: 2042–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schnutgen F, Doerflinger N, Calleja C, Wendling O, Chambon P, Ghyselinck NB (2003) A directional strategy for monitoring Cre‐mediated recombination at the cellular level in the mouse. Nat Biotechnol 21: 562–565 [DOI] [PubMed] [Google Scholar]
- 59. Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 12: 323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Supek F, Bosnjak M, Skunca N, Smuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6: e21800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Oettinghaus B, Schulz JM, Restelli LM, Licci M, Savoia C, Schmidt A, Schmitt K, Grimm A, More L, Hench J et al (2016) Synaptic dysfunction, memory deficits and hippocampal atrophy due to ablation of mitochondrial fission in adult forebrain neurons. Cell Death Differ 23: 18–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, Ozmen L, Bluethmann H, Drose S, Brandt U et al (2009) Amyloid‐beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer's disease mice. Proc Natl Acad Sci USA 106: 20057–20062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vannoni E, Voikar V, Colacicco G, Sanchez MA, Lipp HP, Wolfer DP (2014) Spontaneous behavior in the social homecage discriminates strains, lesions and mutations in mice. J Neurosci Methods 234: 26–37 [DOI] [PubMed] [Google Scholar]
- 67. Weyer SW, Klevanski M, Delekate A, Voikar V, Aydin D, Hick M, Filippov M, Drost N, Schaller KL, Saar M et al (2011) APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. EMBO J 30: 2266–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Madani R, Kozlov S, Akhmedov A, Cinelli P, Kinter J, Lipp HP, Sonderegger P, Wolfer DP (2003) Impaired explorative behavior and neophobia in genetically modified mice lacking or overexpressing the extracellular serine protease inhibitor neuroserpin. Mol Cell Neurosci 23: 473–494 [DOI] [PubMed] [Google Scholar]
- 69. Tremml P, Lipp HP, Muller U, Ricceri L, Wolfer DP (1998) Neurobehavioral development, adult openfield exploration and swimming navigation learning in mice with a modified beta‐amyloid precursor protein gene. Behav Brain Res 95: 65–76 [DOI] [PubMed] [Google Scholar]
- 70. Deacon RM (2006) Assessing nest building in mice. Nat Protoc 1: 1117–1119 [DOI] [PubMed] [Google Scholar]
- 71. Deacon RM (2006) Burrowing in rodents: a sensitive method for detecting behavioral dysfunction. Nat Protoc 1: 118–121 [DOI] [PubMed] [Google Scholar]
- 72. Deacon RM, Rawlins JN (2006) T‐maze alternation in the rodent. Nat Protoc 1: 7–12 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File
Data Availability Statement
RNA‐seq data are available under Gene Expression Omnibus (GEO) GSE101503.