

Abstract
Protein production in Pichia pastoris is often based on the methanol‐inducible P AOX1 promoter which drives the expression of the target gene. The use of methanol has major drawbacks, so there is a demand for alternative promoters with good induction properties such as the glucose‐regulated P GTH1 promoter which we reported recently. To further increase its potential, we investigated its regulation in more details by the screening of promoter variants harboring deletions and mutations. Thereby we could identify the main regulatory region and important putative transcription factor binding sites of P GTH1. Concluding from that, yeast metabolic regulators, monomeric Gal4‐class motifs, carbon source‐responsive elements, and yeast GC‐box proteins likely contribute to the regulation of the promoter. We engineered a P GTH1 variant with greatly enhanced induction properties compared with that of the wild‐type promoter. Based on that, a model‐based bioprocess design for high volumetric productivity in a limited time was developed for the P GTH1 variant, to employ a glucose fed‐batch strategy that clearly outperformed a classical methanol fed‐batch of a P AOX1 strain in terms of titer and process performance.
Keywords: carbon source, Pichia pastoris ( P. pastoris), process engineering, promoter engineering, recombinant protein production, transcription factor binding sites (TFBS)
Abbreviations
- CSRE
carbon source responsive element
- eGFP
enhanced green fluorescent protein
- GCN
gene copy number
- OUR
oxygen uptake rate
- Pp
Pichia pastoris
- Sc
Saccharomyces cerevisiae
- TF
transcription factor(s)
- TFBS
transcription factor binding site(s)
- YDM
yeast dry mass
1. INTRODUCTION
The methylotrophic yeast Pichia pastoris (syn. Komagataella spp.) is a well‐established protein production host. Numerous strain engineering approaches for P. pastoris improved the productivity for various recombinant proteins (Ahmad, Hirz, Pichler, & Schwab, 2014; Byrne, 2015; Puxbaum, Mattanovich, & Gasser, 2015; Zahrl, Pena, Mattanovich, & Gasser, 2017) and effort was also dedicated to establish novel promoters for production purposes (Ahn et al., 2009; Landes et al., 2016; Mellitzer et al., 2014; Prielhofer et al., 2013; Vogl, Ruth, Pitzer, Kickenweiz, & Glieder, 2014). Promoters are key features for the expression of a particular gene such as transcription of RNA of a downstream (3′) ORF is driven by the upstream (5′) promoter sequence. RNA polymerase II (RNAPII) is responsible for transcription of mRNA in eukaryotes. RNAPII promoters consist of a core promoter and several cis‐acting DNA elements: proximal promoter, enhancers, silencers, and boundary/insulator elements (Marsman & Horsfield, 2012; Narlikar & Ovcharenko, 2009; Phillips‐Cremins & Corces, 2013). Yeast core promoters are typically located close (‐75/ +50 bp) to the main transcription start site, frequently containing improper TATA boxes (up to two bases difference to the TATA consensus sequence) and lack promoter elements such as Inr and DPE, which are typically found in other organisms (Lubliner, Keren, & Segal, 2013). Transcriptional regulation responds to different conditions and is conducted through cis‐acting elements and corresponding regulatory proteins (transcription factors [TFs]). A comprehensive review of carbon source‐dependent promoters in yeasts is provided by Weinhandl, Winkler, Glieder, and Camattari (2014).
For biotechnological applications, strong promoters which allow either constitutive or inducible gene expression are commonly used (Mattanovich et al., 2012). Production processes utilizing P. pastoris favorably apply carbon source‐dependent promoters such as the methanol‐inducible alcohol oxidase promoter PAOX1 (Tschopp, Brust, Cregg, Stillman, & Gingeras, 1987). Thereby, the growth phase can be partially separated from the potentially burdening protein production phase. We recently reported another set of promoters, which is also controlled by the carbon source but does not require methanol for induction. These promoters share the features of repression by excess glycerol and induction by limiting glucose (Prielhofer et al., 2013). The promoter of the high‐affinity glucose transporter gene GTH1 (PGTH1 = PG1), which is the strongest of these promoters, is fully induced below 0.05 g/L glucose and repressed at higher glucose concentrations.
P. pastoris promoter studies and random mutagenesis of PAOX1 and of the promoter of glyceraldehyde‐3‐phosphate dehydrogenase PGAP resulted in libraries of promoter variants possessing different activities, altered induction behavior compared with the wild‐type promoter and in the identification of several important transcription factor binding sites ([TFBS]; Ata, Prielhofer, Gasser, Mattanovich, & Calik, 2017; Hartner et al., 2008; Qin et al., 2011). The interesting induction behavior of PGTH1 prompted us to analyze its key promoter features. Rather than to generate libraries by random mutagenesis, we constructed length variants and variants lacking certain TFBS to study their impact on induction and repression. Based on these promoter variants, we were able to identify key regulatory elements and to engineering stronger PGTH1 variants with improved regulation properties.
Productivity is also determined by the process regime and the growth rate (Looser et al., 2015). The model‐based design was previously successfully used to improve PGAP driven P. pastoris protein production processes (Maurer, Kuhleitner, Gasser, & Mattanovich, 2006). Specific productivities at different growth rates (also known as production kinetics) were characterized and successfully used to develop an optimization model. We developed a similar production kinetics‐based modeling to improve PGTH1‐driven processes performance.
2. MATERIALS AND METHODS
2.1. Strains and cultivation
Escherichia coli DH10B (Invitrogen) was used for subcloning. It was routinely cultivated in petri dishes or shake flasks using media supplemented with 25 µg·mL−1 Zeocin for selection. The wild‐type P. pastoris strain CBS2612 (syn. Komagataella phaffii) was used for protein production in this study. The main culture for screenings was either done with buffered M2 minimal‐, yeast extract‐peptone (YP)‐ or BM media and glucose feed beads (12 mm, Kuhner, CH) which provided the carbon source (Prielhofer et al., 2013). YP media contained 20 g·L−1 soy peptone and 10 g·L−1 yeast extract, which can be supplemented with 12.6 g glycerol or 20 g glucose to obtain YPG and YPD, respectively. For cultivation on plates, 20 g·L−1 agar–agar was added to the liquid medium. Buffered medium (BM) contained 13.4 g·L−1 yeast nitrogen base (Cat.No. 291940, Becton Dickinson, FR) with ammonium sulfate, 0.4 mg·L−1 biotin and 100 mM potassium phosphate buffer pH 6.0. Buffered M2 minimal media was also set to pH 6.0.
2.2. Promoter sequence analysis
The PGTH1 promoter sequence (1000 bp upstream of the gene PAS_chr1–3_0011 according to the annotation in the P. pastoris strain GS115) was analyzed for putative TFBS using MatInspector (Cartharius et al., 2005, Genomatix release 8.1, September 2013). The search was based on the MatInspector library Matrix Family Library Version 9.2 (October 2014) carried out with standard search parameters (matrix groups fungi and general core promoter elements, core similarity 0.75, matrix similarity optimized).
2.3. Promoter cloning and transformation into P. pastoris
Cloning and transformation of PGTH1 promoter variants were done as described previously (Prielhofer et al., 2013, see primers in Supporting Information Table S2). Deletions of putative TFBS and TAT15 motif mutations of PGTH1 were cloned using overlap‐extension PCR. Fragment duplications within PGTH1 were cloned using the internal restriction sites PstI and BglII (positions 509–514 and 525–530) to insert fragments amplified from position ‐472 to ‐188 and ‐472 to ‐1, resulting in the variants PGTH1 ‐D1240 and PGTH1 ‐D1427. Promoters and coding sequences (eGFP and i‐body) were ligated into the pPuzzle vector (Stadlmayr et al., 2010) using the ApaI (5′‐GGGCCC‐3′), the SbfI (5′ ‐CCTGCAGG‐3′) and the SfiI (5′ ‐GGCCNNNNNGGCC‐3′) restriction sites, respectively. Genome integration of the expression plasmid was targeted to the 3′‐flanking region of the AOX1 gene of P. pastoris to avoid positional effects on reporter gene expression. Plasmids were linearized within the genome integration region before electroporation (2 kV, 4 ms, GenePulser, BioRad) into electrocompetent P. pastoris. To generate clones with low copy integration, low amounts of DNA were used for the transformation (<1 µg DNA). Multicopy clones were excluded from the screening data, as they can easily be identified by their strongly enhanced eGFP fluorescence (Prielhofer et al., 2013). The gene copy number (GCN) was analyzed with quantitative real‐time polymerase chain reaction (PCR) and resulted in one copy of the expression for PGTH1 #8, PGTH1 ‐∆2 #3, PGTH1 ‐T16 #3 and PGTH1 ‐D1240 #3. P. pastoris cells were first selected and cultivated on YPD agar and then inoculated in liquid YPG medium as preculture for screenings and fermentation. Antibiotic selection by Zeocin was applied on plates and in preculture at a concentration of 25 µg·mL−1.
2.4. Screening, fed‐batch cultivation, and eGFP expression analysis
Expression screenings were done in 24‐deep well plate screenings at 25°C and with shaking at 280 rpm with 2 mL culture per well. Glucose feed beads (6 mm, Kuhner, CH) were used to generate glucose‐limiting growth conditions. Cells were analyzed for eGFP expression during repression (YP + 1% glycerol) and induction (YP + 1 feed bead). Screenings were repeated to verify the reproducibility of the results. For the screening of PGTH1 length variants, two clones each were cultivated in triplicates. Samples were taken at the end of the preculture and after 24 and 48 h of the main culture (a second feed bead was added after 24 h). For analysis of the deletion variants, TAT15 mutants and duplication variants of PGTH1, clones were pool cultivated (a mixed culture of five to nine clones) in three wells.
Fed‐batch bioreactor cultivations were performed as described before (Prielhofer et al., 2013). The batch phase of approximately 25 hr was followed by a fed‐batch phase (glucose fed‐batch media) with constant feed rate for about 100 hr or an optimized feeding rate, according to Maurer et al. (2006) in the fed‐batch process. Samples were taken during the batch and fed‐batch phase and analyzed for eGFP levels or secreted product titers.
Expression of eGFP in screenings was analyzed by flow cytometry. Specific eGFP fluorescence referred to in this study is the fluorescence intensity related to the cell volume for each data point as described by Hohenblum, Borth, and Mattanovich (2003). For all graphs showing specific eGFP fluorescence, the geometric mean of the whole population was used. Please note that the specific eGFP fluorescence of two different screenings cannot be compared.
For bioreactor samples, a plate reader (Infinite 200, Tecan, CH) was used to determine eGFP fluorescence. Samples were diluted to an OD600 of five and fluorescence intensity was related to the bioreactor culture volume. Secreted i‐body titers (single domain antibody‐like molecule of human origin, provided by AdAlta) were determined in the culture supernatant using the HT low MW protein expression kit on the Caliper LabChip® GXII system (PerkinElmer).
3. RESULTS AND DISCUSSION
To identify the most relevant regulatory region of PGTH1, we cloned eight shortened PGTH1 variants starting from the alternative 5′‐positions ‐858, ‐663, ‐492, ‐371, ‐328, ‐283, ‐211 and ‐66 to position ‐1 upstream of the ATG (indicated in Figure 1, numbering based on the positions of the annotation of the GTH1 gene locus PAS_chr1–3_0011 in the P. pastoris strain GS115). These shortened promoter variants were screened for eGFP expression in deep well plates to test for the repression (glycerol) and induction properties (glucose limitation) in comparison with the original 965 bp version of PGTH1 (Figure 2). No difference in eGFP signal was found for all length variants in the repressing condition, showing that promoter repression was not restricted in any of the shortened variants. After 48 h of induction, the expression capacity remained fully functional for the promoter variants down to a length of 328 bp. The 283 bp variant was about two‐thirds as strong as the original PGTH1 promoter. The two shortest length variants (211 and 66 bp) appeared to be almost nonfunctional. These results indicate that the region between the positions −328 and −211 contains important regulatory features.
Figure 1.
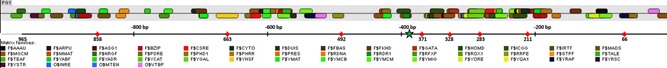
Identification of transcription factor binding sites (TFBS) in the PGTH1 promoter sequence using MatInspector. The PGTH1 sequence (1000 bp upstream of the gene PAS_chr1‐3_0011 according to the annotation in the P. pastoris strain GS115) was analyzed using Matinspector. Matrix families belonging to the matrix groups fungi and general core promoter elements are shown (detailed matrix match table is provided in Supporting InformationTable S1). The green asterisk indicates the position of the prominent TAT15 motif (position ‐390 to ‐374). PGTH1 was initially amplified and cloned from position ‐965 to ‐1 (length of 965 bp). Alternative 5′‐starts of the shortened PGTH1 promoter variants are labeled with red arrows and the length of the corresponding variant [Color figure can be viewed at wileyonlinelibrary.com]
Figure 2.

Screening data of 5′‐shortened PGTH1 promoter variants. The geometric means of the populations’ specific eGFP fluorescence (fluorescence related to cell volume) is shown for clones expressing eGFP under control of PGTH1 (named 965 bp) and the shortened PGTH1 variants (two clones each which were selected in prescreenings were cultivated in triplicates) in repressing and inducing growth conditions. Wild‐type P. pastoris cells were used as negative control. Samples were taken during the repressing preculture and after 24 and 48 h induction with feed beads. eGFP: enhanced green fluorescent protein
3.1. High density of predicted carbon source‐related TFBS marks the main regulatory region of PGTH1
Next, the PGTH1 promoter sequence (1000 bp upstream of the gene PAS_chr1–3_0011) was searched for TFBS matrix families belonging to the matrix groups fungi and general core promoter elements using the MatInspector from Genomatix (Cartharius et al., 2005). One hundred and eleven putative TFBS belonging to 46 different matrix families were found (Figure 1 and Supporting Information Table S1). The most common matrix families in the analyzed sequence were monomeric Gal4‐class motifs (F$MGCM, 12 binding sites), homeo‐domain‐containing transcriptional regulators (F$HOMD, 6 binding sites), fungal basic leucine zipper family (F$BZIP, 5 binding sites) and yeast GC‐Box Proteins (F$YMIG, 5 binding sites). As anticipated from the results obtained with the length variants, we noticed a very high density of predicted TFBS binding sites between position ‐400 and ‐200 with about two‐thirds of the mentioned TFBS (most common matrix families) occurring there (18 out of 28). As for general core promoter elements, no yeast‐ or fungi‐related motifs were identified by the MatInspector, but a TATA box was found starting at position ‐26.
A prominent motif was identified at position ‐390 to ‐375, which we termed TAT15 due to its sequence 5′‐TA(T)15‐3′. Such poly (A/T) regions in promoter regions are known to negatively affect nucleosome binding and to stimulate TF binding at nearby sites in yeast (Weingarten–Gabbay & Segal, 2014).
3.2. Putative binding sites of the carbon source‐related transcription factors Mxr1/Adr1, Rgt1, Cat8/Sip4, and Mig1 were revealed to contribute to the regulatory properties of PGTH1
Putative TFBS with predicted glucose‐ or carbon source dependency were selected for further analyzes (see Figure 3, Table 1, and Supporting Information Table S3). PGTH1 variants with deletions of the respective regions were generated using overlap‐extension PCR. Figure 3 (a) shows all selected TFBS and (b) indicates all predicted TFBS which might (partially) be affected by the deletion (listed in Table 1). For some deletions (e.g. ∆9 and ∆10), some nucleotides of the respective TFBS were left untouched to keep closely neighboring TFBS functional and to separately examine their effect.
Figure 3.
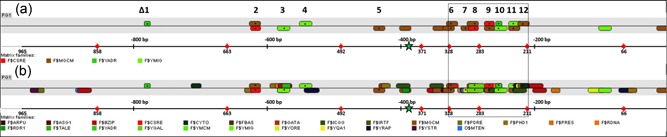
PGTH1 promoter sequence analysis for carbon source‐related transcription factor binding sites (TFBS) using MatInspector and selected TFBS for deletion. Black dots and corresponding numbers indicate TFBS which were selected for deletion (listed in Table 1 and Supporting Information Table S3). Associated matrix families are shown in (a), and (b) illustrates all TFBS which might be affected by the deletions (matrix match detail information is given in Supporting Information Table S1). The black dashed box indicates the main regulatory region of PGTH1 which was identified by the screening of shortened PGTH1 variants. The green asterisk indicates the position of the prominent TAT15 motif which was also selected for deletion and for mutation [Color figure can be viewed at wileyonlinelibrary.com]
Table 1.
Positions and TFBS deletions of PGTH1 TFBS deletion variants
| PGTH1 ‐∆ | Position | TFBS deletions (TF matrices, targets in bold) |
|---|---|---|
| 1 | ‐785 to ‐777 | F$ADR1.01 |
| 2 | ‐628 to ‐612 | F$PHD1.03, F$RGT1.02, F$CSRE.01 |
| 3 | ‐586 to ‐568 | F$REB1.02, F$MIG1.02, F$MSN2.01, F$YAP1.02, F$TOS8.01 |
| 4 | ‐553 to ‐535 | F$MIG1.01, F$RAP1.06, F$AFT2.01 |
| 5 | ‐442 to ‐426 | F$RGT1.02, F$GZF3.01, F$PHD1.01 |
| 6 | ‐337 to ‐316 | F$ASG1.01, F$RGT1.02, F$RGT1.02, F$RDR1.01, F$GATA.01 |
| 7 | ‐310 to ‐299 | F$STE12.01, F$GAT1.01, F$RGT1.02, O$DMTE.01, F$OAF1.01 |
| 8 | ‐293 to ‐285 | F$OAF1.01, F$RGT1.02, F$GAL4.01, F$SIP4.01, F$RDR1.01, F$LAC9.01 |
| 9 | ‐275 to ‐261 | F$LEU3.02, F$CSRE.01, F$RGT1.01, F$TEA1.01 |
| 10 | ‐258 to ‐242 | F$REB1.02, F$MCM1.02, F$MIG1.01, F$ADR1.01 |
| 11 | ‐239 to ‐221 | F$RGT1.02, F$MIG1.01, F$TEA1.01, F$PPR1.01, F$PDRE.01, F$PPR1.01, F$PDRE.01 |
| 12 | ‐220 to ‐209 | F$HAP1.01, F$QA1F.01, F$RGT1.02, F$HAP1.01 |
Note. TF: transcription factor; TFBS: transcription factor binding sites.
All 12 putative TFBS deletions and the TAT15 mutation variants were screened for eGFP expression in repressing (glycerol) and inducing conditions (glucose feed bead; Figure 4). It is important to consider that individual TFs or TFBS are usually not sufficient to fulfill a promoter’s regulation. Also, TFBS deletions may potentially generate unwanted effects by the newly formed adjoined sequence, by altered distances between TFBS or by changes of higher‐order properties (chromatin organization). The same TFBS in different positions of the promoter may have different functions, also because of other adjacent TFBS. At closely neighboring TFBS, TFs might either act synergistically or restrict the binding of other TFs due to steric hindrance.
Figure 4.
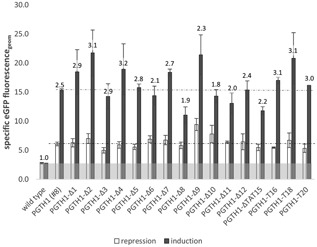
Screening data of the PGTH1 deletion and TAT15 mutation variants. The geometric means of the populations’ specific eGFP fluorescence (fluorescence related to cell volume) are shown for clones expressing eGFP under the control of PGTH1 (clone #8, verified GCN of one) or a PGTH1 variant (up to nine clones were pool cultivated in three wells) in repressing and inducing growth conditions. The numbers above the bars represent the fold‐change of induction versus repression. Wild‐type P. pastoris cells were used as negative control. eGFP: enhanced green fluorescent protein; GCN: gene copy number
Putative TFBS of four different carbon source‐related TF families were deleted in the PGTH1 promoter variants (see Table 1 and Supporting Information Table S3): Yeast metabolic regulator (F$ADR; matrixes: F$ADR1.01), Monomeric Gal4‐class motifs (F$MGCM; matrixes: F$RGT1.01, F$RGT1.02), Carbon source‐responsive elements (F$CSRE, matrixes: F$CSRE.01, F$SIP4.01) and Yeast GC‐Box Proteins (F$YMIG; matrixes: F$MIG1.01 and F$MIG1.02). The corresponding transcription factors in S. cerevisiae are Adr1, Rgt1, Sip4/Cat8, and Mig1, respectively.
3.3. Deletion variants of the PGTH1 promoter reveal putative TFBS responsible for promoter repression and induction
Out of the five deletion variants residing upstream (5′) of the main regulatory region of PGTH1 identified before (see dashed box in Figure 3), the variants PGTH1 ‐∆1, ‐∆2, and ‐∆4 appear to have a beneficial effect on promoter strength whereas the deletion variants PGTH1 ‐∆3 and ∆5 had no effect on GFP expression compared with the original PGTH1 promoter. These results suggest that 5′‐shortening of the promoter might be beneficial for the engineering of PGTH1. TFBS deletions within the main regulatory region of PGTH1 (PGTH1 ‐∆6 to ‐∆12, see Figure 3) had different impacts on eGFP expression, but none showed increased induction without losing the repression properties. Therefore, we assume that the main regulatory region of PGTH1 needs to be maintained in engineered PGTH1 promoter variants to retain its tight regulation. Without this region, also much lower induction in limiting glucose was observed (PGTH1‐328 and PGTH1‐283, Figure 2).
Predicted Mig1 binding sites were deleted in PGTH1 ‐∆3, ‐∆4, ‐∆10 and ‐∆11 (F$MIG1.02 in ∆3, F$MIG1.01 in ∆4, ∆10, and ∆11), whereas PGTH1 ‐∆10 and PGTH1 ‐∆11 also included $ADR1.01 and F$RGT1.02 deletions, respectively. Slightly tighter repression was found for ∆3, whereas ∆4 had unchanged repression but enhanced eGFP levels after induction. Liberated repression seen for ∆10 and weaker promoter induction of ∆10 and ∆11 could also be connected to F$RGT1 sites in this region (F$RGT1.01 and F$RGT1.02 deleted in ∆9 and ∆11). Also, Mig1 could play a bifunctional role in PGTH1 regulation: two MIG1 genes are found in P. pastoris (MIG1‐1, MIG1‐2) and they were shown to be regulated contrariwise upon glucose availability (Prielhofer et al., 2015).
The deletion of F$ADR1.01 sites increased eGFP levels in the variant PGTH1 ‐∆1, although Mxr1 (a positive regulator of methanol metabolism in P. pastoris, homolog of ScAdr1) is supposed to be the main regulator of methanol‐induced genes and was reported to be imported into the nucleus by methanol‐induction (Lin‐Cereghino et al., 2006). Combined deletion of F$ADR1.01 with F$MIG1.01 in PGTH1 ‐∆10 liberated promoter repression on glycerol and weakened its induction, which is a conclusive response for Mig1 TFBS deletion.
The role of the transcriptional regulators Mig1 (F$MIG1) and Mxr1 (F$ADR1) might be more important in other conditions such as excess glucose or methanol induction.
In the main regulatory region, the predicted binding site F$RGT1.02 was deleted in the variants PGTH1 ‐∆6 (two sites), ‐∆7, ‐∆8, ‐∆11 and ‐∆12, and F$RGT1.01 was deleted in ∆9. The variant harboring the deletion of the paired F$RGT1.02 site (∆6, predicted binding sites on opposite strands with a shift of 7 bp) showed a slightly liberated repression and reduced induction. The variants ∆7 and ∆8 contain very close F$RGT1.02 sites, whereas the first lies on the negative‐ and the second on the positive strand; the ∆8 variant also contains the deletion of an F$SIP4.01 site. The first (∆7) showed a slightly liberated repression and increased induction, whereas the second (∆8) was much more weakly induced (but had unchanged promoter repression). This indicates a strong role for the transcriptional activator(s) Cat8‐1 and/or Cat8‐2 (strongest homologs for ScCat8 and ScSip4) for PGTH1 induction. The variant ∆9 was created to delete the closely located F$RGT1.01 and F$CSRE.01 sites (on opposite strands). The drastic loss of repression indicates a strong role of this putative TFBS motif to tightly control PGTH1, most likely through binding of Rgt1, Cat8‐1 and/or Cat8‐2. The deletion of F$RGT1.02 in the variant PGTH1 ‐∆12 did not have an effect on eGFP expression performance. Interestingly, CAT8‐2 transcription is strongly upregulated in limiting glucose compared with glucose surplus, whereas RGT1 and CAT8‐1 were not transcriptionally regulated in the tested conditions (Prielhofer et al., 2015).
Rgt1 can act as a transcriptional activator or repressor in S. cerevisiae (Ozcan, Leong, & Johnston, 1996). Roy, Jouandot, Cho, and Kim (2014) reported that Rgt1 function is controlled by its four phosphorylation sites and that promoter induction does not require its dissociation, as typically seen for transcriptional repressors. Carbon source‐responsive element (CSRE) are bound by the transcriptional activators Sip4 and Cat8 to induce the expression of gluconeogenesis genes in S. cerevisiae (Hiesinger, Roth, Meissner, & Schuller, 2001). Two P. pastoris homologs of ScCat8 can be found: Cat8‐1 (PAS_chr2‐1_0757) and Cat8‐2 (PAS_chr4_0540), both also being the best blastp hits for ScSip4. Based on transcriptomics data Cat8‐2 potentially plays a role during glucose derepression (Prielhofer et al., 2015).
3.4. PGTH1 promoter strength is dependent on the poly (A/T) motif TAT15
The TAT15 (5′‐TA(T)15‐3′) motif was found to be located about 80 bp upstream (5′ position in PGTH1: ‐390 to ‐374) of the main regulatory region of PGTH1. Repeated sequencing of the 5′‐region of GTH1 in P. pastoris GS115, CBS2612 or CBS7435 resulted in the detection of 15 ± 1T’s in the TAT15 motif. To elucidate its impact on promoter performance, the TAT15 motif was selected for deletion (PGTH1 ‐∆TAT15) or exchange to T16 (PGTH1 ‐T16), T18 (PGTH1 ‐T18) and T20 PGTH1 ‐T20). Primers (see primers #37‐42 in Supporting Information Table S2) were initially designed to obtain T18, T20, and T22, but variants with different lengths (T16, T20, and T18, respectively) were obtained and used. Deletion of the TAT15 motif was found to result in lower GFP signals, whereas its prolongation increased the expression strength of PGTH1. This indicates that the use of a prolonged TAT15 motif is potentially beneficial for PGTH1 engineering.
3.5. Partial sequence duplications of the main regulatory region of PGTH1 significantly improve its expression strength
Two duplication variants (PGTH1 ‐D1240 and PGTH1 ‐D1427, the numbers state the lengths of the respective promoter variants) of the PGTH1 promoter were generated by PCR amplification of two sequence fragments (‐472 to ‐188 and ‐472 to ‐1, respectively; see Figure 5) and insertion using the restriction sites PstI and BglII (positions 509‐514 and 525‐530). The duplication sections start upstream of the TFBS deleted in PGTH1 ‐∆5 and end after the main regulatory region of PGTH1 for the first variant (PGTH1 ‐D1240), whereas the second duplication (PGTH1−D1427) reaches until the 3′‐end of the PGTH1 promoter. These variants were screened for eGFP expression in the same way as described for the TFBS deletion and TAT15 mutation variants. Both duplication variants showed a more tight repression in excess glycerol and stronger induction upon limiting glucose (Figure 6).
Figure 5.

Schematic illustration of PGTH1 promoter duplication variants. The initially cloned PGTH1 sequence (a) is 965 bp long (amplified from position 36 to 1000 of the sequence shown here). PstI and BglII restriction sites (located at positions 509‐514 and 525‐530, indicated with scissors) were used to generate PGTH1 variants PGTH1 ‐D1240 and PGTH1 ‐D1426 with duplicate fragments. Sequence fragments corresponding to a,b and a,c of sequence (a) were amplified with primers containing appropriate restriction sites and ligated into the site, thereby generating duplication variants with a length of 1240 bp (b) and 1427 bp (c) [Color figure can be viewed at wileyonlinelibrary.com]
Figure 6.
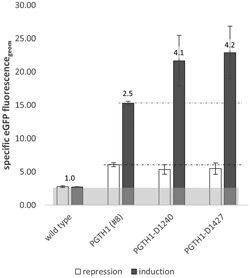
Screening data of the PGTH1 duplication variants. The geometric means of the populations’ specific eGFP fluorescence (fluorescence related to cell volume) are shown for clones expressing eGFP under the control of PGTH1 (clone #8, verified GCN of one) and the two PGTH1 duplication variants (up to nine clones selected in prescreenings were pool cultivated in three wells) in repressing and inducing growth conditions. The numbers above the bars represent the fold‐change of induction versus repression. Wild‐type P. pastoris cells were used as negative control. eGFP: enhanced green fluorescent protein; GCN: gene copy number
We also tested the posttransformational stability of the duplication variant clone PGTH1 ‐D1240 #3 by performing three consecutive batch cultivations without selection pressure, which is equal to about 20 generations. We could verify that enhanced green fluorescent protein (eGFP) expression was stable over the whole cultivation time (data not shown). In comparison, a typical P. pastoris bioreactor process starts with OD600 = 1 (~0.2−0.4 g/L YDM) in the batch phase and ends with ~100 g/L YDM after the fed‐batch phase and thereby takes about 10 generations.
The performance of the generated promoter variants PGTH1 ‐∆2 (deletions of Rgt1,CSRE, and TFBS), PGTH1 ‐T16 (TAT15 motif exchange to T16) and PGTH1 ‐D1240 (duplication variant) were verified in bioreactor cultivation (glycerol batch phase followed by a glucose‐limited fed‐batch as described before; Prielhofer et al., 2013). For each construct, a single copy clone was selected from the screening and compared with the single copy clone PGTH1 #8. Compared with the control strain PGTH1 #8, PGTH1 ‐∆2 #3 had a slightly increased eGFP level at the batch end (t = 0 hr), which was about 10% weaker as the control at the fed‐batch end (Table 2). The clone PGTH1 ‐T16 #3 showed the strongest signal at the batch end, but fell behind the duplication variant PGTH1 ‐D1240 at the fed‐batch end, and reached about 20% improvement compared with the control PGTH1 #8. The duplication variant clone PGTH1‐D1240 #3 showed an increased signal at the batch end and reached 50% increase in GFP fluorescence at the fed‐batch end. Overall, the fed‐batch cultivations could confirm the results obtained in small‐scale screening.
Table 2.
Fed‐batch cultivation of PGTH1 and PGTH1 variants expressing eGFP
| Batch end | Fed batch end | |||||||
|---|---|---|---|---|---|---|---|---|
| Clone | t a (h) | YDM b (g/L) | Relative eGFP fluorescence | % c | t a (h) | YDM b (g/L) | Relative eGFP fluorescence | % c |
| PGTH1 #8 | −5.3 | 9.8 | 44 ± 1 | 100 | 19.5 | 118.6 | 2005 ± 36 | 100 |
| PGTH1 ‐∆2 #3 | −4.6 | 11.0 | 51 ± 1 | 116 | 19.5 | 110.6 | 1819 ± 43 | 91 |
| PGTH1 ‐T16 #3 | −3.0 | 14.2 | 70 ± 1 | 160 | 19.5 | 113.1 | 2383 ± 24 | 119 |
| PGTH1 ‐D1240 #3 | −3.0 | 14.9 | 62 ± 1 | 141 | 19.5 | 113.3 | 2948 ± 33 | 147 |
Fed batch start t = 0.
Yeast dry mass.
GFP signal relative to clone PGTH1 #8.
3.6. Optimized fed‐batch fermentation strategy for superior PGTH1‐driven productivity
PGTH1 ‐D1240 was further evaluated for the secreted production of the industrially relevant single domain antibody‐like protein i‐body (15 kDa, human origin, provided by AdAlta) in comparison with production under PAOX1. A basic fermentation regime using a constant glucose feed for 100 h was applied for the PGTH1‐D1240 process (Prielhofer et al., 2013), and compared with the well‐established three‐stage methanol‐driven process for PAOX1 (Looser et al., 2015; schematic overview see Figure 7a,b). The product titer was 1.2‐fold higher for PGTH1 ‐D1240 compared with P AOX1 (4.3 and 3.6 g·L−1 i‐body, respectively), indicating the high performance of PGTH1 ‐D1240 even with a nonoptimized feeding regime.
Figure 7.
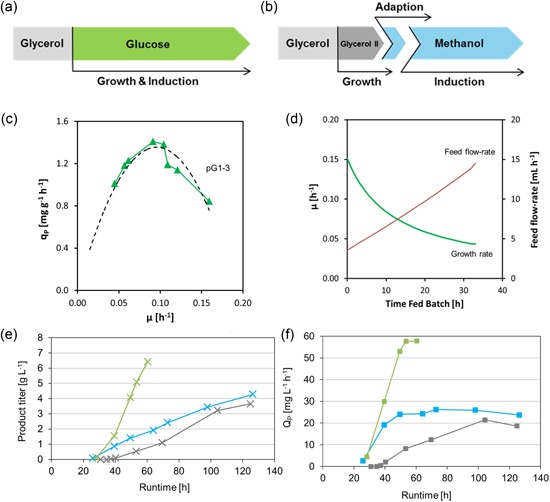
Performance of PGTH1 ‐D1240 in basic and optimized fed‐batch cultivations. A schematic overview of a basic constant glucose‐based process for PGTH1 (a) and a typical three‐stage methanol‐driven process (b) for PAOX1 is shown. Production kinetics (specific productivity qP at different specific growth rates) from fed‐batch cultivations (c) led to the design of an optimized feeding strategy for PGTH1 ‐D1240 (d). The optimized process resulted in increased final supernatant titers and volumetric productivity (QP) for PGTH1 ‐D1240 which was even outperforming the PAOX1‐driven process (Table 3 and e, f: the basic PGTH1 ‐D1240 process (blue), the PGTH1‐optimized process (green) and the standard PAOX1 process (gray) [Color figure can be viewed at wileyonlinelibrary.com]
Next, production kinetics (specific product formation rate qP (mg·g−1·h−1) at different specific growth rates) for PGTH1 ‐D1240 were analyzed in a set of fed‐batch cultivations. This revealed a bell‐shaped relationship for the production of the i‐body with a maximum qP at a growth rate of about 0.1 h−1 (Figure 7c). Based on that, an optimized feeding strategy for P GTH1 ‐D1240 was designed similar to (Maurer et al., 2006; Figure 7d). The optimal feed profile was represented by a linear incremental glucose feed for 32 to 34 h, leading to a decreasing specific growth rate from µ = 0.15 to about 0.05 h−1 throughout the feed phase. The optimized fermentation process resulted in a final titer of 6.4 g·L−1 i‐body in the supernatant after 60 h total fermentation time and thereby highly outperformed the standard process for PGTH1 and PAOX1 in terms of product titer (1.5‐ and 1.8‐fold, respectively) and volumetric productivity (2.2‐ and 2.7‐fold, respectively; data presented in Figure 7e−f and Table 3). The product was also verified by SDS‐PAGE for both process strategies (Supporting Information Figure S1).
Table 3.
Comparison of process and performance parameters
| Parameter | PAOX1 standard | PGTH1 basic | PGTH1 optimized |
|---|---|---|---|
| Feed profile | 3‐stage | Constant | Linear |
| Duration (h) | 124 | 126 | 63 |
| Maximum yeast dry mass YDMmax (g·L−1) | 109 | 90 | 138 |
| Final product concentration (supernatant) (g·L–1) | 3.6 | 4.2 | 6.4 |
| Final product concentration (broth) (g·L–1) | 2.3 | 3.0 | 3.5 |
| Maximal specific secretion rate qP,max (feed phase) (mg·g–1·h–1) | 0.6 | 1.8 | 2.1 |
| Maximal volumetric productivity QP,max (feed phase) (mg·L–1·h–1) | 21 | 26 | 58 |
| Oxygen uptake rate OURmax (mmol·L−1·h−1) | 180 | 100 | 148 |
4. CONCLUSIONS
We could identify the main regulatory region of PGTH1, and important putative TFBS. The analysis of specific deletions indicated that the transcription factors Rgt1 and Cat8‐1 and/or Cat8‐2 likely play an essential role for PGTH1 repression and induction. Interestingly, we found a putative TFBS motif for Rgt1 and Cat8‐1/2 with overlapping binding sites located on + and ‐ strands (variant PGTH1 ‐∆9 with deletion of ‐275 to ‐261, see Table 1), which is obviously important for the glucose‐dependent regulation of PGTH1. We also showed that the motif TA(T)15, which is located upstream of the main regulatory region of PGTH1 (position ‐390 to ‐375), is essential for promoter strength.
A key achievement of this study was the generation of the variant PGTH1 ‐D1240 with the main regulatory region duplicated and greatly enhanced expression capacities compared with the wild‐type PGTH1. It reached up to 50% higher titers in a standard glucose‐limited fed‐batch process.
Furthermore, we successfully applied PGTH1 ‐D1240 in an optimized 60 hr model‐based fed‐batch process to produce an industrially relevant protein at superior titer and volumetric productivity. While typical P. pastoris production processes often take up to one week, this optimized fermentation process reaches high product titers in times comparable with bacterial fermentation processes. In addition, it avoids various disadvantages of using methanol both for the cells and for product quality (Hartner & Glieder, 2006; Schotte et al., 2016), and for production in large scale (e.g., explosion hazard). Furthermore, the significant reduction of the maximum oxygen uptake rate (Table 3) and heat generation (not shown) of a glucose‐limited process compared with a methanol‐induced process, is beneficial for process upscaling.
CONFLICTS OF INTEREST
The authors declare that there is no conflicts of interest. Two patent applications related to the described results have been filed (WO2013050551A1, WO2017021541A1). Some of the authors (R. P, C. K., D. M. and B. G.) are inventors (but not owners) of these patent applications.
AUTHORS CONTRIBUTION
R. P. performed the experimental work, drafted the manuscript and contributed to study design. M. R. and N. W. designed and performed the experimental work for process optimization, drafted part of the manuscript and contributed to study design. K. C. and C. K. contributed to study design. B. G. and D. M. conceived the study, and contributed to data analysis, process design and drafting of the manuscript. All authors read and approved the final manuscript.
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGEMENTS
This project was financially supported by LONZA AG (Visp, CH). We thank AdAlta Limited, Bundoora, Victoria, Australia, for providing the i‐body used in this study.
Prielhofer R, Reichinger M, Wagner N, et al. Superior protein titers in half the fermentation time: Promoter and process engineering for the glucose‐regulated GTH1 promoter of Pichia pastoris . Biotechnology and Bioengineering. 2018;115:2479–2488. 10.1002/bit.26800
Present address
References
REFERENCES
- Ahmad, M. , Hirz, M. , Pichler, H. , & Schwab, H. (2014). Protein expression in Pichia pastoris: Recent achievements and perspectives for heterologous protein production. Applied Microbiology and Biotechnology, 98(12), 5301–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn, J. , Hong, J. , Park, M. , Lee, H. , Lee, E. , Kim, C. , … Lee, H. (2009). Phosphate‐responsive promoter of a Pichia pastoris sodium phosphate symporter. Applied and Environmental Microbiology, 75(11), 3528–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ata, Ö. , Prielhofer, R. , Gasser, B. , Mattanovich, D. , & Çalık, P. (2017). Transcriptional engineering of the glyceraldehyde‐3‐phosphate dehydrogenase promoter for improved heterologous protein production in Pichia pastoris . Biotechnology and Bioengineering, 114(10), 2319–2327. [DOI] [PubMed] [Google Scholar]
- Byrne, B. (2015). Pichia pastoris as an expression host for membrane protein structural biology. Current Opinion in Structural Biology, 32C, 9–17. [DOI] [PubMed] [Google Scholar]
- Cartharius, K. , Frech, K. , Grote, K. , Klocke, B. , Haltmeier, M. , Klingenhoff, A. , … Werner, T. (2005). MatInspector and beyond: Promoter analysis based on transcription factor binding sites. Bioinformatics, 21(13), 2933–2942. [DOI] [PubMed] [Google Scholar]
- Hartner, F. S. , & Glieder, A. (2006). Regulation of methanol utilisation pathway genes in yeasts. Microbial Cell Factories, 5, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner, F. S. , Ruth, C. , Langenegger, D. , Johnson, S. N. , Hyka, P. , Lin‐Cereghino, G. P. , … Glieder, A. (2008). Promoter library designed for fine‐tuned gene expression in Pichia pastoris . Nucleic Acids Research, 36(12), e76–e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiesinger, M. , Roth, S. , Meissner, E. , & Schüller, H. J. (2001). Contribution of Cat8 and Sip4 to the transcriptional activation of yeast gluconeogenic genes by carbon source‐responsive elements. Current Genetics, 39(2), 68–76. [DOI] [PubMed] [Google Scholar]
- Hohenblum, H. , Borth, N. , & Mattanovich, D. (2003). Assessing viability and cell‐associated product of recombinant protein producing Pichia pastoris with flow cytometry. Journal of Biotechnology, 102(3), 281–290. [DOI] [PubMed] [Google Scholar]
- Landes, N. , Gasser, B. , Vorauer‐Uhl, K. , Lhota, G. , Mattanovich, D. , & Maurer, M. (2016). The vitamin‐sensitive promoter PTHI11 enables pre‐defined autonomous induction of recombinant protein production in Pichia pastoris . Biotechnology and Bioengineering, 113(12), 2633–2643. [DOI] [PubMed] [Google Scholar]
- Lin‐Cereghino, G. P. , Godfrey, L. , de la Cruz, B. J. , Johnson, S. , Khuongsathiene, S. , Tolstorukov, I. , … Cregg, J. M. (2006). Mxr1p, a key regulator of the methanol utilization pathway and peroxisomal genes in Pichia pastoris . Molecular and Cellular Biology, 26(3), 883–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looser, V. , Bruhlmann, B. , Bumbak, F. , Stenger, C. , Costa, M. , Camattari, A. , … Kovar, K. (2015). Cultivation strategies to enhance productivity of Pichia pastoris: A review. Biotechnology Advances, 33(6 Pt 2), 1177–1193. [DOI] [PubMed] [Google Scholar]
- Lubliner, S. , Keren, L. , & Segal, E. (2013). Sequence features of yeast and human core promoters that are predictive of maximal promoter activity. Nucleic Acids Research, 41(11), 5569–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsman, J. , & Horsfield, J. A. (2012). Long distance relationships: Enhancer‐promoter communication and dynamic gene transcription. Biochimica et Biophysica Acta, 1819(11‐12), 1217–1227. [DOI] [PubMed] [Google Scholar]
- Mattanovich, D. , Branduardi, P. , Dato, L. , Gasser, B. , Sauer, M. , & Porro, D. (2012). Recombinant protein production in yeasts. Methods in Molecular Biology, 824, 329–358. [DOI] [PubMed] [Google Scholar]
- Maurer, M. , Kühleitner, M. , Gasser, B. , & Mattanovich, D. (2006). Versatile modeling and optimization of fed batch processes for the production of secreted heterologous proteins with Pichia pastoris . Microbial Cell Factories, 5, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellitzer, A. , Ruth, C. , Gustafsson, C. , Welch, M. , Birner‐Grünberger, R. , Weis, R. , … Glieder, A. (2014). Synergistic modular promoter and gene optimization to push cellulase secretion by Pichia pastoris beyond existing benchmarks. Journal of Biotechnology, 191, 187–195. [DOI] [PubMed] [Google Scholar]
- Narlikar, L. , & Ovcharenko, I. (2009). Identifying regulatory elements in eukaryotic genomes. Briefings in Functional Genomics & Proteomics., 8, 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan, S. , Leong, T. , & Johnston, M. (1996). Rgt1p of Saccharomyces cerevisiae, a key regulator of glucose‐induced genes, is both an activator and a repressor of transcription. Molecular and Cellular Biology, 16(11), 6419–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips‐Cremins, J. E. , & Corces, V. G. (2013). Chromatin insulators: Linking genome organization to cellular function. Molecular Cell, 50(4), 461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prielhofer, R. , Maurer, M. , Klein, J. , Wenger, J. , Kiziak, C. , Gasser, B. , & Mattanovich, D. (2013). Induction without methanol: Novel regulated promoters enable high‐level expression in Pichia pastoris . Microbial Cell Factories, 12(1), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prielhofer, R. , Cartwright, S. P. , Graf, A. B. , Valli, M. , Bill, R. M. , Mattanovich, D. , & Gasser, B. (2015). Pichia pastoris regulates its gene‐specific response to different carbon sources at the transcriptional, rather than the translational, level. BMC Genomics, 16(1), 1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puxbaum, V. , Mattanovich, D. , & Gasser, B. (2015). Quo vadis? The challenges of recombinant protein folding and secretion in Pichia pastoris . Applied Microbiology and Biotechnology, 99(7), 2925–2938. [DOI] [PubMed] [Google Scholar]
- Qin, X. , Qian, J. , Yao, G. , Zhuang, Y. , Zhang, S. , & Chu, J. (2011). GAP promoter library for fine‐tuning of gene expression in Pichia pastoris . Applied and Environmental Microbiology, 77(11), 3600–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, A. , Jouandot, D., 2nd , Cho, K. H. , & Kim, J. H. (2014). Understanding the mechanism of glucose‐induced relief of Rgt1‐mediated repression in yeast. FEBS Open Bio, 4, 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotte, P. , Dewerte, I. , De Groeve, M. , De Keyser, S. , De Brabandere, V. , & Stanssens, P. (2016). Pichia pastoris Mut(S) strains are prone to misincorporation of O‐methyl‐L‐homoserine at methionine residues when methanol is used as the sole carbon source. Microbial Cell Factories, 15, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadlmayr, G. , Mecklenbräuker, A. , Rothmüller, M. , Maurer, M. , Sauer, M. , Mattanovich, D. , & Gasser, B. (2010). Identification and characterisation of novel Pichia pastoris promoters for heterologous protein production. Journal of Biotechnology, 150(4), 519–529. [DOI] [PubMed] [Google Scholar]
- Tschopp, J. F. , Brust, P. F. , Cregg, J. M. , Stillman, C. A. , & Gingeras, T. R. (1987). Expression of the lacZ gene from two methanol‐regulated promoters in Pichia pastoris . Nucleic Acids Research, 15(9), 3859–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl, T. , Ruth, C. , Pitzer, J. , Kickenweiz, T. , & Glieder, A. (2014). Synthetic core promoters for Pichia pastoris . ACS Synthetic Biology, 3(3), 188–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten‐Gabbay, S. , & Segal, E. (2014). The grammar of transcriptional regulation. Human Genetics, 133(6), 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhandl, K. , Winkler, M. , Glieder, A. , & Camattari, A. (2014). Carbon source dependent promoters in yeasts. Microbial Cell Factories, 13, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahrl, R. J. , Peña, D. A. , Mattanovich, D. , & Gasser, B. (2017). Systems biotechnology for protein production in Pichia pastoris . FEMS Yeast Research, 17, 7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Supporting information