

Abstract
Visible‐light irradiation (λ=457 nm) enabled the enantioselective ortho photocycloaddition of olefins to phenanthrene‐9‐carboxaldehydes (15 examples, 46–93 % yield, 82–98 % ee). A chiral oxazaborolidine Lewis acid (20 mol %) was employed as the catalyst. It operates by coordination to the aldehyde inducing a bathochromic absorption shift beyond the nπ* absorption of the uncomplexed aldehyde. At long wavelengths the Lewis acid complex is exclusively excited; within the complex, one enantiotopic face of the aromatic aldehyde is efficiently shielded. Lewis acid coordination also alters the type selectivity and the simple diastereoselectivity of the photocycloaddition.
Keywords: arenes, chromophores, cycloaddition, enantioselectivity, Lewis acids, photochemistry
In recent years there has been constantly increasing interest in the enantioselective catalysis of photochemical reactions. Several approaches have been reported to face the challenge of selectivity control in the excited state.1 Among others, it has been shown that acids can be employed to activate an α,β‐unsaturated carbonyl compound by coordination to the carbonyl group and by altering its absorption properties.2 Important contributions include the use of chiral Brønsted acids,3 the use of a chiral Sc‐based Lewis acid in combination with a triplet sensitizer,4, 5 and the use of chelating Rh‐based Lewis acids.6 Chiral AlBr3‐activated oxazaborolidines7 have been introduced by our group to promote enantioselective photocatalysis and have been established as very successful catalysts.8 They operate on α,β‐unsaturated carbonyl compounds by lowering the energy of the allowed (ϵ≥10 000 L mol−1 cm−1) π–π* transition and therefore shift this UV/Vis absorption band, which typically occurs around 250–290 nm, by about 40–50 nm to longer wavelength (bathochromic shift Δλ). Since α,β‐unsaturated carbonyl compounds exhibit a weak (ϵ≤100 L mol−1 cm−1) n–π* transition which occurs also bathochromically relative to the π–π* transition, there is in solution a competition between an excitation of the respective Lewis acid complex and the uncomplexed substrate. As a consequence undesired racemic background reactions occur and the catalyst loading for an effective enantioselective photochemical reaction catalyzed by a chiral oxazaborolidine has been high (50 mol %) so far.8 A possible improvement would be expected if it was possible to shift the above‐mentioned π–π* transition bathochromically beyond the n–π* transition of the respective substrate. Since an identical energy difference ΔE corresponds to a higher difference of Δλ at longer wavelength, we reasoned that chromophores with a π–π* absorption at long wavelength might allow for a selective excitation of the Lewis acid complex without interference of a racemic non‐catalytic reaction. We have now shown that phenanthrene‐9‐carboxaldehydes fulfill the above‐mentioned requirements and we report herein on their enantioselective catalytic photocycloaddition to olefins.
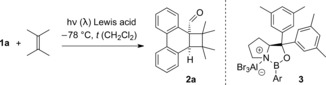
The UV/Vis spectrum of aldehyde9 1 a (Figure 1) revealed an absorption at 316 nm (ϵ=13 780 L mol−1 cm−1), which is assigned to a π–π* transition (1Lb band).10 The shoulder at 361 nm (ϵ=1940 L mol−1 cm−1) has been previously assigned to the phenanthrene 1La band,10 but it seems to partially overlap with the n–π* band of the aldehyde carbonyl group. Indeed, photochemical reactions of 1 a were observed (see below) up to an excitation wavelength of 419 nm. Upon successive addition of the Lewis acid EtAlCl2, a strong absorption became visible with a maximum at λ=387 nm. The absorption reached saturation upon addition of 15 equiv of EtAlCl2 indicating complete complexation of 1 a. The extinction coefficient was determined as ϵ=16 180 L mol−1 cm−1, which supports an assignment of this band to a π–π* transition. The band tails into the long‐wavelength region of the spectrum and there is a significant absorption beyond λ=420 nm. Photochemical experiments with achiral Lewis acids suggested a catalytic photocycloaddition reaction may be possible at λ≥420 nm. Experiments with chiral Lewis acids commenced with the previously reported8a–8c catalyst 3 a. Addition of 2,3‐dimethyl‐2‐butene to substrate 1 a occurred at −78 °C in CH2Cl2 (Table 1) exclusively at the C9/C10 double bond and resulted in product 2 a which is formally the product of an ortho photocycloaddition.11, 12 The highest enantioselectivity was achieved at λ=457 nm (entries 1–3) and the Lewis acid was subsequently optimized by varying the aryl substituent (Ar) at the boron atom (for the optimization studies, see the Supporting Information). Another known13 Lewis acid 3 b with a single substituent in ortho position on the phenyl ring gave a significant increase in ee (entry 4). Eventually, the 2,6‐dimethylphenyl derivative 3 c turned out to be the ideal Lewis acid (entry 5) and it fulfilled our expectations with regard to catalytic turnover (entries 6, 7). A loading of 10 mol % was sufficient to provide a high yield (81 %) and a high enantioselectivity (94 % ee).
Figure 1.
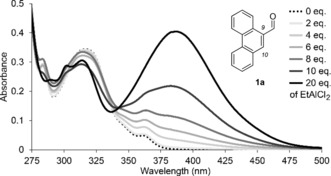
UV/Vis spectrum of compound 1 a (c=0.25 mm in CH2Cl2) in the presence of variable equiv of EtAlCl2.
Table 1.
Photocycloaddition reaction of phenanthrene‐9‐carboxaldehyde (1 a) and 2,3‐dimethyl‐2‐butene in the presence of chiral Lewis acids 3.
| Entry[a] |
λ
[a]
[nm] |
Lewis acid[b] |
Loading [mol %] |
t
[a]
[h] |
Yield[c]
[%] |
ee
[d]
[%] |
|---|---|---|---|---|---|---|
| 1 | 424 | 3 a | 50 | 14 | 53 | 56 |
| 2 | 435 | 3 a | 50 | 9 | 43 | 70 |
| 3 | 457 | 3 a | 50 | 11 | 49 | 79 |
| 4 | 457 | 3 b | 50 | 11 | 54 | 84 |
| 5 | 457 | 3 c | 50 | 11 | 62 | 97 |
| 6 | 457 | 3 c | 25 | 19 | 79 | 94 |
| 7 | 457 | 3 c | 10 | 26 | 81 | 94 |
[a] The reaction was performed at −78 °C with a substrate concentration of c=20 mm in CH2Cl2 at the indicated wavelength (λ) and for the indicated period of time (t). The olefin was used in excess (30 equiv). [b] 3 a: Ar=2,4,6‐trifluorophenyl; 3 b: Ar=2‐(trifluoromethyl)phenyl; 3 c: Ar=2,6‐dimethylphenyl. [c] Yield of isolated product. [d] The enantiomeric excess (ee) was determined by chiral‐phase HPLC analysis.
The substrate scope of the photocycloaddition was explored with some functionalized phenanthrene‐9‐carboxaldehydes 1. Their synthesis was best achieved by linkage of the two arene rings via a Suzuki cross‐coupling and by subsequent introduction of the C9/C10 bond in an aldol condensation (see the Supporting Information). To keep the reaction times low and to allow for complete conversion of all substrates a catalyst loading of 20 mol % was consistently applied (Table 2). Under these conditions the respective products were obtained in yields of 66–93 % and with enantioselectivities of 82–96 % ee. From the preliminary data set, it appears as if substitution at C3 and C6 is beneficial for the enantioselectivity while substituents at positions C2 and C5 (entries 2, 3, 6) lead to a slightly reduced enantioselectivity. Fluoro, chloro, methyl, and trifluoromethyl substitution was tolerated but the study on the functional group tolerance was limited by the availability of the starting materials. In general, compounds with Lewis basic sites (alkoxy, carbonyl) are not recommended owing to competitive binding to the Lewis acid.
Table 2.
Lewis acid catalyzed, enantioselective photocycloaddition reactions of phenanthrene‐9‐carboxaldehydes (1) and 2,3‐dimethyl‐2‐butene.
| Entry[a] | Substrate | X | Y | Product | Yield[b] [%] | ee [c] [%] |
|---|---|---|---|---|---|---|
| 1 | 1 a | H | H | 2 a | 79 | 94 |
| 2 | 1 b | 2‐Me | H | 2 b | 75 | 84 |
| 3 | 1 c | 2‐Cl | H | 2 c | 89 | 88 |
| 4 | 1 d | 3‐F | H | 2 d | 78 | 93 |
| 5 | 1 e | 3‐Me | H | 2 e | 66 | 92 |
| 6 | 1 f | H | 5‐Me | 2 f | 85 | 86 |
| 7 | 1 g | H | 6‐CF3 | 2 g | 92 | 92 |
| 8 | 1 h | H | 6‐F | 2 h | 79 | 82 |
| 9 | 1 i | 2‐Me | 6‐CF3 | 2 i | 89 | 90 |
| 10 | 1 j | 3‐Me | 6‐CF3 | 2 j | 93 | 92 |
| 11 | 1 k | 3‐F | 6‐CF3 | 2 k | 85 | 96 |
[a] The reaction was performed at −78 °C with a substrate concentration of c=20 mm in CH2Cl2 at the indicated wavelength (λ) and for the indicated period of time (t). The olefin was used in excess (30 equiv). [b] Yield of isolated product. [c] The ee was determined by chiral‐phase HPLC analysis.
The absolute configuration of the cyclobutane products was proven by anomalous X‐ray diffraction (Figure 2). Chloro derivative 2 c gave suitable crystals and the configuration at the two newly formed stereogenic centers could be unambiguously assigned. Irrespective of the mechanism of the photocycloaddition, the attack of the olefin at carbon atom C10 occurred from the Si face. It has been previously suggested7, 14 that aldehydes bind to cationic oxazaborolidine Lewis acids by coordination of the carbonyl group to the boron atom and by a second non‐classical hydrogen bond15 of the hydrogen atom to the oxazaborolidine oxygen atom.
Figure 2.
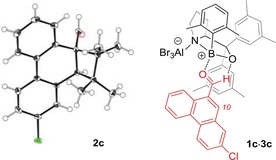
Absolute configuration of product 2 c as determined by anomalous X‐ray diffraction and model for the association of Lewis acid 3 c to phenanthrene‐9‐carboxaldehyde 1 c.
For substrate 1 c, the respective complex 1 c⋅3 c is depicted in Figure 2 and the enantioface differentiation can be explained by an attack of the olefin from the face that is not shielded by the 3,5‐dimethylphenyl ring (in gray) of the oxazaborolidine and that is relative to carbon atom C10 on the Si face. Despite the fact that complex 1 c⋅3 c provides a rationale for the stereochemical outcome of the reaction, it cannot explain at this point the subtle differences which are caused by the choice of aryl (Ar) groups at the boron atom and by the substitution pattern at the phenanthrene substrate.
In a second set of preliminary experiments, we studied the reaction of two phenanthrene‐9‐carboxaldehydes 1 a and 1 d with cyclopentene (Scheme 1). This olefin represents a frequently used cyclic reaction partner in [2+2] photocycloaddition chemistry16 and offers a first indication for the generality of the reaction. To our delight, the enantioselectivity of the Lewis acid catalyzed process remained very high. Apart from the major diastereoisomers 4 a and 5 a in which the carbocyclic rings are positioned on opposite sides of the cyclobutane core (exo), minor endo products were also detected (d.r.=diastereomeric ratio).
Scheme 1.
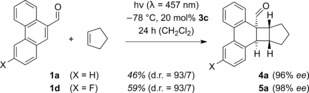
Lewis acid catalyzed, enantioselective photocycloaddition reactions of phenanthrene‐9‐carboxaldehydes 1 a and 1 d to cyclopentene.
Similarly, cyclohexene produced the respective exo product 6 a as the major diastereoisomer which was formed in 90 % ee from aldehyde 1 a (Scheme 2). Apart from the endo diastereoisomer, minimal amounts of a third diastereoisomer were found (see below). The reaction of aldehyde 1 a with isopropylidenecyclohexane gave mainly a single regioisomer 7 (r.r.=regioisomeric ratio), which was obtained in 84 % ee.
Scheme 2.
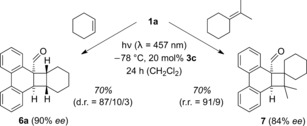
Lewis acid catalyzed, enantioselective photocycloaddition of phenanthrene‐9‐carboxaldehyde (1 a) with cyclohexene and isopropylidenecyclohexane to products 6 a and 7.
As previously noted for [2+2] photocycloaddition reactions,8d the chiral Lewis acid governs not only the enantioselectivity but also has a significant impact on other selectivity parameters. In the current study it was found that the formation of other constitutional isomers was completely suppressed in the presence of the Lewis acid. Irradiation of aldehyde 1 a and 2,3‐dimethyl‐2‐butene at λ=366 nm, 398 nm, and 405 nm led consistently to the formation of oxetane rac‐8 (Figure 3) in a ratio of about 1:5 relative to rac‐2 a. At lower wavelength (λ=254 nm, 350 nm, and 366 nm), decarbonylation reactions were observed. No Paternò–Büchi reaction17 products nor any other side products from carbonyl photochemistry were observed in the presence of the chiral Lewis acid.
Figure 3.
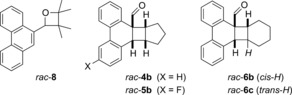
Structure of photocycloaddition products rac‐8, rac‐4 b, rac‐5 b, rac‐6 b, and rac‐6 c.
Even more remarkable was the simple diastereoselectivity of the reaction, which was almost completely inverted compared to the racemic reaction. Direct excitation of 1 a and 1 d in the presence of cyclopentene at λ=366 nm produced predominantly the endo diastereoisomers rac‐4 b (28 %, d.r.=rac‐4 a/rac‐4 b=12:88) and rac‐5 b (40 %, d.r.=9:91). In the case of cyclohexene, direct irradiation at λ=366 nm led in 31 % yield to the endo diastereoisomer rac‐6 b and the trans‐configured product rac‐6 c in almost identical amounts. A small fraction of the exo isomer rac‐6 a was also observed and the diastereomeric ratio (d.r.=rac‐6 a/rac‐6 b/rac‐6 c) was 21:35:44.
Previous work on the ortho photocycloaddition of methyl phenanthrene‐9‐carboxylate had produced evidence that its reaction (λ=350 nm) with 2,3‐dimethyl‐2‐butene proceeds via a triplet intermediate.18 Contrary to our results, it was found that any attempted photocycloaddition in the presence of a Lewis acid was sluggish but it was postulated that the Lewis acid mediated process occurred via a singlet‐state mechanism. Indeed, a change of mechanism could explain the reversal of simple diastereoselectivity in the reaction of phenanthrene‐9‐carboxaldehydes with cyclopentene and cyclohexene. Preliminary fluorescence studies on the complex of 1 a with EtAlCl2 revealed a strong fluorescence at an emission wavelength of λ em=498 nm (excitation wavelength λ exc=400 nm) which was quenched upon addition of 2,3‐dimethyl‐2‐butene. Since fluorescence quenching by energy transfer to an olefin is thermodynamically not feasible,19 it is likely that the excited singlet state of complex 1 a⋅AlEtCl2 reacts with the olefin.
In conclusion, it was found that phenanthrene‐9‐carboxaldehydes undergo an extensive bathochromic shift upon coordination to a Lewis acid. The strong band (λ max=387 nm, ϵ=16 180 L mol−1 cm−1), which is induced upon Lewis acid coordination, stretches beyond all absorption bands of uncomplexed phenanthrene‐9‐carboxaldehydes and thus enables a selective excitation of the Lewis acid complex at long wavelength. In the presence of catalytic amounts of chiral oxazaborolidine Lewis acid 3 c, an enantioselective ortho photocycloaddition was possible and a model for the complexation is suggested based on previous precedence. The result shows for the first time that the aldehyde binding motif is suitable to achieve high enantioface differentiation in photochemical reactions. Mechanistically, there is circumstantial evidence that the catalytic reaction proceeds via an excited singlet state and that this fact influences the selectivity pattern of the photocycloaddition. Further work is required to explore the subtle influence of the substituents at the oxazaborolidine and to understand the change of the selectivity pattern by the Lewis acid.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the European Research Council under the European Union's Horizon 2020 research and innovation programme (grant agreement No 665951—ELICOS) and the Fonds der Chemischen Industrie (Ph. D. fellowship to S.S.) is gratefully acknowledged. We thank O. Ackermann and J. Kudermann for their help with the HPLC and GLC analyses, and Dr. T. S. Chung for the fluorescence measurements.
S. Stegbauer, C. Jandl, T. Bach, Angew. Chem. Int. Ed. 2018, 57, 14593.
Dedicated to all past and present members of the TUM family
Contributor Information
M. Sc. Simone Stegbauer, http://www.oc1.ch.tum.de/home en/
Prof. Dr. Thorsten Bach, Email: thorsten.bach@ch.tum.de.
References
- 1.
- 1a. Silvi M., Melchiorre P., Nature 2018, 554, 41–49; [DOI] [PubMed] [Google Scholar]
- 1b. Garrido-Castro A. F., Maestro M. C., Alemán J., Tetrahedron Lett. 2018, 59, 1286–1294; [Google Scholar]
- 1c. Ramamurthy V., Sivaguru J., Chem. Rev. 2016, 116, 9914–9993; [DOI] [PubMed] [Google Scholar]
- 1d. Brimioulle R., Lenhart D., Maturi M. M., Bach T., Angew. Chem. Int. Ed. 2015, 54, 3872–3890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3944–3963. [Google Scholar]
- 2. Brenninger C., Jolliffe J. D., Bach T., Angew. Chem. Int. Ed. 2018, 10.1002/anie.201804006; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 10.1002/ange.201804006. [DOI] [Google Scholar]
- 3.
- 3a. Vallavoju N., Selvakumar S., Jockusch S., Sibi M. P., Sivaguru J., Angew. Chem. Int. Ed. 2014, 53, 5604–5608; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5710–5714; [Google Scholar]
- 3b. Vallavoju N., Selvakumar S., Jockusch S., Prabhakaran M. T., Sibi M. P., Sivaguru J., Adv. Synth. Catal. 2014, 356, 2763–2768. [Google Scholar]
- 4.
- 4a. Blum T. R., Miller Z. D., Bates D. M., Guzei I. A., Yoon T. P., Science 2016, 354, 1391–1395; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Miller Z. D., Lee B. J., Yoon T. P., Angew. Chem. Int. Ed. 2017, 56, 11891–11895; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12053–12057. [Google Scholar]
- 5.For key publications on visible-light-induced triplet sensitization in [2+2] photocycloaddition reactions, see:
- 5a. Lu Z., Yoon T. P., Angew. Chem. Int. Ed. 2012, 51, 10329–10332; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10475–10478; [Google Scholar]
- 5b. Zou Y.-Q., Duan S.-W., Meng X.-G., Hu X.-Q., Gao S., Chen J.-R., Xiao W.-J., Tetrahedron 2012, 68, 6914–6919; [Google Scholar]
- 5c. Alonso R., Bach T., Angew. Chem. Int. Ed. 2014, 53, 4368–4371; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4457–4460; [Google Scholar]
- 5d. Liu Q., Zhu F.-P., Jin X.-L., Wang X.-J., Chen H., Wu L.-Z., Chem. Eur. J. 2015, 21, 10326–10329; [DOI] [PubMed] [Google Scholar]
- 5e. Mojr V., Svobodová E., Straková K., Neveselý T., Chudoba J., Dvořáková H., Cibulka R., Chem. Commun. 2015, 51, 12036–12039; [DOI] [PubMed] [Google Scholar]
- 5f. Tröster A., Alonso R., Bauer A., Bach T., J. Am. Chem. Soc. 2016, 138, 7808–7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Huang X., Quinn T. R., Harms K., Webster R. D., Zhang L., Wiest O., Meggers E., J. Am. Chem. Soc. 2017, 139, 9120–9123; [DOI] [PubMed] [Google Scholar]
- 6b. Huang X., Li X., Xie X., Harms K., Riedel R., Meggers E., Nat. Commun. 2017, 8, 2245; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Hu N., Jung H., Zheng Y., Lee J., Zhang L., Ullah Z., Xie X., Harms K., Baik M.-H., Meggers E., Angew. Chem. Int. Ed. 2018, 57, 6242–6246; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6350–6354. [Google Scholar]
- 7.Review: Corey E. J., Angew. Chem. Int. Ed. 2009, 48, 2100–2117; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2134–2151. [Google Scholar]
- 8.
- 8a. Guo H., Herdtweck E., Bach T., Angew. Chem. Int. Ed. 2010, 49, 7782–7785; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7948–7951; [Google Scholar]
- 8b. Brimioulle R., Bach T., Science 2013, 342, 840–843; [DOI] [PubMed] [Google Scholar]
- 8c. Brimioulle R., Bauer A., Bach T., J. Am. Chem. Soc. 2015, 137, 5170–5176; [DOI] [PubMed] [Google Scholar]
- 8d. Poplata S., Bach T., J. Am. Chem. Soc. 2018, 140, 3228–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For comparison, the UV/Vis spectrum of a typical α,β-unsaturated aldehyde (cyclohexene-1-carbaldehyde) is depicted in the Supporting Information. The compound displays the π–π* absorption at λ=231 nm (ϵ=15640 L mol−1 cm−1) and the n–π* absorption at λ=315 nm (ϵ=50 L mol−1 cm−1). Upon addition of EtAlCl2, the π–π* absorption is shifted by Δλ=46 nm to λ=277 nm (ϵ=8850 L mol−1 cm−1).
- 10. Akiyama S., Nakagawa M., Nishimoto K., Bull. Chem. Soc. Jpn. 1971, 44, 1054–1062. [Google Scholar]
- 11.Reviews:
- 11a. Remy R., Bochet C. G., Chem. Rev. 2016, 116, 9816–9849; [DOI] [PubMed] [Google Scholar]
- 11b. Hoffmann N., Photochem. Photobiol. Sci. 2012, 11, 1613–1641; [DOI] [PubMed] [Google Scholar]
- 11c. Streit U., Bochet C. G., Beilstein J. Org. Chem. 2011, 7, 525–542; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. Hoffmann N., Synthesis 2004, 481–495. [Google Scholar]
- 12.For recent work on an enantioselective ortho photocycloaddition to benzofurans and benzothiophenes, see Ref. [6c].
- 13. Brimioulle R., Bach T., Angew. Chem. Int. Ed. 2014, 53, 12921–12924; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13135–13138. [Google Scholar]
- 14.
- 14a. Corey E. J., Rohde J. J., Tetrahedron Lett. 1997, 38, 37–40; [Google Scholar]
- 14b. Paddon-Row M. N., Anderson C. D., Houk K. N., J. Org. Chem. 2009, 74, 861–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnston R. C., Cheong P. H.-Y., Org. Biomol. Chem. 2013, 11, 5057–5064. [DOI] [PubMed] [Google Scholar]
- 16. Poplata S., Tröster A., Zou Y.-Q., Bach T., Chem. Rev. 2016, 116, 9748–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Fréneau M., Hoffmann N., J. Photochem. Photobiol. C 2017, 33, 83–108; [Google Scholar]
- 17b. D'Auria M. in CRC Handbook of Organic Photochemistry and Photobiology, 3rd ed. (Eds.: A. G. Griesbeck, M. Oelgemöller, F. Ghetti), CRC, Boca Raton, 2012, pp. 653–681; [Google Scholar]
- 17c. Abe M. in Handbook of Synthetic Photochemistry (Eds.: A. Albini, M. Fagnoni), Wiley-VCH, Weinheim, 2010, pp. 217–239; [Google Scholar]
- 17d. Griesbeck A. G. in Molecular and Supramolecular Photochemistry, Vol. 12 (Eds.: A. G. Griesbeck, J. Mattay), Marcel Dekker, New York, 2005, pp. 89–139; [Google Scholar]
- 17e. Bach T., Synthesis 1998, 683–703. [Google Scholar]
- 18. Lewis F. D., Barancyk S. V., Burch E. L., J. Am. Chem. Soc. 1992, 114, 3866–3870. [Google Scholar]
- 19.The absorption maxima of olefins are typically below 200 nm, corresponding to singlet-state energies above 600 kJ mol−1; see: Klán P., Wirz J., Photochemistry of Organic Compounds, Wiley, Chichester, 2009, pp. 231–232. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary