

Abstract
Balancing selection has been inferred in diverse organisms for nonself recognition genes, including those involved in immunity, mating compatibility, and vegetative incompatibility. Although selective forces maintaining polymorphisms are known for genes involved in immunity and mating, mechanisms of balancing selection for vegetative incompatibility genes in fungi are being debated. We hypothesized that allorecognition and its consequent inhibition of virus transmission contribute to the maintenance of polymorphisms in vegetative incompatibility loci (vic) in the chestnut blight fungus, Cryphonectria parasitica. Balancing selection was demonstrated at two loci, vic2 and vic6, by trans-species polymorphisms in C. parasitica, C. radicalis, and C. japonica and signatures of positive selection in gene sequences. In addition, more than half (31 of 54) of allele frequency estimates at six vic loci in nine field populations of C. parasitica from Asia and the eastern US were not significantly different from 0.5, as expected at equilibrium for two alleles per locus under balancing selection. At three vic loci, deviations from 0.5 were predicted based on the effects of heteroallelism on virus transmission. Twenty-five of 27 allele frequency estimates were greater than or equal to 0.5 for the allele that confers significantly stronger inhibition of virus transmission at three loci with asymmetric transmission. These results are consistent with the allorecognition hypothesis that vegetative incompatibility genes are under selection because of their role in reducing infection by viruses.
Introduction
Polymorphisms for genes controlling nonself recognition in animals, plants, and fungi are maintained by balancing selection (reviewed by Richman 2000). These genes often exhibit high levels of polymorphisms within populations (high allelic richness and even allele frequencies), trans-species polymorphisms, and evidence of positive selection. The two most common mechanisms of balancing selection are overdominance, in which heterozygotes have greater fitness than homozygotes, and frequency-dependent selection, in which rare alleles confer a fitness advantage over more common alleles by virtue of their low frequencies. An example of overdominance can be found in vertebrate immune systems, in which high levels of heterozygosity may be maintained at major histocompatibility complex (MHC) loci by pathogen-mediated selection and mate preferences (Edwards and Hedrick 1998). Polymorphisms in mating recognition loci of plants (Nasrallah 2005) and fungi (May et al. 1999) are maintained by frequency-dependent selection, in which individuals with rare alleles have higher fitness than those with common alleles because they have a greater chance of encountering mates carrying different alleles and are therefore sexually compatible.
Nonself recognition can also occur during the asexual (vegetative) growth phase of fungi, independent of mating. In the Ascomycota, such nonself recognition is controlled by multiple vegetative incompatibility (vic) loci, which are known as heterokaryon incompatibility (het) loci in some species (Glass and Dementhon 2006; Smith and Lafontaine 2013). Ascomycetes typically have 8 to 11 unlinked vic or het loci (Leslie 1993). Hyphal fusions involving individuals with different alleles (heteroallelic) at one or more of these loci are vegetatively incompatible; the fusion cells undergo programmed cell death, preventing the formation of stable heterokaryons (Biella et al. 2002; Jacobson et al. 1998). Indeed, like other nonself recognition genes, vegetative incompatibility loci in Neurospora and Podospora display signatures of balancing selection. These include incompatibility genes that exhibit high levels of allelic diversity (Bastiaans et al. 2014; Hall et al. 2010; Mir-Rashed et al. 2000; Wu et al. 1998), trans-species polymorphisms (Hall et al. 2010; Powell et al. 2001; Powell et al. 2007; Wu et al. 1998), and nucleotide sequences with signatures of positive selection (Bastiaans et al. 2014; Chevanne et al. 2010; Hall et al. 2010; Micali and Smith 2006; Paoletti et al. 2007; Saupe et al. 1995; Wu et al. 1998). However, the underlying mechanisms selecting for polymorphisms at vic loci remain an open question.
Two hypotheses to explain balancing selection for vegetative incompatibility have emerged that are not mutually exclusive. The first is that incompatibility genes have pleiotropic effects, with additional functions unrelated to vegetative incompatibility (reviewed by Smith and Lafontaine 2013). Of particular interest is the suggestion that some het genes may be a subset of genes that function in surveillance and defense against pathogens (Paoletti and Saupe 2009). Under this hypothesis, polymorphisms are maintained because selection favors detection of a greater diversity of pathogens in a manner analogous to polymorphisms at MHC loci in vertebrates and pathogen resistance (R) loci in plants (Dangl and Jones 2001; Edwards and Hedrick 1998). The second hypothesis is that vegetative incompatibility evolved to recognize genetically different conspecific individuals and prevent fusion with them; this is known as the allorecognition hypothesis. Hyphal fusion within a colony is a normal part of development in filamentous fungi as a way of mobilizing resources (Rayner 1996; Simonin et al. 2012). Vegetative incompatibility may be favored by selection because it preserves the genetic integrity of individual genotypes (Todd and Rayner 1980), and prevents the invasion and exploitation by genetically different nuclei (Brusini et al. 2011; Czárán et al. 2014; Debets and Griffiths 1998; Nauta and Hoekstra 1994). In addition, vegetative incompatibility can inhibit the transmission of deleterious cytoplasmic elements, such as viruses, plasmids, or defective mitochondria where hyphal fusion between individuals is required for horizontal transmission to occur (Caten 1972; Cortesi et al. 2001; Debets et al. 1994; Hartl et al. 1975; van Diepeningen et al. 1997). In contrast to the first hypothesis, polymorphism is maintained at vic loci by frequency-dependent selection because an individual with a rare vic genotype is less likely than a common genotype to be exploited by another genotype or infected with cytoplasmic elements. Despite some of the theoretical shortcomings of allorecognition as a selective force maintaining vic gene polymorphism (Aanen et al. 2008), empirical evidence (cited above) is consistent with the hypothesis that allorecognition has the potential to increase fitness by preserving genetic integrity and reducing the risks of infection.
Insights into the structural attributes, gene composition, and function of vegetative incompatibility loci are emerging, as exemplified with Cryphonectria parasitica, the chestnut blight fungus. Allorecognition associated with vegetative incompatibility in C. parasitica has been studied extensively because of its role in the inhibition of virus transmission in relation to biological control of chestnut blight (Milgroom and Cortesi 2004; Zhang and Nuss 2016). Genes at six polymorphic vic loci in C. parasitica were identified by linkage mapping and comparative genomics (Choi et al. 2012; Zhang et al. 2014). Five of these loci (vic1, vic2, vic3, vic6, and vic7) comprise pairs of tightly linked genes controlling heterokaryon incompatibility similar to het loci in Neurospora (Smith and Lafontaine 2013). Heteroallelism at these five vic loci prevents heterokaryon formation and inhibits the transmission of viruses in the laboratory (Choi et al. 2012; Cortesi et al. 2001; Dawe and Nuss 2013; Zhang et al. 2014). The vic4 locus is both structurally and functionally distinct from these other five loci. vic4 activity is modulated by a single gene that is idiomorphic (nonhomologous) in the two “allelic” forms. Differences at vic4 elicit barrage formation between incompatible colonies in culture (mycelial incompatibility) but do not prevent heterokaryon formation (Smith et al. 2006) or virus transmission (Cortesi et al. 2001). Despite the strong evidence from laboratory studies, the degree to which heteroallelism at vic loci inhibits virus transmission in nature is not clear (Milgroom and Cortesi 2004). For example, Carbone et al. (2004) found high levels of migration of Cryphonectria hypovirus 1 (CHV-1) between vegetatively incompatible clones of C. parasitica in two field populations. On the other hand, the fitness of C. parasitica (canker growth on trees, asexual sporulation, and female fertility) may be reduced by 90% or more by infection with some hypoviruses (Nuss 2005; Peever et al. 2000), and therefore inhibition of virus transmission is a potentially strong selective force for maintaining polymorphism at vic loci in C. parasitica.
The allorecognition hypothesis leads to the expectation of even allele frequencies at equilibrium because of frequency-dependent selection. An earlier attempt to test this hypothesis in populations of C. parasitica produced equivocal results. Five of the six known vic loci were polymorphic in almost all of 16 populations sampled (13 in Europe and 3 in the eastern US), but at most loci allele frequencies deviated significantly from 0.5, the expected equilibrium frequency for two alleles at a locus, as found at vic loci in C. parasitica (Milgroom and Cortesi 1999). Two ad hoc explanations for these deviations were proposed (Milgroom and Cortesi 1999). First, C. parasitica and CHV-1 were introduced into Europe and North America from Asia in the first half the 20th century (Dutech et al. 2012; Milgroom et al. 1996), and therefore vic allele frequencies on these continents may not have reached equilibrium. Second, most European populations of C. parasitica are clonal and vic loci are in linkage disequilibrium (LD) (Dutech et al. 2010; Milgroom and Cortesi 1999; Milgroom et al. 2008). Therefore, vic allele frequencies may not evolve independently because they are subject to hitchhiking selection on the entire clone. This lack of independence weakened tests of equilibrium frequencies at vic loci in European populations, and emphasized the need to repeat these analyses on randomly mating populations, especially those in Asia where C. parasitica is native.
In this study, we propose that asymmetric virus transmission is an additional explanation for lack of even allele frequencies reported previously. Inhibition of virus transmission is significantly asymmetric with heteroallelism at three loci (vic1, vic2, and vic7) (Cortesi et al. 2001). For example, when two individuals heteroallelic only at vic1 are paired in culture, transmission occurs in almost 100% of the pairings when the virus-infected donor isolate carries allele vic1-2 and the virus-free recipient carries allele vic1-1. In contrast, transmission is reduced to approximately 10% when the same two individuals are paired in culture but the virus-infected donor carries vic1-1 and the virus-free recipient carries vic1-2. We hypothesize that if inhibition of virus transmission is a selective force maintaining vic gene polymorphisms, then alleles that result in stronger inhibition of virus transmission should be favored by selection. In the example above, allele vic1-2 would be expected to confer greater fitness than vic1-1 by inhibiting transmission into the individual carrying this allele when it encounters a virus-infected individual carrying the alternate allele. This hypothesis leads to the prediction that alleles favored by asymmetric transmission have equilibrium frequencies greater than 0.5. A simple haploid model of frequency-dependent selection that formalizes this prediction, based on a more complicated diploid model (Cockerham et al. 1972), is presented in the Supplementary Information.
The first objective of this study was to look for evidence of balancing selection on vic loci in C. parasitica based on trans-species polymorphisms, positive selection, and even allele frequencies. To avoid some of the shortcomings of testing expectations of the allorecognition hypothesis in European populations (Milgroom and Cortesi 1999), we sampled populations of C. parasitica from Japan and China, where both C. parasitica and CHV-1 are native (Milgroom et al. 1996; Peever et al. 1998) and more likely to be at equilibrium than in Europe or North America. Moreover, populations of C. parasitica in Asia and eastern North America are randomly mating (Dutech et al. 2012; Milgroom and Cortesi 1999; Milgroom et al. 1992), which means that balancing selection can act on each vic locus independently. The second objective was to test the prediction that allele frequencies deviate from 0.5 at vic loci that are known to inhibit virus transmission asymmetrically.
Materials and methods
Sequencing vic alleles to determine trans-species polymorphisms
We tested for trans-species polymorphisms by sequencing genes at vic2 and vic6 loci from C. parasitica (Cp) and two closely related species: C. radicalis (Cr) and C. japonica (Cj; syn. C. nitschkei) (Supplementary Table S1). Genes at these two loci were chosen because they were well characterized functionally and shown to affect vegetative incompatibility and virus transmission in C. parasitica (Choi et al. 2012). As it is unknown whether vic-gene orthologs in other Cryphonectria species function in vegetative incompatibility, isolates of these other species were assigned to allelic classes based on sequence similarities to vic alleles in C. parasitica.
The vic2 locus comprises three genes. Incompatibility is conferred by a patatin-like gene (vic2) and a sec9-like gene (vic2a); alleles for these genes are in complete LD (Choi et al. 2012). The third gene at this locus is located in a 2.7-kb region between vic2 and vic2a and encodes a helicase-like domain (C. parasitica protein ID 332917) that does not function in vegetative incompatibility (Choi et al. 2012). We PCR amplified and sequenced the entire ORF (including introns) of vic2a-2 (~1380 nt) and vic2a-1 (~1289 nt) by Sanger sequencing in both directions. We also sequenced ~534 nt of the helicase-like gene. We did not obtain sequences of vic2. PCR primers used to obtain vic2a amplicons were designed from conserved sequences of vic2a-2 from isolate EP155 (GenBank JN367273.1) and vic2a-1 from isolate EP146 (GenBank JN367274.1). The region of the helicase-like gene sequenced corresponds to scaffold 7:1665487-1666020 of isolate EP155 (C. parasitica EP155 genome v2.0, JGI; http://genome.jgi.doe.gov/Crypa2/Crypa2.home.html).
The vic6 locus comprises two genes (vic6 and pix6) that are adjacent in the C. parasitica genome. Similar to vic2 and vic2a, alleles for these two genes were found previously in complete LD (Choi et al. 2012). We sequenced the entire ORF (including introns) from vic6-2 (~2221 nt) and vic6-1 (~2353 nt). Primer design was based on sequences of vic6-2 from isolate EP155 (GenBank JN256666.1) and vic6-1 from isolate EP146 (GenBank JN367268.1). We also sequenced the entire ORF, including introns, of pix6 alleles (~1043 nt). Primer design was based on pix6-2 from isolate EP155 (GenBank JN367269.1) and pix6-1 from isolate EP146 (GenBank JN367270.1). All sequencing was done by Macrogen USA (Rockville, MD).
Nucleotide sequences of coding regions with introns were analyzed using MEGA 6.2 (Tamura et al. 2013). Sequences were aligned using MUSCLE and maximum likelihood trees were constructed based on the Tamura-Nei model (Tamura and Nei 1993). The tree with the highest log likelihood was chosen as the best inference of evolutionary history for each gene. Using default parameters, a matrix of pairwise distances was estimated using the maximum composite likelihood approach, and initial trees for the heuristic search were obtained by the neighbor-joining method. All nucleotide sites with gaps or missing data were eliminated from the analysis. One thousand bootstrap replications were conducted for each analysis.
Estimation of positive selection
In general, positive selection is inferred when the number of non-synonymous substitutions per potential non-synonymous site (dN) exceeds the number of synonymous substitutions per potential synonymous site (dS), i.e., dN/dS > 1. For our analyses, noncoding regions were removed from nucleotide sequences of vic2a, helicase-like gene, vic6 and pix6 based on GenBank annotations for EP155 and EP146 (Choi et al. 2012). Coding sequences were then aligned using MUSCLE, informed by amino acid sequences with TranslatorX (http://translatorx.co.uk/) (Abascal et al. 2010). Codon-based selection analysis employed a branch-site Random Effects Likelihood test (branch-site REL, Kosakovsky Pond and Frost 2005) to identify branches having evidence of positive selection. For a priori tests of positive selection along branches that separate respective allele 1 and 2 clades, we used branch-site unrestricted statistical test for episodic diversification (BUSTED, Murrell et al. 2015). Branch-site REL and BUSTED are related methods that are used to test for positive selection among different branches and sites (Kosakovsky Pond et al. 2011). These analyses contrast codon dN/dS patterns along specific (foreground) branches with those of all other (background) branches. Evidence for positive selection is provided by a subset of sites with significantly elevated dN/dS ratios in specific lineages. Branch-site REL and BUSTED analyses were performed using an online application (http://www.datamonkey.org/) (Kosakovsky Pond and Frost 2005). Alignments were verified visually using boxshade (http://www.ch.embnet.org/software/BOX_form.html).
Estimation of vic allele frequencies in C. parasitica populations
Before PCR markers were developed for vic genotyping (Short et al. 2015), it was only possible to determine vic genotypes by culturing isolates with each of 64 vic genotype tester strains to assess mycelial compatibility (Cortesi and Milgroom 1998). However, vic genotypes could not be determined for isolates unless they were compatible with a tester isolate. For example, 35 of 176 isolates in the eastern US (Milgroom and Cortesi 1999), and 140 out of 143 isolates in Japan and China (Liu and Milgroom 2007) could not be genotyped by culturing. In this study, we genotyped all six vic loci in the additional 35 isolates from the US by PCR (see below). Data from these 35 isolates were combined with those reported previously (Milgroom and Cortesi 1999) to revise estimates of vic allele frequencies. For additional populations in the eastern US, we calculated allele frequencies at six vic loci using the distributions of multilocus vic genotypes in four populations reported in Fig. 3 of Short et al. (2015). To estimate vic allele frequencies in Asian populations, we genotyped isolates collected in 1992 from Ibaraki Prefecture in Japan, and from Anhui (Xiuning) and Jiangsu (Liang) Provinces in China (Milgroom et al. 1996). C. parasitica was collected from three sites (Chiyoda, Yasato, and Iwama) in Ibaraki Prefecture that are within a 5-km radius and are treated here as a single population. The diversity of vc types was determined previously for samples from Ibaraki and Xiuning; each isolate was vegetatively incompatible with all others in the same population (Liu and Milgroom 2007). No vc type data were available previously for Liang.
Genotyping was done using PCR markers in sequences of vic genes identified in the C. parasitica genome (Choi et al. 2012; Zhang et al. 2014). For a subset of isolates (25 from Japan; 18 from China; Supplementary Table S2), we genotyped alleles at vic2, vic4, vic6, and vic7 using PCR primers designed by Choi et al. (2012) (Supplementary Table S3). For the same subset of isolates we also genotyped the closely linked gene pairs vic2/vic2a and pix6/vic6 which control vegetative incompatibility at the vic2 and vic6 loci, respectively (Choi et al. 2012). For additional isolates and for all genotyping of vic1 and vic3, which were identified later (Zhang et al. 2014), we used PCR primers designed by Short et al. (2015) (Supplementary Table S3).
DNA was extracted from fresh mycelium grown for 4 days on potato dextrose agar overlain with cellophane, using methods described in Kepler et al. (2014). PCR was performed using conditions described previously (Short et al. 2015), except for an annealing temperature of 64 °C for vic1. We used Native Taq DNA polymerase (Thermo Fisher Scientific) for vic1, vic3, vic6, and vic7. We used HiFi platinum Taq (Thermo Fisher Scientific) for vic2 and vic4 to avoid non-specific amplification. All PCRs were done with a Bio-Rad T-100 thermocycler (Bio-Rad Laboratories, Hercules, CA). PCR products were visualized following electrophoresis using 2% TAE agarose gels stained with GelRed (Biotium, Inc., Fremont, CA).
Statistical analysis of vic allele frequencies
We performed binomial exact tests using the statistical software Minitab ver. 17 (Minitab Inc., State College, PA) to test whether vic allele frequencies deviated significantly from 0.5. One-sided tests were used for three loci (vic1, vic2, and vic7) with significant asymmetric virus transmission (Cortesi et al. 2001) because we predicted that equilibrium frequencies for alleles that inhibited transmission more strongly would be greater than 0.5 (Supplementary Information); these alleles included vic1-2, vic2-2, and vic7-1. For loci without asymmetric transmission (vic3, vic4, and vic6) we used two-sided tests for the null hypothesis p = 0.5, where p is the frequency of allele 2 (this is arbitrary; allele 1 has a frequency of q = 1–p and thus the same statistics apply to both alleles). To determine the significance of multiple hypothesis tests, the false discovery rate was set to 0.05 using the method of Benjamini and Hochberg (1995).
We tested for LD among all pairs of vic loci within each population using default parameters in Genepop 4.6 (Rousset 2008). This test calculates the log likelihood ratio statistic (G test) for a contingency table for each locus pair and uses a Markov chain algorithm for significance testing (Raymond and Rousset 1995).
Results
Trans-species polymorphisms
Sequencing revealed trans-species polymorphisms for vic2a, pix6, and vic6 (Supplementary Table S1). Nucleotide sequences for alleles at vic2a, pix6, and vic6 in C. japonica and C. radicalis were similar to known vic alleles in C. parasitica (Choi et al. 2012) and were therefore considered orthologs. Both alleles at vic2a, vic6, and pix6 were sequenced from C. parasitica and C. japonica, whereas only one allele was found for each gene at the vic6 locus (vic6-1 and pix6-1) among 11 isolates of C. radicalis. In phylogenetic reconstructions, nucleotide sequences of vic2a, pix6, and vic6 were more similar within allelic classes between species than they were between alleles within the same species (Fig. 1). The branches separating allelic classes were highly supported by bootstrapping. Phylogenies of pix6 and vic6 were very similar in topology. These findings clearly demonstrate trans-species polymorphism and support the hypothesis that polymorphisms at these vic loci have been maintained by balancing selection.
Fig. 1.

Phylogenetic trees inferred by maximum likelihood analysis and tests of positive selection on the different branches by branch-site REL and BUSTED analyses. Trees are based on nucleotide sequences from Cryphonectria parasitica (Cp), C. japonica (Cj), and C. radicalis (Cr) for a vic2a, b a helicase-like gene located between vic2 and vic2a, c pix6, and d vic6. Bootstrap values (percentages based on 1000 replications) are shown below branches. Average values of ω ( = dN/dS) are shown above each branch. Branches with significant evidence of positive selection (P ≤ 0.01, branch-site REL) are shown with bold lines and designated by lowercase letters in parentheses (see Table 1 for details). A priori tests for positive selection between alleles 1 and 2 (using BUSTED) are shown for dN/dS (ω3), the percentage of nucleotide sites with evidence of positive selection along the branch contributing to the likelihood ratio significance test and the P-value
In contrast to vic2a, vic6, and pix6, the helicase-like gene located between vic2 and vic2a did not show trans-species polymorphism, but had species-specific sequences (Fig. 1b). Within each species, however, the helicase-like gene sequences diverged according to alleles at the vic2 locus, most likely reflecting its close linkage with these genes. Isolate YM2 of C. japonica formed an exception to this pattern, as its sequence was more similar to those in isolates carrying allele vic2-2 than to other isolates carrying allele vic2-1, although these relationships were not well supported by bootstrapping within this species.
Estimation of positive selection from vic gene sequences
We used branch-site REL to test for evidence of positive selection among phylogenetic branches for each of vic2a, helicase-like gene, vic6, and pix6 (Fig. 1). This analysis requires unique sequences, 12 of which were available for vic2a (vic2a-1: isolates CpEP146, CrM2269, CjYM2, CjES19, CjIF6; vic2a-2: isolates CpEP155, Cp09515, CpDU74, CpJA69, Cr09491, CrGM8A, CjOB1-6). For vic2a, branch-site REL identified four branches under positive selection, the most compelling of which linked the clades of vic2a-1 and vic2a-2 (branch a, Fig. 1a), wherein 23% of sites had dN/dS > 5. An a priori test of this “foreground” branch using the BUSTED method further supported the conclusion that alleles vic2a-1 and vic2a-2 diverged under positive selection (P = 2 × 10−16; branch a, Fig. 1a). Divergences in sequence alignments (Supplementary Fig. S1A) were almost exclusively located in the 3′ end of vic2a, between codons 200–390, corresponding to the C-terminus of the VIC2a protein (alignments of vic2a extend to 398 codons, including internal gaps). Evidence for positive selection was also detected along the phylogenetic branch separating pix6-1 and pix6-2 (branch a, Fig. 1c) based on analysis of six unique pix6 sequences available (pix6-1: isolates CpEP146, CrGM8A, Cr09491, CjES19; pix6-2: isolates CpEP155, CjOB1). For this branch, 26% of sites had a dN/dS > 5 based on branch-site REL (Table 1) and positive selection along this branch was supported by BUSTED analysis (P = 1 × 10−8). Similar to vic2a, sequence divergence between pix6-1 and pix6-2 was prominent in the 3′ end of pix6, between codons 137–230 (alignments of pix6 extend to 234 codons, including internal gaps; Supplementary Fig. S1C). These localized substitutions implicate the C-terminal regions of VIC2a and PIX6 in incompatibility specificity.
Table 1.
Branches with evidence of episodic positive selection (P < 0.01) based on branch-site REL and BUSTED analyses
| Gene | Brancha | Percent of sites under positive selectiond | ω +b | P-valuec | ω 3 e | P-value |
|---|---|---|---|---|---|---|
| vic2 | a | 23 | 95 | <0.0001 | 87.7 | 2.0 × 10-16 |
| b | 4 | 5715 | <0.0001 | – | – | |
| c | 9 | 14 | 0.001 | – | – | |
| d | 6 | 19 | 0.007 | – | – | |
| pix6 | a | 26 | 10,000 | <0.0001 | 3330 | 1.0 × 10-8 |
| vic6 | None | – | – | – | 1.3 | >0.5 |
| Helicase-like | None | – | – | – | 1.0 | >0.5 |
a Name of branch refers to those in Fig. 1
b ω+ (dN/dS) for positively selected sites along the branch estimated using branch-site REL (Kosakovsky Pond and Frost 2005)
c P-value for episodic positive selection along the branch corrected by the Holm–Bonferroni method
d Proportion of sites along the branch inferred to show positive selection using branch-site REL
e ω3, tests for positive selection of the branch joining alleles 1 and 2 using BUSTED under the null hypothesis of neutrality (Murrell et al. 2015). For the helicase-like gene, this test is between alleles 1 and 2 only in C. parasitica
In contrast to vic2a and pix6, positive selection was not evident for vic6 based on branch-site REL analyses (Fig. 1d; Table 1) of the 10 unique sequences available (vic6-1: isolates CpEP146, CpYS522, Cr09491, CrGM8A, CjES19; vic6-2: isolates CpEP155, CpBRU17, Cp09515, CpJA69, CjOB1-6). An a priori test using BUSTED also did not provide evidence for positive selection on the branch between vic6-1 and vic6-2 (P > 0.5). Likewise, there was no evidence for positive selection across branches of the helicase-like gene (Fig. 1b, Table 1) based on the nine unique sequences available (isolates CjIF6, CjES19, CpEP155, CpEP146, Cr09491, CrGM8A, CrM2269, CjYM2, CjOB1-6). Altogether, these findings show that branches separating alleles at vic2a and pix6 are under positive selection, but surprisingly vic6 is not, even though it is tightly linked to pix6.
Estimation of LD between vic loci
Alleles at the gene pairs vic2/vic2a and pix6/vic6 were previously shown to be in complete LD for 25 isolates from eastern US populations and three isolates from Italy (Choi et al. 2012). In this study, we genotyped an additional 43 isolates from Japan and China at these gene pairs and confirmed the strict association of alleles at these loci (Supplementary Table S2). These results extend those of the previous study showing these gene pairs are in complete LD and likely act together to effect vegetative incompatibility.
We also tested for LD between alleles at different vic loci to determine whether allele frequencies were independent. Lack of independence was previously considered a reason that vic allele frequencies deviated from 0.5 in other populations (Milgroom and Cortesi 1999). None of the locus pairs showed significant LD in any Asian population, even after pooling the three populations to increase sample sizes and statistical power to detect LD (pooling per se is a potential source of LD when allele frequencies differ among samples). Ten locus pairs showed significant LD in US populations; six of these occurred in the sample from Shenandoah National Park, VA (Supplementary Table S4). LD can arise for a variety of reasons, the most likely of which for these populations is some degree of clonal reproduction (Milgroom et al. 1992). Four additional pairs of loci were in significant LD (with a false discovery rate of 0.10): two pairs in Bartow, WV, and one each in Depot Hill, NY, and Pendleton, WV (Supplementary Table S4) out of 15 possible locus pairs in each population. Based on these findings of LD, we eliminated the sample from Shenandoah National Park from any further analyses, and concluded that LD was low enough in the other populations to treat vic loci as independent.
Estimation of vic allele frequencies in C. parasitica populations
We tested for even allele frequencies as predicted for balancing selection acting on vic genes. All six vic loci were polymorphic in all nine populations (Fig. 2, Supplementary Table S5). Using a false discovery rate of 0.05 for nine estimates for each locus, allele frequencies were considered significantly different from 0.5 when P-values were less than 0.02. In the Asian populations, 13 of the 18 allele frequency estimates (six loci in three populations) were not significantly different from 0.5. In the eastern US, 18 of the 36 estimates were not significantly different from 0.5 (Fig. 2, Table 2). We predicted above (see also the model in Supplementary Information) that equilibrium allele frequencies might deviate from 0.5 at vic1, vic2, and vic7 because of asymmetric virus transmission. For each of these loci, the allele favored by asymmetric virus transmission (vic1-1, vic2-1, and vic7-2) had an estimated frequency significantly greater than 0.5 in 10 of the 12 instances in which it was significantly different from 0.5 (Fig. 2, Table 2). The only discrepancies from these predictions were two US populations (Smokey Mtn. National Park, TN and Tucker Co., WV) in which the alternate allele (vic7-2) was at a frequency significantly greater than 0.5. At the other three loci, which do not exhibit significant asymmetric virus transmission (Cortesi et al. 2001), one of the two alleles (vic3-1, vic4-2, vic6-1) was found at frequencies either equal to or greater than 0.5, with the exception of allele vic6-2 being significantly greater than 0.5 in Smokey Mtn. National Park (Fig. 2).
Fig. 2.
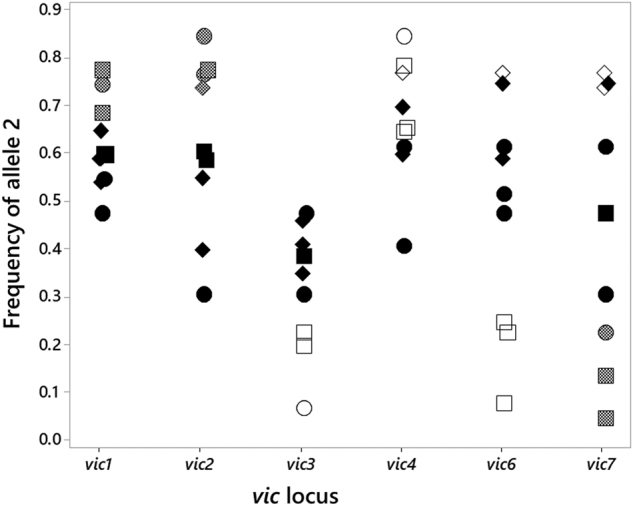
Allele frequencies at vic loci in populations of C. parasitica from Asia and the eastern US. The three Asian populations are indicated by circles (N = 26–44). US populations originally reported in 1999 (Milgroom and Cortesi 1999) are indicated by squares (N = 57–60) and those from 2015 (Short et al. 2015) are indicated by diamonds (N = 20–35). Open symbols are significantly different from 0.5. Solid black symbols are not significantly different from 0.5. Stippled gray symbols are significantly greater (vic1 and vic2) or less (vic7) than 0.5 as predicted by asymmetric virus transmission
Table 2.
Summary of vic loci with allele frequencies significantly different from 0.5 or with the allele favored by asymmetric virus transmission at vic1, vic2, and vic7 significantly greater than 0.5
| Populationa | N | Loci significantly different from any expectations | Loci not significantly different from 0.5 | Loci with allele favored by asymmetric virus transmission significantly greater than 0.5 (vic1, vic2, or vic7 only) |
|---|---|---|---|---|
| Ibaraki, Japan | 44 | – | vic1, vic3, vic4, vic6 | vic2, vic7 |
| Liang, China | 30b | vic3 | vic2, vic4, vic6, vic7 | vic1 |
| Xiuning, China | 26b | vic4 | vic1, vic2, vic3, vic6, vic7 | – |
| Finzel, MD | 57 | vic3, vic4, vic6 | vic1, vic2 | vic7 |
| Bartow, WV | 59 | vic4, vic6 | vic2, vic3 | vic1, vic7 |
| Depot Hill, NY | 61 | vic3, vic4, vic6 | vic7 | vic1, vic2 |
| Pendleton, WV | 20 | – | vic1, vic2, vic3, vic4, vic6, vic7 | – |
| Smokey Mtn. NP, TN | 35 | vic6, vic7 | vic1, vic3, vic4 | vic2 |
| Tucker Co., WV | 22 | vic4, vic7 | vic1, vic2, vic3, vic6 |
a Samples from Japan and China were described in Milgroom et al. (1996); vic genotype data for Finzel, MD, Bartow, WV, and Depot Hill, NY, were taken from Milgroom and Cortesi (1999), plus additional genotyping (described in Materials and methods); vic genotype data for Pendleton, WV, Smokey Mtn. NP, TN, and Tucker Co., WV, were taken from Short et al. (2015). Statistics supporting these results are shown in Supplementary Table S5
b Sample sizes varied slightly among loci in Liang and Xiuning (Supplementary Table S5) because of missing data. Sample sizes shown here are for the maximum in each population
The proportion of allele-frequency estimates deviating from 0.5 or from predictions of asymmetric virus transmission was significantly less in Asia than in the US (P = 0.041; one-sided test). In Asia, 2 out of 18 estimates deviated from expectations; in the eastern US, 12 out of 36 deviated (Table 2). In addition, the number of estimates that deviated significantly from 0.5 was significantly correlated with sample size among all nine populations (r = 0.89, P = 0.001). This correlation does not account for predictions of asymmetric virus transmission but suggests that as sample size, and therefore statistical power, increases, more deviations from expectations can be found.
Discussion
Our data provide several lines of evidence that support the hypothesis of balancing selection on vic loci in C. parasitica. First, a clear pattern of trans-species polymorphism was evident at incompatibility genes vic2a, vic6, and pix6 (Fig. 1). Second, strong evidence of positive selection was found for the divergence of the two allelic classes at vic2a and pix6 (Fig. 1). Third, all six vic loci were polymorphic in all nine populations sampled. More than half (31 of 54) of the allele frequency estimates were not significantly different from 0.5 (Fig. 2), which is the predicted frequency at equilibrium for two alleles at each locus under balancing selection. Our study is unique in relating deviations from even allele frequencies to expectations for asymmetric virus transmission (Supplementary Information). Ten of the 12 allele frequency estimates that deviated significantly from 0.5 at vic1, vic2, and vic7 had an excess of the allele predicted to be favored by asymmetric virus transmission (Fig. 2). All told, these findings support the hypothesis that vic genes in C. parasitica evolved under balancing selection, and that inhibition of virus transmission, as expected under the allorecognition hypothesis, may be a plausible mechanism driving such selection.
Trans-species polymorphisms in vic genes
Trans-species polymorphism has been interpreted as evidence that polymorphism has been maintained at a locus since before speciation, thus providing strong evidence of balancing selection (Richman 2000). In C. parasitica, incompatibility genes vic2a, vic6, and pix6 show clear patterns of trans-species polymorphisms (Fig. 1). These results are similar to the finding of trans-species polymorphism in het genes in Neurospora (Hall et al. 2010; Powell et al. 2001; Powell et al. 2007; Wu et al. 1998). Therefore, our results with Cryphonectria species add to the observation of trans-species polymorphism in genes for vegetative incompatibility in ascomycete fungi.
In contrast to genes that function directly in vegetative incompatibility, the phylogeny inferred from sequences of the helicase-like gene does not show trans-species polymorphism. Sequences of this gene diverged by species (Fig. 1b), with a topology identical to that inferred from ITS sequences (Gryzenhout et al. 2006). This topology would be expected for most of the genome, which did not evolve under balancing selection. For the helicase-like gene, intraspecific variation was associated with alleles of vic2a (Fig. 1a, b), to which it is tightly linked. Purifying selection and LD in the helicase-like gene are discussed further below.
Positive selection acting on vic genes
A second line of evidence for balancing selection besides the presence of trans-species polymorphism is the signature of positive selection in incompatibility gene sequences. Both vic2a and pix6 had significantly elevated dN/dS ratios, indicative of positive selection, along the respective branches between alleles vic2a-1 and vic2a-2, and between pix6-1 and pix6-2 (Fig. 1a, c). We interpret this to mean that positive selection was a driver of divergence between the two alleles of the genes vic2a and pix6. Positive (or diversifying) selection for vegetative incompatibility genes in other fungi has been interpreted as support for balancing selection (Bastiaans et al. 2014; Chevanne et al. 2010; Hall et al. 2010; Micali and Smith 2006; Paoletti et al. 2007; Saupe et al. 1995; Wu et al. 1998). In addition, we detected positive selection between species within allelic classes for vic2a (Fig. 1a). We speculate that further divergence between species could be driven by selection to reduce virus transmission between species, which has been shown to occur between C. parasitica and C. japonica in both the laboratory and in nature (Liu et al. 2003).
We found no evidence for positive selection in vic6. This result is of particular interest since, like vic2a and pix6, vic6 has incompatibility function, is in complete LD with pix6, and exhibits trans-species polymorphism. It should be noted, however, that only one or a few codons may be responsible for allelic specificity in fungal vegetative incompatibility genes (Deleu et al. 1993). In such cases, signatures of positive selection may be difficult to detect across entire coding regions. Therefore, failure to detect positive selection in sequences of vic6 does not rule out the hypothesis that this locus is under positive selection.
Purifying selection and LD in the helicase-like gene
In contrast to the other genes in this study, the helicase-like gene shows no evidence of either trans-species polymorphism or positive selection (Fig. 1b). This gene shows relatively little variation in amino acid sequences (Supplementary Fig. S1B). Moreover, dN/dS estimated among sequences within C. parasitica is 0.05, and among species is 0.23 (results not shown), indicating that this gene is under purifying selection. The helicase-like gene does not function in vegetative incompatibility, even though it is tightly linked to vic2a (Choi et al. 2012), which is a functional incompatibility gene and evolved under balancing selection; no sequence data are available for vic2 for comparison. Thus, markedly different selective regimes have operated on these tightly linked genes. We speculate that purifying selection on the helicase-like gene was sufficiently strong to override hitchhiking selection from vic genes. However, intraspecific variation in helicase-like gene sequences is associated with the vic2 alleles to which they are linked (Fig. 1a, b), demonstrating some effects of this linkage. Overall, our findings indicate a mix of selective effects on the helicase-like gene. Not only is it under purifying selection, as indicated by dN/dS < 1, but also hitchhiking selection because of tight linkage to incompatibility genes vic2 and vic2a.
The difference in selection regimes on vic2a and the helicase-like gene indicates that recombination must have occurred between these two genes. However, in C. parasitica (Choi et al. 2012) and other fungi most vic (or het) loci comprise pairs of genes that function together to effect incompatibility (Smith and Lafontaine 2013). Complete LD between alleles at pairs of vic genes in C. parasitica (this study; Choi et al. 2012) and het genes in Neurospora (Hall et al. 2010; Mir-Rashed et al. 2000; Smith et al. 2000) strongly suggests that recombinants would be selected against. Micali and Smith (2006) demonstrated experimentally that recombinants created by transformation of genes at the het-6 locus in N. crassa experienced markedly reduced fitness, indicating the potential for strong purifying selection. Therefore, we speculate that any recombination between the helicase-like gene and flanking vic genes would have to occur by double crossovers or gene conversion because recombination between vic2 and vic2a is likely to be deleterious.
Even allele frequencies at vic loci
Polymorphisms and even allele frequencies at vic loci provide additional evidence of balancing selection. All six vic loci in C. parasitica were polymorphic in all populations. Moreover, 40 of the 54 allele frequency estimates (six vic loci in nine populations) were not different from 0.5 or were greater than 0.5 for alleles predicted to confer a fitness advantage because of asymmetric virus transmission (Fig. 2, Table 2); the significance of asymmetric virus transmission is discussed in the next section. Previous estimates of vic allele frequencies deviated markedly from 0.5 because of LD caused by the clonal structure of European populations (Milgroom and Cortesi 1999). Inferences from the present study are more robust because we sampled only Asian and US populations, and eliminated one population (Shenandoah National Park, VA) showing high levels of LD; minimal LD was detected in all other populations. Polymorphism and even allele frequencies at vic loci in C. parasitica are consistent with predictions for balancing selection at equilibrium and are similar to observations of het loci in N. crassa and Podospora anserina (Bastiaans et al. 2014; Hall et al. 2010; Mir-Rashed et al. 2000; Wu et al. 1998).
Although all six vic loci were polymorphic in all populations sampled, a potential weakness of this analysis is that small sample sizes limit the statistical power for testing the null hypothesis that allele frequencies are not different from 0.5. In this study, sample sizes were N = 57–61 for three US populations, but as low as N = 20–22 in two of the three US populations for which data were obtained from a published report (Short et al. 2015). Samples sizes for the three Asian and one US population were in the range N = 26–44. The significant positive correlation (r = 0.89) between sample sizes and deviations in allele frequencies from 0.5 (ignoring asymmetric virus transmission) highlights the problem of low statistical power for some populations: larger samples may have more significant differences simply because of greater statistical power. Furthermore, the lack of statistical power is particularly problematic for distinguishing between frequencies not significantly different from 0.5 and those greater than 0.5 predicted for asymmetric virus transmission (see below).
Viruses as a selective factor maintaining polymorphisms at vic loci
We attempted to find support for the role of viruses in balancing selection on vic genes by comparing allele frequencies to expectations for asymmetric virus transmission associated with heteroallelism at vic1, vic2, and vic7 (Cortesi et al. 2001; Huber 1996; Zhang et al. 2014). For these loci, equilibrium allele frequencies are predicted to deviate from 0.5, depending on the strength of the asymmetry and the fitness differential caused by virus infection (see model in Supplementary Information). All allele frequency estimates at vic1 and vic2 were either not significantly different from 0.5 or deviated in accordance with predictions of asymmetric virus transmission. Estimates at vic7 were mixed, however, with three deviations from 0.5 concordant with asymmetric virus transmission predictions, and two discordant. Deviations of estimates from 0.5 at the other three loci also appeared nonrandom (Fig. 2), but heteroallelism at these loci either has no effect on virus transmission (vic4), or does not result in significant asymmetric transmission (vic3 and vic6) (Cortesi et al. 2001). Therefore, there is no a priori reason for predicting the outcomes for these latter loci. Overall, these findings provide support for the hypothesis that asymmetric virus transmission could affect allele frequencies and balancing selection.
The fitness differential in the asymmetric virus transmission model is a function of the probability of virus transmission and deleterious effects of viruses on fitness of the recipients (Supplementary Information). Model predictions, however, depend on equilibrium conditions. C. parasitica is native to east Asia and more likely to have reached equilibrium under balancing selection there than in the US where it was introduced in the early 1900s. Moreover, Cryphonectria hypovirus 1 (CHV-1) has marked effects on the fitness of C. parasitica (Nuss 2005; Peever et al. 2000), and is present throughout Japan and China (Liu et al. 2003; Peever et al. 1998). By contrast, introduced populations in the US are less likely to have reached equilibrium, and, more importantly, the selective pressures from viruses may not be strong. Cryphonectria hypovirus 4 (CHV-4) is the only virus that is commonly found in the eastern US (Hillman and Suzuki 2004; Peever et al. 1997). However, CHV-4 has no measurable effects on the fitness in C. parasitica (Enebak et al. 1994), and therefore it is difficult to ascribe a role to these viruses as a selective force maintaining balanced polymorphisms at vic loci in US populations. The finding of a significantly higher proportion of deviations from model predictions (both even frequencies and those for asymmetric virus transmission; Table 2) in the US than in Asian populations is consistent with US populations not yet having reached equilibrium or not being under as much selection by viruses.
Conclusions
Understanding the selective forces acting on vegetative incompatibility genes has proven to be a difficult challenge (Paoletti and Saupe 2009; Smith and Lafontaine 2013). While data are accumulating that vegetative incompatibility genes evolve under balancing selection, the selective forces have not been identified definitively. The pathogen-surveillance hypothesis for vegetative incompatibility predicts that maintaining polymorphisms increases mean fitness as a result of detecting a more diverse array of pathogens (Paoletti and Saupe 2009). The allorecognition hypothesis predicts a benefit of distinguishing self from nonself by preserving genetic integrity, and thus avoiding resource exploitation by other genotypes (Brusini et al. 2011; Czárán et al. 2014; Debets and Griffiths 1998; Nauta and Hoekstra 1994), or by reducing infection with a pathogen from another individual (Caten 1972; Cortesi et al. 2001; Debets et al. 1994; Hartl et al. 1975; van Diepeningen et al. 1997). Viruses like CHV-1 that markedly reduce the fitness of C. parasitica are logical candidates as agents of balancing selection on vic genes because virus transmission is significantly restricted between vegetatively incompatible individuals. In this study, we tried to test whether inhibition of virus transmission could be a selective factor by comparing vic allele frequencies to those expected from asymmetric virus transmission (Supplementary Information). Our results are mostly, but not completely (e.g., vic7), consistent with predictions of this model, lending support to the hypothesis that viruses may select for polymorphisms at vic loci. Therefore, this study provides evidence in favor of viruses and the allorecognition hypothesis, even though alternative mechanisms or hypotheses were not explicitly tested. The demonstrated role of heteroallelism at vic loci reducing virus transmission is not exclusive of its role in preventing resource exploitation by other conspecific genotypes (Brusini et al. 2011; Czárán et al. 2014; Debets and Griffiths 1998; Nauta and Hoekstra 1994), or the pathogen-surveillance hypothesis (Paoletti and Saupe 2009). At this time, however, we know of no way to distinguish among these hypotheses, and, for the time being, must depend instead on testing specific mechanisms, such as virus transmission, rather than definitive tests of alternative hypotheses.
Data archiving
All nucleotide sequences have been deposited in GenBank; accession numbers are in Supplementary Table S1; sequence alignments are shown in Supplementary Fig. S1. Genotype data for all six vic loci, in all 10 C. parasitica populations are in Supplementary Table S2; allele frequency estimates and statistics are in Supplementary Table S5. Results of LD analyses are in Supplementary Table S4.
Electronic supplementary material
Acknowledgements
We thank Byeong-Jin Cha, Dae-Hyuk Kim, Daniel Rigling, and Kiril Sotirovski for sharing Cryphonectria isolates. Special thank goes to Gil Choi for technical assistance.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41437-018-0060-7) contains supplementary material, which is available to authorized users.
References
- Aanen DK, Debets AJM, de Visser JAGM, Hoekstra RF. The social evolution of somatic fusion. Bioessays. 2008;30(11-12):1193–1203. doi: 10.1002/bies.20840. [DOI] [PubMed] [Google Scholar]
- Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38(suppl_2):W7–W13. doi: 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastiaans E, Debets AJM, Aanen DK, van Diepeningen AD, Saupe SJ, Paoletti M. Natural variation of heterokaryon incompatibility gene het-c in Podospora anserina reveals diversifying selection. Mol Biol Evol. 2014;31(4):962–974. doi: 10.1093/molbev/msu047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc Series B Methodol. 1995;57(1):289–300. [Google Scholar]
- Biella S, Smith ML, Aist JR, Cortesi P, Milgroom MG. Programmed cell death correlates with virus transmission in a filamentous fungus. Proc R Soc B Biol Sci. 2002;269:2269–2276. doi: 10.1098/rspb.2002.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusini J, Robin C, Franc A. Parasitism and maintenance of diversity in a fungal vegetative incompatibility system: the role of selection by deleterious cytoplasmic elements. Ecol Lett. 2011;14(5):444–452. doi: 10.1111/j.1461-0248.2011.01602.x. [DOI] [PubMed] [Google Scholar]
- Carbone I, Liu YC, Hillman BI, Milgroom MG. Recombination and migration of Cryphonectria hypovirus 1 as inferred from gene genealogies and the coalescent. Genetics. 2004;166:1611–1629. doi: 10.1534/genetics.166.4.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caten CE. Vegetative incompatibility and cytoplasmic infection in fungi. J Gen Microbiol. 1972;72:221–229. doi: 10.1099/00221287-72-2-221. [DOI] [PubMed] [Google Scholar]
- Chevanne D, Saupe SJ, Clavé C, Paoletti M. WD-repeat instability and diversification of the Podospora anserina hnwd non-self recognition gene family. BMC Evol Biol. 2010;10:134. doi: 10.1186/1471-2148-10-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi GH, Dawe AL, Churbanov A, Smith ML, Milgroom MG, Nuss DL. Molecular characterization of vegetative incompatibility genes that restrict hypovirus transmission in the chestnut blight fungus Cryphonectria parasitica. Genetics. 2012;190(1):113–127. doi: 10.1534/genetics.111.133983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerham CC, Burrows PM, Young SS, Prout T. Frequency-dependent selection in randomly mating populations. Am Nat. 1972;106(950):493–515. doi: 10.1086/282790. [DOI] [Google Scholar]
- Cortesi P, McCulloch CE, Song H, Lin H, Milgroom MG. Genetic control of horizontal virus transmission in the chestnut blight fungus Cryphonectria parasitica. Genetics. 2001;159:107–118. doi: 10.1093/genetics/159.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortesi P, Milgroom MG. Genetics of vegetative incompatibility in Cryphonectria parasitica. Appl Environ Microbiol. 1998;64:2988–2994. doi: 10.1128/aem.64.8.2988-2994.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czárán T, Hoekstra RF, Aanen DK. Selection against somatic parasitism can maintain allorecognition in fungi. Fungal Genet Biol. 2014;73:128–137. doi: 10.1016/j.fgb.2014.09.010. [DOI] [PubMed] [Google Scholar]
- Dangl JL, Jones JDG. Plant pathogens and integrated defence responses to infection. Nature. 2001;411(6839):826–833. doi: 10.1038/35081161. [DOI] [PubMed] [Google Scholar]
- Dawe AL, Nuss DL. Hypovirus molecular biology: from Koch’s postulates to host self-recognition genes that restrict virus transmission. Adv Virus Res. 2013;86:109–147. doi: 10.1016/B978-0-12-394315-6.00005-2. [DOI] [PubMed] [Google Scholar]
- Debets AJM, Griffiths AJF. Polymorphism of het-genes prevents resource plundering in Neurospora crassa. Mycol Res. 1998;102:1343–1349. doi: 10.1017/S095375629800639X. [DOI] [Google Scholar]
- Debets F, Yang X, Griffiths AJG. Vegetative incompatibility in Neurospora: its effect on horizontal transfer of mitochondrial plasmids and senescence in natural populations. Curr Genet. 1994;26:113–119. doi: 10.1007/BF00313797. [DOI] [PubMed] [Google Scholar]
- Deleu C, Clavé C, Bégueret J. A single amino acid difference is sufficient to elicit vegetative incompatibility in the fungus Podospora anserina. Genetics. 1993;135(1):45–52. doi: 10.1093/genetics/135.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutech C, Barrès B, Bridier J, Robin C, Milgroom MG, Ravigné V. The chestnut blight fungus world tour: successive introduction events from diverse origins in an invasive plant fungal pathogen. Mol Ecol. 2012;21(16):3931–3946. doi: 10.1111/j.1365-294X.2012.05575.x. [DOI] [PubMed] [Google Scholar]
- Dutech C, Fabreguettes O, Capdevielle X, Robin C. Multiple introductions of divergent genetic lineages in an invasive fungal pathogen, Cryphonectria parasitica, in France. Heredity. 2010;105(2):220–228. doi: 10.1038/hdy.2009.164. [DOI] [PubMed] [Google Scholar]
- Edwards SV, Hedrick PW. Evolution and ecology of MHC molecules: from genomics to sexual selection. Trends Ecol Evol. 1998;13(8):305–311. doi: 10.1016/S0169-5347(98)01416-5. [DOI] [PubMed] [Google Scholar]
- Enebak SA, MacDonald WL, Hillman BI. Effect of dsRNA associated with isolates of Cryphonectria parasitica from the central Appalachians and their relatedness to other dsRNAs from North America and Europe. Phytopathology. 1994;84:528–534. doi: 10.1094/Phyto-84-528. [DOI] [Google Scholar]
- Glass NL, Dementhon K. Non-self recognition and programmed cell death in filamentous fungi. Curr Opin Microbiol. 2006;9:553–558. doi: 10.1016/j.mib.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Gryzenhout M, Wingfield BD, Wingfield MJ. New taxonomic concepts for the important forest pathogen Cryphonectria parasitica and related fungi. FEMS Microbiol Lett. 2006;258(2):161–172. doi: 10.1111/j.1574-6968.2006.00170.x. [DOI] [PubMed] [Google Scholar]
- Hall C, Welch J, Kowbel DJ, Glass NL. Evolution and diversity of a fungal self/nonself recognition locus. PLoS ONE. 2010;5(11):14. doi: 10.1371/journal.pone.0014055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL, Dempster ER, Brown SW. Adaptive significance of vegetative incompatibility in Neurospora crassa. Genetics. 1975;81(3):553–569. doi: 10.1093/genetics/81.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman BI, Suzuki N. Viruses of the chestnut blight fungus, Cryphonectria parasitica. Adv Virus Res. 2004;63:423–472. doi: 10.1016/S0065-3527(04)63007-7. [DOI] [PubMed] [Google Scholar]
- Huber DH (1996) Genetic analysis of vegetative incompatibility polymorphisms and horizontal transmission in the chestnut blight fungus Cryphonectria parasitica. Ph.D. thesis, Michigan State University, East Lansing, MI
- Jacobson DJ, Beurkens K, Klomparens KL. Microscopic and ultrastructural examination of vegetative incompatibility in partial diploids heterozygous at het loci in Neurospora crasssa. Fungal Genet Biol. 1998;23(Feb.):45–56. doi: 10.1006/fgbi.1997.1020. [DOI] [PubMed] [Google Scholar]
- Kepler RM, Humber RA, Bischoff JF, Rehner SA. Clarification of generic and species boundaries for Metarhizium and related fungi through multigene phylogenetics. Mycologia. 2014;106(4):811–829. doi: 10.3852/13-319. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005;21(10):2531–2533. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Murrell B, Fourment M, Frost SDW, Delport W, Scheffler K. A random effects branch-site model for detecting episodic diversifying selection. Mol Biol Evol. 2011;28(11):3033–3043. doi: 10.1093/molbev/msr125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie JF. Fungal vegetative compatibility. Annu Rev Phytopathol. 1993;31:127–150. doi: 10.1146/annurev.py.31.090193.001015. [DOI] [PubMed] [Google Scholar]
- Liu YC, Hillman BI, Linder-Basso D, Kaneko S, Milgroom MG. Evidence for interspecies transmission of viruses in natural populations of filamentous fungi in the genus Cryphonectria. Mol Ecol. 2003;12:1619–1628. doi: 10.1046/j.1365-294X.2003.01847.x. [DOI] [PubMed] [Google Scholar]
- Liu YC, Milgroom MG. High diversity of vegetative compatibility types in Cryphonectria parasitica in Japan and China. Mycologia. 2007;99:279–284. doi: 10.1080/15572536.2007.11832587. [DOI] [PubMed] [Google Scholar]
- May G, Shaw F, Badrane H, Vekemans X. The signature of balancing selection: fungal mating compatibility gene evolution. Proc Natl Acad Sci USA. 1999;96(16):9172–9177. doi: 10.1073/pnas.96.16.9172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micali CO, Smith ML. A nonself recognition gene complex in Neurospora crassa. Genetics. 2006;173(4):1991–2004. doi: 10.1534/genetics.106.057562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milgroom MG, Cortesi P. Analysis of population structure of the chestnut blight fungus based on vegetative incompatibility genotypes. Proc Natl Acad Sci USA. 1999;96:10518–10523. doi: 10.1073/pnas.96.18.10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milgroom MG, Cortesi P. Biological control of chestnut blight with hypovirulence: a critical analysis. Annu Rev Phytopathol. 2004;42:311–338. doi: 10.1146/annurev.phyto.42.040803.140325. [DOI] [PubMed] [Google Scholar]
- Milgroom MG, Lipari SE, Powell WA. DNA fingerprinting and analysis of population structure in the chestnut blight fungus, Cryphonectria parasitica. Genetics. 1992;131:297–306. doi: 10.1093/genetics/131.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milgroom MG, Sotirovski K, Spica D, Davis JE, Brewer MT, Milev M, et al. Clonal population structure of the chestnut blight fungus in expanding ranges in southeastern Europe. Mol Ecol. 2008;17:4446–4458. doi: 10.1111/j.1365-294X.2008.03927.x. [DOI] [PubMed] [Google Scholar]
- Milgroom MG, Wang K, Zhou Y, Lipari SE, Kaneko S. Intercontinental population structure of the chestnut blight fungus, Cryphonectria parasitica. Mycologia. 1996;88:179–190. doi: 10.1080/00275514.1996.12026642. [DOI] [Google Scholar]
- Mir-Rashed N, Jacobson DJ, Dehghany MR, Micali OC, Smith ML. Molecular and functional analyses of incompatibility genes at het-6 in a population of Neurospora crassa. Fungal Genet Biol. 2000;30(3):197–205. doi: 10.1006/fgbi.2000.1218. [DOI] [PubMed] [Google Scholar]
- Murrell B, Weaver S, Smith MD, Wertheim JO, Murrell S, Aylward A, et al. Gene-wide identification of episodic selection. Mol Biol Evol. 2015;32(5):1365–1371. doi: 10.1093/molbev/msv035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah JB. Recognition and rejection of self in plant self-incompatibility: comparisons to animal histocompatibility. Trends Immunol. 2005;26(8):412–418. doi: 10.1016/j.it.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Nauta MJ, Hoekstra RF. Evolution of vegetative incompatibility in filamentous ascomycetes. I. Deterministic models. Evolution. 1994;48(4):979–995. doi: 10.1111/j.1558-5646.1994.tb05287.x. [DOI] [PubMed] [Google Scholar]
- Nuss DL. Hypovirulence: mycoviruses at the fungal-plant interface. Nat Rev Microbiol. 2005;3(8):632–642. doi: 10.1038/nrmicro1206. [DOI] [PubMed] [Google Scholar]
- Paoletti M, Saupe SJ. Fungal incompatibility: evolutionary origin in pathogen defense? Bioessays. 2009;31(11):1201–1210. doi: 10.1002/bies.200900085. [DOI] [PubMed] [Google Scholar]
- Paoletti M, Saupe SJ, Clavé C. Genesis of a fungal non-self recognition repetoire. PLoS ONE. 2007;2(3):e283. doi: 10.1371/journal.pone.0000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peever TL, Liu YC, Cortesi P, Milgroom MG. Variation in tolerance and virulence in the chestnut blight fungus-hypovirus interaction. Appl Environ Microbiol. 2000;66:4863–4869. doi: 10.1128/AEM.66.11.4863-4869.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peever TL, Liu YC, Milgroom MG. Diversity of hypoviruses and other double-stranded RNAs in Cryphonectria parasitica in North America. Phytopathology. 1997;87:1026–1033. doi: 10.1094/PHYTO.1997.87.10.1026. [DOI] [PubMed] [Google Scholar]
- Peever TL, Liu YC, Wang KR, Hillman BI, Foglia R, Milgroom MG. Incidence and diversity of double-stranded RNAs occurring in the chestnut blight fungus, Cryphonectria parasitica, in China and Japan. Phytopathology. 1998;88(8):811–817. doi: 10.1094/PHYTO.1998.88.8.811. [DOI] [PubMed] [Google Scholar]
- Powell AJ, Jacobson DJ, Natvig DO. Allelic diversity at the het-c locus in Neurospora tetrasperma confirms outcrossing in nature and reveals an evolutionary dilemma for pseudohomothallic ascomycetes. J Mol Evol. 2001;52(1):94–102. doi: 10.1007/s002390010138. [DOI] [PubMed] [Google Scholar]
- Powell AJ, Jacobson DJ, Natvig DO. Ancestral polymorphism and linkage disequilibrium at the het-6 region in pseudohomothallic Neurospora tetrasperma. Fungal Genet Biol. 2007;44(9):896–904. doi: 10.1016/j.fgb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Raymond M, Rousset F. An exact test for population differentiation. Evolution. 1995;49:1280–1283. doi: 10.1111/j.1558-5646.1995.tb04456.x. [DOI] [PubMed] [Google Scholar]
- Rayner ADM (1996) Interconnectedness and individualism in fungal mycelia. In: Sutton BC (ed) A century of mycology. Cambridge University Press, Cambridge, pp 193–232
- Richman A. Evolution of balanced genetic polymorphism. Mol Ecol. 2000;9:1953–1963. doi: 10.1046/j.1365-294X.2000.01125.x. [DOI] [PubMed] [Google Scholar]
- Rousset F. GENEPOP'007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour. 2008;8(1):103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- Saupe S, Turcq B, Bégueret J. Sequence diversity and unusual variability at the het-c locus involved in vegetative incompatibility in the fungus Podospora anserina. Curr Genet. 1995;27(5):466–471. doi: 10.1007/BF00311217. [DOI] [PubMed] [Google Scholar]
- Short DPG, Double M, Nuss DL, Stauder CM, MacDonald W, Kasson MT. Multilocus PCR assays elucidate vegetative incompatibility gene profiles of Cryphonectria parasitica in the United States. Appl Environ Microbiol. 2015;81(17):5736–5742. doi: 10.1128/AEM.00926-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonin A, Palma-Guerrero J, Fricker M, Glass NL. Physiological significance of network organization in fungi. Eukaryot Cell. 2012;11(11):1345–1352. doi: 10.1128/EC.00213-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ML, Gibbs CC, Milgroom MG. Heterokaryon incompatibility function of barrage-associated vegetative incompatibility genes (vic) in Cryphonectria parasitica. Mycologia. 2006;98(1):43–50. doi: 10.1080/15572536.2006.11832711. [DOI] [PubMed] [Google Scholar]
- Smith ML, Lafontaine DL (2013) The fungal sense of nonself. In: Kasbekar DP, McCluskey K (eds) Neurospora: genomics and molecular biology. Caister Academic Press, Norfolk
- Smith ML, Micali OC, Hubbard SP, Mir-Rashed N, Jacobson DJ, Glass NL. Vegetative incompatibility in the het-6 region of Neurospora crassa is mediated by two linked genes. Genetics. 2000;155(3):1095–1104. doi: 10.1093/genetics/155.3.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd NK, Rayner ADM. Fungal individualism. Sci Prog. 1980;66(263):331–354. [Google Scholar]
- van Diepeningen AD, Debets AJM, Hoekstra RF. Heterokaryon incompatibility blocks virus transfer among natural isolates of black Aspergilli. Curr Genet. 1997;32:209–217. doi: 10.1007/s002940050268. [DOI] [PubMed] [Google Scholar]
- Wu J, Saupe SJ, Glass NL. Evidence for balancing selection operating at the het-c heterokaryon incompatibility locus in a group of filamentous fungi. Proc Natl Acad Sci USA. 1998;95:12398–12403. doi: 10.1073/pnas.95.21.12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DX, Nuss DL. Engineering super mycovirus donor strains of chestnut blight fungus by systematic disruption of multilocus vic genes. Proc Natl Acad Sci USA. 2016;113(8):2062–2067. doi: 10.1073/pnas.1522219113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DX, Spiering MJ, Dawe AL, Nuss DL. Vegetative incompatibility loci with dedicated roles in allorecognition restrict mycovirus transmission in chestnut blight fungus. Genetics. 2014;197(2):701–714. doi: 10.1534/genetics.114.164574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.