

Abstract
Glycans are major nutrients for the human gut microbiota (HGM). Arabinogalactan proteins (AGPs) comprise a heterogenous group of plant glycans in which a β1,3-galactan backbone and β1,6-galactan side chains are conserved. Diversity is provided by the variable nature of the sugars that decorate the galactans. The mechanisms by which nutritionally relevant AGPs are degraded in the HGM are poorly understood. Here we explore how the HGM organism Bacteroides thetaiotaomicron metabolises AGPs. We propose a sequential degradative model in which exo-acting glycoside hydrolase (GH) family 43 β1,3-galactanases release the side chains. These oligosaccharide side chains are depolymerized by the synergistic action of exo-acting enzymes in which catalytic interactions are dependent on whether degradation is initiated by a lyase or GH. We identified two GHs that establish two previously undiscovered GH families. The crystal structures of the exo-β1,3-galactanases identified a key specificity determinant and departure from the canonical catalytic apparatus of GH43 enzymes. Growth studies of Bacteroidetes spp. on complex AGP revealed three keystone organisms that facilitated utilisation of the glycan by 17 recipient bacteria, which included B. thetaiotaomicron. A surface endo-β1,3-galactanase, when engineered into B. thetaiotaomicron, enabled the bacterium to utilise complex AGPs and act as a keystone organism.
The human gut microbiota (HGM) contributes to the physiology and health of its host1. Glycans, the major nutrients for the HGM, are degraded primarily by Bacteroides species within this ecosystem2–4. Understanding glycan utilisation in the HGM underpins prebiotic and probiotic strategies that promote human health. Glycan degradation is mediated by carbohydrate active enzymes (CAZymes), primarily glycoside hydrolases (GHs) and polysaccharide lyases (PLs)5, which are grouped into sequence-based families on the CAZy database (http://www.cazy.org/)6. Although there is structural and catalytic conservation within families, substrate specificity may vary7. Genes encoding glycan degrading systems are up-regulated by the target carbohydrate and are physically linked within polysaccharide utilisation loci (PULs)8,9. Glycan depolymerisation is generally initiated by bacterial surface endo-acting GHs/PLs, and the oligosaccharides generated imported into the periplasm and further metabolised9–11.
A ubiquitous component of the human diet are arabinogalactan proteins (AGPs). These proteoglycans are in every taxonomic plant group12, with high concentrations in processed foods such as red wine and instant coffee13,14. Gum Arabic AGP (GA-AGP) is widely used in the food industry to improve biophysical properties of many products15. AGPs comprise a β1,3-galactan backbone with β1,6-galactan side-chains, which contain carbohydrate decorations (Fig. 1ab). Glycans, comprising 90% of AGPs, are O-linked to hydroxyprolines in the protein component16. AGP utilisation is poorly understood. Oligosaccharide side-chains are released by GH43 subfamily 24 (GH43_24) exo-acting β1,3-galactanases17, however, the mechanism for their unusual substrate specificity remains unclear. Although endo-acting enzymes contribute to glycan degradation, the role of endo-galactanases in AGP metabolism is unknown. While some enzymes that target AGPs have been described18,19, models for the degradation of these glycoproteins are lacking. The prebiotic potential of GA-AGPs is evident20, however, fulfilling the health benefit of these glycans requires a deeper understanding of how these proteoglycans are metabolised by the HGM.
Figure 1. The structure of arabinogalactans, PULs upregulated by the glycans and enzymes that attack these glycans.

Structure of a, larch wood (LA-AGP) and b, gum arabic (GA-AGP) arabinogalactans, and the enzymes that act on these glycans. The enzymes are identified by their locus tag (BTXXXX and BaccellXXXX are derived from B. thetaiotaomicron and B. cellulosilyticus, respectively), assignment to cazy families (GHXX and PLXX indicate glycoside hydrolase and polysaccharide lyase families, respectively) and their predicted cellular location (based on the nature of the signal peptide and, in some cases, cellular location for the observed activity, proteomic analysis or resistance to proteinase K; see Fig. 5aef), in which superscript P and C indicate periplasmic and cytoplasmic location, respectively.and superscript. The black arrows show the linkage cleaved by the enzymes, although the polysaccharide lyase activity of BT0263 is not functionally relevant as it is located in the cytoplasm. We propose that the β-L-arabinofuranose targeted by BT3674 is linked to the β1,3-galactan backbone at O2 or O4. This assumption is based on the observation that the enzyme potentiates the exo-β-1,3-galactosidases that sequentially remove galactose units from the backbone (see Fig. 2a). These galactosidases can target galactose residues decorated at O6 but not at O2 or O4. c, Schematic of B. thetaiotaomicron polysaccharide utilization loci (PULs) upregulated by arabinogalactan degradation.
Here we report a model for simple and complex AGP utilisation by Bacteroides species of the HGM. We reveal mechanisms of substrate specificity and catalysis of exo-acting β1,3-galactanases. Strategies for removing the l-rhamnopyranose (Rhap) cap of complex AGPs were shown to influence synergetic interactions between side-chain degrading GHs and PLs. Critically, the cellular location of the endo-β1,3-galactanase defined whether a bacterium was a keystone organism, or a recipient of AGP-derived oligosaccharides.
Results
Functional significance of PULAGPS and PULAGPL in B. thetaiotaomicron
Previous data identified two PULs (PULAGPL and PULAGPS) upregulated when Bacteroides thetaiotaomicron was cultured on larchwood AGP (LA-AGP) (Fig. 1ac)21. Here we showed that only PULAGPS was substantially activated by GA-AGP (Supplementary Fig. 1a), suggesting that different molecules activate the two PULs. Growth studies of mutants of B. thetaiotaomicron lacking the two AGP PULs showed that ΔPULAGPL failed to grow on LA-AGP but displayed growth on GA-AGP treated with endo-β1,3-galactanases (Supplementary Fig. 2). ΔPULAGPS grew on LA-AGP but poorly on treated GA-AGP (Supplementary Fig. 2). These data suggest that the two PULs orchestrate the degradation of different AGPs. To explore the biochemical basis for these phenotypes, the specificity of the enzymes encoded by these loci were determined (Supplementary Table 1). Models for metabolism of selected AGPs were generated (Fig. 1ab).
Cleavage of the galactan backbone
Known activities within GH families the β-1,3-galactan backbone is depolymerized by GH43 subfamily 24 (GH43_24)22 and/or GH1623 enzymes. Thus, activity of B. thetaiotaomicron GH43_24 enzymes [BT0264, BT0265, BT3683 (also contains a GH16 module) and BT3685] encoded by PULAGPL and PULAGPS were evaluated against d-galactose (Gal) disaccharides, LA-AGP, GA-AGP, and linear β-1,3-galactan. Based on activity against disaccharides (Supplementary Table 2), and an active site pocket (BT3683 and BT0265) in which O3 of bound Gal was not solvent exposed (Fig. 3cd), BT0265, BT3683 and BT3685 are exo-acting β-1,3-galactosidases. BT0265 and BT3683 were active against LA-AGP and GA-AGP releasing oligosaccharide side-chains (Fig. 2abc). Mutational analysis (Supplementary Table 3) showed that only the GH43_24 module contributed to the observed activity of BT3683. Consistent with other GH43_24 β-1,3-galactosidases17, the oligosaccharides generated by BT0265 and BT3683 likely comprise β-1,6-galactooligosaccharide side-chains. This assumption suggests that in BT0265 and BT3683, O6 of the Gal backbone units bound in the active site were solvent exposed enabling side-chain accommodation. BT3685 was more active against β-1,3-galactobiose than the other GH43_24 enzymes, but was inactive against the AGPs tested. The role of the enzyme in degrading AGPs is unclear. The GH43_24 enzyme BT0264 was inactive against galactobiose, released oligosaccharides from LA- and GA-AGP, and generated a range of oligosaccharides from β1,3-galactan with the smaller products increasing with time (Fig. 2d); consistent with endo-activity.
Figure 3. The crystal structure of GH43_24 β1,3-D-galactosidases in complex with ligands.

a, schematic of BT0265 (left) and BT3683 (right) in which the catalytic domains are colour ramped from blue at the N-terminus to red at the C-terminus. The C-terminal β-sandwich domain in BT0265 is coloured cyan. b, shows the solvent exposed surface of BT0265 in complex with the heptasaccharide shown in Supplementary Fig. 3 (terminal α-Gal and α-Rha are not visible). Electron density for the terminal α-Gal was too weak to model the sugar. The red dashes show the polar interactions between the ligand and both side chains and backbone N and O. Residues that make polar contacts with the side chain of the ligand are also shown. c, an overlay of the residues in BT0265 (cyan), BT3683 (green) and the GH43_24 β1,3-galactosidase Cthe_1271 (grey; PDB code 3VSZ) that interact with galactose (yellow) in complex with BT3683. d, BT3683 in complex with galactose (Gal), deoxygalactonojirimycin (DGJ) and galactose-imidazole (Gal-Im). Direct polar interactions between enzyme and ligand are indicated by black dashes and the indirect water-mediated hydrogen bonds in magenta dashes. The red dashed line represents the polar interaction between the catalytic acid (Glu520) and Ser487. The two conformations of Glu520 in the Gal-Im complex is denoted by a and b.
Figure 2. HPAEC analysis of the activity of GH43_24 β1,3-D-galactanases.

The AGPs were at 5 mg/ml for all reactions except BT0264 against LA-AGP and BT3683 against GA-AGP, when substrate concentration was increased to 25 mg/ml, the β-1,3-galactan backbone was at 1.5 mg/ml. Enzyme concentration was 1 µM. Reactions were incubated for 16 h in 20 mM sodium phosphate buffer pH 7.0 containing 150 mM NaCl buffer. The data shown are representative of three independent replicates. a, reveals how the GH127 β-L-arabinofuranosidase BT3674 acts in synergy with the exo-β1,3-galactosidases BT0265 and BT3683 on LA-AGP. The synergy between the endo-β1,3-galactanase with BT0265 and BT3683 acting on LA-AGP and GA-AGP was shown in b and c, respectively. d, shows a time course of BT0264 acting on β-1,3-galactan. Peaks containing a defined galactooligosaccharide are identified by a yellow circle with the degree of polymerization shown in subscript. In b and c the peaks corresponding to β1,6-galactobiose and β1,6-galactotriose were identified by LC-MS (see Supplementary Fig. 1d), and the β1,6 linkage was revealed by sensitivity to the β1,6-galactosidase BT0290.
Synergistic interactions in the degradation of the β-1,3-galactan backbone
In addition to O6-linked side chains, the AGP backbones contain sugar pendants at O2 or O4, commonly β-L-Araf units. These substitutions block progression of the exo-β1,3-galactanases through steric constraints (Fig. 3). Mechanisms for relieving these “roadblocks” include removal of these decorations and/or endo-cleavage of the backbone creating non-reducing termini downstream of O2/O4 decoration. To explore these hypotheses GA- and LA-AGP were incubated with BT3674, which contains an active-site typical of β-L-arabinofuranosidases (Supplementary Fig. 3). The enzyme released arabinose from LA-AGP, mediating an eight-fold increase in oligosaccharides generated by the exo-β1,3-galactanases (Fig. 2a). The endo-β1,3-galactanase BT0264 also increased the activity of the exo-β1,3-galactanases (Fig. 2bc). Thus, B. thetaiotaomicron exploits two mechanisms to reduce stalling of exo-β1,3-galactanases.
Crystal structures of GH43_24 enzymes
The crystal structures of BT0265 and BT3683 revealed that both exo-β-1,3-galactosidases displayed a five-bladed β-propeller fold (Fig. 3a) typical of GH43 enzymes24. Typical of GH43 exo-glycosidases the active-site pocket of BT0265 and BT3683 is in the centre of the β-propeller24. Ligand complexes revealed the polar interactions between Gal, hexasaccharide product and Gal-based inhibitors and the exo-β-1,3-galactosidases, Fig. 3bcd. These polar interactions are augmented by apolar contacts with a hydrophobic platform (Trp261/Trp213 in BT3683/BT0265). Interaction of the essential glutamate, Glu86/Glu87 in BT0265/BT3683 (Supplementary Table 3), with the axial O4 of Gal (Fig. 3bd) confers selectivity for Gal over Glc, and is thus a key specificity determinant. O3 of bound ligands points into the active site pocket explaining the exo- and not endo-activity of the β1,3-galactanases. The lack of interactions with substrate outside of the active site indicates that complementarity of the helical conformation of β1,3-galactan25 and topology of the catalytic centre drives specificity.
The BT0265 hexasaccharide product complex reveals O6 of Gal in the active site is solvent exposed (Fig. 3b). This explains why the enzyme releases backbone Gal residues decorated with oligosaccharides appended at O6. Whether side-chains contribute to specificity is unclear; however, elements of these decorations interact with BT0265 (Fig. 3b),
In GH43 enzymes the catalytic acid (glutamate) and pKa modulator (aspartate) are invariant24. The assignment of Glu240 in BT3683 as the catalytic acid (Fig. 3d) is supported by the reactivity of E240A. This variant did not hydrolyse β1,3-galactobiose but hydrolysed 2,4-dinitrophenyl-β-D-Gal (Supplementary Table 3), consistent with requiring protonation when Gal is the leaving group but not when 2,4-dinitrophenolate (pKa 3.6) is generated. Mutation of the catalytic acid in BT3685 (E225Q) also revealed the expected impact on activity against the two substrates. GH43_24 enzymes lack the aspartate catalytic base that is invariant in other GH43 subfamilies24. In GH43_24 a highly conserved glutamine binds a water molecule (Fig. 3d) that could attack the anomeric carbon of the substrate below the plane of the ring, consistent with the inverting mechanism of BT3685 (Supplementary Fig. 4). Mutation of the glutamine in BT3683 supports a catalytic role for this residue (Supplementary Table 3). The glutamine may form an imidic acid through tautomerization and thus function as the base, as proposed for some inverting enzymes26, or assist in positioning the catalytic water that attacks the anomeric centre of the substrate.
Deconstruction of the AGP side chains
The side-chains, released by exo-β1,3-galactosidases from GA-AGP were characterized by mass spectrometry (Supplementary Fig. 5) and NMR spectroscopy (Supplementary Fig. 6). The major side chains comprised oligosaccharides with a degree of polymerization (DP) of 3 to 7 (Supplementary Fig. 5). The non-reducing terminus of each oligosaccharide comprised Rhap-α1,4-GlcA-β1,6-Gal. Previous studies showed that the B. thetaiotaomicron GH145 α-L-rhamnosidase BT3686 removed Rhap exposing GlcA19. Here we show that the exposed GlcA was removed by the β-glucuronidase BT3677, the founding member of GH154 (Fig. 4, Supplementary Fig. 7a, Supplementary Table 2). BT3677 was only active against oligosaccharides after removal of the terminal Rhap, and is thus exo-acting. The β-glucuronidase hydrolysed the GlcA-β1,6-Gal linkage when Gal was substituted with α-L-Ara at O3 (Fig. 4) but not at O4 (Supplementary Fig. 8).
Figure 4. Degradation of GA-AGP side chains.

The pentasaccharide substrate shown in a grey box was released from GA-AGP by the exo-β1,3-galactosidase BT0265 and then purified by size exclusion chromatography. Individual B. thetaiotaomicron enzymes (1 μM) were incubated with the glycan (5 mM) for 16 h at 37 °C in 20 mM sodium phosphate buffer, pH 7.0. Monosaccharides and oligosaccharides generated were identified by HPAEC-PAD. The data in a and b show that the pentasaccharide could be degraded by the enzymes that comprise the LU and RG pathways, respectively. Note that the enzymes in the two pathways Verification of the degradative pathway was achieved by reconstituting the pathway using the only functioned in the order shown in the figure. The example is representative of independent replicates (n = 3).
B. thetaiotaomicron removes the terminal disaccharide structure of GA-AGP by a rhamnosidase-glucuronidase (RG) pathway, consistent with limited growth of Δbt3686 on GA-AGP (Supplementary Fig. 2b). Cell-free extracts of Δbt3686 cultured on LA-AGP failed to release Rhap from GA-AGP. These data confirm the RG pathway operates in B. thetaiotaomicron and that the side chains in GA-AGP are extensively capped with Rhap. The orthologues of BT3686 in B. cellulosilyticus, and other HGM Bacteroidetes species are not functional rhamnosidases as they lack the catalytic histidine19. B. cellulosilyticus, however, contains a rhamno-glucurono lyase (BACCELL_00875) that cleaved the Rha-α1,4-GlcA linkage, and the resultant 4,5ΔGlcA was released by an unsaturated glucuronidase18. Thus, B. cellulosilyticus releases the capping Rha-GlcA disaccharide through a lyase-unsaturated glucuronidase (LU) pathway. Genomic studies indicate that both routes to removing the capping disaccharide (RG and/or LU pathways) are possible in some Bacteroidetes species. The significance of deploying both pathways is discussed below.
Gal at the base of AGP β-1,6-galactan side-chains can be decorated with Araf that may be capped with α-Gal. No enzyme encoded by B. thetaiotaomicron AGP PULs removed the α-Gal (discussed below). PULAGPS also encodes two arabinofuranosidases; a GH43 enzyme (BT3675) and the non-specific arabinofuranosidase, BT3679, active against wheat AGP (WH-AGP), arabinoxylan and sugar beet arabinan (Supplementary Fig. 7b, Supplementary Table 2). BT3679 establishes a GH family (GH155) exclusive to the Bacteroidetes phylum. Cleavage of 4-nitrophenyl-α-L-arabinofuranoside by BT3679 in the presence of methanol generated methyl-α-arabinofuranoside (Supplementary Fig. 7c), demonstrating a retaining mechanism. In GA-AGP BT3679 cleaved the Araf-α1,3-Gal linkage at the base of the β1,6-galactan backbone irrespective of whether the Gal was decorated at O4 (Supplementary Fig. 8). BT3675 hydrolysed the Araf-α1,3-Gal glycosidic bond, but not when Gal also contained α-L-Araf at O4. The two enzymes and cell-free extracts of B. thetaiotaomicron cultured on AGPs did not cleave the O4-linked Araf. Thus, B. thetaiotaomicron is unable to cleave α-Araf linked O4 to Gal.
The GH35 enzyme BT0290 hydrolysed β-1,6-galactan side-chains in LA-AGP and β-1,6-galactobiose, exhibiting minor activity against β-1,3-galactobiose. The crystal structure of BT0290 revealed a (β/α)8 barrel catalytic module. In the ligand complex Gal is in the active site pocket at the end of the β-barrel (Supplementary Fig. 9), which contains a pair of glutamates that comprise a canonical catalytic apparatus for a retaining enzyme, expected for GH35. The pocket extends onto a planar surface that houses the O6-linked β-Gal in the +1 subsite. Trp215 in the +1 subsite creates a steric block for O3- or O4-linked sugars and provides a hydrophobic platform for an O6-linked β-Gal. This tryptophan is likely a specificity determinant for the β-1,6-galactosidase activity of BT0290.
In vivo degradation of AGPs by HGM Bacteroidetes species
Supplementary Table 4 reports growth profiles of type strains of 20 HGM Bacteroidetes species. All species except Dysgonomonas gadei utilised LA-AGP, while only B. cellulosilyticus, B. caccae and D. gadei grew on GA-AGP or WH-AGP (Supplementary Table 4). This was surprising as B. thetaiotaomicron, at least, degrades side-chains from GA-AGP. The initial depolymerisation of polysaccharides in Bacteroides species occurs at the bacterial surface, generating oligosaccharides suitable for transport into the periplasm10,11. In B. thetaiotaomicron the GH43_24 endo-β1,3-galactanase, BT0264, has a type I signal peptide typical of periplasmic proteins, confirmed by cell localization studies (Fig. 5a, Supplementary Fig. 10). The inability of B. thetaiotaomicron to grow on GA-AGP likely reflects the absence of a surface endo-β1,3-galactanase required to generate the GA-AGP-derived oligosaccharides for import into the periplasm. This was confirmed by growth of B. thetaiotaomicron on GA-AGP and WH-AGP pre-treated with BT0264 (Fig. 5bc, Supplementary Table 4). The BT0264-treated GA-AGP was also a growth substrate for the other 16 Bacteroidetes species unable to utilise intact GA- and WH-AGP (Supplementary Table 4). The inability of the majority of HGM-derived Bacteroidetes species to utilise GA-AGP reflects the lack of an endo-β1,3-galactanase that can degrade extracellular GA-AGP. Growth of these organisms on LA-AGP reflects the low DP of the glycan, enabling direct import into the periplasm.
Figure 5. Cell localization and growth of Bacteroides on complex AGPs.

a, Western blot detection of BT0264 and a known surface enzyme (BT4662)30 in LA-AGP/heparin cultured B. thetaiotaomicron after treatment of the bacterial cells with proteinase K (PK+) or untreated (PK-). Purified recombinant BT0264 was also subjected to proteinase treatment to verify the enzyme is sensitive to the proteinase. The data show that the enzyme is resistant to the proteinase and thus is not located on the cell surface. The blot is an example of biological replicates where n=3. Wild type B. thetaiotaomicron (Bt) and B. thetaiotaomicron expressing Baccell00844 (Bt::Baccell00844) were cultured in 0.2 ml of minimal medium containing AGPs under anaerobic conditions. b, growth was assessed on GA-AGP and GA-AGP pre-treated with BT0264 [GA-AGP(BT0264)] or Baccell00844 [GA-AGP(Baccell00844)]. In c growth was evaluated on wheat AGP (WH-AGP). In b and c error bars report standard errors of the mean of biological replicates (n = 4). d, HPAEC analysis of the products generated by recombinant Baccell00844 (1 μM) incubated with β-1,3-galactan for 16 h using standard conditions. The chromatographs are examples of biological replicates (n = 2). e, Bt, Bt::Baccell00844 and B. cellulosilyticus (Baccell) cells derived from cultured grown on GA-AGP were incubated with 0.5% β1,3-galactan for 16 h in phosphate buffered saline in aerobic conditions for 16 h. Under these conditions substrate is only available to the surface enzymes. Products released from the glycan was evaluated by TLC. The example is from biological replicates n =3. f, Venn diagram of the number of proteins identified in the surfome, the surfome and total proteome, and total proteome. Baccell00844 was unique to the surfome fraction. The 46 proteins detected only in the surfome are described in Supplementary Table 5.
The B. cellulosilyticus genome encodes four GH16 and four GH43_24 enzymes that, potentially, comprise endo-β1,3-galactanases. RT-PCR of SusC genes of three PULs encoding enzymes from these families (Supplementary Fig. 1b), revealed only one locus (contains three susCs) that was significantly upregulated by AGPs (Supplementary Fig. 1c). Of the GH43_24 and GH16 enzymes encoded by these PULs, only Baccell00844 (GH16) degraded β1,3-galactan and is thus an endo-β1,3-galactanase (Fig. 5d). Baccell00844 contains a type II signal peptide, consistent with a surface location. Whole cell assays of B. cellulosilyticus under aerobic conditions, which report only activity of surface proteins11, showed that β1,3-galactan was degraded into numerous oligosaccharides (Fig 5e). This indicates that B. cellulosilyticus displays surface endo-β1,3-galactanase activity, which is likely mediated by Baccell00844. Support for the role played by Baccell00844 is provided by growth of all the Bacteroidetes species on GA-AGP pre-treated with Baccell00844 (Supplementary Table 4). An orthologue to Baccell00844 in B. caccae (BACCAC_03237) may explain its growth on GA-AGP and WH-AGP. Insertion of baccell00844 into B. thetaiotaomicron PULAGPL (B. thetaiotaomicron::baccell00844) enabled the bacterium to grow on intact GA-AGP and WH-AGP (Fig. 5bc). B. thetaiotaomicron::baccell00844, but not wild type B. thetaiotaomicron, degraded β1,3-galactan in aerobic whole cell assays (Fig. 5e) demonstrating acquisition of surface endo-β1,3-galactanase activity. Proteomic analysis of intact cells of B. thetaiotaomicron::baccell00844 revealed tryptic peptides from 46 proteins (Fig 5f) that were detected only on the bacterial surface. These proteins included Baccell00844 (five tryptic peptides identified by MS/MS, Supplementary Fig. 11). Among the 45 B. thetaiotaomicron proteins were a number that have been shown, experimentally, to be surface exposed (SusD/C-like proteins, surface CAZymes and SGBPs; Supplementary Table 5), and all the polypeptides contain canonical type II signal peptides consistent with outer membrane attachment. The presence of Baccell00844 among these 46 proteins supports its proposed surface location in B. thetaiotaomicron::baccell00844. Collectively, the proteomics data and surface endo-β1,3-galactanase activity of B. thetaiotaomicron::baccell00844 demonstrates that growth of the engineered bacterium on intact GA-AGP and WH-AGP is conferred through the surface endo-β1,3-galactanase activity encoded by baccell00844.
Data presented above suggest B. thetaiotaomicron::baccell_00844, in addition to B. cellulosilyticus, B. caccae and D. gadei are keystone organisms for AGP utilisation by Bacteroidetes. To test this hypothesis two of the organisms that cannot grow on untreated GA-AGP, wild type B. thetaiotaomicron and B. ovatus, were co-cultured with B. thetaiotaomicron::baccell_00844, B. cellulosilyticus and B. caccae on the intact glycan, and the bacteria in the co-cultures were quantified by quantitative-PCR of genomic-specific sequences. CFUs of wild type B. thetaiotaomicron and B. ovatus increased (Fig. 6) and thus these organisms grew on GA-AGP in the presence, but not in the absence, of B. cellulosilyticus, B. caccae or B. thetaiotaomicron::baccell_00844. This indicates that B. cellulosilyticus, B. thetaiotaomicron::baccell_00844 or B. caccae provide GA-AGP-derived oligosaccharides as growth substrates for the recipient bacteria. These data establish B. cellulosilyticus, B. thetaiotaomicron::baccell_00844 and B. caccae, and by inference D. gadei, as keystone bacteria in the utilisation of complex AGPs, with B. thetaiotaomicron, B. ovatus, and likely other Bacteroidetes, comprising recipient organisms. B. thetaiotaomicron and B. ovatus demonstrate a preference for products released by B. cellulosilyticus and B. caccae, respectively, providing possible examples of discrete AGP cross-feeding niches provided by each keystone organism.
Figure 6. Growth profile of keystone and recipient Bacteroides species on complex AGPs.

Wild type B. thetaiotaomicron strain VPI-5482 (Bt), B. thetaiotaomicron strain VPI-5482 expressing Baccell00844 (Bt::Baccell00844), B. ovatus strain ATCC8483 (Bo), B. cellulosilyticus strain DSM14838 (Baccell) and B. caccae strain ATCC 43185 (Bcacc) were cultured on nutrient rich (TYG) media overnight. The organisms were then inoculated at ~107 colony forming units (CFUs) per ml into minimal medium containing GA-AGP at 0.5% (w/v), either as a monoculture or in co-culture with one of the other strains. The cultures were incubated in anaerobic conditions and at regular intervals aliquots were removed and plated onto rich (BHI) agar plates to determine the CFUs. The ratio of the strains in the co-cultures were determined by quantitative-PCR with primers that amplify genomic sequences unique to each strain (see Methods for further details). (i) shows the ratio of the organisms in the co-cultures and (ii) the corresponding CFUs for these bacterial strains. Continuous lines correspond to organisms in co-culture and broken lines are monocultures of the bacterial strains. a, Bo and Bt; b, Bo and Baccell; c, Bo and Bcacc; d, Baccell and Bt; e, Bcacc and Bt; f, Bo and Bt::Baccell00844. Error bars represent the s.e.m of biological replicates (n=3).
To establish the extent to which B. thetaiotaomicron utilizes AGP side-chains, limit products generated from growth on BT0264-treated GA-AGP were characterized. The major product was a hexasaccharide derived from a heptasaccharide in which the terminal rhamnose had been removed by BT3686 (Supplementary Fig. 12 and 13). The inability to degrade this oligosaccharide reflects the absence of a α-galactosidase encoded by the AGP-PULs, preventing BT3679 from accessing the 3-linked Araf. The limit product generated by B. cellulosilyticus from GA-AGP was a tetrasaccharide, also derived from the heptasaccharide (Supplementary Fig. 12 and 13). This is consistent with the α-galactosidase gene baccell00859 in the B. cellulosilyticus AGP PUL, and removal of the Rha-GlcA cap by the LU pathway in which the unsaturated glucuronidase can target 4,5ΔGlcA-β1,6-Gal linkages in which the Gal is decorated at O3 and/or O4. Both organisms lacked an α-arabinofuranosidase that targeted O4 linkages.
Analysis of AGP-PULs in HGM Bacteroidetes species
Only B. finegoldii contained a locus equivalent to B. thetaiotaomicron PULAGPL, while PULAGPs was in most species of the Bacteroides genus, with various levels of rearrangements (Supplementary Fig. 14 and 15). No enzyme conservation pattern that correlated with growth on LA-AGP or GA-AGP was identified. For example, B. stercoris grows on LA-AGP but lacks the orthologous enzymes found in its closest relatives. The evolution of AGP-PULs was compared to the (16S-based) phylogenetic tree of the species (Supplementary Table 4). Closely-related species have similar PUL organization, but at the single gene level there are examples of a lack of orthologues. Thus Bacteroidetes AGP PULs are highly dynamic systems that can be rapidly lost, gained, or rearranged between closely related species (see B. massiliensis and B. plebeius in comparison with B. vulgatus and B. dorei; B. cellulosilyticus compared to B. thetaiotaomicron). In consequence 16S-derived taxonomy cannot be used to predict AGP degradation in Bacteoidetes.
Discussion
This study reveals the enzymes required to depolymerise the β1,3-galactan backbone of AGPs, resulting in release of the oligosaccharide side-chains. This diversity likely reflects the substituents at O2 or O4 of the backbone Gals that would limit the progressive action of the critical exo-galactanases. The data also show that the GH43 exo-β1,3-galactanases lack the catalytic base present in all other enzymes of this family. Deviation from conservation of catalytic residues in GH families is rare, although not without precedent27.
Analysis of the enzymes that deconstruct side-chains of two AGPs provides insights into the biological relevance of the AGP PULs in B. thetaiotaomicron. The inability of ΔPULAGPL to grow on LA-AGP reflects the absence of BT0290, the β1,6-galactosidase that hydrolyses the β1,6-galactan side-chains which, in this glycan, are not extensively decorated. BT0290 is less important in degrading complex AGPs, such as GA-AGP, as the decoration of β1,6-galactan side-chains with other sugars represent significant nutrients. The inability of ΔPULAGPS to grow on GA-AGP (endo-β1,3-galactanase pre-treated) reflects extensive capping of the side chains with Rhap. Loss of the rhamnosidase gene bt3686 in PULAGPS greatly restricts further degradation of the side-chains. To summarise, PULAGPS encodes an enzyme consortium that degrades the major side chains in complex AGPs such as GA-AGP, while PULAGPL targets the β-1,6,linked Gal side chains that are important nutrients in simpler glycans such as LA-AGP.
AGPs are diverse and numerous enzymes are required to mediate their deconstruction. Combined with recent reports18,19, four CAZyme families that contribute to AGP degradation were discovered, however, further enzymes likely await discovery. Indeed, in PULAGPL there are 14 genes encoding secreted hypothetical proteins that may contribute to degradation of complex AGPs not investigated here. Unusually, two different pathways remove the disaccharide that caps the side-chains in GA-AGP. Although the more flexible LU pathway should enable more comprehensive degradation, several HGM Bacteroides species utilise the RG pathway that limits downstream processing of the oligosaccharides. The contrasting oligosaccharide utilisation profiles observed between B. thetaiotaomicron and B. cellulosilyticus (Supplementary Fig. 11), and predicted by differences in the AGP PULs in other Bacteroides spp. (Supplementary Fig. 12 and 13), may enable co-existence of species within a common niche targeting different components of the same glycan.
The majority of Bacteroidetes species studied here were unable to utilise GA-AGP, although they grow on the glycan after backbone cleavage. Utilisation of complex AGP by the HGM Bacteroidetes relies on the extracellular endo-activity of a few keystone species. This study in conjunction with recent reports28,29 shows that glycan cross-feeding between HGM Bacteroides species contributes to the ecology of carbohydrate utilisation in this ecosystem. Nevertheless Bacteroides glycan degrading systems generally contain surface endo-acting enzymes that generate fragments which are imported into the periplasm10,11, obviating the requirement for cross-feeding to utilise the polysaccharide.
In conclusion, dissecting mechanisms by which AGPs are degraded by HGM Bacteroidetes species reveals enzyme families of potential biotechnological relevance, and shows how synthetic biology can be used to engineer organisms to degrade AGPs that are abundant in the human diet.
Methods
Cloning, expression and purification of recombinant proteins
DNAs encoding enzymes lacking their signal peptides were amplified by PCR using appropriate primers. The amplified DNAs were cloned into pET28a with an N-terminal His6 tag using NheI and XhoI restriction sites (Table 3SM). The genes were then expressed in E. coli BL21, or Tuner cells, transformed with the appropriate recombinant plasmids. The transformed E. coli strains were cultured in Luria broth (LB) supplemented with 10 μg/ml of kanamycin. Cultured cells were grown at 37 °C to mid-log phase and induced with 1 mM isopropyl β-D-1-thiogalactopyranoside at 16 °C overnight. Cells were pelleted by centrifugation at 5,000 rpm for 10 min and resuspended in 20 mm Tris-HCl buffer, pH 8.0, containing 300 mm NaCl. For selenomethionine-derivatized protein the above procedure was used but adjusted as follows: E. coli B834 cells were transformed with the appropriate recombinant plasmid. Overnight 5-ml cultures, in LB, were then used to inoculate 100 ml of LB culture in a 250-ml flask, which was then grown to an O.D. of 0.4. A methionine-deficient media was prepared using the Molecular Dimensions SelenoMet™ Medium Base (MD12-501) and SelenoMet™ Nutrient mixtures (MD12-502) and was used to wash the cultured B834 cells. The cells were then inoculated into 1 liter of methionine-deficient media to which selenomethione was added to a final concentration of 5 mg/ml. Cells were collected and disrupted by sonication, and the cell-free extract was recovered by centrifugation at 15,000 rpm for 30 min. Recombinant proteins were purified from the cell-free extract using immobilized metal affinity chromatography using Talon™, a cobalt-based matrix. Proteins were eluted from the column in Buffer A containing 100 mm imidazole. For crystallographic studies, BT0265, BT0290, BT3674, BT3679, and BT3683 were further purified by size exclusion chromatography using a Superdex S200 16/600 column equilibrated with Buffer A on a fast protein liquid chromatography system (ÄKTA FPLC; GE Healthcare). All proteins were purified to electrophoretic homogeneity as judged by SDS-PAGE.
Mutagenesis
Site-directed mutagenesis was conducted using the PCR-based QuickChange site-directed mutagenesis kit (Strategene) according to the manufacturer’s instructions, using the appropriate plasmid encoding BT0290, BT3674, BT3683 and BT3685 as the template and appropriate primer pairs.
Large scale purification of oligosaccharides
GA-AGP derived oligosaccharides were generated by incubating 20 g of the glycan with 1 µM of the β1,3-galactosidase BT0265 in 20 mM sodium phosphate buffer pH 7.0 implemented with 150 mM NaCl at 37 °C for 16 h. The oligosaccharide mixture was freeze dried and resuspended in water before being applied to a P2-BioGel (BioRad) column with a 0.22 ml/min flow rate. Fractions were evaluated for oligosaccharide content and purity by TLC. Pure fractions of defined oligosaccharides were pooled and concentrated. Oligosaccharide size was confirmed by Mass Spectrometry and HPAEC.
Chemical synthesis
The synthesis of 2,4-dinitrophenyl-β-d-galactopyranoside was as described previously31
(5R,6S,7S,8R)-5-[(Benzyloxy)methyl]-6,7,8-tri(benzyloxy)-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine
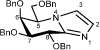 5-Amino-2,3,4,6-tetra-O-benzyl-5-deoxy-1-thio-D-galactono-1,5-lactam32 (61.5 mg, 0.111 mmol) was dissolved in aminoacetaldehyde dimethyl acetal (0.18 mL, 1.652 mmol) and stirred under N2 for 24 h. The mixture was diluted with EtOAc (20 mL) and washed with H2O (2 × 20 mL) and brine (1 × 20 mL). The organic extracts were dried (MgSO4) and then concentrated under reduced pressure. The crude residue was dissolved in toluene (3.2 mL) and H2O (0.3 mL). p-Toluenesulfonic acid monohydrate (54.9 mg, 0.289 mmol) was added to the solution and the reaction mixture was stirred at 65 °C for 18 h. The mixture was diluted with EtOAc (20 mL) and washed with NaHCO3 (2 × 20 mL) and brine (1 × 20 mL). The organic extracts were dried (MgSO4), concentrated and the resulting residue was subjected to flash chromatography (EtOAc/pet. spirits 8:2) to afford the protected galactonoimidazole (49.1 mg, 79% over two steps) as a colourless oil; [α]D26 +73 (c 1.36, CHCl3); 1H NMR (500 MHz, CDCl3): δ 3.74 (1 H, dd, J5,6 = 10.2, J6,7 = 8.3 Hz, H6), 4.02 (2 H, m, H8, H7), 4.34 (1 H, dd, J5,5’ = 1.9, J5’,5’ = 5.8 Hz, CH2(C5)), 4.44 (3 H, m, CH2(C5), H5, CH2Ph), 4.55 (2 H, m, 2 × CH2Ph), 4.62 (2 H, m, 2 × CH2Ph), 4.71 (2 H, m, 2 × CH2Ph), 4.90 (1 H, d, J = 11.9 Hz, CH2Ph), 7.03 (1 H, d, J2,3 = 1.3 Hz, H3), 7.14 (1H, d, J2,3 = 1.3 Hz, H2), 7.18-7.32 (20 H, m, 4 × Ph); 13C NMR (125 MHz, CDCl3): δ 57.5 (1 C, C5), 71.5 (1 C, CH2Ph), 71.7 (1 C, C7), 72.0 (1 C, C6), 71.4 (1 C, CH2Ph), 72.9 (1 C, CH2Ph), 73.5 (1 C, CH2Ph), 73.7 (1 C, C5’), 77.6 (1 C, C8), 119.5 (1 C, C2), 129.2 (1 C, C3), 127.7-138.4 (20 C, 4 × Ph), 142.1 (C8’) ppm; HRMS (ESI)+
m/z 561.2751 [C36H36N2O4 (M+H)+ requires 561.2748].
5-Amino-2,3,4,6-tetra-O-benzyl-5-deoxy-1-thio-D-galactono-1,5-lactam32 (61.5 mg, 0.111 mmol) was dissolved in aminoacetaldehyde dimethyl acetal (0.18 mL, 1.652 mmol) and stirred under N2 for 24 h. The mixture was diluted with EtOAc (20 mL) and washed with H2O (2 × 20 mL) and brine (1 × 20 mL). The organic extracts were dried (MgSO4) and then concentrated under reduced pressure. The crude residue was dissolved in toluene (3.2 mL) and H2O (0.3 mL). p-Toluenesulfonic acid monohydrate (54.9 mg, 0.289 mmol) was added to the solution and the reaction mixture was stirred at 65 °C for 18 h. The mixture was diluted with EtOAc (20 mL) and washed with NaHCO3 (2 × 20 mL) and brine (1 × 20 mL). The organic extracts were dried (MgSO4), concentrated and the resulting residue was subjected to flash chromatography (EtOAc/pet. spirits 8:2) to afford the protected galactonoimidazole (49.1 mg, 79% over two steps) as a colourless oil; [α]D26 +73 (c 1.36, CHCl3); 1H NMR (500 MHz, CDCl3): δ 3.74 (1 H, dd, J5,6 = 10.2, J6,7 = 8.3 Hz, H6), 4.02 (2 H, m, H8, H7), 4.34 (1 H, dd, J5,5’ = 1.9, J5’,5’ = 5.8 Hz, CH2(C5)), 4.44 (3 H, m, CH2(C5), H5, CH2Ph), 4.55 (2 H, m, 2 × CH2Ph), 4.62 (2 H, m, 2 × CH2Ph), 4.71 (2 H, m, 2 × CH2Ph), 4.90 (1 H, d, J = 11.9 Hz, CH2Ph), 7.03 (1 H, d, J2,3 = 1.3 Hz, H3), 7.14 (1H, d, J2,3 = 1.3 Hz, H2), 7.18-7.32 (20 H, m, 4 × Ph); 13C NMR (125 MHz, CDCl3): δ 57.5 (1 C, C5), 71.5 (1 C, CH2Ph), 71.7 (1 C, C7), 72.0 (1 C, C6), 71.4 (1 C, CH2Ph), 72.9 (1 C, CH2Ph), 73.5 (1 C, CH2Ph), 73.7 (1 C, C5’), 77.6 (1 C, C8), 119.5 (1 C, C2), 129.2 (1 C, C3), 127.7-138.4 (20 C, 4 × Ph), 142.1 (C8’) ppm; HRMS (ESI)+
m/z 561.2751 [C36H36N2O4 (M+H)+ requires 561.2748].
(5R,6S,7S,8R)-5-[(Hydroxymethyl]-6,7,8-triol-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine (Galacto-imidazole; Gal-Im)
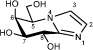 Pd(OH)2/C (20%, 46.2 mg) was added to a solution of EtOAc/MeOH/H2O (5:17:3, 1.0 mL), AcOH (0.44 mL) and the protected imidazole (24.6 mg, 0.044 mmol). The reaction vessel was filled with H2 (34 bar) and agitated for 41 h. The suspension was filtered through a Celite pad and subjected to flash chromatography (EtOAc/MeOH/H2O 8:2:1) to afford the target (8.5 mg, 96%) as an amorphous solid; m.p. 82 °C; [α]D23 +22 (c 0.435, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.88 (1 H, dd, J6,7 = 2.2, J7,8 = 7.7 Hz, H7), 4.05 (2 H, apt. d, CH2(C5)), 4.28 (1 H, m, H5), 4.38 (1 H, dd, J5,6 = 3.4, J6,7 = 2.2 Hz, H6), 4.82 (1 H, d, J2,3 = 7.7 Hz, H8), 7.19 (1 H, d, J = 1.1 Hz, H3), 7.51 (1H, d, J = 1.2 Hz, H2); 13C NMR (125 MHz, CD3OD): δ 61.6 (1 C, C5), 63.1 (1 C, C5’), 67.7 (1 C, C8), 70.5 (1 C, C6), 75.0 (1 C, C7), 119.9 (1 C, C2), 126.4 (1 C, C3), 147.6 (C8’) ppm.
Pd(OH)2/C (20%, 46.2 mg) was added to a solution of EtOAc/MeOH/H2O (5:17:3, 1.0 mL), AcOH (0.44 mL) and the protected imidazole (24.6 mg, 0.044 mmol). The reaction vessel was filled with H2 (34 bar) and agitated for 41 h. The suspension was filtered through a Celite pad and subjected to flash chromatography (EtOAc/MeOH/H2O 8:2:1) to afford the target (8.5 mg, 96%) as an amorphous solid; m.p. 82 °C; [α]D23 +22 (c 0.435, MeOH); 1H NMR (500 MHz, CD3OD): δ 3.88 (1 H, dd, J6,7 = 2.2, J7,8 = 7.7 Hz, H7), 4.05 (2 H, apt. d, CH2(C5)), 4.28 (1 H, m, H5), 4.38 (1 H, dd, J5,6 = 3.4, J6,7 = 2.2 Hz, H6), 4.82 (1 H, d, J2,3 = 7.7 Hz, H8), 7.19 (1 H, d, J = 1.1 Hz, H3), 7.51 (1H, d, J = 1.2 Hz, H2); 13C NMR (125 MHz, CD3OD): δ 61.6 (1 C, C5), 63.1 (1 C, C5’), 67.7 (1 C, C8), 70.5 (1 C, C6), 75.0 (1 C, C7), 119.9 (1 C, C2), 126.4 (1 C, C3), 147.6 (C8’) ppm.
CAZyme Assays
Spectrophotometric quantitative assays for β-d-galactosidase BT0264, BT0290, BT3683 and BT3685; β-l-arabinofuranosidase BT3674; α-l-arabinofuranosidases BT3675 and BT3679 and the β-d-glucuronidase BT3677 were monitored by the formation of NADH, at A340 nm using an extinction coefficient of 6,230 M−1 cm−1, with an appropriately linked enzyme assay system. The assays were adapted from two Megazyme International assay kits; the L-arabinose/D-galactose assay kit (K-ARGA) and the α-glucuronidase assay kit (K-AGLUA). Activity on 4-nitrophenyl-glycosides was monitored at A400nm. The mode of action of enzymes were determined using high performance anion exchange chromatography (HPAEC) or TLC, as appropriate. In brief, aliquots of the enzyme reactions were removed at regular intervals and, after boiling for 10 min to inactivate the enzyme and centrifugation at 13,000g, the amount of substrate remaining or product produced was quantified by HPAEC using standard methodology. The reaction substrates and products were bound to a Dionex CarboPac PA100 (galactooligosaccharides/arabinooligosaccharides), PA1 (monosaccharides) or PA20 (polygalacturonic acid oligosaccharides) column and glycans eluted with an initial isocratic flow of 100 mM NaOH then a 0–200 mM sodium acetate gradient in 100 mM NaOH at a flow rate of 1.0 ml min−1, using pulsed amperometric detection. Linked assays were checked to make sure that the relevant enzyme being analysed was rate limiting by increasing its concentration and ensuring a corresponding increase in rate was observed. A single substrate concentration was used to calculate catalytic efficiency (kcat/KM), and was checked to be markedly less than KM by halving and doubling the substrate concentration and observing an appropriate increase or decrease in rate. The equation V = (kcat/KM)[S][E] where V is the initial rate, [S] and [E] are substrate and enzyme concentration, respectively. All reactions were carried out in 20 mM sodium phosphate buffer, pH 7.0, with 150 mM NaCl (defined as standard conditions) and performed in at least technical triplicates.
Electrospray ionisation mass spectrometry (ESI-MS)
The molecular mass of purified oligosaccharides (in 10 mM ammonium acetate, pH 7.0) were analysed via negative ion mode infusion/offline ESI-MS following dilution (typically 1:1 (v/v)) with 5% trimethylamine in acetonitrile. Electrospray MS data was acquired using an LTQ-FT mass spectrometer (Thermo) with a FT-MS resolution setting of 100,000 at m/z = 400 and an injection target value of 1,000,000. Infusion spray analyses were performed on 5–10 μl of samples using medium ‘nanoES’ spray capillaries (Thermo) for offline nanospray mass spectrometry in negative ion mode at 1 kV.
Liquid chromatography-mass spectrometry
The sample containing the oligosaccharides generated by treatment of LA-AGP with BT0265 was diluted 1:10 (v/v) with Buffer B (85% acetonitrile/15% 50 mM ammonium formate in water, pH 4.7) and 0.5 µL was analysed by LC-MS analysis via elution from a ZIC-HILIC (SeQuant®, 3.5 µm, 200Å, 150 X 0.3 mm, Merck, UK) capillary column. The column was connected to a NanoAcquity HPLC system (Waters, UK) and heated to 35°C with an elution gradient as follows; 100% Buffer B for 5 min, followed by a gradient to 25% Buffer B/75% Buffer A (50 mM ammonium formate in water, pH 4.7) over 40 min. The flow rate was 5 µL/min and 10 column volumes of Buffer B equilibration was performed between injections. MS data was collected using a Bruker Impact II QTof mass spectrometer operated in positive ion mode, 50 – 2000 m/z, with capillary voltage and temperature settings of 2800 V and 200 °C respectively, together with a drying gas flow and nebulizer pressure of 6 L/min and 0.4 Bar. The MS data was analysed using Compass DataAnalysis software (Bruker).
1H-NMR determination of catalytic mechanism
The enzymes BT3685 and BT3679 at ~20 μM were assayed using 2,4-dinitrophenyl-β-D-galactopyranoside (5 mM) and 4-nitrophenyl α-L-arabinofuranoside, respectively. The enzymes were solvent-exchanged three times by ultrafiltration in 20 mM Tris-HCl, 500 mM NaCl, pD 7.5 using D2O as the solvent. Substrates were repeatedly freeze dried using the same buffer and resuspended in D2O. Prior to addition of enzyme an initial 1H-NMR spectrum was obtained. Enzyme was added and spectra were recorded at appropriate time intervals. The emergence of individual monosaccharide product α- and β-anomers in the case of BT3685 was monitored to deduce catalytic mechanism. The reaction catalyzed by BT3679 was carried out in the presence of 2.5 M methanol. The products were freeze-dried and resuspended in D2O. Spectra recorded were analysed for the chemical shift of the anomeric 1H of the methyl L-arabinofuranoside product to determine mechanism.
2D NMR and mass spectrometry of GA-AGP oligosaccharides
1H-NMR
NMR spectra were recorded at 298 K in D2O with a Bruker AVANCE III spectrometer operating at 600 MHz equipped with a TCI CryoProbe. NMR chemical-shift assignments were obtained using 2D 1H-1H TOCSY, ROESY and DQFCOSY alongside 2D 13C HSQC, H2BC, HMBC, HSQC-TOCSY and HSQC-ROESY experiments using established methods33. The mixing times were 70 ms and 200 ms for the TOCSY and ROESY experiments, respectively (data for the tetra- and heptasaccharides are shown in Supplementary Fig. 4). Chemical shifts were measured relative to internal acetone (δH =2.225, δC=31.07 ppm). Data were processed using the Azara suite of programs (v. 2.8, copyright 1993-2017, Wayne Boucher and Department of Biochemistry, University of Cambridge, unpublished) and chemical-shift assignment was performed using Analysis v2.434. The non-reducing-end Rha residue was readily identified from the presence of a methyl group at the 6-position. All the linkages were clear from downfield 13C shifts of the linked atoms, inter-glycosidic crosspeaks in the HMBC spectrum and intense NOE crosspeaks in the ROESY spectrum. The anomeric configurations of the pyranoses were confirmed by measurement of the 1JC-1,H-1 coupling constant (c. 170 and 160 Hz for α- and β-configurations, respectively35) in an F1-coupled 13C HSQC. The assignments were complete and are shown in Supplementary Table 7.
Mass spectroscopy
To confirm the AGP oligosaccharide chain structure suggested by NMR, the sample was per-methylated and analysed by MALDI ToF-MS and MS/MS. A single high intensity peak, with m/z 1393.5 was identified which is consistent with the composition Ara2RhaGal3GlcA. The tandem mass spectrometry (MS/MS) spectrum of this per-methylated oligosaccharide is shown in Supplementary Fig. 5. The presence of Y1 (m/z 259.0) and 1,5X1 (m/z 287.0) indicates the reducing end is Gal. The 0,4A4 (m/z 1217.5) cross-ring fragment indicates the presence of 1,6-linkage onto the reducing end Gal. Y3 (m/z 1205.5) and 1,5X3 (m/z 1233.4) indicate terminal Rha, Y3α (m/z 1175.4) and 1,5X3a (m/z 1203.4) indicate terminal Gal, and Y2β (m/z 1219.5) and 1,4X2 β (m/z 1247.5) terminal Ara residues. Y2 (m/z 987.3) indicates a terminal disaccharide Rha-GlcA. The 1,4-linkage between the terminal Rha and GlcA was confirmed by the cross ring fragments (3,5A2 ion, m/z 313.0; 0,2X2 ion, m/z 1043.3) and elimination ions (G3 ion, m/z 1157.4; E2 ion, m/z 399.0). The non-reducing end 0,4A3 cross-ring fragment (m/z 489.0) and H2 elimination ion (m/z 765.1) suggest the presence of 1,6-linkage between the GlcA and Gal. The 728 Da mass difference between the Y2 and Y1 ions suggests that there are two Gal and two Ara residues between the GlcA and the reducing end Gal. The G2 (m/z 807.1) indicates there is a single backbone residue of Gal. The presence of Y2α ion (m/z 987.3), but absence of an ion corresponding to loss of a dipentose side chain, indicates that the one of the side chains is a disaccharide of Gal linked to Ara. As described above, there is terminal Gal, so this structure is Gal-Ara. Substitution of O3 and O4 but not O2 of the Gal is suggested by the presence of G2 (m/z 807.1) and 0,2X1, (m/z 315.0), ions. The H2 elimination ion, which reflects loss of Rha-GlcA and Ara, suggests an Ara is linked to O4 of the Gal, which is supported by the presence of the 3,5A3 (m/z 677.1).The elimination ions (G2, m/z 807.1; D3, m/z 779.1) suggest that the Gal-Ara disaccharide is linked to the O3 of the Gal on the backbone. The cross-ring fragment 0,2X2α (m/z 1071.3) and elimination ion G3α (m/z 1113.3) suggests that the terminal Gal is not 1,2-linked to the Ara, but we were unable to locate further from the MS/MS the Gal linkage, but the results are consistent with 1,3 linkage to the Ara. The presence of this G3α ion also indicates the furanose form of the Ara.
Growth of Bacteroides and generation of mutants
Bacteroides mutants were generated by deletion of the target gene by counter selectable allelic exchange using the pExchange-tdk plasmid. The full method is described in Ref36. Mutants generated in this study are distinguished by the locus tag of the gene deleted/inactivated (Δbtxxx).
Bacteroides spp. were routinely cultured under anaerobic conditions at 37 °C using an anaerobic cabinet (Whitley A35 Workstation; Don Whitley) in culture volumes of 0.2, 2 or 5 ml) of TYG (tryptone-yeast extract-glucose medium) or minimal medium (MM)31 containing 0.5-1% of an appropriate carbon source and 1.2 mg ml−1 porcine haematin (Sigma-Aldrich) as previously described10. The growth of the cultures was monitored by OD600 nm using a Biochrom WPA cell density meter for the 5 ml cultures or a Gen5 v2.0 Microplate Reader (Biotek) for the 0.2 and 2 ml cultures.
Protein cellular localization of BT0264 using antibodies
Cellular localization of proteins was carried out as described previously37. In brief, B. thetaiotaomicron was grown overnight (OD600 nm value of 2.0) in 5 ml MM containing LA-AGP. The next day, cells were collected by centrifugation at 5,000g for 10 min and resuspended in 2 ml PBS. Proteinase K (0.5 mg ml−1 final concentration) was added to 1 ml of the suspension and the other half left untreated (control). Both samples were incubated at 37 °C for 16 h followed by centrifugation (5,000g for 10 min) to collect cells. To eliminate residual proteinase K activity, cell pellets were resuspended in 1 ml of 1.5 M trichloroacetic acid and incubated on ice for 30 min. Precipitated mixtures were then centrifuged (5,000g, 10 min) and washed twice in 1 ml ice-cold acetone (99.8%). The resulting pellets were allowed to dry in a 40 °C heat block for 5 min and dissolved in 250 μl Laemmli buffer. Samples were heated for 5 min at 98 °C and mixed by pipetting several times before resolving by SDS–PAGE using 12% gels. Electrophoresed proteins were transferred to nitrocellulose membranes by western blotting followed by immunochemical detection using primary rabbit polyclonal antibodies (Eurogentec) generated against BT0264 and secondary goat anti-rabbit antibodies (Santa Cruz Biotechnology).
Proteomics
Cell surface shaving
Bacteriodes cell surface digestion was performed as previously described48, with minor modifications. Briefly, Bacteriodes cells were harvested by centrifugation (3500 g, 15 min, 4 °C) and washed three times with PBS pH 7.4. Cell pellets were subsequently resuspended in surface shaving buffer (PBS pH 7.4 containing 0.25 M Sucrose). Surface shaving was performed using 2 µg trypsin at 37 °C for 30 min with shaking at 300 rpm. Cells in surface shaving buffer without trypsin served as controls. After surface shaving, the cells were pelleted by centrifugation (10000 g, 10 min, room temperature), and the supernatants were filter-sterilized using 0.22 µm spin filters (Corning Incorporated). Sterilized supernatants were subsequently incubated for an additional 16 hours at 37 °C for complete digestion. Trypsin digestion was stopped with the addition of trifluoroacetic acid (TFA) at a final concentration of 1%, and peptides were desalted using Macro C18 Spin Columns (Harvard Apparatus).
Whole-cell lysate preparation
Bacteriodes cells were harvested and washed as described above. Cell pellets were subsequently resuspended in 8 M urea buffer in 50 mM triethylammonium bicarbonate (TEAB), containing 5mM tris(2-carboxyethyl)phosphine. Cells were lysed via sonication using an ultrasonic homogenizer (Hielscher). Proteins were subsequently alkylated for 30 min at room temperature using 10 mM iodoacetamide in the dark. Protein concentration was determined using a Bradford protein assay (Thermo Fisher Scientific). Protein samples, containing 50 µg total protein, was diluted 5 fold with 50 mM TEAB and protein digestion was performed at 37 °C for 18 h with shaking at 300 rpm. A protein to trypsin ratio of 50:1 was used. Trypsin digestion was stopped and peptides were desalted as described above.
Mass spectrometry
Peptides were dissolved in 2% acetonitrile containing 0.1% TFA, and each sample was independently analysed on an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific), connected to a UltiMate 3000 RSLCnano System (Thermo Fisher Scientific). Peptides were injected on an Acclaim PepMap 100 C18 LC trap column (100 μm ID × 20 mm, 3μm, 100Å) followed by separation on an EASY-Spray nanoLC C18 column (75 ID μm × 500 mm, 2μm, 100Å) at a flow rate of 300 nL/min. Solvent A was water containing 0.1% formic acid, and solvent B was 80% acetonitrile containing 0.1% formic acid. The gradient used for analysis of surface-shaved samples was as follows: solvent B was maintained at 3% for 6 min, followed by an increase from 3 to 35% B in 43 min, 35-90% B in 0.5 min, maintained at 90% B for 5.4 min, followed by a decrease to 3% in 0.1 min and equilibration at 3% for 10 min. The gradient used for analysis of proteome samples was as follows: solvent B was maintained at 3% for 6 min, followed by an increase from 3 to 35% B in 218 min, 35-90% B in 0.5 min, maintained at 90% B for 5 min, followed by a decrease to 3% in 0.5 min and equilibration at 3% for 10 min. The Oritrap Fusion Lumos was operated in positive ion data-dependent mode using a modified version of the recently described CHarge Ordered Parallel Ion aNalysis (CHOPIN) method for synchronised use of both the ion trap and the Orbitrap mass analysers49. The CHOPIN method is derived from the “Universal Method” developed by Thermo Fisher, to extend the capabilities of mass analyser parallelization. The precursor ion scan (full scan) was performed in the Orbitrap in the range of 400-1600 m/z with a resolution of 120 000 at 200 m/z, an automatic gain control (AGC) target of 4 x 105 and an ion injection time of 50 ms. MS/MS spectra of doubly charged precursor ions were acquired in the linear ion trap (IT) using rapid scan mode after collision-induced dissociation (CID) fragmentation. A CID collision energy of 32% was used, the AGC target was set to 2 x 103 and a 300 ms injection time was allowed. Precursor ions with charge state 3-7 and with an intensity <5 x 105 were also scheduled for analysis by CID/IT, as described above. Precursor ions with charge state 3-7 and with an intensity > 5 x105 were, however, acquired in the Oritrap (FT) with a resolution of 30 000 at 200 m/z after high-energy collisional dissociation (HCD). An HCD collision energy of 30% was used, the AGC target was set to 1 x 104 and a 40 ms injection time was allowed. The number of MS/MS events between full scans was determined on-the-fly to maintain a 3 s fixed duty cycle. Dynamic exclusion of ions within a ± 10 p.p.m. m/z window was implemented using a 35 s exclusion duration. An electrospray voltage of 2.0 kV and capillary temperature of 275 °C, with no sheath and auxiliary gas flow, was used.
Mass spectrometry data analysis
All tandem mass spectra were analysed using MaxQuant 1.5.1.750, and searched against a combined database of Bacteroides thetaiotaomicron VPI-5482 (containing 4782 entries), B. cellulosilyticus MGS:158 (containing 4369 entries) and the B. cellulosilyticus BACCELL_00844 glycosyl hydrolase family 16 protein. Protein sequences were downloaded from Uniprot on May 10th 2018. Peak list generation was performed within MaxQuant and searches were performed using default parameters and the built-in Andromeda search engine51. The enzyme specificity was set to consider fully tryptic peptides, and two missed cleavages were allowed. Oxidation of methionine, N-terminal acetylation and deamidation of asparagine and glutamine was allowed as variable modifications. No fixed modifications were employed in searches for the surface-shaved samples, whereas carbamidomethylation of cysteine was allowed as fixed modification in proteome searches. A protein and peptide false discovery rate (FDR) of less than 1% was employed in MaxQuant. Proteins were considered confidently identified when they contained at least two unique tryptic peptides. Proteins that contained similar peptides and that could not be differentiated based on tandem mass spectrometry analysis alone were grouped to satisfy the principles of parsimony. Reverse hits and contaminants were removed before downstream analysis. Skyline 4.1.0.11796 was used for extraction of ion chromatograms52. Gene ontology (Ashburner et al. 2000) enrichment was performed using PANTHER53 and subcellular protein localization prediction was performed using LocateP v254. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with data set identifier PXD010274.
Cross-feeding and competition assays
Prior to co-culture each Bacteroides spp. was grown in TYG and washed in PBS before being used to inoculate MM containing 0.5% GA-AGP. Co-cultures were grown in triplicate. Samples of 0.5 ml were taken at regular intervals during growth, which were serially diluted and plated onto Brain-Heart Infusion (BHI, Sigma-Aldrich) with agar and porcine hematin for determination of total CFU/ml of the culture. Mono-cultures of each Bacteroides spp. were also plated for determination of CFU/ml at intervals during the growth. Genomic DNA was purified from the remainder of the co-culture sample (Bacterial genomic DNA purification kit, Sigma-Aldrich). Quantitative PCR (q-PCR) was performed in triplicate on each sample using a ROCHE Lightcycler 96 to determine the ratio of each Bacteroides spp. and mutants in the sample using primers specific for unique regions in each Bacteroides sp. genome. Primers for B. thetaiotaomicron (F:5’-AGGTGCAGGCAACCT-3’, R:5’-AATTCCCGTTCTCCATGTCC-3’); B. ovatus (F:5’-GGAATGAGCATAATCCATATATCAAGATGAAACG-3’, R:5’-TACCTGAAACAATCATCCTTTATTTCTGTAGC-3’); B. cellulsoylticus (F:5’-AGCAGGCGGAATTCGATAAG-3’ R:5’-GTGTACAGTGCCAGGCATAA-3’) and B. caccae (F:5’-GATTATGTGGACAGGTGATCGTGTGATTTC-3’, R:5’-ATTCCACCAAATGTAGGCGGGACGTTTAAT-3’) were used to determine ratio of each species in co-culture and used to calculate the CFU/ml of each organisms in the culture.
Crystal structure determination
Crystallization
BT0290-E182A at 10 mg/ml, was crystallized from the commercial screen Morpheus (Molecular Dimensions, UK) condition D3 (20 mM 1,6-Hexanediol, 20 mM 1-Butanol, 20 mM 1,2-Propanediol (racemic), 20 mM 2-Propanol, 20 mM 1,4-Butanediol, 20 mM 1,3-Propanediol, 100 mM Imidazole-MES pH 6.5, 30% Glycerol and 30% polyethylene glycol 4000). Apo BT0265 was crystallised at 32 mg/ml in 20% PEG 3350 and 0.2 M Sodium/Potassium Tartrate. Crystals were cryoproteted with 20 % glycerol. Crystals of BT0265 Q249A were crystallised at 20 mg/ml, with a 200mg/ml oligosaccharide mixture, in 20 % PEG 3350 and 0.2 M sodium thiocyanate. Crystals were cryo protected with paratone oil.
BT3683 was crystallised at 12.6 mg/ml in 20 % PEG 3350, 0.2 M Ammonium formate and 300 mM L-rhamnose. Crystals formed under these condiotns were then back soaked, in mother liquor overnight to remove the rhamnose. These crystals were then transferred to a fresh drop and soaked with galactose, galactodeoxynorijmycin or galactoimidazole, as desired, at concerntaions in >30 mM. These crystals were left overnight and then cryo protected with paratone oil.
Data collection and processing
Diffraction data for BT0290 and BT3674 were collected at the Diamond Light Source, U.K., on beamline I02, whilst, all other data was collected on bealine IO4-1, at a temperature of 100 °K. Alldata were processed and integrated with XDS and scaled using Aimless38, 39. For all datasets, the space groups were determined using pointless and later confirmed during refinement40. The phase problem was solved by molecular replacement using Phaser41. PDB 3D3A was used as search model for BT0290; BT3674 was solved using 4QJY; BT0265 was solved using 3VSF and a truncated version of BT0265, lacking the C-terminal Ig domain was used to solve BT3683. Additional automated model building for BT0265 was carried out using buccaneer42. Solvent molecules were added using COOT43 and checked manually. All other computing used the CCP4 suite of programs44. Five percent of the observations were randomly selected for the Rfree set. The models were validated using Molprobity45. The data statistics and refinement details are reported in Supplementary Table 6.
Comparative genomics analysis
Using a similar strategy to the identification pectin PULs, AGP PULs were searched for in Bacteroidetes genomes. The identification of similar PULs was based on PUL alignments. Gene composition and order of Bacteroidetes PULs were computed using the PUL predictor described in PULDB46. Then, in a manner similar to amino acid sequence alignments, the predicted PULs were aligned to the appropriate pectin PULs according to their modularity as proposed in the RADS/RAMPAGE method47. Modules taken into account include CAZy families, sensor-regulators and suscd-like genes. Finally, PUL boundaries and limit cases were refined by BLASTP-based analysis. The glycoside hydrolase families discovered in this study are listed in the main text.
Supplementary Material
Acknowledgements
This work was supported in part by an Advanced Grant from the European Research Council (Grant No. 322820) awarded to H.J.G. and B.H. supporting D.N., A.C., J. M.-M., J.B., N.T., a Wellcome Trust Senior Investigator Award to HJG (grant No. WT097907MA) that supported E.C.L. The Biotechnology and Biological Research Council project “Ricefuel” (grant numbers BB/K020358/1) awarded to H.J.G. supported A.L. We thank Diamond Light Source for access to beamline I02, I04-1 and I24 (mx1960, mx7854 and mx9948) that contributed to the results presented here.
Footnotes
Data availability. The data that support the findings of this study are available from the corresponding author upon request. The authors declare that the data supporting the findings of this study are available within the paper and the Supplementary Information. The crystal structure datasets generated (coordinate files and structure factors) have been deposited in the Protein Data Bank (PDB) and are listed in Supplementary Table 6 together with the PDB accession codes.
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article
Author contributions
Enzyme characterisation and oligosaccharide purification were by A.C., D.N. and J.M.-M. Gene deletion strains were constructed by D.N. and A.L. Co-culturing experiments were carried out by J.B. and D.N. Western blots were by D.N. Phylogenetic reconstruction and metagenomic analysis were by N.T. and B.H. Bacterial growth and transcriptomic experiments: E.C.L. and D.N. X-ray protein crystallography was by A.C., A.B. J.M.-M. N.M.R. experiments were by A.C. and K.S. Mass spectrometry was by J.G., L.Y. and P.D. Chemical synthesis was by P.Z.F., S.S. and S.J.W. E.H., M.T. and E.C.L. performed the whole cell proteomics. Experiments were designed by H.J.G. A.C. J.M.-M. and D.N. The manuscript was written by H.J.G. with substantial contributions from N.T., B.H. and S.J.W. Figures were prepared by J.M.-M. and E.C.L.
References
- 1.Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol. 2013;11:497–504. doi: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- 3.Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol. 2012;10:323–335. doi: 10.1038/nrmicro2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter NT, Martens EC. The Critical Roles of Polysaccharides in Gut Microbial Ecology and Physiology. Annu Rev Microbiol. 2017;71:349–369. doi: 10.1146/annurev-micro-102215-095316. [DOI] [PubMed] [Google Scholar]
- 5.Gilbert HJ, Stalbrand H, Brumer H. How the walls come crumbling down: recent structural biochemistry of plant polysaccharide degradation. Curr Opin Plant Biol. 2008;11:338–348. doi: 10.1016/j.pbi.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies G, Henrissat B. Structures and mechanisms of glycosyl hydrolases. Structure. 1995;3:853–859. doi: 10.1016/S0969-2126(01)00220-9. [DOI] [PubMed] [Google Scholar]
- 8.Ndeh D, Gilbert HJ. Biochemistry of complex glycan depolymerisation by the human gut microbiota. FEMS Microbiol Rev. 2018;42:146–164. doi: 10.1093/femsre/fuy002. [DOI] [PubMed] [Google Scholar]
- 9.Martens EC, Koropatkin NM, Smith TJ, Gordon JI. Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J Biol Chem. 2009;284:24673–24677. doi: 10.1074/jbc.R109.022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsbrink J, et al. A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature. 2014;506:498–502. doi: 10.1038/nature12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luis AS, et al. Dietary pectic glycans are degraded by coordinated enzyme pathways in human colonic Bacteroides. Nat Microbiol. 2018;3:210–219. doi: 10.1038/s41564-017-0079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fincher GB, Stone BA, Clarke AE. Arabinogalactan-Proteins - Structure, Biosynthesis, and Function. Annu Rev Plant Phys. 1983;34:47–70. [Google Scholar]
- 13.Vidal S, Williams P, Doco T, Moutounet M, Pellerin P. The polysaccharides of red wine: total fractionation and characterization. Carbohydr Polym. 2003;54:439–447. [Google Scholar]
- 14.Capek P, Matulova M, Navarini L, Suggi-Liverani F. Structural features of an arabinogalactan-protein isolated from instant coffee powder of Coffea arabica beans. Carbohydr Polym. 2010;80:180–185. [Google Scholar]
- 15.Dauqan E, Abdullah A. Utilization of gum arabic for industries and human health. American Journal of Applied Sciences. 2013;10:1270–1279. [Google Scholar]
- 16.McNamara MK, Stone BA. Isolation, characterization and chemical synthesis of a galactosyl-hydroxyproline linkage compound from wheat endosperm arabinogalactan-peptide. Lebensm Wiss Technol. 1981;14:182–187. [Google Scholar]
- 17.Ichinose H, et al. Characterization of an exo-beta-1,3-galactanase from Clostridium thermocellum. Appl Environ Microbiol. 2006;72:3515–3523. doi: 10.1128/AEM.72.5.3515-3523.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munoz-Munoz J, et al. An evolutionarily distinct family of polysaccharide lyases removes rhamnose capping of complex arabinogalactan proteins. J Biol Chem. 2017;292:13271–13283. doi: 10.1074/jbc.M117.794578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munoz-Munoz J, Cartmell A, Terrapon N, Henrissat B, Gilbert HJ. Unusual active site location and catalytic apparatus in a glycoside hydrolase family. Proc Natl Acad Sci U S A. 2017;114:4936–4941. doi: 10.1073/pnas.1701130114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calame W, Weseler AR, Viebke C, Flynn C, Siemensma AD. Gum arabic establishes prebiotic functionality in healthy human volunteers in a dose-dependent manner. Br J Nutr. 2008;100:1269–1275. doi: 10.1017/S0007114508981447. [DOI] [PubMed] [Google Scholar]
- 21.Martens EC, et al. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 2011;9:e1001221. doi: 10.1371/journal.pbio.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mewis K, Lenfant N, Lombard V, Henrissat B. Dividing the Large Glycoside Hydrolase Family 43 into Subfamilies: a Motivation for Detailed Enzyme Characterization. Appl Environ Microbiol. 2016;82:1686–1692. doi: 10.1128/AEM.03453-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kotake T, et al. Endo-beta-1,3-galactanase from winter mushroom Flammulina velutipes. J Biol Chem. 2011;286:27848–27854. doi: 10.1074/jbc.M111.251736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cartmell A, et al. The structure and function of an arabinan-specific alpha-1,2-arabinofuranosidase identified from screening the activities of bacterial GH43 glycoside hydrolases. J Biol Chem. 2011;286:15483–15495. doi: 10.1074/jbc.M110.215962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitazawa K, et al. beta-galactosyl Yariv reagent binds to the beta-1,3-galactan of arabinogalactan proteins. Plant Physiol. 2013;161:1117–1126. doi: 10.1104/pp.112.211722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura A, et al. "Newton's cradle" proton relay with amide-imidic acid tautomerization in inverting cellulase visualized by neutron crystallography. Science Advances. 2015;1 doi: 10.1126/sciadv.1500263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gloster TM, Turkenburg JP, Potts JR, Henrissat B, Davies GJ. Divergence of catalytic mechanism within a glycosidase family provides insight into evolution of carbohydrate metabolism by human gut flora. Chem Biol. 2008;15:1058–1067. doi: 10.1016/j.chembiol.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rakoff-Nahoum S, Coyne MJ, Comstock LE. An ecological network of polysaccharide utilization among human intestinal symbionts. Current biology : CB. 2014;24:40–49. doi: 10.1016/j.cub.2013.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rakoff-Nahoum S, Foster KR, Comstock LE. The evolution of cooperation within the gut microbiota. Nature. 2016;533:255–259. doi: 10.1038/nature17626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cartmell A, et al. How members of the human gut microbiota overcome the sulfation problem posed by glycosaminoglycans. Proc Natl Acad Sci U S A. 2017;114:7037–7042. doi: 10.1073/pnas.1704367114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma SK, Corrales G, Penadés S. Single Step Stereoselective Synthesis of Unprotected 2,4-Dinitrophenyl Glycosides. Tetrahedron Lett. 1995;36:5627–5630. [Google Scholar]
- 32.Vonhoff S, Heightman TD, Vasella A. Inhibition of glycosidases by lactam oximes: Influence of the aglycon in disaccharide analogues. Helvetica Chimica Acta. 1998;81:1710–1725. [Google Scholar]
- 33.Cavanagh J, Fairbrother WJ, Palmer AG, Skelton NJ. Protein NMR Spectroscopy: Principles and Practice. Academic Press; San Diego, CA, USA: 1996. [Google Scholar]
- 34.Vranken WF, et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 35.Bock K, Pedersen C. Study of CH-13 coupling-constants in pentapyranoses and some of their derivatives. Acta Chemica Scandinavica Series B-Organic Chemistry and Biochemistry. 1975;B 29:258–264. [Google Scholar]
- 36.Koropatkin NM, Martens EC, Gordon JI, Smith TJ. Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure. 2008;16:1105–1115. doi: 10.1016/j.str.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cuskin F, et al. Human gut Bacteroidetes can utilize yeast mannan through a selfish mechanism. Nature. 2015;517:165–169. doi: 10.1038/nature13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta crystallographica. Section D, Biological crystallography. 2011;67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kabsch W. XDS. Acta Crystallographica Section D-Biological Crystallography. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans P. Scaling and assessment of data quality. Acta crystallographica. Section D, Biological crystallography. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 41.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta crystallographica. Section D, Biological crystallography. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 43.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica. Section D, Biological crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallographica Section D-Biological Crystallography. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica. Section D, Biological crystallography. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terrapon N, Lombard V, Gilbert HJ, Henrissat B. Automatic prediction of polysaccharide utilization loci in Bacteroidetes species. Bioinformatics. 2015;31:647–655. doi: 10.1093/bioinformatics/btu716. [DOI] [PubMed] [Google Scholar]
- 47.Terrapon N, Weiner J, Grath S, Moore AD, Bornberg-Bauer E. Rapid similarity search of proteins using alignments of domain arrangements. Bioinformatics. 2014;30:274–281. doi: 10.1093/bioinformatics/btt379. [DOI] [PubMed] [Google Scholar]
- 48.Rodriguez-Ortega MJ, et al. Characterization and identification of vaccine candidate proteins through analysis of the group A Streptococcus surface proteome. Nat Biotechnol. 2006;24:191–197. doi: 10.1038/nbt1179. [DOI] [PubMed] [Google Scholar]
- 49.Davis S, et al. Expanding Proteome Coverage with CHarge Ordered Parallel Ion aNalysis (CHOPIN) Combined with Broad Specificity Proteolysis. J Proteome Res. 2017;16:1288–1299. doi: 10.1021/acs.jproteome.6b00915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 51.Cox J, et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 52.MacLean B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mi H, et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45:D183–D189. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou M, Boekhorst J, Francke C, Siezen RJ. LocateP: genome-scale subcellular-location predictor for bacterial proteins. BMC Bioinformatics. 2008;9:173. doi: 10.1186/1471-2105-9-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.