

Abstract
β-sheet proteins carry out critical functions in biology, and hence are attractive scaffolds for computational protein design. Despite this potential, de novo design of all β-sheet proteins from first principles lags far behind the design of all-α or mixed αβ domains due to their non-local nature and tendency of exposed β-strand edges to aggregate. Through study of loops connecting unpaired β-strands (β-arches), we have identified a series of structural relationships between loop geometry, sidechain directionality and β-strand length that arise from hydrogen bonding and packing constraints on regular β-sheet structures. We use these rules to de novo design jelly-roll structures with double-stranded β-helices formed by 8 antiparallel β-strands. The nuclear magnetic resonance structure of a hyperthermostable design closely matched the computational model, demonstrating accurate control over the β-sheet structure and loop geometry. Our results open the door to the design of a broad range of non-local β-sheet protein structures.
INTRODUCTION
β-sheet protein domains are ubiquitous in nature, carrying out a wide range of functions: transporting hydrophobic molecules, recognition and enzymatic processing of carbohydrates, and scaffolding of virus capsids and antibodies, among others. Although β-sheet protein scaffolds are well suited for incorporating new functions, their design from first principles remains an outstanding challenge. Recent progress in de novo protein design has enabled the accurate design of many hyperstable and structurally diverse proteins, but to date other than short β-sheet peptides1–3 all exhibit either all-α or mixed-αβ folds4. The design of the latter has been considerably facilitated by the derivation of a set of rules describing constraints on the backbone geometry of the loops connecting secondary structure elements5, but all-β proteins contain additional features which are less well understood. All β-sheet structures are particularly challenging to design from scratch6 because a larger fraction of the interactions are non-local (between residues distant along the linear sequence) leading to slower folding rates7, and because β-strands, particularly at the edges of β-sheets, can aggregate into amyloid-like structures. Hence, few β-sheet protein design studies have sought to generate new backbone structures8,9 and, except for a recent β-barrel structure with primarily local strand pairings10, those designs confirmed by high resolution structure determination have relied heavily on sequence information11,12 and backbone structures13,14 from naturally occurring β-sheet proteins.
To date, the de novo design of β-sheet loop connections has been limited to β-hairpins (two antiparallel β-strands interacting via backbone hydrogen bonding and connected through a loop) which is the most local strand pairing possible and, in principle, the fastest to fold. However, these structures lack a critical feature of non-local globular all-β structures: loops connecting β-strands not paired to each other, also known as β-arches15. These loops connect distinct β-sheets and pair β-strands with larger sequence separation, and are essential for enabling the protein fold complexity observed in antibodies, β-solenoids, jelly-rolls and greek key containing structures generally. Here we set out to identify the general principles for designing non-local β-sheet structures,
RESULTS
Constraints on β-arch geometry
We undertook the investigation of the constraints on the backbone geometry of β-strands and connecting loops that arise from hydrogen bonding and the requirement for a compact hydrophobic core. We studied sidechain directionality patterns of the two β-strand residues adjacent to β-arch loops (Fig. 1a, left) in naturally occurring protein structures, defining the sidechain orientation of the β-strand residue preceding the loop as concave (represented by “↓”) if its CαCβ vector is parallel to the vector d from the first to the second β-strand, and convex (represented by “↑”) if the CαCβ vector is antiparallel to d. For the residue following the loop the sidechain pattern is described in the same way, but instead using the vector from the second to the first β-strand (-d) as a reference (Fig. 1a). This results in four possible β-arch loop sidechain orientation patterns: “↑↑”, “↑↓”, “↓↑” and “↓↓”. We analyzed the sidechain patterns and the local backbone geometry – as described with ABEGO torsion bins16– of 5,061 β-arch loops from a non-redundant database of natural protein structures (torsion bins “A” and “B” are the α-helix and extended regions, “G” and “E” regions are the positive φ angle equivalents of “A” and “B”; and “O” is the cis peptide bond conformation; Supplementary Fig. 1). We found that all four sidechain orientation patterns frequently occur, and, in contrast to other types of loop connections (i.e. αβ, βα and β-hairpins)5, there was not a correlation between β-arch loop length and sidechain pattern. Instead, each loop ABEGO type, because of the way in which it twists and bends the polypeptide chain16, is associated with a specific flanking residue sidechain pattern (Fig. 1b). The most frequently observed turn types (between 1 and 5 amino acids) for each sidechain pattern are listed in Fig. 1c; for example ABB, BBGB, BABB and BGB are the most frequent loop types for the patterns “↓↓”, “↓↑”,”↑↓” and “↑↑”, respectively.
Fig. 1. Constraints on β-arch geometry.
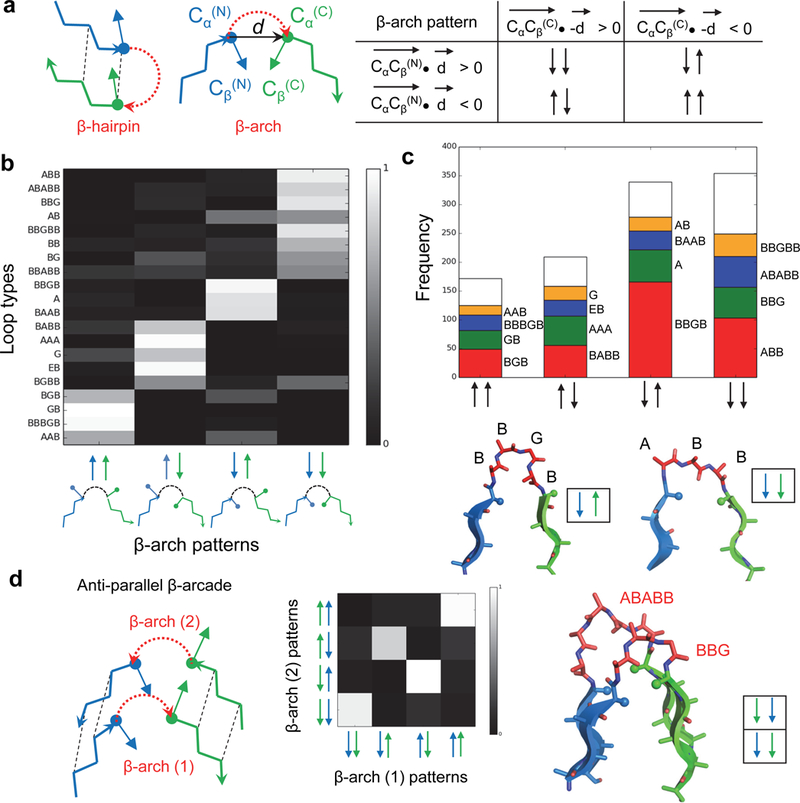
a, Sidechain directionality in the β-arch. Left, comparison between β-hairpin and β-arch; the CαCβ and d vectors used to define the orientation of the two adjacent sidechains are indicated. The four possible sidechain directionality patterns are on the right. b, Turn type dependence of β-arch sidechain patterns. Loops on the y-axis are described by their ABEGO torsion bins (Supplementary Fig. 1). Most of the loops adopt only one of the four possible sidechain patterns. c, Frequency of the most common loops for each of the four β-arch sidechain patterns. There are strong preferences, for example BBGB is strongly associated with the “↓↑” pattern, whereas ABB is strongly associated with the “↓↓” pattern (shown in bottom). Only loops with bending < 120 degrees (Methods) and containing between 1 and 5 amino acids were considered in this analysis. d, Left, two stacked β-arches having in-register strand pairing form β-arcades. Middle, since strand pairs of the β-arcade are in-register, the sidechains adjacent to one β-arch loop must have the same orientation as the paired sidechains that are adjacent to the second β-arch loop, and therefore not all loop pairs are allowed. Right, example of a β-arcade formed by two common β-arches with compatible sidechain patterns.
The next level of non-local interaction complexity in all-β folds involves strand pairing (parallel or antiparallel) between two β-arches forming a β-arcade (Fig. 1d), a common structural motif in naturally occurring β-solenoids15,17. Since the β-arch loops are stacked in-register, the sidechains adjacent to one β-arch loop are likely to have the same orientation as the sidechains adjacent to the second β-arch loop; analysis of naturally occurring β-arcades confirms that the sidechain patterns of the two β-arch loops indeed are correlated (Fig. 1d, middle).
Jelly-roll design principles
The double-stranded β-helix can be regarded as a long β-hairpin wrapped around an axis perpendicular to the direction of β-strands, with β-helical turns formed by the pairing between β-arcades (Fig. 2a). In the compact folded structure, two antiparallel β-sheets pack against each other in a sandwich-like arrangement, with the first strand paired to the last, and all β-strands are connected through β-arch loops except for the central β-hairpin. We aimed at designing β-helices with 3 β-arcades forming two antiparallel 4-stranded β-sheets, with the 8 β-strands connected through 6 β-arches and 1 β-hairpin. The non-local character of the structure grows from the first β-arcade, which starts from the central β-hairpin, to the last one, where the N- and C-termini are paired.
Fig. 2. Double-stranded β-helix topology specification.
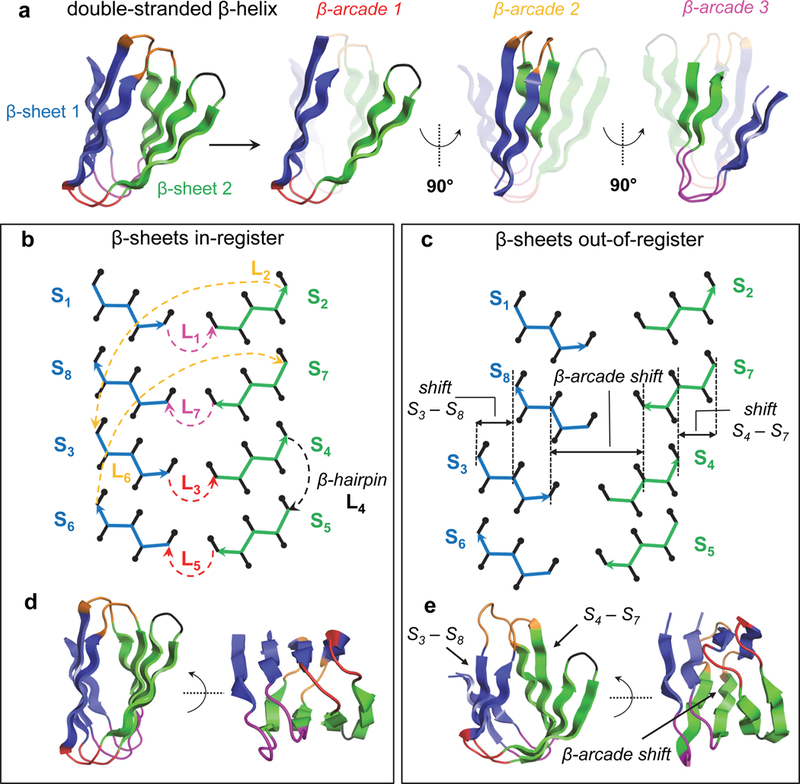
a, The double-stranded β-helix fold consists of two 4-stranded antiparallel β-sheets (in blue and green) with 6 β-arch and 1 β-hairpin connections. Pairs of β-arches forming the three β-arcades are highlighted on the right. β-arch loops belonging to the same β-arcade are displayed with the same color throughout the figure (β-arcades 1, 2 and 3 in red, orange and magenta, respectively). b, Topology diagram of a designed double-stranded β-helix with all β-strand pairs in register. The Cα-traces of the first and second β-sheets are colored in blue and green, respectively. Sidechain Cβ positions oriented toward the inner and outer faces of the β-helix are represented with up and down black arrows with rounded tips, respectively. β-arch loops are colored as in panel a. c, Definition of β-arcade register shift varied during conformational sampling. The β-arcade register shift (between β-arcades 1 and 3) is determined by the register of β-strand pairs S3/S8 and S4/S7, and the lengths of β-strands S3, S4, S8 and S7 (Methods). In this example β-strand pairs S3/S8 and S4/S7 each have a two residue register shift, resulting in an overall β-arcade register shift of 4 residues. Loops are omitted to facilitate visualization. d, Example of a design model with all β-strand pairs in register forming a sandwich-like structure. e, Example of a design model with register shifts between β-arcades 1 and 3 (magenta and red) forming a barrel-like structure.
The analysis from Fig. 1 leads to strong constraints on the construction of β-sheet backbone structures, as the sidechain directionality patterns of the β-strands and loops are coupled in several ways. First, the directionality patterns of the loops preceding and following each β-strand are coupled to the length of the strand (Fig. 2b): for example, a β-strand with an even number of residues that is preceded by a “↑↑” loop must be followed by a “↓↑” or a “↓↓” loop, but not a “↑↑” or “↑↓” loop, due to the alternating pleating of β-strands. Second, since the β-arcades of the β-helix have paired β-strands and β-arch loops, the sidechains adjacent to one β-arch loop must have the same orientation as the paired sidechains adjacent to the second β-arch loop (Fig. 1d). Due to the antiparallel orientation of the β-arcades, “↓↓” and “↑↑” loops are compatible with loops of the same type, but “↑↓” loops are only compatible with “↓↑” (Fig. 1d). Third, the twist and curvature of the two β-sheets of the β-helix is constrained by the hydrogen bonding register between β-arcades 1 and 3 (herein called β-arcade register), and within β-strand pairs S3/S8 and S4/S7, as shown in Fig. 2c.
De novo design of protein structures
We constructed double-stranded β-helix protein backbones by Monte Carlo fragment assembly using blueprints – representations of the target protein topologies specifying the ordering, lengths and backbone torsion bins of secondary structure elements and loop connections5 – in conjunction with backbone hydrogen-bonding constraints specifying all pairings between β-strands. We explored strand lengths between 5 and 7 residues and the most commonly observed β-arch loops between 3 and 5 residues (Fig. 1c). The central β-hairpin was designed with two-residue loops following the ββ-rule5. The register shifts between pairs of β-strands from different β-arcades (1 and 3) were allowed to range from 0 to 2 and the β-arcade register shifts between 0 and 4; strand pairs within the same β-arcade were kept in-register. A total of 3,673 combinations were enumerated, of which 1,853 had mutually compatible strand lengths and loop types consistent with the constraints summarized in the previous paragraph. For each of these internally consistent blueprints, we used Rosetta to build thousands of protein backbones. The resulting ensemble of backbone structures has considerable structural diversity; those with all strands in-register had narrow sandwich-like structures (Fig. 2d), while those with large register shifts had wider barrel-like structures (Fig. 2e).
For each generated backbone, we carried out flexible-sequence design calculations18,19 to identify low-energy amino acid identities and sidechain conformations providing close complementary packing, sidechain-backbone hydrogen bonding in β-arch loops – to pre-organize their conformation and facilitate folding – and high sequence-structure compatibility. We favored inward-pointing charged or polar amino acids at the four edge strands to minimize aggregation propensity20. Loop sequences were designed with consensus profiles obtained from fragments with the same backbone ABEGO torsion bins21. Because the very large size of the space sampled by our design procedure limits convergence on optimal sequence-structure pairs, we carried out a second round of calculations starting from the blueprints yielding the lowest energy designs, intensifying sampling at both the backbone and sequence level. For a subset of designs, we introduced disulfide bonds between paired β-strand positions with high sequence separation (e.g. between the first and last β-strands) and optimal orientation (see Methods) – disulfide bonds distant in primary sequence decrease the entropy of the unfolded state and therefore enhance the thermodynamic stability of the native state. To assess compatibility of the top ranked designed sequences with their structures we characterized their folding energy landscape with biased forward folding simulations21, and those with substantial near-native sampling were subsequently assessed by Rosetta ab initio structure prediction calculations22,23. Designs with funnel-shaped energy landscapes – where the designed structure is at the global energy minima and has a substantial energy gap with respect to alternative conformations – were selected for experimental characterization. Ab initio structure prediction of natural β-sheet proteins tends to oversample local contacts24,25 (i.e. favoring β-hairpins over β-arches), but we succeeded in designing sequences with the β-arches sufficiently strongly encoded that they folded in silico to near the designed target structure.
Experimental characterization
We chose for experimental characterization 19 designs with funnel-shaped energy landscapes ranging between 70 and 94 amino acids (Supplementary Table 1). BLAST searches26,27 indicated that the designed sequences had little or no similarity with native proteins (lowest E-values ranging from 0.003 to > 10; Supplementary Table 2). Synthetic genes encoding the designs (design names are BH_n; where “BH” stands for β-helix and “n” the design number; and a “_ss” suffix if disulfide bonds are present) were obtained, the proteins were expressed in Escherichia coli, and purified by affinity chromatography. 16 of the designs expressed well and were soluble, and two (BH_10 and BH_11) were monomeric (Supplementary Fig. 2) by size-exclusion chromatography coupled with multi-angle light scattering (SEC-MALS) (most of the non-monomeric designs were either dimers or soluble aggregates). Both monomeric designs had far-ultraviolet circular dichroism spectrum (CD) at 25°C characteristic of β proteins, a melting temperature (Tm) above 95°C, and well-ordered structures according to two-dimensional 1H-15N heteronuclear single quantum coherence (HSQC) spectra (Fig. 3a-c and Supplementary Fig. 3). For both designs, the number of NMR peaks matched the number of expected amide resonances based on the protein sequence, but the higher stability of BH_10 in the conditions of the NMR experiments made it a better candidate for NMR structure determination.
Fig. 3. NMR structure of BH_10 is nearly identical to design model.
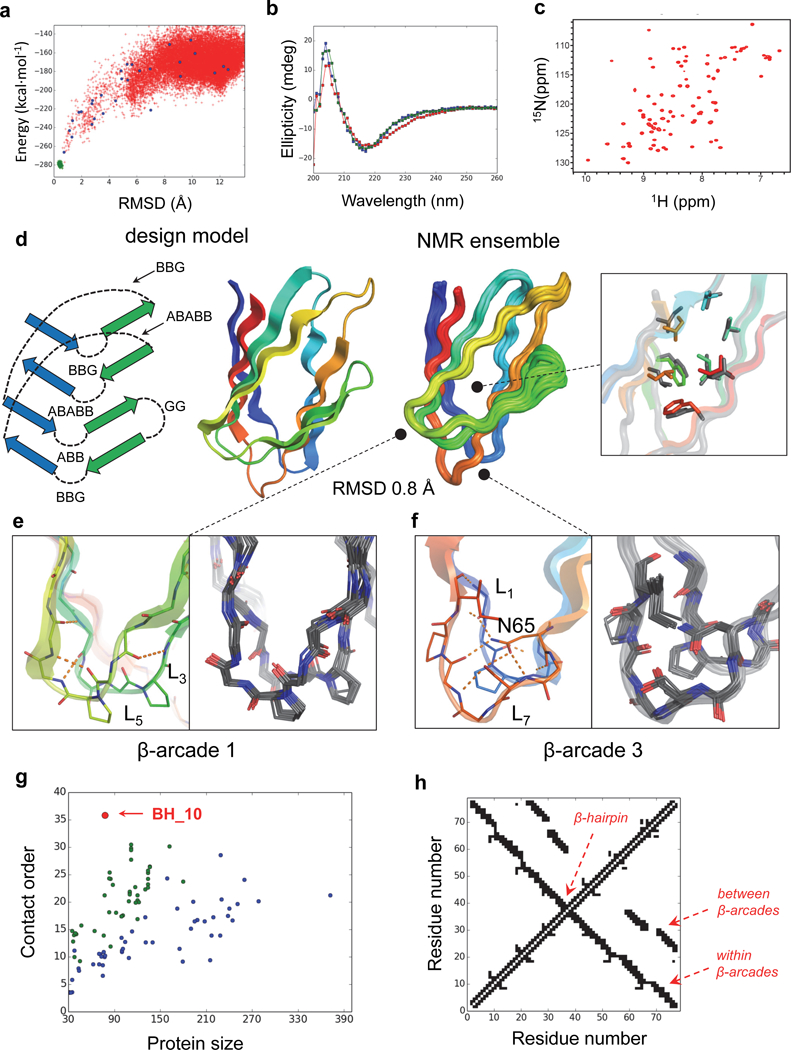
a, Calculated BH_10 folding energy landscape. Each dot represents the lowest energy structure obtained from ab initio folding trajectories starting from an extended chain (red dots), biased forward folding trajectories (blue dots) or local relaxation of the designed structure (green dots); x-axis is the Cα-root mean squared deviation (RMSD) from the designed model; the y-axis, the Rosetta all-atom energy. b, Far-ultraviolet circular dichroism spectra (blue: 25 °C, red: 95 °C, green: 25 °C after cooling). c, 1H-15N HSQC spectra obtained at 37 °C at a 1H field of 800 MHz. d, NMR structure in comparison with the design model. Right inset shows comparison of core side chain rotamers (NMR structure in grey and design in rainbow). The topology scheme of the design model is shown on the left, describing ABEGO torsion bins of all loop connections. Atomic coordinates for design model are in Supplementary Data Set 1. e, Backbone hydrogen bonding of β-arcade 1 is well preserved across the NMR ensemble. f, Sidechain interactions of N65 with backbone and sidechains form a hydrogen-bonded network in β-arcade 3 that is well recapitulated in the NMR ensemble. g, Contact order of de novo protein domains computationally designed to date confirmed by high resolution structure determination; all-α (blue), αβ (green) and all-β (red). BH_10 stands out with a contact order of 35.8 for a chain length of 78 residues. The domains are listed in Supplementary Table 4 and 5. h, Contact map illustrating the large sequence separation of the contacts present in the BH_10 topology.
The two monomeric designs with well-ordered structures were among those with better packed cores and a larger proportion of β-arch loops containing prolines and hydrogen bonding satisfying the backbone polar atoms (Supplementary Table 3). β-arch loops that are structurally pre-organized with the polar groups making internal hydrogen bonding likely favor folding to the correct topology and contribute to stability by compensating for the loss of interactions with water of polar groups in the sidechains and backbone. These interactions could also disfavor the competing local strand pairing arrangement in which the two strands form a β-hairpin – this is a very common pathology in ab initio structure prediction25. For the most stable dimeric design (BH_6) we introduced disulfide bonds to stabilize protein regions having contacts with large sequence separation – e.g. between the N- and C-terminal strands – but this did not succeed in yielding stable monomers. Addition of an α-helix to the C-termini (one of the two extremes of the β-helix) as a capping domain protecting the strand edges from inter-molecular pairing also failed to yield stable monomers, even in combination with disulfide bonds. This suggests that the sequence of the core β-sheet must strongly encode its structure independent of disulfide bonds or protecting domains aimed at increasing stability.
NMR structure of a de novo designed β-helix
We succeeded in solving the structure of BH_10 by 4D NMR spectroscopy (Fig. 3d, Table 1 and Supplementary Fig. 4) – using the 4D-CHAINS/AutoNOE-Rosetta automated pipeline for resonance assignments and structure calculation28 – and found it to be in very close agreement with the computational model (Cα-RMSD 0.84 Å, averaged over 10 NMR models). The overall topology is accurately recapitulated, including all strand pairings, register shifts and loop connections, as supported by 132 long-range nuclear Overhouser effects (NOEs) between backbone amide and sidechain protons (Supplementary Fig. 5). The designed aliphatic and aromatic sidechain packing in the protein core as well as salt bridge interactions across the two β-sheet surfaces were also accurately reproduced – three salt bridges between the two paired β-arcades and one within the third β-arcade are well supported by the observed NOEs (Supplementary Fig. 6). The agreement both in the backbone conformation and hydrogen-bonding interactions of the loops forming the three β-arcades is remarkable, given that these elements are the most flexible parts of the structure and therefore difficult to design due to sampling bottlenecks. The β-arcades were designed with pairs of β-arch loops that mutually interact via backbone-backbone hydrogen bonds – due to the complementarity between their backbone conformations – stabilizing loop pairing and avoiding burial of polar backbone atoms (see Supplementary Fig. 7 for the BH_10 loop sequences and sidechain patterns). For example, β-arcade 1 is formed by ‘BBG’ and ‘ABB’ loops, and the buried backbone NH group of the ‘G’ position in the former makes a hydrogen bond with the buried backbone C=O of the neighboring loop (Fig. 3e). The other two β-arcades were designed with one β-arch loop containing buried and fully hydrogen-bonded asparagines (4 hydrogen bonds in total) that stabilize both loop pairing and the local β-arch conformation (of ‘ABABB’ loops). By design, the asparagine sidechain geometry was further stabilized with hydrophobic stacking interactions from the two β-arch loops of the same arcade. The high degree of convergence of the designed rotamer in the NMR ensemble illustrates the high structural pre-organization of this particular motif (Fig. 3F).
Table 1.
NMR and refinement statistics
| BH_10 (PDB 6E5C) |
|
|---|---|
| NMR distance and dihedral constraints | |
| Distance constraints | |
| Total NOE | 659 |
| Intraresidue | 272 |
| Inter-residue | 387 |
| Sequential (|i – j| = 1) | 222 |
| Medium range (2 ≤ |i – j| ≤ 4) | 33 |
| Long range (|i – j| ≥ 5) | 132 |
| Intermolecular | 0 |
| Hydrogen bonds | 0 |
| Total dihedral-angle restraints | 156 |
| φ | 78 |
| ψ | 78 |
| Structure statistics | |
| Violations (mean ± s.d.) | |
| Distance constraints (Å)a | 0.30 ± 0.46 |
| Dihedral-angle constraints (°)b | 9.30 ± 2.49 |
| Max. dihedral-angle violation (°)b | 47.59 |
| Max. distance-constraint violation (Å)a | 1.32 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 0.00 ± 0.00 |
| Bond angles (°) | 0.00 ± 0.00 |
| Impropers (°) | 0.00 ± 0.00 |
| Average pairwise r.m.s. deviation (Å)c | |
| Heavy | 0.61 ± 0.13 |
| Backbone | 0.51 ± 0.11 |
Distance constraint violations in the structural ensemble were calculated using 7 Å universal upper distance bound for the NOE restraints assigned by AutoNOE-Rosetta.
Dihedral angle restraints were derived from TALOS-N. The violations were calculated for the core secondary structural regions of the ten-lowest energy models using 15° cut-off beyond TALOS-N predicted dihedral angles.
Pairwise r.m.s.d. was calculated among 10 refined models for a core secondary structural region defined by the residues 2–8, 11–18, 21–28, 32–36, 39–43, 46–53, 59–65, and 71–75.
The amino acid sequence of BH_10 is unrelated to any sequence in the NCBI nr database (BLAST found one hit with insignificant sequence similarity; E-value 6.3). We searched the PDB for similarities in structure (using the Dali server29 with the lowest energy NMR model as the query structure) or sequence (with HHpred30 for sensitive profile based sequence search), and identified matches similar in fold but containing additional and irregular secondary structures, and longer loops. These matches are all homodimers with sheet-to-sheet interface packing (Supplementary Fig. 8) or domains integrated in larger structures, in sharp contrast to the BH_10 monomer.
Contact order and sequence determinants of the BH_10 fold
The non-local character of BH_10 is of particular note – a large fraction of the contacting residues are distant along the linear sequence, with extensive strand pairing between the N and C-terminal β-strands. The contact order of the structure – the average separation along the linear sequence of residues in contact in the three dimensional structure – is higher than any previous single-domain protein designed de novo (Fig. 3g-h). High contact order proteins fold more slowly than low contact order proteins as there is a greater loss in chain entropy for forming the first native interactions, and they tend to form long-lived non-native structures that can oligomerize or aggregate31. We have overcome the challenges in designing non-local structures by focusing on backbones lacking internal strain and having maximal internal coherence, and programming β-strand orientation with highly structured loops.
One of the challenges in achieving high contact order through β-arches is to disfavor competing more sequence-local β-hairpins. To evaluate in silico how each of our design features contribute to favoring β-arches over β-hairpins, we generated folding energy landscapes for a series of mutants of BH_10 that disrupt, one at a time, loop hydrogen bonding, sidechain packing of loop neighbors and loop local geometry. For all conformations generated, we classified all the β-strand connections as β-arch or β-hairpin depending on strand pairing formation, and calculated the overall frequency of β-hairpin formation for each pair of consecutive β-strands. As shown in Supplementary Fig. 9, disruption of packing within or between β-arch loops, removal of sidechain-backbone hydrogen bonding interactions and reducing loop geometry encoding by eliminating prolines all increase sampling of competing β-hairpin conformations, and thus substantially decrease sampling of β-arches and the target designed structure.
DISCUSSION
The design of all-β globular proteins from first principles has remained elusive for two decades of protein design research. We have successfully designed a double-stranded β-helix de novo, as confirmed by the NMR structure of the design BH_10, based on a series of rules describing the geometry of β-arch loops and their interactions in more complex β-arcades. Our work also achieves two related milestones: the first accurate design of an all-β globular protein with exposed β-sheet edges, and the most non-local structure yet designed from scratch. Comparison between successful and failed designs suggests folding and stabilization of the monomeric structure (and implicitly, disfavoring of competing topologies with more local strand pairings) is bolstered by loops containing sidechain-backbone and backbone-backbone hydrogen bonds together with well-packed mixed aliphatic/aromatic sidechains in the protein core, inward-pointing polar amino acids at strand edges and salt bridges between paired strands. Previous design studies on β-propellers11 or parallel β-helices12 have used naturally occurring backbone structures and consensus sequence information on the target fold families; this approach while powerful sheds less light on the key principles underlying β-sheet structure construction and does not allow the programming of new backbone geometries. The β-helix fold here designed is well suited for incorporating metal, ligand-binding and active sites, as illustrated by the broad functional diversity of cupin protein domains, which are the closest naturally-occurring structural analogs. With the basic design principles now understood, our de novo design strategy should enable the construction of a wide range of β-helix structures tailored to a broad diversity of target ligands.
Initial advances in protein design were algorithms which allowed rapid identification of a very low energy sequence for a given backbone structure. In recent years, progress has come from the realization that the requirements of burying hydrophobic residues in a core away from solvent, while avoiding the burial of backbone polar groups without compensating hydrogen bonds, together with torsional restrictions on the peptide backbone considerably constrain overall globular protein backbone geometry, particularly for β-sheet containing proteins: it is much harder than originally expected to construct new backbones that have these properties. The de novo design of β-sheet containing proteins advanced considerably following the elucidation of β-sheet design principles for construction of backbones meeting the above constraints while having desired geometries: for example, principles for controlling the chirality of β-hairpins5, reducing strain in β-strands with glycine kinks10, and combining β-bulges and register shifts to curve β-sheets21. The design rules described here are a considerable further advance as they provide control over β-arch connections between distinct β-sheets, and should enable the design of a broad range of β-protein families beyond the β-barrel and β-helix with considerable medical and biotechnological potential; for example the immunoglobulin fold widely utilized for binding and loop scaffolding in nature is topologically very similar to the double-stranded β-helices designed here, with a larger proportion of β-hairpins over β-arches.
METHODS
Loop analysis.
Loop connections between β-strands were collected from a non-redundant database of PDB structures obtained from the PISCES server32 with sequence identity <30% and resolution ≤2 Å. We discarded those loops connecting β-strands with hydrogen bonded pairing (β-hairpins), and the remaining 5,061 β-arch loops were subsequently analyzed. The ABEGO torsion bins of each residue position were assigned based on the definition shown in Supplementary Fig. 1, and the sidechain directionality pattern of neighboring residues was defined according to Fig. 1A. The secondary structure of all residue positions was assigned with DSSP33 and the last β-strand residue preceding and the first β-strand residue following the β-arch loop were chosen as the critical neighboring residues determining the sidechain pattern of the loop. The loop bending was defined as the angle between the loop center of mass and the two strand positions adjacent to the loop. Those loops with bending angles larger than 120 degrees were discarded from the analysis to correctly identify those loops producing a substantial change in the direction of the two connected β-strands. The loop dataset is available in Supplementary Data Set 2.
Backbone generation.
We used the Blueprint Builder mover5 of RosettaScripts34 to build protein backbones by Monte Carlo fragment assembly using 9- and 3-residue fragments compatible with the target secondary structure and torsion bins (ABEGO), as specified in the blueprints of every target topology. We used a poly-valine centroid representation of the protein and a scoring function accounting for backbone hydrogen bonding, Van der Waals interactions (namely to avoid steric clashes), planarity of the peptide bond (omega score term), and compacity of structures (radius of gyration). Thousands of independent folding trajectories are performed and subsequently filtered. Due to the non-local character of β-sheet contacts, we used distance and angle constraints to favor the correct hydrogen bonded pairing between β-strands main chain atoms. For every target topology we automatically set all pairs of residues involved in β-strand pairing to generate all constraints for backbone building. Protein backbones were filtered based on their match with the blueprint specifications (secondary structure, torsion bins and strand pairing), and subsequently ranked based on backbone hydrogen bonding energy (lr_hb score term), and the total energy obtained from one round of all-atom flexible-sequence design (see below)
Flexible sequence design.
Generated protein backbones were subjected to flexible-sequence design calculations with RosettaDesign18,19 using the Rosetta all-atom energy function “Talaris2014”35 to favor amino acid identities and side-chain conformations with low-energy and tight packing. We performed cycles of fixed backbone design followed by backbone relaxation using the FastDesign mover36 of RosettaScripts34. Designed sequences were filtered based on total energy, sidechain packing (measured with RosettaHoles37, packstat and core side-chain average degree21), sidechain-backbone hydrogen bond energy, and secondary structure prediction (match between the designed secondary structure and that predicted by Psipred38 based on the designed sequence). Amino acid identities were restricted based on the solvent accessibility of protein positions, ensuring that hydrophobic amino acids are located in the core and polars in the surface. Further restrictions were imposed to improve sequence-structure compatibility in loop regions. Sequence profiles were obtained for naturally occurring loops with the same ABEGO string sequence, as done previously21.
For those blueprints that yielded the lowest energy designs we performed a second round with ten times more backbone samples. Backbones generated in this second round were subjected to more exhaustive sequence design by running multiple Generic Monte Carlo trajectories optimizing total energy and sidechain average degree simultaneously, and then applied all filters described above.
Design of disulfide bonds and helix capping domain.
We used the Disulfidize mover of RosettaScripts34 to identify pairs of residue positions able to form disulfide bonds with a good scoring geometry. We searched for disulfide bonds between residues distant in primary sequence and with a disulfide score < −1.0. We designed a C-terminal helix capping domain (followed with a β-strand pairing with the first β-strand) using the backbone generation protocol described above but starting from design BH_6. The structure of BH_6 was kept fixed during fragment assembly and the C-terminal domain was generated. Then sequence design was performed for the C-terminal domain and those neighboring residues within 10 Å.
Sequence-structure compatibility.
For assessing the local compatibility between designed sequences and structures we picked 200 naturally occurring fragments (9- and 3-mers) with sequences similar to the design and evaluated the structural similarity (by RMSD) between the ensemble of picked fragments and the local designed structure. Those with overall low RMSD fragments, and therefore with high fragment quality, were subsequently assessed by Rosetta folding simulations using the Rosetta energy function “ref2015”39. First, biased forward folding simulations21 (using the three-lowest RMSD fragments and 40 folding trajectories) were used to quickly identify those designs more likely to have funnel-shaped energy landscapes. Those designs achieving near-native sampling (RMSD to target structure below 1.5 Å) were then assessed by standard Rosetta ab initio structure prediction22,23.
To evaluate the amount of β-hairpin sampling in each loop connection during ab initio structure prediction we first detected all strand pairings formed in each generated decoy and then mapped the residues involved in those strand pairings to the secondary structure elements of the designed structure. After secondary structure mapping, pairings between strands consecutive in the sequence were counted as β-hairpins. The total count of β-hairpins sampled in each loop over the total number of generated decoys is a relative quantity of hairpin sampling that allowed to compare the β-hairpin propensity of different loops and mutants, as shown in Supplementary Fig. 8.
Contact order.
To evaluate the non-local character of protein structures we computed contact order as the average sequence separation between pairs of Cα atoms within a distance of 8 Å and with a sequence separation of 3 residues at least.
Protein expression and purification.
Genes encoding the designed sequences were obtained from Genscript and cloned into the pET-28b+ (with N-terminal 6×His tag and a thrombin cleavage site) expression vectors. Plasmids were transformed into Escherichia coli BL21 Star (DE3) competent cells, and starter cultures were grown at 37°C in Luria-Bertani (LB) medium overnight with kanamycin. Overnight cultures were used to inoculate 500 ml of LB medium supplemented with antibiotic and cells were grown at 37 °C and 225 r.p.m until an optical density (OD600) of 0.5–0.7 was reached. Protein expression was induced with 1mM of isopropyl β-D-thiogalactopyranoside (IPTG) at 18 °C and, after overnight expression, cells were collected by centrifugation (at 4 °C and 4400 r.p.m for 10 minutes) and resuspended in 25 ml of lysis buffer (20 mM imidazole and phosphate buffered saline, PBS). Resuspended cells were lysed in the presence of lysozyme, DNAse and protease inhibitors. Lysates were centrifuged at 4 °C and 18,000 r.p.m. for 30 minutes; and the supernatant was loaded to a nickel affinity gravity column pre-equilibrated in lysis buffer. The column was washed with three column volumes of PBS+30 mM imidazole and the purified protein was eluted with three column volumes of PBS+250 mM imidazole. The eluted protein solution was dialyzed against PBS buffer overnight. The expression of purified proteins was assessed by SDS-polyacrylamide gel electrophoresis and mass spectrometry; and protein concentrations were determined from the absorbance at 280 nm measured on a NanoDrop spectrophotometer (ThermoScientific) with extinction coefficients predicted from the amino acid sequences using the ProtParam tool (https://web.expasy.org/protparam/). Proteins were further purified by FPLC size-exclusion chromatography using a Superdex 75 10/300 GL (GE Healthcare) column.
Circular dichroism (CD).
Far-ultraviolet CD measurements were carried out with the AVIV 420 spectrometer. Wavelength scans were measured from 260 to 195 nm at temperatures between 25 and 95 °C, using a 1 mm path-length cuvette. Protein samples were prepared in PBS buffer (pH 7.4) at a concentration of 0.2–0.4 mg/mL.
Size exclusion chromatography combined with multiple angle light scattering (SEC-MALS).
SEC-MALS experiments were performed using a Superdex 75 10/300 GL (GE Healthcare) column combined with a miniDAWN TREOS multi-angle static light scattering detector and an Optilab T-rEX refractometer (Wyatt Technology). One hundred microliter protein samples of 1–3 mg/ml were injected to the column equilibrated with PBS (pH 7.4) or TBS (pH 8.0) buffer at a flow rate of 0.5 ml/min. The collected data was analyzed with ASTRA software (Wyatt Technology) to estimate the molecular weight of the eluted species.
Protein expression of isotopically labeled proteins for NMR structure determination.
Plasmids were transformed using standard heat shock transformation into Lemo21 expression strain of E. coli (NEB) and plated onto a minimal M9 media containing glucose and kanamycin to maintain tight control over expression. A single colony was selected, inoculated into 50 mL of Luria Broth containing 50 ug/mL of kanamycin and grown at 37°C with shaking overnight. After approximately 18 hours, the 50 mL starter culture was removed and 25 mL was used to inoculate 500mL of Terrific Broth containing 50 ug/mL kanamycin and mixed mineral salts40. The Terrific Broth (TB) culture was grown at 37°C with shaking at 250 rpm until OD600 reached a value of 1.0. At this time the culture was removed and the cells were pelleted by centrifugation at 4000 rpm for 15 minutes. The TB broth was removed and the pelleted cells were resuspended gently with 50 mL of 20 mM NaPO4 150 mM NaCl pH 7.5. The resuspended cells were transferred into minimal labeling media, containing N15 labelled Ammonium Chloride at 50mM and C13 glucose to 0.25% (w/v), as well as trace metals, 25 mM Na2HPO4, 25 mM KH2PO4, and 5 mM Na2SO4. The culture was returned to 37°C, at 250 rpm for 1 hour in order to replace unlabelled Nitrogen and Carbon with labelled Nitrogen and Carbon. After 1 hour, IPTG was added to 1mM, the temperature was reduced to 25°C and the culture allowed to express overnight. The following morning the culture was removed and the cells were pelleted by centrifugation at 4000 rpm for 15 minutes. The cells were resuspended with 40 mL of Lysis Buffer (20 mM Tris 250 mM NaCl 0.25% Chaps pH 8) and lysed with a Microfluidics M110P Microfluidizer at 18000 psi. The lysed cells were clarified using centrifugation at 24000×g for 30 minutes. The labelled protein in the soluble fraction was purified using Immobilized Metal Affinity Chromatography (IMAC) using standard methods (QIagen Ni-NTA resin). The purified protein was then concentrated to 2 mL and purified by FPLC size-exclusion chromatography using a Superdex 75 10/300 GL (GE Healthcare) column into 20 mM NaPO4 150 mM NaCl pH 7.5. The efficiency of labelling was confirmed using mass spectrometry.
1H-15N heteronuclear single quantum coherence spectra (HSQC).
0.81 mM BH_10 and 0.64 mM BH_11 were exhaustively buffer exchanged into NMR buffer (50 mM NaCl, 20 mM sodium phosphate pH 6.5, 0.01% (v/v) NaN3, 4 mM EDTA and 1 U Roche protease inhibitor cocktail) in 95% H2O/5% D2O. Two-dimensional 1H-15N HSQC experiments were acquired at 37ºC with 4 scans, acquisition times of 72 ms (15N) in the indirect dimension and recycle delay of 2 s.
Chemical shift assignment.
For chemical shift assignment of BH_10, a set of two non-uniformly sampled (NUS) 4D NMR experiments, a 4D HC(CC-TOCSY(CO))NH and 4D 13C,15N edited HMQC-NOESY-HSQC, were acquired at 800 MHz at 37ºC as previously described28. For the 4D HC(CC-TOCSY(CO))NH experiment, spectra widths were set to 12,500 (acquisition dimension) × 2100 (15N) × 8000 (13C) × 7300 (1H) Hz and acquisition times in the indirect dimensions of 60 ms (15N), 8 ms (13C) and 8 ms (1H) using 16 scans and a recycle delay of 1 s. Spectra were acquired with 2000 hypercomplex NUS points distributed over the indirectly detected dimensions (0.38% sparsity). For the 4D 13C,15N edited HMQC-NOESY-HSQC, spectra widths were set to 12,500 (acquisition dimension) × 1000 (15N) × 8000 (13C) × 10,000 (1H) Hz, respectively and acquisition times in the indirect dimensions of 38 ms (15N), 10 ms (13C) and 20 ms (1H) using 8 scans, a recycle delay of 1 s and a NOESY mixing time of 120 ms. Spectra were acquired with 4000 hypercomplex NUS points distributed over the indirectly detected dimensions (0.32% sparsity). 4D NUS spectra were processed in NMRPipe41 using SMILE reconstruction42 and analyzed using NMRFAM-SPARKY43. For every 1H-15N HSQC peak the corresponding planes in 4D-HCNH TOCSY and 4D-HCNH NOESY spectra were inspected and peaks were picked manually. The 4D peaklists were used as input for the 4D-CHAINS algorithm28 to obtain sequence specific resonance assignments of backbone and sidechain atoms automatically. The overall assignment completeness reached 92%. No aromatic resonances were assigned. 4D-CHAINS assignments together with the 4D-HCNH NOESY peaklist were used in AutoNOE-Rosetta for structure determination.
NOE assignment and structure determination using AutoNOE-Rosetta.
To determine the structural models of the BH_10 target protein, we used CS-Rosetta44 that provides AutoNOE-Rosetta45 and RASREC-Rosetta46 protocols. AutoNOE-Rosetta is an iterative NOE assignment method, that utilizes RASREC-Rosetta to model protein structures de novo. These methods make use of valuable information contained within NMR chemical shifts about secondary and tertiary structures, and dynamics of proteins to model targets of interest accurately44,47. The primary aim of AutoNOE-Rosetta is to label proton atoms to the unassigned NOESY cross-peaks by mapping their resonance frequencies to the assigned chemical shift frequencies. The resulting assignments can be utilized to create NOE-based distance restraints that aid the structure calculation process. The method begins by creating an initial mapping between assigned chemical shift list and unassigned NOESY cross-peak list. This mapping produces ambiguous assignments due to possible noise in the NOESY spectra48,49. These assignments undergo evaluation and filtering. The evaluation criteria rely on the symmetry of cross-peaks, chemical shift compatibility, intermediate structural model compatibility (in the subsequent stages of the protocol), and the participation of any NOE in a network of NOEs (network anchoring)50. The cross-peaks are eliminated if they lie along the diagonal in the NOESY spectra or their contribution to some of the evaluation criteria (such as network anchor score) is insignificant. The intensities of high-scoring NOE peaks are calibrated to produce distance restraints. These restraints are used to calculate structures within the highly parallel RASREC-Rosetta, which performs fragment assembly44 using Monte Carlo methods and additional optimized algorithms46. This process of assigning NOEs and calculating structures is carried out iteratively across eight distinct stages. The final stage retains well-converged structural models alongside generated NOE restraints used for their calculation.
The process of setting up AutoNOE-Rosetta calculations is highly automated and accessible via Python interface within the toolbox available at the CS-Rosetta website (https://csrosetta.chemistry.ucsc.edu). Prior to setting up NOE assignment and structure calculation runs, (i) NMR chemical shifts and target sequence are used to predict secondary structure (rigid regions) and flexible end regions from TALOS-N51, (ii) the flexible end regions are trimmed from sequence and chemical shift files since they deteriorate the performance of structure determination methods by inducing large number of degrees of freedom, and (iii) the predicted secondary structure together with trimmed chemical shift files are used to select 200 structural fragments of amino acid lengths three and nine, for each position in the target sequence. Upon completion of previous steps, AutoNOE-Rosetta calculations are setup with target sequence, structural fragments, chemical shifts, and unassigned NOESY cross-peak lists. For the BH_10 target protein, we obtained NMR chemical shifts from 4D-CHAINS28 and additionally utilized amide to aliphatic (HCNH) unassigned NOESY cross-peak list. Thereafter, we performed four rounds of AutoNOE-Rosetta calculations, where each round was supplied with a different restraint weight (standard restraint weights of 5, 10, 25 and 50 were used). All the calculation runs were evaluated using a function that assesses all-atom energies (“ref2015”39) of the structural models and their convergence. After selecting the best-scoring restraint weight run, ten models that exhibit lowest energy within this run were selected. Commands to setup the calculations were used exactly as provided in the Supplementary Methods of a previous work28. Molprobity52 was used to compute Ramachandran statistics for the ten-lowest energy structural models (100% of residues in favored regions of Ramachandran space, and 99% in favored regions) and deviations from the ideal geometry (Table 1).
Salt bridges.
We used ESBRI53 to predict salt-bridges in the ten lowest-energy structural models. Out of 19 salt-bridges predicted using ESBRI, AutoNOE-Rosetta recovers four salt-bridges in the form of NOE contacts on the surface of the BH_10 protein. To identify salt-bridges, we examined the NOE restraints assigned by AutoNOE-Rosetta, between negatively charged glutamic acid or aspartic acid and positively charged arginine or lysine. We further filtered the restraints based on the upper distance bound of 4 Å in the ten lowest-energy structures. From these filters, we found that the salt-bridges recapitulated by the NOE assignment module between the residue pairs are: (15, 62), (23, 78), (33,64) and (35, 62).
Hydrophobic core of BH_10.
Buried residues were selected from the ten lowest-energy structural models using a 10 Å2 solvent accessible surface area (SASA) threshold. There are 18 buried residues that contribute up to 70% of the total NOEs assigned by AutoNOE-Rosetta, and two of them are aromatic residues (F34 and F73). While 4D-CHAINS does not assign chemical shifts of sidechain groups of aromatic residues (specifically aromatic rings), it provides respective chemical shift assignments of backbone atoms (Cα, Hα, N, H) and the β-carbon and -proton (Cβ, Hβ) atoms. AutoNOE-Rosetta assigned a total of nine NOEs for the aromatic residues in the hydrophobic core, and the placement of aromatic sidechains was optimized by the Rosetta’s packer algorithm. Upon close examination of the BH_10 structures, we found that the geometry of the two aromatic sidechains was constrained by neighboring residues with methyl groups placed based on NOEs; supporting the accuracy of the aromatic sidechain placement.
Visualization of protein structures and image rendering.
Images of protein structures were created with PyMOL54.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Rettie for mass spectrometry assistance and the rest of Baker lab members for discussion. We acknowledge computing resources provided by the Hyak supercomputer system funded by the STF at University of Washington, and Rosetta@Home volunteers in ab initio structure prediction calculations. Work carried out at the Baker laboratory was supported by the Howard Hughes Medical Institute, Open Philanthropy, and the Defense Threat Reduction Agency. E.M. was supported by a Marie Curie International Outgoing Fellowship (FP7-PEOPLE-2011-IOF 298976). G.O. was supported by a Marie Curie International Outgoing Fellowship (FP7-PEOPLE-2012-IOF 332094). This research was financially supported by Ministry of Education, Youths and Sports of the Czech Republic within CEITEC 2020 (LQ1601) project, and from Grant Agency of Masaryk University (GAMU) to K.T. This research was supported by an R35 Outstanding Investigator Award to N.G.S. through NIGMS (1R35GM125034–01), and by the Office of the Director, NIH, under High End Instrumentation (HIE) Grant S10OD018455, which funded the 800 MHz NMR spectrometer at UCSC.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
DATA AVAILABILITY
NMR chemical shifts and NOESY cross-peak lists used to determine structures of BH_10 have been deposited in the BMRB with accession code 30495. Coordinates of ten lowest-energy structures and the restraint lists have been deposited in the wwPDB as PDB 6E5C. The design model of BH_10 is available as Supplementary Data Set 1, and the loop dataset used to analyze the sidechain patterns of naturally occurring β-arches in Supplementary Data Set 2. Other data are available from the corresponding authors upon request.
CODE AVAILABILITY
The Rosetta macromolecular modelling suite (http://www.rosettacommons.org) is freely available to academic and non-commercial users. Scripts and protocols used in this article for generating protein backbone blueprints, performing Rosetta design calculations and analyzing protein structures are all available on github (https://github.com/emarcos/beta_sheet).
REFERENCES
- 1.Kortemme T, Ramírez-Alvarado M & Serrano L Design of a 20-amino acid, three-stranded beta-sheet protein. Science 281, 253–256 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Searle MS & Ciani B Design of beta-sheet systems for understanding the thermodynamics and kinetics of protein folding. Curr. Opin. Struct. Biol. 14, 458–464 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Hughes RM & Waters ML Model systems for beta-hairpins and beta-sheets. Curr. Opin. Struct. Biol. 16, 514–524 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Marcos E & Adriano-Silva D Essentials of de novo protein design: Methods and applications. WIREs Comput Mol Sci e1374 (2018). [Google Scholar]
- 5.Koga N et al. Principles for designing ideal protein structures. Nature 491, 222–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hecht MH De novo design of beta-sheet proteins. Proceedings of the National Academy of Sciences 91, 8729–8730 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plaxco KW, Simons KT & Baker D Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 277, 985–994 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Quinn TP, Tweedy NB, Williams RW, Richardson JS & Richardson DC Betadoublet: de novo design, synthesis, and characterization of a beta-sandwich protein. Proc. Natl. Acad. Sci. U. S. A. 91, 8747–8751 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nanda V et al. De novo design of a redox-active minimal rubredoxin mimic. J. Am. Chem. Soc. 127, 5804–5805 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Dou J et al. De novo design of a fluorescence-activating β-barrel. Nature (2018). doi: 10.1038/s41586-018-0509-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voet ARD et al. Computational design of a self-assembling symmetrical β-propeller protein. Proceedings of the National Academy of Sciences 111, 15102–15107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacDonald JT et al. Synthetic beta-solenoid proteins with the fragment-free computational design of a beta-hairpin extension. Proceedings of the National Academy of Sciences 113, 10346–10351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ottesen JJ & Imperiali B. Design of a discretely folded mini-protein motif with predominantly beta-structure. Nat. Struct. Biol. 8, 535–539 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Hu X, Wang H, Ke H & Kuhlman B Computer-based redesign of a beta sandwich protein suggests that extensive negative design is not required for de novo beta sheet design. Structure 16, 1799–1805 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hennetin J, Jullian B, Steven AC & Kajava AV Standard conformations of beta-arches in beta-solenoid proteins. J. Mol. Biol. 358, 1094–1105 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Lin Y-R et al. Control over overall shape and size in de novo designed proteins. Proc. Natl. Acad. Sci. U. S. A. 112, E5478–85 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kajava AV, Baxa U & Steven AC β arcades: recurring motifs in naturally occurring and disease-related amyloid fibrils. The FASEB Journal 24, 1311–1319 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuhlman B & Baker D Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. U. S. A. 97, 10383–10388 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuhlman B et al. Design of a novel globular protein fold with atomic-level accuracy. Science 302, 1364–1368 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Richardson JS & Richardson DC Natural -sheet proteins use negative design to avoid edge-to-edge aggregation. Proceedings of the National Academy of Sciences 99, 2754–2759 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marcos E et al. Principles for designing proteins with cavities formed by curved β sheets. Science 355, 201–206 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rohl CA, Strauss CEM, Misura KMS & Baker D Protein structure prediction using Rosetta. Methods Enzymol. 383, 66–93 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Bradley P Toward High-Resolution de Novo Structure Prediction for Small Proteins. Science 309, 1868–1871 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Kuhn M, Meiler J & Baker D Strand-loop-strand motifs: prediction of hairpins and diverging turns in proteins. Proteins 54, 282–288 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Bradley P & Baker D Improved beta-protein structure prediction by multilevel optimization of nonlocal strand pairings and local backbone conformation. Proteins: Struct. Funct. Bioinf. 65, 922–929 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Altschul SF et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camacho C et al. BLAST : architecture and applications. BMC Bioinformatics 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evangelidis T et al. Automated NMR resonance assignments and structure determination using a minimal set of 4D spectra. Nat. Commun. 9, 384 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holm L. & Laakso LM Dali server update. Nucleic Acids Res. 44, W351–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zimmermann L et al. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 430, 2237–2243 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Clark P. Protein folding in the cell: reshaping the folding funnel. Trends Biochem. Sci. 29, 527–534 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Wang G. & Dunbrack RL Jr. PISCES: a protein sequence culling server. Bioinformatics 19, 1589–1591 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Kabsch W. & Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983). [DOI] [PubMed] [Google Scholar]
- 34.Fleishman SJ et al. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS One 6, e20161 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Meara MJ et al. Combined covalent-electrostatic model of hydrogen bonding improves structure prediction with Rosetta. J. Chem. Theory Comput. 11, 609–622 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhardwaj G. et al. Accurate de novo design of hyperstable constrained peptides. Nature 538, 329–335 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheffler W. & Baker D. RosettaHoles2: a volumetric packing measure for protein structure refinement and validation. Protein Sci. 19, 1991–1995 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones DT Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Alford RF et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 13, 3031–3048 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Studier FW Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Delaglio F et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 42.Ying J, Delaglio F, Torchia DA & Bax A Sparse multidimensional iterative lineshape-enhanced (SMILE) reconstruction of both non-uniformly sampled and conventional NMR data. J. Biomol. NMR 68, 101–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee W, Tonelli M & Markley JL NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nerli S, McShan AC & Sgourakis NG Chemical shift-based methods in NMR structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 106-107, 1–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lange OF Automatic NOESY assignment in CS-RASREC-Rosetta. J. Biomol. NMR 59, 147–159 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Lange OF & Baker D Resolution-adapted recombination of structural features significantly improves sampling in restraint-guided structure calculation. Proteins 80, 884–895 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berjanskii MV & Wishart DS Unraveling the meaning of chemical shifts in protein NMR. Biochim. Biophys. Acta 1865, 1564–1576 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Nilges M A calculation strategy for the structure determination of symmetric dimers by 1H NMR. Proteins 17, 297–309 (1993). [DOI] [PubMed] [Google Scholar]
- 49.Nilges M Ambiguous distance data in the calculation of NMR structures. Fold. Des. 2, S53–7 (1997). [DOI] [PubMed] [Google Scholar]
- 50.Herrmann T, Güntert P & Wüthrich K Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Shen Y & Bax A Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Costantini S, Colonna G & Facchiano AM ESBRI: a web server for evaluating salt bridges in proteins. Bioinformation 3, 137–138 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.