

SUMMARY
Trypanosoma brucei is a neglected tropical disease endemic to Africa. The polyamine spermidine is essential for post-translational hypusine modification of eukaryotic initiation factor 5A (eIF5A), which is catalyzed by deoxyhypusine synthase (TbDHS). In trypanosomatids, deoxyhypusine synthase (DHS) activity is dependent on heterotetramer formation between two paralogs, DHSc and DHSp, both with minimal activity on their own due to missing catalytic residues. We determined the X-ray structure of TbDHS showing a single functional shared active site is formed at the DHSc/DHSp heterodimer interface, with deficiencies in one subunit complemented by the other. Each heterodimer contains two NAD+ binding sites, one housed in the functional catalytic site and the second bound in a remnant dead site that lacks key catalytic residues. Functional analysis of these sites by site-directed mutagenesis identified long-range contributions to the catalytic site from the dead site. Differences between trypanosomatid and human DHS that could be exploited for drug discovery were identified.
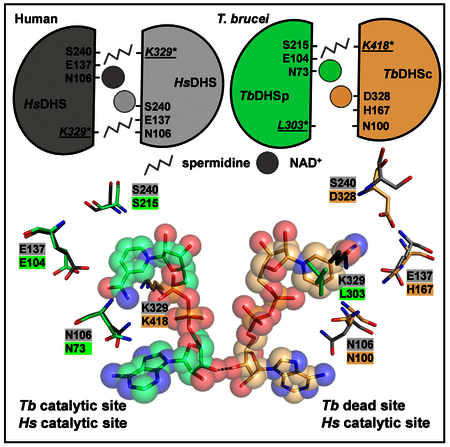
Graphical Abstract
In Brief
Afanador et al. determined the X-ray structure of an essential enzyme for survival of the eukaryotic pathogen Trypanosoma brucei. This enzyme is composed of two inactive paralogs, together forming an active heterotetramer via active-site complementation. This structure differs from the human homolog, highlighting the potential for species-selective inhibition.
INTRODUCTION
The trypanosomatids Trypanosoma brucei, Trypanosoma cruzi, and Leishmania species are eukaryotic single-cell parasites that cause human African trypanosomiasis (HAT), American trypanosomiasis (Chagas disease), and leishmaniasis, respectively. Collectively they infect 18–20 million people worldwide, yet drug therapies remain inadequate (Buscher et al., 2017; Field et al., 2017). Current treatments for HAT include Eflornithine (2-(difluoromethyl)-L-ornithine), a suicide inhibitor of the polyamine biosynthetic enzyme ornithine decarboxylase (Figure 1A). As an extracellular parasite, T. brucei requires the complete polyamine biosynthetic pathway for survival (Willert and Phillips, 2012). Trypanosomatids synthesize a unique spermidine-glutathione conjugate termed trypanothione, which is essential for redox balance in the cell (Figure 1A). They have also evolved a distinct polyamine regulatory mechanism, namely the requirement for two enzymes in the polyamine pathway to be activated by oligomerization with paralogous pseudoenzymes (Nguyen et al., 2013; Willert and Phillips, 2012).
Figure 1. Polyamine Pathway and Reaction Mechanism of DHS.

(A) Polyamine and hypusine biosynthetic pathways in trypanosomatids. Ornithine decarboxylase (ODC), S-adenosylmethionine decarboxylase (AdoMetDC), composed of a heterodimer of AdoMetDCc and AdoMetDCp, spermidine synthase (SpdSyn), eukaryotic initiation factor 5A (eIF5A), deoxyhypusine synthase (DHS), and deoxyhypusine hydroxylase (DOHH).
(B) DHS catalyzed reaction mechanism. AdoMet, S-adenosylmethionine; Put, putrescine; Spd, spermidine; DAP, diaminopropane.
(C) Chemical structure of GC7, DHS inhibitor, and non-hydrolysable spermidine analog.
Despite unique metabolic and regulatory strategies, as for all eukaryotes the polyamine spermidine is essential in trypanosomatids as a substrate for the modification of eukaryotic initiation factor 5A (eIF5A) with hypusine (Nguyen et al., 2013,2015). eIF5A is a translation elongation factor that is required to alleviate ribosome stalling in nascent proteins in all eukaryotes, including playing a critical role in translation termination (Dever et al., 2018). This activity is also present in bacteria, but it is carried out by the elongation factor P (EF-P) with a β-lysylation modification replacing hypusine (Ude et al., 2013).
Modification of eIF5A with either deoxyhypusine or hypusine is critical for its function, and this modification is universally essential in all eukaryotes (Park et al., 2010). Two enzymes are required to catalyze hypusine synthesis: nicotinamide adenine dinucleotide (NAD+)-dependent deoxyhypusine synthase (DHS) and deoxyhypusine hydroxylase (Figures 1A and1B). Inhibitors of human DHS (hDHS), including the spermidine analog 1-(7-aminoheptyl)guanidine (GC7) (Figure 1C), have been shown to affect tumor formation (Nakanishi and Cleveland, 2016) and viral replication (Mounce et al., 2017). DHS is essential for T. brucei growth and for virulence in a mouse model of HAT (Nguyen et al., 2013) and for growth of Leishmania (Chawla et al., 2010).
DHS catalyzes a four-step reaction to produce deoxyhypusine-eIF5A (Figure 1B) (Park et al., 2010). In the first step spermidine is oxidized in an NAD+-dependent reaction to form dehydrospermidine, which is attacked by an essential catalytic Lys (K329 in hDHS, Figure 2) to form a covalent intermediate. Mutation of K329 in hDHS leads to complete loss of activity (Joe et al., 1997). Transfer of this intermediate (4-aminobutyl-DHS) to a Lys on eIF5A (K50-eIF5A) followed by oxidation leads to formation of deoxyhypusine-eIF5A and regeneration of NAD+. Structural and mechanistic studies have shown that hDHS is a homotetramer, where each dimer houses two active sites at the dimer interface for a total of four NAD+ binding sites per tetramer (Umland et al., 2004).
Figure 2. Structure-Based Sequence Alignment of hDHS (PDB: 1DHS), TbDHSc, and TbDHSp.

Residues highlighted show the NAD+ binding site (yellow), spermidine/GC7 binding site (cyan), and both ligands (green) from hDHS (PDB: 1RQD). The catalytic Lys is identified with the symbol ¥. Residues targeted for TbDHSc:DHSp mutagenesis are indicated with arrows. Helices are labeled with “H” and α sheets with “B.” B1* and B3* are found only in DHSc, H1’ is only in hDHS, and H6Λ^ is in both hDHS and DHSp. Structural alignment by PROMALS3D (Pei et al., 2008). Residues not resolved in the structure are underlined.
In contrast to other eukaryotes, the genomes of both the trypanosomatid and Entamoeba species encode two DHS paralogs (Nguyen et al., 2013). In T. brucei, we showed that the active form of TbDHS is a heterotetramer formed between a catalytically impaired gene product, DHSc (c for catalytic), and a catalytically dead paralog, DHSp (p for prozyme). We coined the term prozyme (p) to describe a catalytically dead pseudoenzyme that is required to activate its catalytically impaired paralog. DHSc contains the catalytic Lys (K418) residue, while DHSp does not (replaced by L303) (Table 1 and Figure 2). Oligomerization between DHSc and DHSp resulted in a >1,000-fold increase in catalytic activity compared with DHSc activity alone. Two paralogous subunits are also required to form active S-adenosylmethionine decarboxylase (AdoMetDC), which is essential for the synthesis of spermidine (Figure 1A) (Willert and Phillips, 2012). Recently we determined the X-ray crystal structure of the inactive and active forms of T. brucei AdoMetDC, demonstrating that heterodimerization between the two AdoMetDC paralogs leads to enzyme activation through an allosteric mechanism leading to relief of autoinhibition (Volkov et al., 2016). By contrast, the structural basis for why TbDHS requires heterotetramer formation for activity has not been previously elucidated.
Table 1.
Active-Site Amino Acid Comparison between hDHS and TbDHSc:DHSp
| NAD 4-Å Shell |
TbDHSc:DHSp |
|
| hDHS | Dead Site | Catalytic Site |
| F54hʺ | L40p’ | F49c’ |
| T104h | T98c | T71p |
| S105h | A99c | S72p |
| N106ha | N100ca | N73pa |
| L107h | L101c | L74p |
| S109h | G103c | S76p |
| T131h | S161c | T98p |
| A132h | G162c | A99p |
| G133ha | G163ca | G100pa |
| E136h | E166c | E103p |
| E137ha | H167ca | E104pa |
| A235h | S323c | A210p |
| D238h | D326c | D213p |
| G239h | G327c | G214p |
| S240ha | D328ca | S215pa |
| G282h | G371c | G260p |
| G283h | G372c | A261p |
| I306h | L395c | V280p |
| N307h | N396c | T281p |
| T308h | N397c | T282p |
| A309h | A398c | G283p |
| S317h | A406c | S291p |
| A341h | S430c | G315p |
| D342h | E431c | D316p |
| A343h | V432c | A317p |
| G284h’ | G262p | G373c |
| V285h’ | L263p | V374c |
| H288h’a | H266pa | H377ca |
| D313h’ | D287p | D402c |
| S315h’ | C289p | S404c |
| D316h’ | E290p | D405c |
| S317h’ | S291p | A406c |
| GC7 4-Å Shell |
TbDHSc:DHSp |
|
| hDHS | Dead Site | Catalytic Site |
| N106h | N100c | N73p |
| R165h | H207c | R132p |
| I166h | F208c | V133p |
| G167h | G209c | G134p |
| V171h | Y213c | V138p |
| S240h | D328c | S215p |
| D243h | E331c | D218p |
| H288h’ | H266p | H377c |
| N292h’ | R270p | N381c |
| L295h’ | a | L384c |
| G314h’ | G288p | G403c |
| S315h’ | C289p | S404c |
| D316h’ | E290p | D405c |
| E323h’ | A297p | E412c |
| W327h’ | N301p | W416c |
| K329h’b | L303pa,b | K418cb |
Residues from the 4-Å shell around NAD+ or GC7 within a heterodimer. Numbering is based on the TbDHSc:DHSp structure reported herein or for hDHS, PDB: 1DHS (NAD+), or PDB: 1RQD (GC7/NAD+). Residues in bold and underlined vary between TbDHSc:DHSp and hDHS.
Each residue is followed by a letter indicating the subunit: c for DHSc, p for DHSc, h or h’ for hDHS; c’, p’, and hʺ indicate contribution from bottom dimer.
Residues contributing to the nicotinamide flip in the dead site.
The catalytic Lys or changed residue to Leu.
Herein, we report the X-ray structure of heterotetrameric T. brucei DHS (TbDHSc:DHSp), which shows that TbDHSc and TbDHSp together form a shared active site with a single functional catalytic site per heterodimer. The tetrameric enzyme is then formed from two such heterodimers. A remnant of the second active site that is observed at the dimer interface of homotetrameric DHSsfrom other species is also present at each TbDHSheterodimer interface (defined as the “dead site”), and while the dead site is missing key catalytic residues it still binds NAD+. The activesite configuration was also probed by site-directed mutagenesis. We found that DHSc contributes most of the key residues required for spermidine binding, whereas DHSp provides most of the residues required for NAD+ binding. NAD+ bound in the dead site packs against the NAD+ in the catalytic site, similarly to the hDHS structure (Umland et al., 2004). However, extensive differences in the amino acid composition of the TbDHSc:DHSp NAD+ pocket in the dead site suggest it may be possible to identify non-competitive inhibitors that would bind in the dead site and disrupt binding of NAD+ to the catalytic site. This mode of inhibition would be unique to the trypanosomatids and would provide the potential for selective inhibitor binding versus the human enzyme that could be exploited for drug discovery.
RESULTS
Modeling the TbDHSc:DHSp Active-Site Structure by Sequence Analysis
Based on the hDHS X-ray structure (PDB: 1RQD) (Umland et al., 2004), two possible models for the configuration of the TbDHSc:DHSp heterotetramer are consistent with the requirement of both DHSc and DHSp to form the active enzyme. In model 1, DHSc and DHSp form a heterodimer that contains one fully functional shared catalytic site and one “catalytically dead” site (defined as the dead site) at the dimer interface (Figure 3A). The tetramer would be composed of two such heterodimers, but could be formed by interactions between the same subunit or different subunits. In model 2 (not shown), DHSc forms a homodimer with two functional catalytic sites at the dimer interface. The tetramer would then be composed of one functional DHSc dimer and one catalytically inactive homodimeric DHSp that would be required for allosteric activation of the DHSc homodimer.
Figure 3. Tetrameric and Monomeric Configurations of TbDHSc:DHSp.

(A) Cartoon representation of hDHSand TbDHSc:DHSp. hDHS (gray)forms two equivalent functional active sites (catalytic site) in each of the two dimer interfaces of the homotetramer. For TbDHSciDHSp one catalytic site and one dead site are formed between DHSc (orange) and DHSp (green) in the dimer interface; the tetramer is composed of a dimer of heterodimers.
(B)Heterotetrameric structure of TbDHSc:DHSp as a ribbon diagram with the four NAD+ shown as spheres. Helices found only in DHSc are labeled H7 and H8.
(C) Alignment of TbDHSc and TbDHSp monomers: 1,575 atoms aligned, RMSD of 1.1 Å.
(D) Alignment of hDHS (PDB: 1DHS) and TbDHSc monomers: 1,795 atoms aligned, RMSD of 0.8 Å.
(E) Alignment of hDHS (PDB: 1DHS) and TbDHSp monomers: 1,804 atoms aligned, RMSD of 0.8 Å. See also Figure S1.
To distinguish between these models, we evaluated the amino acid sequence conservation between the trypanosomatid, human and other eukaryotic DHSs, and compared these sequences with the hDHS structure (PDB: 1RQD) to identify key residues that are essential for binding of spermidine (represented by GC7 in the crystal structure) and NAD+ (Table 1 and Figure 2). This analysis shows that amino acid residues in the hDHS NAD+ binding site are mostly conserved in DHSp but not DHSc, while the opposite is true for spermidine. We therefore hypothesized that model 1 is correct and that trypanosomatid DHSc/DHSp will form a shared active site with only a single functional catalytic site per dimer interface. Furthermore, the model predicts that the NAD+ binding site will be contributed primarily from DHSp and the spermidine binding site largely contained within DHSc. X-ray structure determination was then undertaken to provide a detailed understanding of the active-site structure in TbDHSc:DHSp.
X-Ray Crystal Structure of TbDHSc:DHSp Shows a Shared Active Site between Paralogs
The heterotetramer, TbDHSc:DHSp, was crystallized as described in STAR Methods, yielding crystals that diffracted (space group P212121) to 3.5-Å resolution (Table 2). The structure was solved by molecular replacement using the program Phaser, yielding an Rwork = 21.7% and Rfree = 26.7%. A total of three heterotetramers are present in the asymmetric unit (Figure S1A) with a root-mean-square deviation (RMSD) of <0.5 Å. Each tetramer contains two DHSc and two DHSp arranged in the shared active-site configuration (Figures 3A, 3B, and S1B). The heterotetramer is formed from a dimer of TbDHSc:DHSp heterodimers with a single catalytic site and a remnant dead site formed at each dimer interface. The structure also shows that four molecules of NAD+ are bound per tetramer, demonstrating that both the catalytic site and the dead site are able to bind NAD+. This arrangement has significant implications for the function of the catalytic site and for understanding differences with the human enzyme (discussed below).
Table 2.
Data Collection and Refinement Statistics for TbDHSciDHSp
| Data Collection | |
|---|---|
| Crystal | Native |
| PDB ID | 6DFT |
| Space group | P212121 |
| Cell constants (Å) | 64.36, 242.16, 266.30 |
| Wavelength (Å) | 0.97918 |
| Resolution range (Å) | 48.75–3.50 (3.56–3.50) |
| Unique reflections | 52,848 (2,556) |
| Multiplicity | 4.1 (3.4) |
| Data completeness (%) | 97.2 (94.4) |
| Rmerge (%)a | 18.8 (79.6) |
| Rpim (%)b | 10.1 (45.5) |
| CC1/2 (last resolution shell) | 0.65 |
| I/σ(I) | 6.4 (1.4) |
| Wilson B value (Å2) | 64.6 |
| Refinement Statistics | |
| Resolution range (Å) | 48.75–3.50 (3.59–3.50) |
| No. of reflections Rwork/Rfree | 49,148/1,956 (1,790/72) |
| Data completeness (%) | 91.5 (49.0) |
| Atoms (non-H protein) | 32,372 |
| Rwork (%) | 22.2 (30.2) |
| Rfree (%) | 26.2 (33.7) |
| RMSD bond length (Å) | 0.002 |
| RMSD bond angle (°) | 0.52 |
| Mean B value (Å2) (protein chains, A/B/C/D/E/F/G/H/I/J/K/L) | 55.6/54.6/55.8/56.7/63.7/ 62.4/60.3/58.7/68.2/64.7/ 60.1/68.1 |
| Mean B value (Å2) (NAD, A/B/C/D/E/F/G/H/I/J/K/L) | 45.6/43.5/45.7/52.6/50.5/ 53.2/51.8/46.2/59.6/55.9/ 54.9/53.2 |
| Ramachandran plot (%) (favored/additional/disallowed)c | 93.9/5.8/0.34 |
| Clashscore/overall scorec | 3.88/1.58 |
| Maximum likelihood coordinate | 0.46 |
| Error | |
Data for the outermost shell are given in parentheses.
Rmerge= 100 ΣhΣi|Ih,i - ⟨Ih⟩|/ΣhΣi ⟨Ih,i⟩, where the outer sum (h) is over the unique reflections and the inner sum (i) is over the set of independent observations of each unique reflection.
Rpim = 100 ΣhΣi[1/(nh - 1)]1/2|Ih,i - ⟨Ih⟩|/ΣhΣi ⟨Ih,i⟩, where nh is the number of observations of reflections h.
As defined by the validation suite MolProbity (Chen et al., 2010).
The overall fold of TbDHSc:DHSp is similar to hDHS (PDB: 1DHS) with an RMSD of 2.1 Å, using chains A–D from our structure. Each subunit contains the characteristic nucleotide binding (or Rossmann) fold, which houses the NAD+ binding site (Figures 3C–3E). The cores of both TbDHSc and DHSp subunits are well conserved and are formed by five parallel b sheets, similarly to hDHS. In the hDHS structure with only NAD+ bound (PDB: 1DHS) (Liao et al., 1998), the entrance of the active site is blocked by the N-terminal α helix, and this helix has been suggested to be involved in a ball-and-chain mechanism, where it moves out of the active site upon substrate binding (PDB: 1RQD, a spermidine analog was used in the structure) (Umland et al., 2004). In our TbDHSc:DHSp structure, the N terminus of DHSc is disordered and no density is observed for the first 20–23 residues (Figures 2 and 3C–3E). DHSp is shorter than DHSc and hDHS, and does not contain the residues that form the first a helix in those structures (Figures 2 and 3C–3E). DHSc is in turn 134 residues longer than hDHS with one extended helix (helix 7) and one additional helix (helix 8) accommodating the extra sequence (Figures 2, 3C, and 3D). Several flexible loops connecting secondary structural elements are not resolved in the structure (Figure 2) due to missing electron density.
The TbDHSciDHSp Heterodimer Forms a Shared Active Site with One Catalytic Site and One Dead Site at the Dimer Interface
Our structure shows that the functional unit is formed from a TbDHSc:DHSp heterodimer with the active sites located at the dimer interface. Two NAD+ molecules are found at the dimer interface: one NAD+ primarily coordinated by DHSp is bound in the catalytic site, and a second NAD+ binding site with most residues contributed from DHSc is bound in the dead site (Figures 3B and 4A–4C). The finding that NAD+ is bound to the dead site was unexpected given the observation that many residues in the dead site differ from those found in the NAD+ binding site of the TbDHSc:DHSp catalytic site and from hDHS, including several key catalytic residues (Table 1 and Figure 2). However, the NAD+ molecules within a dimer in both hDHS and TbDHSc:DHSp are positioned in close proximity (2.5 Å at the 3’-OH) making contacts between the ribose rings at the two sites (Figure 4C). Preserving NAD+ binding in the TbDHSc:DHSp dead site was likely required to maintain good NAD+ binding in the catalytically functional site. The catalytic site contains the essential catalytic Lys residue (K418-DHSc:DHSp), as well as DHSc:DHSp-E104p and D405-DHSc:DHSp (Figure 4C), which have been shown to be essential for activity in hDHS (Lee et al., 2001). These residues are contributed across the dimer interface from both DHSc and DHSp subunits to form the catalytic site (Figures 4A–4C). By contrast, in the catalytically dead site the essential Lys residue has been replaced by DHSc:DHSp-L303, and additionally many other key catalytic residues have also been substituted with other amino acids. These include the conserved paralogous residues for DHSc:DHSp-E104p and D405-DHSc:DHSp listed above for the catalytically functional active site, which have been substituted with H167-DHSc:DHSp, and DHSc:DHSp-E290, respectively, in the dead site (Table 1, Figures 2, 4B, and 4C). Interestingly, a conserved residue contributed from the opposite dimer (A’) (F54-hDHS), and positioned nearby to the NAD+ ring in monomer A, is only found in DHSc (F49-DHSc:DHSp), and is mutated to Leu in DHSp (DHSc:DHSp-L40) (Figure 4C).
figure 4. TbDHSc:DHSp Contains One Catalytic Site and One Dead Site at the Dimer Interface.

(A) Heterodimer of TbDHSc (orange) and TbDHSp (green) (ribbon diagram) with two NAD+ (spheres).
(B) Dimer cartoon showing NAD+ sites and the putative spermidine binding site (based on the hDHS structure). Amino acid residues targeted for site-directed mutagenesis (Table 2) are shown. Residues conserved in both subunits are marked by the same symbol (# or Λ). The catalytic Lys (K418) and the corresponding residue in the dead site (L303) are labeled with an asterisk (*) and underlined.
(C) NAD+ binding sites at the dimer interface of TbDHSc:DHSp. Two residues (marked by ‘) that are contributed from the opposite dimer are also shown (F49-DHSc [blue] and DHSp-L40 [magenta]).
TbDHSciDHSp Catalytic Site Configuration Is Similar to hDHS
The catalytic site composition of TbDHSc:DHSp was compared with hDHS (PDB: 1DHS). NAD+ in the catalytic site of TbDHSc:DHSp overlays closely with the position of NAD+ in hDHS (Figure 5A). Most residues in the NAD+ binding site of the catalytic site are contributed from DHSp. Residues that contact the nicotinamide ring of NAD+ are strongly conserved with hDHS including H377-DHSc:DHSp, DHSc:DHSp-N73, DHSc:DHSp-G100, and DHSc:DHSp-E104 (Table 1 and Figure 5A). Similarly to hDHS, the nicotinamide ring also makes an H bond with the backbone of DHSc:DHSp-N73 and DHSc:DHSp-G100 (Figures 5A and 6A). However, in the adenine binding site, five differences in amino acid sequence between the TbDHSc:DHSp catalytic site and hDHS were identified, including TbDHSc:DHSp-A261 (Gly282-hDHS), TbDHSc:DHSp-G283 (A309-hDHS), TbDHSc: DHSp-G315 (A341-hDHS), TbDHSc:DHSp-T281 (N307-hDHS), and TbDHSc:DHSp-V280 (I306-hDHS) (Figure 5A).
Figure 5. Comparison of the TbDHSc:DHSp and hDHS.

(A and B) Comparison of TbDHSc:DHSp and hDHS catalytic sites. (A) NAD+ binding site, in the same orientation as the catalytic site in Figure 4C. TbDHSc:DHSp and hDHS (PDB: 1DHS) tetramers were superimposed in PyMol using only TbDHSp with 283 atoms aligned to an RMSD = 0.74 Å. Amino acid differences between hDHS and TbDHSare underlined. (B) GC7 binding site. This view is rotated around the vertical axis by approximately 215° and translated approximately 6 Å from (A), as indicated. TbDHSc:DHSp and hDHS (PDB: 1RQD) were superimposed as above to RMSD = 0.8 Å. hDHS is in gray, TbDHSc is in orange, and TbDHSp is in green. GC7 is displayed as gray sticks. The NAD+ is displayed as spheres.
(C) Comparison of the TbDHSc:DHSp dead site with the hDHS catalytic site. The hDHS spermidine binding pocket was aligned with the TbDHSc:DHSp dead-site pocket. Select residues within the 4-Å shell of bound ligands are shown.
Figure 6. Comparison of NAD+ and GC7 Binding Sites between TbDHSc and TbDHSp Sites.

(A) NAD+ binding pocket in the catalytic site.
(B) NAD+ binding pocket in the dead site. NAD+ (spheres and lines), TbDHSc (orange sticks), and TbDHSp (green sticks). F49-DHSc (blue) and DHSp-L40 (magenta) are from the opposite dimer.
(C) Overlay of the catalytic and dead sites highlighting the nicotinamide flip (red arrows).
(D) Polder omit map (blue mesh) for NAD (Liebschner et al., 2017) contoured at 4× the RMS level. The map was calculated in the program suite Phenix (Adams et al., 2010) by omitting the coordinates for NAD+.
(E) GC7 binding pocket in the catalytic site. GC7 (gray sticks) is from hDHS (PDB: 1RQD) based on the superimposition described in Figure 5B.
(F) Dead-site amino acids in the region of the GC7 site showing the highly variable amino acid composition relative to the catalytic site. Orientation is similar to Figure 5B and (E).
To understand the binding of spermidine in the catalytic site, we superimposed the hDHS structure in ternary complex with NAD+ and GC7 (PDB: 1RQD, Figure 5B) with our structure. We found that the TbDHSc:DHSp catalytic site retains the residues needed to bind GC7/spermidine. Similar to hDHS, DHSc: DHSp-D218 is predicted to make a salt bridge with the amino group of GC7 and an H bond with N381-DHSc:DHSp. A conserved Trp residue (W416-DHSc:DHSp) sits near the backbone of GC7. The GC7 guanyl is predicted to form an H bond with E412-DHSc:DHSp, G403-DHSc:DHSp, and S404-DHSc: DHSp, and the catalytic Lys (K418-DHSc:DHSp) is positioned in the correct orientation pointing toward GC7 (Figure 5B). Only one species amino acid difference is observed in the GC7 site, where DHSc:DHSp-V133 is substituted for I166-hDHS.
The Dead-Site NAD+-Binding Pocket: Variable Amino Acids Alter the Conformation of the Nicotinamide Ring
NAD+ in the dead site assumes a more elongated configuration than in the TbDHSc:DHSp catalytic site or the active sites in hDHS (PDB: 1DHS) due to rotation of the nicotinamide relative to its position in the catalytic site (Figures 6A–6D). These conformational differences in the NAD+ binding mode are conserved across all six TbDHSc:DHSp dead sites and all six catalytic sites in the asymmetric unit. The average distance between the adenine-6-amino and the nicotinamide amide carbon in the dead site is 13.1 ± 0.3 Å, compared with an average distance in the catalytic site of 10.5 ± 0.5 Å, similar to the hDHS distance of 10.1 Å (Figure 6D). Amino acid differences between the two sites contribute to the repositioning of NAD+ in the dead site (Table 1; Figures 6B and 6C). First, an invariant Glu (E137-hDHS and TbDHSc:DHSp-E104) residue found in the catalytic site has been replaced with His (H167-DHSc:DHSp) in the dead site. The side chain of H167-DHSc:DHSp occupiesthe position of the amide portion of the nico-tinamide in the catalytic site (Figure 6B) causing the nicotinamide ring to flip in the dead site. H167-DHSc:DHSp is stabilized in this position by H-bonding with the side chain of N100-DHSc:DHSp, which flipped toward the NAD+ relative to the position of this residue in the catalytic site (e.g., TbDHSc:DHSp-N73 or N106-FhDHS) (Figures 6B and6C). The dead-site position of the nicotinamide ring is further stabilized by H-bonding with G163-DHSc: DHSp, and the side chain of D328-DHSc:DHSp (which has replaced DHSc:DHSp-S215 and S240-hDHS in the catalytic site) and DHSc:DHSp-H266. It is likely that these changes in amino acid composition in the NAD+ binding pocket ofthe dead site will translate to differences in binding affinity of NAD+ in comparison with the catalytic site. The observed reorientation of the nicotinamide might also contribute to the lack of activity of the dead site, although loss of the catalytic Lys (K418-DHSc:DHSp is replaced by DHSc:DHSp-L303) at the dead site is sufficient to account for the inactivity (Figures 5B, 5C, 6E, and 6F).
The TbDHSc:DHSp Dead Site Is Unlikely to Bind Spermidine
While it is clear that the dead site will be unable to catalyze for-mation of the required covalent enzyme-substrate intermediate because of the lack of the catalytic Lys residue, we sought to un-derstand whether the dead site might still bind a ligand in the spermidine site. Structural overlay of the TbDHSc:DHSp dead site with the GC7/spermidine binding site of hDHS (PDB: 1RQD) (Table 1 and Figure 5C) or with the TbDHSc:DHSp cata-lytic site (Figures 6E and 6F) shows that while a potential ligand binding cavity is present in the dead site it has a significantly altered amino acid composition. The observed amino acid changes in dead-site pocket are likely to be incompatible with the binding of spermidine. These differences include introduce-tion of a positive charge (DHSc:DHSp-R270) near where the amino group of GC7 binds in hDHS, the replacement of a conserved Asp (D243-hDHS) with a larger residue (E331-DHSc:DHSp) that would restrict the pocket size, replacement of the conserved Glu reside (E323-hDHS) with an Ala (DHSc:DHSp-A297) that is predicted to eliminate a key salt bridge interaction with the a primary amino group of spermidine (guanyl in GC7), and replacement of an invariant Trp residue (W327-hDHS) that stacks against the aliphatic portion of GC7/spermidine with Asn (DHSc:DHSp-N301) (Figures 5C, 6E, and 6F). This Asn residue H-bonds with DHSc:DHSp-E290 (Figure 6F), which is an invariant Asp residue in the catalytic sites (D405-DHSc:DHSp and D316-hDHS) that H-bonds to the catalytic Lys (K418-DHSc:DHSp and K329-hDHS) (Figures 5B and 6E).
Functional Studies to Assess the Contributions of Amino Acid Residues in DHSc and DHSp to Catalysis
To provide functional evidence that TbDHSc:DHSp form a shared active site and to probe the role of NAD+ binding to the dead site in the function ofthe catalytic site, we designed a series of Ala mutagenesis experiments. Given that TbDHSc:DHSp still binds four NAD+ molecules, we wanted to assess whether the naturally occurring amino acid changes in the dead site impair enzyme activity. To this end we targeted residues that have previously been shown to be essential for hDHS activity (Lee et al., 2001) and that spanned residues in both the NAD+ and putative spermidine binding sites (Table S1). Two sets of matched pairs where the same residue was mutated in both the catalytic (DHSc:DHSp-E103” and H377A-DHSc:DHSp#) and dead site (E166-DHSc: DHSpA and DHSc:DHSp-H266#) were selected, the symbols indicating the structurally homologous partner residue (Figure 4B). Additional residues that were mutated only in the cata-lytic site were D405-DHSc:DHSp, DHSc:DHSp-D218A, and DHSc:DHSp-E104. Mutant enzymes were copurified in complex with wild-type (WT) counterparts so that each heterotetramer contained a single mutation per dimer. TbDHSc:DHSp complex formation was confirmed for each mutant based on the finding that DHSc and DHSp copurified over the Ni+2-agarose column despite the fact that only DHSc contains a His-6 tag. The pres-ence of both proteins in the complex was verified by SDS-PAGE analysis. We determined the melting temperature (Tm) of all mutants using a thermal shift assay and found that the Tm ranged between 51.1°C and 57.6°C, with that of WT being 56.0°C (Table S2). While most mutant enzymes had a Tm similar to that of WT, E166A-DHSc:DHSp had a DTm of 4.9°C less than that of WT. While this is a relatively large shift in Tm that suggests the E166A mutant has impaired stability, this mutant showed cat-alytic efficiency similar to that of WT, so the reduced stability did not translate to an effect on catalytic efficiency (see below).
Single-Turnover Fluorescence Assay for NADH Formation
DHS catalyzes a reaction that proceeds through four catalytic steps (Figure 1B). The formation of NADH in the first step can be monitored by a single-turnover fluorescence assay (Wolff et al., 2000). NADH buried in the active site is fluorescent at the wavelength used (excitation λ 350 nm; emission l 441 nm), whereas NAD+ is not. Addition of 1 mM spermidine to a mixture of WT TbDHSc:DHSp complex (1 μM) in the presence of 1 mM NAD+ led to a rapid burst of fluorescence due to formation of NADH (Figure 7A). For the WT enzyme the reaction appears to be 100% complete in the dead time (~30 s) of the experiment. GC7 was previously shown to inhibit the TbDHSc:DHSp hetero-tetramer with an IC50 of 1.5 mM (Nguyen et al., 2013). GC7 (10 mM) inhibited the conversion of NAD+ to NADH in the fluores-cence assay as measured by incubation of the ternary complex of TbDHSc:DHSp/NAD+/GC7 for 5 min prior to addition of sper-midine (Figure 7A). To follow the reoxidation of NADH, we subsequently added 15 mM eIF5Ato both the uninhibited and inhibited TbDHSc:DHSp/NADH/spermidine mixture, which led to a rapid decrease in fluorescence due to the oxidation of NADH to NAD+ (Figure 7A). However, because all substrates are now pre-sent, the enzyme immediately converts NAD+ back to NADH, reaching equilibrium. The magnitude of the change was greater for the uninhibited reaction, whereas in the presence of GC7 much less NADH had formed to be converted back to NAD+ over the time course of our observation.
Figure 7. Single-Turnover and Steady-State Kinetic Analysis of TbDHSc:DHSp WT and Mutant Enzymes.

(A) Single-turnover NADH formation for WT TbDHSc:DHSp. NADH formation was monitored by fluorescence under single-turnover conditions for excitation at 350 nm and emission at 441 nm. Spermidine (Spd) (1 mM) was added to enzyme (1 μM) and NAD+ (1 mM) or eIF5A (15 μM) at the indicated time (orange line). Pre-incubation of TbDHSc:DHSp with 10 μM GC7 inhibits the reaction (black line). Experiments were done in duplicate and traces represent the average of the duplicate.
(B) Single-turnover NADH formation for TbDHSc:DHSp mutant enzymes. Mutant enzyme pairs are identified with symbols (Λ and #) as shown in Figure 4C.
(C) Steady-state kinetic analysis of WT and mutant DHS activity. For WT and active mutants, [DHS] = 5 nM, while inactive mutants were assayed at 1 μM. Substrate concentrations were 1 mM NAD+, 78 μM spermidine, and 15 μM eIF5A (KM = 0.7 μM), which represent saturating concentrations for the WT enzyme (Nguyen et al., 2013). Data were collected in triplicate, and error bars represent SEM. See also Tables S1 and S2.
Effects of Mutations on Single-Turnover Formation of NADH
Analysis of the mutant enzymes using the aforementioned sin-gle-turnover fluorescent assay showed that both enzymes that contained mutations in the dead site, E166A-DHSc:DHSp and DHSc:DHSp-H266A, displayed activity similar to that of WT TbDHSc:DHSp (Figure 7B), confirming that the dead site does not catalyze the single-turnover reaction on its own. Both mutant enzymes showed a burst of fluorescence in the dead time of the reaction (~30 s). These mutants reached a maximum fluorescence at a different value than the WT enzyme, likely due to differences in the hydration of NADH which could affect the fluorescence signal intensity. By contrast, all enzymes that con-tained a mutation in the catalytic site were impaired in the single-turnover reaction. DHSc:DHSp-D218A (Figure 6C, left panel) was unable to catalyze the single-turnover formation of NADH, consistent with its known role in binding and orienting spermidine in hDHS (Lee et al., 2001). The other mutant enzymes (D405A-DHSc:DHSp, DHSc:DHSp-E103A, DHSc:DHSp-E104A, and H377A-DHSc:DHSp), while impaired, retained some ability to slowly catalyze the single-turnover reaction (Figure 7B). The mutated residues in DHSc:DHSp-E103A, DHSc:DHSp-E104A, and H377A-DHSc:DHSp are in the NAD+ site, with DHSc: DHSp-E104A H-bonding with the nicotinamide in NAD+. Of these, D405A-DHSc:DHSp showed the most significant NADH turnover, albeit at a slower rate (discussed below).
Steady-State Kinetic Analysis of the Mutant Enzymes
To quantify the effects of the mutations, we next determined the specific activity of the mutant enzymes by steady-state kinetics (Figure 1B). For WT TbDHSc:DHSp the KM values for spermidine and NAD+ were determined to be 9.8 μM and 1.3 μM, respectively (Figure 7C and Table 3). Amino acid mutations in the dead site (E166A-DHSc:DHSp and DHSc:DHSp-H266A) had no impact on the specific activity relative to WT TbDHSc:DHSp, again confirming that the site lacks a catalytic function, whereas the mutation of residues in the catalytic site led to minimally a 1,000-fold reduction in specific activity compared with WT TbDHSc:DHSp. Since the mutants DHSc:DHSp-E103A, H377A-DHSc:DHSp, D405A-DHSc:DHSp, and DHSc:DHSp-E104A retained some activity in the fluorescence assay, the complete loss of activity in the steady-state assay suggests that these residues play a role in later reaction steps that occur after the initial oxidation of NADH (Figure 1B), e.g., transfer of the 4-aminobutyl group to eIF5A, or formation of deoxyhypusine-eIF5A coupled with NADH reoxidation. For D405A-DHSc:DHSp, an H bond with the catalytic Lys (K418-DHSc:DHSp) is observed (Figure 6E). In hDHS, these analogous mutant enzymes retained partial binding of NAD+, but were unable to cleave spermidine (step 2 in Figure 1B; formation of diaminopropane) (Lee et al., 2001).
Table 3.
Steady-State Kinetic Parameters for TbDHSc:DHSp and Mutants
| NAD+ |
Spermidine |
|||
|---|---|---|---|---|
| kcat (s−1) | KM (μM) | kcat(s−1) | KM (μM) | |
| WT | 0.15 ± 0.0045 | 1.3 ± 0.18 | 0.18 ±0.010 | 9.8 ± 1.2 |
| E166A-DHSc:DHSp | 0.16 ± 0.00057 | 1.2 ±0.20 | 0.12 ± 0.010 | 11 ± 1.5 |
| DHSc:DHSp-H266A | 0.16± 0.010 | 98 ± 26 | 0.081 ± 0.01 | 12 ± 1.8 |
Experiments were conducted at 5 nM DHS and 15 μM TbeIF5A. Substrate concentrations were: (1) NAD+ KM, NAD+ (1.3–1,000 μM) and [14C] spermidine (78 μM); (2) spermidine KM, NAD+ (1 mM) and [14C]spermidine (0.12–78 μM). Error represents the SE of the fit for triplicate data.
For mutant enzymes that showed activity in the steady-state assay similar to WT, we performed a substrate titration to determine the Michaelis-Menten parameters (Table 3). The kcat values for E166A-DHSc:DHSp and DHSc:DHSp-H377A were similar to that of WT using either spermidine or NAD+ as the titrated substrate, which was likewise true for the KM of spermidine. By contrast, while the KM for E166A-DHSc:DSHp versus NAD+ was similar to WT, the KM for NAD+ of DHSc:DHSp-H266A increased 80-fold from 1.3 μM to 98 μM. These data suggest that binding of NAD+ to the dead site (which contains DHSc:DHSp-H266A) plays a significant role in promoting binding of NAD+ to the catalytic site.
DISCUSSION
Eukaryotic genomes contain many examples of pseudoenzymes, which are enzyme paralogs that lack catalytic residues (Murphy et al., 2017a, 2017b). Gene duplication provides the drive for pseudoenzyme formation. Pseudoenzyme functions continue to be discovered, but many have been found to regulate their paralogous enzymes, and these mechanisms are particularly common in the kinase and protease enzyme families (Kung and Jura, 2016; Reynolds and Fischer, 2015). Our finding that two enzymes in the trypanosomatid polyamine pathway require oligomerization with catalytically dead paralogous pseudoenzymes for activity represents one of the most interesting examples of pseudoenzyme regulation (Nguyen et al., 2013; Volkov et al., 2016).
Here, we demonstrate by structural and mutagenesis studies that TbDHSc:DHSp enzymatic activity is dependent on formation of a heterodimer between the pseudoenzyme paralogs TbDHSc and TbDHSp, which restores one functional shared catalytic site across the dimer interface. The tetrameric enzyme is then formed as a dimer of the functional heterodimer. The TbDHSc subunit primarily contributes residues to form the spermidine binding site, including the catalytic Lys residue, while TbDHSp contributes most of the residues needed for NAD+ binding within the catalytic site. The second adjacent site (dead site) is still able to bind NAD+, but it is a remnant of the catalytic site that lacks essential residues and is inactive, as confirmed by our mutagenesis studies. However, binding of NAD+ to the dead site is required to promote NAD+ binding to the catalytic site. The dead site has many amino acid differences in the NAD+ binding site that would be predicted to alter affinity for the cofactor relative to the active site, and these differences could contribute to a strategy to identify species-specific inhibitors of the trypanosomatid enzymes for drug discovery.
The structural basis for pseudoenzyme regulation has been studied in several systems, uncovering an array of strategies for both activation and inhibition of their paralogous counterpart. Most examples studied to date describe allosteric activation by heteromerization, such as is the case for T. brucei AdoMetDC, where heterodimerization with the pseudoenzyme causes structural changes that relieve autoinhibition (Volkov et al., 2016). However, other pseudoenzyme functions include protein localization/scaffolding, substrate competition (e.g., the chitinase pseudoprotein YKL-39) (Murphy et al., 2017a, 2017b), or dominant negative interactions (e.g., pseudokinases JAK2 and TYK2 [Saharinen and Silvennoinen, 2002]). The mechanism of psuedoenzyme regulation in TbDHSc:DHSp differs from these examples, and instead of the situation where one paralog retained catalytic residues while the other lost them, each TbDHS paralog has lost catalytic residues, and thus they depend on each other to retain catalytic activity through formation of a shared active site. Another example of shared active-site complementation has been described for the AdoMetDC/SpdSyn gene fusion protein from Tetrahymena thermophile, although structural data is not yet available (Li et al., 2015).
Shared active sites are a common structural feature of many enzymes in addition to DHS (Grishin and Phillips, 1994), and this feature lends itself to evolution of the type of regulation observed for TbDHS. Starting from the shared active-site structure of an ancestral DHS, a simple case of gene duplication followed by evolution of point mutations could have led to the requirement for a heterotetrameric DHS in trypanosomatids to retain enzyme activity, despite the fact that each gene copy had lost the ability to encode an active enzyme on its own. Retention of defective gene duplicates with different amino acid substitution due to mutual codependence to form a shared active site is a process that has been termed gene subfunctionalization (Force et al., 1999). This mechanism is in contrast to the more complex evolutionary pathway that was required to generate the allosteric regulation uncovered for trypanosomatid AdoMetDC (Volkov et al., 2016).
The shared active-site configuration observed in the TbDHSc:DHSp structure was also confirmed by mutagenesis. Residues that were mutated in the catalytic site led to loss of activity, while those mutated in the dead site did not affect activeity, with the exception of DHSc:DHSp-H266A, which affected the KM for NAD+ (discussed below). In the catalytic site, the equivalent residue to H377-DHSc:DHSp in the human structure has been proposed to be the hydride acceptor in the first reaction step (Umland et al., 2004) (Table S1), and our data support this role (Figure 1B). DHSc:DHSp-E103, DHSc:DHSp-E104A, DHSc:DHSp-D218A, and D405A-DHSc:DHSp are all predicted to be in the spermidine/GC7 binding site of the TbDHSc:DHSp catalytic site based on overlaying the GC7 position from the human structure. Mutation of all four of these residues leads to loss of catalytic efficiency in the steady-state reaction and to either a slowed or complete loss of NAD+ reduction in the first step. In our structure D405-DHSc:DHSp forms an H bond with the catalytic Lys (K418-DHSc:DHSp), implying that it could be acting as the catalytic base.
The structure of TbDHSc:DHSp also reveals key differences in the active site relative to the human enzyme that might be exploited for the development of species-selective inhibitors. In both human and T. brucei DHS the two NAD+ binding sites are in van der Waals contact with each via the ribose ring. However, in TbDHSc:DHSp, the dead site has many amino acid differences when compared with hDHS and or the T. brucei catalytic site. These differences alter the NAD+ site as well as the spermidine/GC7 site, making it unlikely to bind spermidine. The amino acid differences in residues that interact with NAD+ in the dead site have caused a rotation of the nicotinamide portion of the cofactor. This rotation is stabilized by a different H-bond network than observed in the active sites of TbDHSc:DHSp and hDHS. Based on these differences it is unlikely that the affinity for NAD+ at the catalytic site is the same as the dead site, and consequently both sites will likely bind smallmolecule inhibitors with different affinity and, thus, selectively. The spermidine/GC7 binding site in the dead site is almost completely gone. While there is a small cavity, most residues needed for binding and catalysis are missing, with several changes to invariant residues (Figure 5C).
Our data also show that perturbations in the T. brucei DHS dead site can affect the functioning of the catalytic site. Mutation of DHSc:DHSp-H266 to Ala in the dead site led to an 80-fold increase in the KM for NAD+. DHSc:DHSp-H266 forms an H bond with the carboxyl group of the nicotinamide ring in the dead site, an interaction that is lost in the mutant enzyme and which would be predicted to disrupt dead site NAD+ binding. The finding that this mutation alters the KM for NAD+ suggests that reduced affinity for NAD+ at the dead site also affects binding of NAD+ in the catalytic site. These data provide a clear path forward for the identification of species-selective inhibitors through the identification of small molecules that bind to the dead site and affect the ability of the catalytic site to bind NAD+. Whether it will be sufficient to identify compounds that are purely non-competitive (binding only to the dead site), or whether hybrid molecules that span both pockets will be needed, remains to be determined. However, either approach can exploit the species selectivity that will be gained from binding at the dead site. Comparison of the TbDHSc:DHSp catalytic site with the human active site also identified five differences in the NAD+ binding site near the adenine ring. These changes would be predicted to alter the volume of the pocket and could provide the potential for selective competitive inhibitors to be identified in addition to the strategy described above for identification of non-competitive inhibitors.
Because DHS is essential in all eukaryotes, the potential for selective inhibition is a key criterion to demonstrate the feasibility of targeting this enzyme for drug discovery. While DHS from most eukaryotes share the same homotetrameric structure as the human enzyme, suggesting that this may be a difficult task in these cases, the enzymes from all three pathogenic trypanosomatids and from Entamoeba share the heterotetrameric structure where active DHS is formed by two catalytically impaired paralogs (Nguyen et al., 2013). Thus, our data suggest that DHS from these parasitic species will be amenable to the identification of species-selective inhibitors, and provide the potential for the identification of a pan-trypanosomatid drug. The X-ray structure of TbDHSc:DHSp reported here provides a structural basis for such an effort.
STAR★METHODS
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Margaret Phillips (margaret.phillips@utsouthwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
T. brucei Genes
T. brucei genes used in this work can be found in TriTrypDB: Tb927.10.2750 (TbDHSc); Tb927.1.870 (TbDHSp); and Tb11.03.0410 (TbeIF5A).
Cloning
The final plasmid for the co-expression of T. brucei DHSc and DHSp in E. coli, pETDuet-TbDHSc:DHSp was generated by excising His-SUMO-DHScfrom pE-SUMO-TbDHSc (Nguyen et al., 2013) and cloning of this fragment into the pETDuet1 (EMD Millipore) multiple cloning site 1 (MCS1) using NcoI and HindIII restriction sites; DHSp was PCR amplified (see primers in Table 2) from pE-SUMO-TbDHSp and ligated into the MCS2 using NdeI and XhoI. The final construct was confirmed by sequencing. In this construct DHSc contains a His-SUMO N-terminal tag, whereas DHSp has no tag. The DHS sequences were previously codon optimized. TbeIF5A was PCR amplified from T. brucei single marker genomic DNA and cloned into pET28b (Novagen) using BamHI and SalI. The final plasmid, pET28b-TbeIF5A, was verified by sequencing and contains a N-terminal His-tag.
Plasmids encoding mutant DHSs were generated by Quick Change site-directed mutagenesis of pETDuet-TbDHSc:DHSp (primers listed in Table 2), except for H377A-DHSc:DHSp, for which the full gene was synthesized by Genscript, pUC57-His-SUMO-H377A-DHSc:DHSp. The insert was excised using NcoI and HindIII, and ligated to MCS1 of the original plasmid. All mutations were confirmed by sequencing.
METHOD DETAILS
Multiple Sequence Alignment
TbDHS amino acid sequences were obtained from TriTrypDB using the above accession numbers. hDHS (GenBank: AAA96151.1) sequence was obtained from PDB: 1RQD. Yeast DHS (GenBank: AJU19156.1) was obtained by NCBI BlastP using hDHS as input sequence. Entamoeba hystolitica (DHSc: NCBI: XP_653614; DHSp: NCBI: XP_653426) sequences were obtained by NCBI BlastP using TbDHSc and TbDHSp as input. Sequences were aligned with Clustal Omega (version 1.2.2).
Protein Expression and Purification
Expression plasmids were transformed into T1 phage resistant E. coli BL21 (DE3) cells for expression of the various TbDHSc:DHSp constructs. For expression of the TbDHSc:DHSp complex, and mutants, cells were selected with ampicillin (100 μg/mL) and grown to A600 of 0.6 at 37°C. Protein expression was induced with 0.5 mM IPTG and grown for 3 hr at 37°C. Cells were harvested by centrifugation and pellets frozen at −80°C. For expression of the substrate, TbeIF5A, cells were selected with kanamycin (50 μg/mL) and grown to A600 of 0.6 at 37°C. Protein expression was induced with 0.2 mM IPTG and cells were grown for 16 h at 16°C. Cells were harvested by centrifugation and frozen at −80°C.
TbDHSc:DHSp expressing bacterial pellets were resuspended in Buffer A (25 mM HEPES, 200 mM NaCl, 10 mM imidazole, 5% glycerol, 0.5% CHAPS, pH 8.0) with 2 mM phenylmethylsulfonyl fluoride (PMSF) and lysed by high pressure disruption (EmulsiFlex-C5, Avestin). Lysate was clarified by centrifugation (15,000xg for 0.5 hours) and the soluble fraction was purified by Ni2+-affinity column chromatography (HiTrap Chelating HP Column, GE Healthcare). TbDHSc:DHSp complex was eluted using a linear gradient of 50 mM to 500 mM of imidazole in Buffer A. The His-SUMO N-terminal tag on DHSc was removed by proteolysis with the yeast SUMO protease, Ulp1 by overnight incubation at 4°C using 5 μg/mL Ulp1 (Nguyen et al., 2013). The mixture was diluted in Buffer A to reduce the imidazole concentration and the cleaved protein was applied to a second Ni2+-column and the flow through collected. Proteins were visualized by SDS-PAGE for purity, and to assess the extent of complex formation. WT TbDHSc:DHSp complex was further purified by gel filtration chromatography on a Superdex 200 Prep Grade (GE Healthcare) using 10 mM HEPES, 100 mM NaCl, pH 7.5.
TbeIF5A expressing bacterial pellets were resuspended in Buffer B (25 mM HEPES, 150 mM NaCl, 0.5% IGEPAL, 5% glycerol, 10 mM imidazole, pH 8.0) with 2 mM PMSF, and lysed and clarified as described for TbDHSc:DHSp. The soluble fraction was run through a Ni2+-affinity chromatography column and protein eluted using a linear gradient of 50 mM to 500 mM of imidazole in Buffer B. TbeIF5A was further purified by anion exchange chromatography using a Q-column (HiTrap Q HP, GE Healthcare). Buffer used for this purification was 25 mM HEPES, 0.5% IGEPAL, 5% glycerol, pH 8.0; and protein was eluted from this column with a salt gradient, 0-500 mM NaCl in the same buffer. Finally, the protein was purified by gel filtration chromatography in 25 mM HEPES, 150 mM NaCl, 5% glycerol, pH 8.0 using the column described above.
TbDHSc:DHSp was quantified at A280 using heterotetrameric values for TbDHSc:DHSp and an extinction coefficient of 144.6 cm−1mM−1 as calculated using ProtParam on ExPASy. TbeIF5A was quantified using a Bradford assay (Bio-Rad).
Crystallization, Data Collection and Structure Determination
The WT TbDHSc:DHSp heterotetramer was crystallized at 20°C using the sitting drop vapor-diffusion method with a reservoir solution containing 0.1 M HEPES (pH 7.0), 18% PEG6000, and the drop was at a 1:1 ratio (2 μL total) of reservoir solution and protein. The protein, 22 mg/mL, was pre-equilibrated with 2 mM NAD+ and 10 mM TCEP for 10 minutes at room temperature, and the resulting crystal form was plate cluster, which were crushed and used for microseeding. A new reservoir containing 0.1 M HEPES (pH 7.0), 8% PEG6000 was used. The drop contained 1 μL of protein as described above and 1 μL of reservoir solution, and was equilibrated for 48 hours at 20°C at which time 0.2 μL of seeds were added. Crystals were cryo-protected with reservoir solution supplemented with 20% ethylene glycol and then flash-cooled in liquid nitrogen.
Diffraction data were collected at beamline 19-ID (SBC-CAT) at the Advanced Photon Source (Argonne National Laboratory, Argonne, Illinois, USA). Native crystals diffracted to a minimum Bragg spacing (dmin) of 3.50 Å and exhibited the symmetry of space group P212121 with cell dimensions of a = 64.4 Å, b = 242.2 Å, c = 266.3 Å and contained three heterotetramers (i.e., six TbDHSc and six TbDHSp) per asymmetric unit. Data were processed in the program HKL-3000 (Minor et al., 2006) with applied corrections for the effects resulting from absorption in a crystal and for radiation damage (Borek et al., 2003; Otwinowski et al., 2003), the calculation of an optimal error model, and corrections to compensate the phasing signal for a radiation-induced increase of non-isomorphism within the crystal (Borek et al., 2010, 2013). These corrections were crucial for successful phasing. Crystals of TbDHSc:DHSp displayed mildly anisotropic diffraction and while 98% complete to 4.2 Å resolution, diffraction rapidly fell off in intensity to the highresolution limit of 3.50 Å resolution. Phases for TbDHSc:DHSp were obtained by molecular replacement in the program Phaser (McCoy et al., 2007). The search model was generated by formation of a heterotetramer TbDHSc:DHSp based on the crystal structure of homodimeric human DHS (PDB id code 1DHS) (Liao et al., 1998). First, we used the Sculptor program in the CCP4 suite (Winn et al., 2011) to generate a model for each monomer, DHSc and DHSp, based on the human structure. The resulting models were aligned to the homotetrameric human structure using the program PyMol (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.). Several iterative rounds of density modification in the program Parrot (Cowtan, 2010) followed by automated model building in the program Buccaneer (Cowtan, 2006) yielded a model for TbDHSc:DHSp with 4,011 residues (83.3% of total) built and primary sequence assigned. The overall fold and secondary structural elements of TbDHSp strongly match that of the human DHS monomer. Confirmation of the structure and sequence assignment of TbDHSc was determined by comparing the secondary structure of TbDHSc to TbDHSp, and inspection of difference electron density maps. Completion of this model was performed by manual rebuilding in the program Coot (Emsley et al., 2010). Refinement was performed in the program Phenix (Adams et al., 2010) with non-crystallographic symmetry and secondary structure restraints in the initial rounds of refinement, as well as individual coordinate and isotropic atomic displacement parameter refinement. In the final rounds of refinement, the non-crystallographic symmetry and secondary structure restraints were removed. The final model for TbDHSc:DHSp (Rwork = 21.7%, Rfree = 26.7%) contained twelve protein chains, 4,149 residues (86.1% of total) and twelve molecules of NAD+. Fourteen residues (0.34%) are outliers in a Ramachandran plot as defined in the program MolProbity (Chen et al., 2010). The rather high Rfree value is likely due to the large percentage (~14%) of disordered protein chains in the crystalline lattice. Data collection and structure refinement statistics are summarized in Table 2.
Structural Comparisons
Structural RMSD of the tetramers was calculated using TM-Align (Zhang and Skolnick, 2005). The structural alignments for active site figures were performed in PyMol using only TbDHSp. The number of atoms used in the alignment and the RMSD as calculated by PyMol is listed in each figure legend.
Steady-State Kinetic Assays for DHS Activity
Formation of modified eIF5A was measured via incorporation of 14C transferred from radiolabeled spermidine in a filter binding assay (Wolff et al., 2011). The previously published protocol was followed with minor modifications. Preliminary studies were done to determine both the enzyme concentration and reaction time required to yield linear reaction kinetics. [14C]-spermidine (78 μM for a total of 25 nCi), NAD+ (1 mM), 15 μM eIF5Aand 5 nM TbDHSc:DHSp (tetrameric concentration) were used in all radioactivity assays, except for determination of Km’s where NAD+ (1.3 – 1000 μM) and spermidine (0.12 – 78 μM) were assayed over a concentration range as indicated. Reactions were allowed to proceed for 20 min. Initial rates of velocity data were fitted to the Michaelis-Menten equation using GraphPad Prism version 7.01 for Windows. The catalytic rate, kcat, was calculated based on heterotetramer concentration. All experiments were conducted in triplicates.
Fluorescence Assay
Assays were conducted as previously described for hDHS with minor modifications (Wolff et al., 2000). Fluorescence was measured using a Synergy H1 microplate reader (Biotek) at an excitation of 350 nm and emission of 441 nm for 5 or 10 minutes. Reactions were set in Fluotrac 200 plates (Greiner) with a final volume of 100 ± 3 μL. Enzyme concentration was calculated for heterotetrameric TbDHSc:DHSp and mutants at 1 μM. All reactions contained 1 mM spermidine, 1 mM NAD+, 1 mM DTT and 15 μM eIF5A. GC7 (N1-guanyl-1,7-diaminoheptane) was used at 10 μM for inhibition of DHS; GC7 was present during the pre-incubation and prior to spermidine addition to ensure binding to the enzyme/NAD+ complex.
Thermal Shift Assay
We followed published protocols (Afanador et al., 2013) for this assay with some modifications. TbDHSc:DHSp heterotetramer (1 μM)was used for all measurements. SYPRO Orange (1 μL of) (Sigma, Product Number S-5692 at a final concentration of 5X) was added to each reaction. Reactions were set up in real-time PCR Tube Strips with Masterclear Cap Strips (Eppendorf) with a final volume of 30 μL. The reaction mixture was incubated in the RT-PCR machine (CFX96 Real-Time System (Bio-Rad)) for 2 min at 20°C followed by 0.2°C increments in increasing temperature every 5 seconds until a final temperature of 80°C was reached. A pre-defined HEX filter was used to monitor SYPRO Orange signal upon protein thermal unfolding. The derivative of the fluorescence curve, as calculated by the instrument software (Bio-Rad CFX Manager 3.1), was used to determine the Tm.
QUANTIFICATION AND STATISTICAL ANALYSIS
Steady-State Kinetic Analysis
Triplicate data were collected for each substrate concentration used to determine the kinetic parameters (Km, kcat, specific activity). To determine Km and kcat, data were then fitted to the Michaelis-Menten equation in Prism (v 7.0 Graph-Pad Software Inc, San Diego, CA) and error presented represents standard error of the fit as calculated by Prism.
Thermal Shift Assay
Curves were fit to determine the derivative of the fluorescence curve using Bio-Rad CFX Manager 3.1 software to determine the Tm. The displayed results represent the average ± standard deviation over 6 replicates.
DATA AND SOFTWARE AVAILABILITY
Atomic coordinates and structure factors for the TbDHS structure reported in the manuscript have been deposited in the Protein Data Bank under accession code PDB: 6DFT.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus
Strains | ||
| E. coli BL21 (DE3) | Invitrogen | n/a |
|
Chemicals, Peptides, and Recombinant Proteins | ||
| Phenylmethylsulfonyl fluoride (PMSF) | Sigma | Cat#P7627 |
| Yeast SUMO protease - Ulp1 | (Nguyen et al., 2013) | n/a |
| IGEPAL | Sigma | Cat#I8896 |
| CHAPS | Research Product International | Cat#C41010 |
| PEG6000 | Molecular Dimensions | Cat#MD2–100-12 |
| [14C]-spermidine | American Radiolabeled Chemicals | Cat#ARC 3138 |
| GC7 (N1-guanyl-1,7-diaminoheptane) | Biosearch Technologies | Cat#G-1000–10 |
| SYPRO Orange | Sigma | Cat#S-5692 |
|
Deposited Data | ||
| Structure factors/coordinates for
TbDHS bound to NAD |
This paper | PDB: 6DFT |
| Structure used for phasing: human DHS bound to NAD | Liao et al., 1998 | PDB: 1DHS |
| Structure used for comparative alignments: hDHS bound to NAD and GC7 | Umland et al., 2004 | PDB: 1RQD |
|
Oligonucleotides | ||
| Primer for PCR amplification of
TbDHSp 5’-GGCGGCCATATGAGCGGTGTGCCGTTTCCG 5’-GGCGGCCTCGAGTTAGTTACCGTCTTCACGCAC |
Sigma | n/a |
| Primer for PCR amplification of
TbeIF5A 5’-GGCGGCGGATCCATGTCTGACGATGAGGGACAG 5’-CCGCCGCTCGAGTTATCGCTCGGCTGCATTCTTG |
Sigma | n/a |
| Primer for mutagenesis
E166A-DHSc:DHSp 5’-GAACCGCTATTCTACCGTGGGCGCTCAGGCGAGTAATC 5’-CACGTGCCAGATTACTCGCCTGAGCGCCCACGGTAG |
Sigma | n/a |
| Primer for mutagenesis
E405A-DHSc:DHSp 5’-GAACAATGCGCAAGAATTCGATGGTAGTGCCGCAGGTGCTCG 5’-CACCCGGGCGAGCACCTGCGGCACTACCATCGAATTC |
Sigma | n/a |
| Primer for mutagenesis
DHSc:DHSp-E103A 5’-GGTTACCACGGCCGGCGGTGTTGCGGAAGATGTCATC 5’-GTATCGCCCAGACATTTGATGACATCTTCCGCAACACCGCCGGC |
Sigma | n/a |
| Primer for mutagenesis
DHSc:DHSp-E104A 5’-GTTACCACGGCCGGCGGTGTTGAAGCGGATGTCATC 5’-GGGTATCGCCCAGACATTTGATGACATCCGCTTCAACACCGCCGG |
Sigma | n/a |
| Primer for mutagenesis
DHSc:DHSp-D218A 5’-CTTTCACCGACGGCTCTATGGGTGCTATGATCTATTTC 5’-CTTTACGGCTATAGTTGTAGAAATAGATCATAGCACCCATAGAG |
Sigma | n/a |
| Primer for mutagenesis
DHSc:DHSp-H266A 5’-CTGGGCGCGGGTCTGCCGAAAGCTCACCTGCTGC 5’-CTGCTTGAACATTGCGCAGCAGGTGAGCTTTCGGCAGAC |
Sigma | n/a |
|
Recombinant DNA | ||
| pUC57-His-SUMO-H377A-DHSc:DHSp | Genscript | n/a |
| pE-SUMO-TbDHSc | (Nguyen et al., 2013) | |
| pETDuet1 | EMD Millipore | n/a |
| pE-SUMO-TbDHSp | (Nguyen et al., 2013) | |
| pET28b | Novagen | n/a |
|
Software and Algorithms | ||
| TriTrypDB – Kinetoplastid genomic resource | http://tritrypdb.org/tritrypdb/ | |
| Clustal Omega |
https://www.ebi.ac.uk/Tools/msa/clustalo/ |
|
| NCBI BlastP | https://blast.ncbi.nlm.nih.gov/Blast.cgi | |
| ProtParam on ExPASy | https://web.expasy.org/protparam/ | |
| HKL-3000 | (Minor et al., 2006) | http://www.hkl-xray.com/hkl-3000 |
| Phaser | (McCoy et al., 2007) | http://www.phaser.cimr.cam.ac.uk/index.php/Phaser_Crystallographic_Software |
| Sculptor | (Winn et al., 2011) | https://www.phenix-online.org/ |
| Parrot | (Cowtan, 2010) | http://www.ccp4.ac.uk/html/parrot.html |
| Buccaneer | (Cowtan, 2006) | https://www.ccp4.ac.uk/newsletters/newsletter44/articles/buccaneer.html |
| Phenix | (Adams et al., 2010) | https://www.phenix-online.org/ |
| MolProbity | (Chen et al., 2010) | http://molprobity.biochem/duke.edu/. |
| Coot | (Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PyMol | The PyMOL Molecular
Graphics System |
https://pymol.org/2/ |
| TM-align | (Zhang and Skolnick, 2005) | https://zhanglab.ccmb.med/umich.edu/TM-align/. |
| Prism 7 | GraphPad Software | https://www.graphpad.com |
| Bio-Rad CFX Manager 3.1 software | http://www.bio-rad.com/en-us/product/cfx-manager-software?ID=aed9803d-cb4d-4ecb-9263-3efc9e650edb | |
| Others | ||
| T. brucei single marker genomic DNA | This work | n/a |
| Sequencing was performed at the
Sanger Sequencing Core |
UTSW | https://www.utsouthwestern.edu/labs/sanger-sequencing-core/ |
| HiTrap Chelating HP Column | GE Healthcare | Cat#17040903 |
| Superdex 200 Prep Grade | GE Healthcare | Cat#28989335 |
| HiTrap Q HP | GE Healthcare | Cat#17115401 |
| Synchrotron | APS ANL | 19-ID (SBC-CAT) |
Highlights.
Crystal structure of Trypanosoma brucei deoxyhypusine synthase (DHS) is reported
Obligate heterotetrameric is formed between two paralogous pseudoenzymes
Enzyme activity is restored by active-site complementation
NAD+ binding sites show potential for selectivity compared with human DHS
ACKNOWLEDGMENTS
Research was supported by NIH NIAID grant R37AI034432 (to M.A.P.). M.A.P. acknowledges the support of the Welch Foundation grant I-1257. Results are derived from work at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. SBC-CAT is operated by UChicago Argonne, for the US Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. M.A.P. holds the Sam G. Winstead and F. Andrew Bell Distinguished Chair in Biochemistry.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes one figure and two tables and can be found with this article online at https://doi.org/10.1016/j.str.2018.07.012.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular struc-ture solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afanador GA., Muench SP., McPhillie M., Fomovska A., Schon A., Zhou Y., Cheng G., Stec J., Freundlich JS., Shieh HM., et al. (2013). Discrimination of potent inhibitors of Toxoplasma gondii enoyl-acyl carrier protein reductase by a thermal shift assay. Biochemistry 52, 9155–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borek D, Cymborowski M, Machius M, Minor W, and Otwinowski Z (2010). Diffraction data analysis in the presence of radiation damage. Acta Crystallogr. D Biol. Crystallogr 66, 426–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borek D, Dauter Z, and Otwinowski Z (2013). Identification of patterns in diffraction intensities affected by radiation exposure. J. Synchrotron Radiat 20, 37–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borek D, Minor W, and Otwinowski Z (2003). Measurement errors and their consequences in protein crystallography. Acta Crystallogr. D Biol. Crystallogr 59,2031–2038. [DOI] [PubMed] [Google Scholar]
- Buscher P, Cecchi G, Jamonneau V, and Priotto G (2017). Human African trypanosomiasis. Lancet 390, 2397–2409. [DOI] [PubMed] [Google Scholar]
- Chawla B, Jhingran A, Singh S, Tyagi N, Park MH, Srinivasan N, Roberts SC, and Madhubala R (2010). Identification and characterization of a novel deoxyhypusine synthase in Leishmania donovani. J. Biol. Chem 285, 453–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K (2006). The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr 62, 1002–1011. [DOI] [PubMed] [Google Scholar]
- Cowtan K. (2010). Recent developments in classical density modification. Acta Crystallogr. D Biol. Crystallogr 66, 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Dinman JD, and Green R (2018). Translation elongation and re-coding in eukaryotes. Cold Spring Harb. Perspect. Biol 10.1101/cshperspect.a032649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MC., Horn D., Fairlamb AH., Ferguson MAJ., Gray DW., Read KD., De Rycker M., Torrie LS., Wyatt PG., Wyllie S., et al. (2017). Anti-try-panosomatid drug discovery: an ongoing challenge and a continuing need. Nat. Rev. Microbiol 15, 447. [DOI] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, and Postlethwait J (1999). Preservation of duplicate genes by complementary, degenerative mu-tations. Genetics 151, 1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin NV, and Phillips MA (1994). The subunit interfaces of oligomeric enzymes are conserved to a similar extent to the overall protein sequences. Protein Sci 3, 2455–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe YA, Wolff EC, Lee YB, and Park MH (1997). Enzyme-substrate in-termediate at a specific lysine residue is required fordeoxyhypusine synthesis. The role of Lys329 in human deoxyhypusine synthase. J. Biol. Chem 272, 32679–32685. [DOI] [PubMed] [Google Scholar]
- Kung JE, and Jura N (2016). Structural basisforthe non-catalyticfunctions of protein kinases. Structure 24, 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH., Um PY., and Park MH. (2001). Structure-function studies of human deoxyhypusine synthase: identification of amino acid residues critical for the binding of spermidine and NAD. Biochem. J 355, 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Kim SH., Zhang Y., Hanfrey CC., Elliott KA., Ealick SE., and Michael AJ. (2015). Different polyamine pathways from bacteria have replaced eukaryotic spermidine biosynthesis in ciliates Tetrahymena thermo-phila and Paramecium tetaurelia. Mol. Microbiol 97, 791–807. [DOI] [PubMed] [Google Scholar]
- Liao DI, Wolff EC, Park MH, and Davies DR (1998). Crystal structure of the NAD complex of human deoxyhypusine synthase: an enzyme with a ball-and-chain mechanism for blocking the active site. Structure 6, 23–32. [DOI] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, and Adams PD (2017). Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Struct. Biol 73,148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, and Chruszcz M (2006). HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Mounce BC, Olsen ME, Vignuzzi M, and Connor JH (2017). Polyamines and their role in virus infection. Microbiol. Mol. Biol. Rev 81, 10.1128/MMBR.00029-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Farhan H, and Eyers PA (2017a). Bio-Zombie: the rise of pseudoenzymes in biology. Biochem. Soc. Trans 45, 537–544. [DOI] [PubMed] [Google Scholar]
- Murphy JM, Mace PD, and Eyers PA (2017b). Live and let die: insights into pseudoenzyme mechanisms from structure. Curr. Opin. Struct. Biol 47, 95–104. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, and Cleveland JL (2016). Targeting the polyamine-hypusine circuitforthe prevention and treatment ofcancer. AminoAcids 48,2353–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen S, Jones DC, Wyllie S, Fairlamb AH, and Phillips MA (2013). Allosteric activation of trypanosomatid deoxyhypusine synthase by a catalytically dead paralog. J. Biol. Chem 288, 15256–15267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen S, Leija C, Kinch L, Regmi S, Li Q, Grishin NV, and Phillips MA (2015). Deoxyhypusine modification of eukaryotic translation initiation factor 5A (eIF5A) is essential for Trypanosoma brucei growth and for expres-sion of polyprolyl-containing proteins. J. Biol. Chem 290, 19987–19998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Borek D, Majewski W, and Minor W (2003). Multiparametric scaling of diffraction intensities. Acta Crystallogr. A 59, 228–234. [DOI] [PubMed] [Google Scholar]
- Park MH, Nishimura K, Zanelli CF, and Valentini SR (2010). Functional significance of eIF5A and its hypusine modification in eukaryotes. Amino Acids 38, 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J, Kim BH, and Grishin NV (2008). PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res 36, 2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds SL, and Fischer K (2015). Pseudoproteases: mechanisms and function. Biochem. J 468, 17–24. [DOI] [PubMed] [Google Scholar]
- Saharinen P, and Silvennoinen O (2002). The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J. Biol. Chem 277, 47954–47963. [DOI] [PubMed] [Google Scholar]
- Ude S, Lassak J, Starosta AL, Kraxenberger T, Wilson DN, and Jung K (2013). Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science 339, 82–85. [DOI] [PubMed] [Google Scholar]
- Umland TC, Wolff EC, Park MH, and Davies DR (2004). A new crystal structure of deoxyhypusine synthase reveals the configuration of the active enzyme and of an enzyme.NAD.inhibitor ternary complex. J. Biol. Chem 279, 28697–28705. [DOI] [PubMed] [Google Scholar]
- Volkov OA, Kinch L, Ariagno C, Deng X, Zhong S, Grishin N, Tomchick DR, Chen Z, and Phillips MA (2016). Relief of autoinhibition by conformational switch explains enzyme activation by a catalytically dead paralog. Elife 5, 10.7554/eLife.20198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert E, and Phillips MA (2012). Regulation and function of polyamines in African trypanosomes. Trends Parasitol 28, 66–72. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff EC, Lee SB, and Park MH (2011). Assay of deoxyhypusine synthase activity. Methods Mol. Biol 720, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff EC, Wolff J, and Park MH (2000). Deoxyhypusine synthase generates and uses bound NADH in a transient hydride transfer mechanism. J. Biol. Chem 275, 9170–9177. [DOI] [PubMed] [Google Scholar]
- Zhang Y, and Skolnick J (2005). TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 33, 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.