

Abstract
Epigenetic control of regulatory networks is only partially understood. Expression of Insulin-like growth factor-II (IGF2) is controlled by genomic imprinting, mediated by silencing of the maternal allele. Loss of imprinting of IGF2 (LOI) is linked to intestinal and colorectal cancers, causally in murine models and epidemiologically in humans. However, the molecular underpinnings of the LOI phenotype are not clear. Surprisingly, in LOI cells, we find a reversal of the relative activities of two canonical signaling pathways triggered by IGF2, causing further rebalancing between pro- and anti-apoptotic signaling. A predictive mathematical model shows that this network rebalancing quantitatively accounts for the effect of receptor tyrosine kinase inhibition in both WT and LOI cells. This mechanism also quantitatively explains both the stable LOI phenotype and the therapeutic window for selective killing of LOI cells, and thus, prevention of epigenetically controlled cancers. These findings suggest a framework for understanding epigenetically modified cell signaling.
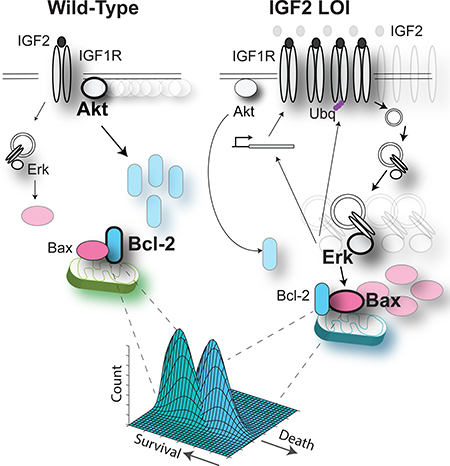
Graphical Abstact
Introduction
Epigenetic control of cell regulation has been increasingly seen as a critical contributor to diverse physiological and pathological cell and tissue behaviors (Feinberg, 2013, Timp and Feinberg, 2013, Timp et al., 2009). In particular, genomic imprinting regulates the expression of various genes in a parent-oforigin-specific manner, so that the loss of imprinting can alter the effective dosage of these genes, leading to profound changes in tissue structure and function (Timp et al., 2009). A striking example of how loss of imprinting can cause a critical alteration in tissue homeostasis is provided by regulation of the maternally imprinted Insulin like growth factor-2 (IGF2) (Holm et al., 2005, Kaneda and Feinberg, 2005, Cui et al., 2003, Kaneda et al., 2007, Steenman et al., 1994, Ferron et al., 2015, Venkatraman et al., 2013). Loss of imprinting of IGF2 leads to an approximately 2-fold increase in IGF2 expression and results in abnormally elongated intestinal crypts, and a 5-fold increased frequency of intestinal and colorectal neoplasias in humans (Sakatani et al., 2005). Epigenetic changes at this locus are also associated with a number of other syndromes (Weksberg et al., 2010, Anderson et al., 1999, Bartholdi et al., 2009), and in particular, with Wilms tumor, a common childhood kidney cancer (Ogawa et al., 1993, Steenman et al., 1994). Clinical relevance of loss of IGF2 imprinting (LOI) thus makes understanding of the associated deregulation of cell proliferation and survival a significant and urgent task (Vu et al., 2010). In spite of the increased recognition of epigenetic control, we still lack mechanistic understanding of the consequences of this regulation and LOI in particular for cell function.
Intriguingly, in an in vivo rodent intestinal tumor model, LOI mice display enhanced responsiveness to a drug targeting the kinase function of the cognate receptor of IGF2 – IGF1R (Kaneda et al., 2007). When treated with azoxymethane, the animals develop an increased number of aberrant crypt foci (ACF), the precursor lesion of intestinal carcinoma, when compared to the matched wild-type (WT) mice. Subsequent treatment with NVP-AEW541, a potent inhibitor of the IGF1R kinase activity, triggered a significant decrease in the number of ACF in LOI mice, while the WT mice showed no change in the number of ACF. Although this study revealed differential kinetics of Akt activation at different IGF2 doses in LOI cells, the relevance of these findings to the LOI phenotype and the associated potential therapeutic window, has remained largely unexplored.
Here, we find that LOI can induce a dramatic and surprising re-balancing of the regulatory network canonically triggered by IGF2. In particular, compared to WT cells, the same doses of IGF2 in LOI cells trigger a weaker activation of Akt1 (Akt) and stronger activation of Erk1/2 (Erk) signaling pathways. This result is particularly surprising, since both pathways are thought to be upregulated by IGF1R in both WT and LOI cells. We identified the mechanism of this counter-intuitive network re-balancing, ascribing it to a series of molecular feedback loops, and showing the re-wired signaling network activity then propagates downstream to the level of the anti- and pro-apoptotic determinants, and, in turn rebalancing their activity. Using a combination of computational modeling and experimental analysis culminating in the new single cell-level phenotypic ‘death plane’ approach, we show that the molecular re-wiring in signaling networks and apoptotic regulation can account for the therapeutic window of receptor kinase inhibition and that the molecular correlates of the LOI phenotype are self-sustained and driven by IGF2 overexpression. This analysis unraveled how a common loss of epigenetic control can translate into extensive changes in intracellular networks and enable development of a therapeutically relevant phenotypic alteration.
Results
Elevated expression of both the ligand, IGF2, and its primary cognate receptor, IGF1R, in LOI vs. WT cells (Kaneda et al., 2007) suggests that two canonical pathways activated by this receptor, targeting Akt and Erk respectively, would be coordinately upregulated in LOI vs. WT cells in response to elevated IGF2 doses. Surprisingly, when stimulated with 100ng/ml of IGF2, the activation of these two pathways in mouse embryonic fibroblast (MEF) cell lines were divergent: whereas the relative fold activation levels of Akt were lower, the fold activation of Erk was higher in LOI cells vs. WT cells. This result was seen for both the peak values of the temporal activity profiles and for the signaling activities integrated over 3 hr. time period (Fig. 1A inset). We verified this result in 3 different LOI and WT MEF cell lines (Supplementary Fig. S1A, S1B). We also observed this change in the balance of Erk and Akt activities for different doses of IGF2 (Supplementary Fig. S1C). This finding raised the following questions: 1) what is the mechanism of the differential regulation of Akt and Erk pathways, both of which are activated by the same receptor-ligand complex?; 2) does the differential change in the pathway activities in LOI vs. WT cells lead to the elevated cell sensitivity to IGF1R kinase inhibition displayed by LOI cells? In the following analysis we set out to answer these questions.
Fig. 1 : Analysis of the effects of Loss of Imprinting of IGF2 on the IGF1R pathway dynamics and IGF1R trafficking:

A) The fold increase in pAkt and pErk signaling was measured in WT and LOI cells by stimulating with 100ng/ml of IGF2 for the indicated time intervals and analzyed by immunoblotting. The fold increase in pAkt and pErk signaling over the baseline at time t=0 was computed and plotted as an average of three independent experiments with error bars for standard error of the means (Inset: Peak values at t=10 min); B) Schematic of the re-balancing of the IGF2 signaling network. C) Internalization rate of IGF1R was measured in a receptor biotinylation assay with IGF2 stimulation. The fold increase in total internalized receptors was quantified and plotted as a function of time; (D) Ligand mediated receptor degradation in LOI and WT cells was analyzed by using inhibitors of lysosomal (Chloroquine) and proteasomal (Mg132) receptor degradation; (E) IGF1R mRNA levels in LOI and WT cells measured after stimulation with IGF2(100ng/ml) for the indicated time points and analyzed by qRT-PCR. IGF-1R expression was normalized against β-Actin in each well and plotted as a fold increase over the baseline. Atleast 3 biological replicates were used in (D) and (E). Raw images are available at the Mendeley link.
Signaling from growth factor receptors is tightly linked to receptor trafficking, which might lead to differential downstream kinase activation (Sorkin and von Zastrow, 2009). We observed significant ligand-induced internalization and concomitant degradation of IGF1R over 3 hrs in LOI but not WT cells, following IGF2 exposure (Fig. 1C, 1D). Supporting this result was an increased expression of βArrestin1/2 and Mdm2, which are known to mediate ligand induced receptor internalization (Supplementary Fig. S1D) in LOI cells. Receptor degradation was completely abrogated by inhibitors of lysosomal (Chloroquine) and proteasomal (MG132) degradation (Fig. 1D). Surprisingly, longer-term stimulation of LOI but not WT cells also revealed an increase in transcriptional activation of Igf1r (Fig. 1E). These results suggested that both amount and localization of IGF1R can be controlled by the IGF2 mediated signaling in LOI cells.
Does the differential receptor internalization affect Erk and Akt signaling? We addressed this question through incubating cells with an inhibitor of receptor endocytosis Monodansylcadaverine (MDC)(McMahon and Boucrot, 2011, Schutze et al., 1999, Marnell et al., 1982). We found that the use of this inhibitor completely abolished Erk signaling in LOI cells (Fig. 2A) and strongly inhibited it in WT cells (Supplemental Fig. S2). The stronger effect of MDC on LOI vs. WT cells was consistent with a much more substantial IGF1R internalization in LOI cells following IGF2 stimulation (Fig. 1C). On the other hand, we found that Akt signaling in LOI cells, although mildly attenuated, occurred with dynamics similar to those in the absence of the receptor internalization (Fig. 2B). This result supported the importance of receptor trafficking in differential stimulation of the branches of the signaling network, but also raised the question of what might account for partial attenuation of Akt activity when receptor internalization was impaired. Potential mechanisms include reduction of active receptors due to receptor degradation (Fig. 1D)(Girnita et al., 2003) or other aspects of receptor trafficking, and phosphatase activity at the plasma membrane. To explore these possibilities, we used a cocktail of inhibitors of receptor degradation (MG132+Chloroquine) and vesicle recycling (Monensin) (Marnell et al., 1982). This cocktail completely abolished the attenuation in Akt response (Fig. 2C), whereas either the inhibitor of degradation or inhibitor of recycling alone failed to do so (Fig. 2C). The effect of the cocktail was replicated when Okadaic acid -- an inhibitor of cell surface phosphatase PP2A(Liu et al., 2003) was used instead of Monensin (Fig. 2D). Okadaic acid alone or the receptor degradation inhibitors used separately each failed to abrogate the Akt activity reduction in MDC treated cells (Fig. 2D). These results, in combination, suggested that increased receptor internalization in LOI cells can lead to preferential activation of Erk by the internalized receptor, whereas the activity of Akt is primarily regulated by receptors at plasma membrane modulated by the plasma membrane localized PP2A.
Fig 2 : Analysis of the effect of receptor trafficking on Akt and Erk signaling:

(A,B) Erk and Akt signaling were analyzed in WT and LOI cells in the presence of an inhibitor of receptor internalization Monodansylcadaverine. The immunoblots were quantified and results plotted after normalizing against the peak values; (C) Effect of receptor degradation and recycling on phosphorylated Akt (pAkt) was analyzed in LOI cells by inhibition of receptor recycling (Monensin Sodium), and of lysosomal (Chloroquine) and proteasomal (Mg132) receptor degradation. As in (A,B), the immunoblots were quantified and results were normalized to the peak values, and then plotted as a function of time; (D) The effects of receptor degradation and phosphatase PP2A activity on pAkt were analyzed in the presence of Okadoic Acid, an inhibitor of PP2A. The immunoblot results were quantified and plotted as a function of time after normalizing to the peak value.
The observation of a long term increase in Igf1r transcription and preferential activation of the Erk pathway in IGF2 stimulated LOI cells suggested that Erk might influence IGF1R expression in a feedback fashion. Indeed, we found that the inhibitor of Mek1/2 (U0126), the upstream kinase activating Erk, abrogated the increase in Igf1r mRNA levels, after LOI cell stimulation with IGF2 over a period of 24 hours (Fig. 3A). Although it was somewhat counter-intuitive that a transient activation of Erk can lead to long-term stimulation of receptor transcription, it was possible that downstream Erk targets could have much longer activation time scales, integrating transient Erk signaling activity over time. Indeed, we found that cMyc – a central homeostatic regulator activated by transient Erk pulse, showed a much longer activity increase in LOI cells (Supplementary Fig. S3A,B). This integration of Erk activity over time is further explored experimentally and computationally below. Interestingly, when analyzed over a shorter term (15 min.), LOI cells pre-treated with U0126 and stimulated with IGF2 also showed a significant increase in IGF1R protein levels, and a corresponding increase in Akt and Mek1/2 signaling (Fig. 3B). This result suggested that the Erk pathway also controls the short term receptor degradation following IGF2 mediated cell stimulation. Indeed, we observed that, in the presence of a proteasomal inhibitor MG-132 used to prevent the degradation of ubiquitinated proteins and to enhance their detection, incubation of cells with U0126 decreased ubiquitination of IGF1R (Fig. 3C). Since ubiquitination of IGF1R is a necessary step for its internalization (Sehat et al., 2008) and subsequent degradation, these results suggest that enhanced activity of the Erk signaling pathway in LOI cells can regulate both the short-term receptor internalization and degradation, and a longer-term receptor synthesis.
Fig 3: Quantifying the effect of Erk mediated feedback loops:

(A) Effect of Erk on Igf1r transcription was measured in LOI cells stimulated with IGF2, using the Mek1,2 inhibitor U0126. The Igf1r expression was averaged over 3 experiments and plotted as a fold increase over the baseline. Error bars represent standard error of the mean; (B) Effect of Erk on IGF1R degradation in LOI and WT cells was assessed in the presence of Mek1,2 inhibitor U0126. pMek and pAkt were also measured to confirm downstream effects of IGF1R levels. The immunoblots were quantified and plotted as an average of 3 experiments; (C) Ubiquitination of IGF1R in LOI cells was measured and quantified (right panel) with or without the U0126-mediated Mek1,2 inhibition, by immunoprecipitating IGF1R and immunoblotting with an antibody against Ubiquitin. The proteasomal inhibitor MG132 was used to preserve the ubiquitinated IGF1R against degradation; (D) A computational model incorporating signaling pathway elements, i.e., receptor trafficking, Akt/Erk activation and Erk-driven feedback loops, was implemented as shown in the schematic and described in Supp. Materials; (E) Computational model training and prediction: the 3 sets of data for pAkt and pErk dynamics used to train the computational model are shown in symbols in the left hand panel. The computational model output covering the training set (the model space), was obtained by allowing parameter variation, and is shown as a shaded region. The model was then further tested (right panel) by generating the predictions for the average signaling dynamics based on the training sets (red curves) along with the average pAkt and pErk signaling shown in red symbols. The computational model prediction for WT behavior simulated by reduced IGF1R and internalization rate is shown in blue curves while the corresponding experimental data for the WT cells is shown in blue symbols; (F) A threshold for signaling in LOI cells : The time integral of the Erk signaling as predicted by the computational model was plotted as a function of IGF2 concentration which was then extended to show a mapping for time integrated Erk and a regime of transcriptional regulation of IGF1R. The red regime indicates a 2-fold increased transcriptional increase of IGF1R in the LOI cells.
The results presented above suggested the existence of a complex feedback mechanism in LOI cells, whereby the Erk pathway can enhance its own signaling activity by increasing short-term internalization and longer-term synthesis of IGF1R. The resulting putative positive feedback is consistent with elevated levels of IGF1R in LOI cells and enhanced Erk-dependent proliferation of these cells (Kaneda et al., 2007). To explore this mechanism further, we developed a computational model of the IGF2-IGF1R-Akt-Erk signaling network (Fig. 3D, see Supplementary Modeling File), which we first trained for LOI cells on three separate repeats of experiments summarized in Fig. 1, treating each experiment separately (as shown in left hand panels of Figs. 3E, see details of the model in Supplementary Data)(D. R. Jones, 1993). The parameters of the model for each repeat were estimated separately, by minimizing the mean square error between the experimental Akt and Erk profiles vs. the corresponding activation profiles predicted by the model (Barnett et al., 2005, Surinya et al., 2008, Forbes et al., 2002, Danielsen et al., 1990, Wiley et al., 2003, Hatakeyama et al., 2003). This defined the model space, capturing experimental variability and thus parameter uncertainty (shaded areas in Fig. 3E). The model was then tested by matching the means of the experimentally obtained Akt and Erk activation profiles in LOI cells with the profiles generated by the ‘consensus’ model, for which each parameter was estimated as the mean of the three available parameter values (red curves in the right-hand side of Fig. 3E). We then used the same parameter values (determined for LOI cells) to also fit the data for WT cells, except accounting for experimentally determined differences in the IGF1R expression and rate of its internalization (see Supplementary Modeling File for details).
Predictions for the WT dynamics of both Akt and Erk qualitatively matched the experimental data (in blue, top and bottom right panels Fig. 3E), suggesting that receptor expression and internalization rates indeed are the key factors in establishing the LOI phenotype. However, the predictions of the WT responses could be further quantitatively improved by allowing other parameters to vary between the WT and LOI models (Supplementary Fig. S3C). In particular, we found that the improved fit for Akt dynamics critically depended on a change in the value of another parameter: the rate of Akt dephosphorylation in WT vs. LOI cells. Interestingly, this prediction was consistent with the differential Akt-specific phosphatase activity, revealed in experiments shown in Fig. 2D. This consistence of the model prediction and experimental observation provided further validation for the predictive power of the model. A similar attempt to improve the fit for Erk WT data however revealed a more complex parameter inter-dependence and little overall improvement. We therefore used the model in which LOI signaling network was different from WT in only three parameter values, characterizing receptor expression, receptor trafficking and Akt dephosphorylation. Importantly, this computational model further predicted that the Erk mediated positive feedback impinging on receptor regulation could lead to a sustained increase in both the receptor expression and Erk signaling (Fig. 3F). This result suggested that the enhanced IGF2 abundance can ultimately trigger a new stable state of the cell, characterized by an enhanced Erk signaling, decreased Akt signaling, and elevated IGF1R expression. This state can lead to a stable LOI phenotype, and account for its emergence following the epigenetic alteration.
What are the consequences of altered balancing of the Erk and Akt signaling pathway activities in LOI vs. WT cells, particularly in terms of regulation of cell survival when cells are challenged with the IGF1R kinase inhibitors? Both of these signaling pathways have the potential to control cell survival, in part through regulation of the balance of abundance of pro- and anti-apoptotic Bcl-2 family proteins(Kennedy et al., 1997, Scheid et al., 1999). We indeed found that Bax, a pro-apoptotic member of the Bcl-2 family, was overexpressed in LOI cells vs. WT cells following IGF2 stimulation (Fig. 4A). Conversely, Bcl-2, an anti-apoptotic member of the Bcl-2 family was expressed at relatively lower levels in LOI cells under the same conditions (Fig. 4B). We also confirmed these findings in the colons of LOI and WT mice (Supplementary Fig S4A,B). The change in the balance between Bax and Bcl-2 in WT vs. LOI cells was further confirmed in individual cells double-stained using antibodies against these proteins (Fig. 4F). Examination of expression of other members of the Bcl-2 protein family suggested the roles of other pro- and anti–apoptotic proteins, such as Bak-1 and Bcl-Xl, although at a lower level of significance (Fig. 4C).
Fig 4 : Re-balancing of abundance of anti and pro-apoptotic Bcl family proteins:

(A,B) Anti- and proapoptotic signaling was measured in WT and LOI cells by immunoblotting for Bax and Bcl-2 proteins. Blots are shown for biological triplicates; (C) Anti and pro-apoptotic transcript levels of Bcl-2 and Bcl-Xl along with Bax and Bak-1 were measured in LOI and WT cells; (D,E) Effect of Erk inhibition on Bax and Bcl-2 levels was measured in LOI cells treated with increasing dose of the Mek1,2 inhibitor U0126 in 3 biological replicates; (F) Joint distribution of Bax and Bcl-2 in the same individual WT and LOI cells were measured by immunofluorescence staining. The marginal distributions, the sample mean and variance for the distributions were estimated (shown at the bottom); (G) The computational model of IGF2 signaling was extended to include Bax, Bcl-2 levels as proportional to the time integrals of the Akt, Erk signaling. H) The predicted Akt, Erk signaling of the computational model was integrated over time to obtain estimates of Bax and Bcl-2 levels.
To explore a possible relationship between regulation of pro- and anti-apoptotic proteins and differential signaling activity in LOI cells, we explored whether Bax or Bcl-2 expression levels were affected by the perturbation of the IGF-2 signaling network. We found that LOI cells pretreated with the Mek1/2 inhibitor U0126 at various doses, followed by IGF2 stimulation for 24 hours, showed a dose dependent response of Bax protein levels(Fig 4D). On the other hand, we found that the Bcl-2 levels showed dose-dependent sensitivity to a PI3K inhibitor LY294002 under the same IGF2 stimulation (Supplementary Fig. S4D). Furthermore, we found that inhibition of Erk activity did not affect Bcl-2 levels (Fig. 4E, Supplementary Fig S4C). These results suggested that both canonical IGF2-activated signaling pathways can impinge on regulation of pro- and anti-apoptotic processes, through regulation of the Bcl2 family proteins. To provide quantitative support for this hypothesis, we expanded the computational model developed above, with a simple additional assumption, i.e., postulating that the abundance of Bax and Bcl-2 are functions of Erk and Akt activities respectively, integrated over time (Fig. 4G)(Pugazhenthi et al., 2000). Using this assumption (see Supp. Materials for other details on the model expansion) we could predict the relative approximately 2-fold differences in the abundances of Bax and Bcl-2 proteins in LOI and WT cells (Lindner et al., 2013, Raychaudhuri and Das, 2013) (Fig. 4H), which showed a very close agreement with the experimental data (Fig. 4A). This result further suggested that integration of transient Erk and Akt activities can indeed have profound longer term consequences. In combination, these results provided support for the hypothesis that re-balancing of signaling pathways in the IGF2 stimulated signaling network can lead downstream to re-balancing of Bcl-2 family proteins resulting in differential propensity of LOI and WT cells to undergo apoptosis. We provide further evidence for this hypothesis below.
The relative imbalance of Bcl-2 and Bax abundances can lead to differential sensitivity of LOI vs. WT cells to agents affecting the components of the signaling network (Oltvai et al., 1993, Perlman et al., 1999). Indeed, our prior observations suggested that LOI but not WT cells are highly sensitive to inhibition of the IGF1R kinase activity by NVPAEW-541. This inhibitor of the receptor kinase activity preferentially deactivates Akt, while having only a moderate effect on Erk (Fig. 5A, Supplementary Fig. S5A) (Kaneda et al., 2007). Thus, this drug, by virtue of differential effect on Akt vs. Erk, may also alter the balance between Bax and Bcl-2, and sensitize cells to apoptotic death (Supplementary Fig S5B). Indeed, the activities of Caspase-9 and −3, the primary targets of pro-apoptotic regulators Bax and Bak-1 (Kluck et al., 1997), were dramatically upregulated in LOI vs. WT cells within a few hours of exposure to NVPAEW-541 (Figs. 5B, 5C). This was consistent with the increased levels of activated Bax in LOI cells at 5 hrs. following treatment with the drug (Fig. 5D). Annexin-V staining provided further support for regulated cell death in LOI cells triggered by NVPAEW-541 (Supplementary Fig. S5C). Overall, these results were consistent with the apoptotic regulation of cell death in LOI cells treated with the IGF-1R kinase inhibitor, as a result of differential Bax/Bcl-2 expression controlled by Erk/Akt signaling network that is differentially balanced in LOI vs. WT cells.
Fig 5 : Analysis of the increased susceptibility of LOI cells to IGF-1R kinase inhibition:

(A) WT and LOI cells were treated with indicated doses of IGF1R inhibitor NVPAEW-541 and analyzed by immunoblotting for pAkt and pErk; (B) Caspase activation in NVPAEW-541 was detected by probing for activated Caspase-9 and Caspase-3 via immunoblotting; (C) Activated Caspase-9 and Caspase-3 were also measured by cleavage of fluorogenic substrates and plotted as a function of fluorescence readout with time; (D) Detection of activated Bax in LOI and WT cells treated with NVPAEW-541 was performed by immunocytochemistry. Activated Bax is shown in green. The cells were counterstained with nuclear DAPI (blue) and β-Actin staining (red); (E) Gaussian distributions of Bax and Bcl-2 were generated with means (see Supp. Material) and standard deviations computed from the joint distributions (cf. Fig. 4F) for both WT and LOI cells; (F) Utilizing the IGF2 computational model a relationship between Akt and Bcl-2 as a function of Akt inhibition was generated and plotted; (G) By defining survival as the codition: [Bcl-2]>=[Bax], the Akt inhibition vs. Bcl-2 relationship from (B) along with the Bax/Bcl-2 distributions in (A) were used to compute and plot the cell survival as a percentage of Akt inhibition; (H) WT and LOI cells treated with NVPAEW-541 were assayed for cell survival using Alamar blue dye over a period of 24 hours. The survival was plotted as a percentage of the cells surviving as a function of the dose ; (I) A schematic for the relative positions of WT and LOI cells in the survival-death space was developed and shown as a function of Bax and Bcl-2 concentrations. In the bottom panel is shown the effect of Bcl-2 inhibition on the cells and their positions;
Given these results, we explored computationally whether the expanded mathematical model of the signaling network would predict higher sensitivity of LOI cells vs. WT cells to NVPAEW-541. Intuitively, our analysis suggests that a given level of IGF1R inhibition can lead to distinct levels of Akt activity in WT vs. LOI cells and, as a consequence, allow for a greater of tolerance to the perturbation under which Bcl2 levels would still exceed the levels of Bax. Since the relative excess of Bax vs. Bcl-2 (or vice versa) is thought to lead to the pro-apoptotic (vs. anti-apoptotic) response, this result suggested that a much greater dose of the kinase inhibitor would be required to induce the death response in WT vs. LOI cells. We first quantified this prediction in the mathematical model by taking into account the variability in the levels of Bax and Bcl-2 expression on the individual cell level in the LOI and WT cell populations (Fig. 5E) and by accounting for the relationship between the degree of Akt activity and Bcl-2 abundance (Fig. 5F, inferred from Fig. 4H) in WT and LOI cells. Given these model refinements and the experimental data in Fig. 5E, the computational model predicted the % survival of the cellular population following exposure to NVPAEW-541 (Fig. 5G). We then tested this prediction by experimentally analyzing % cell survival in response to NVPAEW-541 in WT and LOI cells using an Alamar Blue assay normalized to untreated cells. The results yielded excellent match with the computational model predictions, providing additional support for the model assumptions and for the proposed mechanism for the differential sensitivity of LOI and WT cells to the IGF1R kinase inhibitor (Fig. 5H).
Overall, our data argue that an epigenetic alteration (LOI) leading to a doubling of the expression of a growth factor, by virtue of its effect on the signaling and apoptotic pathways, can potentiate apoptosis in response to inhibition of the receptor kinases activity. One convenient and instructive way to qualitatively and quantitatively model this phenomenon is to ‘map’ the cell populations onto a parametric space, defined by the relative abundances of pro- and anti-apoptotic molecules (e.g., Bax and Bcl-2), with each cell population in this space defined by the ranges of expression of these molecules (Fig. 5I, inferred from Fig. 4F). Embedded within this space is a ‘Death Boundary’ defined as a line separating the cellular states dominated by either Bcl-2 or Bax, as illustrated in Fig. 5I. The epigenetic change would then be interpreted as a stable shift of the LOI population closer to this ‘Death Boundary’ vs. the WT population due to differences in the expression of Bcl-2 and Bax. The application of the receptor kinase inhibitor can additionally shift both WT and LOI cell populations closer to the Death Boundary due to the pro-apoptosis effect of inhibition of Akt. However, for a range of drug doses, only cells within the LOI but not WT population would ‘cross’ this boundary and die, thus accounting for the experimentally observed therapeutic window. This analysis, mapping the cell populations experimentally characterized on the single cell level onto to a ‘phenotypic plane’ incorporating the mechanistic details of the regulatory process of interest (here: the apoptosis/survival phenotypic domains separated by the ‘Death Boundary’) can serve as a convenient quantitative tool to explore the effect of various perturbations affecting cell populations in this and other settings.
Discussion
The results of this study uncover complex changes in cell regulation that can be triggered by a subtle epigenetic alteration — loss of imprinting of a gene. The immediate consequence of the loss of imprinting of IGF2 is doubling of the gene dosage and the level of expression of the growth factor the gene encodes. These effects of LOI lead to a paradoxical change in the signaling makeup of the affected cells: continuous exposure to higher IGF2 levels results in adaptive signaling network re-balancing. Whereas the fold-change in the IGF2-stimulated activation of Erk becomes more pronounced vs. that in cells unaffected by LOI, the fold-change in activation of Akt becomes diminished. The results further suggest that LOI can result in a stable cellular phenotype emerging through a feedback mediated enhancement of Erk signaling at the expense of Akt signaling, again resulting from an increased level of expression of IGF2. The key mechanism underlying re-balancing of signaling activity in LOI vs. the WT cells is due to differential coupling of Erk and Akt signaling to plasma membrane vs. early endosome localized receptor complexes. Experimental and modeling analysis also revealed a key effect of differential Akt inactivation kinetics in LOI vs. WT cells. The differential activation of Erk and Akt leads to re-balancing of expression of the apoptosis controlling proteins, resulting in a critically important phenotypic outcome: an increased propensity of LOI cells to undergo regulated cell death in response to inhibition of receptor kinase activity. The proposed mechanism regulating this phenotype provides a quantitative explanation for the previously identified putative therapeutic window, specifying the ranges of the dose of the IGF1R kinase inhibitor that can kill most of LOI cells, while leaving most of the WT cells alive. More generally, this study establishes a framework for analysis of how epigenetic and genetic alterations of the signaling networks can sensitize cells to therapeutic interventions. This framework, quantitatively introduced in our results is further discussed below.
There is an increasing amount of evidence suggesting that mitogenic signals can also sensitize cells to pro-apoptotic signals (Tentner et al., 2012, Evan and Littlewood, 1998). It is a sensible protective mechanism ensuring that unwarranted or excessive cell proliferation can be balanced by regulated cell death. An important mechanism connecting apoptosis regulation to signaling activated by mitogenic stimuli is the control of the relative abundance of the members of the Bcl-2 family of proteins(Kennedy et al., 1997, Scheid et al., 1999). Changes in the stoichiometry can either protect a cell from or predispose it to apoptotic death, thus defining the natural ‘Death Boundary’ in a parametric plane, as shown in Fig. 5H. Cells expressing different combinations of pro- and anti-apoptotic proteins can be mapped onto the same plane, thus underscoring both the average distance of the population from the Death Boundary, and the inherent variability within this population. Mitogenic stimuli might transiently shift the cell population closer to the Death Boundary. Here we show that this shift can become stable in response to an epigenetic LOI transition, thus increasing both Erk-dependent proliferation and enhanced sensitivity of cell survival to receptor mediated activation of Akt. However, a stable shift of a population with respect to the Death Boundary can occur in many other ways, including genetic mutations leading to what has been referred to as oncogene addiction, particularly through the socalled oncogenic shock (Sharma and Settleman, 2007). Furthermore, this analysis can be extend to other processes, e.g., the effects of genetic alterations decoupling mitogenic signaling from the apoptosis control and thus allowing increased proliferation without the inherent trade-off of enhanced apoptosis sensitivity. As shown in this study, the mapping of cell populations onto the parametric apoptosis plane can be done quantitatively, opening the path to predictive modeling of the effects of therapeutic interventions. Indeed, we show that we can describe the effects of a drug by shifts of both WT and LOI populations closer to the Death Boundary, which accounts for experimentally observed therapeutic window.
Interestingly, our results may also provide a novel insight into the role of IGF2 in enhanced regeneration of normal adult livers(Bohm et al., 2010). In contrast to the fetal liver, where IGF2 is imprinted, in the adult liver, the expression of this factor is bi-allelic, creating a potential for an LOI-like phenotype(Davies, 1994). Of interest, it has been observed that, although growth factors can promote proliferation of liver cells, simultaneous inhibition of Erk activation instead potentiates pro-survival/pro-differentiation phenotype(Fremin et al., 2012). Our results argue that this paradoxical observation is a consequence of the pathway re-balancing, with Erk inhibition leading to a relative increase in Akt-mediated pro-survival signaling (cf. Fig. 3B). The LOI-like state of the liver can thus promote proliferation of liver cells, whereas an additional relative reduction in Erk activation (possibly due to other signaling inputs) can promote differentiation and survival, thus supplying two important aspects of successful regeneration process.
Differential balancing between Erk and Akt has been observed before in response to specific mutations as well as kinome-wide genetic perturbations(Lu et al., 2011). Furthermore, Cavin-3, a tumor suppressor protein, facilitates Erk signal transduction at the expense of Akt signaling. Conversely, a loss of Cavin-3 thus leads to increase in Akt signaling at expense of Erk, and is then manifested in enhanced aerobic glycolysis and resistance to apoptosis(Hernandez et al., 2013). Forced pharmacological changes in the balancing between Erk and Akt signaling pathways can also lead to selection of alternative differentiation phenotypes, as in differentiation of smooth muscle cells(Hayashi et al., 1999). Our study suggests that Erk and Akt re-balancing can occur as a consequence of an epigenetic rather than genetic change. Generally, there might be multiple mechanisms of mutual Erk and Akt regulation, leading to differential balancing of their activities. Although our study suggests that this can happen through the control of receptor complex trafficking, a more direct crosstalk between these pathways can also affect the relative signaling activities(Lu et al., 2011, Wang et al., 2009). Whatever the mechanism, our analysis further suggests that signaling pathways triggered by the same chemical input can be linked to alternative phenotypic outcomes, with potentially dramatic consequences for how we view tissue homeostasis and various pathologies, including cancer.
Contact For Reagent And Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by Andre Levchenko (andre.levchenko@yale.edu)
Experimental Model And Subject Details
Mouse Embryonic Fibroblasts
IGF2 LOI and WT mice pregnant at days between 14 to 16 days were used to extract embryos to derive MEFs. Sex of the embryos were determined and male and female MEFs were separately numbered and stored. Matched LOI and WT MEFs were used in all the experiments. Cells were used in early passage and were maintained at 37degC, 5% CO2 in DMEM and 10% FBS.
IGF2/LOI mice
Mice with C56BL/6J background carrying a deletion in the H19 gene (3 kb) and 10 kb of the upstream region including the differentially methylated region (DMR) regulating IGF-II silencing were obtained from S. Tilghman (Princeton University, Princeton, NJ) (Leighton et al., 1995). We maintained the strain by breeding female wild-type C56BL/6J and male H19 +/−, keeping the maternal Igf2 allele silenced. In experimental crosses, mice with biallelic Igf2 expression and control littermates were isolated by crossing female H19 +/− with male wild-type C56BL/6J mice. The animals were maintained according to Yale Institutional protocol number 2014–11609.
Method Details
Immuno-Blotting:
Mouse colons and mouse embryonic fibroblasts (MEFs) were used as the source of the material for immune-blotting (western blotting). Animals were euthanized, colons extracted and pulverized by sonication in a commercially available animal tissue extraction buffer following the protocols in (Kaneda et al., 2007). The cells were cultured following the protocols from the same study. Briefly, cells were serum starved overnight, treated to experimental conditions and harvested in denaturing SDS based lysis buffers, suspended in 4x loading buffer and heated to 95 degC for 5 minutes and stored for archival at −20degC. Lysates (about 20μg) were reduced with DTT and subjected to gelelectrophoresis in precast Bis-Tris gels under reduced conditions and transferred to 0.2μm Nitrocellulose membrane in a dry-transfer iBlot apparatus. Membranes were blocked with 5%BSA in PBS and then incubated with the relevant primary antibody followed by the secondary with 5 to 6 washes in washing buffer in between. The membranes were developed using PicoWest chemiluminescence substrate and imaged using a BioRad imaging station. The images of the blots were then quantified using the gel band intensity analysis macro in ImageJ. Several independent experiments were performed, as indicated, to assure biological reproducibility and statistical power of analysis.
Immunostaining analysis:
Cells were seeded on 35 mm Mattek dishes and allowed to attach for 10 to 12 hours. Cells were then serum starved overnight in DMEM media with 0%FBS and stimulated with IGF2 of indicated concentration for the indicated period of time. The cells are then washed in ice-cold PBS and fixed in 4% Fomaldehyde. The cells are then permeabilized with 0.1% TritonX-100 in PBS and then blocked with 10% goat serum for 1 hour. The cells were incubated with the primary antibody at the specified concentration in blocking buffer and incubated overnight at 4degC. After washing thoroughly, the cells were incubated with the appropriate secondary antibody, Hoechst dye and CellMask Deep Red at the specified concentrations for 1hr at room temperature. The cells were then washed thoroughly in PBS and imaged on a Zeiss AxioObserver Z1 inverted microscope with DAPI, GFP/FITC, Cy3/Rhodamine and Cy5 excitation and emission filter sets. The fluorescence signal from the target protein was quantified using custom Matlab code based on the Watershed algorithm. Using the Hoechst stain for the nucleus and the CellMask stain for the cytoplasm, these cellular compartments were identified and the fluorescence signal quantified as mean signal / cell.
Cell survival assay:
Cells were seeded at a density of 2000 cells/well in a dark walled, transparent bottom 96 well assay plate overnight and then serum starved for 24 hours followed by 24 hours of NVPAEW541 treatment. 10% Alamar blue was added to each well and fluorescence measured at 590nm in a plate reader. No drug control was used to express the percentage of cell survival.
Annexin V analysis:
Cells were grown to 95% confluency, serum starved overnight, treated with NVPAEW541 where indicated, trypsinized to achieve single cell suspension, washed and incubated with AnnexinV:PE as indicated by the BD protocol. The cells were also counter-stained with 7-AAD to exclude dead cells. The cells were analyzed by flow cytometry, gated and plotted as scatter diagrams.
Cell surface biotinylation and receptor internalization rate:
Cells are grown in flasks to 95% confluency and serum starved overnight. Cells are washed in ice-cold PBS and incubated with SuphoNHS-SS-Biotin to label all cell surface receptors. The cells are washed and stimulated with IGF-2 at 37degC to aid internalization of the receptors. After stimulation the remaining biotin on the receptors on the surface are cleaved with glutathione reducing agent and the cells are lysed in IP buffer. The lysate is then incubated with streptavidin coated dynabeads to pull down biotin labeled receptors and analyzed by Western blotting. The blots are then probed with Anti-IGF1R antibodies to quantify internalized IGF1R.
Internalization rate with FM143FX:
Cells were seeded on Mattek dishes and left overnight for attachment. They were then serum starved overnight and incubated with 100uM FM143FX dye for 15 minutes at 4degC for the dye to stain the cell surface. The dye is then removed, cells washed to remove excess of the dye and the DMEM medium (without phenol red) replaced back. The cells were incubated with IGF-2 at 37degC for various times. After the set time points the cells were washed, fixed and imaged for the internalized dye. Cells were also counterstained with Hoechst 33342 and HCS CellMask™ Deep Red (Life Technologies, CA) to stain the nucleus and the cytoplasm. The internalized dye was then quantified by using the nuclear and cytoplasmic stain as a mask to delineate the cell using custom Matlab code based on the Watershed algorithm.
qRT-PCR assays:
Cells were grown to confluence and collected as pellets in RNAprotect cell reagent (Qiagen, CA). According to instructions of the RNEasy kit (Qiagen, CA) total RNA was extracted from the cells. The RNA was then converted to cDNA according to the instructions of the QuantiTec Rev. Transcription kit (Qiagen,CA). Using custom Taqman assays (AppliedBiosystems, CA) for the gene targets indicated in corresponding figures, the cDNA was added to the Taqman probes in triplicates in a PCR mix in 384 well plates. Using multiple dyes for the gene of interest and β-actin in the same well, the fold expression of the gene of interest, normalized to β-actin, was analyzed..
Quantification And Statistical Analysis
Pairwise t-test:
Pairwise t-test was used to test statistical significance in all cases and was implemented in Matlab as ttest2.
Standard Error of the Means:
Standard error of the means (S.E.M) was used to depict the variance of the data in the figures as Mean+/−SEM.
Methods File S1
Computational model of the IGF2-IGF1R-Akt-Erk pathway related to Figures 3, 4 and 5.
Supplementary Material
Highlights.
IGF2 LOI is an epigenetic change leading to increased cancer predisposition
IGF2 LOI leads to rebalancing of activities of canonical AKT and ERK pathways
Altered signaling balance leads to rebalancing of pro- and anti-apoptotic control
This mechanism quantitatively accounts for a possible therapeutic window
Vanaja et.al delineate a mechanism in which a subtle epigenetic modification results in altered balance of the two arms of a signaling network. This causes increased growth at the cost of enhanced sensitivity to receptor inhibition, providing a mechanism for a therapeutic intervention specifically affecting epigenetic ally modified cells.
Acknowledgements:
The work was supported by NIH grants: CA209992 and GM072024 (K.G.V. and A.L.)
Footnotes
Declaration of Interests : The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- ANDERSON J, GORDON A, MCMANUS A, SHIPLEY J & PRITCHARD-JONES K 1999. Disruption of imprinted genes at chromosome region 11p15.5 in paediatric rhabdomyosarcoma. Neoplasia, 1, 340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARNETT SF, DEFEO-JONES D, FU S, HANCOCK PJ, HASKELL KM, JONES RE, KAHANA JA, KRAL AM, LEANDER K, LEE LL, MALINOWSKI J, MCAVOY EM, NAHAS DD, ROBINSON RG & HUBER HE 2005. Identification and characterization of pleckstrin-homology-domaindependent and isoenzyme-specific Akt inhibitors. Biochem J, 385, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARTHOLDI D, KRAJEWSKA-WALASEK M, OUNAP K, GASPAR H, CHRZANOWSKA KH, ILYANA H, KAYSERILI H, LURIE IW, SCHINZEL A & BAUMER A 2009. Epigenetic mutations of the imprinted IGF2-H19 domain in Silver-Russell syndrome (SRS): results from a large cohort of patients with SRS and SRS-like phenotypes. J Med Genet, 46, 192–7. [DOI] [PubMed] [Google Scholar]
- BOHM F, KOHLER UA, SPEICHER T & WERNER S 2010. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol Med, 2, 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUI H, CRUZ-CORREA M, GIARDIELLO FM, HUTCHEON DF, KAFONEK DR, BRANDENBURG S, WU Y, HE X, POWE NR & FEINBERG AP 2003. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science, 299, 1753–5. [DOI] [PubMed] [Google Scholar]
- JONES DR, C. D. P., STUCKMAN BE 1993. Lipschitzian Optimization Without the Lipschitz Constant. Journal of Optimization Theory and Applications, 79, 157–181 [Google Scholar]
- DANIELSEN A, LARSEN E & GAMMELTOFT S 1990. Chromaffin cells express two types of insulin-like growth factor receptors. Brain Res, 518, 95–100. [DOI] [PubMed] [Google Scholar]
- DAVIES SM 1994. Developmental regulation of genomic imprinting of the IGF2 gene in human liver. Cancer Res, 54, 2560–2. [PubMed] [Google Scholar]
- EVAN G & LITTLEWOOD T 1998. A matter of life and cell death. Science, 281, 1317–22. [DOI] [PubMed] [Google Scholar]
- FEINBERG AP 2013. The epigenetic basis of common human disease. Trans Am Clin Climatol Assoc, 124, 84–93. [PMC free article] [PubMed] [Google Scholar]
- FERRON SR, RADFORD EJ, DOMINGO-MUELAS A, KLEINE I, RAMME A, GRAY D, SANDOVICI I, CONSTANCIA M, WARD A, MENHENIOTT TR & FERGUSON-SMITH AC 2015. Differential genomic imprinting regulates paracrine and autocrine roles of IGF2 in mouse adult neurogenesis. Nat Commun, 6, 8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FORBES BE, HARTFIELD PJ, MCNEIL KA, SURINYA KH, MILNER SJ, COSGROVE LJ & WALLACE JC 2002. Characteristics of binding of insulin-like growth factor (IGF)-I and IGF-II analogues to the type 1 IGF receptor determined by BIAcore analysis. Eur J Biochem, 269, 961–8. [DOI] [PubMed] [Google Scholar]
- FREMIN C, EZAN F, GUEGAN JP, GAILHOUSTE L, TROTARD M, LE SEYEC J, RAGEUL J, THERET N, LANGOUET S & BAFFET G 2012. The complexity of ERK1 and ERK2 MAPKs in multiple hepatocyte fate responses. J Cell Physiol, 227, 59–69. [DOI] [PubMed] [Google Scholar]
- GIRNITA L, GIRNITA A & LARSSON O 2003. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor 1 receptor. Proc Natl Acad Sci U S A, 100, 8247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HATAKEYAMA M, KIMURA S, NAKA T, KAWASAKI T, YUMOTO N, ICHIKAWA M, KIM JH, SAITO K, SAEKI M, SHIROUZU M, YOKOYAMA S & KONAGAYA A 2003. A computational model on the modulation of mitogen-activated protein kinase (MAPK) and Akt pathways in heregulininduced ErbB signalling. Biochem J, 373, 451–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAYASHI K, TAKAHASHI M, KIMURA K, NISHIDA W, SAGA H & SOBUE K 1999. Changes in the balance of phosphoinositide 3-kinase/protein kinase B (Akt) and the mitogen-activated protein kinases (ERK/p38MAPK) determine a phenotype of visceral and vascular smooth muscle cells. J Cell Biol, 145, 727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERNANDEZ VJ, WENG J, LY P, POMPEY S, DONG H, MISHRA L, SCHWARZ M, ANDERSON RG & MICHAELY P 2013. Cavin-3 dictates the balance between ERK and Akt signaling. Elife, 2, e00905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLM TM, JACKSON-GRUSBY L, BRAMBRINK T, YAMADA Y, RIDEOUT WM 3RD & JAENISCH R 2005. Global loss of imprinting leads to widespread tumorigenesis in adult mice. Cancer Cell, 8, 275–85. [DOI] [PubMed] [Google Scholar]
- KANEDA A & FEINBERG AP 2005. Loss of imprinting of IGF2: a common epigenetic modifier of intestinal tumor risk. Cancer Res, 65, 11236–40. [DOI] [PubMed] [Google Scholar]
- KANEDA A, WANG CJ, CHEONG R, TIMP W, ONYANGO P, WEN B, IACOBUZIO-DONAHUE CA, OHLSSON R, ANDRAOS R, PEARSON MA, SHAROV AA, LONGO DL, KO MS, LEVCHENKO A & FEINBERG AP 2007. Enhanced sensitivity to IGF-II signaling links loss of imprinting of IGF2 to increased cell proliferation and tumor risk. Proc Natl Acad Sci U S A, 104, 20926–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENNEDY SG, WAGNER AJ, CONZEN SD, JORDAN J, BELLACOSA A, TSICHLIS PN & HAY N 1997. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev, 11, 701–13. [DOI] [PubMed] [Google Scholar]
- KLUCK RM, BOSSY-WETZEL E, GREEN DR & NEWMEYER DD 1997. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science, 275, 1132–6. [DOI] [PubMed] [Google Scholar]
- LEIGHTON PA, INGRAM RS, EGGENSCHWILER J, EFSTRATIADIS A & TILGHMAN SM 1995. Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature, 375, 34–9. [DOI] [PubMed] [Google Scholar]
- LINDNER AU, CONCANNON CG, BOUKES GJ, CANNON MD, LLAMBI F, RYAN D, BOLAND K, KEHOE J, MCNAMARA DA, MURRAY F, KAY EW, HECTOR S, GREEN DR, HUBER HJ & PREHN JH 2013. Systems analysis of BCL2 protein family interactions establishes a model to predict responses to chemotherapy. Cancer Res, 73, 519–28. [DOI] [PubMed] [Google Scholar]
- LIU W, AKHAND AA, TAKEDA K, KAWAMOTO Y, ITOIGAWA M, KATO M, SUZUKI H, ISHIKAWA N & NAKASHIMA I 2003. Protein phosphatase 2A-linked and -unlinked caspase-dependent pathways for downregulation of Akt kinase triggered by 4-hydroxynonenal. Cell Death Differ, 10, 772–81. [DOI] [PubMed] [Google Scholar]
- LU Y, MULLER M, SMITH D, DUTTA B, KOMUROV K, IADEVAIA S, RUTHS D, TSENG JT, YU S, YU Q, NAKHLEH L, BALAZSI G, DONNELLY J, SCHURDAK M, MORGAN-LAPPE S, FESIK S, RAM PT & MILLS GB 2011. Kinome siRNA-phosphoproteomic screen identifies networks regulating AKT signaling. Oncogene, 30, 4567–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARNELL MH, STOOKEY M & DRAPER RK 1982. Monensin blocks the transport of diphtheria toxin to the cell cytoplasm. J Cell Biol, 93, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCMAHON HT & BOUCROT E 2011. Molecular mechanism and physiological functions of clathrinmediated endocytosis. Nat Rev Mol Cell Biol, 12, 517–33. [DOI] [PubMed] [Google Scholar]
- OGAWA O, BECROFT DM, MORISON IM, ECCLES MR, SKEEN JE, MAUGER DC & REEVE AE 1993. Constitutional relaxation of insulin-like growth factor II gene imprinting associated with Wilms’ tumour and gigantism. Nat Genet, 5, 408–12. [DOI] [PubMed] [Google Scholar]
- OLTVAI ZN, MILLIMAN CL & KORSMEYER SJ 1993. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell, 74, 609–19. [DOI] [PubMed] [Google Scholar]
- PERLMAN H, ZHANG X, CHEN MW, WALSH K & BUTTYAN R 1999. An elevated bax/bcl-2 ratio corresponds with the onset of prostate epithelial cell apoptosis. Cell Death Differ, 6, 48–54. [DOI] [PubMed] [Google Scholar]
- PUGAZHENTHI S, NESTEROVA A, SABLE C, HEIDENREICH KA, BOXER LM, HEASLEY LE & REUSCH JE 2000. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem, 275, 10761–6. [DOI] [PubMed] [Google Scholar]
- RAYCHAUDHURI S & DAS SC 2013. Low probability activation of Bax/Bak can induce selective killing of cancer cells by generating heterogeneity in apoptosis. J Healthc Eng, 4, 47–66. [DOI] [PubMed] [Google Scholar]
- SAKATANI T, KANEDA A, IACOBUZIO-DONAHUE CA, CARTER MG, DE BOOM WITZEL S, OKANO H, KO MS, OHLSSON R, LONGO DL & FEINBERG AP 2005. Loss of imprinting of Igf2 alters intestinal maturation and tumorigenesis in mice. Science, 307, 1976–8. [DOI] [PubMed] [Google Scholar]
- SCHEID MP, SCHUBERT KM & DURONIO V 1999. Regulation of bad phosphorylation and association with Bcl-x(L) by the MAPK/Erk kinase. J Biol Chem, 274, 31108–13. [DOI] [PubMed] [Google Scholar]
- SCHUTZE S, MACHLEIDT T, ADAM D, SCHWANDNER R, WIEGMANN K, KRUSE ML, HEINRICH M, WICKEL M & KRONKE M 1999. Inhibition of receptor internalization by monodansylcadaverine selectively blocks p55 tumor necrosis factor receptor death domain signaling. J Biol Chem, 274, 10203–12. [DOI] [PubMed] [Google Scholar]
- SEHAT B, ANDERSSON S, GIRNITA L & LARSSON O 2008. Identification of c-Cbl as a new ligase for insulin-like growth factor-I receptor with distinct roles from Mdm2 in receptor ubiquitination and endocytosis. Cancer Res, 68, 5669–77. [DOI] [PubMed] [Google Scholar]
- SHARMA SV & SETTLEMAN J 2007. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev, 21, 3214–31. [DOI] [PubMed] [Google Scholar]
- SORKIN A & VON ZASTROW M 2009. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol, 10, 609–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEENMAN MJ, RAINIER S, DOBRY CJ, GRUNDY P, HORON IL & FEINBERG AP 1994. Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms’ tumour. Nat Genet, 7, 433–9. [DOI] [PubMed] [Google Scholar]
- SURINYA KH, FORBES BE, OCCHIODORO F, BOOKER GW, FRANCIS GL, SIDDLE K, WALLACE JC & COSGROVE LJ 2008. An investigation of the ligand binding properties and negative cooperativity of soluble insulin-like growth factor receptors. J Biol Chem, 283, 5355–63. [DOI] [PubMed] [Google Scholar]
- TENTNER AR, LEE MJ, OSTHEIMER GJ, SAMSON LD, LAUFFENBURGER DA & YAFFE MB 2012. Combined experimental and computational analysis of DNA damage signaling reveals context-dependent roles for Erk in apoptosis and G1/S arrest after genotoxic stress. Mol Syst Biol, 8, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIMP W & FEINBERG AP 2013. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer, 13, 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIMP W, LEVCHENKO A & FEINBERG AP 2009. A new link between epigenetic progenitor lesions in cancer and the dynamics of signal transduction. Cell Cycle, 8, 383–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VENKATRAMAN A, HE XC, THORVALDSEN JL, SUGIMURA R, PERRY JM, TAO F, ZHAO M, CHRISTENSON MK, SANCHEZ R, YU JY, PENG L, HAUG JS, PAULSON A, LI H, ZHONG XB, CLEMENS TL, BARTOLOMEI MS & LI L. 2013. Maternal imprinting at the H19-Igf2 locus maintains adult haematopoietic stem cell quiescence. Nature, 500, 345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VU TH, NGUYEN AH & HOFFMAN AR 2010. Loss of IGF2 imprinting is associated with abrogation of long-range intrachromosomal interactions in human cancer cells. Hum Mol Genet, 19, 901–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG CC, CIRIT M & HAUGH JM 2009. PI3K-dependent cross-talk interactions converge with Ras as quantifiable inputs integrated by Erk. Mol Syst Biol, 5, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEKSBERG R, SHUMAN C & BECKWITH JB 2010. Beckwith-Wiedemann syndrome. Eur J Hum Genet, 18, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILEY HS, SHVARTSMAN SY & LAUFFENBURGER DA 2003. Computational modeling of the EGFreceptor system: a paradigm for systems biology. Trends Cell Biol, 13, 43–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.