

SUMMARY
Circadian clock dysfunction is a common symptom of aging and neurodegenerative diseases, though its impact on brain health is poorly understood. Astrocyte activation occurs in response to diverse insults and plays a critical role in brain health and disease. We report that the core circadian clock protein BMAL1 regulates astrogliosis in a synergistic manner via a cell-autonomous mechanism and a lesser non-cell-autonomous signal from neurons. Astrocyte-specific Bmal1 deletion induces astrocyte activation and inflammatory gene expression in vitro and in vivo, mediated in part by suppression of glutathione-S-transferase signaling. Functionally, loss of Bmal1 in astrocytes promotes neuronal death in vitro. Our results demonstrate that the core clock protein BMAL1 regulates astrocyte activation and function in vivo, elucidating a mechanism by which the circadian clock could influence many aspects of brain function and neurological disease.
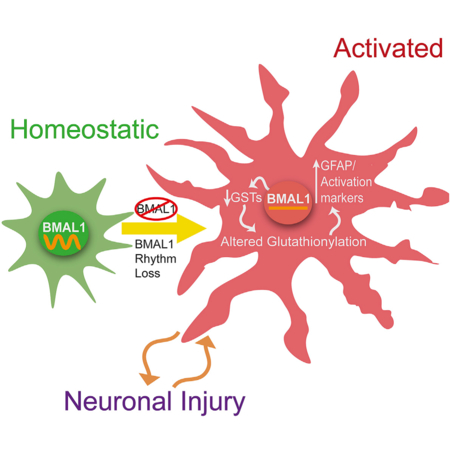
In Brief
Lananna et al. show that the circadian clock protein BMAL1 regulates astrocyte activation via a cell-autonomous mechanism involving diminished glutathione-S-transferase signaling. This finding elucidates a function of the core circadian clock in astrocytes and reveals BMAL1 as a modulator of astrogliosis.
INTRODUCTION
Changes in behavioral circadian rhythms are common in many neurodegenerative diseases (Breen et al., 2014; Hatfield et al., 2004; Musiek and Holtzman, 2016) and can occur early in disease progression (Musiek et al., 2018). Circadian rhythms are generated by the suprachiasmatic nucleus of the hypothalamus (SCN) (Mohawk et al., 2012), which synchronizes cellular clocks throughout the body to the light-dark (L:D) cycle. The core molecular clock consists of a transcriptional-translational feedback loop with a positive transcriptional limb, consisting of heterodimers of the basic-helix-loop-helix (bHLH) transcription factor BMAL1 (also known as ARNTL [aryl hydrocarbon receptor nuclear translocator-like protein 1]) with either CLOCK (circadian locomotor output cycles kaput) or NPAS2 (neuronal PAS domain protein 2). The negative limb includes Period (PER), cryptochrome (CRY), and REV-ERB genes, which are transcriptional targets of BMAL1 and provide negative feedback inhibition of BMAL1 function (Mohawk et al., 2012). The core clock regulates transcription of thousands of genes in a highly tissue-specific manner, regulating cellular functions including metabolism, inflammation, and redox homeostasis (Bass and Takahashi, 2010). In addition to disrupted activity and sleep rhythms, dysregulated circadian gene expression patterns are also observed in neurodegenerative disease models and patients (Song et al., 2015; Breen et al., 2014; Cronin et al., 2017). However, the impact of clock dysfunction on neurological diseases of the brain remains poorly understood.
Astrocytes play a critical role in brain health and neurodegenerative disease, as dysfunctional astrocytes can drive neurodegeneration (Lian et al., 2015; Macauley et al., 2011; Yamanaka et al., 2008). Astrocyte activation is a ubiquitous response to brain injury, from neurodegeneration to trauma, which has historically been characterized by increased expression of the cytoskeletal protein glial fibrillary acidic protein (GFAP) (Sofroniew, 2014). Recent work has begun to elucidate the diversity of astrocyte activation phenotypes beyond GFAP, as astrocytes activated by different stimuli express unique transcriptional profiles that can be associated with divergent phenotypes, ranging from neurotoxic to neurotrophic (Liddelow et al., 2017; Zamanian et al., 2012). However, a further understanding of astrocyte activation mechanisms is needed in order to effectively target astrocyte function therapeutically.
Astrocyte activation and circadian clock dysfunction are two pervasive, often coexistent features of neurological diseases, though their interaction is unknown. We previously observed that deletion of the core clock gene Bmal1, which abrogates all circadian clock function, caused severe, spontaneous astrogliosis throughout the mouse brain, which was accompanied by increased oxidative stress, synaptic damage, and inflammation (Musiek et al., 2013). However, the cellular and molecular mechanisms linking BMAL1 to astrocyte activation and function remain uncertain. Thus, we sought to characterize the astrocyte activation induced by circadian clock disruption and evaluate whether the clock protein BMAL1 might regulate astrogliosis in a cell-autonomous manner.
RESULTS
We first asked if nongenetic disruption of circadian rhythms could influence Gfap levels in the brain. Exposure of wild-type (WT) C57BL/6 mice to a 10 hr:10 hr L:D cycle induces circadian desynchrony and eventually arrhythmicity, as well as loss of detectable circadian transcript oscillations in the cerebral cortex (Figures 1A and S1A). After 6 weeks of circadian disruption, Gfap transcript levels were increased throughout the circadian cycle (Figure S1B), with an average increase of 19.7% (Figure 1B). Thus, light-induced behavioral circadian rhythm disruption promotes astrocyte Gfap expression.
Figure 1. Light-Induced Circadian Disruption or Neuron-Specific Bmal1 Deletion Partially Recapitulates the Astrocyte Activation Phenotype Observed following Brain-Specific Bmal1 Deletion.
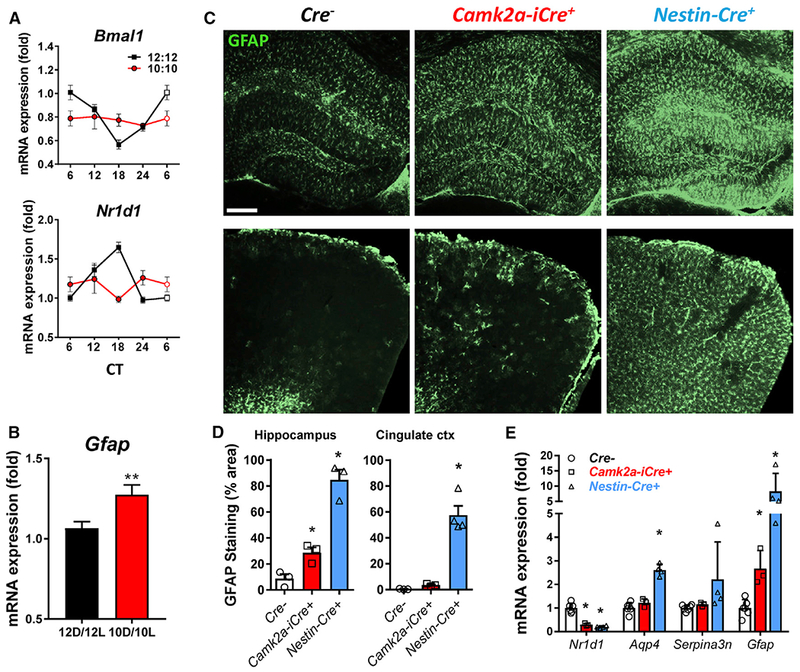
(A) Cortical qPCR from 6-week-old WT mice housed in 12 hr:12 hr or 10 hr:10 hr light-dark cycles for 6 weeks, harvested every 6 hr from CT6 to CT24, reveals blunting of circadian gene oscillations in 10 hr:10 hr mice.
(B) Total Gfap mRNA levels for mice from (A). **p < 0.01 by 2-tailed t test.
(C) Representative images showing GFAP immunostaining of hippocampus (top) and cingulate cortex (bottom) of neuron-specific Bmal1 KO mice (Cam2a-iCre+;Bmal1f/f) and brain-specific (neurons+astrocytes) Bmal1 KO mice (Nestin-Cre+;Bmal1f/f) at 4 months. Scale bar, 100 μm.
(D) Quantification of GFAP immunoreactivity in two brain regions of Cre−, Cam2a-iCre+;Bmal1f/f, and Nestin-Cre+;Bmal1f/f mice. n = 3–4 mice per genotype. Each data point represents 1 mouse. *p < 0.05 by 1-way ANOVA.
(E) Quantification of astrocyte activation transcripts by qPCR in the cortex of Cre−;Bmal1f/f, Cam2a-iCre+;Bmal1f/f, and Nestin-Cre+;Bmal1f/f mice. The clock-controlled gene Nr1d1 is shown to illustrate loss of BMAL1-mediated transcription. *p < 0.05 by 2-way ANOVA with Dunnett’s post-test for multiple comparisons. All data represent mean ± SEM.
We previously observed that abrogation of circadian function by deletion of Bmal1 in mice induced striking increases in GFAP+ astrocytes throughout the brain (Musiek et al., 2013; Yang et al., 2016). We asked if the astrocyte activation observed following Bmal1 deletion was simply a response to neuronal injury or a cell-autonomous process regulated by the core clock in astrocytes. To address this, we examined the brains of neuron-specific Bmal1 knockout (KO) mice (Camk2a-iCre;Bmal1f/f). These mice express Cre recombinase in a pan-neuronal manner under the control of a BAC-CaMKIIa promoter, yielding wide-spread neuronal deletion of Bmal1 (Figure S1C) and loss of behavioral circadian rhythms (Izumo et al., 2014). At 4 months, Camk2a-iCre+;Bmal1f/f mice exhibited a modest increase in astrocyte activation, as assessed by GFAP immunostaining (Figures 1C and 1D) and qPCR (Figure 1E), in the cortex and hippocampus. However, similarly aged Nestin-Cre+;Bmal1f/f (referred to as NBmal1 KO) mice, in which Bmal1 is deleted in neurons and astrocytes, have much more prominent astrogliosis throughout the brain and higher expression of the activation-related transcripts Aqp4 and Serpina3n (Figures 1C–1E). While NBmal1 KO mice exhibit large increases in GFAP+ astrocytes in the cortex, this did not appear to be due to increased astrocyte division, as NBmal1 KO mice had no significant increase in dividing Ki67+, GFAP+ double-positive cells (Figure S1D) or immunoreactivity for the pan-astrocyte marker S100B (Figure S1E). This finding suggests that while a small component of astrogliosis observed following Bmal1 deletion may be a response to neuronal injury, BMAL1 within astrocytes appears to play a critical cell-autonomous role in astrocyte activation.
To assess the potential cell-autonomous effects of Bmal1 on astrocyte activation, we generated primary astrocyte-enriched cultures from Bmal1 KO mice and WT littermates and examined activation markers in vitro. Bmal1 KO astrocytes were viable and appeared to have normal morphology but had significantly elevated expression of Gfap and Aqp4 mRNA and GFAP protein (Figures 2A and 2B), suggesting spontaneous activation in the absence of neuronal influence. In order to exclude a developmental influence of BMAL1, we used small interfering RNA (siRNA) to knock down Bmal1 expression in WT primary astrocyte cultures. This method resulted in a 95% decrease in BMAL1 protein (Figures 2C and 2D) and a 66% decrease in expression of the BMAL1 target Nr1d1 (which encodes REV-ERBa) (Figure S2A). Bmal1 knockdown (KD) in astrocytes induced a highly consistent (across 14 independent experiments) 36% increase in Gfap mRNA expression (Figure S2A) as well as a 68% increase in GFAP protein (Figures 2C and 2D). We also saw significant increases in the astrocyte activation marker S100a6 (1.7-fold), the cytokine Il6 (2.4-fold), and Il33 (1.6-fold) (Figure S2A), an astrocytic cytokine implicated in astrocyte-microglia signaling (Wicher et al., 2017). Bmal1 KD in primary astrocytes from Per2-luciferase reporter mice caused a loss of circadian Per2 rhythms (Figure 2E). Thus, disruption of circadian function via KD of Bmal1 in WT astrocytes induces a cell-autonomous reactive phenotype in vitro.
Figure 2. Bmal1 Deficiency Induces Cell-Autonomous Astrocyte Activation In Vitro and In Vivo.
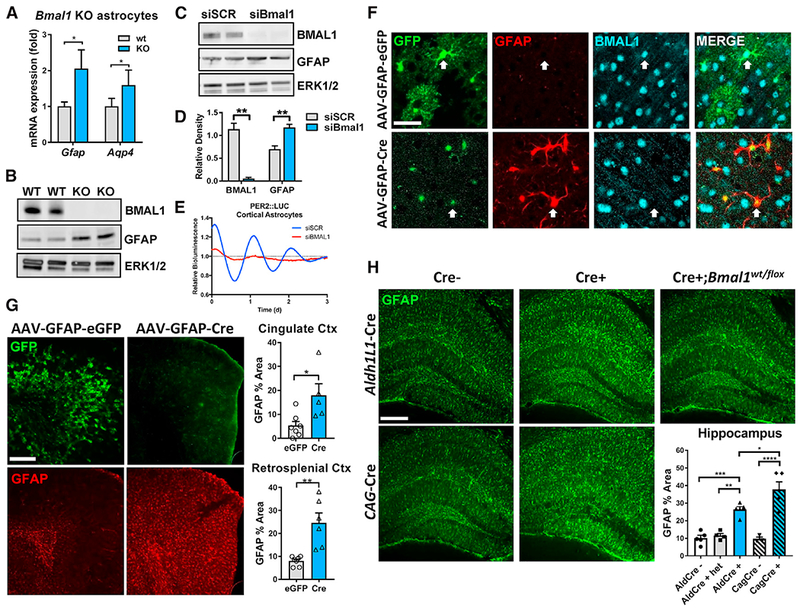
(A) qPCR of Gfap and Aqp4 mRNA in primary astrocyte-enriched cultures from WT or global Bmal1 KO mice. n = 8–12 samples from 3 independent experiments.
(B) Western blot showing increased GFAP protein levels in cultured astrocyte cell lysates from Bmal1 KO mice. p42/44 ERK protein is shown as a loading control.
(c) Western blot showing knockdown of BMAL1 and increased GFAP protein in WT astrocyte cultures 7 days after treatment with non-targeting siRNA (siSCR) or siRNA targeting Bmal1 (siBmal1). p42/44 ERK protein is shown as a loading control.
(D) Densitometric quantification of blots in (C). n = 4 samples/group.
(E) Representative trace depicting detrended circadian oscillations in luminescence seen in primary astrocyte cultures from Per2-Luc reporter mice treated with siRNA (siSCR, blue; or siBmal1, red). Similar results were obtained in 3 separate cultures.
(F) Representative confocal images from cerebral cortex of Bmal1f/f mice 5 months after intracerebroventricular (i.c.v.) injection at age P2 (postnatal day 2) with viral vectors inducing astrocyte-specific expression of EGFP (AAV8-GFAP-EGFP) or Cre (AAV8-GFAP-CreGFP). AAV8-GFAP-GFP-infected cells show whole-cell GFP expression and persistent colocalized nuclear BMAL1 staining, while AAV8-GFAP-Cre infected cells show nuclear GFP expression, increased GFAP expression, and loss of nuclear BMAL1. Scale bar, 30 μm.
(G) Representative images (left) and quantification (right) illustrating increased percent area covered by GFAP+ astrocytes in cortex in micefrom (F) injected with AAV8-GFAP-Cre, as compared to AAV8-GFAP-GFP-injected controls. Note that AAV8-GFAP-Cre virus encodes a CreGFP fusion protein that is nuclear localized and more difficult to see (F). Scale bar, 100 μm. Each data point represents 1 mouse.
(H) Representative images and quantification of percent area of GFAP staining from hippocampus of Aldh1l1-CreERT2+;Bmal1f/f and Aldh1 l1-CreERT2+;Bmal1wt/flox mice at 3 months(2 months after tamoxifen treatment), CAG-CreERT2+;Bmal1f/f mice at 4 months (2 months after tamoxifen treatment), and Bmal1f/f controls. Scale bar, 100 μm.
All data represent mean + SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p<0.0001 by 2-tailed t tests(C) and (G) or 1-way ANOVA(H) with Holm-Sidak correction for multiple comparisons when applicable.
We next sought to confirm these findings by specifically deleting Bmal1 in astrocytes in vivo. We found that intracerebroventricular injection of an AAV8 viral vector expressing EGFP under a Gfap promoter (AAV8-GFAP-EGFP) on postnatal day 2 led to widespread EGFP expression in resting astrocytes throughout the cerebral cortex, with almost no neuronal labeling (Figures 2F, 2G, and S2B). Thus, we injected P2 Bmal1f/f mice either with AAV8-GFAP-GFP or with an identical virus expressing a Cre-GFP fusion protein (AAV8-GFAP-CreGFP). At 5 months, we observed Bmal1 deletion and increased GFAP expression specifically in astrocytes expressing AAV8-GFAP-CreGFP (Figure 2F). The GFAP-CreGFP vector exhibits very weak nuclei-restricted fluorescence, making it more difficult to see than that of the AAV8-GFAP-EGFP vector. Mice treated with the EGFP control vector had wide-spread astrocyte labeling throughout the cingulate and retrosplenial cortices, intact BMAL1 expression, and very little astrocyte activation as defined by GFAP immunoreactivity (Figures 2F and 2G). Mice injected with the CreGFP vector showed astrocytic nuclear GFP expression and 3.4- and 3.1-fold increases in GFAP immunoreactivity in the cingulate and retrosplenial cortices, respectively (Figure 2G). Finally, we generated tamoxifen-inducible astrocyte-specific Bmal1 KO mice using an Aldh1l1-CreERT2 driver line (Aldh1l1-CreERT2;Bmal1f/f mice) (Srinivasan et al., 2016). We treated Cre+ and Cre− mice with tamoxifen at 1 month and examined astrocyte activation at 3 months. We observed specific deletion of Bmal1 in astrocyte nuclei of the hippocampus and cortex (Figures S2D and S2E). Aldh1l1-Cre+ mice had significant increases in astrogliosis, as assessed by GFAP immunostaining in the hippocampus (Figure 2H), and a nearly significant (p = 0.055) increase in the retrosplenial cortex (Figure S2C). While it is difficult to directly compare tamoxifen-inducible and constitutive Cre lines, Aldh1l1-Cre+ mice had similar levels of astrogliosis to tamoxifen inducible global Bmal1 KO mice (CAG-CreERT2+;Bmal1f/f) (Yang et al., 2016) also harvested 2 months after TAM treatment (Figures 2H and S2C). Astrogliosis was not caused by Cre expression, as Cre+;Bmal1flox/wt mice had no phenotype (Figures 2H and S2C). Ald1l1-Cre+ mice also had spontaneous and significant increases in a number of additional astrocyte activation transcripts including S100a6 (2.5-fold), C4b (2.6-fold), Cxcl5 (7.5-fold), and Mmp14 (3.1-fold) (Figure S2F). In total, these experiments demonstrate that Bmal1 deletion in astrocytes can induce activation in a cell-autonomous manner.
While activated astrocytes generally exhibit morphologic changes and increased GFAP expression, the transcriptional profile of activated astrocytes is heterogeneous and can define their function (Liddelow et al., 2017; Zamanian et al., 2012). In order to examine the transcriptional profile in the brain following Bmal1 deletion, we performed microarray analysis of cortical tissue from 11-month-old NBmal1 KO mice, Cre− controls, and Per1Brdm/Per2Brdm (Per1/2mut) mice placed in constant dark conditions for 36 hr and then harvested at clock time (CT) 6:00 and 18:00. Per1/2mut mice were included because they have a dysfunctional circadian clock but no astrogliosis phenotype as well as tonically increased levels of BMAL1 transcriptional targets (Figures 3A and S3A). To understand astrocyte transcriptional changes in NBmal1 KO brain, we examined the overlap between the 200 most upregulated transcripts in NBmal1 KO with the 200 most astrocyte-enriched transcripts according to the Barres lab RNA-sequencing (RNA-seq) database (Zhang et al., 2014) (Figure 3A). Thirty genes overlapped, including Gfap, Aqp4 (encoding the astrocytic water channel aquaporin-4), and Megf10, a gene involved in astrocyte-mediated synaptic elimination (Chung et al., 2013). C4b, a complement cascade component also implicated in synapse elimination and a marker of astrocyte activation and aging (Boisvert et al., 2018; Clarke et al., 2018), was strongly upregulated (4-fold) in NBmal1 KO brain. Several other disease-associated astrocytic genes were increased in NBmal1 KO brain, including inflammatory mediators such as Cxcl5 and Ccr7 and the matrix metalloproteinases Mmp14 and Mmp2. Nearly all of these transcripts were either unchanged or decreased in Per1/2mut mice, demonstrating the key role of decreased BMAL1 function in this phenotype. We next performed microfluidic qPCR profiling of global Bmal1 KO hippocampal samples for markers of astrocyte activation and polarization (Liddelow et al., 2017), which demonstrated that Bmal1 deletion increased most pan-reactive markers but did not clearly polarize astrocytes toward a neurotoxic or neurotrophic profile (Figure 3B). These genetic profiling studies suggest that Bmal1 deletion induces a unique state of astrocyte activation.
Figure 3. BMAL1 Regulates Astrocyte Activation via a Glutathionylation-Dependent Mechanism.
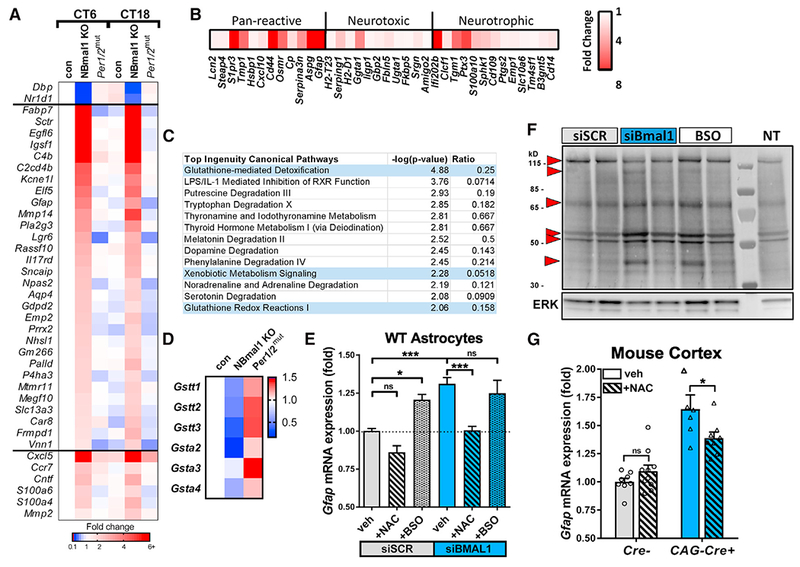
(A) Astrocyte-specific genes upregulated in Nestin-Cre+;Bmal1f/f(NBmal1 KO) cortexand changes to the same genes in Per1/2mut mice. Transcripts upregulated at least 2-fold in NBmal1 KO were compared to the top 200 astrocyte-specific genes (Zhang et al., 2014), and overlapping genes are displayed. Other notable genes implicated in astrocyte activation and function were upregulated in NBmal1 KO cortex (bottom). The clock-controlled genes Dbp and Nr1d1 are direct Bmal1 transcriptional targets.
(B) Expression of neurotoxic, neurotrophic, and pan-reactive astrocyte activation genes (Liddelow et al., 2017) in 5-month-old global Bmal1 KO versus WT littermate hippocampus assayed by microfluidic qPCR. Each data point represents the average of 4 Bmal1 KO mice normalized to the average of 4 WT littermates.
(C) Results from Ingenuity Pathway Analysis canonical pathway analysis of NBmal1 KO microarray data. Pathways related to glutathione homeostasis are colored blue.
(D) Expression levels for downregulated glutathione-S-transferases in NBmal1 KO cortex versus Bmal1f/fcontrols. Expression levels in Per1/2mut cortex included for comparison (based on microarray data).
(E) qPCR illustrating effect of glutathione manipulation on Gfap transcript levels following Bmal1 knockdown. WT primary astrocytes treated with control siRNA (siSCR, gray bars) or siBmal1 (blue bars), with or without N-acetyl-cysteine (NAC; striped bars) to increase glutathione levels, or buthionine sulfoxime (BSO; speckled bars) to deplete glutathione. n = 9 samples/condition from 3 separate experiments.
(F) WT astrocytes transfected with siSCR, siBmal1, or siSCR +BSO (BSO) were treated with biotin-linked glutathione ethyl ester (bioGEE) to assess glutathionylation. Increased band intensity indicates decreased endogenous glutathionylation. Arrows denote altered bands. NT, not treated with bioGEE, showing nonspecific labeling.
(G) qPCR depicting effects of NAC treatment on Gfap transcript levels in cortex of inducible Bmal1 KO mice (CAG-CreERT2+;Bmal1f/f). Inducible Bmal1 KO mice and Cre− control littermates were treated with NAC before, during, and after 5-day tamoxifen treatment to delete Bmal1. Mice were harvested 9 days after the start of tamoxifen. n = 6–10 mice per group.
All data represent mean + SEM. *p < 0.05, ***p < 0.001 by t test with Holm-Sidak correction for multiple comparisons.
Although Per1/2mut and NBmal1 KO mice are both behaviorally arrhythmic, expression of BMAL1 transcriptional targets Nr1d1 and Dbp are elevated solely in Per1/2muu mice because of loss of inhibition of BMAL1 (Figures 3A and S3A). Gfap is not elevated in Per1/2mut mice (Figures 3A and S3A), suggesting that loss of BMAL1 activity, or at least blunting of BMAL1 oscillation (as in Figure 1A), is needed to induce astrocyte activation. In order to determine if Gfap and other selected activation markers are clock-controlled genes, we harvested cortex from 4-month-old NBmal1 KO and Cre- controls at 4-hr intervals across one circadian cycle in constant darkness. Gfap mRNA levels showed no circadian oscillation in control or Cre+ mice and were elevated across the circadian cycle in Cre+ mice. These findings were duplicated in other markers of astrocyte activation, including Aqp4 and Mmp14 (Figure S3B). Thus, the astrogliosis phenotype in Bmal1 KOs is not likely due to direct regulation of Gfap or other activation markers by the clock.
To address potential mechanisms linking BMAL1 to astrocyte activation, we performed pathway analysis on our NBmal1 KO transcriptomic data using two bioinformatics tools: Ingenuity Pathway Analysis (IPA) and Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Resources 6.8 (Huang et al., 2009). We analyzed an inclusive list consisting of all transcripts that differed between control and NBmal1 KO at 6 p.m. with uncorrected p values <0.01 (Table S1). IPA canonical pathway analysis identified glutathione-mediated detoxification as the top hit (Figure 3C; p = 0.000013). Functional annotation clustering with DAVID showed that the most enriched gene cluster focused on glutathione-S-transferase (GST) activity, and 4 of the top 5 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways identified were related to glutathione metabolism (Figure S3C). GST enzymes catalyze the phase II detoxification of reactive intermediates in cells via adduction to glutathione and can alter cellular signaling via glutathionylation of proteins (Grek et al., 2013). We found that expression of several GST isoforms was suppressed in NBmal1 KO cortex and increased in Per1/2mut cortex (Figure 3D), suggesting reciprocal regulation by the positive and negative limbs of the circadian clock. In support of our microarray analysis, we observed significant decreases in 2 of the 3 GST isoforms probed in cortex from 3-month-old CAG-Cre+;Bmal1f/f mice 9 days after tamoxifen treatment (Figure S3D) and in 1 of the 3 GST isoforms probed in hippocampus from 3-month-old Aldh1l1-Cre+ mice 2 months after tamoxifen treatment (Figure S3E). We next asked if altered glutathione homeostasis within astrocytes linked Bmal1 deficiency with astrocyte activation. Our data clearly show that Bmal1 deletion induced changes in both Gfap mRNA and GFAP protein, which are very closely correlated across all models (see Figure S4A), so Gfap mRNA was used as a marker for astrogliosis in subsequent experiments. Treatment with N-acetyl cysteine (NAC), a glutathione precursor, completely prevented increased Gfap expression in astrocytes following Bmal1 KD (Figure 3E). Conversely, incubation of WT astrocytes with buthionine sulfoxime (BSO), which blocks glutathione production, lowered cellular glutathione levels (Figure S3F) and caused an increase in Gfap mRNA similar to that observed with Bmal1 KD (Figure 3E). Treatment of Bmal1-deficient astrocytes with BSO did not induce a further increase in Gfap, suggesting that BSO and Bmal1 KD are causing increases in Gfap through overlapping pathways. Interestingly, neither Bmal1 KO nor KD of Bmal1 in cultured primary astrocytes altered glutathione levels (Figure S3F). Additionally, the oxidation state of glutathione (ratio of reduced GSH to oxidized GSSG) was not altered in cortex from NBmal1 KO or Per1/2mut mice (Figure S3G). As glutathione levels and oxidation state remained unchanged with loss of Bmal1, we next examined glutathione signaling via glutathionylation, which has been shown to oscillate in a circadian manner in the SCN (Wang et al., 2012). Western blot analysis of WT astrocytes treated with biotin-linked glutathione ethyl ester (bioGEE) revealed wide-spread changes in the pattern of S-glutathionylated proteins in Bmal1 KD cells, which were similar to changes seen in BSO-treated controls (Figure 3F). In general, numerous bands showed increased biotin labeling, indicating decreased glutathionylation, as would be expected with decreased GST expression. These data suggest that the observed astrogliosis may be due at least in part to altered protein glutathionylation resulting from impaired expression of specific GSTs.
We next examined whether bolstering glutathione signaling can prevent astrogliosis following Bmal1 deletion in vivo. We previously showed that tamoxifen-inducible global Bmal1 deletion in mice (CAG-Cre+;Bmal1f/f mice) causes astrocyte activation (Yang et al., 2016). We treated CAG-Cre+;Bmal1f/f mice with NAC in their drinking water (40 mM beginning 5 days before tamoxifen) and via intraperitoneal injection (150 mg/kg daily beginning 2 days before tamoxifen) and induced Bmal1 deletion via tamoxifen administration for 5 days. Both administrations of NAC were continued for the duration of the experiment. Cortex was harvested 9 days after the start of tamoxifen treatment and analyzed by qPCR. Tamoxifen treatment induced an 87% loss in Bmal1 and a 79% loss in expression of the Bmal1 target Nr1d1 (Figure S3H), as well as a 64% increase if Gfap expression (Figure 3G). Administration of NAC was able to provide a significant but partial rescue of the Gfap increase seen in CAG-Cre+;Bmal1f/f animals (Figure 3G). These results suggest that Bmal1 regulates Gfap expression at least in part through modulation of glutathione signaling in vivo.
Finally, we examined potential pathogenic consequences of Bmal1 deletion in astrocytes. We generated primary astrocyte-enriched cultures from CAG-Cre+;Bmal1f/f mice and Cre− littermates and treated them with tamoxifen in vitro to delete Bmal1. These cultures are primarily astrocytes, but this methods yields cultures which contain ~2% microglia and/or oligodendrocyte precursor cells (Schildge et al., 2013). Primary WT cortical neurons were then co-cultured on these astrocytes for up to 12 days, and a subset was subjected to oxidative stress by hydrogen peroxide exposure (100 μM H2O2) for 24 hr (Figure 4A). We found no difference in the number of neurons surviving on Cre− and Cre+ astrocyte cultures 1 day after plating (as quantified by staining for neuronal nuclei with NeuN, as these cells do not yet have many MAP2 positive neurites) (Figure 4B). At day 7 after plating, there was a 45% decrease in NeuN+ nuclei (Figures 4B and 4C), a trend toward a decrease in MAP2 staining, and a trend toward an increase in cleaved-caspase-3 staining (Figures S4B and S4C) in the Cre+ astrocyte condition. The decrease in MAP2 staining in cultures containing Cre+ astrocytes became significant after 12 days (Figures 4D and 4E). H2O2 exposure at day 12 decreased MAP2 levels in Cre− cultures, and the combination of Bmal1 deletion and H2O2 killed nearly all neurons (Figures 4D and 4E). This finding shows that Bmal1-deficient astrocytes are less able to sustain neuronal viability, particularly in the face of oxidative stress.
Figure 4. Loss of Bmal1 Impairs Astrocytic Support of Neurons.

(A) Diagram depicting co-culture experimental design. DIV, days in vitro; TAM, tamoxifen.
(B) Representative images showing NeuN+ WT neuronal nuclei at DIV 1 and 7, grown on primary astrocytes from Cre− control mice, or inducible Bmal1 kO mice (iCAG-Cre+) treated with tamoxifen in vitro. Scale bar, 100 μm.
(C) Quantification of NeuN+ nuclei from (B) normalized to NeuN counts from Cre− condition, n = 8 replicates from 2 independent experiments.
(D) Representative images of MAP2+ WT neurons grown as in (B) at DIV 12. Lower panels show neurons from adjacent wells treated for 24 hr with hydrogen peroxide at DIV 11 (H2O2, 100 μM). Scale bar, 100 μm.
(E) Quantification of MAP2 immunoreactivity (percent area) from (D). n = 9 replicates from 3 independent experiments.
All data represent mean ± SEM. *p < 0.05, **p < 0.01 by 2-tailed t test.
DISCUSSION
Despite a large body of evidence describing circadian disruptions in both aging and neurodegenerative disease, little is known about the implications of circadian dysfunction on the brain. Here, we provide evidence that behavioral circadian disruption or disruption of the astrocytic molecular clock via manipulations of the master clock gene Bmal1 induce astrogliosis and astrocyte dysfunction. The critical but complex role of astrocytes in brain pathologies and neuroinflammation underscores the importance of elucidating mechanisms of astrocyte activation, and our data illuminate BMAL1 as a potent regulator of astrocyte activation by both cell-autonomous and non-autonomous mechanisms.
The existence of the astrocyte circadian clock has been well documented (Prolo et al., 2005), and the critical role of the astrocyte clock in maintaining circadian rhythms in the SCN has recently been described (Barca-Mayo et al., 2017; Brancaccio et al., 2017; Tso et al., 2017). Clock genes have been shown to regulate astrocytic glutamate uptake and ATP release (Beaulé et al., 2009; Marpegan et al., 2011), but the function of the core clock in astrocytes is otherwise unexplored. We have shown here, using several in vitro and in vivo methods, that loss of Bmal1 function in astrocytes causes cell-autonomous astrocyte activation. However, deletion of Bmal1 specifically in neurons also induces a partial astrocyte activation phenotype, suggesting a coexistent cell-non-autonomous mechanism. Bmal1 deletion in cultured neurons causes toxicity (Musiek et al., 2013), implicating a possible damage signal from Bmal1-deficient neurons that activates nearby astrocytes. Alternatively, the neuron-specific Bmal1 KO mice used in this study are behaviorally arrhythmic (Izumo et al., 2014), and this loss of rhythms could induce astrocyte activation, as seen in 10 hr:10 hr L:D-exposed animals (Figure 1). Thus, cell-non-autonomous influences on glial activation in the brain must be examined in the future.
One important question is whether the loss of rhythmic function of the circadian clock plays a key role in this phenotype or if this is a “non-circadian” function of BMAL1. Our data suggest that arrhythmicity in the setting of increased BMAL1 expression, as in Per1/2mut mice, does not induce astrocyte activation, as these mice express increased levels of GSTs. Thus, astrocyte activation appears to be dependent on suppression of BMAL1-mediated transcription. However, BMAL1 DNA binding is highly rhythmic and regulated by the clock (Koike et al., 2012), making it difficult to separate entirely from circadian rhythms. Bmal1 KD in astrocytes abrogates circadian Per2-luc oscillations, which could contribute to increased Gfap expression in cell culture. Moreover, we show in Figure 1 that circadian disruption by exposure of mice to 10 hr:10 hr L:D conditions, which blunts Bmal1 oscillations in the brain, can induce Gfap expression. Thus, dampening the rhythmicity of BMAL1 expression through circadian clock disruption likely plays a role in mediating astrogliosis, perhaps by restricting BMAL1 levels at certain key times of day. BMAL1 levels can be suppressed by several factors such as aging (Wyse and Coogan, 2010), inflammation (Curtis et al., 2015), or amyloid-beta (Song et al., 2015), all of which could potentially influence astrocyte activation and function in a BMAL1-dependent manner.
The activation profile induced in Bmal1-deficient astrocytes encompasses upregulation of a variety of genes across the pre-defined neurotoxic, neurotrophic, and pan-reactive categories (Liddelow et al., 2017). However, it is important to remember that these astrocyte phenotypes are thought to be induced by cytokine release from microglia, whereas activation due to loss of Bmal1 can be cell autonomous. Our findings contrast with those of Nakazato et al., who found that S100b-Cre+;Bmal1f/f mice did not develop astrocyte activation and attributed the astrocyte activation phenotype of Nestin-Cre+;Bmal1f/f mice to pericyte dysfunction (Nakazato et al., 2017). While our data do not exclude a contribution of blood-brain barrier dysfunction to the Nestin-Bmal1 phenotype, we provide multiple lines of evidence demonstrating cell-autonomous astrocyte activation. While Bmal1-deficient astrocytes do not assume a clear neurotoxic (“A1”) phenotype, they are less able to support neuronal survival in a co-culture system and express numerous transcripts associated with inflammation and neurodegeneration. These include C4b, a complement component and marker of astrocyte activation and aging (Boisvert et al., 2018; Clarke et al., 2018), Megf10, which mediates astrocytic synapse elimination (Chung et al., 2013), and Pla2g3, a phospholipase implicated in ROS-induced neuronal damage (Martínez-García et al., 2010). Activation marker Lcn2 (lipocalin 2), an anti-inflammatory mediator in the brain (Kang et al., 2018), was not increased, again suggesting a pro-inflammatory state.
Our data support a mechanism in which loss of BMAL1 induces astrogliosis through disruption of GST-mediated protein glutathionylation. In addition to its role in quenching oxidative stress, glutathione can form adducts on cysteine residues in a process termed glutathionylation, which is catalyzed by GSTs and can serve a signaling function (Grek et al., 2013; Mailloux and Treberg, 2016). Circadian rhythms of protein S-glutathionylation in the SCN may regulate neuronal excitability (Wang et al., 2012). Because Bmal1-deficient astrocytes have normal total glutathione levels, it is likely that disruption of GST-mediated glutathione signaling or utilization, but not glutathione depletion, mediates BMAL1-controlled astrogliosis. Accordingly, we observed an altered pattern of protein S-glutathionylation in Bmal1-deficient astrocytes, which resembled that seen in BSO-treated cells. Thus, supplementing Bmal1-deficient astrocyte cultures or mice with NAC presumably prevents astrogliosis by promoting nonenzymatic glutathionylation of protein targets, thereby circumventing the loss of GSTs. Further studies are needed to identify specific glutathionylation targets that mediate astrocyte activation and understand regulation of astrocyte glutathione signaling in health and disease.
In summary, our data identify the circadian clock protein BMAL1 as a cell-autonomous regulator of astrocyte activation and neurotrophic function and posit altered GST-mediated protein glutathionylation as a mediator of this effect. Our findings suggest that BMAL1 serves as a link between the core circadian clock and astrogliosis and provide insights into how the circadian clock might influence neurodegeneration.
STAR★Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Gfap – Rabbit polyclonal, 1:5000 | Dako/Agilent | Cat# Z0334; RRID:AB_10013482 |
| Gfap- Mouse monoclonal, 1:1000 (Co-stain only) | Novus | Cat# NBP1-05197; RRID:AB_1555288 |
| Bmal1- Rabbit Monoclonal, 1:2000 (IF/IHC only) | Novus | Cat# NB100-2288; RRID:AB_10000794 |
| Bmal1- Rabbit polyclonal, 1:1000 (WB only) | Bethyl | Cat# A302-616A; RRID:AB_10555918 |
| MAP2- Mouse monoclonal, 1:1000 | EMD/Milipore | Cat# AB5622; RRID:AB_91939 |
| Ki67- Rat monoclonal, SolA15, 1:500 | ThermoFisher | Cat# 14-5698-82; RRID:AB_2016711 |
| NeuN- Mouse monoclonal, 1:1000 | EMD/Millipore | Cat# MAB377; RRID:AB_2298772 |
| P42/44 MAPK ERK 1/2- Rabbit monoclonal, 1:1000 (WB) | Cell Signaling Technology | Cat# 9102; RRID:AB_330744 |
| Bacterial and Virus Strains | ||
| AAV8-GFAP-eGFP | UNC Viral Vector Core | N/A |
| AAV8-GFAP-GFP-Cre | UNC Viral Vector Core | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine RNAiMAX transfection reagent | ThermoFisher/Life | Cat# 13778075 |
| N-acetyl-L-cysteine | Sigma | Cat# A7250 |
| Tamoxifen | Sigma | Cat# T5648 |
| L-Buthionine sulfoxime | Sigma | Cat# 5082280001 |
| Poly-D-lysine hydrobromide | Sigma | Cat# P6407 |
| Critical Commercial Assays | ||
| BioGEE Glutathionylation assay kit | ThermoFisher | Cat# G36000 |
| High Capacity RNA-to-cDNA Kit | Applied Biosystems | Cat# 4387406 |
| GSH/GSSG Ratio Detection Assay Kit | Abcam | Cat# ab138881 |
| Taqman Universal PCR Master Mix | Applied Biosystems | Cat# 4304437 |
| PureLink RNA Mini Kit | Ambion/Life | Cat# 12183025 |
| Deposited Data | ||
| Agilent 4x44k mouse microarray v2 data from Nestin-Bmal1, Per1;Per2 double mutant, and control cortex at CT 6 and 18. | This paper | ArrayExpress (EMBL-EBI) Accession#: E-MTAB-7151 |
| Experimental Models: Organisms/Strains | ||
| B6.Cg-Tg(CAG-cre/Esr1*)5Amc/J (CAG-CreERT2 mice) | Jackson Labs | Cat# 004682 |
| B6;FVB-Tg(Aldh1l1-cre/ERT2)1Khakh/J (Aldh1l1-CreERT2 mice) | Jackson Labs | Cat# 029655 |
| B6.Cg-Tg(Nes-cre)1Kln/J (Nestin-Cre mice) | Jackson Labs | Cat# 003771 |
| B6.129S4(Cg)-Arntltm1Weit/J (Bmal1f/f mice) | Jackson Labs | Cat# 007668 |
| CamKIIalpha iCre BAC (CamK2a-Cre mice) | Casanova et al., 2001 | N/A |
| Oligonucleotides | ||
| SMARTpool ON-TARGETplus Arntl siRNA | Dharmacon | Cat# L-040483-01-0005 |
| ON-TARGETplus Non-targeting siRNA1 | Dharmacon | Cat# D-001810-02-05 |
| Gfap Taqman Gene expression assay | Applied Biosystems | Mm01253033_m1 |
| Actb Taqman Gene expression assay | Applied Biosystems | Mm02619580_g1 |
| Bmal1 (Arntl) Taqman Gene expression assay | Applied Biosystems | Mm00500223_m1 |
| Aqp4 Taqman Gene expression assay | Applied Biosystems | Mm00802131_m1 |
| Nr1d1 Taqman Gene expression assay | Applied Biosystems | Mm00520708_m1 |
| Serpina3n Taqman Gene expression assay | Applied Biosystems | Mm00776439_m1 |
| S100a6 Taqman Gene expression assay | Applied Biosystems | Mm00771682_g1 |
| Il6 Taqman Gene expression assay | Applied Biosystems | Mm00446190_m1 |
| Il33 Taqman Gene expression assay | Applied Biosystems | Mm00505403_m1 |
| C4b Taqman Gene expression assay | Applied Biosystems | Mm00437893_g1 |
| Cxcl5 Taqman Gene expression assay | Applied Biosystems | Mm00436451_g1 |
| Mmp14 Taqman Gene expression assay | Applied Biosystems | Mm00485054_m1 |
| Timp1 Taqman Gene expression assay | Applied Biosystems | Mm01341361_m1 |
| Dbp Taqman Gene expression assay | Applied Biosystems | Mm00497539_m1 |
| Gsta2 Taqman Gene expression assay | Applied Biosystems | Mm03019257_g1 |
| Gsta4 Taqman Gene expression assay | Applied Biosystems | Mm00494803_m1 |
| Gstt3 Taqman Gene expression assay | Applied Biosystems | Mm00519528_m1 |
| Software and Algorithms | ||
| Ingenuity Pathway Analysis | QIAGEN | Build Version: 400896M; Content version 28820210 |
| Imaris | Bitplane | Version 9 |
| Prizm | GraphPad | Version 7 |
| DAVID Bioinformatics Resource | https://david.ncifcrf.gov/ | Version 6.8 |
| Fiji | https://fiji.sc/ | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Erik Musiek (musieke@wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Nestin-Cre+, CAG-CreERT2+, Global Bmal1 KO, Aldh1L1-CreERT2+, and Bmal1f/f mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and were bred at Washington University. Camk2a-iCre+;Bmal1f/f mice were bred at University of Texas Southwestern. All cohorts of mice were mixed sex with ages specified in figure legends. All mice were backcrossed at least 10 generations and were maintained on a C57Bl\6 background and housed under 12-hour light/12-hour dark conditions. In all cases, a Cre+; Bmal1f/f male mouse was bred with Bmal1f/f females to generate an equal mix of Cre− and Cre+ mice, all on the Bmal1f/f background. All littermates were thus used for experiments. Aldh1L1-CreERT2+ and CAG-CreERT2+ mice (Cre− and Cre+ littermates from several litters) were given tamoxifen (Sigma, dissolved in corn oil, 2mg/mouse/day for 5 days) by oral gavage at 1mo or 2mo, respectively, to induce Bmal1 deletion in the applicable tissue. Behavioral circadian desynchronization experiments were performed at Washington State University, using adult male C57BL/6N mice (age 5-6 weeks on arrival, Harlan Envigo). All mice were group housed for gene expression experiments (N = 4-5 per cage), or singly housed for actigraphy, with food and water available ad libitum. All mouse experiments were conducted in accordance with protocols approved by the Washington University Department of Comparative Medicine, or by the IACUCs at UTSW or WSU.
Primary cell cultures
Primary cells were extracted from mixed sex P1-3 (astrocytes) or E16-17 (neurons) CD1 WT, Bmal1f/f, or iCAG-Cre+;Bmal1f/f mice per experimental designation. Wild-type (WT) CD1 mice for culture experiments were obtained from Charles River Laboratories (Wilmington, MA). Mice were briefly anesthetized by cooling on ice, cleansed with ethanol, then decapitated with scissors, and the brain placed in ice-cold DMEM (Life Technologies). Cortices were dissected, stripped of meninges in ice-cold DMEM, and incubated in 0.05% Trypsin-EDTA (Life Technologies) at 37°C for 15 (neurons) or 20 (astrocytes) minutes. 37°C DMEM plus 10% FBS (GIBCO) was then added to neutralize trypsin, cortices were spun down for 5 minutes at ~300 g and gently triturated in 37°C DMEM plus 10% FBS (GIBCO). For astrocytes cultures, cells were then plated on 10cm or T75 tissue culture plates (coated for 2 hours at 37°C with 50 μg/mL poly-D-lysine (PDL- MP Biosciences, Santa Ana, CA), then rinsed twice with ddH2O) at a concentration of 2 cortices (1 brain) per plate. Astrocytes were then grown to confluence (~10 days), then shaken at 37°C in a heated shaker at 225 rpm for 2 hours, followed by media change to remove adherent microglia. Cells were later lifted using Trypl-E dissociation reagent (Life Technologies) for 5 minutes at 37°C, washed with DMEM plus 10% FBS, and replated on 12-well (mRNA experiments), 48-well (neuronal co-cultures), or 96-well (GSH experiments) plates (coated with PDL as above) at a 1:2 dilution. Astrocyte-enriched cultures were then grown in DMEM+ 10% FBS + penicillin/streptomycin until used for experiments.
METHOD DETAILS
Behavioral Desynchronization
Activity was monitored by implanted Mini-mitter telemetry device (Star BioSciences). For circadian desynchronization, mice were transferred into a 20h light/dark cycle, with 10h of light (~200 Lux) and 10h of dark for 6 weeks then harvested as described below at CT0, 6, 12, and 18; N = 4-5/group/time.
Intracerebroventricular injections
Bmal1f/f mice were injected on postnatal day 2 (P2) with either AAV8-GFAP-GFP or AAV8-GFAP-CreGFP viral vectors (2 μL, bihemispheric, from UNC viral vector core), using a 30 g Hamilton syringe (10ul, 45 degree bevel) with a cuff to ensure proper depth (1.5mm). The injection site was 1mm lateral and 1mm posterior to bregma. Mice were housed as described above, harvested at 5 months of age and analyzed as described in the immunohistochemisty section.
Immunohistochemistry
Mice were anesthetized by i.p. pentobarbital (150mg/kg), followed by perfusion for 3 minutes with ice-cold Dulbecco’s modified PBS (DPBS) containing 3 g/l heparin. One hemisphere was post-fixed in 4% paraformaldehyde for 24 hours (4°C), then cryoprotected with 30% sucrose in PBS (4°C) for 48 hours. 50-micron serial coronal sections were then cut on a freezing sliding microtome and stored in cryoprotectant solution (30% ethylene glycol, 15% sucrose, 15% phosphate buffer in ddH20). Sections were washed in Tris buffered saline (TBS), blocked for 30 minutes in TBSX (TBS with 0.25% Triton X-100 (Sigma-Aldrich, St. Louis, MO)) containing 3% goat serum, then incubated overnight in TBSX containing 1% goat serum plus primary antibody at 4°C. Sections were then washed in TBS, incubated for 1 hour at room temperature in TBSX with 1:1000 fluorescent secondary antibody, and mounted on slides using Fluoromount G. Epiflourescent images were obtained on a Nikon Eclipse 80i fluorescent microscope. A Nikon A1Rsi scanning confocal microscope was used at 20× for Ki67/GFAP co-localization, at 40× for Aldh1L1-Cre BMAL1/GFAP quantification, and at 60× for viral injection and representative Aldh1L1-Cre co-localization pictures.
Culture experiments
For neuronal cultures, triturated cells were transferred to a second tube to remove debris, then diluted in Neurobasal+B27+glutamine (Life Technologies) and counted prior to plating on astrocyte beds. For co-culture experiments, astrocytes were grown to confluence after replating in 48-well plates and neurons were added immediately following primary dissection at a concentration of 37,500 cells/well in Neurobasal media plus B-27 supplement and glutamine. After 48 hours, 50% of the media was changed to Neurobasal plus anti-oxidant-free B-27 (AOF B-27, Life Technologies) and glutamine. Subsequently, 50% media changes were performed using Neurobasal plusAOF B-27 and glutamine every 3 days until neuronal DIV 7 or 11. In cultures grown to DIV 11, half of the wells received 100 μM H2O2 for 24 hours. All wells were then fixed with 4% paraformaldehyde for 15 minutes, washed 3× with TBS and stained for MAP2, Cleaved-Caspase-3 or NEUN and quantified using protocol in immunoreactivity quantification section.
Immunoreactivity quantification
GFAP and MAP2 staining were quantified from 10× images using ImageJ software (NIH). GFAP 8-bit images were thresholded by hand to encompass all GFAP immunoreactivity, and a threshold value was set and maintained for all sections. GFAP percent area coverage was then calculated for each image. In general, 1 image from each section and 2-3 sections/mouse were averaged, and the average from each mouse used as a data point. For Ki67/GFAP co-localization, puncta were hand-counted from maximal intensity projections of z stacks in each region of interest. GFAP, BMAL1, and DAPI co-localizations were hand counted by 2 independent, blinded experimenters in 6 images/mouse (3 hippocampus, 3 cortex) with the average for each mouse reported. Cleaved-Caspase-3 puncta co-localizing with MAP2 were hand counted by 2 independent, blinded experimenters in 3-5 images/well over 11 wells from 3 independent experiments with averages/well reported.
qPCR
Flash-frozen brain tissue was homogenized with a mechanical homogenizer for 20 s in RNA kit lysis buffer (PureLink™ RNA Mini Kit, Life Technologies, Carlsbad CA) plus 1% β-mercaptoethanol. RNA was then purified using the kit protocol. Cell lysates were homogenized in TRIzol (Life Technologies). The aqueous layer was then utilized, after chloroform extraction (added 1:5, sample then spun at 13000xg for 15 minutes), in RNA isolation using the kit protocol. RNA concentration was then measured by Nanodrop spectrophotometer and cDNA was made using a high capacity RNA-cDNA reverse transcription kit (Applied Biosystems/Life Technologies) with 1 mg RNA used per 20 μL reaction. Real-time quantitative PCR was performed with ABI TaqMan primers and ABI PCR Master Mix buffer on either the ABI StepOnePLus or QuantStudio 12k Real-Time PCR thermocyclers. During analysis, mRNA measurements were normalized to β-actin (Actb) mRNA levels. All Taqman qPCR primers were obtained from Thermo/Fisher.
Microfluidic qPCR was performed on flash frozen hippocampal tissue from 6mo Global Bmal1 KO and WT littermates mice at Stanford University using a BioMark Real-Time PCR system (Fluidigm, South San Francisco, CA) and analyzed as previously described (Liddelow et al., 2017).
Western blotting
Tissue samples were homogenized by sonication radioimmunoprecipitation (RIPA) buffer (Cell Signaling Technologies, Danvers MA) containing cOmplete protease inhibitors (Roche Lifesceinces USA, Indianapolis IN). Invitrogen nuPAGE Novex gels, SDS running buffer, and an iBlot transfer device were utilized per the manufacturer’s instructions (Life Technologies). Bands were visualized using Lumigen TMA-6 chemiluminescence reagents (Lumigen, Southfield MI) on a Syngene GBOX imaging system. Band density was calculated using ImageJ software and normalized to ERK as a loading control.
Microarray
11mo NBmal1 KO, Cre- littermate, and Per1/2mut mice were placed in constant darkness for 36 hours, then anesthetized with i.p. pentobarbital in the dark, followed by perfusion. Mice were harvested at CT 6 and 18 (6am and 6pm), 2-4 mice/genotype/time point. RNA was isolated from flash frozen cortex samples as above and submitted to the Genome Technology Access Center at Washington University for quality control, MessageAmp RNA library preparation, and Agilent 4x44k mouse v2 microarray. Raw data was normalized and analyzed using Partek Genomics Suite v6.6. For DAVID analysis, version 6.8 was used. The gene list from Table S1 was uploaded using Official Gene Symbol as the identifier, species = Mus musculus, and 692/842 genes were recognized. Two analyses were run: Functional Annotation Clustering (stringency: high), and KEGG pathway analysis. For Ingenuity Pathway Analysis (QIAGEN, Venlo, Netherlands), content version 28820210, build version 400896 was used, and the same gene list was employed (with ratios and p values). Cannonical Pathway analysis was performed. Dataset will be uploaded to GEO Datasets website (https://www.ncbi.nlm.nih.gov/gds/).
siRNA transfections
Astrocytes were transfected with siRNA using lipofectamine RNAiMAX (Life Technologies) in Optimem (LifeTechnologies) according to manufacturer’s instructions 24 hours after replating. After 48 hours, media was changed to DMEM plus 10% FBS. SiRNAs targeting mouse Bmal1 or non-targeting (designed to target no known mouse gene) were obtained from Dharmacon (Lafayette, CO). ON-TARGETplus SmartPool siRNA was used, which contains 5 separate siRNAs targeting the gene of interest which are pooled together (Bmal1: product L040483-01-005, non-targeting: D-001210-01-05). An siRNA:RNAiMAX ratio of 1:1.25) was used, and 40 pmol of siRNA (2uL of 20uM stock) was added to each well, using 12-well plates. Media was changed 24-48 hours later.
BSO and NAC experiments
For NAC experiments, astrocytes were incubated in growth media supplement with 1mM N-acetylcysteine (Sigma) starting 24 hours after transfection with siRNA, and for the remainder of the experiment. 72 hours before harvest, media was changed to DMEM plus 2% FBS and, where indicated, containing 250 μM buthionine sulfoxime (BSO, Sigma) and cells were harvested as described above at day 8 after siRNA treatment.
CAG-Cre+;Bmal1f/f mice and Cre- littermates (age 2-3 mo) used for in vivo NAC experiments were given either control drinking water or water containing 40mM NAC starting day 0 until sacrifice. Beginning on day 3 and continuing until sacrifice, mice were also given daily intraperitoneal injections of NAC (150mg/kg) or sterile DPBS for controls. Tamoxifen (Sigma, dissolved in corn oil, 2mg/mouse/day for 5 days) was administered by oral gavage to all mice beginning day 5 after water treatment. Water was refreshed once at day 7. Mice were perfused at day 14 (4 days after completion of tamoxifen) and tissue was prepared as described above.
Per2:luciferase Bioluminescence Recording
Primary astrocyte cultures were prepared from P1-2 PER2::LUC transgenic mice, and transfected with siRNA as above. 48 hours later, cells were synchronized by media change, and PER2::LUC activity was continuously monitored from a photomultiplier tube (HC135-11MOD, Hamamatsu) from transfected cortical astrocytes plated on a sealed 35mm Primaria dish (BD Biosystems, San Diego, CA) with 1 mL of serum-free DMEM supplemented with 2% B27 (Invitrogen), pen/strep, 10 mM HEPES, 0.35 g/L NaHCO3, 4.5 g/L D-glucose, 2% L-glutamine, and 0.1 mM beetle luciferin (Promega,Madison,WI). Acquired in vitro bioluminescence traces were detrended by division over a 24h running average and fitted with a damped sine function (Chronostar 2.0, PMID: 19299560). Only traces with a coefficient of correlation > 0.80 and period between 18-30h were considered circadian and rhythmic.
BioGEE glutathionylation assay
Primary astrocyte cultures were prepared and transfected with siBmal1 and siSCR siRNA as described above. At day 5 post-transfection media was changed to DMEM, +2% FBS, +penicillin/streptomycin. Also at day 5,250uM BSO (Sigma) was added to a subset of wells to block glutathione synthesis. At day 7 post-transfection all media was refreshed (DMEM, +2% FBS, +penicillin/streptomycin, +250uM BSO where appropriate) and 100uM BioGEE Glutathiolation Detection Reagent (Invitrogen, G36000) was added to each well except for one (non-treated control). After 3 hours, cells were lysed in RIPA buffer and processed as in western blotting section. As the entire size range of membrane was needed to blot for glutathione ethyl ester linked biotin, a parallel gel was run using the same sample mix in order to blot for loading control (p42/44 ERK). After transfer, the loading control membrane was blotted and imaged as above in the western blotting section. The membrane for detection of glutathionylation was washed and blocked as above except that TBS-T (tris buffered saline +0.125 tween) was replaced with PBS-T (phosphate buffered saline +0.125% tween) per method recommendation. After blocking, membrane was placed in 5% milk in PBS-T +streptavidin poly-HRP40 conjugate (Fitzgerald, Boston, MA, 65R-S104PHRP, 1:2000) for 1 hour, washed 5 times in PBS-T and imaged as in western blotting section.
Glutathione Measurements
GSH and GSSG levels were quantified in mouse cortex samples using the Glutathione Assay Kit (Cayman Chemical, Ann Arbor, MI). In cell culture experiments, glutathione measurements were obtained at day 7 using the Intracellular Glutathione Detection Assay Kit (Abcam, Cambridge, MA) by kinetic read (beginning immediately upon adding reagent and then every 5 minutes for 60 minutes) on a Tecan Infinite M1000 Pro fluorescent plate reader (Tecan, Switzerland).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prizm v7.02. In general, data are represented by mean ± SEM. In mouse experiments, “n” represents the number of mice and in cell culture experiments “n” represents the number of biological replicates. Specific statistical tests used, p value level definitions, and additional details are listed in figure legends. When multiple t test were performed, Holm-Sidak correction test for multiple comparisons was applied unless otherwise noted.
DATA AND SOFTWARE AVAILABILITY
The accession number for the microarray data reported in this paper is ArrayExpress: E-MTAB-7151. Array available at https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-7151/
Supplementary Material
Highlights.
Circadian disruption promotes astrocyte activation
Astrocyte-specific deletion of the circadian clock gene BMAL1 induces activation
BMAL1 regulates astrocyte activation by altering glutathione-S-transferase signaling
Loss of astrocyte BMAL1 enhances neuronal cell death in a co-culture system
ACKNOWLEDGMENTS
This study was funded by NINDS grant K08NS079405 (E.S.M.), NIA grants R01AG054517 (E.S.M.) and R21AG050054 (I.N.K.), NSF CAREER award 1553067 (I.N.K.), and an Alzheimer’s Association New Investigator Award (E.S.M.). J.S.T. is an Investigator at the Howard Hughes Medical Institute. S.A.L. was supported by the Australian National Health and Medical Research Council (GNT1052961), the Glenn Foundation Glenn Award, an anonymous donation, and V. and S. Coates.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Barca-Mayo O, Pons-Espinal M, Follert P, Armirotti A, Berdondini L, and De Pietri Tonelli D (2017). Astrocyte deletion of Bmal1 alters daily locomotor activity and cognitive functions via GABA signalling. Nat. Commun 8, 14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass J, and Takahashi JS (2010). Circadian integration of metabolism and energetics. Science 330, 1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaule C, Swanstrom A, Leone MJ, and Herzog ED (2009). Circadian modulation of gene expression, but not glutamate uptake, in mouse and rat cortical astrocytes. PLoS ONE 4, e7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert MM, Erikson GA, Shokhirev MN, and Allen NJ (2018). The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 22, 269–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancaccio M, Patton AP, Chesham JE, Maywood ES, and Hastings MH (2017). Astrocytes control circadian timekeeping in the suprachiasmatic nucleus via glutamatergic signaling. Neuron 93, 1420–1435.e5.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen DP, Vuono R, Nawarathna U, Fisher K, Shneerson JM, Reddy AB, and Barker RA (2014). Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol. 71, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova E, Fehsenfeld S, Mantamadiotis T, Lemberger T, Greiner E, Stewart AF, and Schütz G. (2001). A CamKIIalpha iCre BAC allows brain-specific gene inactivation. Genesis 31, 37–42. [DOI] [PubMed] [Google Scholar]
- Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, et al. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, and Barres BA (2018). Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 115, E1896–E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin P, McCarthy MJ, Lim ASP, Salmon DP, Galasko D, Masliah E, De Jager PL, Bennett DA, and Desplats P (2017). Circadian alterations during early stages of Alzheimer’s disease are associated with aberrant cycles of DNA methylation in BMAL1. Alzheimers Dement. 13, 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis AM, Fagundes CT, Yang G, Palsson-McDermott EM, Wochal P, McGettrick AF, Foley NH, Early JO, Chen L, Zhang H, et al. (2015). Circadian control of innate immunity in macrophages by miR-155 targeting Bmal1. Proc. Natl. Acad. Sci. USA 112, 7231–7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grek CL, Zhang J, Manevich Y, Townsend DM, and Tew KD (2013). Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem 288, 26497–26504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield CF, Herbert J, van Someren EJ, Hodges JR, and Hastings MH (2004). Disrupted daily activity/rest cycles in relation to daily cortisol rhythms of home-dwelling patients with early Alzheimer’s dementia. Brain 127, 1061–1074. [DOI] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Izumo M, Pejchal M, Schook AC, Lange RP, Walisser JA, Sato TR, Wang X, Bradfield CA, and Takahashi JS (2014). Differential effects of light and feeding on circadian organization of peripheral clocks in a forebrain Bmal1 mutant. eLife 3, e04617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SS, Ren Y, Liu CC, Kurti A, Baker KE, Bu G, Asmann Y, and Fryer JD (2018). Lipocalin-2 protects the brain during inflammatory conditions. Mol. Psychiatry 23, 344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, and Takahashi JS (2012). Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, Shim DJ, Rodriguez-Rivera J, Taglialatela G, Jankowsky JL, et al. (2015). NFkBactivated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 85, 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley SL, Pekny M, and Sands MS (2011). The role of attenuated astrocyte activation in infantile neuronal ceroid lipofuscinosis. J. Neurosci. 31, 15575–15585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailloux RJ, and Treberg JR (2016). Protein S-glutathionlyation links energy metabolism to redox signaling in mitochondria. Redox Biol. 8, 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marpegan L, Swanstrom AE, Chung K, Simon T, Haydon PG, Khan SK, Liu AC, Herzog ED, and Beaulé C. (2011). Circadian regulation of ATP release in astrocytes. J. Neurosci. 31, 8342–8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-García A, Sastre I, Recuero M, Aldudo J, Vilella E, Mateo I, S´nchez-Juan P, Vargas T, Carro E, Bermejo-Pareja F, et al. (2010). PLA2G3, a gene involved in oxidative stress induced death, is associated with Alzheimer’s disease. J. Alzheimers Dis. 22, 1181–1187. [DOI] [PubMed] [Google Scholar]
- Mohawk JA, Green CB, and Takahashi JS (2012). Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 35, 445–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, and Holtzman DM (2016). Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 354, 1004–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, Lim MM, Yang G, Bauer AQ, Qi L, Lee Y, Roh JH, Ortiz-Gonzalez X, Dearborn JT, Culver JP, et al. (2013). Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Invest. 123, 5389–5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, Bhimasani M, Zangrilli MA, Morris JC, Holtzman DM, and Ju YS (2018). Circadian rest-activity pattern changes in aging and preclinical Alzheimer disease. JAMA Neurol. 75, 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazato R, Kawabe K, Yamada D, Ikeno S, Mieda M, Shimba S, Hinoi E, Yoneda Y, and Takarada T (2017). Disruption of Bmal1 impairs blood-brain barrier integrity via pericyte dysfunction. J. Neurosci. 37, 10052–10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prolo LM, Takahashi JS, and Herzog ED (2005). Circadian rhythm generation and entrainment in astrocytes. J. Neurosci. 25, 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildge S, Bohrer C, Beck K, and Schachtrup C. (2013). Isolation and culture of mouse cortical astrocytes. J. Vis. Exp (Published online January 19, 2013) 10.3791/50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV (2014). Astrogliosis. Cold Spring Harb. Perspect. Biol. 7, a020420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Moon M, Choe HK, Han DH, Jang C, Kim A, Cho S, Kim K, and Mook-Jung I (2015). Aβ-induced degradation of BMAL1 and CBP leads to circadian rhythm disruption in Alzheimer’s disease. Mol. Neurodegener. 10, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Lu TY, Chai H, Xu J, Huang BS, Golshani P, Coppola G, and Khakh BS (2016). New transgenic mouse lines for selectively targeting astrocytes and studying calcium signals in astrocyte processes in situ and in vivo. Neuron 92, 1181–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tso CF, Simon T, Greenlaw AC, Puri T, Mieda M, and Herzog ED (2017). Astrocytes regulate daily rhythms in the suprachiasmatic nucleus and behavior. Curr. Biol. 27, 1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TA, Yu YV, Govindaiah G, Ye X, Artinian L, Coleman TP, Sweedler JV, Cox CL, and Gillette MU (2012). Circadian rhythm of redox state regulates excitability in suprachiasmatic nucleus neurons. Science 337, 839–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicher G, Wallenquist U, Lei Y, Enoksson M, Li X, Fuchs B, Abu Hamdeh S, Marklund N, Hillered L, Nilsson G, and Forsberg-Nilsson K (2017). Interleukin-33 promotes recruitment of microglia/macrophages in response to traumatic brain injury. J. Neurotrauma 34, 3173–3182. [DOI] [PubMed] [Google Scholar]
- Wyse CA, and Coogan AN (2010). Impact of aging on diurnal expression patterns of CLOCK and BMAL1 in the mouse brain. Brain Res. 1337, 21–31. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, and Cleveland DW (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci 11, 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Chen L, Grant GR, Paschos G, Song WL, Musiek ES, Lee V, McLoughlin SC, Grosser T, Cotsarelis G, and FitzGerald GA (2016). Timing of expression of the core clock gene Bmal1 influences its effects on aging and survival. Sci. Transl. Med 8, 324ra16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, and Barres BA (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.