

Abstract
Background
Sudden cardiac arrest (CA) often results in severe injury to the brain, and neuroprotection after CA has proved to be difficult to achieve. Herein, we sought to investigate the effects of metformin pretreatment on brain injury secondary to CA and cardiopulmonary resuscitation.
Methods and Results
Rats were subjected to 9‐minute asphyxial CA after receiving daily metformin treatment for 2 weeks. Survival rate, neurologic deficit scores, neuronal loss, AMP‐activated protein kinase (AMPK), and autophagy activation were assessed at indicated time points within the first 7 days after return of spontaneous circulation. Our results showed that metformin pretreatment elevated the 7‐day survival rate from 55% to 85% and significantly reduced neurologic deficit scores. Moreover, metformin ameliorated CA‐induced neuronal degeneration and glial activation in the hippocampal CA1 region, which was accompanied by augmented AMPK phosphorylation and autophagy activation in affected neuronal tissue. Inhibition of AMPK or autophagy with pharmacological inhibitors abolished metformin‐afforded neuroprotection, and augmented autophagy induction by metformin treatment appeared downstream of AMPK activation.
Conclusions
Taken together, our data demonstrate, for the first time, that metformin confers neuroprotection against ischemic brain injury after CA/cardiopulmonary resuscitation by augmenting AMPK‐dependent autophagy activation.
Keywords: AMP‐activated protein kinase signal transduction, autophagy, cardiac arrest, diabetic therapy/glitazones, metformin, neuroprotection
Subject Categories: Cardiopulmonary Resuscitation and Emergency Cardiac Care, Cardiopulmonary Arrest
Clinical Perspective
What Is New?
Metformin confers neuroprotection against ischemic brain injury induced by cardiac arrest/cardiopulmonary resuscitation.
AMP‐activated protein kinase–mediated autophagy activation mediates metformin's neuroprotection.
What Are the Clinical Implications?
Metformin is a potential neuroprotectant for post‐resuscitated patients.
This study not only extends the clinical application of metformin, but also provides a basis for finding the strategy of preventing and/or treating brain injury after cardiopulmonary resuscitation.
Introduction
Sudden cardiac arrest (CA) is one of the major causes of death and disability.1 The annual incidence of CA involves ≈320 000 out‐of‐hospital patients and 200 000 in‐hospital patients in the United States.2 Currently, the overall survival rate for CA patients remains low, with a rate of <15% for out‐of‐hospital CA3 and <20% for in‐hospital CA.4 Severe energy depletion is one of the main characteristics after CA. During ischemia, the decrease of ATP synthesis leads to decrease of mitochondrial membrane potential and influx of calcium to induce neuron injury. After return of spontaneous circulation (ROSC), reperfusion triggers excessive generation of oxygen‐free radicals that directly damage the cell membrane and promote inflammation.5 Because brain tissue is exquisitely susceptible to ischemia injury, CA affects most profoundly on the neurologic dysfunction, which contributes significantly to mortality and morbidity among survivors achieving ROSC. Currently, there is no effective medicine intervention to reduce neuronal death and neurologic deficits in patients with CA.
Autophagy, a highly conserved cellular catabolic process for degradation and recycling of misfolded or damaged proteins and organelles,6 is considered to be a cytoprotective mechanism under most circumstances, although pathologically increased autophagy may lead to cell death.7 A large pool of evidence has demonstrated that autophagy activation is neuroprotective in ischemic stroke.8, 9, 10 Despite the growing attention on autophagy as a novel target for ischemic brain injury, studies on agents that modulate autophagy and could be used clinically are still limited.
Metformin is the first‐line hypoglycemic agent for treating type 2 diabetes mellitus. Emerging clinical and experimental studies have demonstrated that metformin possesses a variety of other beneficial effects beyond its glucose‐lowering effect. The UK Prospective Diabetes Study has shown that long‐term metformin treatment can effectively reduce stroke incidence and cardiovascular mortality.11 Recent animal studies have further demonstrated that long‐term metformin pretreatment can obviously alleviate the damage of ischemic stroke to brain tissue.12, 13, 14 In addition, metformin is also effective in treating global cerebral ischemia,15 intracerebral hemorrhage,16 epilepsy,17 Parkinson disease,18, 19 and Huntington disease.20 However, whether and how metformin treatment is effective in reducing brain injury after CA remain uninvestigated.
AMP‐activated protein kinase (AMPK) serves as a cellular energy sensor and is activated in response to a series of metabolic stresses, such as hypoxia, oxidative stress, and glucose deprivation.21, 22 Once activated, AMPK promotes catabolic pathways to generate more ATP, and inhibits anabolic pathways, thus allowing for adaptive changes in growth and metabolism under low energy conditions.23 Emerging evidence indicates that AMPK can be activated by metformin in the brain, exerting a neuroprotective role in response to energy depletion through maintaining cellular energy homeostasis,24, 25, 26 promoting mitochondrial biogenesis,27 and increasing brain‐derived neurotrophic factor expression to promote neuronal survival.28 Recently, autophagy has been proposed as a downstream target of AMPK, and AMPK‐induced autophagy activation protects against ischemic injury to peripheral tissue injury29, 30, 31 as well as ischemic brain injury.32 These studies have prompted us to hypothesize that metformin pretreatment may prime the brain to better stand ischemic damage resulting from asphyxial CA and cardiopulmonary resuscitation (CA/CPR) through promoting AMPK‐mediated autophagy activation.
In the present study, we tested the above hypothesis by investigating the effects of long‐term metformin pretreatment on neuroprotection, AMPK activation, and autophagy induction in a rat model of asphyxial CA/CPR. Our data showed that long‐term metformin pretreatment significantly ameliorated CA/CPR‐induced neuronal injury, profoundly facilitated functional recovery. Furthermore, AMPK‐dependent autophagy activation played an important role in mediating metformin's neuroprotection.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Animal Preparation
This study was approved by the Animal Care and Use Committee of the Nanfang Hospital, Southern Medical University (Guangdong, China) and followed the National Guidelines for Animal Experimentation (No. L2016053). All procedures were in accordance with institutional guidelines. Male Sprague‐Dawley rats weighing between 300 and 320 g were obtained from the Experimental Animal Center of Southern Medical University and housed in a 12‐hour light and dark cycle with free access to water and food.
Rat Model of Asphyxial CA
Asphyxial CA/CPR was performed as previously described, with modification.33 Briefly, rats were anesthetized with pentobarbital sodium (40 mg/kg), orotracheally intubated, and connected to a ventilator (RWD). Intravascular catheters (PE50; Smiths Medical, Ashford, UK) were inserted into the right femoral artery and vein for blood pressure monitoring and drug delivery. After 5‐minute stabilization, rats were chemically paralyzed by IV vecuronium (2 mg/kg) and the endotracheal tube was disconnected for 9 minutes, leading to circulatory arrest in ≈5 minutes, which was confirmed by a gradual decrease in mean arterial pressure decreased to <20 mm Hg. At the end of 9‐minute asphyxia, CPR was initiated by injection of epinephrine (0.01 mg/kg), effective ventilation with 100% oxygen, and finger chest compressions (200 compressions/min). ROSC was defined as increase of mean arterial pressure >60 mm Hg lasting at least 5 minutes. Rats that failed to ROSC within 5 minutes and unable to be weaned from ventilator after 1‐hour observation were excluded from the continuing experiments.
To avoid spontaneous hypothermia, rectal temperatures were monitored and maintained at 37.0±0.5°C with a temperature feedback system (RWD) during the surgery. Blood glucose level was intermittently detected using a glucometer (Johnson, USA). After spontaneous breathing was confirmed, rats were weaned from ventilator. Then, after 1‐hour observation, all catheters were removed and surgical wounds were sutured. At the end of each experimental period, rats were returned to their cages with easily accessible food and water.
Drug Administration and Experimental Groups
Rats were randomized to various treatment groups by using random number table. Metformin hydrochloride (Aladdin, Shanghai, China) was dissolved in sterile saline at a concentration of 30 mg/mL, and 200 mg/kg was administered intragastrically once daily for 2 weeks, while rats in the vehicle group received equivalent volume of saline. The last gavage was performed 24 hours before induction of CA/CPR model (Figure S1A). For postarrest metformin treatment, metformin was administered intragastrically after ROSC and administered daily until the animals were euthanized. Compound C (Cc; 100 nmol/rat), an AMPK inhibitor, obtained from MCE in the form of liquid (10 mmol/L in dimethyl sulfoxide) was administered intracerebroventricularly (i.c.v.). Chloroquine (25 mg/kg, IP), an autophagy inhibitor, was purchased from Sigma‐Aldrich (St Louis, MO). The dose and administration route were chosen according to a previous study.34
The present study included 3 parts (Figure S1B). In part 1, the effects of metformin pretreatment in the CA/CPR model were assessed. Forty rats that successfully resuscitated (n=20 for each group) were followed up for 7 days, and survival, neurologic outcome, and histological injury were evaluated. In parts 2 and 3, AMPK activation and autophagy biomarkers were examined at indicated time points after the indicated treatments.
Survival Study and Neurologic Function Evaluation
Rats subjected to CA/CPR were followed up to 7 days, and survival rate was recorded daily. A previously validated scale of neurologic deficit score (NDS)35 was used to assess the neurologic outcome at 24, 48, and 72 hours and 7 days after ROSC, which is a composite of consciousness, motor, sensory, reflex, and balance tests, and was conducted by 2 investigators (K.L., K.H.) who were blinded to animal grouping. An NDS of 80 was considered neurologically normal, whereas an NDS of 0 indicated brain death.
Histological Examination
Brains were fixed with 4% paraformaldehyde overnight and embedded in paraffin, and 4‐μm‐thick sections were cut and stained with cresyl violet (Beyotime, Hangzhou, China) to assess the number of surviving neurons. The primary antibodies used were microtubule‐associated protein 2 (MAP2; Abcam, Cambridge, UK) for dendrite, glial fibrillary acidic protein (GFAP; Abcam) for astrocyte, and ionized calcium‐binding adapter molecule‐1 (Iba‐1; Wako, Osaka, Japan) for microglia. Cell counting was performed on 3 randomly selected nonoverlapping fields in the hippocampal CA1 region per slide by 3 independent observers (K.L., K.H., Y.G.) blinded to the experimental groups. The number of Nissl‐positive neurons and the relative intensity of MAP2, Iba‐1, and GFAP staining were determined with ImageJ software, 1.49v (National Institutes of Health, Bethesda, MD).
For immunofluorescence, brain sections were incubated in specific primary antibodies as follows: Microtubule‐associated protein 1 light chain 3 (LC3;1:300; Abcam) and Neuron (NeuN;1:200; Abcam). Bound primary antibodies were detected with Alexa Fluor dye of donkey antimouse or antirabbit (Abcam) and observed with a confocal microscope (FluoView; Olympus, Tokyo, Japan). Digital images were recorded and analyzed using ImageJ.
Transmission Electron Microscopic Examination
Twenty‐four hours after CA/CPR, rats were perfused with precooled PBS, followed by PBS containing 2% paraformaldehyde and 2% glutaraldehyde after anesthetization. The brains were removed and cut into small sections, and they were kept overnight in 2.5% glutaraldehyde in 0.1 mol/L PBS. The next day, the sections were immersed in 1% osmium tetroxide for 1 hour, dehydrated in graded ethanol, and embedded in epoxy resin. Polymerization was performed at 80°C for 24 hours. Sections were cut into ultrathin sections (60–70 nm) with an ultramicrotome, poststained with uranyl acetate and lead citrate, and viewed under a transmission electron microscope.
Western Blot Analysis
Brain tissues underwent lysis and were used for immunoblotting, as previously reported.36 Antibodies against Phospho‐AMPK (p‐AMPK;1:1000; Cell Signaling Technology, Beverly, MA), AMPK (1:1000; Cell Signaling Technology), LC3 (1:1000; Abcam), p62 (1:1000; Abcam), and mouse anti–β‐actin (1:10 000; CWBIO, Beijing, China) were used. The densities of protein bands were quantified by using ImageJ and normalized to β‐actin.
Statistical Analysis
All data were presented as means±SDs. Continuous data were analyzed with unpaired t tests or one‐way analysis of variance, followed by post hoc multiple comparison tests (least significant difference test). Physiological variables and NDSs were examined by 2‐factor (group×time) repeated‐measures ANOVA. Difference in survival rate was analyzed by the log‐rank test. SPSS 20.0 (IBM, Armonk, NY) and GraphPad Prism 6.0 (GraphPad, La Jolla, CA) were used for statistical analyses. P<0.05 was considered statistically significant.
Results
Metformin Pretreatment Improves Animal Survival and Neurologic Recovery in Post‐CA/CPR Rats
A total of 242 rats were prepared for the study (Figure S1B). Among them, 222 underwent asphyxial CA/CPR, whereas the remaining 20 rats received sham operation. Thirty‐eight rats failed to ROSC, randomly distributed in the metformin group (14/74), the vehicle group (12/74), the metformin+Compound C group (3/19), the metformin+vehicle 1 group (3/18), the metformin+chloroquine group (4/20), and the metformin+vehicle group (2/17), which were excluded from the continuing experiments. The success rates of CPR modeling in these groups were compared, and no significant differences were observed among groups. For animals that successfully resuscitated (n=184), 13 rats (3 in the metformin group, 5 in the vehicle group, 2 in the metformin+Cc group, 1 in the metformin+vehicle 1 group, and 2 in the metformin+chloroquine group) resuscitated from CA but were unable to be weaned from ventilator, which were also excluded according to our predefined exclusion criteria. There were no significant differences in the fail rate of removing ventilator among these groups. There was no significant difference in body weight, time from asphyxia to CA, and time required for ROSC among the 4 experimental groups (Table 1). As shown in Table 2, there were also no statistically significant differences in physiological variables (mean arterial pressure, heart rate, rectal temperature, glucose level, and lactate level) among the 4 experimental groups.
Table 1.
Baseline Characteristics Among the 4 Experimental Groups
| Parameters | Vehicle (n=20) | Metformin (n=20) | Metformin+Vehicle (n=10) | Metformin+Cc (n=10) |
|---|---|---|---|---|
| Body weight, g | 403±18 | 394±26 | 391±16 | 392±19 |
| Time from asphyxia to CA, s | 222.0±26 | 213.9±17 | 232.8±37 | 237.0±27 |
| Time required for ROSC, s | 98.77±32 | 96.67+31 | 91.0±19 | 92.6±42 |
CA indicates cardiac arrest; Cc, compound C; ROSC, return of spontaneous circulation.
Table 2.
Physiological Parameter Comparison Across the Groups
| Parameters | Time Points | Vehicle | Metformin | Metformin+Vehicle | Metformin+Cc |
|---|---|---|---|---|---|
| Mean arterial pressure, mm Hg | Baseline | 127±9 | 128±10 | 127±11 | 126±11 |
| 30 min | 84±13 | 82±13 | 80±10 | 82±15 | |
| 60 min | 80±9 | 81±12 | 89±3 | 80±11 | |
| Heart rate, beats/min | Baseline | 429±39 | 421±32 | 423±37 | 418±24 |
| 30 min | 355±33 | 352±26 | 365±27 | 347±28 | |
| 60 min | 380±33 | 375±20 | 379±25 | 360±22 | |
| Rectal temperature, °C | Baseline | 36.9±0.4 | 37.0±0.3 | 36.9±0.2 | 36.9±0.2 |
| 30 min | 36.8±0.3 | 37.0±0.2 | 36.9±0.3 | 36.9±0.2 | |
| 60 min | 36.8±0.2 | 37.1±0.2 | 37.0±0.3 | 37.0±0.2 | |
| Glucose, mmol/L | Baseline | 5.8±0.7 | 5.8±0.8 | 5.6±0.4 | 5.7±0.5 |
| 30 min | 6.0±0.6 | 6.0±0.5 | 5.8±0.3 | 5.7±0.4 | |
| 60 min | 6.0±0.6 | 5.8±0.4 | 5.8±0.6 | 6.0±0.5 | |
| Lactate, mmol/L | Baseline | 1.71±0.14 | 1.72±0.16 | 1.74±0.13 | 1.73±0.13 |
| 30 min | 3.64±0.68 | 3.68±0.54 | 3.63±0.50 | 3.65±0.55 | |
| 60 min | 3.33±0.46 | 3.29±0.35 | 3.07±0.35 | 3.24±0.39 |
Physiological variables were measured at baseline and at 30 and 60 minutes after return of spontaneous circulation. The values are expressed as mean±SD. Cc indicates compound C.
The 7‐day survival experiment showed that 55% of the rats (11 of 20) in the vehicle group survived at day 7, which was much higher than that in the metformin group (85%; 17 of 20) (P<0.05) (Figure 1A). In addition, we also assessed the NDS that represents the neurologic deficiency, and found that the NDS at 24, 48, and 72 hours after ROSC was significantly higher in the metformin group than those in the vehicle group (P<0.05) (Figure 1B). No significant difference was found at 7 days, although an improved trend was observed in the metformin group. Together, these results suggest that long‐term metformin pretreatment improves 7‐day survival and neurologic outcome after CA/CPR.
Figure 1.
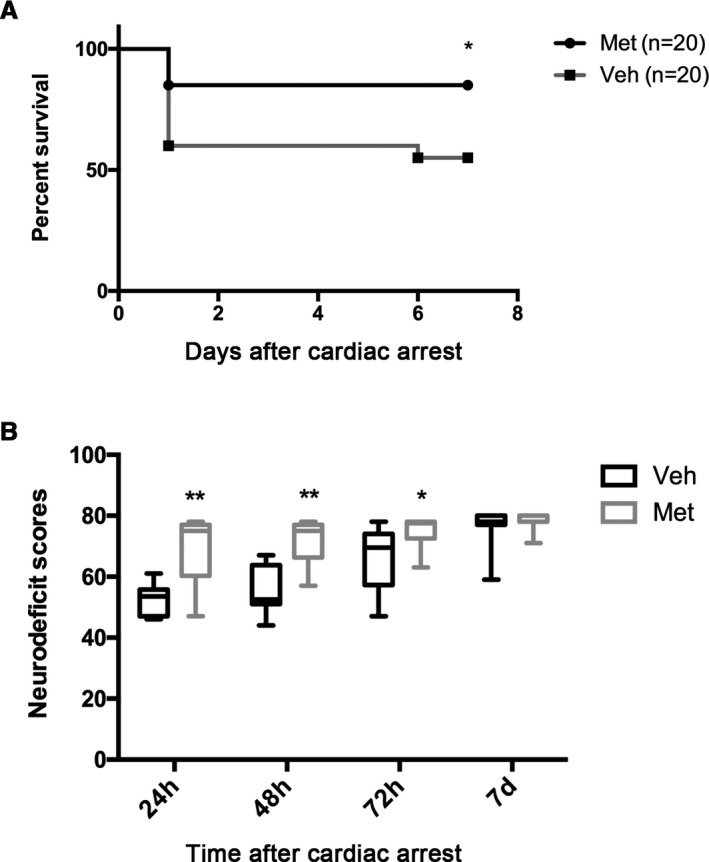
Effects of metformin (Met) pretreatment on survival and neurologic outcome after cardiac arrest and cardiopulmonary resuscitation. A, Kaplan‐Meier analyses of cumulative survival during 7‐day follow‐up after return of spontaneous circulation (ROSC). B, Neurologic deficit scores (0=brain death; 80=normal) of survived rats at 24, 48, and 72 hours and at 7 days after ROSC. *P<0.05, **P<0.01 vs vehicle (Veh) group.
Metformin Pretreatment Attenuates Neuronal Death and Suppresses Microglia and Astrocyte Activation in the Hippocampal CA1 Region
Transient global cerebral ischemia and reperfusion after CA can lead to delayed neuronal death and activation of microglia and astrocytes in selectively vulnerable areas, such as the hippocampal CA137; we, therefore, conducted histological analysis of the number of surviving neurons in hippocampal CA1 region. As shown in Figure 2A and 2E, the surviving neurons assessed by Nissl staining were significantly reduced in the vehicle‐treated CA/CPR group compared with sham‐operated post‐CA/CPR rats, and this reduction was significantly attenuated by metformin pretreatment. Similar results were observed for MAP2, a protein that is enriched in neuronal dendrites and acts as a stabilizing molecule for the dendritic cytoskeletal integrity. Extensive loss of MAP2‐immunoreactive dendrites was clearly observed in hippocampus CA1 region of vehicle‐treated CA/CPR rats, and this dendritic loss was significantly attenuated by metformin pretreatment (Figure 2B and 2F).
Figure 2.
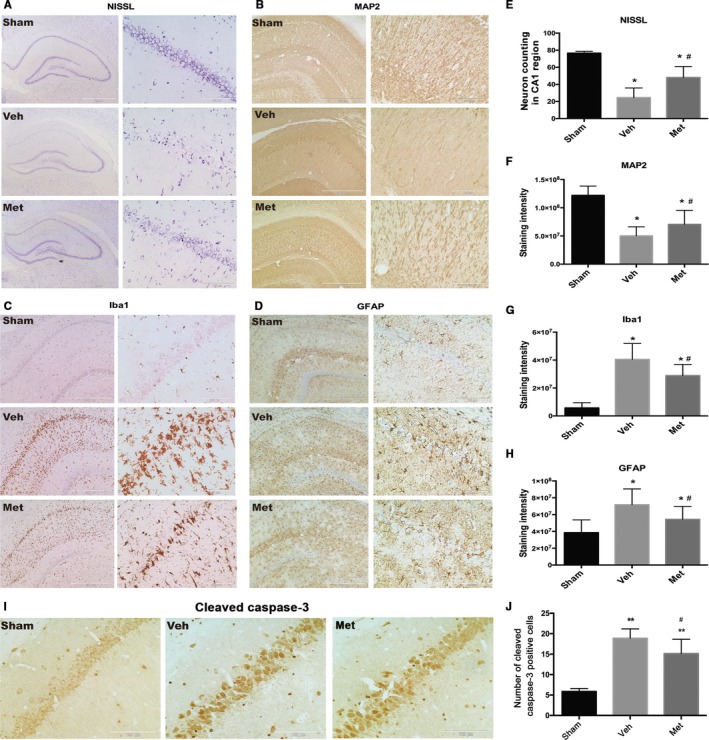
Effects of metformin (Met) pretreatment on neuronal death and activation of microglia and astrocytes after cardiac arrest and cardiopulmonary resuscitation. Representative photomicrographs of immunohistochemistry for neuron loss (Nissl; A), microtubule‐associated protein 2 (MAP2; B), ionized calcium‐binding adapter molecule 1 (Iba‐1; C), and glial fibrillary acidic protein (GFAP; D) in the hippocampal CA1 region of the sham‐operated and the experimental groups (vehicle‐treated group [Veh] and Met‐treated group) at 7 days after return of spontaneous circulation. Scale bar: 1 mm (A) and 500 μm (B through D) for lower magnification and 100 μm (A through D) for higher magnification. E through H, The semiquantitative results of Nissl, MAP2, Iba‐1, and GFAP, respectively. I and J, Representative photomicrographs of immunohistochemistry of cleaved caspase 3 antibody (brown granules) and quantification of cleaved caspase 3–positive cells. Scale bar: 100 μm (I). *P<0.05, **P<0.01 vs the sham‐operated group; # P<0.05 vs the Veh group.
In addition, neuroinflammation was activated in hippocampus CA1 region after CA/CPR, evidenced by significantly increased numbers of Iba1‐positive microglia and GFAP‐positive astrocytes in this area of vehicle‐treated rats subjected to CA/CPR (Figure 2C and 2D). Metformin pretreatment significantly suppressed the activation of microglia and astrocytes compared with the vehicle‐treated group (Figure 2G and 2H). Taken together, metformin pretreatment significantly ameliorated histological injury in post‐CA rats.
Metformin Attenuates CA/CPR‐Induced Apoptosis of Hippocampal Neurons at CA1 Region
To investigate whether metformin pretreatment could ameliorate neuronal apoptosis at the hippocampal CA1 region in the post‐CA/CPR rats, we conducted immunostaining of cleaved caspase 3 to determine the apoptosis of neurons at 24 hours after CA/CPR. Compared with the sham group, a robust degenerative reaction was detected in the hippocampal CA1 region of the vehicle group (P<0.01). Furthermore, the administration of metformin significantly ameliorated the neuroapoptosis induced by CA/CPR (P<0.05) (Figure 2I and 2J).
Metformin Activates AMPK in Hippocampal Neurons After CA/CPR
To further study the mechanism underlying the neuroprotective effects of metformin, we examined the phosphorylation levels of AMPK in hippocampal tissue to determine whether the dose of metformin used in this study could activate AMPK in the brain. As shown in Figure 3A and 3B, a significant increase in the phosphorylation of AMPK was observed at 48 hours after CA/CPR (P<0.05 versus sham). In metformin‐pretreated group, AMPK activation occurred as early as 6 hours after CA/CPR and remained elevated for up to 48 hours (P<0.01 versus sham). Moreover, pretreatment with metformin significantly augmented the CA/CPR‐induced increase in the phosphorylation and activation of AMPK (P<0.05 versus vehicle). These results indicate that AMPK activation occurred earlier and in an augmented amplitude in the hippocampus of metformin‐pretreated rats subjected to CA/CPR compared with vehicle‐treated group.
Figure 3.
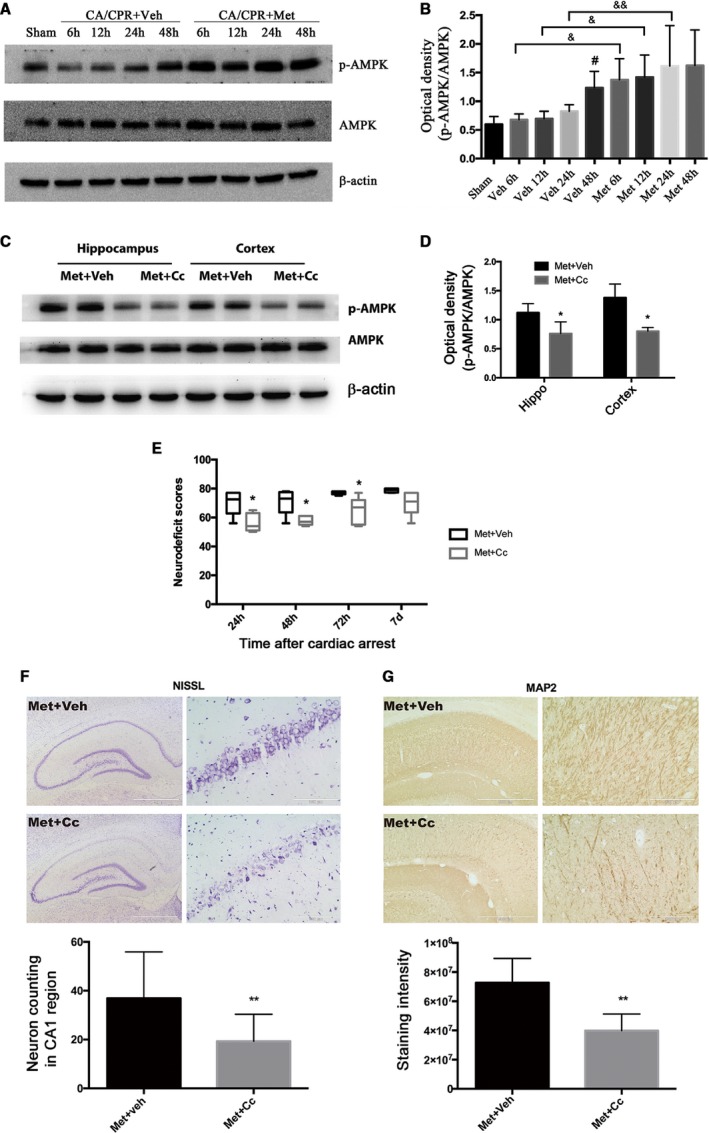
Metformin (Met)–stimulated AMP‐activated protein kinase (AMPK) phosphorylation and the effects of inhibition of AMPK on Met‐afforded neuroprotection in cardiac arrest and cardiopulmonary resuscitation (CA/CPR)–treated rats. A, Representative immunoblots of p‐AMPK, AMPK and β‐actin. β‐Actin served as a loading control. B, Optical densities of p‐AMPK and AMPK protein bands were quantitated under different treatments detected at different time points after return of spontaneous circulation (ROSC). C, Representative immunoblots of p‐AMPK and AMPK expression in hippocampal (Hippo) and cortical brain tissue showed that compound C (Cc) administration significantly inhibited Met‐induced AMPK activation. D, The optical densities of AMPK and p‐AMPK protein bands were quantitated and expressed as ratio of p‐AMPK/AMPK. E, Neurologic deficit scores (0 indicates brain death; 80 indicates normal) were assessed at 24, 48, and 72 hours and at 7 days in rats subjected to the indicated treatments after ROSC. F and G, Representative photomicrographs of immunohistochemistry for Nissl staining (neuron loss) and microtubule‐associated protein 2 (MAP2) staining (dendritic injury) and semiquantitative results in the Hippo CA1 region of Met+vehicle (Veh)– or Met+Cc‐treated rats at 7 days after ROSC. Scale bar: 1 mm (F) and 500 μm (G) for lower magnification and 100 μm (F and G) for higher magnification. *P<0.05, **P<0.01 vs the Met+Veh group; # P<0.05 vs the sham‐operated group; & P<0.05, && P<0.01 vs the relative time point groups.
Inhibition of AMPK by Cc Reversed the Neuroprotection of Metformin Against CA/CPR‐Induced Brain Injury
To determine whether AMPK activation was critical for the neuroprotective effects of metformin, we blocked AMPK signaling pathway by administration of AMPK‐specific inhibitor Cc (10 μL) 1 hour before the onset of CA/CPR. As shown in Figure 3C and 3D, administration of Cc significantly inhibited CA/CPR‐induced AMPK phosphorylation in hippocampal and cortical brain tissue, demonstrating the inhibitory effect of Cc on AMPK activation under our experimental conditions.
Then, we wondered whether blockage of AMPK pathway has impact on long‐term survival protection of metformin in CA/CPR. We next assessed the effect of AMPK inhibition with Cc on metformin‐afforded neuroprotection at 7 days after CA/CPR. As shown in Figure 3E, rats in the metformin+Cc group showed significantly lower NDS than those in the metformin+vehicle group at 24, 48, or 72 hours after CA/CPR, indicating that metformin‐induced amelioration in neurologic deficits was abolished by Cc. Similarly, Nissl staining showed that Cc treatment significantly decreased the number of surviving neurons in hippocampal CA1 region compared with vehicle treatment (Figure 3F). Moreover, dendritic injury assessed by MAP2 staining showed that significantly more dendritic damage was observed in hippocampal CA1 region of metformin+Cc‐treated rats than that of metformin+vehicle‐treated rats (Figure 3G). These results suggest that inhibition of AMPK abolished the neuroprotection against CA/CPR‐induced injury by long‐term metformin pretreatment.
Autophagy Is Activated in the Hippocampus of Post‐CA/CPR Rats
To examine the induction of autophagy after CA/CPR, we measured the levels of autophagy marker proteins of LC3 and p62 in the hippocampal tissue. The conversion of LC3‐I to LC3‐II that marks the formation of autophagosomes was detected by Western blot analysis. We found that CA/CPR induced a time‐dependent accumulation of LC3‐II, which started to increase at 12 hours and peaked between 24 and 48 hours after ROSC (Figure 4A and 4B). Autophagy activation was further verified by the change of p62 protein that started to decline at 12 hours and further attenuated at 24 and 48 hours after ROSC, because p62 expression is negatively correlated to the level of autophagy activation (Figure 4A and 4C). These results demonstrate that autophagy is induced in a time‐dependent manner after CA/CPR.
Figure 4.
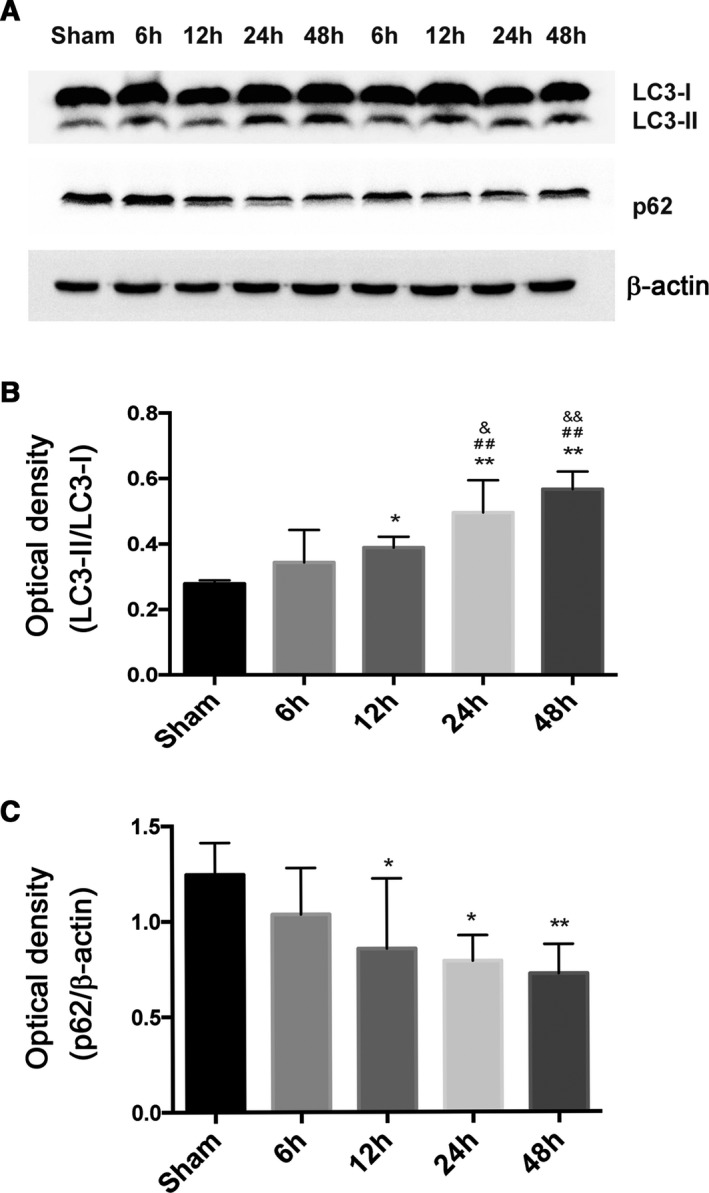
Autophagy induction in hippocampal brain tissue after cardiac arrest and cardiopulmonary resuscitation (CA/CPR). Rats were subjected to sham operation or CA/CPR. The levels of autophagy marker proteins were detected at different time points (6, 12, 24, and 48 hours) after return of spontaneous circulation (ROSC). A, Representative immunoblots of Microtubule‐associated protein 1 light chain 3 (LC3) and p62 in hippocampal brain tissue after CA/CPR. β‐Actin served as a loading control. B and C, Optical densities of LC3‐II/I and p62 protein bands were quantitated at different time points after ROSC. *P<0.05, **P<0.01 vs the sham‐operated group; ## P<0.01 vs the 6‐hour group; & P<0.05, && P<0.01 vs the 12‐hour group.
Metformin Enhances CA/CPR‐Induced Activation of Autophagy
To determine the influence of metformin on neuronal autophagy activation in the neurons of rats subjected to CA/CPR, we evaluated the LC3 punctuate, a marker of autophagy, using double immunostaining for the neuron marker NeuN and LC3‐II at 24 hours after CA/CPR. As shown in Figure 5A and 5B, the LC3‐positive punctuate could be obviously detected in the cortex of vehicle‐treated group compared with sham group (P<0.01 versus sham). Intriguingly, metformin pretreatment led to a significant increase in the number of neurons with LC3‐positive punctuate in the cortex of rats subjected to CA/CPR (P<0.01 versus vehicle), indicating that metformin pretreatment could further enhance CA/CPR‐induced activation of autophagy.
Figure 5.
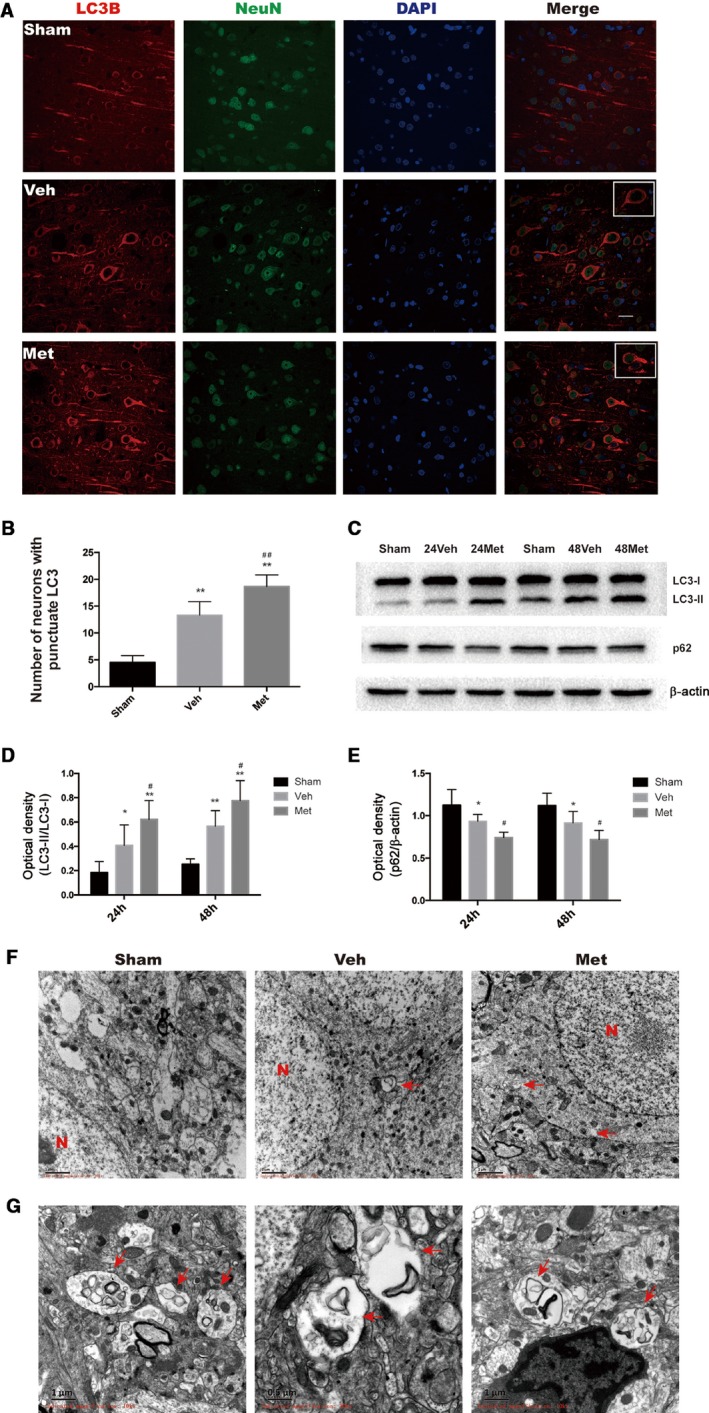
Metformin (Met) enhances autophagy activation induced by cardiac arrest and cardiopulmonary resuscitation (CA/CPR). A, Representative Microtubule‐associated protein 1 light chain 3 (LC3)/Neuron (NeuN)/4′,6‐diamidino‐2‐phenylindole (DAPI) photomicrographs of the cortex in different groups. Met increased LC3‐II immunofluorescence‐positive neurons. Scale bar: 20 μm. B, Quantitative analysis showing that Met increased the number of NeuN‐positive cells expressing LC3‐II punctuate staining. C, Representative photographs of Western blot analysis showing the expression of LC3‐II and p62 in the hippocampal tissue at 24 and 48 hours after CA/CPR. D and E, Quantitative analysis showing that Met further increased the LC3‐II/LC3‐I ratio and decreased the expression of p62. F, Representative electron micrographs showing autophagic vacuoles in hippocampal brain tissue. G, High magnification of electron micrographs showing structures of autophagosomes. The red arrows point to autophagosomes. *P<0.05, **P<0.01 vs sham group; # P<0.05, ## P<0.01 vs vehicle (Veh)–treated group.
Then, we next analyzed the changes of LC3 and p62 proteins in hippocampal tissue at 24 and 48 hours after CA/CPR in rats treated with or without metformin. A significant increase in LC3‐II formation was observed in the hippocampal tissue at 24 hours (P<0.05 versus sham) and 48 hours (P<0.01 versus sham) after CA/CPR (Figure 5C and 5D). Moreover, this increase was augmented in rats pretreated by metformin (P<0.05 versus vehicle). Accordingly, the protein level of p62 was significantly decreased in the hippocampus of rats subjected to CA/CPR (Figure 5C and 5E, P<0.05 versus sham), and metformin pretreatment augmented this decrease (P<0.05 versus vehicle).
Last, we examined autophagy with electron microscopy in brain tissue at 24 hours after CA/CPR, in which autophagosomes and related autophagic vacuoles were monitored. As shown in Figure 5F and 5G, autophagic vacuoles were clearly seen in hippocampal neurons of vehicle‐ and metformin‐pretreated rats subjected to CA/CPR, but not in sham‐operated rats, at 24 hours after ROSC.
Induction of Autophagy Contributes to Metformin‐Afforded Neuroprotection
To determine whether the induction of autophagy contributes to neuroprotection elicited by long‐term metformin pretreatment, we administered chloroquine, a pharmacological inhibitor of autophagy, 1 hour before the onset of CA/CPR. We first verified the inhibitory effect of chloroquine on autophagy at 24 hours after CA/CPR. As shown in Figure 6A, by blocking the last step of autophagic flux, chloroquine prevented the lysosomal degradation of LC3‐II and thus led to increased accumulation of LC3‐II protein in both hippocampus (P<0.05 versus vehicle) and cortex (P<0.05 versus vehicle) (Figure 6B). Moreover, chloroquine treatment significantly reversed metformin‐induced reduction of p62 protein levels (Figure 6C), further supporting the inhibitory role of chloroquine in autophagy (because p62 level is negatively correlated to the extent of autophagy activation).
Figure 6.
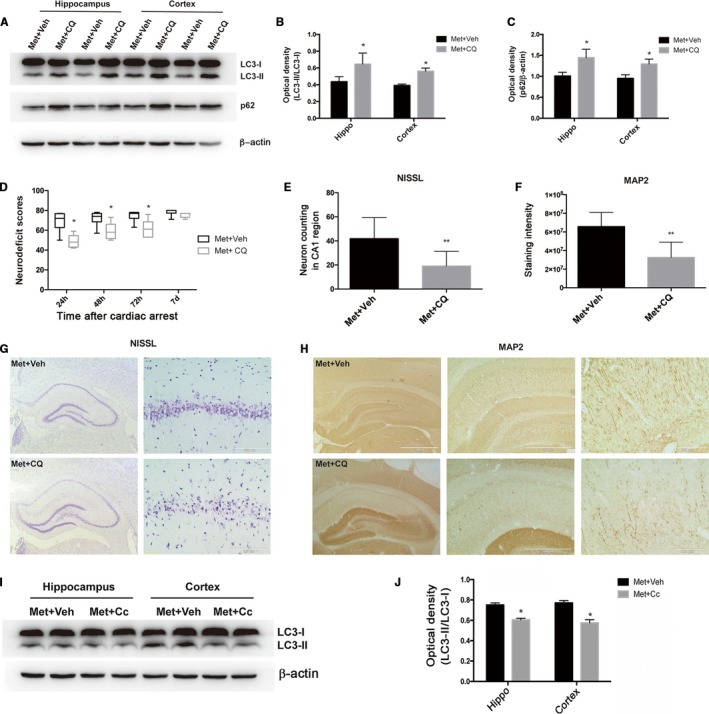
Inhibition of autophagy abolished the metformin (Met)–afforded neuroprotection in cardiac arrest and cardiopulmonary resuscitation (CA/CPR)–treated rats. A, Representative immunoblots of LC3‐II and p62 expression in hippocampal (Hippo) and cortical brain tissue showed that chloroquine (CQ) administration significantly inhibited Met‐induced autophagy activation. B and C, The optical densities of LC3‐II and p62 protein bands were quantitated and expressed as ratio of LC3‐II/I and p62/β‐actin, which showed that CQ administration significantly inhibited Met‐induced autophagy induction. D, Neurologic deficit scores in rats subjected to the indicated treatments after return of spontaneous circulation (ROSC). E and F, Semiquantitative results of Nissl and microtubule‐associated protein 2 (MAP2) staining, respectively. G and H, Representative photomicrographs of immunohistochemistry for Nissl staining (neuron loss) and MAP2 staining (dendritic injury) in the Hippo CA1 region of Met+vehicle (Veh)– or Met+CQ‐treated rats at 7 days after ROSC. Scale bar: 1 mm (left panels of G and H) and 500 μm (middle panels of H) for lower magnification and 100 μm (right panels of G and H) for higher magnification. I, Representative immunoblots of LC3 in Hippo and cortical brain tissue after CA/CPR. β‐Actin served as a loading control. J, Optical densities of LC3‐II and LC3‐I protein bands were quantitated. *P<0.05, **P<0.01 vs the Met+Veh group. Cc indicates compound C.
Then, we investigated the influence of chloroquine on neuroprotective effect of metformin. As shown in Figure 6D, metformin+chloroquine group showed significantly lower NDS than those in the metformin+vehicle group at 24, 48, or 72 hours after CA/CPR, indicating that metformin‐induced amelioration in neurologic deficits was abolished by chloroquine. Moreover, Nissl staining and MAP2 staining further showed that chloroquine treatment significantly decreased the number of surviving neurons and aggravated dendritic injury in hippocampal CA1 region compared with vehicle‐treated rats (Figure 6G, 6H, 6E and 6F).
Taken together, chloroquine abolished the neuroprotective effect of metformin, indicating the induction of autophagy may mediate, at least partly, the neuroprotecion of metformin against CA‐induced brain injury.
Inhibition of AMPK by Cc Reduced Cerebral Autophagy Activation Induced by Metformin in the Brain of Rats Subjected to CA/CPR
The above data have demonstrated that metformin augmented AMPK activation and autophagy induction in the brain tissue of rats subjected to CA/CPR. To determine whether there was an intrinsic link between these 2 changes, we assessed the effect of AMPK inhibitor Cc on autophagy induction in the brain of the rats subjected to CA/CPR. As shown in Figure 6I and 6J, pretreatment with Cc significantly attenuated the protein levels of LC3‐II in hippocampal and cortical brain tissue at 24 hours after CA/CPR in metformin‐pretreated rats compared with vehicle treatment (P<0.05, versus metformin+vehicle). These data indicate that metformin pretreatment may augment autophagy induction in the brain through promoting AMPK activation during CA/CPR injury.
Postarrest Metformin Treatment Ameliorated Histological Injury in the Hippocampal CA1 Region
To determine whether postarrest metformin treatment could also reduce neurologic injury after CA/CPR, we intragastrically administered metformin to rats (5 or 6 rats per group) daily after ROSC and assessed its effect on the histological injury in the hippocampal CA1 region. As shown in Figure 7, chloroquine or Cc alone did not ameliorate the neuron loss and dendritic injury comparing the vehicle group, and as expected, metformin alone did not affect the basal neuron numbers in hippocampal CA1 region in sham‐operated rats (Figure 7A and 7B). Postarrest metformin treatment significantly prevented the extensive loss of MAP2‐immunoreactive dendrites as well as neuronal loss assessed by Nissl staining in hippocampus CA1 region, and this protection was attenuated by pretreatment with Cc or chloroquine (Figure 7A, 7B, 7E and 7F). Moreover, Iba1‐positive microglia and GFAP‐positive astrocytes were significantly increased in rats subjected to CA/CPR, and this neuroinflammatory response was partly suppressed by postarrest metformin treatment (Figure 7C, 7D, 7G and 7H). Again, pretreatment with Cc or chloroquine abolished metformin‐afforded inhibition on neuroinflammation, evidenced by more Iba1‐positive microglia and GFAP‐positive astrocytes in the metformin+Cc‐ and metformin+chloroquine‐treated groups than the metformin‐treated group. Taken together, postarrest metformin treatment significantly ameliorated histological injury and neuroinflammation in rats subjected to CA/CPR, and inhibition of AMPK or autophagy effectively abolished metformin‐afforded neuroprotection.
Figure 7.
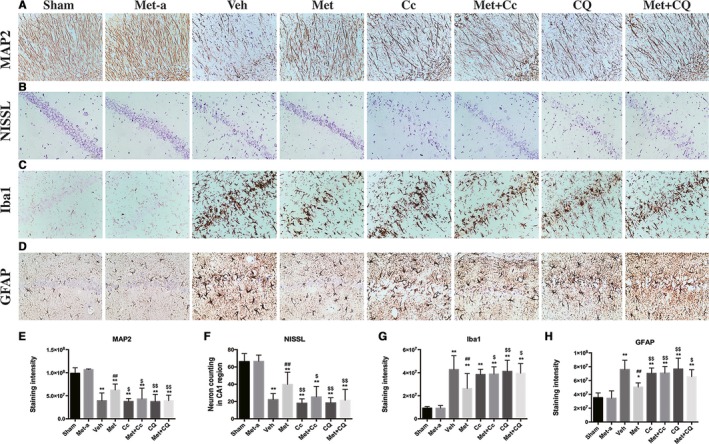
Effects of postarrest metformin (Met) treatment on neuronal death and activation of microglia and astrocytes after cardiac arrest and cardiopulmonary resuscitation (CA/CPR). Representative photomicrographs of immunohistochemistry for microtubule‐associated protein 2 (MAP2; A), neuron loss (Nissl; B), ionized calcium‐binding adapter molecule 1 (Iba‐1; C), and glial fibrillary acidic protein (GFAP; D) in the hippocampal CA1 region of the sham‐operated, sham‐treated with Met (Met‐a), and the experimental groups (vehicle‐treated group [Veh], Met‐treated group, compound C [Cc]–treated group, Met+Cc‐treated group, chloroquine [CQ]–treated group, and Met+CQ‐treated group) at 7 days after return of spontaneous circulation. Scale bar: 50 μm. E through H, The semiquantitative results of MAP2, Nissl, Iba‐1, and GFAP, respectively. *P<0.05, **P<0.01 vs the sham‐operated group; ## P<0.01 vs the Veh group; § P<0.05; §§ P<0.01 vs the Met group.
Discussion
Metformin is the first‐line hypoglycemic agent for the treatment of type 2 diabetes mellitus. Accumulating evidence has demonstrated that metformin possesses a variety of pleiotropic effects beyond its glucose‐lowering effect. In this study, we investigated the effect of long‐term metformin treatment on the survival and neurologic outcome of rats subjected to CA/CPR. The major findings are as follows: (1) pretreatment by metformin significantly improves the neurologic outcome and survival rate in rats subjected to CA/CPR; (2) metformin reduces neuronal death and suppresses the activation of microglia and astrocytes in the hippocampal CA1 region; (3) metformin pretreatment results in an early and sustained AMPK activation in hippocampal brain tissue; (4) autophagy is induced in a time‐dependent manner after CA/CPR, and long‐term metformin pretreatment further enhances autophagy induction; and (5) inhibition of AMPK or autophagy abolishes metformin‐afforded neuroprotection, and AMPK activation contributes to the augmentation of autophagy induction by metformin. Our results indicate that long‐term metformin treatment is effective in protecting the brain against CA/CPR‐induced injury.
To our knowledge, this is the first report demonstrating the neuroprotection of metformin on the rat model of CA/CPR. A large‐scale clinical trial has revealed that metformin can reduce the risk of all‐cause mortality and stroke independent of its hypoglycemic effects.11 Since then, a great deal of attention has been attracted to the study of metformin's “off‐target” (ie, beyond its glucose‐lowering effect) protection. Several studies have shown that metformin is effective in reducing ischemic injury.12 The mechanisms underlying this protection include the following: inflammation inhibition,14 blood‐brain barrier protection,13 promotion of neurogenesis and enhancement of spatial memory formation,38 promotion of the growth of nerve and recovery,39, 40 and improvement of poststroke angiogenesis and recovery.40 Consistent with previous studies, herein, our data demonstrate that long‐term metformin pretreatment affords strong neuroprotection and can be lifesaving under the conditions of CA/CPR. Moreover, our data show that metformin pretreatment suppresses the activation of microglia and astrocytes, which may represent part of the mechanism accounting for metformin‐mediated neuroprotection in CA/CPR because microgliosis and astrocytosis are well known to contribute to delayed neuronal loss and mitochondria dysfunction in ischemic brain injuries.41, 42, 43
Unexpected CA occurs in patients with cardiovascular diseases or clinically not recognized, occasionally in healthy people during exercise or as a consequence of accidents such as trauma, near drowning, electrocution, and strangulation.44 A frequent lack of prediction of CA onset makes its prevention difficult, which thus emphasizes the importance of effective therapeutic intervention after CA occurs. Herein, our data show that daily administration of metformin for 14 days before CA occurs can drastically attenuate brain injury induced by CA/CPR, supporting that long‐term daily use of metformin may serve as a promising strategy to limit brain injury in CA/CPR. This intervention could be practicable in the clinical “real‐world” setting because metformin is well tolerated and safe and is routinely applied to patients with prediabetic condition, diabetes mellitus, or metabolic disorders, such as obesity, that are all conditions with increased risk for sudden CA.45 Our data indicate that long‐term metformin pretreatment appears to generate a prolonged neuroprotection against CA/CPR injury. However, whether this observed neuroprotection is because metformin‐pretreated rats become more resistant to acute ischemic injury during the phase of CA (resulting in less severe initial injury) or because they have acquired a machinery that could limit reperfusion injury or facilitate recovery during the phase of CPR remains to be determined in future work. In addition to the preventive effect of metformin, we also found that daily administration of metformin for 7 days after CA occurs significantly attenuated brain injury induced by CA/CPR, which further supports metformin as a neuroprotective agent on CA/CPR‐induced brain injury. In addition, short‐term intervention of metformin has been shown to be neuroprotective in focal cerebral ischemia and reperfusion46 however, further study is required to determine whether and to what extent short‐term metformin administration is effective in reducing brain injury under CA condition.
One mechanism by which metformin protects the post‐CA brain injury is via activation of AMPK. CA results in a rapid and complete loss of oxygen and severe energy deficit, which leads to interruption of cellular function, membrane and pump dysfunction, and increased free radical generation to induce neuronal injury.5, 47, 48 The resultant energy deficiency will elevate the AMP/ATP ratio to activate AMPK in the brain.21 AMPK activation is well known to be neuroprotective under conditions of cerebral ischemia, hypoxia, and oxidative damage.49, 50 The results of the current study support a protective role for the activation of AMPK after CA/CPR. Herein, we observed a delayed AMPK activation (48 hours after CA) in rats subjected to CA/CPR. Long‐term metformin pretreatment significantly induces a much earlier (6 hours after CA) and stronger activation of AMPK in hippocampus tissue after CA/CPR. These data suggest that metformin pretreatment may enlarge AMPK‐mediated neuroprotection to help affected neurons to survive through the conditions of CA/CPR. Indeed, our data show that inhibition of AMPK with Cc abolishes the neuroprotective role of metformin, supporting an important role of AMPK in mediating metformin's neuroprotection in the condition of CA/CPR. However, how metformin pretreatment alters the pattern of AMPK activation, in particular the speeded activation, remains to be studied in future studies.
The other exciting finding in this study is that metformin enhances cerebral autophagic activities in rats after CA/CPR, which may prevent the development of brain injury. Autophagy is an essential degraded process that removes the damaged organelles and toxic proteins and subsequently recycles amino acids to facilitate cellular energy production, limit endoplasmic reticulum stress, and promote cell survival under subsequent ischemic exposure.8, 51, 52 Although controversies remain about the role of autophagic activation in stressful conditions, a great deal of studies have demonstrated that induction of autophagy contributes to the neuroprotection in response to cerebral ischemia8, 10, 52 or focal brain damage.9 As an AMPK activator, metformin has been revealed to induce autophagy through an AMPK‐dependent manner in heart tissue, which subsequently provides protection in response to stress.31 Similarly, our data herein demonstrated that autophagy induction in the brain is downstream of AMPK activation in response to CA/CPR, and metformin treatment further augments this induction. With the presence of AMPK inhibitor, metformin can no longer increase autophagic activation, suggesting that metformin‐induced autophagy activation is AMPK dependent. In addition, the neuroprotection induced by long‐term metformin pretreatment is abolished when autophagy is inhibited.
McCullough et al have reported that long‐term metformin treatment effectively reduces infarct volume through downregulating AMPK in a rat model of ischemic stroke, and short‐term treatment with metformin enhances brain injury through activating AMPK,12 supporting a deleterious effect of AMPK activation in ischemic brain injury. These contradictory results suggest that AMPK activation may act as a double‐edged sword, and its downstream effect may depend on the stimulus, the extent, and the duration of its activation. There could also be another possibility that short‐term AMPK activation and long‐term activation may act on different downstream targets, thus resulting in a different effect.
Being a glucose‐lowering drug, metformin can be naturally linked to glucose decrease and lactic acidosis, especially when an overdose is given. Fortunately, a nonclinical report at the 75th Scientific Sessions of the American Diabetes Association that studied the relationship between systemic metformin exposure and increased plasma lactate concentrations shows that intragastric administration of metformin (300 mg/kg) does not increase blood levels of lactic acid in normoglycemic rats.53 Two other large clinical studies have also demonstrated that there is no difference in the incidence of lactic acidosis between metformin‐ and non–metformin‐treated groups.54, 55 Herein, our data also demonstrate that the dosage of metformin used in the study is safe and does not cause hypoglycemia and lactic acidosis.
In conclusion, our present study demonstrates, for the first time, that long‐term metformin pretreatment confers neuroprotection in a rat model of CA/CPR, and this neuroprotective effect is associated with enhanced autophagy induction by sustained AMPK activation. Because metformin is widely used for the treatment of diabetes mellitus and other metabolic disorders, our data support that long‐term metformin administration may represent a novel and important preventive strategy to reduce CA‐induced brain injury.
Sources of Funding
This work was supported by the National Natural Science Foundation of China (No. 81471339) and President Foundation of Southern Medical University Nanfang Hospital (No. 2016L010).
Disclosures
None.
Supporting information
Figure S1. Experimental procedures and flow diagram of the Met‐pretreated study.
(J Am Heart Assoc. 2018;7:e008389 DOI: 10.1161/JAHA.117.008389.)
Contributor Information
Suyue Pan, Email: pansuyue82@126.com.
Zhong Ji, Email: jizhong@fimmu.com.
References
- 1. Field JM, Hazinski MF, Sayre MR, Chameides L, Schexnayder SM, Hemphill R, Samson RA, Kattwinkel J, Berg RA, Bhanji F, Cave DM, Jauch EC, Kudenchuk PJ, Neumar RW, Peberdy MA, Perlman JM, Sinz E, Travers AH, Berg MD, Billi JE, Eigel B, Hickey RW, Kleinman ME, Link MS, Morrison LJ, O'Connor RE, Shuster M, Callaway CW, Cucchiara B, Ferguson JD, Rea TD, Vanden Hoek TL. Part 1: executive summary: 2010 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2010;122:S640–S656. [DOI] [PubMed] [Google Scholar]
- 2. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER III, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–e322. [DOI] [PubMed] [Google Scholar]
- 3. Chan PS, McNally B, Tang F, Kellermann A. Recent trends in survival from out‐of‐hospital cardiac arrest in the United States. Circulation. 2014;130:1876–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Girotra S, Nallamothu BK, Spertus JA, Li Y, Krumholz HM, Chan PS. Trends in survival after in‐hospital cardiac arrest. N Engl J Med. 2012;367:1912–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mongardon N, Dumas F, Ricome S, Grimaldi D, Hissem T, Pene F, Cariou A. Postcardiac arrest syndrome: from immediate resuscitation to long‐term outcome. Ann Intensive Care. 2011;1:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self‐digestion. Nature. 2008;451:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci. 2006;29:528–535. [DOI] [PubMed] [Google Scholar]
- 8. Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy. 2010;6:482–494. [DOI] [PubMed] [Google Scholar]
- 9. Viscomi MT, D'Amelio M, Cavallucci V, Latini L, Bisicchia E, Nazio F, Fanelli F, Maccarrone M, Moreno S, Cecconi F, Molinari M. Stimulation of autophagy by rapamycin protects neurons from remote degeneration after acute focal brain damage. Autophagy. 2012;8:222–235. [DOI] [PubMed] [Google Scholar]
- 10. Wang P, Guan YF, Du H, Zhai QW, Su DF, Miao CY. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy. 2012;8:77–87. [DOI] [PubMed] [Google Scholar]
- 11. UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood‐glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 1998;352:854–865. [PubMed] [Google Scholar]
- 12. Li J, Benashski SE, Venna VR, McCullough LD. Effects of metformin in experimental stroke. Stroke. 2010;41:2645–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Y, Tang G, Li Y, Wang Y, Chen X, Gu X, Zhang Z, Wang Y, Yang GY. Metformin attenuates blood‐brain barrier disruption in mice following middle cerebral artery occlusion. J Neuroinflammation. 2014;11:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu XC, Jiang T, Zhang QQ, Cao L, Tan MS, Wang HF, Ding ZZ, Tan L, Yu JT. Chronic metformin preconditioning provides neuroprotection via suppression of NF‐kappaB‐mediated inflammatory pathway in rats with permanent cerebral ischemia. Mol Neurobiol. 2015;52:375–385. [DOI] [PubMed] [Google Scholar]
- 15. Ashabi G, Khalaj L, Khodagholi F, Goudarzvand M, Sarkaki A. Pre‐treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab Brain Dis. 2015;30:747–754. [DOI] [PubMed] [Google Scholar]
- 16. Qi B, Hu L, Zhu L, Shang L, Wang X, Liu N, Wen N, Hong Y, Fang D. Metformin attenuates neurological deficit after intracerebral hemorrhage by inhibiting apoptosis, oxidative stress and neuroinflammation in rats. Neurochem Res. 2017;42:2912–2920. [DOI] [PubMed] [Google Scholar]
- 17. Zhao RR, Xu XC, Xu F, Zhang WL, Zhang WL, Liu LM, Wang WP. Metformin protects against seizures, learning and memory impairments and oxidative damage induced by pentylenetetrazole‐induced kindling in mice. Biochem Biophys Res Commun. 2014;448:414–417. [DOI] [PubMed] [Google Scholar]
- 18. Kang H, Khang R, Ham S, Jeong GR, Kim H, Jo M, Lee BD, Lee YI, Jo A, Park C, Kim H, Seo J, Paek SH, Lee YS, Choi JY, Lee Y, Shin JH. Activation of the ATF2/CREB‐PGC‐1alpha pathway by metformin leads to dopaminergic neuroprotection. Oncotarget. 2017;8:48603–48618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Patil SP, Jain PD, Ghumatkar PJ, Tambe R, Sathaye S. Neuroprotective effect of metformin in MPTP‐induced Parkinson's disease in mice. Neuroscience. 2014;277:747–754. [DOI] [PubMed] [Google Scholar]
- 20. Jin J, Gu H, Anders NM, Ren T, Jiang M, Tao M, Peng Q, Rudek MA, Duan W. Metformin protects cells from mutant huntingtin toxicity through activation of AMPK and modulation of mitochondrial dynamics. Neuromolecular Med. 2016;18:581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hardie DG. AMP‐activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. [DOI] [PubMed] [Google Scholar]
- 22. Kim EK, Miller I, Aja S, Landree LE, Pinn M, McFadden J, Kuhajda FP, Moran TH, Ronnett GV. C75, a fatty acid synthase inhibitor, reduces food intake via hypothalamic AMP‐activated protein kinase. J Biol Chem. 2004;279:19970–19976. [DOI] [PubMed] [Google Scholar]
- 23. Kahn BB, Alquier T, Carling D, Hardie DG. AMP‐activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. [DOI] [PubMed] [Google Scholar]
- 24. Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S. AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem. 2009;109(suppl 1):17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sasaki H, Asanuma H, Fujita M, Takahama H, Wakeno M, Ito S, Ogai A, Asakura M, Kim J, Minamino T, Takashima S, Sanada S, Sugimachi M, Komamura K, Mochizuki N, Kitakaze M. Metformin prevents progression of heart failure in dogs: role of AMP‐activated protein kinase. Circulation. 2009;119:2568–2577. [DOI] [PubMed] [Google Scholar]
- 26. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk‐Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP‐activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ashabi G, Khodagholi F, Khalaj L, Goudarzvand M, Nasiri M. Activation of AMP‐activated protein kinase by metformin protects against global cerebral ischemia in male rats: interference of AMPK/PGC‐1alpha pathway. Metab Brain Dis. 2014;29:47–58. [DOI] [PubMed] [Google Scholar]
- 28. Yoon H, Oh YT, Lee JY, Choi JH, Lee JH, Baik HH, Kim SS, Choe W, Yoon KS, Ha J, Kang I. Activation of AMP‐activated protein kinase by kainic acid mediates brain‐derived neurotrophic factor expression through a NF‐kappaB dependent mechanism in C6 glioma cells. Biochem Biophys Res Commun. 2008;371:495–500. [DOI] [PubMed] [Google Scholar]
- 29. Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP‐activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. [DOI] [PubMed] [Google Scholar]
- 30. Wang LT, Chen BL, Wu CT, Huang KH, Chiang CK, Hwa Liu S. Protective role of AMP‐activated protein kinase‐evoked autophagy on an in vitro model of ischemia/reperfusion‐induced renal tubular cell injury. PLoS One. 2013;8:e79814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiang T, Yu JT, Zhu XC, Zhang QQ, Tan MS, Cao L, Wang HF, Shi JQ, Gao L, Qin H, Zhang YD, Tan L. Ischemic preconditioning provides neuroprotection by induction of AMP‐activated protein kinase‐dependent autophagy in a rat model of ischemic stroke. Mol Neurobiol. 2015;51:220–229. [DOI] [PubMed] [Google Scholar]
- 33. Huang K, Gu Y, Hu Y, Ji Z, Wang S, Lin Z, Li X, Xie Z, Pan S. Glibenclamide improves survival and neurologic outcome after cardiac arrest in rats. Crit Care Med. 2015;43:e341–e349. [DOI] [PubMed] [Google Scholar]
- 34. An JY, Zhou LL, Sun P, Pang HG, Li DD, Li Y, Zhang M, Song JN. Role of the AMPK signaling pathway in early brain injury after subarachnoid hemorrhage in rats. Acta Neurochir (Wien). 2015;157:781–792. [DOI] [PubMed] [Google Scholar]
- 35. Geocadin RG, Ghodadra R, Kimura T, Lei H, Sherman DL, Hanley DF, Thakor NV. A novel quantitative EEG injury measure of global cerebral ischemia. Clin Neurophysiol. 2000;111:1779–1787. [DOI] [PubMed] [Google Scholar]
- 36. Huang K, Wang Z, Gu Y, Hu Y, Ji Z, Wang S, Lin Z, Li X, Xie Z, Pan S. Glibenclamide is comparable to target temperature management in improving survival and neurological outcome after asphyxial cardiac arrest in rats. J Am Heart Assoc. 2016;5:e003465 DOI: 10.1161/JAHA.116.003465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hazelton JL, Balan I, Elmer GI, Kristian T, Rosenthal RE, Krause G, Sanderson TH, Fiskum G. Hyperoxic reperfusion after global cerebral ischemia promotes inflammation and long‐term hippocampal neuronal death. J Neurotrauma. 2010;27:753–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang J, Gallagher D, DeVito LM, Cancino GI, Tsui D, He L, Keller GM, Frankland PW, Kaplan DR, Miller FD. Metformin activates an atypical PKC‐CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell. 2012;11:23–35. [DOI] [PubMed] [Google Scholar]
- 39. Liu Y, Tang G, Zhang Z, Wang Y, Yang GY. Metformin promotes focal angiogenesis and neurogenesis in mice following middle cerebral artery occlusion. Neurosci Lett. 2014;579:46–51. [DOI] [PubMed] [Google Scholar]
- 40. Venna VR, Li J, Hammond MD, Mancini NS, McCullough LD. Chronic metformin treatment improves post‐stroke angiogenesis and recovery after experimental stroke. Eur J Neuorsci. 2014;39:2129–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Di Filippo M, Chiasserini D, Tozzi A, Picconi B, Calabresi P. Mitochondria and the link between neuroinflammation and neurodegeneration. J Alzheimers Dis. 2010;20(suppl 2):S369–S379. [DOI] [PubMed] [Google Scholar]
- 42. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tang M, Alexander H, Clark RS, Kochanek PM, Kagan VE, Bayir H. Minocycline reduces neuronal death and attenuates microglial response after pediatric asphyxial cardiac arrest. J Cereb Blood Flow Metab. 2010;30:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kirkham F. Cardiac arrest and post resuscitation of the brain. Eur J Paediatr Neurol. 2011;15:379–389. [DOI] [PubMed] [Google Scholar]
- 45. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res. 2015;116:1887–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jiang T, Yu JT, Zhu XC, Wang HF, Tan MS, Cao L, Zhang QQ, Gao L, Shi JQ, Zhang YD, Tan L. Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre‐activation of AMPK‐dependent autophagy. Br J Pharmacol. 2014;171:3146–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adrie C, Adib‐Conquy M, Laurent I, Monchi M, Vinsonneau C, Fitting C, Fraisse F, Dinh‐Xuan AT, Carli P, Spaulding C, Dhainaut JF, Cavaillon JM. Successful cardiopulmonary resuscitation after cardiac arrest as a “sepsis‐like” syndrome. Circulation. 2002;106:562–568. [DOI] [PubMed] [Google Scholar]
- 48. Huet O, Dupic L, Batteux F, Matar C, Conti M, Chereau C, Lemiale V, Harrois A, Mira JP, Vicaut E, Cariou A, Duranteau J. Postresuscitation syndrome: potential role of hydroxyl radical‐induced endothelial cell damage. Crit Care Med. 2011;39:1712–1720. [DOI] [PubMed] [Google Scholar]
- 49. Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP‐activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. [DOI] [PubMed] [Google Scholar]
- 50. Spasic MR, Callaerts P, Norga KK. AMP‐activated protein kinase (AMPK) molecular crossroad for metabolic control and survival of neurons. Neuroscientist. 2009;15:309–316. [DOI] [PubMed] [Google Scholar]
- 51. Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. [DOI] [PubMed] [Google Scholar]
- 52. Sheng R, Liu XQ, Zhang LS, Gao B, Han R, Wu YQ, Zhang XY, Qin ZH. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy. 2012;8:310–325. [DOI] [PubMed] [Google Scholar]
- 53. DeFronzo R, Fleming GA, Chen K, Bicsak TA. Metformin‐associated lactic acidosis: current perspectives on causes and risk. Metabolism. 2016;65:20–29. [DOI] [PubMed] [Google Scholar]
- 54. Bodmer M, Meier C, Krahenbuhl S, Jick SS, Meier CR. Metformin, sulfonylureas, or other antidiabetes drugs and the risk of lactic acidosis or hypoglycemia: a nested case‐control analysis. Diabetes Care. 2008;31:2086–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Salpeter SR, Greyber E, Pasternak GA, Salpeter EE. Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitus. Cochrane Database Syst Rev. 2010;14(4):CD002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Experimental procedures and flow diagram of the Met‐pretreated study.