

Abstract
A series of 2-(1H-indol-3-yl)ethylthiourea derivatives were prepared by condensation of 2-(1H-indol-3-yl)ethanamine with appropriate aryl/alkylisothiocyanates in anhydrous media. The structures of the newly synthesized compounds were confirmed by spectroscopic analysis and the molecular structures of 8 and 28 were confirmed by X-ray crystallography. All obtained compounds were tested for antimicrobial activity against Gram-positive cocci, Gram-negative rods and for antifungal activity. Microbiological evaluation was carried out over 20 standard strains and 30 hospital strains. Compound 6 showed significant inhibition against Gram-positive cocci and had inhibitory effect on the S. aureus topoisomerase IV decatenation activity and S. aureus DNA gyrase supercoiling activity. Compounds were tested for cytotoxicity and antiviral activity against a large panel of DNA and RNA viruses, including HIV-1 and other several important human pathogens. Interestingly, derivative 8 showed potent activity against HIV-1 wild type and variants bearing clinically relevant mutations. Newly synthesized tryptamine derivatives showed also a wide spectrum activity, proving to be active against positive- and negative-sense RNA viruses.
Keywords: antibacterial activity, anti-HIV activity, antiviral activity, thiourea derivatives of indole, topoisomerase
1. Introduction
Indole derivatives have attracted a great deal of attention due to their wide therapeutic applications (Figure 1). Tryptamine-containing arylpiperazine derivatives (e.g., delavirdine, ateviridine) belong to the group of non-nucleoside reverse transcriptase inhibitors (NNRTIs), used in the treatment of AIDS that represents the major health problem worldwide, with 1.0 million deaths and 36.7 million people living with HIV in 2016 [1]. Anti-retroviral therapy, nowadays based on combinations of drugs belonging to the NNRTI, nucleoside reverse transcriptase (NRTI), protease (PR) and, more recently, integrase (IN) inhibitor classes, has improved the quality of life of AIDS patients. However, due to a side effect and rapid emergence of drug-resistant viruses drug [2], a continuous development of new anti-HIV compounds is needed to overcome these limitations.
Figure 1.

Pharmacologically active indole ring-containing derivatives.
Indole-derived azonine compounds, such as pericine, are CNS-active alkaloids, ligands of opioid receptors. Gliotoxin and its analogues act as antimicrobials and have antiproliferative activity in lymphosarcoma cells. Indometacin is a non-steroidal anti-inflammatory agent that reduces fever, pain or swelling. Current research on panobinostat is focused on its antiproliferative effects in various cancer cell lines [3].
In order to search for potent inhibitors of hepatitis B virus (HBV) and hepatitis C virus (HCV), a series of N-substituted tryptamine analogues were synthesized [4,5]. What is more, series of 1,3,5-triazine derivatives of tryptamine show anti-HIV activity both by inhibiting cell-free virus infection and cell-to-cell virus transmission [6]. Furthermore, various families of indole-derived compounds exhibit antimicrobial activity against Gram-positive [7,8,9,10,11] and Gram-negative bacteria [7,9,10], as well as fungi [7,12]. The cytotoxic [13,14,15,16] and anticancer potency of substituted indole compounds was also proven. In this field they act as inhibitors of serine/threonine kinase (AKT) [17], tyrosinase [18], human sirtuins SIRT1-3 [19] or transcription factors NF-kB activation [20].
The thiourea moiety is an important synthon responsible for numerous biological activities, such as antimicrobial [21,22,23] antiviral [24,25], antiproliferative [26] and cytotoxic effects [22,27]. The 1,3-disubstituted thiourea scaffold is a structure with a huge potential for multi-direction activity. Through rationally planned modifications of thiourea derivative structures, a wide range of compounds with high pharmacological activity and low side-effects can be obtained. The literature indicates that including an electron acceptor group (e.g., a halogen atom) in a strategic part of a molecule significantly influences its biological activity, leading to an increase in lipophilicity, and as a result in the speed of absorption and transport of substances in vivo. The presence of a strongly electronegative substituent also changes the properties of the adjacent functional groups, which influences the reactivity, metabolic stability and chemical nature of the molecule [28,29,30]. High stability, in turn, increases the bioavailability of the compound, which improves its pharmacokinetic parameters. For these reasons, halogen derivatives constitute ca. 20% of the total number of medical drugs currently available on the world market [29]. Encouraged by the above observations and considering the interesting pharmacological profile of indole derivatives, we have synthesized new tryptamines incorporating the 1,3-disubstituted thiourea scaffold as promising antimicrobial and antiviral agents.
2. Results and Discussion
2.1. Chemistry
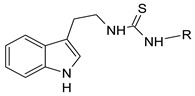
The synthesis of 28 thiourea derivatives is described. Sixteen of them had not been described previously in the literature. To prepare the thiourea derivatives 2-(1H-indol-3-yl)ethanamine was condensed with the corresponding isothiocyanate (Scheme 1 and Table 1).
Scheme 1.

Synthesis of thiourea derivatives of 2-(1H-indol-3-yl)ethanamine.
Table 1.
Structure and molar weight of the investigated compounds 1–28.
| Number | R | Mol. Weight (g/mol) |
|---|---|---|
| 1 | Phenyl | 295.40 |
| 2 | 4-Methoxyphenyl | 325.43 |
| 3 | 4-Methylphenyl | 309.43 |
| 4 | 4-Chlorophenyl | 329.85 |
| 5 | Benzoyl | 323.41 |
| 6 | 3,4-Dichlorophenyl | 364.29 |
| 7 | 3-Bromophenyl | 374.30 |
| 8 | 4-Bromophenyl | 374.30 |
| 9 | Cyclohexyl | 301.45 |
| 10 | Benzyl | 309.43 |
| 11 | 2-Fluorophenyl | 313.39 |
| 12 | 3-Fluorophenyl | 313.39 |
| 13 | 2-Chlorophenyl | 329.85 |
| 14 | 3-Chlorophenyl | 329.85 |
| 15 | 2-Bromophenyl | 374.30 |
| 16 | Phenethyl | 323.46 |
| 17 | 4-Fluorophenyl | 313.39 |
| 18 | Allyl | 259.37 |
| 19 | Methallyl | 273.40 |
| 20 | Ethoxycarbonyl | 291.37 |
| 21 | 4-Iodophenyl | 421.30 |
| 22 | Ethyl | 247.36 |
| 23 | 3-Chloro-4-methylphenyl | 343.87 |
| 24 | 3-Trifluoromethylphenyl | 363.40 |
| 25 | Methyl | 233.33 |
| 26 | 3-Chloro-6-methylphenyl | 343.87 |
| 27 | 4-Butyl-2-methylphenyl | 365.53 |
| 28 | 3-Chloro-4-fluorophenyl | 347.84 |
Obtained compounds were purified by column chromatography and/or crystallized from a nonpolar solvent. MS, 1H-NMR and 13C-NMR spectra confirmed the identity of the products. The molecular structures of 8 and 28 (Supplementary Material Figure S1) were also determined by an X-ray crystal structure analysis.
2.2. Microbiology
Twenty eight thiourea derivatives of tryptamine were examined in vitro against Gram-positive cocci, Gram-negative rods and Candida albicans. The microorganisms used in this study have universal utility in the antimicrobial tests in the research for new active agents [21]. All tested compounds were screened by disc diffusion method [31], next these showing significant activity, were examined for their minimal inhibitory concentration (MIC) [32]. The results of antimicrobial activity are summarized in Table 2. In Table 2 only the results for active derivatives are included.
Table 2.
Activities against Gram-positive bacteria—minimal inhibitory concentrations (MIC, µg/mL) and diameter of growth inhibitory zone (GIZ, mm, applied 400 µg per disc).
| Strains | 6 | 14 | 15 | 19 | Ref. 1 |
|---|---|---|---|---|---|
| S. aureus NCTC 4163 | 6.25 (10) | 50 (13) | 50 (11) | 400 (12) | 0.25 (26) |
| S. aureus ATCC 25923 | 6.25 (15) | 50 (14) | 100 (11) | 400 (12) | 0.5 (26) |
| S. aureus ATCC 6538 | 12.5 (16) | 50 (13) | 50 (12) | 200 (11) | 0.25 (28) |
| S. aureus ATCC 29213 | 6.25 (15) | 50 (15) | 50 (11) | - | 0.5 (22) |
| S. epidermidis ATCC 12228 | 12.5 (16) | 50 (12) | - | - | 0.25 (30) |
| B. subtilis ATCC 6633 | 12.5 (15) | 50 (12) | 50 (15) | 400 (13) | <0.12 (38) |
| B. cereus ATCC 11778 | 6.25 (15) | 25 (15) | 50 (14) | 200 (13) | 0.25 (26) |
| M. luteus ATCC 9341 | 12.5 (15) | 200 (12) | 200 (13) | 200 (13) | 0.25 (26) |
| M. luteus ATCC 10240 | 6.25 (23) | 12.5 (20) | 50 (14) | 50 (14) | 1 (25) |
1 Ref.—ciprofloxacin (GIZ—5 µg/9 mm disc). Lack of the growth inhibition area.
Preliminary tests by disc-diffusion method indicated antimicrobial activity against standard Gram-positive cocci. The next step was estimation of MIC values for not only standard but also hospital strains. The study was carried out over 20 standard, 20 hospital of Staphylococcus aureus and 10 hospital of Staphylococcus epidermidis strains used for routine antimicrobial media susceptibility testing. Hospital strains were isolated from different biological materials of the patients hospitalized in the Warsaw Medical University hospitals. MIC values for the standard Gram-positive strains were in the range 400–6.25 μg/mL (Table 3). For the hospital Staphylococcus epidermidis (MSSA) rods the MIC value ranged from 400 to 6.25 μg/mL and the average value was 25 μg/mL.
Table 3.
Activity of compounds against hospital methicillin-susceptible strains of S. aureus (MSSA), methicillin-resistant strains of S. aureus (MRSA) and S. epidermidis (MRSE)—minimal inhibitory concentrations (MIC, µg/mL).
| S. aureus MSSA | 440/11 | 441/11 | 442/11 | 443/11 | 444/11 | 445/11 | 446/11 | 447/11 | 448/11 | 449/11 |
| 6 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 |
| 14 | 25 | 25 | 25 | 25 | 12,5 | 25 | 25 | 25 | 25 | 25 |
| Ciprofloxacin | 0.4 | 0.4 | 0.4 | 0.4 | 0.8 | 0.4 | 0.4 | 0.8 | 0.8 | 0.4 |
| S. aureus MRSA | 389/10 | 390/10 | 391/10 | 392/10 | 393/10 | 394/10 | 399/10 | 450/11 | 451/11 | 452/11 |
| 6 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 |
| 14 | 25 | 25 | 25 | 25 | 25 | 25 | 25 | 25 | 25 | 25 |
| Ciprofloxacin | 25 | 100 | 100 | 100 | 50 | 25 | 200 | 100 | 100 | 200 |
| S. epidermidis MRSE | 430/11 | 431/11 | 432/11 | 433/11 | 434/11 | 435/11 | 436/11 | 437/11 | 438/11 | 439/11 |
| 6 | 6.25 | 6.25 | 6.25 | 6.25 | 12.5 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 |
| 14 | 25 | 6.25 | 25 | 25 | 25 | 25 | 50 | 25 | 50 | 25 |
| 15 | 400 | 50 | 50 | 50 | 50 | 100 | 100 | 100 | 200 | 50 |
| 19 | 12.5 | 100 | 12.5 | 50 | 12.5 | 50 | 100 | 200 | 100 | 50 |
| Ciprofloxacin | 0.8 | 6.25 | 50 | 50 | 50 | 0.4 | 0.4 | 0.8 | 0.4 | 50 |
On the other hand, the MIC values obtained for the hospital Staphylococcus aureus (MSSA) for the derivatives 6 and 14 varied in the range 25–6.25 μg/mL. MRSA strains were also susceptible to these compounds, and the MIC value range changed from 25 to 6.25 μg/mL. The comparison between the chemical structure and the biological activity led to the conclusion that thiourea-derived tryptamines with halogen substituents at the ring are more potent antimicrobials than alkylthioureas. Analysis of a position and a type of an electronegative element at a benzene ring showed that chlorine at the position C-3 promotes antimicrobial activity (derivatives 6 and 14). Substitution of the benzene ring with a second chlorine atom in the 4-position increases antimicrobial activity (compound 6). The introduction of another substituent in the 4-position (fluorine, methyl) causes the disappearance of antimicrobial activity (compounds 23, 28). Other halogens (F and Br) in position 3 do not affect antimicrobial activity. On the other hand, the substitution of the benzene ring with bromine at position 2 implies the moderate antimicrobial activity, as in the previous derivatives, only for Gram-positive strains (compound 15). The methallyl indole derivative 19 expressed a moderate antibacterial activity. These results are in the general agreement with our previous research concerning the activity of heterocyclic thiourea compounds [33]. To assess the mechanism of antimicrobial activity, S. aureus topoisomerase IV decatenation assay and S. aureus DNA gyrase supercoiling assay were performed. Ciprofloxacin (CFX) was used as a positive control. The decatenation activity of S. aureus topoisomerase IV was almost completely inhibited by CFX at 32 µg/mL and compound 6 has an inhibitory activity against S. aureus topoisomerase with IC50 = 28.9 ± 0.3 µg/mL (Table 4). Compound 6 shows inhibition of the supercoiling activity of S. aureus gyrase but weaker than that of S. aureus topoisomerase, and the IC50 for inhibition of DNA gyrase supercoiling activity for compound 6 was 72.6 ± 1.2 µg/mL (Table 4).
Table 4.
Inhibitory activities against DNA topoisomerase IV and DNA gyrase, expressed as IC50 ± SEM (µg/mL).
| Compounds | S. aureus Topoisomerase IV | S. aureus DNA Gyrase |
|---|---|---|
| 6 | 28.90 ± 0.30 | 72.60 ± 1.2 |
| Ciprofloxacin | 1.70 ± 0.15 | 3.55 ± 0.13 |
IC50—half of the maximal inhibitory concentration.
On the contrary, compound 14 was not significantly effective against DNA topoisomerase IV (Figure 2) and DNA gyrase (data not shown).
Figure 2.

Effect of the tested compounds on S. aureus topoisomerase IV activity. Decreasing amounts of compounds 14 and 6 were incubated with 200 ng kinetoplast DNA and run on agarose gel. Lane 1: incubation mixture without enzyme (negative control). Lane 2: S. aureus topoisomerase IV assay with dilution buffer (control). Lane 3: S. aureus topoisomerase IV with DMSO (control). Lane: 4: Ciprofloxacin at concentration 32 μg/mL. Lane: 5–11: compound 14 at concentrations 64, 32, 16, 8, 4, 2, 0.5 μg/mL, respectively. Lane: 12–18: compound 6 at concentrations 64, 32, 16, 8, 4, 2, 0.5 μg/mL, respectively.
2.3. Antiviral and Antiproliferative Evaluation
All twenty-eight thiourea derivatives were tested in cell-based assays against the Human Immunodeficiency Virus type-1 (HIV-1) and against representative of several RNA and DNA virus families. The results are reported in the Table 5 and Table 6.
Table 5.
Antiviral activity of thiourea derivatives against HIV-1 and ssRNA+ (BVDV, YFV, CV-B5, Sb-1) viruses and cytotoxicity against the cell lines used in the assays.
| Compounds | MT-4 a CC50 | HIV-1 b EC50 | MDBK c CC50 | BVDV d EC50 | BHK e CC50 | YFV f EC50 | Vero-76 g CC50 | CV-B5 h EC50 | Sb-1 i EC50 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 31 | >31 | >100 | >100 | 48 | >48 | 64 | >64 | >64 |
| 2 | 32 | >32 | >100 | >100 | 44 | >44 | 56 | >56 | >56 |
| 3 | 33 | >33 | >100 | ≥100 | 16 | >16 | 75 | >75 | >75 |
| 4 | 45 | >45 | >100 | >100 | 47 | >47 | 55 | >55 | >55 |
| 5 | 38 | >38 | 67 | 20 ± 1.5 | 34 | >34 | 29 | >29 | >29 |
| 6 | 45 | >45 | 27 | >27 | 10 | >10 | 13 | >13 | >13 |
| 7 | 45 | >45 | 55 | 20 ± 2.0 | 10 | >10 | 12 | >12 | >12 |
| 8 | 45 | 8.7 ± 0.4 | 52 | >52 | 10 | >10 | 13 | >13 | >13 |
| 9 | 49 | >49 | 55 | >55 | 13 | >13 | 14 | >14 | >14 |
| 10 | 78 | >78 | >100 | >100 | 52 | >52 | >100 | 30 ± 2.4 | >100 |
| 11 | 48 | >48 | 69 | >69 | 34 | >34 | 73 | >73 | >73 |
| 12 | 47 | >47 | 49 | >49 | 16 | >16 | 60 | >60 | >60 |
| 13 | 47 | >47 | 49 | >49 | 11 | >11 | 53 | 12 ± 0.9 | >53 |
| 14 | 42 | >42 | 32 | >32 | 6.0 | >6.0 | 32 | 9.0 ± 1.2 | >32 |
| 15 | 46 | >46 | 48 | >48 | 28 | >28 | 46 | >46 | >46 |
| 16 | 49 | >49 | >100 | 46 | 18 | >18 | >100 | 18 ± 1.7 | >100 |
| 17 | 44 | >44 | 53 | >53 | 30 | >30 | 53 | >53 | >53 |
| 18 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 19 | >100 | >100 | >100 | ≥100 | 35 | >35 | 89 | >89 | >89 |
| 20 | >100 | >100 | >100 | 35 ± 2.8 | >100 | >100 | >100 | >100 | >100 |
| 21 | 2.4 ± 0.2 | >2.4 | >100 | >100 | 11 | >11 | 11 | >11 | >11 |
| 22 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 23 | 44 | >44 | 25 | >25 | 10 | >10 | 17 | >17 | >17 |
| 24 | 44 | >44 | 23 | >23 | 9.0 | >9.0 | 16 | >16 | >16 |
| 25 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 26 | 45 | >45 | 42 | 15 ± 1.2 | 12 | >12 | 17 | >17 | >17 |
| 27 | 45 | >45 | 11 | >11 | 9.5 | >9.5 | 12 | >12 | >12 |
| 28 | 45 | >45 | 37 | 13 ± 1.2 | 30 | >30 | 32 | 20 ± 2.1 | >32 |
| Efavirenz | 40 | 0.002 ± 0.0002 | |||||||
| 2′-C-methyl-guanosine | >10 | 1.1 ± 0.1 | >10 | 1.9 ± 0.1 | |||||
| 2′-C-methyl-cytidine | >100 | 18 ± 0.9 | 7.3 ± 0.4 | ||||||
Data represent mean values for three independent determinations. Standard deviations are reported for the more active compounds. Also for the others values the variation was less than 15%. a Compound concentration (µM) required to reduce the proliferation of mock-infected MT-4 cells by 50%. b Compound concentration (µM) required to achieve 50% protection of MT-4 cells from HIV-1 induced cytopathogenicity. c Compound concentration (µM) required to reduce the viability of mock-infected MDBK cells by 50%. d Compound concentration (µM) required to achieve 50% protection of MDBK cells from BVDV-induced cytopathogenicity. e Compound concentration (µM) required to reduce the viability of mock-infected BHK cells by 50%. f Compound concentration (µM) required to achieve 50% protection of BHK cells from YFV-induced cytopathogenicity. g Compound concentration (µM) required to reduce the viability of mock-infected VERO-76 cells by 50%. h,i Compound concentration (µM) required to reduce the plaque number of CV-B5 h, Sb-1 i by 50% in VERO-76 monolayers.
Table 6.
Antiviral activity of thiourea derivatives against ssRNA-(RSV, VSV), dsRNA (Reo-1) and dsDNA (VV, HSV-1) viruses and cytotoxicity against the cell lines used in the assays.
| Compounds | BHK a CC50 | Reo-1 b EC50 | Vero-76 c CC50 | RSV d EC50 | VSV e EC50 | VV f EC50 | HSV-1 g EC50 |
|---|---|---|---|---|---|---|---|
| 1 | 48 | >48 | 64 | >64 | >64 | >64 | >64 |
| 2 | 44 | >44 | 56 | >56 | >56 | >56 | >56 |
| 3 | 16 | >16 | 75 | 20 ± 3.0 | >75 | >75 | >75 |
| 4 | 47 | >47 | 55 | >55 | >55 | >55 | >55 |
| 5 | 34 | >34 | 29 | >29 | >29 | >29 | >29 |
| 6 | 10 | >10 | 13 | >13 | >13 | >13 | >13 |
| 7 | 10 | >10 | 12 | >12 | >12 | >12 | >12 |
| 8 | 10 | >10 | 13 | >13 | >13 | >13 | >13 |
| 9 | 13 | >13 | 14 | >14 | >14 | >14 | >14 |
| 10 | 52 | >52 | >100 | >100 | >100 | >100 | >100 |
| 11 | 34 | >34 | 73 | >73 | >73 | >73 | >73 |
| 12 | 16 | >16 | 60 | >60 | >60 | >60 | >60 |
| 13 | 11 | >11 | 53 | >53 | >53 | >53 | >53 |
| 14 | 6.0 | >6.0 | 32 | >32 | >32 | >32 | >32 |
| 15 | 28 | >28 | 46 | 20 ± 2.5 | >46 | >46 | >46 |
| 16 | 18 | >18 | >100 | 30 ± 2.6 | >100 | >100 | >100 |
| 17 | 30 | >30 | 53 | >53 | >53 | >53 | >53 |
| 18 | >100 | 61 | >100 | >100 | >100 | >100 | >100 |
| 19 | 35 | >35 | 89 | >89 | >89 | >89 | >89 |
| 20 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 21 | 11 | >11 | 11 | >11 | >11 | >11 | >11 |
| 22 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 23 | 10 | >10 | 17 | >17 | >17 | >17 | >17 |
| 24 | 9.0 | >9.0 | 16 | >16 | >16 | >16 | >16 |
| 25 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 26 | 12 | >12 | 17 | >17 | >17 | >17 | >17 |
| 27 | 9.5 | >9.5 | 12 | >12 | >12 | >12 | >12 |
| 28 | 30 | >30 | 32 | >32 | >32 | >32 | >32 |
| 2′-C-methyl-cytidine | >100 | 10 ± 1.2 | |||||
| 6-Aza-uridine | 12 | 1.4 ± 0.06 | |||||
| Mycophenolic acid | 13 | 1.5 ± 0.2 | |||||
| Acycloguanosine | >100 | 3.0 ± 0.2 | |||||
Data represent mean values for three independent determinations. Standard deviations are reported for the more active compounds. Also for the others values the variation was less than 15%. a Compound concentration (µM) required to reduce the proliferation of mock-infected BHK cells by 50%. b Compound concentration (µM) required to achieve 50% protection of BHK cells from Reo-1-induced cytopathogenicity. c Compound concentration (µM) required to reduce the viability of mock-infected VERO-76 cells by 50%. d–g Compound concentration (µM) required to reduce the plaque number of RSV d, VSV e, VV f, HSV-1 g by 50% in VERO-76 monolayers.
Interestingly, fourteen derivatives exhibited an antiviral activity, although in some cases not very potent, against one or more viruses. None of them turned out to be active against YFV, Sb-1, VSV, VV and HSV-1; also the activity against Reo-1 of derivative 18 was not particularly significant. In general, tryptamine derivatives showed different degree of cytotoxicity for the cell monolayers used to support the multiplication of different viruses (MDBK, BHK, Vero-76) in stationary growth; in particular, sixteen compounds turned out to be cytotoxic against all cell lines used, four compounds turned out to be not cytotoxic.
Only derivative 8 showed interesting activity (EC50 = 8.7 μM) against HIV-1, associated with a moderate cytotoxicity (CC50 = 45 µM). As far as the antiviral activity is concerned, six compounds (5, 7, 16, 20, 26, 28) showed activity against BVDV, a member of Pestiviruses that have a serious impact on livestock [34], associated in two cases (16, 20) with lack of cytotoxicity for MDBK cell lines. Five compounds (10, 13, 14, 16, 28) exhibited activity (EC50 from 9.0 up to 30 μM) against CV-B5 virus, a member of Enterovirus genus that are important human pathogens that cause both acute and chronic diseases in infants, young children and immunocompromised individuals [35]. Finally, three derivatives (3, 15, 16) resulted moderately active against RSV, the most common respiratory pathogen in infants and young children worldwide [36].
Since a critical issue in the long-term clinical management of HIV disease is the development of drug resistance, compound 8 was further tested against a set of viruses possessing mutations that confer selective resistance to NRTI and NNRTI inhibitors, and that often appear during HAART therapy, reducing its effectiveness [37,38]. Interestingly, the activity against A17, AZTR and MDR strains (Table 7) is comparable with those of HIV-1 wild-type, demonstrating its activity against variants carrying clinically relevant NRTI and NNRTI mutations.
Table 7.
Cytotoxicity and antiviral activity of tryptamine derivative 8 against HIV-1 and its NRTI- and NNRTI-resistant mutants.
| Compounds | CC50 a | EC50 b | |||||
|---|---|---|---|---|---|---|---|
| MT | HIV-1 | N119 | A17 | EFVR | AZTR | MDR | |
| 8 | 45 | 8.7 ± 0.8 | >45 | 12 ± 1.5 | >45 | 9.0 ± 1.0 | 9.0 ± 0.8 |
| Efavirenz | 40 | 0.002 ± 0.0003 | 0.018 ± 0.002 | 0.075 ± 0.009 | 7.0 ± 0.8 | 0.001 ± 0.0002 | NA |
| AZT | 45 | 0.022 ± 0.004 | 0.025 ± 0.004 | 0.01 ± 0.001 | 0.02 ± 0.001 | 0.35 ± 0.04 | 0.08 ± 0.006 |
| Nevirapine | >100 | 0.080 ± 0.005 | 6.3 ± 0.05 | 80 | 100 | 0.078 ± 0.008 | 5.0 ± 0.6 |
Data represent mean values for three independent determinations. a Compound concentration (µM) required to reduce the proliferation of mock-infected MT-4 cells by 50%. b Compound concentration (µM) required to achieve 50% protection of MT-4 cells from HIV-1 induced cytopathogenicity.
Generally, no correlation could be found between the type of substituent in position 3 of the thiourea and antiviral activity. It can only be seen that aliphatic substituents (cyclohexyl, ethyl, methyl) block both antimicrobial and antiviral activity (compounds 9, 22, 25). Other derivatives, those with an aromatic ring or a double bond (allyl, methallyl) are characterized by activity against various viruses. Many of the tested compounds showed a moderate cytotoxicity against the MT4 cells, with the compound 21 interestingly turning out to be cytotoxic at a very low value (CC50 = 2.4 µM). Its antiproliferative properties were further evaluated against different cell lines derived from human haematological tumours (Table 8), showing potent activities at micromolar level (CC50 = 5.0–12 µM) and confirming its promising potential.
Table 8.
Cytotoxicity of tryptamine derivative 21 against human leukaemia/lymphoma cell lines.
| Compounds | CC50 a | ||
|---|---|---|---|
| CCRF-CEM b | WIL-2NS c | CCRF-SB d | |
| 21 | 5.0 ± 0.8 | 5.8 ± 1.0 | 12 ± 1.6 |
| Doxorubicin | 0.02 ± 0.002 | 0.02 ± 0.003 | 0.03 ± 0.003 |
Data represent mean values for three independent determinations. a Compound concentration (µM) required to reduce cell proliferation by 50%, under conditions allowing untreated controls to undergo at least three consecutive rounds of multiplication. b CD4+ human acute T-lymphoblastic leukaemia. c Human splenic B-lymphoblastoid cells. d Human acute B-lymphoblastic leukemia.
Biological data suggested that tryptamine derivatives proved to be active against different viruses. Their wide spectrum of activity against different RNA viruses and their different degree of cytotoxicity offer interesting indications for SAR studies, with the aim to design and develop more potent derivatives. Of particular interest are the derivative 8, strongly active against HIV-1 and variants carrying clinically relevant NRTI and NNRTI mutations, and the derivative 16 that showed activity, although moderate, against three different RNA viruses, belonging to both ssRNA+ (BVDV, CV-B5) and ssRNA-(RSV), with lack of cytotoxicity (CC50 > 100 µM) for MDBK and Vero-76 cell lines.
3. Materials and Methods
3.1. General Information
1H-NMR and 13C-NMR spectra were recorded on a model Avance DMX 300 spectrometer (Bruker, Billerica, MA, USA, 1H at 300 MHz and 13C at 75, MHz respectively). The chemical shift values are expressed in ppm relative to TMS as an internal standard. Mass spectral ESI measurements were carried out on a ZQ Micromass instrument (Waters, Milford, MA, USA) equipped with a quadrupole mass analyzer. The spectra were recorded in the positive ion mode at a declustering potential of 40–60 V. The samples were previously separated on a UPLC column (C18) using an ACQUITY UPLC system by Waters connected with a DPA detector. Flash chromatography was performed on silica gel 60 (200–400 mesh, Merck, Kenilworth, NJ, USA) using chloroform/methanol (19:1 vol) mixture as eluent. Analytical TLC was carried out on silica gel F254 (Merck) plates (0.25 mm thickness).
The diffraction data for the X-ray crystal structure analysis were collected for 8 and 28 at 200 K and 150 K, respectively, with an Xcalibur CCD diffractometer (Oxford Diffraction Ltd, Abingdon, UK), using graphite monochromated MoKα radiation. Crystal structures were solved by direct methods using the SHELXS-97 program and refined by full-matrix least squares method on F2 using the SHELXL-97 program [39]. All ordered non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were positioned geometrically and allowed to ride on their parent atoms, with Uiso(H) = 1.2 Ueq(C). The experimental details and final atomic parameters for 8 and 28 have been deposited with the Cambridge Crystallographic Data Centre as supplementary material (CCDC ID: 992,428 and 992,429). Copies of the data can be obtained free of charge on request by e-mailing data_request@ccdccam.ac.uk or via www.ccdc.cam.ac.uk/data_request/cif.
3.2. Chemistry
General Procedure for the Synthesis of Thiourea Derivatives of 2-(1H-indol-3-yl)ethanamine
A solution of 2-(1H-indol-3-yl)ethanamine (0.0038 mol, 0.61 g) in anhydrous acetonitrile (25 mL) was treated with an appropriate isothiocyanate (0.0042 mol) and the mixture was refluxed for 8 h. Then solvent was removed on rotary evaporator. The residue was purified by column chromatography (chloroform/methanol; 9.8:0.2 vol.). The compound was crystallized from acetonitrile or another appropriate solvent.
1-(2-(1H-Indol-3-yl)ethyl)-3-phenylthiourea (1). This compound was synthesized as described previously [40].
1-(2-(1H-Indol-3-yl)ethyl)-3-(4-methoxyphenyl)thiourea (2). Yield 87%. m.p. 146–148 °C. 1H-NMR (DMSO-d6) δ (ppm): 3.07 (t, 2H, CH2, J = 6.6 Hz); 3.77 (s, 3H, CH3); 3.89 (t, 2H, CH2, J = 6.3 Hz); 6.42 (s, 1H, NH); 6.71 (d, 1H,CHarom., J = 8.7 Hz); 6.83 (d, 2H, CHarom., J = 9.3 Hz); 7.02 (bs, 1H, NH); 7.35–7.41 (m, 3H, CHarom.); 7.55–7.67 (m, 3H, CHarom.); 7.99 (bs, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 24.6 (CH2), 44.6 (CH2), 55.2 (CH3), 111.3 (C), 111.6 (CH), 113.9 (CH, CH), 118.2 (CH), 118.5 (CH), 120.9 (CH), 122.7 (CH), 125.8 (CH), 127.3 (C), 128.6 (C), 131.5 (C), 136.3 (C), 156.5 (CH), 180.4 (C). HRMS (ESI) calc. for C18H19N3OS [M − H]−: 324.4279, found: 324.4298.
1-(2-(1H-Indol-3-yl)ethyl)-3-(4-methylphenyl)thiourea (3). This compound was synthesized as described previously [41].
1-(2-(1H-Indol-3-yl)ethyl)-3-(4-chlorophenyl)thiourea (4). This compound was synthesized as described previously [39].
1-(2-(1H-Indol-3-yl)ethyl)-3-benzoylthiourea (5). This compound was synthesized as described previously [42].
1-(2-(1H-indol-3-yl)ethyl)-3-(3,4-dichlorophenyl)thiourea (6). Yield 81%. m.p. 168–169 °C. 1H-NMR (DMSO-d6) δ (ppm): 2.99 (t, 2H, CH2, J = 7.2 Hz); 3.76 (q, 2H, CH2, J = 5.4 Hz); 6.98 (t, 1H, CHarom., J = 7.8 Hz); 7.08 (t, 1H,CHarom., J = 7.2 Hz); 7.18 (d, 1H, CHarom., J = 2.4 Hz); 7.30–7.36 (m, 2H, CHarom.); 7.52 (d, 1H,CHarom., J = 8.7 Hz); 7.62 (d, 1H, CHarom., J = 7.8 Hz); 7.85 (s, 1H, CHarom.); 7.99 (bs, 1H, NH); 9.72 (s, 1H, NH); 10.84 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 24.3 (CH2), 44.5 (CH2), 111.4 (C), 111.4 (CH), 118.3 (CH), 118.4 (CH), 120.9 (CH), 122.5 (CH), 122.8 (CH), 123.7 (CH), 125.3 (C), 127.2 (C), 130.2 (C), 130.5 (C), 136.3 (C), 139.7 (CH), 180.2 (C). HRMS (ESI) calc. for C17H15 Cl2N3S [M − H]−: 363.2921, found: 363.2943.
1-(2-(1H-Indol-3-yl)ethyl)-3-(3-bromophenyl)thiourea (7). Yield 79%. m.p. 152–154 °C. 1H-NMR (DMSO-d6) δ (ppm): 3.09 (t, 2H, CH2, J = 6.6 Hz); 3.97 (q, 2H, CH2, J = 5.7 Hz); 6.06 (s, 1H, NH); 6.81 (d, 1H, CHarom., J = 7.2 Hz); 7.00 (s, 1H, CHarom.); 7.05 (d, 1H, CHarom., J = 8.1 Hz); 7.08 (d, 1H, CHarom., J = 9.0 Hz); 7.13 (s, 1H, CHarom.); 7.20 (t, 1H, CHarom., J = 7.2 Hz); 7.29 (d, 1H,CHarom., J = 8.1 Hz); 7.36 (d, 1H, CHarom., J = 8.1 Hz); 7.40 (bs, 1H, NH); 7.57–7.60 (d, 1H, CHarom., J = 8.7 Hz); 8.03 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 24.3 (CH2), 44.5 (CH2), 111.4 (C), 111.5 (CH), 118.3 (CH), 118.5 (CH), 121.0 (CH), 121.1 (CH), 121.3 (CH), 122.8 (CH), 124.9 (CH), 126.3 (C), 127.2 (C), 130.4 (C), 136.3 (C), 141.2 (CH), 180.1 (C). HRMS (ESI) calc. for C17H16BrN3S [M − H]−: 373.2980, found: 373.2978.
1-(2-(1H-Indol-3-yl)ethyl)-3-(4-bromophenyl)thiourea (8). Yield 81%. m.p. 156–158 °C. 1H-NMR (DMSO-d6) δ (ppm): 3.09 (t, 2H, CH2, J = 6.6 Hz); 3.95 (t, 2H, CH2, J = 6.6 Hz); 6.24 (bs, 1H, NH); 6.71 (d, 1H, CHarom., J = 8.7 Hz); 6.97 (s, 1H, CHarom.); 7.11 (t, 1H, CHarom., J = 7.8 Hz); 7.17–7.29 (m, 3H, CHarom.); 7.39 (d, 1H,CHarom., J = 8.1 Hz); 7.56 (d, 1H, CHarom., J = 8.1 Hz); 7.62 (bs, 1H, NH); 8.01 (bs, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 24.4 (CH2), 44.5 (CH2), 111.4 (C), 111.5 (CH), 115.8 (CH), 118.2 (CH, CH), 118.5 (CH), 121.0 (CH), 122.8(CH), 124.7 (CH), 127.2 (CH), 131.3(C, C), 136.3 (C), 138.7, 180.1 (C). HRMS (ESI) calc. for C17H16BrN3S [M − H]−: 373.2980, found: 373.2978. Crystal data: crystal system monoclinic, space group Cc, unit cell dimensions a = 35.17(2), b = 10.863(4), c = 8.798(3) Å, β = 100.44(5) °, V = 3306(3) Å3; Z = 8, dc = 1.504 g/cm3, μ = 2.613 mm−1, F(000) = 1520. A crystal of dimensions 0.20 × 0.15 × 0.04 mm was used for intensity measurements. Within the θ range 2.98–26.31° (−43 ≤ h ≤ 33, −13 ≤ k ≤ 13, −7 ≤ l ≤ 10), 9510 reflections were collected. The 4871 unique reflections [R(int) = 0.0699] were used for the refinement of 397 parameters. Final R indices on F2 for 2599 observed reflections [I > 2σ(I)] were: R1 = 0.0615, wR2 = 0.0825, goodness-of-fit = 0.941, Δρmax/min = 0.57/−0.44 e Å−3.
1-(2-(1H-Indol-3-yl)ethyl)-3-cyclohexylthiourea (9). This compound was synthesized as described previously [40].
1-(2-(1H-Indol-3-yl)ethyl)-3-benzylthiourea (10). This compound was synthesized as described previously [43].
1-(2-(1H-Indol-3-yl)ethyl)-3-(2-fluorophenyl)thiourea (11). Yield 90%. m.p. 167–168 °C. 1H-NMR (DMSO-d6) δ (ppm): 3.09 (t, 2H, CH2, J = 6.6 Hz); 3.98(t, 2H, CH2, J = 6.3 Hz); 6.06 (bs, 1H, NH); 6.88–6.98 (m, 3H, CHarom.); 7.04–7.17 (m, 3H, CHarom.); 7.35 (d, 2H,CHarom., J = 8.1 Hz); 7.58 (d, 1H, CHarom., J = 7.5 Hz); 7.62 (bs, 1H, NH); 7.99 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 24.5 (CH2), 44.7 (CH2), 111.3 (C), 111.5 (CH), 115.6 (CH), 115.9 (d, J = 31.2 Hz, CH), 118.2 (CH), 118.5 (CH), 121.0 (CH), 122.8 (CH), 124.0 (d, J = 3.7 Hz, CH), 126.6 (d, J = 11.8 Hz, CH), 127.2 (C), 127.9 (d, J = 7.8 Hz, C), 136.3 (C), 157.5 (d, J = 245.3Hz, CH), 181.1 (C). HRMS (ESI) calc. for C17H16FN3S [M − H]−: 312.3924, found: 312.3921.
1-(2-(1H-Indol-3-yl)ethyl)-3-(3-fluorophenyl)thiourea (12). Yield 88%. m.p. 162–164 °C. 1H-NMR (DMSO-d6) δ (ppm): 3.10 (t, 2H, CH2, J = 6.6 Hz); 3.98 (q, 2H, CH2, J = 5.7 Hz); 6.13 (s, 1H, NH); 6.65 (d, 2H, CHarom., J = 6.9 Hz); 6.87 (d, 1H, CHarom., J = 6.9 Hz); 6.98 (s, 1H, CHarom.); 7.05–7.23 (m, 3H, CHarom.); 7.37 (d, 2H, CHarom., J = 8.1 Hz); 7.49 (s, 1H, NH); 7.58–7.60 (d, 2H, CHarom., J = 7.8 Hz); 8.01 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 24.4 (CH2), 44.5 (CH2), 109.2 (C), 110.3 (d, J = 24,9 Hz, CH), 111.4 (d, J = 21,3 Hz, CH), 111.5 (CH), 118.3 (CH), 118.5 (q, J = 3.9 Hz, CH), 121.0 (CH), 122.8 (CH), 127.2 (CH), 130.1 (q, J = 32 Hz, CH), 136.3 (C), 141.3 (d, J = 10.8 Hz, C), 160.3 (d, J = 242 Hz, C), 163.5 (CH), 180.1 (C). HRMS (ESI) calc. for C17H16FN3S [M − H]−: 312.3924, found: 312.3921.
1-(2-(1H-Indol-3-yl)ethyl)-3-(2-chlorophenyl)thiourea (13). Yield 79%. m.p. 170–172 °C. 1H-NMR (DMSO-d6) δ (ppm): 2.97 (t, 2H, CH2, J = 7.5 Hz); 3.75 (q, 2H, CH2, J = 5.7 Hz); 6.98 (t, 1H,CHarom., J = 7.5 Hz); 7.07 (t, 1H, CHarom., J = 6.9 Hz); 7.16–7.35 (m, 4H, CHarom.); 7.47 (d, 1H,CHarom., J = 7.8 Hz); 7.63 (d, 2H, CHarom., J = 6.9 Hz); 7.95 (bs, 1H, NH); 9.16 (s, 1H, NH); 10.83 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 24.5 (CH2), 44.7 (CH2), 111.3 (C), 111.5 (CH), 118.2 (CH), 118.5 (CH), 120.9 (CH), 122.7 (CH), 126.9 (CH), 127.1 (CH), 127.2 (CH), 128.9 (C), 129.1 (C), 129.4(C), 136.0(C), 136.2 (CH), 181.1 (C). HRMS (ESI) calc. for C17H16ClN3S [M − H]−: 328.8470, found: 328.8467.
1-(2-(1H-Indol-3-yl)ethyl)-3-(3-chlorophenyl)thiourea (14). Yield 85%. m.p. 169–171 °C. 1H-NMR (DMSO-d6) δ (ppm): 2.98 (t, 2H, CH2, J = 7.5 Hz); 3.77 (q, 2H, CH2, J = 5.7 Hz); 6.98 (t, 1H, CHarom., J = 8.1 Hz); 7.04–7.14 (m, 2H, CHarom.); 7.18 (d, 1H, CHarom., J = 2.4 Hz); 7.23–7.36 (m, 3H, CHarom.); 7.63 (d, 2H, CHarom., J = 8.4 Hz); 7.91 (bs, 1H, NH); 9.65 (s, 1H, NH); 10.84 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 24.4 (CH2), 44.5 (CH2), 111.4 (C), 111.5 (CH), 118.2 (CH, CH), 118.5 (CH), 120.96 (CH), 122.0 (CH), 122.8 (CH), 123.4 (CH), 127.2 (C), 130.1 (C), 132.6 (C), 136.3 (C), 140.9 (CH), 180.1 (C). HRMS (ESI) calc. for C17H16ClN3S [M − H]−: 328.8470, found: 328.8468.
1-(2-(1H-Indol-3-yl)ethyl)-3-(2-bromophenyl)thiourea (15). Yield 83%. m.p. 150–152 °C. 1H-NMR (DMSO-d6) δ (ppm): 2.98 (t, 2H, CH2, J = 7.2 Hz); 3.75 (q, 2H, CH2, J = 5.4 Hz); 6.98 (t, 1H,CHarom., J = 7.8 Hz); 7.07 (t, 1H, CHarom., J = 6.6 Hz); 7.12–7.17 (m, 2H, CHarom.); 7.32–7.38 (m, 2H, CHarom.); 7.57 (d, 1H, CHarom., J = 7.2 Hz); 7.62–7.66 (m, 2H, CHarom.); 7.91 (bs, 1H, NH); 9.11 (s, 1H, NH); 10.83 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 24.5 (CH2), 44.7 (CH2), 111.3 (C), 111.5 (CH), 118.2 (CH), 118.5 (CH), 120.2 (CH), 120.9 (CH), 122.8 (CH), 127.2 (CH), 127.4 (CH), 127.7 (C), 129.7 (C), 132.5 (C), 136.2 (C), 137.4 (CH), 181.1 (C). HRMS (ESI) calc. for C17H16BrN3S [M − H]−: 373.2980, found: 373.2978.
1-(2-(1H-Indol-3-yl)ethyl)-3-phenethylthiourea (16). This compound was synthesized as described previously [44].
1-(2-(1H-Indol-3-yl)ethyl)-3-(4-fluorophenyl)thiourea (17). This compound was synthesized as described previously [42].
1-(2-(1H-Indol-3-yl)ethyl)-3-allylthiourea (18). This compound was synthesized as described previously [40].
1-(2-(1H-Indol-3-yl)ethyl)-3-(2-methylallyl)thiourea (19). Yield 76%. m.p. 141–143 °C. 1H-NMR (DMSO-d6) δ (ppm): 1.67 (s, 3H, CH3); 2.90 (t, 2H, CH2, J = 7.2 Hz); 3.67 (bs, 2H, CH2); 3.98 (bs, 2H, CH2); 4.77 (s, 2H, =CH2); 6.98 (t, 1H, CHarom., J = 7.8 Hz); 7.08 (t, 1H, CHarom., J = 6.9 Hz); 7.14 (d, 1H, CHarom., J = 2.4 Hz); 7.33 (d, 1H, CHarom., J = 8.1 Hz); 7.42 (bs, 1H, NH); 7.56 (bs, 1H, NH); 7.59 (d, 1H, CHarom., J = 7.8 Hz); 10.82 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 20.3 (CH3), 24.9 (CH2), 44.4 (C), 48.7 (CH2), 109.9 (CH2), 111.3 (CH2), 111.6 (C), 118.2 (CH), 118.46 (CH), 120.9 (CH), 122.7 (CH), 127.3 (C), 136.2 (C), 142.4 (CH), 182.5 (C). HRMS (ESI) calc. for C15H19N3S [M − H]−: 272.3964, found: 272.3486.
1-(2-(1H-Indol-3-yl)ethyl)-3-ethoxycarbonylthiourea (20). This compound was synthesized as described previously [43].
1-(2-(1H-Indol-3-yl)ethyl)-3-(4-iodophenyl)thiourea (21). Yield 88%. m.p. 162–165 °C. 1H-NMR (DMSO-d6) δ (ppm): 2.97 (t, 2H, CH2, J = 7.5 Hz); 3.75 (q, 2H, CH2, J = 6.0 Hz); 6.98 (t, 1H, CHarom., J = 6.9 Hz); 7.07 (t, 1H,CHarom., J = 6.9 Hz); 7.20 (m, 3H, CHarom.); 7.34 (d, 1H, CHarom., J = 8.1 Hz); 7.61 (m, 3H, CHarom.); 7.83 (bs, 1H, NH); 9.57 (s, 1H, NH); 10.84 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 24.39 (CH2), 44.53 (CH2), 111.34 (C), 111.48 (CH), 118.22 (CH, CH), 118.46 (CH), 120.95 (CH, CH), 122.81 (CH), 124.92 (CH), 127.21 (CH), 136.26 (C), 137.13 (C, C), 139.18 (CH), 180.03 (C). HRMS (ESI) calc. for C17H16IN3S [M − H]−: 420.2985, found: 420.2987.
1-(2-(1H-Indol-3-yl)ethyl)-3-ethylthiourea (22). This compound was synthesized as described previously [45].
1-(2-(1H-Indol-3-yl)ethyl)-3-(3-chloro-4-methylphenyl)thiourea (23). Yield 83%. m.p. 161–163 °C. 1H-NMR (DMSO-d6) δ (ppm): 2.28 (s, 3H, CH3); 2.98 (t, 2H, CH2, J = 7.5 Hz); 3.76 (q, 2H, CH2, J = 5.7 Hz); 6.98 (t, 1H, CHarom., J = 8.1 Hz); 7.08 (t, 1H, CHarom., J = 6.9 Hz); 7.13–7.18 (m, 2H, CHarom.); 7.25 (d, 1H, CHarom., J = 8.4 Hz); 7.34 (d, 1H,CHarom., J = 7.8 Hz); 7.57 (d, 1H,CHarom., J = 2.1 Hz); 7.62 (d, 1H, CHarom., J = 7.5 Hz); 7.81 (bs, 1H, NH); 9.55 (s, 1H, NH); 10.84 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 18.9 (CH3), 24.4 (CH2), 44.5 (CH2), 111.3 (C), 111.5 (CH), 118.2 (CH), 118.5 (CH), 120.9 (CH), 121.7 (CH), 122.8 (C), 123.0 (CH), 127.2 (CH), 130.7 (C), 130.9 (C), 132.6 (C), 136.3 (C), 138.4 (CH), 180.2 (C). HRMS (ESI) calc. for C18H18ClN3S [M − H]−: 342.8736, found: 342.8769.
1-(2-(1H-Indol-3-yl)ethyl)-3-(3-(trifluoromethyl)phenyl)thiourea (24). Yield 92%. m.p. 169–171 °C. 1H-NMR (DMSO-d6) δ (ppm): 2.99 (t, 2H, CH2, J = 7.2 Hz); 3.79 (q, 2H, CH2, J = 5.7 Hz); 6.98 (t, 1H, CHarom., J = 7.8 Hz); 7.08 (t, 1H, CHarom., J = 7.8 Hz); 7.18 (d, 1H,CHarom., J = 2.1 Hz); 7.35 (d, 1H, CHarom., J = 8.1 Hz); 7.40 (d, 1H, CHarom., J = 7.8 Hz); 7.50 (d, 1H, CHarom., J = 7.8 Hz); 7.64 (d, 1H, CHarom., J = 7.8 Hz); 7.98 (s, 1H, CHarom.); 7.98 (bs, 1H, NH); 9.79 (s, 1H, NH); 10.85 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 24.4 (CH2), 44.4 (CH2), 79.2 (C), 111.4 (C), 111.4 (CH), 118.2 (CH), 118.4 (CH), 118.6 (CH), 121.0 (q, 3.8 Hz, CH), 122.8 (q, J = 3.8 Hz, CH), 125.8 (q, J = 272 Hz, C), 126.1 (CH), 127.2 (CH), 128.8 (C), 129.5 (q, J = 33.6 Hz, C), 136.3 (C), 140.4, 180.4 (C). HRMS (ESI) calc. for C18H16F3N3S [M − H]−: 362.3999, found: 362.3940.
1-(2-(1H-Indol-3-yl)ethyl)-3-methylthiourea (25). This compound was synthesized as described previously [46].
1-(2-(1H-Indol-3-yl)ethyl)-3-(3-chloro-6-methylphenyl)thiourea (26). Yield 87%. m.p. 163–165 °C. 1H-NMR (DMSO-d6) δ (ppm): 2.13 (s, 3H, CH3); 2.96 (t, 2H, CH2, J = 7.2 Hz); 3.73 (q, 2H, CH2, J = 5.1 Hz); 6.98 (t, 1H,CHarom., J = 6.3 Hz); 7.07 (t, 1H, CHarom., J = 6.9 Hz); 7.15–7.26 (m, 3H, CHarom.); 7.34 (d, 1H, CHarom., J = 8.1 Hz); 7.37 (s, 1H, CHarom.); 7.63 (d, 1H, CHarom., J = 7.8 Hz); 7.71 (bs, 1H, NH); 9.11 (s, 1H, NH); 10.82 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 17.2 (CH3), 24.6 (CH2), 44.8 (CH2), 111.3 (C), 111.6 (CH), 118.2 (CH), 118.5 (CH), 120.9 (CH), 122.7 (CH), 125.8 (C), 127.0 (CH), 127.3 (CH), 129.7 (C), 131.8 (C), 133.1 (C), 136.2 (C), 138.6 (CH), 180.9 (C). HRMS (ESI) calc. for C18H18ClN3S [M − H]−: 342.8736, found: 342.8772.
1-(2-(1H-Indol-3-yl)ethyl)-3-(4-butyl-2-methylphenyl)thiourea (27). Yield 77%. m.p. 172–174 °C. 1H-NMR (DMSO-d6) δ (ppm): 0.8 (t, 3H, CH3, J = 7.5 Hz); 1.25–1.37 (m, 2H, CH2, J = 7.5 Hz); 1.49–1.59 (m, 2H, CH2, J = 7.2 Hz); 2.11 (s, 3H, CH3); 2.49 (s, 2H, CH2); 2.93 (t, 2H, CH2, J = 7.5 Hz); 3.69 (q, 2H, CH2, J = 6.0 Hz); 6.94–7.11 (m, 6H, CHarom.); 7.25 (bs, 1H, NH); 7.33 (d, 1H, CHarom., J = 8.1 Hz); 7.63 (d, 1H, CHarom., J = 7.8 Hz); 9.01 (s, 1H, NH); 10.79 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 13.8 (CH3), 17.6 (CH2), 21.8 (CH2), 24.8 (CH3), 33.0 (CH2), 34.3 (CH2), 40.3 (CH2), 111.3 (C), 111.6 (CH), 118.1 (CH), 118.5 (CH), 120.9 (CH, CH), 122.6 (C), 126.2 (C), 127.3 (CH), 127.6 (CH), 130.4 (C), 134.5 (C), 136.2 (C), 140.6, 180.8 (C). HRMS (ESI) calc. for C22H27N3S [M − H]−: 364.5348, found: 364.5360.
1-(2-(1H-Indol-3-yl)ethyl)-3-(3-chloro-4-fluorophenyl)thiourea (28). Yield 91%. m.p. 162–164 °C. 1H-NMR (DMSO-d6) δ (ppm): 2.98 (t, 2H, CH2, J = 7.2 Hz); 3.76 (q, 2H, CH2, J = 5.7 Hz); 6.98 (t, 1H, CHarom., J = 8.1 Hz); 7.08 (t, 1H,CHarom., J = 7.2 Hz); 7.18 (d, 1H, CHarom., J = 2.1 Hz); 7.24–7.30 (m, 1H, CHarom.); 7.34 (d, 2H,CHarom., J = 8.7 Hz); 7.62 (d, 1H,CHarom., J = 7.8 Hz); 7.72 (d, 1H, CHarom., J = 6.6 Hz); 7.87 (bs, 1H, NH); 9.60 (s, 1H, NH); 10.84 (s, 1H, NH). 13C-NMR (DMSO-d6) δ (ppm): 24.4 (CH2), 44.5 (CH2), 111.4 (C), 111.5 (CH), 116.6 (d, J = 22 Hz, CH), 118.2 (CH), 118.4 (d, J = 18.6 Hz, CH), 121.0 (CH), 122.8 (CH), 123.6 (CH), 124.9 (d, J = 7.2 Hz, C), 127.2 (C), 136.3 (C), 136.5 (d, J = 3.3 Hz, C), 152.2 (C), 155.5 (d, J = 242.6 Hz, CH), 180.4 (C). HRMS (ESI) calc. for C17H15ClFN3S [M − H]−: 346.8375, found: 346.8378. Crystal data: crystal system triclinic, space group P-1, unit cell dimensions a = 8.592(2) Å, b = 12.4914(3) Å, c = 15.309(3) Å, α = 85.32(2)°, β = 81.17(2)°, γ = 88.87(2)°, V = 1678.1(6) Å3; Z = 4, dc = 1.428 g/cm3, μ = 0.377 mm−1, F(000) = 720. A crystal of dimensions 0.40 × 0.20 × 0.03 mm was used for intensity measurements. Within the θ range 2.04–28.54° [−11 ≤ h ≤ 11, −16 ≤ k ≤ 16, −20 ≤ l ≤ 20], reflections collected 22058. The 5859 unique reflections [R(int) = 0.0360] were used for the refinement of 427 parameters. Final R indices on F2 for 5698 observed reflections [I > 2σ(I)] were: R1 = 0.0483, wR2 = 0.1019, goodness-of-fit 1.018, Δρmax/min = 0.76/−0.80 e Å−3.
3.3. Biology
3.3.1. In Vitro Evaluation of Antimicrobial Activity
The antibacterial activity of compounds was tested against a series of Gram-positive bacteria: Staphylococcus aureus ATCC 4163, Staphylococcus aureus ATCC 25923, Staphylococcus aureus ATCC 29213, Staphylococcus aureus ATCC 6538,Staphylococcus epidermidis ATCC 12228, Bacillus subtilis ATCC 6633, Bacillus cereus ATCC 11778, Enterococcus hirae ATCC 10541, Micrococcus luteus ATCC 9341, Micrococcus luteus ATCC 10240 and Gram-negative rods: Escherichia coli ATCC 10538, Escherichia coli ATCC 25922, Escherichia coli NCTC 8196, Proteus vulgaris NCTC 4635, Pseudomonas aeruginosa ATCC 15442, Pseudomonas aeruginosa NCTC 6749, Pseudomonas aeruginosa ATCC 27853, Bordetella bronchiseptica ATCC 4617. Antifungal activity was tested against yeasts: Candida albicans ATCC 10231, Candida albicans ATCC 90028, Candida parapsilosis ATCC 220191. Microorganisms used in this study were obtained from the collection of the Department of Pharmaceutical Microbiology, Medical University of Warsaw, Poland.
3.3.2. Media, Growth Conditions and Antimicrobial Activity Assays
Antimicrobial activity was examined by the disc diffusion and Minimal Inhibitory Concentration (MIC) method under standard conditions, using Mueller–Hinton II agar medium (Becton Dickinson, Franklin Lakes, NJ, USA) for bacteria and RPMI agar with 2% glucose (Sigma, St. Louis, MO, USA) for yeasts, according to CLSI (previously NCCLS) guidelines [31]. Solutions containing the tested agents were prepared in methanol or DMSO. For the disc diffusion method, sterile paper discs (9 mm diameter, Whatman No. 3 chromatography filter paper) were dripped with the compound solutions tested to obtain 400 μg of substance per disc. Dry discs were placed on the surface of an appropriate agar medium. The results (diameter of the growth inhibition zone) were read after 18 h of incubation at 35 °C. MIC’s were examined by the twofold serial agar dilution technique [32]. Concentrations of the tested compounds in solid medium ranged from 3.125 to 400 μg/mL. The final inoculum of studied organisms was 104 CFU/mL (colony forming units per mL), except the final inoculum for E. hirae ATCC 10541, which was 105 CFU/mL. Minimal inhibitory concentrations were read off after 18 h of incubation at 35 °C.
3.3.3. Inhibitory Activities against DNA Topoisomerase IV and DNA Gyrase
S. aureus topoisomerase IV decatenation assay was performed with 200 ng of kinetoplast DNA (Inspiralis Limited, Norwich, UK) as a substrate. S. aureus DNA gyrase supercoiling assay was performed with 0.5 µg of relaxed pBR322 DNA (Inspiralis) as a substrate. Enzymes activity was detected by incubation for 30 min at 37 °C in a total reaction volume of 30 µL and in the presence of different concentrations of tested compounds and the reference ciprofloxacin at a concentration of 32 µg/mL. The reactions were terminated by adding an equal volume of STEB buffer (40% sucrose, 100 mM Tris-HCl pH 8, 1 mM EDTA, 0.5 mg/mL bromophenol blue), followed by extraction with 1 volume of chloroform/isoamyl alcohol (24:1). Then, 20 µL of the aqueous phase of each sample was loaded onto a 1% agarose gel. Following electrophoresis, gels were stained with ethidium bromide, visualized under UV light in a transilluminator (ChemiDoc MP, BioRad, Hercules, CA, USA) and analyzed by the Image Lab 6.0 software (Image Lab’s, Cleveland, OH, USA).
3.3.4. Cells and Viruses
Cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Cell lines supporting the multiplication of RNA and DNA viruses were the following: CD4+ human T-cells containing an integrated HTLV-1 genome (MT-4); Madin Darby Bovine Kidney (MDBK) [ATCC CCL 22 (NBL-1) Bos taurus]; baby hamster kidney (BHK-21) [ATCC CCL 10 (C-13) Mesocricetus auratus] and monkey kidney (Vero-76) [ATCC CRL 1587 Cercopithecus aethiops]. Human Immunodeficiency Virus type-1 (HIV-1) IIIB laboratory strain was obtained from the supernatant of the persistently infected H9/IIIB cells (NIH 1983). Viruses representative of ssRNA+ were: (i) Flaviviridae: yellow fever virus (YFV) [strain 17-D vaccine (Stamaril Pasteur J07B01)], bovine viral diarrhoea virus (BVDV) [strain NADL (ATCC VR-534)]; (ii) Picornaviridae: enterovirus B [coxsackievirus B5 (CV-B5), strain Ohio-1 (ATCC VR-29)], and enterovirus C [poliovirus type-1 (Sb-1), Sabin strain Chat (ATCC VR-1562)]. Viruses representative of ssRNA- were: (iii) Pneumoviridae: human respiratory syncytial virus (RSV) [strain A2 (ATCC VR-1540)]; (iv) Rhabdoviridae: vesicular stomatitis virus (VSV) [lab strain Indiana (ATCC VR 1540)]. The virus representative of dsRNA was: (v) Reoviridae: reovirus type-1 (Reo-1) [simian virus 12, strain 3651 (ATCC VR-214)]. DNA virus representatives were: v) Poxviridae: vaccinia virus (VV) [vaccine strain Elstree-Lister (ATCC VR-1549)]; (vi) Herpesviridae: human herpes 1 (HSV-1) [strain KOS (ATCC VR-1493)]. Mutants carrying NNRTI mutations used: Y181C mutant (NIH-N119) of HIV-1 derives from an AZT-sensitive clinical isolate passaged initially in CEM and then in MT-4 cells, in the presence of nevirapine (up to 10 µM); K103N + Y181C mutant (NIH A17) derives from an IIIB strain passaged in H9 cells in the presence of BI-RG 587 (up to 1 µM); K103R + V179D + P225H mutant (EFVR) derives from an IIIB strain passaged in MT-4 cells in the presence of efavirenz (up to 2 µM). N119, A17 and EFVR stock solutions had titres of 1.2 × 108, 2.1 × 107, and 4.0 × 107 CCID50/mL, respectively. Mutants carrying NRTI mutations used: AZTR strain (67N, 70R, 215F, 219Q); MDR strain (74V, 41L, 106A, 215Y).
3.3.5. Cytotoxicity Assays
Exponentially growing MT-4 cells were seeded at an initial density of 4 × 105 cells/mL in 96-well plates in RPMI-1640 medium, supplemented with 10% fetal bovine serum (FBS), 100 units/mLpenicillin G and 100 μg/mL streptomycin. MDBK and BHK cells were seeded in 96-well plates at an initial density of 6 × 105 and 1 × 106 cells/mL, respectively, in minimum essential medium with Earle’s salts (MEM-E), l-glutamine, 1 mM sodium pyruvate and 25 mg/L kanamycin, supplemented with 10% horse serum (MDBK) or 10% foetal bovine serum (FBS) (BHK). Vero-76 cells were seeded in 96-well plates at an initial density of 4 × 105 cells/mL, in Dulbecco’s Modified Eagle Medium (D-MEM) with l-glutamine and 25 mg/L kanamycin, supplemented with 10% FBS. Cell cultures were then incubated at 37 °C in a humidified, 5% CO2 atmosphere, in the absence or presence of serial dilutions of test compounds. Cell viability was determined after 48–96 h at 37 °C by MTT method for MT-4, Vero-76, MDBK and BHK [47].
Cell lines derived from human haematological tumours [CD4+ human acute T-lymphoblastic leukaemia (CCRF-CEM), human splenic B-lymphoblastoid cells (WIL-2NS), human acute B-lymphoblastic leukaemia (CCRF-SB)] were seeded at an initial density of 1 × 105 cells/mL in 96 well plates in RPMI-1640 medium supplemented with 10% foetal calf serum (FCS), 100 units/mL penicillin G and 100 μg/mL streptomycin. Cell viability was determined after 96 h at 37 °C by the MTT method. All cell cultures were then incubated at 37 °C in a humidified, 5% CO2 atmosphere, in the absence or presence of serial dilutions of test compounds in culture medium. Before dilutions, compounds were dissolved in DMSO at 100 mM.
3.3.6. Antiviral Assays
Activity against HIV-1 wt and mutant strains (N119, A17, EFVR, AZTR, MDR) was based on inhibition of virus-induced cytopathogenicity in exponentially growing MT-4 cell acutely infected with a multiplicity of infection (m.o.i.) of 0.01. Briefly, 50 μL of RPMI containing 1 × 104 MT-4 cells were added to each well of flat-bottom microtitre trays, containing 50 μL of RPMI without or with serial dilutions of extracts or fractions. Then, 20 μL of a HIV-1 suspension containing 100 CCID50 were added. After 4-days incubation at 37 °C, cell viability was determined by the MTT method.
Compound’s activity against YFV and Reo-1 was based on inhibition of virus-induced cytopathogenicity in BHK-21 cells acutely infected with a m.o.i. of 0.01. Compound’s activity against BVDV was based on inhibition of virus-induced cytopathogenicity in MDBK cells acutely infected at a m.o.i. of 0.01. After a 3 or 4-days incubation at 37 °C, cell viability was determined by the MTT method, as described earlier [47]. Compound’s activity against CV-B5, Sb-1, VSV, VV, HSV-1 and RSV was determined by plaque reduction assays in infected Vero-76 cell monolayers, as described earlier [48,49]. Concentrations resulting in 50% inhibition (CC50 or EC50) were determined by linear regression analysis. Efavirenz, 2′-C-methylguanosine, 2′-C-methylcytidine, 6-azauridine, mycophenolic acid and acycloguanosine were used as reference inhibitors.
Acknowledgments
We gratefully acknowledge the Sardinia Regional Government for the financial support of Silvia Madeddu through his scholarship (P.O.R. Sardegna F.S.E. Operational Programme of the Autonomous Region of Sardinia, European Social Fund 2007–2013).
Supplementary Materials
The following are available online.
Author Contributions
Conceptualization, D.S. and M.S. (Marta Struga); Formal Analysis, S.M.; J.S., G.K.-T.; Investigation, G.S., S.M., G.G., M.W., A.E.K., O.S., T.L. and J.S.; Resources, G.K.-T.; Data Curation, G.S., S.P., D.S.; Writing—Original Draft Preparation, G.S.; Writing—Review & Editing, G.G.; Supervision, D.S.; Project Administration, M.S. (Marta Struga); Funding Acquisition, M.S. (Marta Struga), P.T., M.S. (Michał Skrzycki). All authors approved the final version of the manuscript.
Funding
This work was supported by the Medical University of Warsaw and carried out with the use of CePT infrastructure financed by the European Union—the European Regional Development Fund within the Operational Programme Innovative Economy for 2007–2013.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds are available from the authors.
References
- 1.UNAIDS DATA 2017. [(accessed on 20 July 2017)]; Available online: www.unaids.org/en/resources/documents/2017/2017_data_book.
- 2.Richman D.D. Antiviral drug resistance. Antivir. Res. 2006;71:117–121. doi: 10.1016/j.antiviral.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Kaushik N.K., Kaushik N., Attri P., Kumar N., Kim C.H., Verma A.K., Choi E.H. Biomedical Importance of Indoles. Molecules. 2013;18:6620–6662. doi: 10.3390/molecules18066620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qu S.J., Wang G.F., Duan W.H., Yao S.Y., Zuo J.P., Tan C.H., Zhu D.Y. Tryptamine derivatives as novel non-nucleosidic inhibitors against hepatitis B virus. Bioorg. Med. Chem. 2011;19:3120–3127. doi: 10.1016/j.bmc.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Zhang N., Zhang X., Zhu J., Turpoff A., Chen G., Morrill C., Huang S., Lennox W., Kakarla R., Liu R., et al. Structure-activity relationship (SAR) optimization of 6-(indol-2-yl)pyridine-3-sulfonamides: Identification of potent, selective, and orally bioavailable small molecules targeting hepatitis C (HCV) NS4B. J. Med. Chem. 2014;57:2121–2135. doi: 10.1021/jm401621g. [DOI] [PubMed] [Google Scholar]
- 6.Lozano V., Aguado L., Hoorelbeke B., Renders M., Camarasa M.J., Schols D., Balzarini J., San-Félix A., Pérez-Pérez M.J. Targeting HIV entry through interaction with envelope glycoprotein 120 (gp120): Synthesis and antiviral evaluation of 1,3,5-triazines with aromatic amino acids. J. Med. Chem. 2011;54:5335–5348. doi: 10.1021/jm200560r. [DOI] [PubMed] [Google Scholar]
- 7.Bouchikhi F., Rossignol E., Sancelme M., Aboab B., Anizon F., Fabbro D., Prudhomme M., Moreau P. Synthesis and biological evaluation of diversely substituted indolin-2-ones. Eur. J. Med. Chem. 2008;43:2316–2322. doi: 10.1016/j.ejmech.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 8.Huang M.L., Benson M.A., Shin S.B.Y., Torres V.J., Kirshenbaum K. Amphiphilic Cyclic Peptoids That Exhibit Antimicrobial Activity by Disrupting Staphylococcus aureus Membranes. Eur. J. Org. Chem. 2013;17:3560–3566. doi: 10.1002/ejoc.201300077. [DOI] [Google Scholar]
- 9.Williams J.D., Nguyen S.T., Gu S., Ding X., Butler M.M., Tashjian T.F., Opperman T.J., Panchal R.G., Bavari S., Peet N.P., et al. Potent and broad-spectrum antibacterial activity of indole-based bisamidine antibiotics: Synthesis and SAR of novel analogs of MBX 1066 and MBX 1090. Bioorg. Med. Chem. 2013;21:7790–7806. doi: 10.1016/j.bmc.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minvielle M.J., Eguren K., Melander C. Highly active modulators of indole signaling alter pathogenic behaviors in Gram-negative and Gram-positive bacteria. Chem. Eur. J. 2013;19:17595–17602. doi: 10.1002/chem.201303510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh K., Singh K., Wan B., Franzblau S., Chibale K., Balzarini J. Facile transformation of Biginelli pyrimidin-2(1H)-ones to pyrimidines. In vitro evaluation as inhibitors of Mycobacterium tuberculosis and modulators of cytostatic activity. Eur. J. Med. Chem. 2011;46:2290–2294. doi: 10.1016/j.ejmech.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 12.El-Sawy E.R., Mandour A.H., Mahmoud K., Islam I.E., Abo-Salem H.M. Synthesis, antimicrobial and anti-cancer activities of some new N-ethyl, N-benzyl and N-benzoyl-3-indolyl heterocycles. Acta Pharm. 2012;62:157–179. doi: 10.2478/v10007-012-0020-3. [DOI] [PubMed] [Google Scholar]
- 13.Rezaei Z., Firouzabadi H., Iranpoor N., Ghaderi A., Jafari M.R., Jafari A.A., Zare H.R. Design and one-pot synthesis of alpha-aminophosphonates and bis(alpha-aminophosphonates) by iron(III) chloride and cytotoxic activity. Eur. J. Med. Chem. 2009;44:4266–4275. doi: 10.1016/j.ejmech.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 14.Kumar R., Gupta L., Pal P., Khan S., Singh N., Katiyar S.B., Meena S., Sarkar J., Sinha S., Kanaujiya J.K., et al. Synthesis and cytotoxicity evaluation of (tetrahydro-beta-carboline)-1,3,5-triazine hybrids as anticancer agents. Eur. J. Med. Chem. 2010;45:2265–2276. doi: 10.1016/j.ejmech.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Akkoç M.K., Yüksel M.Y., Durmaz İ., Atalay R.C. Design, synthesis, and biological evaluation of indole-based 1,4-disubstituted piperazines as cytotoxic agents. Turk. J. Chem. 2012;36:515–525. doi: 10.3906/kim-1111-5. [DOI] [Google Scholar]
- 16.Ji Y.Y., Zhu Y.M., Wang J.W. GS-2, a pyrazolo[1,5-a]indole derivative with inhibitory activity of topoisomerases, exerts its potent cytotoxic activity by ROS generation. Environ. Toxicol. Pharmacol. 2013;36:1186–1196. doi: 10.1016/j.etap.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 17.Yang D., Wang P., Liu J., Xing H., Liu Y., Xie W., Zhao G. Design, synthesis and evaluation of novel indole derivatives as AKT inhibitors. Bioorg. Med. Chem. 2014;22:366–373. doi: 10.1016/j.bmc.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 18.Yamazaki Y., Kawano Y., Yamanaka A., Maruyama S. N-[(Dihydroxyphenyl)acyl]serotonins as potent inhibitors of tyrosinase from mouse and human melanoma cells. Bioorg. Med. Chem. Lett. 2009;19:4178–4182. doi: 10.1016/j.bmcl.2009.05.115. [DOI] [PubMed] [Google Scholar]
- 19.Mellini P., Kokkola T., Suuronen T., Salo H.S., Tolvanen L., Mai A., Lahtela-Kakkonen M., Jarho E.M. Screen of Pseudopeptidic Inhibitors of Human Sirtuins 1-3: Two Lead Compounds with Antiproliferative Effects in Cancer Cells. J. Med. Chem. 2013;56:6681–6695. doi: 10.1021/jm400438k. [DOI] [PubMed] [Google Scholar]
- 20.Xu L., Russu W.A. Molecular docking and synthesis of novel quinazoline analogues as inhibitors of transcription factors NF-κB activation and their anti-cancer activities. Bioorg. Med. Chem. 2013;21:540–546. doi: 10.1016/j.bmc.2012.10.051. [DOI] [PubMed] [Google Scholar]
- 21.Struga M., Rosolowski S., Kossakowski J., Stefanska J. Synthesis and microbiological activity of thiourea derivatives of 4-azatricyclo[5.2.2.0(2,6)]undec-8-ene-3,5-dione. Arch. Pharm. Res. 2010;33:47–54. doi: 10.1007/s12272-010-2223-9. [DOI] [PubMed] [Google Scholar]
- 22.Vega-Pérez J.M., Periñán I., Argandoña M., Vega-Holm M., Palo-Nieto C., Burgos-Morón E., López-Lázaro M., Vargas C., Nieto J.J., Iglesias-Guerra F. Isoprenyl-thiourea and urea derivatives as new farnesyl diphosphate analogues: Synthesis and in vitro antimicrobial and cytotoxic activities. Eur. J. Med. Chem. 2012;58:591–612. doi: 10.1016/j.ejmech.2012.10.042. [DOI] [PubMed] [Google Scholar]
- 23.Modh R.P., Patel A.C., Mahajan D.H., Pannecouque C., De Clercq E., Chikhalia K.H. Synthesis and evaluation of novel 4-substituted styrylquinazolines as potential antimicrobial agents. Arch. Pharm. 2012;345:964–972. doi: 10.1002/ardp.201200291. [DOI] [PubMed] [Google Scholar]
- 24.Ranise A., Spallarossa A., Schenone S., Bruno O., Bondavalli F., Vargiu L., Marceddu T., Mura M., La Colla P., Pani A. Design, synthesis, SAR, and molecular modeling studies of acylthiocarbamates: A novel series of potent non-nucleoside HIV-1 reverse transcriptase inhibitors structurally related to phenethylthiazolylthiourea derivatives. J. Med. Chem. 2003;46:768–781. doi: 10.1021/jm0209984. [DOI] [PubMed] [Google Scholar]
- 25.Karakuş S., GünizKüçükgüzel S., Küçükgüzel I., De Clercq E., Pannecouque C., Andrei G., Snoeck R., Sahin F., Bayrak O.F. Synthesis, antiviral and anticancer activity of some novel thioureas derived from N-(4-nitro-2-phenoxyphenyl)-methanesulfonamide. Eur. J. Med. Chem. 2009;44:3591–3595. doi: 10.1016/j.ejmech.2009.02.030. [DOI] [PubMed] [Google Scholar]
- 26.Saeed S., Rashid N., Jones P.G., Ali M., Hussain R. Synthesis, characterization and biological evaluation of some thiourea derivatives bearing benzothiazole moiety as potential antimicrobial and anticancer agents. Eur. J. Med. Chem. 2010;45:1323–1331. doi: 10.1016/j.ejmech.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 27.Kumbhare R.M., Dadmal T., Kosurkar U., Sridhar V., Rao J.V. Synthesis and cytotoxic evaluation of thiourea and N-bis-benzothiazole derivatives: A novel class of cytotoxic agents. Bioorg. Med. Chem. Lett. 2012;22:453–455. doi: 10.1016/j.bmcl.2011.10.106. [DOI] [PubMed] [Google Scholar]
- 28.Böhm H.J., Banner D., Bendels S., Kansy M., Kuhn B., Müller K., Obst-Sander U., Stahl M. Fluorine in medicinal chemistry. ChemBioChem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 29.Müller K., Faeh C., Diederich F. Fluorine in pharmaceuticals: Looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 30.Hagmann W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 31.Clinical and Laboratory Standards Institute . Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically. Clinical and Laboratory Standards Institute; Wayne, PA, USA: 2006. Approved Standard M7-A7. [Google Scholar]
- 32.Clinical and Laboratory Standards Institute . Performance Standards for Antimicrobial Disc Susceptibility Tests. Clinical and Laboratory Standards Institute; Wayne, PA, USA: 2006. Approved Standard M2-A9. [Google Scholar]
- 33.Stefanska J., Szulczyk D., Koziol A.E., Miroslaw B., Kedzierska E., Fidecka S., Busonera B., Sanna G., Giliberti G., La Colla P., et al. Disubstituted thiourea derivatives and their activity on CNS: Synthesis and biological evaluation. Eur. J. Med. Chem. 2012;55:205–213. doi: 10.1016/j.ejmech.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 34.Houe H. Economic impact of BVDV infection in dairies. Biologicals. 2003;31:137–143. doi: 10.1016/S1045-1056(03)00030-7. [DOI] [PubMed] [Google Scholar]
- 35.Kemball C.C., Alirezaei M., Whitton J.L. Type B coxsackieviruses and their interactions with the innate and adaptive immune systems. Future Microbiol. 2010;5:1329–1347. doi: 10.2217/fmb.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wright M., Piedimonte G. Respiratory syncytial virus prevention and therapy: Past, present, and future. Pediatr. Pulmonol. 2011;46:324–347. doi: 10.1002/ppul.21377. [DOI] [PubMed] [Google Scholar]
- 37.Van Laethem K., De Luca A., Antinori A., Cingolani A., Perno C.F., Vandamme A.M. A genotypic drug resistance interpretation algorithm that significantly predicts therapy response in HIV-1-infected patients. Antivir. Ther. 2002;7:123–129. [PubMed] [Google Scholar]
- 38.Tang M.W., Shafer R.W. HIV-1 antiretroviral resistance: Scientific principles and clinical applications. Drugs. 2012;72:e1–e25. doi: 10.2165/11633630-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheldrick G.M. A short history of SHELX. Acta Crystal. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 40.Novak L., Hanania M., Kovacs P., Kolonits P., Szantay C. Preparation of novel cyanoguanidine derivatives of tryptamines. Synthesis. 2001;1:0108–0118. doi: 10.1055/s-2001-9752. [DOI] [Google Scholar]
- 41.Aychiluhim T.M., Rao V.R. Multi-compnent synthesis of 3-{3-[2-(1H-Indol-3-yl)ethyl]}-2,3-dihydro-2-(aryliminothiazol-4-yl)-2H-chromen-2-ones. Org. Prep. Proced. Int. 2014;46:66–75. doi: 10.1080/00304948.2014.866469. [DOI] [Google Scholar]
- 42.Kumbhare R.M., Kumar V.K., Dadmal T.L., Kosurkar U.B., Appalanaidu K. Synthesis and Study the Antimicrobial Activity of Novel 2-(1H-indol-3-yl)-N-(3,4-diphenylthiazol-2(3H)-ylidene) Ethanamine Derivatives. Med. Chem. 2013;9:287–293. doi: 10.2174/1573406411309020011. [DOI] [PubMed] [Google Scholar]
- 43.Szulczyk D., Kędzierska E., Fidecka S., Koziol A.E., Struga M. 5-HT2 receptor affinity, docking studies and pharmacological evaluation of novel indole-derived thioureas. Farmacol. Rep. in press. [Google Scholar]
- 44.National Center for Biotechnology Information PubChem Substance Database; SID=337029832. [(accessed on 29 June 2018)]; Available online: https://pubchem.ncbi.nlm.nih.gov/substance/337029832.
- 45.Rogers S.A., Whitehead D.C., Mullikin T., Melander C. Synthesis and bacterial biofilm inhibition studies of ethyl N-(2-phenethyl) carbamate derivatives. Org. Biomol. Chem. 2010;8:3857–3859. doi: 10.1039/c0ob00063a. [DOI] [PubMed] [Google Scholar]
- 46.Gaspari P., Banerjee T., Malachowski W.P., Muller A.J., Prendergast G.C., DuHadaway J., Bennett S., Donovan A.M. Structure-activity study of brassinin derivatives as indoleamine 2,3-dioxygenase inhibitors. J. Med. Chem. 2006;49:684–692. doi: 10.1021/jm0508888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pauwels R., Balzarini J., Baba M., Snoeck R., Schols D., Herdewijn P., Desmyter J., De Clercq E. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J. Virol. Methods. 1988;20:309–321. doi: 10.1016/0166-0934(88)90134-6. [DOI] [PubMed] [Google Scholar]
- 48.Sanna G., Farci P., Busonera B., Murgia G., La Colla P., Giliberti G. Antiviral properties from plants of the Mediterranean flora. Nat. Prod. Res. 2015;29:2065–2070. doi: 10.1080/14786419.2014.1003187. [DOI] [PubMed] [Google Scholar]
- 49.Carta A., Sanna G., Briguglio I., Madeddu S., Vitale G., Piras S., Corona P., Peana A.T., Laurini E., Fermeglia M., et al. Quinoxaline derivativesas new inhibitors of coxsackievirus B5. Eur. J. Med. Chem. 2018;145:559–569. doi: 10.1016/j.ejmech.2017.12.083. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.