

Chromodomain-peptide recognition specificity is decided by physiochemical properties defined by posttranslational modifications.
Abstract
Posttranslational modifications (PTMs) play critical roles in regulating protein functions and mediating protein-protein interactions. An important PTM is lysine methylation that orchestrates chromatin modifications and regulates functions of non-histone proteins. Methyllysine peptides are bound by modular domains, of which chromodomains are representative. Here, we conducted the first large-scale study of chromodomains in the human proteome interacting with both histone and non-histone methyllysine peptides. We observed significant degenerate binding between chromodomains and histone peptides, i.e., different histone sites can be recognized by the same set of chromodomains, and different chromodomains can share similar binding profiles to individual histone sites. Such degenerate binding is not dictated by amino acid sequence or PTM motif but rather rooted in the physiochemical properties defined by the PTMs on the histone peptides. This molecular mechanism is confirmed by the accurate prediction of the binding specificity using a computational model that captures the structural and energetic patterns of the domain-peptide interaction. To further illustrate the power and accuracy of our model, we used it to effectively engineer an exceptionally strong H3K9me3-binding chromodomain and to label H3K9me3 in live cells. This study presents a systematic approach to deciphering domain-peptide recognition and reveals a general principle by which histone modifications are interpreted by reader proteins, leading to dynamic regulation of gene expression and other biological processes.
INTRODUCTION
Protein-protein interactions, particularly those involving posttranslational modifications (PTMs), play critical roles in the majority of biological processes, and many diseases are caused by disruption of these interactions (1). In particular, PTMs on histone tail peptides, such as lysine methylation (2, 3), dictate chromatin structure remodeling and orchestrate gene expression, which is at the heart of epigenetics (4, 5). These PTMs are normally recognized by modular domains (6, 7). For instance, methylated peptides can be recognized by chromodomains (8, 9), and acetylated peptides by bromodomains (10). Understanding the recognition mechanisms and building models to predict the binding specificity of domain-peptide interactions is crucial for revealing the biophysical principles governing protein-protein interactions in general. Furthermore, mechanistic characterization could allow for engineering useful mutants of protein domains for various applications, such as proteomics (11) or live-cell imaging of histone modifications (12).
Computationally predicting the specificity of domain-peptide recognition remains a great challenge because of the weak binding and peptide flexibility. In particular, the recognition of methylated peptides is largely understudied owing to the lack of high-throughput binding data in vitro and the difficulty of proteomic experiments to identify methylation events in vivo (13). Methylation has been recognized as an important PTM not only on histone proteins but also on non-histone proteins as illustrated by their critical roles in epigenetics [e.g., presence of monomethylation of H3 Lys4 (H3K4me1) and lack of H3 Lys27 trimethylation (H3K27me3) mark active enhancers; (14)] and tumorigenesis [e.g., the tumor suppressor p53 is methylated at Lys372, which restricts p53 to the nucleus and enhances p53 stability; (15)]. Furthermore, it has been recognized that histone PTM–specific antibodies suffer from significant off-target binding defects and lot-to-lot variability (16) and that histone reader domains can serve as legitimate alternatives to histone PTM antibodies in chromatin immunoprecipitation–sequencing applications (17). There is an urgent need to understand the recognition mechanisms of methylated peptides and a computational model to engineer probes for use in epigenetics research.
We chose the chromodomain-methyllysine recognition as a model system to investigate the underlying mechanisms (Fig. 1A). In human and other mammalian cells, conserved chromodomains have been identified across a wide range of proteins involved in chromatin remodeling, and they exhibit diverse binding preferences toward methylated lysine sites on the histone tails (3, 8, 9, 18). Chromodomains are 50 to 70 amino acid residues long and frequently recognize histone peptides with methylated lysine, such as K4, K9, K27, and K36 on H3 and K20 on H4 (2). We have generated a large amount of binding data between 29 chromodomains encoded in the human proteome and 457 methylated peptides (including 232 non-histone peptides). To our knowledge, this is the first large-scale binding specificity study of chromodomains against a significant number of potential non-histone targets. We have developed a computational model to accurately predict the binding specificity between chromodomains and methylated peptides. Furthermore, we used the model to engineer the chromodomain of CBX1 to bind to the H3K9me3 mark with significantly higher affinity than the wild-type (WT) domain without sacrificing its specificity. The engineered domain labeled H3K9me3 in living cells more effectively than the WT domain.
Fig. 1. Pipeline of the chromodomain-peptide interaction screening experiments.
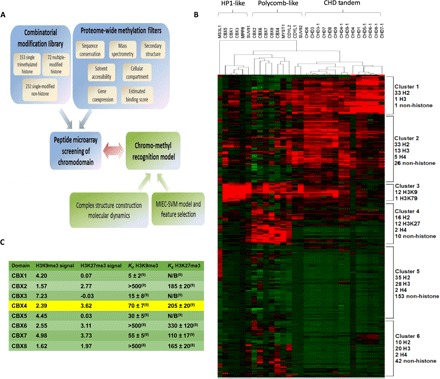
(A) Workflow of comprehensive screening for chromodomain recognition profiling. The screened peptides (combinatorial modification library) were composed of 153 possible single trimethylated histone peptides in the human proteome and 72 histone peptides that include up to three modifications at sites K4, K9, K27, K36, K56, and K79 of the H3 histone protein and 232 non-histone peptides in the human proteome (selected by various filters) that are likely bound by the chromodomains [proteome-wide methylation filters; see the Supplementary Methods and Materials and details in (19, 20)]. The peptides were printed on a microarray to test their binding to 29 human chromodomains (peptide microarray screening of chromodomain). The chromodomain-peptide recognition specificity can be predicted using a quantitative model (chromo-methyl recognition model) that captures the structural and energetic features at the binding interface (complex structure construction/MD, MIEC-SVM model, and feature selection). (B) Chromodomain-peptide binding intensities on the microarray (shown as z scores; red, binding; green, nonbinding). The z scores were calculated by standard normalization using all the signal intensities for each chromodomain. For any data point x, z score = (x − mean)/SD. Three categories of chromodomains and six clusters of peptides with different chromodomain binding preferences (the size and the composition of peptide sequences for each peptide cluster are shown on the right) were identified by hierarchical clustering. (C) Literature comparison of H3K9me3 and H3K27me3 z scores to reported Kd values (in μM) of interactions of the CBX chromodomains. Green color indicates consistent with previous study, and yellow is inconsistent.
MATERIALS AND METHODS
Peptide microarray experiments, data acquisition, and analysis
A total of 29 WT chromodomains and two CBX1 mutants were expressed as glutathione S-transferase (GST) fusion proteins using pGEX-KG vector in Escherichia coli strain BL21(DE3) based on a previously described protocol (19). A total of 467 unmodified and modified peptides [selected by a bioinformatics pipeline previously developed and described in (19, 20) and in the Supplementary Methods and Materials] were synthesized by Sigma-Aldrich (desalted, mass spectrometry checked). The peptides were then printed as triplets onto glass slides (ArrayIt). The CBX1 mutant was screened against an Active Motif MODified Histone Peptide Array. Binding of the peptide microarray and the chromodomain proteins was performed and analyzed using a previously described protocol [see (19) and Supplementary Methods and Materials for details].
Fluorescence polarization (FP)
Fluorescein-labeled methylated histone peptides (Karebay) were used for dissociation constant (Kd) determination by serial dilution in triplicate. The peptides used were H3K9me3 [NH2-ARTKQTARK(me3)STGG-minipeg-K(5-fam)-NH2] or H3K27me3 [Ac-QLATKAARK(me3)SAPA-minipeg-K(5-fam)-NH2] at a concentration of 1 nM. Serial dilutions were prepared in tris-buffered saline buffer [25 mM tris and 125 mM NaCl (pH 8)]. FP (fluorescence polarization) values were read on a DTX 880 Multimode Detector Beckman Coulter plate reader with excitation filter at 485 nm and two emission filters at 535 nm equipped with polarizers. Data were fit to a nonlinear regression equation using GraphPad Prism 4 software.
Building the molecular interaction energy component–support vector machine model
Template construction and conformational sampling
The template chromo-peptide complex structures for 13 chromodomains (CBX1–8, MPP8, CDYL1, CDYL2, SUV91, and SUV92) were obtained from either the Protein Data Bank or homology modeling based on a previously described method (19, 20). The peptide in each chromo-peptide structure contains nine residues (truncated if more than nine) with trimethylated lysine on the eighth position. For each chromo-peptide template, molecular dynamics (MD) simulation was performed for conformational optimization and sampling. Binding interface residue backbone root mean square deviation (RMSD) was evaluated for the 13 chromodomains to verify the equilibrium. After the production run, eight snapshots were evenly selected from the trajectory between 3 and 5 ns as chromo-peptide binding complex templates for each system. The chromo-peptide binding complex templates were mutated in silico to each of the 457 peptides. Restrained by the computational cost, we performed 5000 steps of energy minimization instead of MD simulation to optimize all the complex structures obtained through mutation (see Supplementary Methods and Materials for details).
Calculation of molecular interaction energy components
Residue pairwise energy decomposition on minimized structures was performed by mm_pbsa.pl in the Assisted Model Building with Energy Refinement (AMBER) package (21–23) (see Supplementary Methods and Materials for details), and a molecular interaction energy component (MIEC) profile was generated on the basis of the energy decomposition result consisting of chromo-peptide MIEC profile and peptide internal MIEC profile. For chromo-peptide MIEC profile, the energy contributions from eight snapshots are averaged, and all residue pairs less than 10 Å were included to reflect binding characteristics of the chromo-peptide interactions. The sequences of chromodomains were aligned using sequence and structural information to match residues from different domains. For peptide internal MIEC profile, MIECs of the adjacent peptide residue pairs were calculated to represent the conformational preference of the peptide. MIEC profile for each chromo-peptide interaction contains 158 chromo-peptide residue pairs and eight peptide-peptide pairs (664 energy components in total) (see Supplementary Methods and Materials for details).
Construction, training, and testing of the MIEC–support vector machine model
LASSO (least absolute shrinkage and selected operator) logistic regression method (24) was applied to the MIEC profile to select informative features to construct the MIEC–support vector machine (SVM) model (see table S4). All SVM training and tests were conducted using the Library for Support Vector Machines (LIBSVM) package (25). The polynomial kernel function was used. A nested cross-validation was performed to exclude an overfitting issue in the training process (see the Supplementary Material for details and table S5). Both threefold cross-validation and leave-one-domain-out (LODO) test were performed to evaluate the prediction accuracy of the MIEC-SVM model (see Supplementary Methods and Materials for details).
Selection of candidate sites to randomize on the CBX1 chromodomain
To select for sites to randomize in the CBX1 library to enhance its binding affinity toward H3K9me3, we used two different strategies. The first was to identify residues important for binding H3K9me3 compared with nonbinding peptides. Comparison of MIEC components of H3K9me3 binding to nonbinder components, along with avoiding choosing residues that were too conserved among chromodomains, we constructed a ranked list of candidate sites that showed the largest energetic difference between binders and nonbinders. The second strategy was essentially the same as the first strategy except that we selected sites generally important for CBX1 binding by comparing all CBX1 binder MIECs to a representative nonbinder (H3K27me3). Again, a conservation filter was applied to prevent selection of structurally important residues on the chromodomain (see Supplementary Methods and Materials for a more detailed description along with table S6).
Yeast surface display library construction and screening
The five sites selected for randomization on the CBX1 chromodomain [residues 22, 25, 59, 60, and 62 of National Center for Biotechnology Information (NCBI) P83916.1] were fully randomized using primers containing NNK degenerate codons, where the WT domain was used as template for polymerase chain reaction (forward: 5′-acaattcgtctcggtaccagaatatNNKgtggaaNNKgttctcgaccgtcgagtggtaaagggcaaagtggagtac-3′; reverse: 5′-ataattcgtctcctcgagtgttttctgtgactgcagaaactcagcaatgagMNNgggMNNMNNcaggttctcttctggc-3′ (N is any base, K is g/t, and M is c/a). The library was ligated into a modified pYD1 vector, followed by transformation into EBY100 yeasts using the LiAc/ssDNA/PEG-3350 method (26), resulting in approximately 106 transformants. The yeast surface display library was screened using biotin-Y-ahx-RTKQTARK(me3)S-NH2 (ahx, aminohexanoic acid) as the antigen. Yeast was sorted on a BD FACSAria II (Moores Cancer Center, University of California San Diego). Random hits were sequenced by dideoxy sequencing.
Live-cell imaging of the H3K9me3 mark
Molecular cloning, cell culture, transfection, and immunocytochemistry
The CBX1 chromodomain (either WT or the triple mutant) was cloned into a pcDNA3 vector and fused to the N terminus of the photoactivatable mCherry (PAmCherry) protein. HeLa and mouse embryonic fibroblast (MEF) cells were cultured using standard protocols and plated on 3.5-cm glass-bottom dishes for imaging. For live-cell photoactivated localization microscopy (PALM) imaging, HeLa and MEF cells were transiently transfected using either Lipofectamine 2000 (Thermo Fisher Scientific) or Polyjet (SignaGen). For fixed-cell stochastic optical reconstruction microscopy (STORM) imaging, cells were fixed with 4% paraformaldehyde (PFA) for 20 min and then washed with 100 mM glycine in Hanks’ balanced salt solution (HBSS) to quench the free PFA. Cells were permeabilized and blocked in a permeabilization solution with 0.1% Triton X-100, 0.2% bovine serum albumin, 5% goat serum, and 0.01% sodium azide in HBSS. The cells were then incubated overnight at 4°C with an anti-H3K9me3 antibody (Abcam, ab8898) at a 1:500 dilution, followed by 1 to 2 hours with goat anti-rabbit Alexa 647–conjugated antibodies at 1:1000 dilution. The cells were then postfixed again in 4% PFA, quenched with 100 mM glycine in HBSS, and washed with HBSS to prepare for imaging. Immediately before imaging, the medium was changed to STORM-compatible buffer [50 mM tris-HCl (pH 8.0), 10 mM NaCl, and 10% glucose) with glucose oxidase (560 μg/ml), catalase (170 μg/ml), and mercaptoethylamide (7.7 mg/ml).
For the two-color STORM imaging, the cells were incubated overnight at 4°C with a mouse anti-FLAG antibody (for detection of FLAG-tagged CBX1; Sigma, F1804) at a 1:200 dilution and with a rabbit anti-H3K9me3 antibody (Abcam, 8898) at a 1:500 dilution. They were then incubated 1 to 2 hours with goat anti-rabbit and anti-mouse antibodies, labeled with Alexa 647 and Alexa 568, respectively, at a 1:1000 dilution.
Super-resolution imaging and image analysis
STORM and PALM images were obtained using a Nikon Ti total internal reflection fluorescence (TIRF) microscope with N-STORM, an Andor IXON3 Ultra DU897 EMCCD, and a 100× oil immersion TIRF objective. Photoactivation was driven by a Coherent 405-nm laser, while excitation was driven either with a Coherent 561-nm laser or a 647-nm laser. Illumination was done in a “near-TIRF” format, in which the TIRF angle was adjusted so that molecules in the nucleus were illuminated. All image analysis and image reconstruction were performed using the Localizer software (27) in the Igor Pro 6.3 environment. STORM and PALM images were segmented using the GLRT algorithm (27), and localizations were fit using Gaussian fitting. Reconstructed bitmap images were created in which intensity corresponds to the number of localizations in each box in a 0.2 pixel wide grid. For two-color STORM imaging colocalization analysis, the protocol can be found in the Supplementary Methods and Materials.
RESULTS
To have a comprehensive view of how modified histone peptides are recognized by chromodomains, we have studied the binding between 29 chromodomains encoded in the human proteome (table S1) and 153 nine–amino acid–long histone peptides with a single trimethylated lysine. To examine the combinatorial effect of PTMs, we studied an additional 72 H3 histone peptides with multiple modifications, which includes up to three possible combinations of modifications adjacent to K4, K9, K27, K36, K56, and K79 of the H3 histone protein. The sequences were designed to include reported modifications of adjacent arginine (symmetric or asymmetric dimethylarginine), serine (phosphoserine), threonine (phosphothreonine), and lysine (monomethyl or trimethyl). Because sites surrounding K4, K9, and K27 are known to be heavily modified, up to four modifications in total were included in the peptides, while for the other sites located in less modified regions of the histones, up to two modifications are included in the sequence. To further illuminate the importance of PTMs that encode the recognition specificity beyond amino acid sequence, we also included 232 non-histone peptides from the human proteome with a single trimethylated lysine but otherwise diverse sequences (table S2). The inclusion of diverse non-histone peptides provides a unique opportunity to examine whether there exist sequence motifs dominating the recognition of histone modifications by the reader proteins, which cannot be studied using histone peptides only due to their sequence similarity. We also included 10 proline-rich peptides without any modifications that do not bind to chromodomains as negative controls on the microarray to bring the total number of peptides to 467.
Validation of the binding detected by microarray
To investigate binding interactions between the 29 chromodomains and 467 peptides, a high-throughput assay was needed, and we chose to use peptide microarray technology (19, 20). The detection accuracy and efficiency of the peptide microarray platform were confirmed by two independent lines of evidence (Fig. 1, A to C).
Overall microarray performance: Correct categorization of chromodomains
The 29 chromodomains encoded in the human proteome can be classified into three categories based on their methyllysine-binding mode (Fig. 1B). The available crystal or solution structures show that chromodomains have two general types of structures: (i) a single binding mode where one single chromodomain forms a binding pocket to accommodate the histone peptide (such as CBX proteins), and (ii) a potential tandem binding mode in which two chromodomains are linked through a loop and both domains may interact with the peptide [such as chromodomain-helicase DNA binding (CHD) proteins]. For the first binding mode, the chromodomains can be further divided into two subgroups: HP1-like proteins and Polycomb-like proteins (from Drosophila melanogaster HP1 and Pc chromodomains).
We performed hierarchical clustering on the fluorescence intensities (represented as z scores) detected by the peptide microarray (Fig. 1B and fig. S2) and found that the overall binding profiles can correctly group chromodomains with similar structural features: Polycomb-like CBX members (CBX2, CBX4, CBX6, CBX7, and CBX8) were clustered together with some other single chromodomains such as MYST1 and CDYL2, but separated from the HP1-like CBX members CBX1, CBX3, and CBX5, and further separated from CHD proteins. This clustering result is consistent with the known or possible chromodomain binding modes (Fig. 1B), suggesting that our microarray can correctly discriminate different structural characteristics and binding modes of the chromodomains. Although the clustering of the domains was mostly consistent, we observed certain noticeable outliers. Notably, SUV92 and CDYL1 seemed to cluster somewhat with the CHD tandem domains. This may suggest a CHD tandem-like binding mode for these domains, where SUV92 and CDYL1 could form dimers/multimers, thereby allowing them to use more than one chromodomain to recognize their targets. It has been reported that CDYL1 forms multimers to recognize heterchromatin in vivo (28). Another outlier was MS3L1, which unlike the other HP1-like chromodomains lacked significant interaction with cluster 3 yet showed some interaction with cluster 5. It has been shown that binding to double-stranded DNA (dsDNA) facilitates the binding of the MS3L1 chromodomain to H4K20me1 (29). Our data did not show interaction with this mark, but our array screens did not include dsDNA in the binding buffer. It would be interesting in a future study to include dsDNA when screening chromodomains against arrays to see if that could alter or enhance their binding specificity/affinity for the array peptides.
Further validation with literature evidence
Literature-reported H3K9 and H3K27 binding by CBX1–8 were satisfyingly reproduced on the microarray (see comparison summary in Fig. 1C and table S3): CBX1, 3, and 5 behaved like HP1 and bound predominantly to all three methylated H3K9 peptides (9, 18); much worse binding of the HP1-like CBXs toward H3K27 was observed. For Polycomb-like CBX2, 4, 6, 7, and 8, binding to both methylated H3K9 and H3K27 was confirmed, with trimethylation as the state most favorable for binding. CBX2 was more Polycomb-like with higher H3K27 signal intensity compared with those of H3K9. For CBX6 and CBX8, reported as the weakest binders in the literature (8, 9), the peptide microarray showed signals with di- and trimethylated peptides at both sites. Contrary to previous reports, we found that the CBX4 chromodomain seemed to prefer H3K27me3 over H3K9me3. One possible explanation is that the binding studies in (8, 9) used longer peptides (at least 15 residues), whereas H3K9/H3K27 on our array were only nine residues, suggesting that CBX4 may make binding contacts to residues C-terminal to S10 of H3K9 to define its specificity for this mark. Together, the microarray results of the control peptides show that the array data are largely consistent with previous findings in the literature.
Chromodomain-methyllysine recognition is degenerate in two dimensions
Obviously, there is significant redundancy of chromodomain-methylated peptide recognition (columns and rows in Fig. 1B and fig. S2), i.e., one peptide may be recognized by different chromodomains, and one chromodomain may recognize a variety of methylated sequences regardless of the surrounding context. The 457 peptides can be clustered into six groups based on their chromodomain binding preferences. Notably, the histone and non-histone peptides are mixed in five of six clusters, suggesting that the recruitment of a reader protein (complex) to a certain histone site may be facing not only competition from other histone reader proteins but also competition from binding of the reader domain toward other modified non-histone proteins, especially those component proteins in the same complex or neighboring complexes within spatial proximity. For example, the histone acetyltransferase MYST1 and RBBP5 co-occur in some MLL1/MLL complexes to methylate and acetylate histones H3 and H4 (30), and binding between the MYST1 chromodomain and a methylated RBBP5 sequence was indeed observed in our result. Similarly, H3K9 methyltransferase SUV91 chromodomain showed binding to a methylated SIRT1 sequence, both of which serve as core component proteins in the eNoSC complex that represses ribosomal RNA transcription for cellular survival (31).
Recognition mechanism is beyond sequence or modification motif
When investigating the mechanisms governing the chromodomain recognition, we could not find any sequence motif in any of the six peptide clusters. No significant amino acid enrichment was observed at any of the eight nonmethyllysine positions in the peptides, except for the special case of cluster 3 composed of a set of H3K9 peptides with different PTMs that strongly prefer the HP1-like chromodomains (Fig. 2A). This observation suggests that there is no definitive sequence motif for methyllysine recognition by different chromodomains. Consistently, the binding peptides identified for each individual chromodomain contain no significant sequence motif either (Fig. 2B and table S3).
Fig. 2. Chromodomain binding sequence motif analysis from the peptide array screens.

(A) No significant sequence motif was observed in the sequences for each peptide cluster. Peptides were aligned such that methylated lysine was set as the eighth amino acid position. (B) Alignment of sequence motifs for each individual chromodomain shows a lack of any strong binding motif, consistent with (A).
Furthermore, no definitive PTM combination patterns could be found in the 72 H3 multiply modified peptides to dictate the chromodomains’ binding preference. The peptides with multiple modifications were integrated in the microarray, including methylated arginine, phosphorylated serine, and threonine in addition to the target methyllysine, in order to study the impact of their coexistence in the chromodomain recognition sequence (table S2 and fig. S2). We observed that the same histone peptides with different PTM combinations often distribute to several clusters that exhibit different preferences to chromodomains (e.g., multiply modified H3K9 peptides were found in clusters 2, 3, and 6). This phenomenon also holds for different histone peptides with the same PTM combination pattern (e.g., H3R26me2/K27me3 in cluster 4 versus H3R8me2/K9me3 in cluster 3, H3T22ph/K27me3 in cluster 4 versus H3S31ph/K36me3 in cluster 6). Together, these observations suggest an absence of PTM patterns that govern chromodomains’ binding specificity.
A quantitative model to predict the chromo-peptide binding specificity
The absence of either primary sequence or modification motif pattern for the chromodomain-peptide recognition, together with the fact that many histone peptides show similar binding behavior to non-histone peptides, strongly suggests that the multiple modifications have altered the property of the peptide sequence in a profound way. Therefore, the commonly used sequence-based bioinformatics methods are unable to delineate the binding specificity. To reveal the underlying mechanisms, we constructed a MIEC-SVM model (19, 20, 32) to capture the structural and energetic patterns critical for the chromodomain-peptide binding (Fig. 3A). We focused on 13 HP1- and Polycomb-like proteins that bind to peptides with a single chromodomain as their binding modes are similar (fig. S3). The model showed excellent performance in predicting the binding specificity between these 13 chromodomains and all the 457 peptides, as indicated by the average area under the receiver characteristic curve (AUC-ROC) of 0.832 in 500 threefold cross-validations. Furthermore, satisfactory results of LODO (AUCLODO = 0.766) tests demonstrate MIEC-SVM’s generalization ability, i.e., MIEC-SVM can be used to predict interactions of new chromodomains or new peptides (Fig. 3B). This observation confirmed that the binding specificity of chromodomain-peptide interactions is encoded in the structural and energetic characteristics despite the lack of sequence or PTM motifs.
Fig. 3. Construction and validation of the chromodomain MIEC-SVM model.
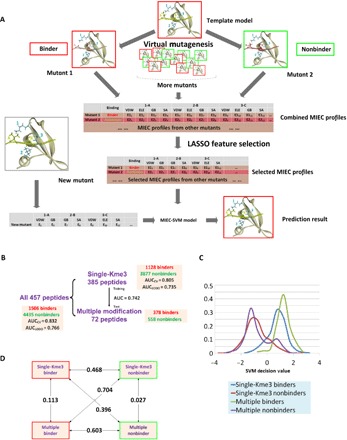
(A) Flowchart of MIEC-SVM that predicts binding specificity between chromodomains and methyllysine peptides. Complex structures between 13 chromodomains and 457 peptides were constructed by computationally mutating peptide sequence from a template complex for each chromodomain (virtual mutagenesis). From the modeled complex structures, MIEC terms between peptide-protein residues at the binding interface were computed. The MIECs and the binding/nonbinding label (obtained from microarray experiments) for each domain-peptide pair were input to a LASSO logistic regression model to select most predictive MIECs (LASSO feature selection). These selected MIEC features were then used to train an SVM model to discriminate binding from nonbinding events. VDW, Van der Waals forces; ELE, electrostatic forces; GB, polar contribution to the desolvation energy; SA, nonpolar contribution to the desolvation energy. (B) Performance of MIEC-SVM model on three different peptide groups (all peptides, singly modified peptides, and multiply modified peptides). The MIEC-SVM model showed consistent performance regardless of the number of modifications on the peptides, indicating that chromodomain-peptide recognition share the same MIEC features for singly and multiply modified peptides. (C) SVM decision value distribution of the four classes of peptides (binders/nonbinders with single or multiple modifications). Binders and nonbinders are well separated regardless of the modification number. (D) Pairwise Jensen-Shannon (JS) divergences between the SVM decision value distributions of the four classes. The differences between any binder class and nonbinder class (regardless of the PTM number) are large (larger JS divergence value) singly modified binder–singly modified nonbinder, JS = 0.468 (P < 1.0 × 10−20); singly modified binder–multiply modified nonbinder, JS = 0.396 (P < 1.0 × 10−19); multiply modified binder–single modified nonbinder, JS = 0.704 (P < 1.0 × 10−20); and multiply modified binder–multiply modified nonbinder, JS = 0.603 (P < 1.0 × 10−20). In contrast, binder (or nonbinder) peptides are similar to each other regardless of the PTM numbers: JS values of 0.113 for binders (P = 7.0 × 10−15 for statistical similarity) and 0.027 for nonbinders (P = 6.1 × 10−10). All P values were calculated on the basis of the background distributions of JS divergence of randomly selected decision values for the same number of binders or nonbinders as the foreground.
To further illuminate this point, we assessed the prediction performance of models trained on either singly or multiply modified peptides and predicted the remaining peptides. There were 385 single-Kme3 peptides with only trimethylated lysine on positions 8 and 72 non–single-Kme3 peptides including multiple modifications and unmodified lysine on position 8. Since the 385 single-Kme3 peptides provided sufficient training data, we tested whether MIEC-SVM model trained from these single-Kme3 peptides would be able to predict the binding of multiply/nonmodified Kme3 peptides (Fig. 3B). The satisfactory performance (AUC-ROC = 0.742) indicated that the structural and energetic characteristics of chromodomain-peptide recognition are shared between singly and multiply modified peptides. As the number of multiply modified peptides tested in the microarray was small, it was insufficient to train the MIEC-SVM model on them to predict singly modified peptides.
Furthermore, we observed similar distribution of singly and multiply modified binders in the MIEC-SVM feature space. We extracted the SVM decision values for each of the following four classes: single-Kme3 binders, multiply modified binders, single-Kme3 nonbinders, and multiply modified nonbinders. An obvious separation between binders and nonbinders was observed, which is not unexpected given the satisfactory performance of MIEC-SVM (Fig. 3C). Furthermore, the decision value distributions of the singly and multiply modified binders were significantly overlapped (Fig. 3, C and D), which confirmed the similar binding patterns of the singly and multiply modified peptides. This observation further confirmed that PTMs provide additional diversity of physiochemical properties at the binding interface but all binders share the same MIEC patterns.
MIEC-SVM model applied to chromodomain engineering
We next sought to use our trained model to enhance the binding affinity of the chromodomain of human CBX1 toward its preferred binder, the H3K9me3 mark. We chose the CBX1 chromodomain over the other chromodomains for several reasons. First, CBX1 showed a good MIEC-SVM prediction performance. Second, its chromodomain already binds fairly tightly and selectively to the H3K9me3 peptide [Kd of 5 μM; (9)]. Last, the other two chromodomains we felt were good candidates (SUV92 and MPP8) showed poor expression on the yeast surface.
After selection of the appropriate chromodomain, we performed computational analysis to select potentially important sites on the CBX1 chromodomain to enhance the binding affinity to H3K9me3 (see Fig. 4A). We constructed a list of sites that were most important for recognition of H3K9me3 compared with nonbinding peptides. Second, we constructed a list of sites important for binding to all CBX1 binders compared with H3K27me3 (a nonbinder) to select residues generally important for binding. Residues that were too conserved among chromodomains or contributed little to the binding energy difference of binding versus nonbinding peptides were filtered out. From the two lists of sites, we selected three sites from the K9me3 list (sites 59, 60, and 62) and three from the general list (sites 22, 25, and 59; see table S6 and the Supplementary Methods and Materials for a more detailed description of the site selection) of the CBX1 protein (UniProt ID: P83916).
Fig. 4. Application of the chromodomain MIEC-SVM model to engineering the CBX1 chromodomain.
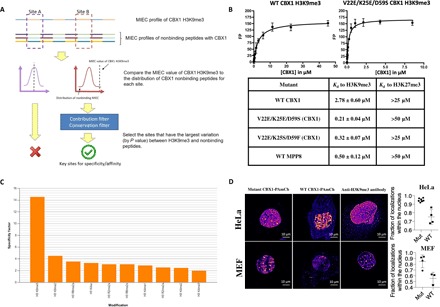
(A) Selection of key sites to randomize for the yeast display experiment with the CBX1 chromodomain. Sites “A” and “B” are just two representative residues being analyzed for site selection for demonstration purposes. The other strategy of selecting residues to randomize on CBX1 (comparing H3K27me3 to CBX1 binders) used the same procedure. (B) Kd values obtained from fluorescence polarization binding studies between the WT CBX1 and MPP8 chromodomains, along with the V22E mutants isolated from yeast surface display selections. The Kd values were derived from a nonlinear regression equation after performing experiments in triplicate against 1 nM H3K9me3 [NH2-ARTKQTARK(me3)STGG-mini-PEG-K(5-fam)-NH2] and 1 nM H3K27me3 [Ac-QLATKAARK(me3)SAPA-mini-PEG-K(5-fam)-NH2] peptides. Kd values toward H3K27me3 are based on visual approximation from unsaturated binding curves; for the V22E mutants, up to 180 μM of each protein was used in an attempt to get a binding curve to H3K27me3. (C) Peptide array screening of the V22E/K25E/D59S CBX1 chromodomain mutant. An Active Motif histone peptide array, containing 384 peptide spots printed in duplicate, was screened against 1 nM GST-CBX1 mutant. Spots were visualized by chemiluminescence, and the spot intensities were analyzed by Active Motif array analysis software. The height of the y axis (specificity factor) represents the ratio of the average intensity of all array spots containing the mark (listed on the x axis) over the average intensity of spots not containing the mark. (D) Left: Representative reconstructed PALM image of HeLa cells (top row) and MEF cells (bottom row) transiently transfected with CBX1 (V22E/K25E/D59S)–PAmCherry (left) WT CBX1-PAmCherry (center), or immunostained with Alexa 647–labeled anti-H3K9me3 antibody (right). Scale bars, 10 μm. Right: Quantification of fraction of PALM localizations located within the nuclear region for HeLa cells (top) and MEF cells (bottom) (unpaired two-tailed t test). Mut: V22E/K25E/D59S triple mutant.
Upon selection of the positions deemed most critical for enhancing binding affinity toward H3K9me3, we randomized CBX1 chromodomain positions 22, 25, 59, 60, and 62 using degenerate codons (NNK) and introduced the DNA library into the pYD1 vector for yeast surface display library selections (26). A fairly low diversity library (about 106 clones) was screened using a biotin-labeled H3K9me3 peptide. The selected CBX1 chromodomain mutants, upon purification as GST fusions from E. coli, were tested for their binding affinity toward H3K9me3 by fluorescence polarization binding assay (see Fig. 4B). The two mutants selected from the H3K9me3 sorts (V22E/K25S/D59F and V22E/K25E/D59S) showed superior binding to H3K9me3 than the WT domain (Kd values of 0.32 and 0.21 μM versus 2.78 μM of the WT domain), yet showed very weak binding to the similar H3K27me3 peptide. To our knowledge, the V22E/K25E/D59S mutant has the strongest binding affinity of any human chromodomain toward H3K9me3, even over that of MPP8. To verify that the mutant binds specifically toward H3K9me3, we screened it against a MODified Histone Peptide Array (Active Motif), which contains 384 various histone peptide marks. Analysis of the developed array revealed that the mutant retained the specificity of WT CBX1 toward H3K9me3 (Fig. 4C).
Application of the engineered CBX1 chromodomain in live-cell histone imaging
We aimed to test whether we could use the V22E/K25E/D59S chromodomain to label the H3K9me3 modification in the nuclei of living cells. Using PALM imaging (33), we used PAmCherry (34)–chromodomain fusions to visualize H3K9me3 in live cells, compared with either the WT CBX1 chromodomain or an anti-H3K9me3 antibody (Abcam, ab8898). Upon transient expression in either HeLa or MEF cells, we observed punctate structures with very dense labeling as well as a meshwork pattern of moderately high localization density interspersed with regions with lower labeling (Fig. 4D).
Overall, the mutant showed a very high level of nuclear localization (approximately 85% in MEF and 95% in HeLa cells) (Fig. 4D), which is impressive given the lack of a nuclear localization sequence (NLS) tag in the construct. This was an obvious improvement over the WT domain and comparable to fixed-cell images created using Abcam (ab8898). Furthermore, we tested the degree of colocalization of the triple mutant with the antibody in two-color fixed STORM imaging in HeLa cells (fig. S4). The extent of colocalization was determined using a published method based on Getis and Franklin’s local point pattern analysis (35). We measured that an average of 63% of localizations of the CBX1 mutant coclustered with the H3K9me3 antibody, and 94% of H3K9me3 antibody localizations coclustered with mutant CBX1 (V22E/K25E/D59S). These data are consistent with the peptide array data and confirm the CBX1 mutant’s specificity for the H3K9me3 mark within the cellular environment. We also created movies using PALM imaging of both the WT and mutant CBX1-PAmCherry labeling H3K9me3 in both HeLa and MEF cells (movies S1 to S4). Again, the mutant showed greater nuclear localization than the WT and resulted in a noticeably better signal-to-noise ratio in the images.
CONCLUSION
We conducted the first comprehensive survey of chromodomain-methyllysine recognition using an integrated experimental and computational procedure. We found great degeneracy of chromodomain proteins binding to modified histone peptides in that one chromodomain can bind to diverse modifications at different histone sites, while one histone modification pattern can be bound by different reader proteins. We found this recognition degeneracy is not associated with amino acid sequence or PTM patterns, but rather is rooted in the same physiochemical properties of the binding interface that are defined by the PTMs.
Our studies have demonstrated that the MIEC-based model can accurately characterize chromodomain-peptide recognition by capturing the physiochemical features rather than the amino acid sequence or PTM pattern of the binding peptides. We also demonstrated the model to be applicable in the prediction of interaction between new peptides and chromodomains. Our finding suggests a general principle governing the recognition of histone modifications by reader proteins and provides a model to quantify such recognition. It not only extends our knowledge of the role of chromodomains in protein-protein interaction networks but also can be applied to engineering chromodomains to perform useful functions. In this study, we demonstrated the power of our model by accurately predicting residues to be randomized in a small chromodomain library and enhanced the binding affinity of the CBX1 chromodomain by 13-fold compared with the WT domain without sacrificing its binding specificity. The improved CBX1 chromodomain performed significantly better at labeling H3K9me3 in live HeLa and MEF cells and could be used to image the dynamics of H3K9 methylation in real time, which is not possible with anti-histone antibodies.
Supplementary Material
Acknowledgments
We are grateful to C. Arrowsmith for providing us with the CBX1–8 constructs, which we modified for cloning. Funding: This work was supported by NIH (R01GM085188 to W.W. and R35CA197622 to J.Z.). Author contributions: R.H., W.H., G.C.H.M., and B.R. performed all the experiments, analyzed the data, and wrote the paper. N.L. conducted all the computational simulations, analyzed the data, and wrote the paper. R.S.L.S. contributed to computational simulations. Q.P. and Y.W. contributed to the yeast display experiments. E.K. contributed to the peptide microarray experiments. J.Z. designed, supervised the imaging experiments, and analyzed the data. W.W. conceived and designed the study, analyzed the data, and wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The MIEC-SVM code can be downloaded at http://wanglab.ucsd.edu/MIEC-SVM/. The CBX1 constructs used for imaging can be provided by the laboratory of J.Z., the GST fusion constructs of CBX1 can be provided by the laboratory of W.W., and the pYD1 vector used for constructing the yeast display library can be provided by the laboratory of Y.W., pending scientific review and a completed material transfer agreement.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/11/eaau1447/DC1
Supplementary Methods and Materials
Fig. S1. Hierarchical clustering of chromodomains based upon z scores.
Fig. S2. Two-dimensional hierarchical clusters of peptides binding to chromodomains based on z scores (as separate PDF file).
Fig. S3. Multiple sequence alignment of chromodomains.
Fig. S4. Getis-Franklin single-molecule coclustering analysis (35) of H3K9me3 and the CBX1 (V22E/K25E/D59S) chromodomain.
Table S1. List of chromodomains screened by microarray.
Table S2. Four hundred sixty-seven peptides printed on the histone microarray (as separate Excel file).
Table S3. Sequence and averaged signal intensities of identified binders from the peptide microarray for all 29 chromodomains (as separate Excel file).
Table S4. Receptor-ligand residue pairs after LASSO feature selection (as separate Excel file).
Table S5. Nested cross-validation performed to evaluate overfitting in the training process (as separate Excel file).
Table S6. List of ranked sites for the CBX1 chromodomain that were considered for randomization (as separate Excel file).
Movie S1. WT CBX1-PAmCherry in MEF cells.
Movie S2. V22E/K25E/D59S CBX1-PAmCherry in MEF cells.
Movie S3. WT CBX1-PAmCherry in HeLa cells.
Movie S4. V22E/K25E/D59S CBX1-PAmCherry in HeLa cells.
REFERENCES AND NOTES
- 1.Deribe Y. L., Pawson T., Dikic I., Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 17, 666–672 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Eissenberg J. C., Structural biology of the chromodomain: Form and function. Gene 496, 69–78 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Martin C., Zhang Y., The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 6, 838–849 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Kouzarides T., Chromatin modifications and their function. Cell 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Ruthenburg A. J., Li H., Patel D. J., Allis C. D., Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 8, 983–994 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taverna S. D., Li H., Ruthenburg A. J., Allis C. D., Patel D. J., How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14, 1025–1040 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yun M., Wu J., Workman J. L., Li B., Readers of histone modifications. Cell Res. 21, 564–578 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernstein E., Duncan E. M., Masui O., Gil J., Heard E., Allis C. D., Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol. Cell. Biol. 26, 2560–2569 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaustov L., Ouyang H., Amaya M., Lemak A., Nady N., Duan S., Wasney G. A., Li Z., Vedadi M., Schapira M., Min J., Arrowsmith C. H., Recognition and specificity determinants of the human Cbx chromodomains. J. Biol. Chem. 286, 521–529 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filippakopoulos P., Picaud S., Mangos M., Keates T., Lambert J.-P., Barsyte-Lovejoy D., Felletar I., Volkmer R., Müller S., Pawson T., Gingras A.-C., Arrowsmith C. H., Knapp S., Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214–231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bian Y., Li L., Dong M., Liu X., Kaneko T., Cheng K., Liu H., Voss C., Cao X., Wang Y., Litchfield D., Ye M., Li S. S.-C, Zou H., Ultra-deep tyrosine phosphoproteomics enabled by a phosphotyrosine superbinder. Nat. Chem. Biol. 12, 959–966 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Sanchez O. F., Mendonca A., Carneiro A. D., Yuan C., Engineering recombinant protein sensors for quantifying histone acetylation. ACS Sens. 2, 426–435 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Moore K. E., Gozani O., An unexpected journey: Lysine methylation across the proteome. Biochim. Biophys. Acta 1839, 1395–1403 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S. A., Flynn R. A., Wysocka J., A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chuikov S., Kurash J. K., Wilson J. R., Xiao B., Justin N., Ivanov G. S., McKinney K., Tempst P., Prives C., Gamblin S. J., Barlev N. A., Reinberg D., Regulation of p53 activity through lysine methylation. Nature 432, 353–360 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Nishikori S., Hattori T., Fuchs S. M., Yasui N., Wojcik J., Koide A., Strahl B. D., Koide S., Broad ranges of affinity and specificity of anti-histone antibodies revealed by a quantitative peptide immunoprecipitation assay. J. Mol. Biol. 424, 391–399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kungulovski G., Kycia I., Tamas R., Jurkowska R. Z., Kudithipudi S., Henry C., Reinhardt R., Labhart P., Jeltsch A., Application of histone modification-specific interaction domains as an alternative to antibodies. Genome Res. 24, 1842–1853 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shanle E. K., Shinsky S. A., Bridgers J. B., Bae N., Sagum C., Krajewski K., Rothbart S. B., Bedford M. T., Strahl B. D., Histone peptide microarray screen of chromo and Tudor domains defines new histone lysine methylation interactions. Epigenetics Chromatin 10, 12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li N., Stein R. S., He W., Komives E., Wang W., Identification of methyllysine peptides binding to chromobox protein homolog 6 chromodomain in the human proteome. Mol. Cell. Proteomics 12, 2750–2760 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stein R. S. L., Li N., He W., Komives E., Wang W., Recognition of methylated peptides by Drosophila melanogaster polycomb chromodomain. J. Proteome Res. 12, 1467–1477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko, and P. A. Kollman, AMBER 11 (University of California, San Francisco, 2010). [Google Scholar]
- 22.Duan Y., Wu C., Chowdhury S., Lee M. C., Xiong G., Zhang W., Yang R., Cieplak R., Luo R., Lee T., Caldwell J., Wang J., Kollman P., A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comp. Chem. 24, 1999–2012 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A., Development and testing of a general amber force field. J. Comp. Chem. 25, 1157–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 24.T. Hastie, R. Tibshirani, J. H. Friedman, The Elements of Statistical Learning Data Mining, Inference, and Prediction (Springer, ed. 2, 2009), pp. xxii, 745 pp. [Google Scholar]
- 25.Chang C. C., Lin C. J., LIBSVM—A library for support vector machines. ACM TIST 2, 27:1–27:27 (2011). [Google Scholar]
- 26.Gietz R. D., Schiestl R. H., High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Dedecker P., Mo G. C. H., Dertinger T., Zhang J., Widely accessible method for superresolution fluorescence imaging of living systems. Proc. Natl. Acad. Sci. U.S.A. 109, 10909–10914 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franz H., Mosch K., Soeroes S., Urlaub H., Fischle W., Multimerization and H3K9me3 binding are required for CDYL1b heterochromatin association. J. Biol. Chem. 284, 35049–35059 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim D., Blus B. J., Chandra V., Huang P., Rastinejad F., Khorasanizadeh S., Corecognition of DNA and a methylated histone tail by the MSL3 chromodomain. Nat. Struct. Mol. Biol. 17, 1027–1029 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dou Y., Milne T. A., Tackett A. J., Smith E. R., Fukuda A., Wysocka J., Allis C. D., Chait B. T., Hess J. L., Roeder R. G., Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 121, 873–885 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Murayama A., Ohmori K., Fujimura A., Minami H., Yasuzawa-Tanaka K., Kuroda T., Oie S., Daitoku H., Okuwaki M., Nagata K., Fukamizu A., Kimura K., Shimizu T., Yanagisawa J., Epigenetic control of rDNA loci in response to intracellular energy status. Cell 133, 627–639 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Li N., Ainsworth R., Wu M., Ding B., Wang W., MIEC-SVM: Automated pipeline for protein peptide/ligand interaction prediction. Bioinformatics 32, 940–942 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Betzig E., Patterson G. H., Sougrat R., Lindwasser O. W., Olenych S., Bonifacino J. S., Davidson M. W., Lippincott-Schwartz J., Hess H. F., Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Subach F. V., Patterson G. H., Manley S., Gillette J. M., Lippincott-Schwartz J., Verkhusha V. V., Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat. Methods 6, 153–159 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossy J., Cohen E., Gaus K., Owen D. M., Method for co-cluster analysis in multichannel single-molecule localisation data. Histochem. Cell Biol. 141, 605–612 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Kabsch W., A solution for best rotation to relate 2 sets of vectors. Acta Crystallogr. A 32, 922–923 (1976). [Google Scholar]
- 37.M. J. Frisch, G. W. T., H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09 Revision A.1 (Gaussian Inc., 2009).
- 38.Bayly C. I., Cieplak P., Cornell W. D., Kollman P. A., A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 97, 10269–10280 (1993). [Google Scholar]
- 39.Wang J., Wang W., Kollmann P., Case D., Antechamber, an accessory software package for molecular mechanical calculation. J. Comput. Chem. 25, 1157–1174 (2005). [Google Scholar]
- 40.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). [Google Scholar]
- 41.Ryckaert J.-P., Ciccotti G., Berendsen H. J. C., Numerical-integration of cartesian equations of motion of a system with constraints: Molecular-dynamics of n-alkanes. J. Comp. Phys. 23, 327–341 (1977). [Google Scholar]
- 42.Krivov G. G., Shapovalov M. V., Dunbrack R. L. Jr., Improved prediction of protein side-chain conformations with SCWRL4. Proteins 77, 778–795 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Onufriev A., Bashford D., Case D. A., Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55, 383–394 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Friedman J., Hastie T., Tibshirani R., Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010). [PMC free article] [PubMed] [Google Scholar]
- 45.Rossy J., Owen D. M., Williamson D. J., Yang Z., Gaus K., Conformational states of the kinase Lck regulate clustering in early T cell signaling. Nat. Immunol. 14, 82–89 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/11/eaau1447/DC1
Supplementary Methods and Materials
Fig. S1. Hierarchical clustering of chromodomains based upon z scores.
Fig. S2. Two-dimensional hierarchical clusters of peptides binding to chromodomains based on z scores (as separate PDF file).
Fig. S3. Multiple sequence alignment of chromodomains.
Fig. S4. Getis-Franklin single-molecule coclustering analysis (35) of H3K9me3 and the CBX1 (V22E/K25E/D59S) chromodomain.
Table S1. List of chromodomains screened by microarray.
Table S2. Four hundred sixty-seven peptides printed on the histone microarray (as separate Excel file).
Table S3. Sequence and averaged signal intensities of identified binders from the peptide microarray for all 29 chromodomains (as separate Excel file).
Table S4. Receptor-ligand residue pairs after LASSO feature selection (as separate Excel file).
Table S5. Nested cross-validation performed to evaluate overfitting in the training process (as separate Excel file).
Table S6. List of ranked sites for the CBX1 chromodomain that were considered for randomization (as separate Excel file).
Movie S1. WT CBX1-PAmCherry in MEF cells.
Movie S2. V22E/K25E/D59S CBX1-PAmCherry in MEF cells.
Movie S3. WT CBX1-PAmCherry in HeLa cells.
Movie S4. V22E/K25E/D59S CBX1-PAmCherry in HeLa cells.