

Abstract
Background
Prior studies have shown that nutrient excess induces endoplasmic reticulum (ER) stress in nonvascular tissues from patients with diabetes mellitus (DM). ER stress and the subsequent unfolded protein response may be protective, but sustained activation may drive vascular injury. Whether ER stress contributes to endothelial dysfunction in patients with DM remains unknown.
Methods and Results
To characterize vascular ER stress, we isolated endothelial cells from 42 patients with DM and 37 subjects without DM. Endothelial cells from patients with DM displayed higher levels of ER stress markers compared with controls without DM. Both the early adaptive response, evidenced by higher phosphorylated protein kinase–like ER eukaryotic initiation factor‐2a kinase and inositol‐requiring ER‐to‐nucleus signaling protein 1 (P=0.02, P=0.007, respectively), and the chronic ER stress response evidenced by higher C/EBPα‐homologous protein (P=0.02), were activated in patients with DM. Higher inositol‐requiring ER‐to‐nucleus signaling protein 1 activation was associated with lower flow–mediated dilation, consistent with endothelial dysfunction (r=0.53, P=0.02). Acute treatment with liraglutide, a glucagon‐like peptide 1 receptor agonist, reduced p‐inositol‐requiring ER‐to‐nucleus signaling protein 1 (P=0.01), and the activation of its downstream target c‐jun N‐terminal kinase (P=0.025) in endothelial cells from patients with DM. Furthermore, liraglutide restored insulin‐stimulated endothelial nitric oxide synthase activation in patients with DM (P=0.019).
Conclusions
In summary, our data suggest that ER stress contributes to vascular insulin resistance and endothelial dysfunction in patients with DM. Further, we have demonstrated that liraglutide ameliorates ER stress, decreases c‐jun N‐terminal kinase activation and restores insulin‐mediated endothelial nitric oxide synthase activation in endothelial cells from patients with DM.
Keywords: endothelium, nitric oxide, vascular function
Subject Categories: Endothelium/Vascular Type/Nitric Oxide
Clinical Perspective
What Is New?
We have demonstrated that liraglutide ameliorates endoplasmic reticulum stress and restores insulin‐mediated eNOS activation in endothelial cells from patients with diabetes mellitus.
What Are the Clinical Implications?
This study provides evidence for an acute beneficial effect of liraglutide on endoplasmic reticulum stress and endothelial phenotype in patients with diabetes mellitus.
Our findings support the possibility that endoplasmic reticulum stress may be a mechanism for the cardiovascular effects of liraglutide in the vasculature, and treatments with endoplasmic reticulum stress inhibitors may be a novel strategy to restore endothelial function in human diabetes mellitus.
Introduction
Diabetes mellitus (DM) type 2 is a key public health problem worldwide, predicted to affect more than 500 million people by 2035.1 DM is a major risk for cardiovascular diseases (CVDs) including coronary heart disease, stroke, peripheral artery disease, heart failure, and atrial fibrillation; and DM was listed as the primary or secondary cause of >200 000 deaths in the United States in 2014.2 The dramatic increase in the incidence of DM has resulted in a critical need for novel therapies to protect the vasculature and prevent the prevalence of cardiovascular disease in DM.
Endothelial dysfunction has been described as a hallmark and a predictor of CVD.3, 4, 5 However, our understanding the molecular mechanisms underlying insulin resistance and endothelial dysfunction in the vasculature of patients with DM is still uncertain. A greater understanding of the mechanisms that lead to an impairment in endothelial function and the subsequent development of CVDs is necessary to identify new targets for the treatment of CVD in DM.
Over the past decade, the endoplasmic reticulum (ER) has emerged as an important regulator of metabolic processes and recently has been implicated in the pathogenesis of endothelial dysfunction.6, 7, 8 The ER is a large, membrane‐enclosed cellular compartment, critical to the physiologic regulation of many cellular processes, including protein folding, lipid biosynthesis and redox homeostasis.9, 10, 11 Under physiologic conditions, the ER's protein load and folding capacity are in balance; however, ER overload produces an accumulation of misfolded proteins in the ER, leading to the activation of the unfolded protein response (UPR), a process that is known as ER stress.12 UPR is initiated by the activation of 3 transmembrane proteins: (1) RNA‐dependent protein kinase‐like ER eukaryotic initiation factor‐2a kinase (PERK); (2) inositol‐requiring ER‐to‐nucleus signaling protein 1 (IRE1α); and (3) activating transcription factor 6 (ATF6).13, 14 Acute ER stress activation has protective properties; however, chronic activation of the UPR leads to adverse cellular consequences. Specifically, chronic activation of the UPR promotes increased oxidative stress and inflammation and often results in cell death in endothelial cells.15, 16 Emerging preclinical and clinical evidence support the notion that pharmacologic modulators of ER stress have therapeutic potential as novel treatments for metabolic diseases.10 Experimental studies have demonstrated that the glucagon‐like peptide 1 (GLP‐1) receptor agonist, liraglutide, downregulates high glucose‐induced UPR activation and ER stress in cultured endothelial cells.17 Further, liraglutide improves cardiovascular function in animal models.18 However, limited translational research has addressed the involvement of ER stress activation in the vasculature, and the contribution of ER stress to endothelial dysfunction in human DM is not known.
In the present study, we explore the activation of ER stress in the vasculature of DM patients, and investigate whether chronic ER stress activation is involved in vascular endothelial dysfunction and insulin resistance in human DM. We hypothesize that ER stress activation could function as a central regulatory node mediating abnormal endothelial phenotype in human DM that might serve as a novel treatment target for vascular protection.
Methods
Beyond the descriptions provided here, data, analytical methods, and study materials will not be available.
Reagents
Insulin (#I9278), Glucose (#G8769), Exendin fragment 9–39 (E7269), Tunicamycin (#11089‐65‐9) were from Sigma, and liraglutide from American Peptide Company (#46‐1‐48).
Study Participants
Adult patients with type 2 DM were enrolled from the clinical practices at Boston Medical Center. Controls were enrolled through advertisement or through participation in prior research studies. DM was defined as fasting glucose ≥126 mg/dL and glycated hemoglobin A1c ≥6.5%, or ongoing pharmacologic treatment for type 2 DM. Patients with DM treated with GLP‐1 receptor agonists or dipeptidyl peptidase‐4 (DPP4) inhibitors were excluded from the study. Control individuals without DM were defined as fasting glucose <100 mg/dL. Fasting glucose and lipids were measured in the Boston Medical Center Clinical Laboratory.
Vascular Function Testing
We measured brachial artery flow‐mediated dilatation in each patient as previously described.19, 20 Briefly, high‐resolution ultrasound was used to measure brachial artery diameter before and 1 minute after induction of hyperemic response by 5‐minute cuff occlusion of the upper arm. Doppler flow signals were recorded from the brachial artery before and after cuff occlusion to measure reactive hyperemia.21 Afterwards, we measured nitroglycerin‐mediated dilatation, which reflects non–endothelium‐dependent dilatation.
Fresh Isolation of Human Endothelial Cells
Peripheral venous endothelial cell biopsies were performed as previously described.22, 23 Briefly, a 20‐gauge intravenous catheter was placed in a superficial forearm vein using aseptic technique. Endothelial cells were collected by gentle abrasion of the vessel wall with a 0.018‐inch J‐wire introduced through the catheter. Endothelial cells were recovered from the wire by centrifugation in a dissociation buffer and plated on poly‐l‐lysine–coated microscope slides. Once plated, cells were either directly fixed with 4% paraformaldehyde immediately, or fixed after insulin, liraglutide, or exendin stimulation. Slides were then washed with phosphate‐buffered saline, fixed, and stored at −80°C until further processing. Acute treatments were up to 1.5 hours to preserve the in vivo phenotype.
Endothelial Cell Culture and Treatment
Human aortic endothelial cells (HAECs) were purchased from Lonza and maintained with endothelial growth medium‐2 containing 5 mmol/L glucose in a standard incubator (37°C, 5% CO2). Cells from passages 4 to 7 and 90% confluence were used for the experiments after being starved for 3 hours in serum‐free medium. For drug treatment, cultured endothelial cells were incubated with 100 nmol/L liragutide for 15 minutes, which is within the therapeutic range achieved in humans with 1.8 μg/day treatment with liraglutide, and a standard dose used in different types of endothelial cells including human umbilical vein endothelial cells,17 HAECs,24 and cardiac microvascular endothelial cells.25 To investigate the role of GLP‐R1, the GLP‐1R inhibitor Exendin fragment (9–39) (10 nmol/L) was incubated for 30 minutes before liraglutide treatment, as previously described.25
Western Blot Analysis
After drug treatment, endothelial cells were washed in phosphate‐buffered saline, trypsinized, and the pellet was lysed in ice‐cold lysis buffer (20 mmol/L Tris‐HCl, pH 7.4, 150 Na2EDTA, 1% Triton) and protease/phosphatase inhibitor cocktail (following the instructions provided by the supplier). Protein content in the samples was quantified by bicinchonic acid protein assay (Pierce), and 20 to 35 μg protein was subjected to electrophoresis in 4% to15% sodium dodecyl sulfate–polyacrylamide gels, under reducing conditions, and then transferred to a polyvinylidene difluoride membrane using the Bio‐Rad Transblot Turbo Transfer System. Membranes were blocked with 3% bovine serum albumin for 1 hour at room temperature, and incubated overnight with the respective primary antibody at 4°C (1:1000–1:5000). Blots were probed with anti‐eNOS (BD Transduction, #610296); antiphospho‐eNOS (Ser1177, #9571), CCAAT/enhancer binding protein homologous protein (#28945) and IRE1α from Cell Signaling (#3294) and p‐IRE1α (ab124945) from Abcam. Immunoblots were normalized to monoclonal β‐actin antibody (Sigma‐Aldrich, #A1978). Horseradish peroxidase–conjugated secondary antibodies were incubated for 1 hour at room temperature (antirabbit #HAF008 or antimouse #HAF007, R&D Systems). Immune complexes were visualized with Amersham ECL Western Blotting Detection Reagents. Membranes were stripped with Restore Western Blot Stripping Buffer (ThermoFisher) for 10 minutes at 37°C, and reprobed with either total protein or housekeeping proteins to verify equal loading. Resulting bands were quantified by densitometry.
Quantitative Immunofluorescence Staining
Fixed samples were thawed and rehydrated with 50 mmol/L glycine in phosphate‐buffered saline. Cells were permeabilized with 0.1% Triton X‐100, and nonspecific binding sites were blocked in 0.5% bovine serum albumin. Slides were incubated overnight at 4°C with primary antibodies (1:100) against the following targets: phospho‐eNOS (Ser1177, Abcam #ab184154), phospho‐c‐jun N‐terminal kinase (JNK) (Thr183/Tyr185, Cell Signaling #9255), CHOP (Cell Signaling #2895); pIRE1 (Abcam #ab48187); IRE1 (Novus Biologicals #NB100‐2324); p‐PERK (SantCruz #sc‐32577); PERK (ThermoFisher #PA5‐38811); ATF4 (Abcam #ab50546); ATF6 (Abcam #122897); GRP78 (Abcam #108615). All cells were also stained with anti–von Willebrand factor antibody (1:200) for endothelial cell identification. Slides were incubated with corresponding Alexa Fluor‐488 and Alexa‐Fluor‐594 antibodies (1:200) for 45 minutes at 37°C. Coverslips were mounted with Vectashield containing 4′,6‐diaminido‐2‐phenylindole for nuclear identification. Slides were imaged with a confocal microscope (Leica SP5) at 63× magnification. All images were captured at the same exposure time and corrected for background fluorescence. Fluorescence intensity was quantified with NIS Element AR Software. The fluorescence intensity of at least 20 cells from each patient and protein of interest was averaged and normalized to the intensity of HAEC, simultaneously stained to adjust for any variation in antibody and staining conditions. We stain the same batch and passage of cultured aortic endothelial cells to normalize for each protein of interest. Intensity quantification was performed blinded to participant identity and DM status.
Statistical Analysis
Statistical analyses were performed using SPSS version 20.00 (IBM Corporation, Armonk, NY). Data are expressed as mean±standard deviation, unless otherwise noted. Variables were evaluated for normality by the Shapiro‐Wilk test. Participants with DM and controls were compared using the unpaired Student t test for normally distributed continuous variables or Mann‐Whitney U test for those not normally distributed; chi‐squared tests were used for discrete variables. Paired (pre/post) samples were compared using the paired Student t test for normally distributed continuous variables or the Wilcoxon signed rank test for variables not normally distributed.
Spearman correlation coefficients were used to correlate measures of vascular function and expression of proteins of interest. A 2‐tailed P<0.05 was considered to be statistically significant. To understand the magnitude of the differences found, the estimated effect size (Cohen's d) was calculated using an online tool that takes into account unpaired and paired data (https://memory.psych.mun.ca/models/stats/effect_size.shtml). Cohen's d effect size >0.5 was considered a moderate difference, and >0.8 was considered a large difference.26
Study Approval
The study protocol was approved by the Boston Medical Center Institutional Review Board (H‐26605), and all participants provided written informed consent before inclusion in the study.
Results
Study Participants and Vascular Function
We enrolled 42 subjects with DM and 37 subjects without DMof similar age and sex. The clinical characteristics and measures of vascular function are shown in the Table. As expected, patients with DM had clinical parameters consistent with metabolic abnormalities including higher fasting glucose, higher body mass index, and higher hemoglobin A1c. Likewise, patients with DM had lower low‐density lipoprotein levels, likely reflecting the use of cholesterol‐lowering medications. Endothelial‐dependent flow‐mediated dilatation of the brachial artery was significantly lower in the patients with DM, consistent with the presence of endothelial dysfunction. There were no differences in nitroglycerin‐mediated dilatation among groups, suggesting the absence of smooth muscle dysfunction.
Table 1.
Clinical and Vascular Characteristics
| Nondiabetic (n=37) | Diabetic (n=42) | P Value | |
|---|---|---|---|
| Clinical characteristics | |||
| Age, y | 49±8 | 51±9 | 0.34 |
| Female sex, % | 32 | 55a | 0.048 |
| Black race, % | 73 | 76 | 0.795 |
| Body mass index, kg/m2 | 30.9±7.9 | 37.3±8.9a | 0.001b |
| Total cholesterol, mg/dL | 189±44 | 190±51 | 0.942 |
| LDL cholesterol, mg/dL | 125±31 | 119±41 | 0.471 |
| HDL cholesterol, mg/dL | 52±15 | 44±13a | 0.06b |
| Triglycerides, mg/dL | 93.9±43 | 133±86a | 0.07b |
| Fasting glucose, mg/dL | 86.7±11 | 190±99a | <0.001b |
| Hemoglobin A1c, % | 5.4±0.3 | 8.5±2.5a | <0.001b |
| Systolic blood pressure, mm Hg | 133±17 | 133±17 | 0.777b |
| Diastolic blood pressure, mm Hg | 83±13 | 81±13 | 0.383b |
| Hypertension, % | 13 | 67a | 0.000b |
| Smoking, % | 38 | 38 | 0.981 |
| Antiplatelet therapy, % | 3 | 17a | 0.038 |
| Lipid‐lowering therapy, % | 8 | 45a | <0.001 |
| ACE inhibitor or ARB therapy, % | 5 | 50a | <0.001 |
| Metformin, % | 0 | 69a | <0.001 |
| Sulfonylureas, % | 0 | 24a | 0.002 |
| Insulin therapy, % | 0 | 26a | 0.001 |
| Vascular function | |||
| Baseline diameter, mm | 4.20±1.1 | 4.32±0.8 | 0.93b |
| Baseline flow, mL/min | 150±149 | 172±104 | 0.079b |
| Hyperemic flow, mL/min | 1206±797 | 1055±594 | 0.461b |
| Flow‐mediated dilation, % | 9.8±5.7 | 6.1±3.3a | 0.03b |
| Flow‐mediated dilation, mm | 0.41±0.05 | 0.25±0.03 | 0.004b |
| Nitroglycerin‐mediated dilation | 11.7±7.6 | 11.7±5.9 | 0.975 |
| Baseline flow velocity | 15.2±10.6 | 22.3±34.5 | 0.185b |
| Hyperemic flow velocity | 110.8±52.7 | 91.8±70.9 | 0.118b |
Data are expressed as mean±SD. ACE indicates angiotensin‐converting enzyme; ARB, angiotensin receptor blocker; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein.
A two‐tailed P<0.05 was considered to be statistically significant.
Mann‐Whitney test to compare patients with diabetes mellitus and controls; others by chi‐square test or t test as appropriate.
DM Is Associated With Elevated ER Stress in the Vascular Endothelium
To characterize vascular ER stress, we studied the acute and chronic ER stress response in freshly isolated venous endothelial cells from patients with DM and patients without DM. We first evaluated total protein expression of the 3 ER membrane–localized proteins considered to be “acute sensors” of ER stress: IRE1α, PERK, and ATF6. As shown in Figure S1, expression of these proteins was similar between the patients without DM and participants with DM.
To evaluate IRE1α and PERK activation, we performed quantitative immunofluorescence to measure the phosphorylation at the activation sites (Serine (Ser) 724 and Threonine (Thr) 981, respectively). Endothelial cells from patients with DM had elevated levels of activated IRE1α (**P=0.007) and PERK (*P=0.02) compared with controls without DM, suggesting an elevated ER stress response in patients with DM (Figure 1). Notably, we found a positive correlation between IRE1α and PERK activation in the endothelium (r=0.54, *P=0.03). Interestingly, higher levels of activated IRE1α were associated with lower flow‐mediated dilatation, consistent with a link between ER stress activation and impaired endothelial vasodilator function (r=0.53, *P=0.02). (Figure 1C). Although acute ER stress activation could be beneficial for the cell, chronic ER stress results in an increased translation of ATF4, which promotes the upregulation of genes involved in oxidative stress and apoptosis. Chronic ER stress results in an elevated expression of the pro‐apoptotic transcription factor CHOP.27 Endothelial cells from patients with DM showed higher CHOP expression compared with controls without DM (*P=0.02) (Figure 2), suggesting that DM is associated with a chronic ER stress activation and the activation of apoptotic signaling pathways in the vascular endothelium. Moreover, higher CHOP expression was associated with higher activation of IRE1α (r=0.82, *P=0.04) and PERK (r=0.59, *P=0.03).
Figure 1.
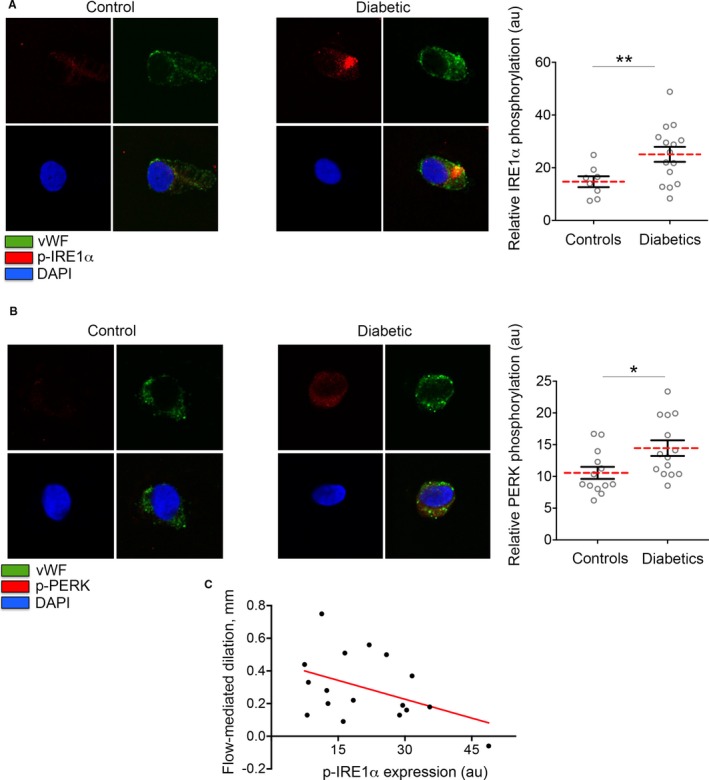
Activation of the unfolded protein response in vascular endothelial cells of patients with diabetes mellitus (DM). Venous endothelial cells patients with and without DM were freshly isolated as described in the Materials and Methods section of this article. Endothelial cells were identified by von Willebrand factor staining and nuclear morphology. A, Inositol‐requiring ER‐to‐nucleus signaling protein 1 (IRE1α) activation was evaluated as IRE1α phosphorylation at Ser 724. Representative cell from a patient with DM (right) shows higher activation of IRE1α when compared with a representative cell from a controls without DM (left). Pooled data showed that the IRE1α phosphorylation at its activatory site is enhanced in patients with DM (n=15) compared with the controls without DM (n=8) (**P=0.007, Cohen's d=1.08). B, RNA‐dependent protein kinase–like ER eukaryotic initiation factor‐2a kinase (PERK) activation was evaluated as PERK phosphorylation at Thr 981. Representative cells from patients without DM (left) and patients with DM (right) are shown. Pooled data showed that PERK phosphorylation is higher in patients with diabetes mellitus compared with controls without diabetes mellitus (n=14/13, *P=0.020, Cohen's d=0.954). Variables were compared using the t test. C, Higher IRE1a activation in freshly isolated venous endothelial cells from patients were associated with lower mediated dilation (r=0.53, *P=0.02). DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
Figure 2.
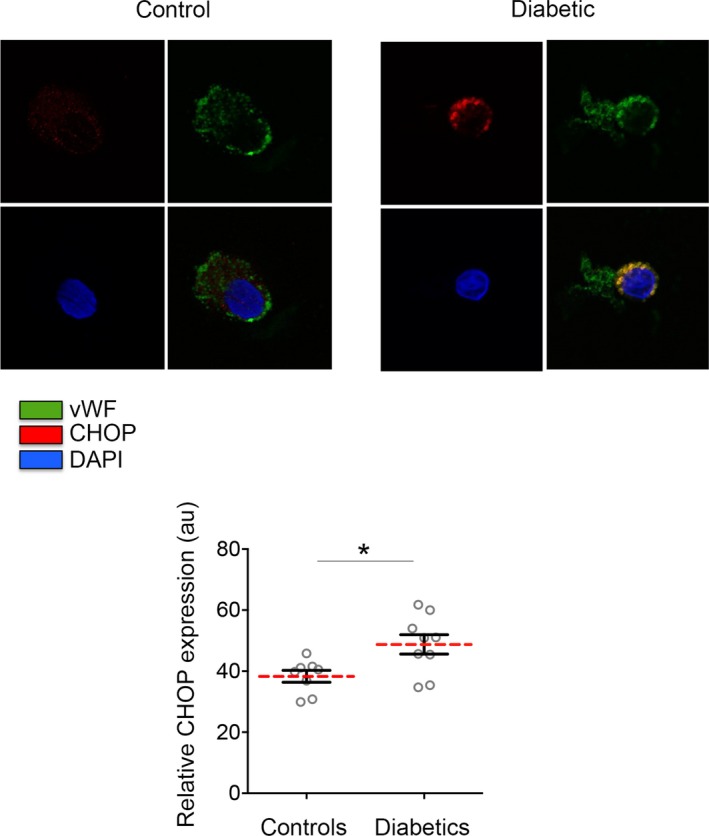
Diabetes mellitus (DM) is associated with a chronic activation of endoplasmic reticulum (ER) stress in the vasculature. Venous endothelial cells from patients with and without DM were freshly isolated and identified by von Willebrand factor staining and nuclear morphology. Representative cell from a diabetic patient (right) shows higher expression of the chronic ER stress marker CCAAT/enhancer binding protein homologous protein (CHOP) compared with a representative cell from a control without DM (left). Pooled data showed that CHOP is higher expressed in endothelial cells from patients with DM (n=9) when compared with patients without DM (n=8) (*P=0.015; Cohen's d=1.322). Variables were compared using the t test. DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
Collectively, our findings suggest that DM is associated with an aberrant activation of ER stress in the vascular endothelium.
ER Stress Activation Induces Endothelial Dysfunction
A number of experimental studies in cultured endothelial cells and in animal models suggest a role for ER stress in endothelial dysfunction.7 We were interested in whether acute stimulation of ER stress would produce similar effects in a healthy endothelium as the phenotype observed in the endothelium of patients with DM. To study the relationship between ER stress and endothelial dysfunction in our model, we conducted in vitro studies with commercially available HAECs in the presence and absence of an ER stress activator, tunicamycin. Tunicamycin treatment–induced ER stress as demonstrated by enhanced activation of IRE‐1α and increased expression of CHOP (Figure S2A). To investigate the importance of ER stress activation in endothelial dysfunction and insulin resistance, we treated HAECs with insulin in the presence or absence of tunicamycin and quantified eNOS phosphorylation at its activation site, Ser 1177. As shown in Figure S2B, ER stress activation by tunicamycin treatment impairs insulin‐induced eNOS phosphorylation at its activation site in endothelial cells in culture.
GLP‐1 Analogue, Liraglutide, Reduces Acute ER Stress in EC from Patients With DM
To determine whether ER stress contributes to endothelial dysfunction in the setting of DM, we utilized liraglutide, a GLP‐1 analogue, which has been demonstrated to reduce ER stress in cultured endothelial cells exposed to high glucose17 and to exert favorable effects on cardiovascular function in both preclinical and clinical studies.18, 28, 29
To gain insight into the potential role of liraglutide in the vasculature of patients with DM, we evaluated the acute effect of liraglutide on ER stress activation. We treated freshly isolated endothelial cells from patients with DM, treated with liraglutide for 45 minutes, and evaluated acute ER stress activation by PERK and IRE1α activation. As shown in Figure S3, acute treatment with liraglutide did not induce a change in PERK activation; however, we observed a decrease not only in IRE1α activation but also in the activation of its downstream target, JNK (Figure 3). Interestingly, we have previously demonstrated the important role of JNK in endothelial dysfunction and insulin resistance in human DM.30
Figure 3.
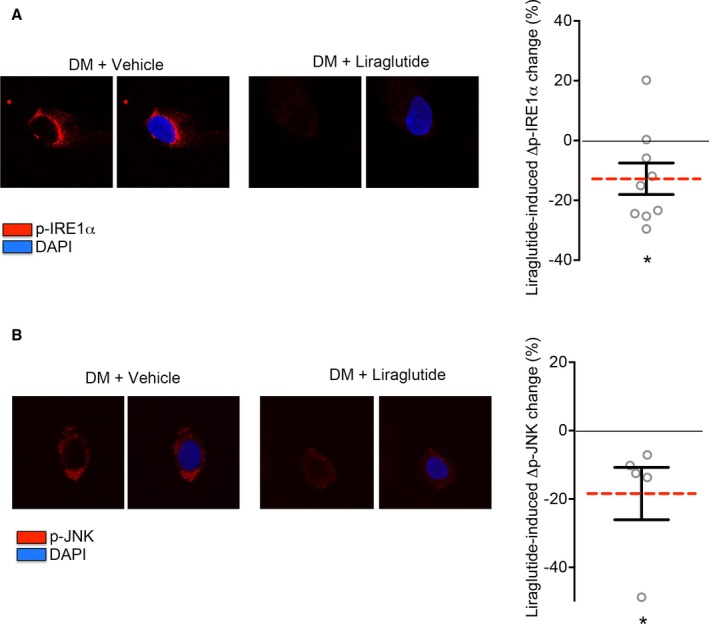
Liraglutide reduces endoplasmic reticulum (ER) stress acute activation and the subsequent inflammatory response in endothelial cells from patients with diabetes mellitus (DM). Venous endothelial cells were isolated from patients with DM and acute ER stress activation was studied in the presence and absence of liraglutide. A, Inositol‐requiring ER‐to‐nucleus signaling protein 1 (IRE1α) phosphorylation was studied in endothelial cells from patients with DM in the presence of liraglutide (0–100 nmol/L). Representative cells from the same patient are shown. Pooled data showed that the IRE1α activation decrease after liraglutide treatment (n=9; *P=0.01; Cohen's d=1.125). B, phospho‐c‐jun N‐terminal kinase (JNK) activation was evaluated as JNK phosphorylation at Thr183/Tyr185. Representative cells from a patient with DM treated with liraglutide (0–100 nmol/L) are shown. Pooled data demonstrated a reduction in JNK activation in endothelial cells from patients with DM in the presence of liraglutide (n=5; *P=0.025; Cohen's d=1.563). Variables were compared using the paired t test. DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
Collectively, the findings in freshly isolated endothelial cells from patients with DM suggest that liraglutide reduces acute ER stress activation in the endothelium of diabetic patients with DM.
Liraglutide Restores Insulin Response and Endothelial Function in Patients With DM
To determine whether liraglutide improves endothelial function in DM, we first studied the effect of liraglutide on eNOS activation. GLP‐1 analogues have been described previously to induce a transient phosphorylation of eNOS at Ser1177.31 As shown in Figure S4, and concordant with our previous reports, patients with DM had higher basal levels of activated eNOS at Ser1177.32 We found that liraglutide treatment induced an increase in eNOS phosphorylation at its activation residue (Ser1177) in endothelial cells isolated from patients without DM. However, liraglutide did not promote eNOS activation in endothelial cells freshly isolated from patients with DM.
Next, we evaluated whether liraglutide and ER stress inhibition ameliorate endothelial dysfunction by studying insulin‐mediated eNOS activation in endothelial cells from patients with DM in the presence and absence of the ER stress inhibitor. As shown in Figure 4 and consistent with our previous reports, insulin stimulation did not induce eNOS phosphorylation in patients with DM.32 Pretreatment with liraglutide for 15 minutes before insulin stimulation (10 nmol/L, 30 minutes) restored eNOS activation and endothelial function in endothelial cells isolated from patients with DM (n=10; P=0.019).
Figure 4.
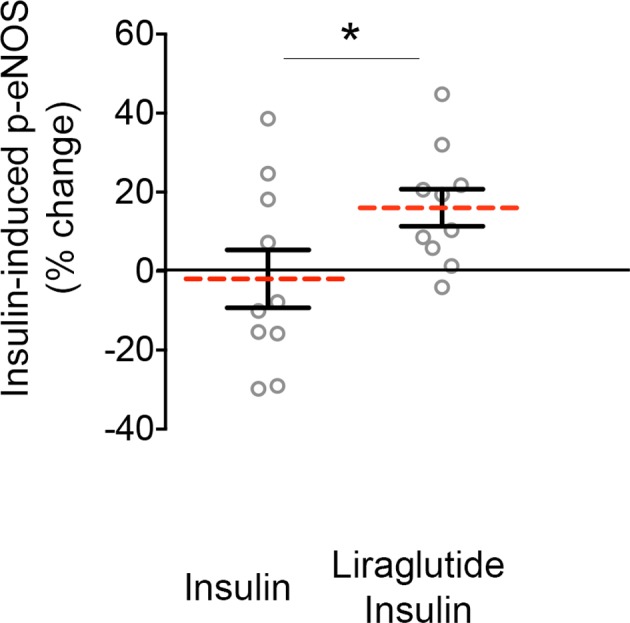
Liraglutide treatment restores endothelial function and insulin response in endothelial cells from patients with diabetes mellitus (DM). Freshly endothelial cells isolated from patients with DM were treated with 0 or 10 nmol/L insulin for 30 minutes in the absence or in the presence of liraglutide and then fixed. Endothelial nitric oxide (eNOS) activation was evaluated as eNOS phosphorylation at Ser1177. Pooled data demonstrated that liraglutide restores insulin‐induced eNOS phosphorylation in endothelial cells isolated from patients with DM (n=10, *P=0.041; Cohen's d=0.752). Variables were compared using the paired t test.
Collectively, our results suggest that liraglutide treatment restores vascular insulin sensitivity and this effect was independent of a direct activation of eNOS by the GLP‐1 analogue.
Liraglutide‐Induced Amelioration of Endothelial Dysfunction in DM is Dependent on GLP‐1 Receptor
Liraglutide is a GLP‐1 receptor agonist described to activate different signaling pathways via GLP‐1R‐dependent or independent signals.18 To determine if the effects of liraglutide on the vascular endothelium are mediated through GLP‐1R, freshly isolated endothelial cells from patients with DM were pretreated for 30 minutes with the GLP‐1R antagonist exendin fragment (9–39) before liraglutide and insulin sequential stimulation. Exendin fragment (9–39) abolished liraglutide‐driven restoration of insulin sensitivity in endothelial cells from patients with DM (n=5; P=0.02) (Figure 5).
Figure 5.
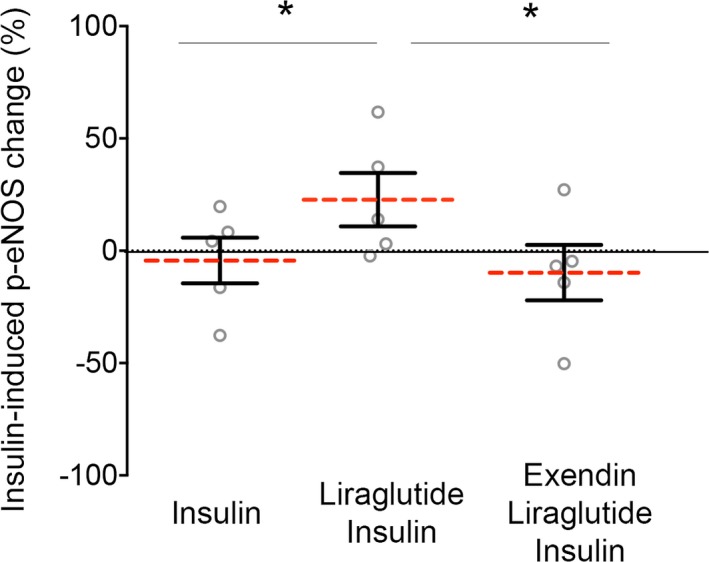
Liraglutide effect on endothelial function in diabetes mellitus (DM) is dependent on the glucagon‐like peptide 1 receptor (GLP‐1R). Venous endothelial cells from patients with DM were freshly isolated as described in the Materials and Methods section of this article and were treated with GLP‐1R antagonist, exendin fragment (9–39) (10 nmol/L, 30 minute), before the addition of liraglutide (100 nmol/L, 15 minutes) and insulin (10 nmol/L 30 minutes) sequential treatment. Pooled data demonstrated that exendin (9–39) impaired liraglutide‐driven restoration of insulin sensitivity in diabetic patients (n=5; *P=0.020; Cohen's d=1.676). Variables were compared using the paired t test.
Our findings indicate that the beneficial effect of liraglutide on DM induced ER stress in endothelial cells is dependent on GLP‐1R.
Discussion
In the present study, we demonstrated the involvement of ER stress in endothelial dysfunction and insulin resistance in patients with DM. Using the venous endothelial biopsy procedure, we first demonstrated an enhanced and aberrant ER stress activation in the vasculature of diabetic patients when compared with controls without DM. Further, we found that IRE1α activation was associated with endothelial dysfunction in patients with DM. We demonstrated that ER stress activation induced insulin resistance in commercially available endothelial cells, recapitulating the phenotype we observed in the endothelial cells from patients with DM. Furthermore, the GLP‐1 analogue liraglutide ameliorated ER stress and JNK activation in endothelial cells from patients with DM. Liraglutide treatment also restored insulin‐mediated eNOS activation in endothelial cells freshly isolated from patients with DM in a GLPR‐1 dependent manner. Taken together, our findings suggest that aberrant ER stress activation contributes to insulin resistance and endothelial dysfunction in the vasculature of patients with DM and that ER stress inhibition has potential as a strategy to improve vascular health in patients with DM.
Prior studies have demonstrated that ER overload produces an accumulation of misfolded proteins, UPR activation, and ER stress, which contributes to the development and progression of chronic disorders including neurodegeneration, atherosclerosis, liver disease, and cancer, among others.33, 34 In the past decade, the ER has been identified as an important regulator of metabolic processes,35 and Gargalovic et al were among the first to directly link ER stress to endothelial disturbances.36 Experimental studies in cultured endothelial cells have demonstrated that chemically induced ER stress causes endothelial dysfunction and insulin resistance.37, 38 Further, a number of studies in animal models have provided additional insights into the molecular mechanisms linking ER stress induction and endothelial dysfunction.39, 40 Nevertheless, limited translational research has addressed the impact of ER stress in the vasculature,41, 42 and the contribution of ER stress to endothelial dysfunction in human DM is not known. Our finding that ER stress is aberrantly activated in endothelial cells from patients with DM lends additional support for the concept that ER stress plays a role in vascular endothelial dysfunction in patients with DM.
Currently, much of the data on the regulation of ER stress in human metabolic diseases and endothelial function have been focused on the involvement of the IRE1α and PERK branches of the UPR.35 In this study, we have demonstrated an enhanced activation of acute ER stress markers IRE1α and PERK, and a lack of differential expression in ATF6 in patients with DM when compared with controls without DM. However, further studies focused in the activation or subcellular location of ATF6 would be necessary to discard the activation of ATF6 branch in the vasculature of patients with DM. During ER stress, IRE1α and PERK‐ATF4 trigger the activation of JNK, a key inflammatory signaling pathway.43, 44, 45 Interestingly, we have previously reported an enhanced activation of JNK in patients with DM, that contributes to reduced nitric oxide production and lower flow‐mediated dilatation.30 In the present study, we develop evidence that ER stress pathways may be an important regulator of JNK activation in the vasculature in patients with DM by demonstrating increased activation of both the IRE1α and PERK pathways. Prolonged ER stress results in the induction of the pro‐apoptotic transcription factor CHOP, regarded as a key mediator of cell death, endothelial impairment, and disease in response to ER overload.27, 46, 47, 48 In the present study, we showed an enhanced expression of CHOP in the vascular endothelium of patients with DM when compared with controls without DM. Furthermore, we found that CHOP abundance was associated with UPR activation in the vasculature.
GLP‐1 is a gut hormone secreted in a nutrient‐dependent manner that stimulates insulin secretion and inhibits glucagon secretion, thereby reducing postprandial glycemia. Native GLP‐1 has limited therapeutic potential because it is rapidly degraded by DPP4, and truncated GLP‐1 is unable to interact with its receptor and exert its function. Two strategies have been developed to sustain GLP‐1–mediated effects over a longer period: inhibition of DPP4 and the development of GLP‐1 receptor agonists.49 Clinical trials have investigated the efficacy of agents altering GLP‐1 signaling in DM to reduce cardiovascular complications.50, 51 DPP4 inhibitors were predicted to have beneficial effects on the cardiovascular system; however, clinical trials with several DPP4 inhibitors have not shown reduction of cardiovascular events.52, 53, 54, 55 However, there is evidence that GLP‐1 receptor agonists have cardiovascular protective effects.50, 56, 57 In the recent LEADER (Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results) trial, liraglutide significantly reduced the risk of the 3 major adverse cardiovascular events (death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke) in patients with DM who were at high risk for cardiovascular events.58 Furthermore, the addition of liraglutide to antidiabetic therapies improved several cardiovascular risk markers in clinical studies.59, 60, 61, 62, 63, 64, 65 However, the mechanism by which liraglutide confers cardiovascular benefits has not been fully elucidated. Experimental studies have demonstrated that GLP‐1 agonists have anti‐inflammatory effects on endothelial cells. Treatment with the GLP‐1 receptor agonist liraglutide inhibited tumor necrosis factor‐α, intracellular adhesion molecule‐1 and vascular cell adhesion molecule‐1 in human endothelial cells.66, 67 In animal models, liraglutide has been shown to reduce serum levels of several inflammatory markers such us C‐reactive protein, interleukin‐6, tumor necrosis factor‐α and the plasmin plasminogen activator inhibitor‐1.68
Interestingly, liraglutide was recently shown to downregulate ER stress in cultured endothelial cells and in animal diabetic models.17, 69, 70 In the present study, we provide evidence that liraglutide reduced acute ER stress activation (IRE1α) and JNK phosphorylation in endothelial cells freshly isolated from patients with DM. A previous study in patients with DM showed a trend in liraglutide‐mediated endothelium‐dependent vasodilation.71 Here, we demonstrate that liraglutide has the potential to restore insulin action in the endothelium of patients with DM, probably due to the lower variability in the response in our ex vivo study. Moreover, in the present study we demonstrate that the beneficial effect on the vasculature is dependent on the GLP‐1 receptor.
Some limitations of our study should be addressed. This study was performed in venous endothelial cells; arterial endothelial cells may be more directly relevant to the development of cardiovascular disease. However, both venous and arterial endothelial cells are exposed to similarly abnormal metabolic abnormalities in DM. Patients with DM have multiple systemic risk factors that likely contribute together to the endothelial phenotype observed, including differences in lipid profile. Prior studies have shown that administration of GLP‐1 agonists has an impact on lipid homeostasis.72, 73, 74, 75 Liraglutide administration appears to have beneficial effects on plasma lipids and lipoproteins, including a reduction in total cholesterol, triglycerides, and low‐density lipoprotein cholesterol, and an increase in high‐density lipoprotein cholesterol.76, 77, 78 Moreover, we have demonstrated that intravenous lipid infusion induces ER stress in endothelial cells.41 Additional studies will be required to determine the complete set of regulators of ER stress involved in its abnormal activation that lead to endothelial dysfunction and cardiovascular events in patients with DM.
Although recent trials have described the beneficial role of novel drugs in the treatment of DM in reducing the risk of cardiovascular complications in patients with DM, the mechanism remains incompletely elucidated. Our findings provide evidence for an acute beneficial effect of liraglutide on endothelial phenotype including ER stress activation in cells isolated from patients with DM. Our work supports further studies to determine whether vascular ER stress activation may be a mechanism for the cardiovascular benefit of liraglutide and a therapeutic target in DM.
Author Contributions
RBR performed the experiments, analyzed the data and wrote the manuscript. RMW, BF and DK collected human samples. MH recruited participants. MMS performed vascular function analysis in patients. JLF and JZ contributed to review the manuscript. NMH directed the study and reviewed the manuscript.
Sources of Funding
This project was supported by National Heart, Lung and Blood Institute grants HL081587, HL115391, and HL102299. Bretón‐Romero was supported by American Heart Association (AHA) Founders Affiliate Postdoctoral Grant 6POST27260178. Zhang was supported by NIH multidisciplinary training grant in cardiovascular research (T32 HL0007224). Fetterman was supported by AHA‐AWRP Summer 2016 Mentored Clinical & Population Research Award 17MCPRP32650002.
Disclosures
None.
Supporting information
Figure S1. DM is not associated with changes in the expression of acute ER stress markers. Venous endothelial cells from diabetic and non‐diabetic patients were freshly isolated as described in the Materials and Methods section of this article. Endothelial cells were identified by van W illebrand factor staining and nuclear morphology. Total protein expression was determined by quantitative fluorescence and the use of specific antibodies against IRE1α (A), PERK (B), and ATF6 (C). Variables were compared using the t test.
Figure S2. ER stress induction by tunicamycin impairs insulin‐induced eNOS phosphorylation in endothelial cells in culture. A, Human aortic endothelial cells (HAECs) were incubated with tunicamycin for the indicated time points and ER stress activation were evaluated by an enhanced IRE1α phosphorylation and higher expression of CHOP. The blots shown are representative of at least 4 independent experiments that yielded equivalent results. The bar graph represents the mean±SEM of at least 4 independent experiments (n=9 for IRE1α phosphorylation, and n=4 for CHOP). B, Insulin‐mediated endothelial nitric oxide (eNOS) activation was evaluated as eNOS phosphorylation at Ser1177, ER stress activation by tunicamycin impaired insulin‐mediated changed in eNOS phosphorlation at Ser1177 (n=5). Variables were compared vs controls using paired t test.
Figure S3. Liraglutide does not decrease PERK activation in endothelial cell from patients with DM. Venous endothelial cells were isolated from patients with DM, and PERK activation was studied in the presence and absence of liraglutide. Pooled data showed that the PERK activation did not decrease after acute liraglutide treatment (n=4; P=0.81; Cohen's d=0.133). Variables were compared using the paired t test.
Figure S4. GLP‐1 analogue, liraglutide, activated endothelial nitric oxide synthase in venous endothelial cells from patients without DM. Venous endothelial cells were freshly isolated from patients with DM and controls without DM, and eNOS activation was studied in the presence and absence of liragultide. Pooled data showed that the liraglutide enhanced eNOS activation in endothelial cells isolated from patients without DM (n=8; P=0.006; Cohen's d=1.362), but it did not modify eNOS activation at its activation residue in endothelial cells isolated from patients with DM (n=10; P=0.33; Cohen's d=0.324). Variables were compared using the paired t test.
(J Am Heart Assoc. 2018;7:e009379 DOI: 10.1161/JAHA.118.009379.)
References
- 1. Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. 2014;103:137–149. [DOI] [PubMed] [Google Scholar]
- 2. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P; American Heart Association Statistics Committee and Stroke Statistics Subcommittee . Heart disease and stroke statistics‐2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD. The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J. 2010;4:302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Widlansky ME, Gokce N, Keaney JF Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003;42:1149–1160. [DOI] [PubMed] [Google Scholar]
- 5. Tabit CE, Chung WB, Hamburg NM, Vita JA. Endothelial dysfunction in diabetes mellitus: molecular mechanisms and clinical implications. Rev Endocr Metab Disord. 2010;11:61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in PERK‐/‐ mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–1163. [DOI] [PubMed] [Google Scholar]
- 7. Lenna S, Han R, Trojanowska M. Endoplasmic reticulum stress and endothelial dysfunction. IUBMB Life. 2014;66:530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner‐Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. [DOI] [PubMed] [Google Scholar]
- 9. Battson ML, Lee DM, Gentile CL. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2017;312:H355–H367. [DOI] [PubMed] [Google Scholar]
- 10. Lee J, Ozcan U. Unfolded protein response signaling and metabolic diseases. J Biol Chem. 2014;289:1203–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci. 2016;73:79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. [DOI] [PubMed] [Google Scholar]
- 13. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic‐reticulum‐resident kinase. Nature. 1999;397:271–274. [DOI] [PubMed] [Google Scholar]
- 14. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. [DOI] [PubMed] [Google Scholar]
- 15. Hummasti S, Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res. 2010;107:579–591. [DOI] [PubMed] [Google Scholar]
- 16. Salvado L, Palomer X, Barroso E, Vazquez‐Carrera M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol Metab. 2015;26:438–448. [DOI] [PubMed] [Google Scholar]
- 17. Schisano B, Harte AL, Lois K, Saravanan P, Al‐Daghri N, Al‐Attas O, Knudsen LB, McTernan PG, Ceriello A, Tripathi G. GLP‐1 analogue, liraglutide protects human umbilical vein endothelial cells against high glucose induced endoplasmic reticulum stress. Regul Pept. 2012;174:46–52. [DOI] [PubMed] [Google Scholar]
- 18. Ban K, Noyan‐Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon‐like peptide 1 receptor are mediated through both glucagon‐like peptide 1 receptor‐dependent and ‐independent pathways. Circulation. 2008;117:2340–2350. [DOI] [PubMed] [Google Scholar]
- 19. McMackin CJ, Vita JA. Update on nitric oxide‐dependent vasodilation in human subjects. Methods Enzymol. 2005;396:541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vita JA, Keaney JF Jr. Endothelial function: a barometer for cardiovascular risk? Circulation. 2002;106:640–642. [DOI] [PubMed] [Google Scholar]
- 21. Mitchell GF, Parise H, Vita JA, Larson MG, Warner E, Keaney JF Jr, Keyes MJ, Levy D, Vasan RS, Benjamin EJ. Local shear stress and brachial artery flow‐mediated dilation: the Framingham heart study. Hypertension. 2004;44:134–139. [DOI] [PubMed] [Google Scholar]
- 22. Colombo PC, Ashton AW, Celaj S, Talreja A, Banchs JE, Dubois NB, Marinaccio M, Malla S, Lachmann J, Ware JA, Le Jemtel TH. Biopsy coupled to quantitative immunofluorescence: a new method to study the human vascular endothelium. J Appl Physiol. 2002;92:1331–1338. [DOI] [PubMed] [Google Scholar]
- 23. Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, Duess MA, Levit A, Kim B, Hartman ML, Joseph L, Shirihai OS, Vita JA. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011;124:444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krasner NM, Ido Y, Ruderman NB, Cacicedo JM. Glucagon‐like peptide‐1 (GLP‐1) analog liraglutide inhibits endothelial cell inflammation through a calcium and AMPK dependent mechanism. PLoS One. 2014;9:e97554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y, Zhou H, Wu W, Shi C, Hu S, Yin T, Ma Q, Han T, Zhang Y, Tian F, Chen Y. Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the Sr‐Ca(2+)‐XO‐ROS axis via activation of the GLP‐1R/PI3K/Akt/survivin pathways. Free Radic Biol Med. 2016;95:278–292. [DOI] [PubMed] [Google Scholar]
- 26. Sullivan GM, Feinn R. Using effect size–or why the P value is not enough. J Grad Med Educ. 2012;4:279–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Urra H, Dufey E, Lisbona F, Rojas‐Rivera D, Hetz C. When ER stress reaches a dead end. Biochem Biophys Acta. 2013;1833:3507–3517. [DOI] [PubMed] [Google Scholar]
- 28. Forst T, Michelson G, Ratter F, Weber MM, Anders S, Mitry M, Wilhelm B, Pfutzner A. Addition of liraglutide in patients with type 2 diabetes well controlled on metformin monotherapy improves several markers of vascular function. Diabet Med. 2012;29:1115–1118. [DOI] [PubMed] [Google Scholar]
- 29. Marso SP, Daniels GH, Brown‐Frandsen K, Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, Steinberg WM, Stockner M, Zinman B, Bergenstal RM, Buse JB, LEADER Steering Committee, LEADER Trial Investigators . Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Breton‐Romero R, Feng B, Holbrook M, Farb MG, Fetterman JL, Linder EA, Berk BD, Masaki N, Weisbrod RM, Inagaki E, Gokce N, Fuster JJ, Walsh K, Hamburg NM. Endothelial dysfunction in human diabetes is mediated by Wnt5a‐JNK signaling. Arterioscler Thromb Vasc Biol. 2016;36:561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ding L, Zhang J. Glucagon‐like peptide‐1 activates endothelial nitric oxide synthase in human umbilical vein endothelial cells. Acta Pharmacol Sin. 2012;33:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tabit CE, Shenouda SM, Holbrook M, Fetterman JL, Kiani S, Frame AA, Kluge MA, Held A, Dohadwala MM, Gokce N, Farb MG, Rosenzweig J, Ruderman N, Vita JA, Hamburg NM. Protein kinase C‐beta contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation. 2013;127:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ozcan L, Tabas I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu Rev Med. 2012;63:317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gargalovic PS, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Baruch‐Oren T, Berliner JA, Kirchgessner TG, Lusis AJ. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2490–2496. [DOI] [PubMed] [Google Scholar]
- 37. Galan M, Kassan M, Kadowitz PJ, Trebak M, Belmadani S, Matrougui K. Mechanism of endoplasmic reticulum stress‐induced vascular endothelial dysfunction. Biochem Biophys Acta. 2014;1843:1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou QG, Fu XJ, Xu GY, Cao W, Liu HF, Nie J, Liang M, Hou FF. Vascular insulin resistance related to endoplasmic reticulum stress in aortas from a rat model of chronic kidney disease. Am J Physiol Heart Circ Physiol. 2012;303:H1154–H1165. [DOI] [PubMed] [Google Scholar]
- 39. Park SW, Zhou Y, Lee J, Lee J, Ozcan U. Sarco(endo)plasmic reticulum Ca2+‐ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc Natl Acad Sci U S A. 2010;107:19320–19325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. [DOI] [PubMed] [Google Scholar]
- 41. Tampakakis E, Tabit CE, Holbrook M, Linder EA, Berk BD, Frame AA, Breton‐Romero R, Fetterman JL, Gokce N, Vita JA, Hamburg NM. Intravenous lipid infusion induces endoplasmic reticulum stress in endothelial cells and blood mononuclear cells of healthy adults. J Am Heart Assoc. 2016;5:e002574. doi: 10.1161/JAHA.115.002574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaplon RE, Chung E, Reese L, Cox‐York K, Seals DR, Gentile CL. Activation of the unfolded protein response in vascular endothelial cells of nondiabetic obese adults. J Clin Endocrinol Metab. 2013;98:E1505–E1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. [DOI] [PubMed] [Google Scholar]
- 44. Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K. Obesity‐induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep. 2012;2:799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, Klein S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brunton S. GLP‐1 receptor agonists vs. DPP‐4 inhibitors for type 2 diabetes: is one approach more successful or preferable than the other? Int J Clin Pract. 2014;68:557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tomlinson B, Hu M, Zhang Y, Chan P, Liu ZM. Effects of glucose‐lowering drugs on cardiovascular outcomes in patients with type 2 diabetes. Expert Opin Drug Metab Toxicol. 2016;12:1267–1271. [DOI] [PubMed] [Google Scholar]
- 51. Ferrannini E, DeFronzo RA. Impact of glucose‐lowering drugs on cardiovascular disease in type 2 diabetes. Eur Heart J. 2015;36:2288–2296. [DOI] [PubMed] [Google Scholar]
- 52. Green JB, Bethel MA, Armstrong PW, Buse JB, Engel SS, Garg J, Josse R, Kaufman KD, Koglin J, Korn S, Lachin JM, McGuire DK, Pencina MJ, Standl E, Stein PP, Suryawanshi S, deVan Werf F , Peterson ED, Holman RR; TECOS Study Group . Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:232–242. [DOI] [PubMed] [Google Scholar]
- 53. White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Bakris GL, Perez AT, Fleck PR, Mehta CR, Kupfer S, Wilson C, Cushman WC, Zannad F; EXAMINE Investigators . Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327–1335. [DOI] [PubMed] [Google Scholar]
- 54. Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, Cavender MA, Udell JA, Desai NR, Mosenzon O, McGuire DK, Ray KK, Leiter LA, Raz I; SAVOR‐TIMI 53 Steering Committee and Investigators . Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317–1326. [DOI] [PubMed] [Google Scholar]
- 55. Zannad F, Cannon CP, Cushman WC, Bakris GL, Menon V, Perez AT, Fleck PR, Mehta CR, Kupfer S, Wilson C, Lam H, White WB; EXAMINE Investigators . Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in examine: a multicentre, randomised, double‐blind trial. Lancet. 2015;385:2067–2076. [DOI] [PubMed] [Google Scholar]
- 56. Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA, Lingvay I, Rosenstock J, Seufert J, Warren ML, Woo V, Hansen O, Holst AG, Pettersson J, Vilsboll T; SUSTAIN‐6 Investigators . Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834–1844. [DOI] [PubMed] [Google Scholar]
- 57. Tomlinson B, Hu M, Zhang Y, Chan P, Liu ZM. An overview of new GLP‐1 receptor agonists for type 2 diabetes. Expert Opin Investig Drugs. 2016;25:145–158. [DOI] [PubMed] [Google Scholar]
- 58. Kalra S. Follow the leader‐liraglutide effect and action in diabetes: evaluation of cardiovascular outcome results trial. Diabetes Ther. 2016;7:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Russell‐Jones D, Vaag A, Schmitz O, Sethi BK, Lalic N, Antic S, Zdravkovic M, Ravn GM, Simo R; Liraglutide Effect and Action in Diabetes 5 (LEAD‐5) met+SU Study Group . Liraglutide vs insulin glargine and placebo in combination with metformin and sulfonylurea therapy in type 2 diabetes mellitus (LEAD‐5 met+SU): a randomised controlled trial. Diabetologia. 2009;52:2046–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nauck M, Frid A, Hermansen K, Shah NS, Tankova T, Mitha IH, Zdravkovic M, During M, Matthews DR; LEAD‐2 Study Group . Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (Liraglutide Effect and Action in Diabetes)‐2 study. Diabetes Care. 2009;32:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pratley R, Nauck M, Bailey T, Montanya E, Cuddihy R, Filetti S, Garber A, Thomsen AB, Hartvig H, Davies M; 1860‐LIRA‐DPP‐4 Study Group . One year of liraglutide treatment offers sustained and more effective glycaemic control and weight reduction compared with sitagliptin, both in combination with metformin, in patients with type 2 diabetes: a randomised, parallel‐group, open‐label trial. Int J Clin Pract. 2011;65:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ma Z, Chen R, Liu Y, Yu P, Chen L. Effect of liraglutide vs. NPH in combination with metformin on blood glucose fluctuations assessed using continuous glucose monitoring in patients with newly diagnosed type 2 diabetes. Int J Clin Pharmacol Ther. 2015;53:933–939. [DOI] [PubMed] [Google Scholar]
- 63. Dungan KM, Povedano ST, Forst T, Gonzalez JG, Atisso C, Sealls W, Fahrbach JL. Once‐weekly dulaglutide versus once‐daily liraglutide in metformin‐treated patients with type 2 diabetes (award‐6): a randomised, open‐label, phase 3, non‐inferiority trial. Lancet. 2014;384:1349–1357. [DOI] [PubMed] [Google Scholar]
- 64. Davies M, Pratley R, Hammer M, Thomsen AB, Cuddihy R. Liraglutide improves treatment satisfaction in people with type 2 diabetes compared with sitagliptin, each as an add on to metformin. Diabet Med. 2011;28:333–337. [DOI] [PubMed] [Google Scholar]
- 65. Brady EM, Davies MJ, Gray LJ, Saeed MA, Smith D, Hanif W, Khunti K. A randomized controlled trial comparing the GLP‐1 receptor agonist liraglutide to a sulphonylurea as add on to metformin in patients with established type 2 diabetes during ramadan: the treat 4 ramadan trial. Diabetes Obes Metab. 2014;16:527–536. [DOI] [PubMed] [Google Scholar]
- 66. Liu H, Dear AE, Knudsen LB, Simpson RW. A long‐acting glucagon‐like peptide‐1 analogue attenuates induction of plasminogen activator inhibitor type‐1 and vascular adhesion molecules. J Endocrinol. 2009;201:59–66. [DOI] [PubMed] [Google Scholar]
- 67. Shiraki A, Oyama J, Komoda H, Asaka M, Komatsu A, Sakuma M, Kodama K, Sakamoto Y, Kotooka N, Hirase T, Node K. The glucagon‐like peptide 1 analog liraglutide reduces TNF‐alpha‐induced oxidative stress and inflammation in endothelial cells. Atherosclerosis. 2012;221:375–382. [DOI] [PubMed] [Google Scholar]
- 68. Guo N, Sun J, Chen H, Zhang H, Zhang Z, Cai D. Liraglutide prevents diabetes progression in prediabetic OLETF rats. Endocr J. 2013;60:15–28. [DOI] [PubMed] [Google Scholar]
- 69. Ji Y, Zhao Z, Cai T, Yang P, Cheng M. Liraglutide alleviates diabetic cardiomyopathy by blocking CHOP‐triggered apoptosis via the inhibition of the IRE‐alpha pathway. Mol Med Rep. 2014;9:1254–1258. [DOI] [PubMed] [Google Scholar]
- 70. Zhao L, Guo H, Chen H, Petersen RB, Zheng L, Peng A, Huang K. Effect of liraglutide on endoplasmic reticulum stress in diabetes. Biochem Biophys Res Commun. 2013;441:133–138. [DOI] [PubMed] [Google Scholar]
- 71. Nandy D, Johnson C, Basu R, Joyner M, Brett J, Svendsen CB, Basu A. The effect of liraglutide on endothelial function in patients with type 2 diabetes. Diab Vasc Dis Res. 2014;11:419–430. [DOI] [PubMed] [Google Scholar]
- 72. Lutz TA, Osto E. Glucagon‐like peptide‐1, glucagon‐like peptide‐2, and lipid metabolism. Curr Opin Lipidol. 2016;27:257–263. [DOI] [PubMed] [Google Scholar]
- 73. Rizzo M, Rizvi AA, Patti AM, Nikolic D, Giglio RV, Castellino G, Li Volti G, Caprio M, Montalto G, Provenzano V, Genovese S, Ceriello A. Liraglutide improves metabolic parameters and carotid intima‐media thickness in diabetic patients with the metabolic syndrome: an 18‐month prospective study. Cardiovasc Diabetol. 2016;15:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rondinelli M, Rossi A, Gandolfi A, Saponaro F, Bucciarelli L, Adda G, Molinari C, Montefusco L, Specchia C, Chiara Rossi M, Scardapane M, Arosio M, Genovese S. Use of liraglutide in the real world and impact at 36 months on metabolic control, weight, lipid profile, blood pressure, heart rate, and renal function. Clin Ther. 2017;39:159–169. [DOI] [PubMed] [Google Scholar]
- 75. Chen J, Zhao H, Ma X, Zhang Y, Lu S, Wang Y, Zong C, Qin D, Wang Y, Yingfeng Yang Y, Wang X, Liu Y. GLP‐1/GLP‐1R signaling in regulation of adipocyte differentiation and lipogenesis. Cell Physiol Biochem. 2017;42:1165–1176. [DOI] [PubMed] [Google Scholar]
- 76. Astrup A, Carraro R, Finer N, Harper A, Kunesova M, Lean ME, Niskanen L, Rasmussen MF, Rissanen A, Rossner S, Savolainen MJ, Van Gaal L; NN8022‐1807 Investigators . Safety, tolerability and sustained weight loss over 2 years with the once‐daily human GLP‐1 analog, liraglutide. Int J Obes (Lond). 2012;36:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Vilsboll T, Zdravkovic M, Le‐Thi T, Krarup T, Schmitz O, Courreges JP, Verhoeven R, Buganova I, Madsbad S. Liraglutide, a long‐acting human glucagon‐like peptide‐1 analog, given as monotherapy significantly improves glycemic control and lowers body weight without risk of hypoglycemia in patients with type 2 diabetes. Diabetes Care. 2007;30:1608–1610. [DOI] [PubMed] [Google Scholar]
- 78. Zinman B, Gerich J, Buse JB, Lewin A, Schwartz S, Raskin P, Hale PM, Zdravkovic M, Blonde L; Investigators LEAD‐4 Study Investigators . Efficacy and safety of the human glucagon‐like peptide‐1 analog liraglutide in combination with metformin and thiazolidinedione in patients with type 2 diabetes (LEAD‐4 MET+TZD). Diabetes Care. 2009;32:1224–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. DM is not associated with changes in the expression of acute ER stress markers. Venous endothelial cells from diabetic and non‐diabetic patients were freshly isolated as described in the Materials and Methods section of this article. Endothelial cells were identified by van W illebrand factor staining and nuclear morphology. Total protein expression was determined by quantitative fluorescence and the use of specific antibodies against IRE1α (A), PERK (B), and ATF6 (C). Variables were compared using the t test.
Figure S2. ER stress induction by tunicamycin impairs insulin‐induced eNOS phosphorylation in endothelial cells in culture. A, Human aortic endothelial cells (HAECs) were incubated with tunicamycin for the indicated time points and ER stress activation were evaluated by an enhanced IRE1α phosphorylation and higher expression of CHOP. The blots shown are representative of at least 4 independent experiments that yielded equivalent results. The bar graph represents the mean±SEM of at least 4 independent experiments (n=9 for IRE1α phosphorylation, and n=4 for CHOP). B, Insulin‐mediated endothelial nitric oxide (eNOS) activation was evaluated as eNOS phosphorylation at Ser1177, ER stress activation by tunicamycin impaired insulin‐mediated changed in eNOS phosphorlation at Ser1177 (n=5). Variables were compared vs controls using paired t test.
Figure S3. Liraglutide does not decrease PERK activation in endothelial cell from patients with DM. Venous endothelial cells were isolated from patients with DM, and PERK activation was studied in the presence and absence of liraglutide. Pooled data showed that the PERK activation did not decrease after acute liraglutide treatment (n=4; P=0.81; Cohen's d=0.133). Variables were compared using the paired t test.
Figure S4. GLP‐1 analogue, liraglutide, activated endothelial nitric oxide synthase in venous endothelial cells from patients without DM. Venous endothelial cells were freshly isolated from patients with DM and controls without DM, and eNOS activation was studied in the presence and absence of liragultide. Pooled data showed that the liraglutide enhanced eNOS activation in endothelial cells isolated from patients without DM (n=8; P=0.006; Cohen's d=1.362), but it did not modify eNOS activation at its activation residue in endothelial cells isolated from patients with DM (n=10; P=0.33; Cohen's d=0.324). Variables were compared using the paired t test.