

Abstract
Signaling by the transforming growth factor‐β (TGF‐β) is an essential pathway regulating a variety of cellular events. TGF‐β is produced as a latent protein complex and is required to be activated before activating the receptor. The mechanical force at the cell surface is believed to be a mechanism for latent TGF‐β activation. Using β‐actin null mouse embryonic fibroblasts as a model, in which actin cytoskeleton and cell‐surface biophysical features are dramatically altered, we reveal increased TGF‐β1 activation and the upregulation of TGF‐β target genes. In β‐actin null cells, we show evidence that the enhanced TGF‐β signaling relies on the active utilization of latent TGF‐β1 in the cell culture medium. TGF‐β signaling activation contributes to the elevated reactive oxygen species production, which is likely mediated by the upregulation of Nox4. The previously observed myofibroblast phenotype of β‐actin null cells is inhibited by TGF‐β signaling inhibition, while the expression of actin cytoskeleton genes and angiogenic phenotype are not affected. Together, our study shows a scenario that the alteration of the actin cytoskeleton and the consequent changes in cellular biophysical features lead to changes in cell signaling process such as TGF‐β activation, which in turn contributes to the enhanced myofibroblast phenotype.
Keywords: mouse embryonic fibroblast (MEF), myofibroblast, NADPH oxidase 4 (Nox4), reactive oxygen species (ROS), TGF‐β, β‐actin
1. INTRODUCTION
The transforming growth factor‐β (TGF‐β) family consists of various signaling proteins with roles in development, cell differentiation, and pathophysiological processes (Boudreau, Casterline, Rada, Korzeniowska, & Leto, 2012; Cong, Iwaisako, Jiang, & Kisseleva, 2012; Gordon & Blobe, 2008; Sampson, Plas, & Berger, 2009; Zinski, Tajer, & Mullins, 2017). In canonical pathways, TGF‐β proteins signal through two classes of serine–threonine kinase receptors to phosphorylate the effector SMAD proteins, which then translocate into the nucleus and regulate transcription (Zi, Chapnick, & Liu, 2012). TGF‐β is secreted in the form of latent complexes containing latent TGF‐β‐binding proteins (LTBPs) and latency‐associated peptides. This latent complex remaining in the extracellular matrix (ECM) is inactive and needs to be processed to release active TGF‐β (Buscemi et al., 2011; Shi et al., 2011). At least two different mechanisms were reported to activate latent TGF‐β. One seems to rely on the proteases in the ECM (Jenkins, 2008; Nishimura, 2009), and the other relies on the conformational change induced by mechanical force at cell surface to liberate the active TGF‐β (Annes, Chen, Munger, & Rifkin, 2004; Buscemi et al., 2011; Wipff, Rifkin, Meister, & Hinz, 2007). The latent TGF‐β in the ECM is believed to serve as a mechanosensor molecule, which transduces mechanical cues at cell surface into intracellular signals to control cellular differentiation (Hinz, 2009).
The role of latent TGF‐β as a mechanosensor is best illustrated in wound healing process. After tissue injury, latent TGF‐β can be activated to drive tissue remodeling (Hinz, 2015; Wan et al., 2012). Specifically, TGF‐β promotes the differentiation of precursor cells into fibrogenic myofibroblasts (Amara et al., 2010; Hecker et al., 2009; Meyer‐Ter‐Vehn, Katzenberger, Han, Grehn, & Schlunck, 2008). Myofibroblasts function to secrete ECM to promote fibrosis at the wound and contract the ECM to accelerate wound closure (Darby, Laverdet, Bonté, & Desmoulière, 2014; Hecker et al., 2009). Intravenous injection of TGF‐β1 can accelerate the wound healing in rat’s tongue (El Gazaerly, Elbardisey, Eltokhy, & Teaama, 2013), while in the absence of TGF‐β1 or in the presence of TGF‐β inhibition the wound healing process is impaired (Crowe, Doetschman, & Greenhalgh, 2000; Wang et al., 2017).
TGF‐β‐induced reactive oxygen species (ROS) have been functionally implicated in different cellular processes, such as myofibroblast differentiation under fibrogenic conditions (Amara et al., 2010; Jiang, Liu, Dusting, & Chan, 2014), epithelial–mesenchymal transition (EMT; Boudreau et al., 2012; Rhyu et al., 2005), and cytoskeleton reorganization (Hu et al., 2005). Although in some studies, TGF‐β may promote ROS production in mitochondria by affecting electron transport complex activity (Casalena, Daehn, & Bottinger, 2012; Jain et al., 2013), NADPH oxidases (Nox) are responsible for TGF‐β‐induced cellular ROS in other cases (Boudreau et al., 2012; Hecker et al., 2009; Hu et al., 2005). Nox4 gene expression is found to be induced by TGF‐β and is required for TGF‐β‐mediated myofibroblast activation and fibrogenesis (Hecker et al., 2009; Sampson et al., 2009). Therefore, TGF‐β‐dependent ROS production can be a therapeutic target for pathophysiological fibrosis.
Our recent study demonstrates a direct role of endogenous β‐actin level in regulating the biophysical features at the cell surface (Xie, Deliorman, Qasaimeh, & Percipalle, 2018). We wondered whether the altered biophysical feature affects TGF‐β signaling in β‐actin null cells, since mechanical force at the cell surface can activate latent TGF‐β. Indeed, when we compared the transcriptomes between wild‐type (WT) β‐actin+/+ and knockout (KO) β‐actin−/− mouse embryonic fibroblasts (MEFs), TGF‐β1 signaling pathway was identified as an upstream activator for expression changes of certain genes in the KO cells. Different classes of TGF‐β1 target genes were upregulated in the KO cells. The elevated TGF‐β1 signaling activation in the KO cells seems to be caused by the efficient activation and utilization of latent TGF‐β1, while Tgfb1 gene was expressed at a comparable level between WT and KO cells. We further demonstrated that TGF‐β‐dependent cellular ROS production is required for the enhanced myofibroblast features of the KO cells, which is likely to be mediated by the upregulation of Nox4 gene. However, other gene programs and angiogenic features of the KO cells were not affected after TGF‐β inhibition. Our data, together with our recent finding that WT and KO cells exhibit major differences in cell surface mechanical properties (Xie et al., 2018), suggest that the enhanced TGF‐β1 activation in the KO cells is likely due to the altered biophysical properties at the cell surface. The elevated TGF‐β activation contributes to the enhanced myofibroblast features of the β‐actin null cells, without affecting other gene programs or cellular features. Collectively, our study shows a scenario that after genetic reprograming of the actin cytoskeleton, the alterations in cellular biophysical features result in the change in signaling process to affect cellular phenotype.
2. MATERIALS AND METHODS
2.1. Antibodies and reagents
Antibodies of myosin light chain 9 (Myl9; ab64161), Itga11 (ab198826) were from Abcam (Cambridge, UK). Antibodies against β‐actin (clone AC‐74), diphenyleneiodonium chloride (DPI; D2926), SB431542 hydrate (S4317), N‐acetyl‐l‐cysteine (A7250), Dulbecco’s modified Eagle’s medium (DMEM) high glucose (D5671), fetal bovine serum (FBS; F0804), and penicillin–streptomycin (P0781) were from Sigma‐Aldrich (Taufkirchen, Germany). Antibodies of mouse immunoglobulin G (IgG) horseradish peroxidase (HRP; 62‐6520), rabbit IgG HRP (65‐6120), smooth muscle actin antibody (PA5‐19465), Maxima SYBR Green qPCR Master Mix (K0252), RevertAid First Strand cDNA Synthesis Kit (K1622), Pierce™ ECL western blotting substrate (32106), Pierce protein assay kit (23225), TRIzol® reagent (15596‐018), CellROX™ Deep Red reagent (C10422), MitoSOX™ Red mitochondria superoxide indicator (M36008), and Pierce Renilla‐Firefly Luciferase Dual Assay Kit (16185) were purchased from Thermo Fisher Scientific (Waltham, MA). Anti‐GAPDH–HRP (HRP‐60004) was from ProSci (Fort Collins, CO). Mouse TGF Beta 1 PicoKine™ ELISA Kit was from BosterBio (Pleasanton, CA).
2.2. Cell culture
β‐Actin+/+ WT MEFs and β‐actin−/− KO MEFs were maintained in DMEM with high glucose (Sigma‐Aldrich), supplemented with 10% FBS (Sigma‐Aldrich) and 100 U/ml penicillin and 100 µg/ml streptomycin (Sigma‐Aldrich), in a humidified incubator with 5% CO2 at 37°C. For certain experiments, serum‐free DMEM was used as specified.
2.3. CellROX staining analysis using high‐content screening (HCS) platform
MEFs, cultured in 96‐ or 384‐well clear‐bottom, black polystyrene plate (Corning, Oneonta, NY), were stained with 5 µM of CellROX Deep Red Reagent in the medium for 30 min. After two washes with phosphate‐buffered saline (PBS), stained cells were washed fixed by 3.7% formaldehyde for 15 min, then further stained with Hoechst 43222 (1:6000) in PBS for 10 min. After two washes with PBS, cells in the plate were scanned via Cellomics ArrayScan™ XTI High‐Content Analysis (HCS) Platform (Thermo Fisher Scientific, Waltham, MA), with a ×10 objective. Individual cells were identified based on Hoechst DNA staining of single nuclei. Compartment Analysis Bio Application software (Cellomics, Pittsburgh, PA) was applied to quantitatively analyze the CellROX‐stained spots in the cytoplasm in single cells. For each experiment, at least 500 valid single cells per culture well were quantified and 10 independent culture wells (10 biological replicates) were analyzed. For chemical treatment, cultured cells were preincubated with SB431542 (16 µM) and DPI (1 µM) for 6 hr, before staining and fixation.
2.4. Flow cytometry analysis
MEF cells cultured in 24‐well plates were stained with 5 µM MitoSOX Red mitochondrial superoxide indicator in the medium for 30 min. Stained cells were washed twice with PBS and trypsinized cells resuspended in PBS were immediately analyzed by flow cytometer BD FACSAria II (BD Bioscience, San Jose, CA). FlowJo software (FlowJo, LLC, Ashland, OR) was used for data analysis.
2.5. RNA‐seq analysis data
Detailed RNA‐seq analysis was described in Xie et al. (2017). RNA‐seq data were deposited in GEO repository, and the GEO accession number is GSE95830. Ingenuity pathway analysis (Qiagen Bioinformatics, Redwood City, CA) was used to predict the upstream regulators based on the changes of differentially expressed genes (adjusted p < 0.05, with fold change ≥2) between WT and KO MEFs.
2.6. Western blot analysis
The cell lysate was harvested using RIPA buffer and protein concentrations were quantified by BCA Protein Assay Kit (Thermo Fisher Scientific). Samples were mixed with 2× Laemmli sample buffer (Sigma‐Aldrich) and heated at 95°C for 5 min. Immunoblotting was done using anti‐Myl9 (1:1000), anti‐Itga11 (1:800), anti‐α‐smooth muscle actin (α‐SMA; 1:1000), anti‐β‐actin (1:1500), anti‐GAPDH–HRP (1:1000), goat anti‐mouse IgG HRP (1:2500), and goat anti‐rabbit IgG HRP (1:2500) antibodies. Protein band development was performed using Pierce ECL western blotting substrate (Thermo Fisher Scientific) and was imaged using ChemiDoc MP Imaging System (Bio‐Rad, Hercules, CA).
2.7. RNA isolation and quantitative real‐time quantitative polymerase chain reaction (qPCR)
Total RNA was purified using RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Isolated RNA was then reverse‐transcribed to cDNA by RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Quantitative real‐time PCR was performed using Maxima SYBR Green qPCR Mix (Thermo Fisher Scientific) on Stratagene 3005 qPCR System (Agilent Technology, Santa Clara, CA). Gene expression level was normalized to the expression of Nono reference gene based on method. Primers of qPCR are listed below:
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| Actb | TATCGCTGCGCTGGTCG | CCCACGATGGAGGGGAATAC |
| Tgfb1 | CCACCTGCAAGACCATCGAC | CTGGCGAGCCTTAGTTTGGAC |
| Tgfbr1 | TCCCAACTACAGGACCTTTTTCA | GCAGTGGTAAACCTGATCCAGA |
| Nox4 | AAACACCTCTGCCTGCTCAT | CAGGACTGTCCGGCACATAG |
| Col3a1 | ACGTAGATGAATTGGGATGCAG | GGGTTGGGGCAGTCTAGTG |
| Fn1 | GCTCAGCAAATCGTGCAGC | CTAGGTAGGTCCGTTCCCACT |
| Smoc1 | AATCCACAGGCTACTGTTGGT | CATCGGCCTCTATGCTCTTGG |
| Lama4 | ATGAGCTGCAAGGAAAACTATCC | CTGTTTCGTTGGCTTCACTGA |
| Acta2 | GGACGTACAACTGGTATTGTGC | TCGGCAGTAGTCACGAAGGA |
| Tagln | CAACAAGGGTCCATCCTACGG | ATCTGGGCGGCCTACATCA |
| Actg1 | AATCGCCGCACTCGTCATT | GCCCTACGATGGAAGGGAA |
| Itgb8 | AGTGAACACAATAGATGTGGCTC | TTCCTGATCCACCTGAAACAAAA |
| Eng | TGCACTTGGCCTACGACTC | TGGAGGTAAGGGATGGTAGCA |
| Hpse | ACCGACGACGTGGTAGACTT | GCAGGAGATAAGCCTCTAGCC |
| Nono | GCCAGAATGAAGGCTTGACTAT | TATCAGGGGGAAGATTGCCCA |
2.8. Angiogenesis assay
Angiogenesis assay was performed using In vitro Angiogenesis Assay Kit (ab204726; Abcam). ECM gel solution (50 µl) was added to a prechilled 96‐well plate and solidified at 37°C for 20 min. Cells (2 × 104) in DMEM with 10% FBS were seeded to each well and cultured for 18 hr. Images were taken by phase‐contrast microscope (DMI6000B; Leica, Allendale, NJ).
2.9. TGF‐β1 enzyme‐linked immunosorbent assay (ELISA)
MEF cells (2.5 × 105) were cultured at confluence in 350 µl serum‐free DMEM or DMEM containing 10% FBS for 24 hr. TGF‐β1 levels in the medium were determined using Mouse TGF Beta 1 PicoKine™ ELISA Kit (BosterBio) according to the manufacturer’s protocol. Briefly, 100 µl cell culture supernatant was activated with 20 µl of 1 M HCl for 10 min at room temperature, followed by pH neutralization with 1.2 M NaOH–0.5 M HEPES. The activated supernatant (100 µl) was added to 96‐well plate precoated with anti‐TGF‐β1 antibody and incubated at 37°C for 90 min. The supernatant was discarded and 100 µl of biotinylated anti‐TGF‐β1 antibody was added to each sample for 60 min incubation at 37°C. Contents in the plate were discarded and the plate was washed three times with PBS, followed by incubation with 100 µl ABC working solution for 30 min at 37°C. After five times wash with PBS, 90 µl TMB color‐developing agent was added. After 30 min of incubation at 37°C, the reaction was terminated by the addition of 100 µl stop solution. Absorbance at 450 nm was recorded using Synergy H1 microplate reader (BioTek, Dubai, U.A.E). TGF‐β1 concentration was extracted based on the standard curve of recombinant TGF‐β1.
2.10. Floating collagen gel contraction assay
Cells were preincubated with chemical inhibitors as indicated for 24 hr before the experiment. Rat collagen I solution (200 µl of 3 mg/ml) was mixed with 2.5 × 105 cells in 400 µl DMEM supplemented with 10% FBS (R&D Systems, Minneapolis, MN). NaOH (6 µl) at 1 M was added to the cell mixture to initiate gel polymerization. Immediately after NaOH addition, the mixture was homogenized by pipetting up and down and then transferred to a 24‐well plate to allow the collagen gel to solidify at 37°C for 15 min. Fresh DMEM (500 µl; 10% FBS) with chemical inhibitors at the same concentrations as preincubation stage was added to each well and the solidified gel was detached from the well using sterile pipette tips, to allow the gel to float in the culture medium. After 48 hr, the whole well image of the gel was collected by Nikon SMZ18 stereomicroscope (Nikon Instruments, Cusago MI, Italy). The morphology of the cells in the gel matrix was collected by Olympus FV1000 confocal microscope (Olympus Corporation, Shinjuku, Tokyo, Japan) after 24 hr. The relative gel surface area was measured by ImageJ software (National Institute of Health, Bethesda, MD).
2.11. Total collagen assay
Sirius Red Total Collagen Detection Kit (Chondrex, Redmond, WA) was used to quantify the total collagen according to the manufacturer’s instructions, with slight modification. MEF cells (2.5 × 105) were seeded and allowed to be grown confluent in 24‐well plate for 24 hr. 0.1 M acetic acid (250 µl) was added to each well to solubilize the extracellular collagen, with a gentle shake for 30 min at room temperature. Both supernatant and cell debris were collected and centrifuged at 10,000 rpm for 5 min. Then, 100 µl supernatant was mixed with 500 µl Sirius Red dye for 20 min at room temperature. After centrifugation at 10,000 rpm for 5 min, the red pellet was dissolved in 250 µl extraction buffer and measured at optical density 530 nm using Synergy H1 microplate reader (BioTek).
2.12. Dual luciferase assay
Plasmids pRL‐SV40P (control vector), SBE4‐Luc, and FBE/SBE‐Luc were purchased from Addgene (Cambridge, MA). 1 × 104 MEF cells were seeded in a 96‐well plate. Cells were transfected with 100 ng pRL‐SV40P and 200 ng of SBE4‐Luc or 200 ng of FBE/SBE‐Luc using TurboFect transfection reagent (Thermo Fisher Scientific) in a serum‐free medium. Forty‐eight hours posttransfection in the serum‐free medium, the dual luciferase assay was performed using Pierce Renilla‐Firefly Luciferase Dual Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Cell lysis buffer (100 µl) was added to each well and the plate was shaken for 15 min. Cell lysate (20 µl) was mixed with 50 µl working solution in a white 96‐well plate for luminescence. Red Firefly luciferase signal was captured by integrating the luminescence signals from 640 to 680 nm, and green Renilla luciferase signal was recorded by integrating the signals from 500 to 540 nm, using Synergy H1 microplate reader (BioTek). The integration time is 300 ms, step: 2 nm, gain: 200, normal speed, with 100 ms delay. The relative TGF‐β activity was calculated by normalize the firefly luciferase signal to Renilla luciferase signal.
2.13. 3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5 ‐diphenyltetrazolium bromide (MTT) assay
MTT assay was performed as described in Xie, Wang, Wong, and Fung (2013). Six thousand cells per well were seeded in the 96‐well plate. Cells were treated with chemicals at varying concentration for 18 hr. MTT reagent (16 μl, 5 mg/ml; Sigma‐Aldrich) was added to each well and incubated at 37°C for 3 hr. Culture medium was aspirated and 120 μl dimethyl sulfoxide was added to each well for 10‐min incubation at room temperature. Absorbance was measured at a wavelength of 570 nm by Synergy H1 microplate reader (BioTek, Dubai, U.A.E).
2.14. Statistical analysis
All experimental data are expressed as mean ± SEM. Statistical difference and significance between the groups were determined by either Student’s t test or one‐way analysis of variance using GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA).
3. RESULTS
3.1. In β‐actin KOs, elevated TGF‐β signaling results from the active utilization of latent TGF‐β1
We have recently shown that the transcriptome of β‐actin−/− KO MEFs exhibit significant changes of gene expression programs in comparison with the β‐actin+/+ WT MEFs (Xie et al., 2017). To identify the potential upstream regulators that may be responsible for controlling subsets of genes or gene programs, we analyzed differentially expressed genes (at least two‐fold change, adjusted p < 0.05) between WT and KO cells using an Ingenuity Pathway Analysis software. The implemented upstream analysis tool predicts activation or inhibition of upstream transcription factors or signaling pathways based on the gene expression changes (Krämer, Green, Pollard, & Tugendreich, 2014). Among the top 10 upstream regulators identified, TGF‐β1 (TGFB1), SMAD3, TGF‐β receptor 1 (TGFBR1) and SMAD4 are predicted to be activated in the KO cells when compared to WT cells based on the activation Z score and p value (Figure 1a, highlighted in blue). These four regulators function at different steps of TGF‐β signaling cascades, indicating that TGF‐β signaling is likely to be activated in β‐actin null cells. Indeed, different classes of genes that were previously known to be induced by TGF‐β1 were upregulated in the KO cells when compared to WT cells (Figure 1b; Datto et al., 1995; Fang et al., 2016; O’Callaghan & Williams, 2000; Ranganathan et al., 2007; Thuault et al., 2006; Xu, Lamouille, & Derynck, 2009; Yu et al., 2008). These genes include Tmsb4x, Tagln, Actn1, and Msn that are involved in cytoskeleton function (Ranganathan et al., 2007; Yu et al., 2008); Fn1, Ctcf, Col1a1, and Lox that are components or regulators in ECM formation (Fang et al., 2016; O’Callaghan & Williams, 2000; Ranganathan et al., 2007); Hmga2, Snai1, and Vim that are regulator or markers in EMT (Thuault et al., 2006; Xu et al., 2009); and Gsk3b, Plcb4, Wnt5a, and Cdk1a (p21) which function in different cellular signaling processes (Datto et al., 1995; Ranganathan et al., 2007). To confirm the TGF‐β pathway was activated to a higher level in β‐actin−/− cells, we used a previous published TGF‐β luciferase reporter FBE/SBE‐Luc, which contains FAST‐1‐binding element (FBE) adjacent to Smad‐binding element (SBE; Zhou, Zawel, Lengauer, Kinzler, & Vogelstein, 1998). The SBE‐Luc serves as a negative control as it was unable to confer response to TGF‐β signaling (Zhou et al., 1998). We observed that the KO cells activated FBE/SBE‐Luc construct to a significantly higher level in comparison to WT MEFs, while no difference was observed for the SBE‐Luc construct (Figure 1c). Therefore, the KO cells show increased level of TGF‐β signal activation.
Figure 1.
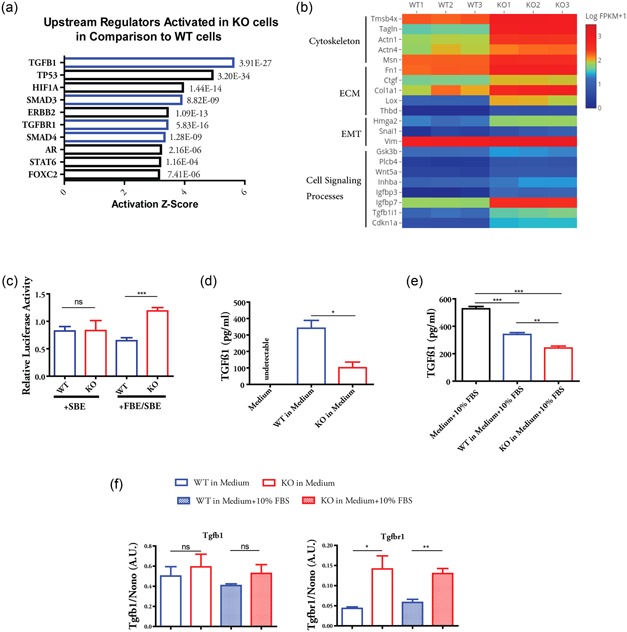
Cells lacking β‐actin shows elevated activation of TGF‐β signaling by actively consuming latent TGF‐β1. (a) Genes differentially expressed by at least two‐fold between β‐actin+/+ WT and β‐actin−/− KO MEFs were subjected to Ingenuity Pathway Analysis (Qiagen Bioinformatics). Top 10 upstream regulators predicted to be activated were shown, with their corresponding activation Z score and p value. (b) Heatmap of the expression level of TGF‐β1‐targeted genes between WT and KO MEFs. Three biological replicates of RNA‐seq data are shown. (c) WT and KO cells were cotransfected with plasmid expressing Renilla luciferase and plasmid containing FBE–SBE‐driven firefly luciferase or SBE‐driven firefly luciferase. Luciferase assay was performed after 48 hr culture in serum‐free medium. Firefly luciferase activity was normalized to Renilla luciferase activity. n = 4 independent experiments. (d) Quantification of TGF‐β1 level in WT and KO cell‐cultured serum‐free DMEM by ELISA. (e) Quantification of TGF‐β1 level in WT and KO cells cultured in DMEM (with 10% FBS) by ELISA. (f) Relative expression of Tgfb1 and Tgfbr1 between WT and KO cells cultured in DMEM or DMEM + 10% FBS for 24 hr. Data in (d)–(f) are summary of three biological replicates. Student’s t test was used in (c), (d), and (f) and one‐way analysis of variance with Tukey’s post hoc test was used in (e); Values are shown as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001. DMEM, Dulbecco’s modified Eagle’s medium; ECM, extracellular matrix; ELISA, enzyme‐linked immunosorbent assay; EMT, epithelial–mesenchymal transition; FBE–SBE, fast‐1‐binding element–Smad‐binding element; FBS, fetal bovine serum; KO, knockout; MEF, mouse embryonic fibroblast; ns, nonsignificant; TGF‐β, transforming growth factor β; WT, wild type
To explore the possible mechanism, we quantified the TGF‐β1 level in the serum‐free medium with cultured cells by ELISA. To our surprise, the overall TGF‐β1 level is much lower in the medium with the KO cells than that of the WT cells (Figure 1d). The lower level of TGF‐β1 is not due to the reduced Tgfb1 gene expression in the KO cells (Figure 1f and Supporting Information Figure S1A). Although TGF‐β1 receptor gene (Tgfbr1) is upregulated in the KO cells (Figure 1f and Supporting Information Figure S1B), it is well established that TGF‐β is produced as latent, high‐molecular‐weight complex that needs to be activated before receptor binding (Hinz, 2015; Shi et al., 2011). Therefore, we hypothesized that the KO cells may have an enhanced ability to activate and utilize the latent TGF‐β1. As the serum contains high level of latent TGF‐β (Oida & Weiner, 2010), we cultured the cells in medium with 10% FBS to investigate the consumption rate of latent TGF‐β1 between the WT and KO cells. The results from ELISA showed that the KO cells significantly decreased the TGF‐β1 level in the medium to a greater extent in comparison to WT cells (Figure 1e). The physiological–mechanical force at the cell surface has been recognized as the major contributor to the release and activation of latent TGF‐β, which is mediated via integrins (Buscemi et al., 2011; Hinz, 2015; Maeda et al., 2011; Shi et al., 2011; Wipff et al., 2007). We recently reported major alterations in the cell surface mechanical properties of β‐actin−/− KO cells in comparison to WT cells (Xie et al., 2018). The KO cells, with reorganized actin cytoskeleton, also upregulate several integrin genes (Xie et al., 2017). Therefore, we conclude that the KO cells activate and consume the latent TGF‐β in the medium more efficiently than WT cells, leading to elevated TGF‐β signaling and the upregulation of its target genes. In further support of this, the ECM components involved in the process of TGF‐β activation, such as fibronectin (Fn1; Fontana et al., 2005), Ltbp1 (Buscemi et al., 2011), and thrombospondin‐1 (Thbs1; Murphy‐Ullrich & Poczatek, 2000), were also found to express at significantly higher level in the KO cells (Supporting Information Figure S1C–E).
3.2. TGF‐β‐dependent Nox4 upregulation contributes to ROS production in the absence of β‐actin
It is known that TGF‐β1 induces ROS production and ROS mediates some of TGF‐β effects such as TGF‐β‐induced fibrosis (Liu & Desai, 2015). We, therefore, analyzed the oxidative status between WT and KO cells by CellROX Deep Red dye, which exhibits fluorescence upon ROS oxidation and can be quantified after fixation. The HCS platform was used for automated image quantification. The nuclear region of each cell was identified by Hoechst staining (blue circles; Figure 2a), and the cytoplasmic region of each individual nucleus was computationally inferred based on the proximity of neighboring nuclei (green boundaries; Figure 2a). The CellROX staining in the cytoplasm (red spots; Figure 2a) was quantified in single cells. On average, the KO cells displayed significantly higher cellular ROS levels when compared with WT cells (Figure 2b). As the TGF‐β‐mediated ROS can come from mitochondria (Jain et al., 2013), we stained the WT and KO cells with MitoSOX Red, a mitochondria‐targeted superoxide indicator in live cells. Flow cytometry analysis demonstrated a lower level of mitochondrial superoxide in the KO cells (Figure 2c). Therefore, the elevated cellular ROS levels in the KO cells are not due to the increased mitochondrial superoxide production.
Figure 2.
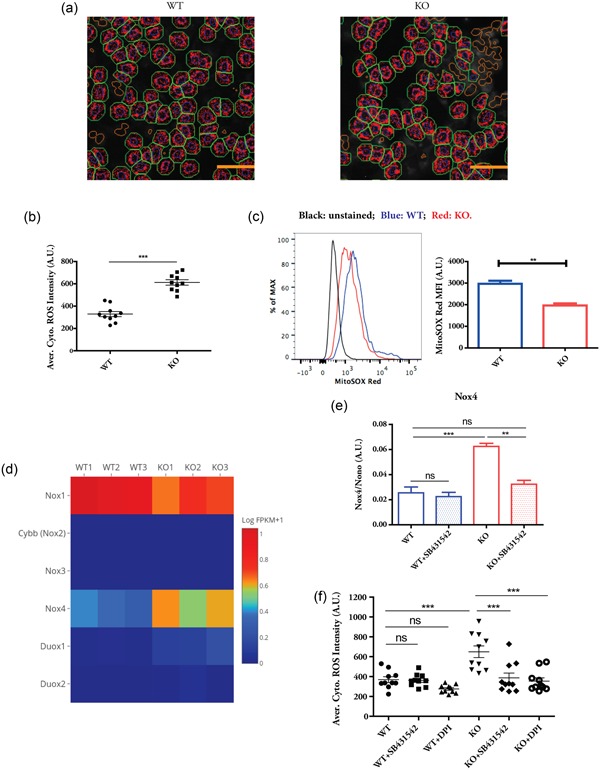
TGF‐β‐dependent upregulation of Nox4 gene contributes to increased ROS production in β‐actin KO cells. (a) High content image analysis of CellROS staining between WT and KO cells. Blue circles: Valid single nuclei; green boundary: Simulated cytoplasm; red spots: Identified CellROS staining. Scale bar: 50 µm. (b) Quantification of average cytoplasmic ROS intensity. Each data point represents the mean of at least 500 single cells in an independent culture well. Data show the summary of 10 biological replicates, representative of two independent experiments. (c) FACS analysis of MitoSOX staining. Data are summary of three biological replicates, representative of three independent experiments. (d) Heatmap showing the expression levels of Nox gene family between WT and KO cells. (e) Quantitative polymerase chain reaction analysis of Nox4 expression in the absence or in the presence of SB431542 for 24 hr. n = 3 biological replicates. (f) High content image analysis of CellROS staining in the absence or in the presence of SB431542 and diphenyleneiodonium chloride for 6 hr. Each data point represents the mean of at least 500 single cells in an independent well. Data are representative of three independent experiments. Student’s t test was used in (b) and (c), and one‐way analysis of variance with Tukey’s post hoc test was used in (e) and (f); values are given as mean ± SEM; **p < 0.01, ***p < 0.001. Nox4, NADPH oxidase 4; ns, not significant; KO, knockout; ROS, reactive oxygen species; TGF‐β, transforming growth factor‐β; WT, wild type
Nox are a family of enzymes responsible for intracellular ROS production under a variety of pathophysiological conditions (Guichard et al., 2006). Nox4 gene is found to be induced by TGF‐β in different cell types (Boudreau et al., 2012; Hecker et al., 2009; Yan et al., 2014). We, therefore, asked whether Nox4 gene is upregulated and contributes to elevated ROS in the KO cells. Indeed, RNA‐seq data showed that among all the members of Nox gene family, Nox4 showed clear upregulation in the KO cells (Figure 2d). The upregulation of Nox4 in the KO cells was dependent on TGF‐β signaling, since the treatment of a specific inhibitor of TGF‐β receptor kinase SB431542 (Halder, Beauchamp, & Datta, 2005) abrogated its increased expression level (Figure 2e). We further applied SB431542 and Nox inhibitor DPI to WT and KO cells to study their effects on cellular ROS. The inhibition of TGF‐β receptor signaling and Nox activity reduced the cellular ROS level in the KO cells, without significant changes observed in WT cells. Taken together, these results indicate that in β‐actin null cells activated TGF‐β signaling induces Nox4 upregulation and contributes to elevated ROS production.
3.3. TGF‐β signaling contributes to myofibroblast feature in the absence of β‐actin
Our recent study shows that β‐actin KOs display myofibroblast features, including enhanced abilities to produce and contract ECM (Xie et al., 2017). Since Nox4 induced by TGF‐β mediates myofibroblast differentiation (Hecker et al., 2009; Jiang et al., 2014), we next investigated whether the inhibition of TGF‐β signaling affects the acquired myofibroblast features in the KO cells by applying a gel contraction assay. As shown in Figure 3a, the results show that KO cells have enhanced ability to contract the gel in comparison to WT cells. After preincubation with subtoxic dosage of SB431542 (8 µM) and DPI (0.5 µM; see Supporting Information Figure S2 for MTT cytotoxicity assay), there was no difference of gel contraction ability between WT and KO cells although the contractility of both WT and KO cells were impaired (Figure 3a,b). After the treatment with antioxidant N‐acetyl cysteine (NAC), the contractility ability of WT cells seemed not to be affected, while NAC greatly impaired the contractility of KO cells (Figure 3a,b). These data suggest the enhanced contractility of the KO cells depends on TGF‐β signaling and ROS production. Noticeably, inhibition of Nox oxidase activity by DPI totally abrogated the ability to contract the gel in both WT and KO cells (Figure 3a,b), implying the essential role of Nox oxidases in controlling cellular contractility. Interestingly, inhibition of Nox activity by DPI prevents the generation of cell protrusion in 3D collagen gel matrix, and the majority of WT and KO cells remain round shaped (Supporting Information Figure S3). The TGF‐β inhibition also inhibited cell protrusion in WT cells and impaired cellular interconnections of KO cells within the gel matrix (Supporting Information Figure S3). The cellular morphology in collagen matrix did not seem to be affected by NAC treatment.
Figure 3.
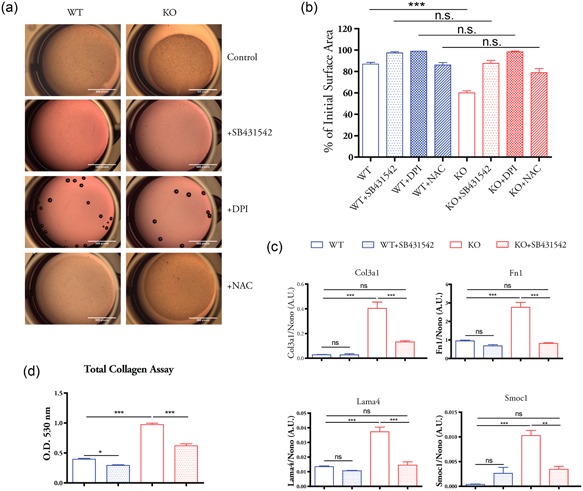
TGF‐β signaling contributes to enhanced myofibroblast features in β‐actin KO cells. (a) Cells were preincubated with SB431542 (8 µM), DPI (0.5 µM), NAC (2 mM), or DMSO solvent (control) for 24 hr. Float collagen gel assay was performed in the presence of same concentration of each compound. (b) Quantification of gel surface area in each experiment condition. (c) Expression of selected ECM genes were analyzed by quantitative polymerase chain reaction with or without SB431542 (8 µM, 24 hr) treatment. (d) Total extracellular collagen of WT and KO cells with or without SB431542 (8 µM, 24 hr) treatment were extracted and determined using Sirius red dye. Statistics in (b)–(d): n = 3 biological replicates, one‐way analysis of variance with Tukey’s post hoc test; values are given as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001. DMSO, dimethyl sulfoxide; DPI, diphenyleneiodonium chloride; ECM, extracellular matrix; KO, knockout; NAC, N‐acetyl cysteine; ns, not significant; OD, optical density; TGF‐β, transforming growth factor‐β; WT, wild type
We further studied the effect of TGF‐β signaling inhibition on ECM genes that are upregulated in the KO cells. The treatment of SB431542 significantly reduced the expression of collagen type III α1 (Col3a1), fibronectin (Fn1), laminin α4 (Lama4), and SPARC‐related modular calcium binding 1 (Smoc1) in the KO cells, with little effect on WT cells (Figure 3c). Quantitative analysis of total extracellular collagen showed that the KO cells produced much higher level of total collagen than WT cells (Figure 3d). Although TGF‐β inhibition reduced the collagen level in both WT and KO cells, the extent of decrease is more significant in the KO cells (Figure 3d). This is consistent with the established role of TGF‐β signaling in inducing ECM gene expression (Verrecchia & Mauviel, 2002). Together, our data showed that TGF‐β signaling contributes to the enhanced myofibroblast features in the KO cells, such as augmented ability to contract ECM and elevated ECM gene expression.
3.4. TGF‐β inhibition does not affect the expression of actin cytoskeleton genes and the angiogenic property of β‐actin KOs
Since in β‐actin null MEFs we observed changes in the expression of genes programs related to actin cytoskeleton, integrins, and angiogenesis, and, correspondingly, the acquisition of an angiogenic phenotype (Xie et al., 2017), we tested whether the elevated TGF‐β signaling in the KO cells also contributes to those gene programs. qPCR analysis demonstrated that actin cytoskeleton and integrin genes upregulated in the KO cells, such as α‐SMA (Acta2), gamma actin (Actg1), transgelin (Tagln), integrin α11 (Itga11), and integrin β8 (Itgb8) were not suppressed after treatment with the TGF‐β inhibitor SB431542 (Figure 4a). The same results were observed at protein level for α‐SMA, Myl9, and Itga11 that were expressed at relatively higher level in the KO cells (Figure 4b). Likewise, genes that are downregulated in the KO cells such as endoglin (Eng) and heparanase (Hpse) were not affected by SB431542 treatment (Figure 4a). We also applied in vitro angiogenesis assay and showed that the KO cells maintained the enhanced ability to form capillary‐like tubes even after SB431542 treatment. Therefore, the altered gene programs related to major components of actin cytoskeleton and the angiogenic phenotype were not dependent on the elevated TGF‐β signaling in β‐actin KO cells.
Figure 4.
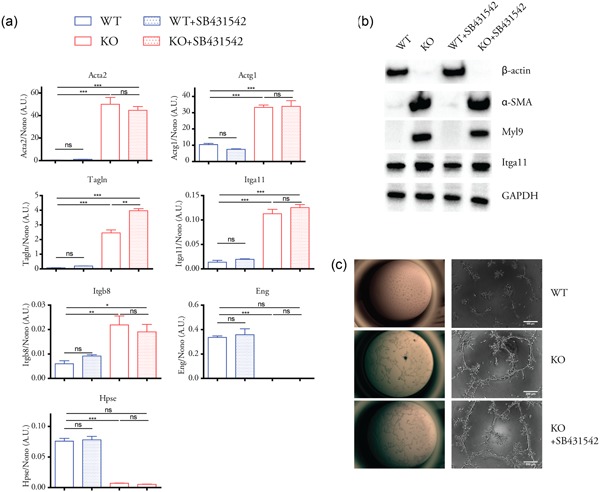
TGF‐β signaling inhibition did not affect the genetic reprogramming related to actin cytoskeleton and angiogenesis in β‐actin KO cells. (a) Expression of selected genes (previously shown to be significantly changed between WT and KO cells) were analyzed by quantitative polymerase chain reaction with or without SB431542 (8 µM, 24 hr) treatment. n = 3 biological replicates, one‐way analysis of variance with Tukey’s post hoc test; values are shown as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001. (b) Western blot analysis of selected proteins with or without SB431542 (8 µM, 24 hr) treatment. (c) In vitro angiogenesis assay. For KO cells + SB431542 group, KO cells were preincubated with SB431542 (8 µM, 24 hr), and then plated on extracellular matrix gel in complete DMEM in the presence of SB431542 (8 µM, 24 hr). DMEM, Dulbecco’s modified Eagle’s medium; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; Itga11, integrin α11; KO, knockout; Myl9, myosin light chain 9; ns, not significant; TGF‐β, transforming growth factor‐β; WT, wild type; α‐SMA, α‐smooth muscle actin
4. DISCUSSION
TGF‐β signaling pathway has been implicated in a variety of developmental and pathophysiological processes (Cong et al., 2012; Hinz, 2009; Zinski et al., 2017). In this study, through a comparative analysis of the transcriptomes of WT (β‐actin+/+) and KO (β‐actin−/−) MEFs, we identified TGF‐β1 signaling pathway as upstream regulator which is activated in the absence of β‐actin to induce the upregulation of a group of its target genes. This finding is consistent with a proteomic study in which TGF‐β1 is predicted to be a potential upstream activator for overexpressed actin‐binding proteins in the β‐actin KOs (Ampe, Libbrecht, & van Troys, 2013). However, our results indicate that the enhanced TGF‐β activation observed in the KO cells is caused by the active utilization of the latent TGF‐β1 in the culture medium instead of upregulating Tgfb1 gene. TGF‐β is secreted as part of a latent protein complex and acts as an extracellular mechanosensor to switch on fibrosis (Duscher et al., 2014; Hinz, 2015). Latent TGF‐β is tethered to the ECM by LTBPs, which can be released and activated by the cell surface mechanical force transmitted via integrins (Annes et al., 2004; Buscemi et al., 2011; Shi et al., 2011). We recently reported that β‐actin KO cells exhibit dramatic changes in their biophysical properties at cell surface, including increased elasticity and adhesion to the probe as revealed by atomic force microscopy (Xie et al., 2018). We, therefore, conclude that in the absence of β‐actin the augmented activation and utilization of latent TGF‐β is likely due to the altered biomechanical properties at the cell surface.
In many fibrogenic responses and EMT processes, ROS production plays an important role in mediating the effect of TGF‐β signaling (Hagler et al., 2013; Jiang et al., 2014; Liu & Desai, 2015; Rhyu et al., 2005). The origin of ROS induced by TGF‐β activation may differ under different conditions (Liu & Desai, 2015). In some studies, ROS generated by mitochondrial electron transport chain complexes are reported to be responsible for TGF‐β‐mediated effects (Casalena et al., 2012; Jain et al., 2013). In other cases, ROS generated by Nox contributes to TGF‐β‐dependent cellular processes (Amara et al., 2010; Hiraga, Kato, Miyagawa, & Kamata, 2013; Hu et al., 2005; Liu & Desai, 2015). In the β‐actin null cells with an overall higher level of cellular ROS, we observed a relatively lower level of mitochondrial superoxide than WT cells. We, therefore, conclude that the increased cellar ROS in the absence of β‐actin is not a consequence of impaired mitochondrial function. Our results further show that Nox4 is upregulated in the KO cells, while other Nox members do not show significant expression change. The upregulation of Nox4 expression is dependent on TGF‐β signaling in the KO cells. Moreover, we show evidence that the treatment of TGF‐β receptor inhibitor and Nox inhibitor can reduce cellular ROS levels, and lower the ECM gene expression and impair the ability to contract ECM in the KO cells. Overall, these results are consistent with previous studies in which Nox4 expression is found to be upregulated by TGF‐β and where Nox4‐derived ROS levels are found to mediate fibroblast to myofibroblast transdifferentiation (Hecker et al., 2009; Jiang et al., 2014; Sampson et al., 2009). Therefore, our study supports the model whereby TGF‐β signaling activation in the KO cells induces Nox4 expression and Nox4‐derived ROS contributes to the enhanced myofibroblast features.
Seeing that in the absence of β‐actin several gene programs were found to exhibit altered expression (Xie et al., 2017), we further investigated whether the activated TGF‐β signaling contributes to other differentially expressed genes between WT and KO cells. We found that inhibition of TGF‐β signaling does not affect the previously observed upregulation of actin cytoskeleton and integrin genes in the KO cells. For example, α‐SMA is a marker in myofibroblast differentiation which can be rapidly induced by TGF‐β (Hecker et al., 2009; Thannickal et al., 2003). However, in the KO cells, the heavily upregulated α‐SMA (Acta2) is not affected after TGF‐β inhibitor treatment. Moreover, the augmented angiogenic properties are not affected after inhibition of TGF‐β signaling. These results indicate that in the absence of β‐actin, TGF‐β signaling activation does not contribute to the core gene programs implicated in the actin cytoskeleton and integrin genes or in angiogenesis. These core gene programs altered in the KO cells are due to the global chromatin change in the absence of nuclear β‐actin (Xie et al., 2017). Collectively, our data support the idea that the observed activation of TGF‐β signaling in the KO cells is a consequence of the altered biophysical features at cell surface after actin isoform switching and the actin cytoskeleton reorganization (Xie et al., 2018). The enhanced TGF‐β signaling and elevated cellular ROS production then contribute to augmenting the myofibroblast features in the β‐actin KO cells.
In summary, we found that in the cells lacking β‐actin, elevated TGF‐β activation promotes Nox4 gene expression, resulting in higher levels of cellular ROS production. The enhanced TGF‐β activation is due to the active utilization of the latent TGF‐β by the KO cells, which is likely due to the altered biomechanical properties observed at the cell surface. The activated TGF‐β signaling contributes to enhanced myofibroblast features but is not necessary for other gene expression programs. Therefore, we conclude that in the absence of β‐actin, the TGF‐β signaling activation is a downstream effect of the genetic reprogramming and actin cytoskeleton reorganization. These results, together with recently published work further emphasize a primary function for nuclear β‐actin in the regulation of core gene programs. After genetic reprogramming, actin isoform switching and the resulted changes in biophysical properties in turn lead to downstream effects such as the elevated activation of latent TGF‐β, which ultimately affects certain cellular phenotype.
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Xie X, Percipalle P. Elevated transforming growth factor β signaling activation in β‐actin‐knockout mouse embryonic fibroblasts enhances myofibroblast features. J Cell Physiol. 2018;233:8884–8895. 10.1002/jcp.26808
References
REFERENCES
- Amara, N. , Goven, D. , Prost, F. , Muloway, R. , Crestani, B. , & Boczkowski, J. (2010). NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGF beta 1‐induced fibroblast differentiation into myofibroblasts. Thorax, 65, 733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ampe, C. , Libbrecht, J. , & Van Troys, M. (2013). Beta‐actin knock‐out mouse embryonic fibroblasts show increased expression of LIM‐, CH‐, EFh‐domain containing proteins with predicted common upstream regulators. Cytoskeleton, 70, 766–774. [DOI] [PubMed] [Google Scholar]
- Annes, J. P. , Chen, Y. , Munger, J. S. , & Rifkin, D. B. (2004). Integrin alphaVbeta6‐mediated activation of latent TGF‐beta requires the latent TGF‐beta binding protein‐1. The Journal of Cell Biology, 165, 723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau, H. E. , Casterline, B. W. , Rada, B. , Korzeniowska, A. , & Leto, T. L. (2012). Nox4 involvement in TGF‐beta and SMAD3‐driven induction of the epithelial‐to‐mesenchymal transition and migration of breast epithelial cells. Free Radical Biology & Medicine, 53, 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscemi, L. , Ramonet, D. , Klingberg, F. , Formey, A. , Smith‐Clerc, J. , Meister, J. J. , & Hinz, B. (2011). The single‐molecule mechanics of the latent TGF‐beta1 complex. Current Biology, 21, 2046–2054. [DOI] [PubMed] [Google Scholar]
- Casalena, G. , Daehn, I. , & Bottinger, E. (2012). Transforming growth factor‐beta, bioenergetics, and mitochondria in renal disease. Seminars in Nephrology, 32, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, M. , Iwaisako, K. , Jiang, C. , & Kisseleva, T. (2012). Cell signals influencing hepatic fibrosis. International Journal of Hepatology, 2012, 158547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe, M. J. , Doetschman, T. , & Greenhalgh, D. G. (2000). Delayed wound healing in immunodeficient TGF‐beta 1 knockout mice. Journal of Investigative Dermatology, 115, 3–11. [DOI] [PubMed] [Google Scholar]
- Darby, I. A. , Laverdet, B. , Bonté, F. , & Desmoulière, A. (2014). Fibroblasts and myofibroblasts in wound healing. Clinical, Cosmetic & Investigational Dermatology, 7, 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datto, M. B. , Li, Y. , Panus, J. F. , Howe, D. J. , Xiong, Y. , & Wang, X. F. (1995). Transforming growth factor beta induces the cyclin‐dependent kinase inhibitor p21 through a p53‐independent mechanism. Proceedings of the National Academy of Sciences of the United States of America, 92, 5545–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duscher, D. , Maan, Z. N. , Wong, V. W. , Rennert, R. C. , Januszyk, M. , Rodrigues, M. , … Gurtner, G. C. (2014). Mechanotransduction and fibrosis. Journal of Biomechanics, 47, 1997–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Gazaerly, H. , Elbardisey, D. M. , Eltokhy, H. M. , & Teaama, D. (2013). Effect of transforming growth factor beta 1 on wound healing in induced diabetic rats. International Journal of Health Sciences, 7, 160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, Y. , Chang, H. M. , Cheng, J. C. , Klausen, C. , Leung, P. C. K. , & Yang, X. (2016). Transforming growth factor‐beta1 increases lysyl oxidase expression by downregulating MIR29A in human granulosa lutein cells. Reproduction, 152, 205–213. [DOI] [PubMed] [Google Scholar]
- Fontana, L. , Chen, Y. , Prijatelj, P. , Sakai, T. , Fässler, R. , Sakai, L. Y. , & Rifkin, D. B. (2005). Fibronectin is required for integrin alpha v beta 6‐mediated activation of latent TGF‐beta complexes containing LTBP‐1. FASEB Journal, 19, 1798–1808. [DOI] [PubMed] [Google Scholar]
- Gordon, K. J. , & Blobe, G. C. (2008). Role of transforming growth factor‐beta superfamily signaling pathways in human disease. Biochimica et Biophysica Acta, 1782, 197–228. [DOI] [PubMed] [Google Scholar]
- Guichard, C. , Pedruzzi, E. , Fay, M. , Ben Mkaddem, S. , Coant, N. , Daniel, F. , & Ogier‐Denis, E. (2006). The Nox/Duox family of ROS‐generating NADPH oxidases. Medecine Sciences, 22, 953–959. [DOI] [PubMed] [Google Scholar]
- Hagler, M. A. , Hadley, T. M. , Zhang, H. , Mehra, K. , Roos, C. M. , Schaff, H. V. , … Miller, J. D. (2013). TGF‐beta signalling and reactive oxygen species drive fibrosis and matrix remodelling in myxomatous mitral valves. Cardiovascular Research, 99, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder, S. K. , Beauchamp, R. D. , & Datta, P. K. (2005). A specific inhibitor of TGF‐beta receptor kinase, SB‐431542, as a potent antitumor agent for human cancers. Neoplasia, 7, 509–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker, L. , Vittal, R. , Jones, T. , Jagirdar, R. , Luckhardt, T. R. , Horowitz, J. C. , … Thannickal, V. J. (2009). NADPH oxidase‐4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nature Medicine, 15, 1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz, B. (2009). Tissue stiffness, latent TGF‐beta1 activation, and mechanical signal transduction: Implications for the pathogenesis and treatment of fibrosis. Current Rheumatology Reports, 11, 120–126. [DOI] [PubMed] [Google Scholar]
- Hinz, B. (2015). The extracellular matrix and transforming growth factor‐beta1: Tale of a strained relationship. Matrix Biology, 47, 54–65. [DOI] [PubMed] [Google Scholar]
- Hiraga, R. , Kato, M. , Miyagawa, S. , & Kamata, T. (2013). Nox4‐derived ROS signaling contributes to TGF‐beta‐induced epithelial‐mesenchymal transition in pancreatic cancer cells. Anticancer Research, 33, 4431–4438. [PubMed] [Google Scholar]
- Hu, T. , Ramachandrarao, S. P. , Siva, S. , Valancius, C. , Zhu, Y. , Mahadev, K. , … Sharma, K. (2005). Reactive oxygen species production via NADPH oxidase mediates TGF‐beta‐induced cytoskeletal alterations in endothelial cells. American Journal of Physiology. Renal Physiology, 289, F816–F825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, M. , Rivera, S. , Synenki, L. , Zirk, A. , Eisenbart, J. , Feghali‐Black, C. , … Chandel, N. S. (2013). Mitochondrial reactive oxygen species regulate TGF‐beta signaling. Journal of Biological Chemistry, 288, 770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins, G. (2008). The role of proteases in transforming growth factor‐beta activation. The International Journal of Biochemistry & Cell Biology, 40, 1068–1078. [DOI] [PubMed] [Google Scholar]
- Jiang, F. , Liu, G. S. , Dusting, G. J. , & Chan, E. C. (2014). NADPH oxidase‐dependent redox signaling in TGF‐beta‐mediated fibrotic responses. Redox Biology, 2, 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer, A. , Green, J. , Pollard, J., Jr. , & Tugendreich, S. (2014). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics, 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, R. M. , & Desai, L. P. (2015). Reciprocal regulation of TGF‐beta and reactive oxygen species: A perverse cycle for fibrosis. Redox Biology, 6, 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda, T. , Sakabe, T. , Sunaga, A. , Sakai, K. , Rivera, A. L. , Keene, D. R. , … Sakai, T. (2011). Conversion of mechanical force into TGF‐beta‐mediated biochemical signals. Current Biology, 21, 933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer‐Ter‐Vehn, T. , Katzenberger, B. , Han, H. , Grehn, F. , & Schlunck, G. (2008). Lovastatin inhibits TGF‐beta‐induced myofibroblast transdifferentiation in human tenon fibroblasts. Investigative Ophthalmology & Visual Science, 49, 3955–3960. [DOI] [PubMed] [Google Scholar]
- Murphy‐Ullrich, J. E. , & Poczatek, M. (2000). Activation of latent TGF‐beta by thrombospondin‐1: Mechanisms and physiology. Cytokine & Growth Factor Reviews, 11, 59–69. [DOI] [PubMed] [Google Scholar]
- Nishimura, S. L. (2009). Integrin‐mediated transforming growth factor‐beta activation, a potential therapeutic target in fibrogenic disorders. American Journal of Pathology, 175, 1362–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan, C. J. , & Williams, B. (2000). Mechanical strain‐induced extracellular matrix production by human vascular smooth muscle cells: Role of TGF‐beta(1). Hypertension, 36, 319–324. [DOI] [PubMed] [Google Scholar]
- Oida, T. , & Weiner, H. L. (2010). Depletion of TGF‐beta from fetal bovine serum. Journal of Immunological Methods, 362, 195–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan, P. , Agrawal, A. , Bhushan, R. , Chavalmane, A. K. , Kalathur, R. , Takahashi, T. , & Kondaiah, P. (2007). Expression profiling of genes regulated by TGF‐beta: Differential regulation in normal and tumour cells. BMC Genomics, 8, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhyu, D. Y. , Yang, Y. , Ha, H. , Lee, G. T. , Song, J. S. , Uh, S. T. , & Lee, H. B. (2005). Role of reactive oxygen species in TGF‐beta1‐induced mitogen‐activated protein kinase activation and epithelial‐mesenchymal transition in renal tubular epithelial cells. Journal of the American Society of Nephrology, 16, 667–675. [DOI] [PubMed] [Google Scholar]
- Sampson, N. , Plas, E. , & Berger, P. (2009). NADPH oxidase 4 (NOX4) derived ROS mediate fibroblast to myofibroblast transdifferentiation in the diseased prostatic stroma. FASEB Journal, 25, 503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, M. , Zhu, J. , Wang, R. , Chen, X. , Mi, L. , Walz, T. , & Springer, T. A. (2011). Latent TGF‐beta structure and activation. Nature, 474, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal, V. J. , Lee, D. Y. , White, E. S. , Cui, Z. , Larios, J. M. , Chacon, R. , … Thomas, P. E. (2003). Myofibroblast differentiation by transforming growth factor‐beta 1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. Journal of Biological Chemistry, 278, 12384–12389. [DOI] [PubMed] [Google Scholar]
- Thuault, S. , Valcourt, U. , Petersen, M. , Manfioletti, G. , Heldin, C. H. , & Moustakas, A. (2006). Transforming growth factor‐beta employs HMGA2 to elicit epithelial‐mesenchymal transition. The Journal of Cell Biology, 174, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrecchia, F. , & Mauviel, A. (2002). Transforming growth factor‐beta signaling through the Smad pathway: Role in extracellular matrix gene expression and regulation. Jounal of Investigative Dermatology, 118, 211–215. [DOI] [PubMed] [Google Scholar]
- Wan, M. , Li, C. , Zhen, G. , Jiao, K. , He, W. , Jia, X. , … Cao, X. (2012). Injury‐activated transforming growth factor beta controls mobilization of mesenchymal stem cells for tissue remodeling. Stem Cells, 30, 2498–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Qin, W. , Zhou, Y. , Chen, B. , Zhao, X. , Zhao, H. , … Ning, J. (2017). Transforming growth factor beta plays an important role in enhancing wound healing by topical application of povidone‐iodine. Scientific Reports, 7, 991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipff, P. J. , Rifkin, D. B. , Meister, J. J. , & Hinz, B. (2007). Myofibroblast contraction activates latent TGF‐beta1 from the extracellular matrix. The Journal of Cell Biology, 179, 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, X. , Almuzzaini, B. , Drou, N. , Kremb, S. , Yousif, A. , Farrants, A. Ö. , … Percipalle, P. (2017). Beta‐actin‐dependent global chromatin organization and gene expression programs control cellular identity. FASEB Journal, 32, 1296–1314. [DOI] [PubMed] [Google Scholar]
- Xie, X. , Deliorman, M. , Qasaimeh, M. A. , & Percipalle, P. (2018). The relative composition of actin isoforms regulates cell surface biophysical features and cellular behaviors. Biochimica et Biophysica Acta, 1862, 1079–1090. [DOI] [PubMed] [Google Scholar]
- Xie, X. , Wang, S. , Wong, T. C. , & Fung, M. (2013). Genistein promotes cell death of ethanol‐stressed HeLa cells through the continuation of apoptosis or secondary necrosis. Cancer Cell International, 13, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J. , Lamouille, S. , & Derynck, R. (2009). TGF‐beta‐induced epithelial to mesenchymal transition. Cell Research, 19, 156–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, F. , Wang, Y. , Wu, X. , Peshavariya, H. M. , Dusting, G. J. , Zhang, M. , & Jiang, F. (2014). Nox4 and redox signaling mediate TGF‐beta‐induced endothelial cell apoptosis and phenotypic switch. Cell Death & Disease, 5, e1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H. , Königshoff, M. , Jayachandran, A. , Handley, D. , Seeger, W. , Kaminski, N. , & Eickelberg, O. (2008). Transgelin is a direct target of TGF‐beta/Smad3‐dependent epithelial cell migration in lung fibrosis. FASEB Journal, 22, 1778–1789. [DOI] [PubMed] [Google Scholar]
- Zhou, S. , Zawel, L. , Lengauer, C. , Kinzler, K. W. , & Vogelstein, B. (1998). Characterization of human FAST‐1, a TGF beta and activin signal transducer. Molecular Cell, 2, 121–127. [DOI] [PubMed] [Google Scholar]
- Zi, Z. , Chapnick, D. A. , & Liu, X. (2012). Dynamics of TGF‐beta/Smad signaling. FEBS Letters, 586, 1921–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinski, J. , Tajer, B. , & Mullins, M. C. (2017). TGF‐beta family signaling in early vertebrate development. Cold Spring Harbor Perspectives in Biology. 10.1101/cshperspect.a033274 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Supporting information