

Abstract
Glycans can be directly labeled using unnatural monosaccharide analogs, termed metabolic chemical reporters (MCRs). These compounds enable the secondary visualization and identification of glycoproteins by taking advantage of bioorthogonal reactions. Most widely used MCRs have azides or alkynes at the 2-N-acetyl position but are not selective for one class of glycoprotein over others. To address this limitation, we are exploring additional MCRs that have bioorthogonal functionality at other positions. Here, we report the characterization of 2-azido-2-deoxy-glucose (2AzGlc). We find that 2AzGlc selectively labels intracellular O-GlcNAc modifications, which further supports a somewhat unexpected, structural flexibility in this pathway. In contrast to the endogenous modification N-acetyl-glucosamine (GlcNAc), we find that 2AzGlc is not dynamically removed from protein substrates and that treatment with higher concentrations of per-acetylated 2AzGlc is toxic to cells. Finally, we demonstrate that this toxicity is an inherent property of the small-molecule, as removal of the 6-acetyl-group renders the corresponding reporter nontoxic but still results in protein labeling.
Graphical Abstract

Metabolic chemical reporters (MCRs) of glycans are typically monosaccharide analogs that contain bioorthogonal functional groups, such as an alkyne or azide.1–3 Once living cells are treated with one of these analogs, the MCRs can be metabolically transformed into at least one, if not more, high-energy sugar donors that can be subsequently utilized by glycosyltransferases resulting in the incorporation of MCRs into glycans. An ever growing number of bioorthogonal reactions can then be employed for the selective installation of tags for the visualization, characterization, and identification of glycoconjugates, most notably glycoproteins.4 In the past five years, we and others have demonstrated that the majority of glycoprotein MCRs are not specific for one type of glycosylation.5–8 For example, the original MCR for intra-cellular O-GlcNAc modification, N-azidoacetyl-glucosamine (GlcNAz),9 labels both O-GlcNAcylated proteins and cell-surface glycans (i.e., N-linked and mucin O-linked).6,8 Additionally, we have shown that MCRs that have their bioorthogonal functionality at the N-acetyl position, which makes up the vast majority of glycan reporters, have the potential to be de-N-acetylated, resulting in labeling of the protein acetylation pathway.10 These issues result in a lack of specificity that can limit the information that can be gained in glycoprotein visualization experiments or the number of a specific type of glycoprotein that can be identified using enrichment and proteomics.
We recently demonstrated that changes to the MCR structure resulted in the first selective reporter of O-GlcNAcylation, 6-azido-6-deoxy-N-acetyl-glucosamine (6AzGlcNAc).8 O-GlcNAcylation is the addition of the monosaccharide N-acetyl-glucosamine to serine and threonine side chains of proteins located in the cytosol, nucleus, and mitochondria.11–13 O-GlcNAc transferase (OGT) uses the uridine diphosphate sugar donor UDP-GlcNAc to add O-GlcNAc to protein substrates, and these modifications can be subsequently removed by O-GlcNAcase (OGA), rendering O-GlcNAcylation dynamic.14 Genetic knockout of either of these enzymes is lethal in mammals and insects,15–18 and the modification plays key roles in human disease, including cancer and neurodegeneration.19,20 Here, we demonstrate that 2-azido-2-deoxy-glucose (2AzGlc) is also a selective MCR for O-GlcNAc modifications. Treatment of cells with the per-acetylated reporter Ac42AzGlc (Figure 1A) resulted in the labeling of a variety of proteins by 2AzGlc in mammalian cells but no detectable incorporation of 2AzGlc into cell-surface glycoproteins. Additionally, bioorthogonal enrichment of proteins that were labeled with Ac42AzGlc followed by proteomics identified many known O-GlcNAcylated proteins. In contrast to 6AzGlcNAc, 2AzGlc appears to not be removed from substrate proteins by OGA, consistent with the enzymatic mechanism that requires anchimeric assistance of the N-acetyl group.21 Additionally, treatment of cells with Ac42AzGlc at a high concentration (200 μM) was toxic to cells after overnight exposure. Interestingly, this toxicity is inherent to the structure of the small molecule, as deprotection of the 6-O-acetate to yield Ac32AzGlc removed all cell-death but retained similar levels of protein labeling. Importantly, this observation has been made on other MCRs by the Yarema lab, which they attribute to changing the balance between the productive biosynthetic transformation of reporters to their corresponding donor sugars versus toxic side-reactions.22
Figure 1.
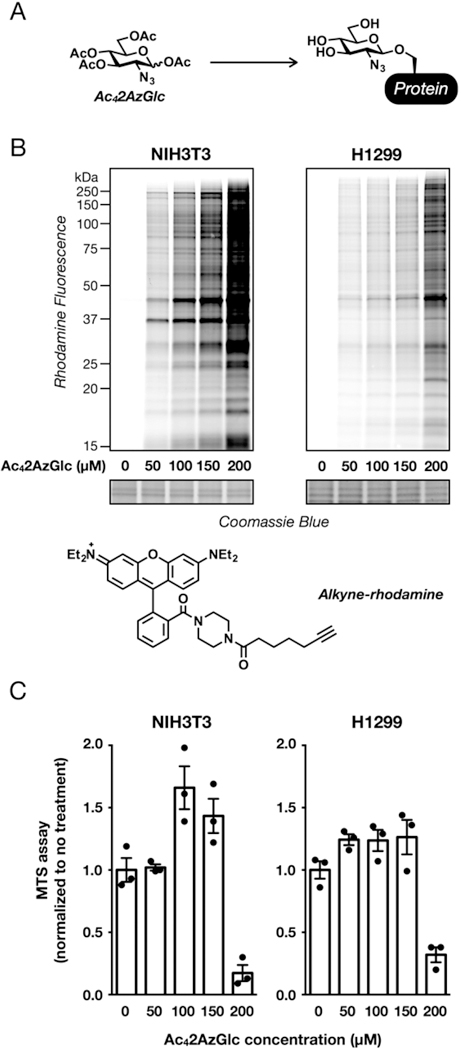
Evaluation of per-acetylated-2-azido-glucose (Ac42AzGlc) as a metabolic chemical reporter (MCR). (A) Treatment of living cells with MCRs, like Ac42AzGlc, results in modification of proteins that can then be visualized using bioorthogonal reactions. (B) Ac42AzGlc treatment results in labeling of a wide range of proteins. NIH3T3 or H1299 cells were treated with the indicated concentrations of Ac42AzGlc for 6 h, followed by CuAAC with alkyne-rhodamine and visualization of labeled proteins by in-gel fluorescence scanning. (C) High concentrations of Ac42AzGlc are toxic to mammalian cells. NIH3T3 or H1299 cells were treated with the indicated concentrations of Ac42AzGlc for 16 h, and their viability was then measured using an MTS assay. Quantitated data is are average of three separate biological experiments, and error bars represent ± SEM from the mean of biological replicates (n = 3).
RESULTS AND DISCUSSION
Given our interesting previous results that structural changes in the chemical structure of MCRs can change their selectivity, we set out to explore 2AzGlc, as it lacks the acetamido functionality that is common to the vast majority of monosaccharide positions found in glycoproteins and that has been a consistent feature of all previous MCRs. Therefore, we rationalized that it may have interesting properties concerning its metabolism and glycoprotein selectivity. Toward this goal, we prepared the per-O-acetylated compound, Ac42AzGlc, in two steps from glucosamine. These acetates enable diffusion of the reporter through cellular membranes where they are removed by lipases and/or hydrolases. NIH3T3 and H1299 cells were then treated with increasing concentrations of Ac42AzGlc for 6 h, and the corresponding cell lysates were subjected to the copper(I)-catalyzed azide−alkyne cyclo-addition (CuAAC) with an alkyne-rhodamine tag. In-gel fluorescence scanning showed a range of labeled proteins (Figure 1B). In both cell lines, we observed a large increase in labeling when the concentration of Ac42AzGlc was increased from 150 to 200 μM. Notably, this higher concentration also resulted in toxicity when the cells were treated for longer periods of time (i.e., 16 h). This observation is consistent with previous experiments by the Bertozzi lab, who commented on the toxicity of Ac42AzGlc but did not characterize its labeling.23 To explore this toxicity further, the same cell lines were treated with a range of Ac42AzGlc concentrations for 16 h, and their NAD(P)H-dependent reductase activity was measured using a commercially available MTS assay (Figure 1C). Interestingly, an increase in activity was observed for lower concentrations of Ac42AzGlc, indicating additional metabolic flux into reductive equivalents. However, the cells displayed a dramatic loss of activity between 150 and 200 μM Ac42AzGlc, consistent with our morphological observations.
Despite this toxicity, we next set out to characterize the selectivity of Ac42AzGlc as an MCR. To determine if 2AzGlc is incorporated into cell-surface glycans, H1299 cells were treated with Ac42AzGlc (200 μM) for 6 h, which is before the onset of obvious cellular toxicity. The intact cells were then subjected to a strain-promoted azide−alkyne cycloaddition (SPAAC) with DBCO-biotin, which can only label extracellular azides, followed by incubation with FITC-avidin and analysis by flow-cytometry (Figure 2A). As controls, we also simultaneously treated cells with either Ac4GalNAz (200 μM), which does label cell-surface glycoproteins,8,24 or Ac36AzGlcNAc (200 μM), which does not.8 We observed essentially no fluorescence signal from cells treated with either Ac42AzGlc or Ac36AzGlcNAc, despite a strong signal from the Ac4GalNAz-treated cells, indicating that Ac42AzGlc does not label cell-surface glycans. We next used Ac42AzGlc in an unbiased proteomics experiment in NIH3T3 cells, as we have utilized these cells in the past for the identification of labeled proteins with other MCRs. Specifically, NIH3T3 cells were treated in triplicate with Ac42AzGlc or Ac4GlcNAc as a control (both at 200 μM) for 6 h, again before any signs of toxicity. The cells were then lysed under denaturing conditions (4% SDS), and the labeled proteins underwent CuAAC with an alkyne biotintag. The labeled proteomes were then reduced and alkylated with iodoacetamide, followed by incubation with streptavidin-conjugated beads. After extensive washing, the enriched proteins were subjected to on-bead trypsinolysis, and the resulting peptides were analyzed using LC-MS/MS and identified using Proteome Discoverer and Mascot. Spectral counting was used to quantify the proteins enriched in either sample.
Figure 2.
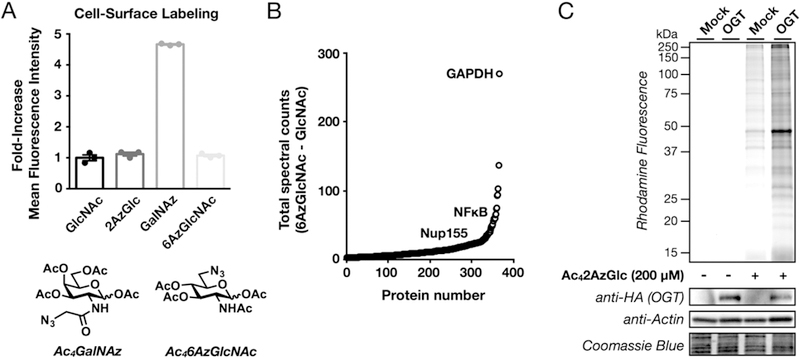
Ac42AzGlc, an O-GlcNAc metabolic chemical reporter. (A) Cell surface glycoproteins are not labeled after treatment with Ac42AzGlc. H1299 cells were treated with the indicated metabolic chemical reporters (200 μM) for 6 h, at which time the cells were collected and subjected to copper-free click chemistry with DBCO-biotin (Click Chemistry Tools). After incubation with FITC-avidin, surface glycoprotein labeling was analyzed by flow cytometry. (B) Unbiased identification of O-GlcNAcylated proteins using 2AzGlc. NIH3T3 cells were treated in triplicate with Ac42AzGlc (200 μM) or Ac4GlcNAc (200 μM) vehicle for 6 h, followed by CuAAC with alkyne-biotin, enrichment with streptavidin-coated beads, and on-bead trypsinolysis. Proteins that were identified using LC-MS/MS are represented as the total number of positive spectral counts minus the total number of control spectral counts. Representative known O-GlcNAcylated proteins are shown. (C) O-GlcNAc transferase (OGT) overexpression increases 2AzGlc labeling. NIH3T3 cells that stably express either HA-tagged OGT or an empty plasmid were treated with Ac42AzGlc (200 μM) or DMSO vehicle for 6 h. At this time, the cell lysates were subjected to CuAAC with alkyne-rhodamine, and labeled proteins were visualized using in-gel fluorescence.
Proteins were considered to have been labeled by 2AzGlc if they met certain criteria. First, the proteins must have been identified by at least one peptide in each of the three Ac42AzGlc-treated samples. Second, the sum of the spectral counts from the Ac42AzGlc-treated samples must be 3-times greater than the sum of the spectral counts for the same protein in the Ac4GlcNAc-treated sample. Finally, the spectral counts in the MCR-treated samples must be statistically significantly different from the control samples (P < 0.05, t test). With these requirements, we identified 361 proteins (Figure 2B and Table S1). Importantly, in this list were 265 proteins that had been previously identified in other proteomics experiments as being potentially O-GlcNAcylated. We then annotated these proteins based on their subcellular localization, since O-GlcNAcylated proteins will be intracellular while cell-surface glycoproteins will be extra-cellular/lumenal or have a transmembrane domain. Using the Uniprot database, we found that Ac42AzGlc labeled essentially exclusively intracellular or transmembrane proteins and only resulted in the enrichment of one exclusively extracellular protein, EGF-containing fibulin-like extracellular matrix protein, that could not bear O-GlcNAcylation.
Next, we used retroviral transformation to generate NIH3T3 cells that stably overexpress an HA-tagged OGT or were transduced with an empty plasmid as a negative control. These cells were treated with Ac42AzGlc (200 μM) for 6 h, followed by CuAAC with alkyne-rhodamine and analysis using in-gel fluorescence (Figure 2C). Notably, we observed significantly more labeling in the OGT overexpressing cells, further indicating that 2AzGlc is an MCR for O-GlcNAcylation. To explore the generality of 2AzGlc labeling, we next treated a variety of mammalian cell lines with Ac42AzGlc (200 μM) for 6 h. In-gel fluorescence scanning, after CuAAC with alkyne-rhodamine, showed strong labeling in all of the cell lines tested (Figure 3), indicating that 2AzGlc is a general MCR for the visualization and identification of O-GlcNAcylated proteins. We next explored the reversibility of 2AzGlc labeling. As stated in the introduction, the enzymatic mechanism of OGA involves anchimeric assistance of the N-acetyl group to generate an oxazoline intermediate. A wealth of carbohydrate chemistry has demonstrated that a 2-azido group is incapable of participating in this fashion,25 raising the likely possibility that once 2AzGlc is installed by OGT, it cannot be removed by OGA. To directly test this hypothesis, we performed a pulse-chase experiment. Briefly, HeLa cells were treated with Ac42AzGlc (200 μM) for 6 h. At this time the medium was exchanged for fresh medium containing Ac4GlcNAc (200 μM) and either the OGA inhibitor Thiamet-G (10 μM)26 or DMSO vehicle. After an additional 12 h, the corresponding cell lysates were subjected to CuAAC with alkyne-rhodamine and analysis by in-gel fluorescence (Figure 4A). In contrast to the 6AzGlcNAc labeling that we previously demonstrated is maintained by Thiamet-G treatment,8 we observed no difference in the reduction of 2AzGlc labeling, indicating that it is not a substrate for OGA. Interestingly, we did, however, observe a relatively rapid loss in 2AzGlc labeling over 12 h, suggesting that the labeled proteins were being turned over by another mechanism. Proteasomal degradation is one of the major pathways by which proteins are targeted for turnover. To determine if 2AzGlc-labled proteins are being destroyed by this pathway, we performed another pulse-chase experiment. In this case, the pulse was identical to the one above, but in the chase, we added either the proteasome inhibitor MG-132 (10 μM) or DMSO control. In-gel fluorescence scanning following CuAAC with alkyne-rhod-amine only showed a slight stabilization of the fluorescent signal (Figure 4B), indicating that the majority of the 2AzGlc-dependent labeling is being removed by an unknown mechanism.
Figure 3.
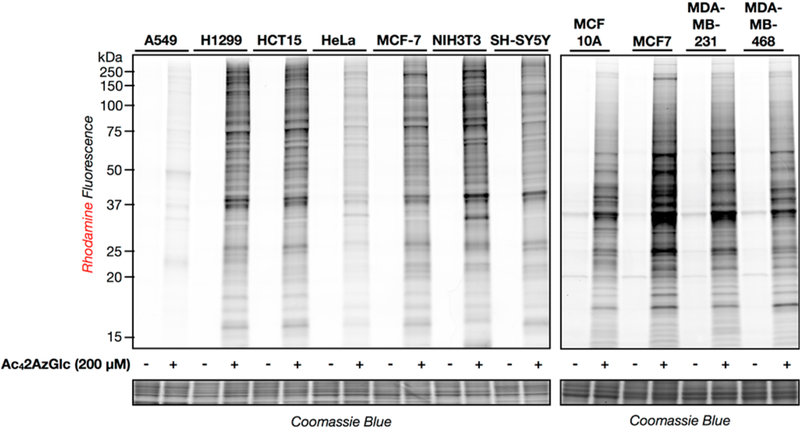
Ac42AzGlc, a general chemical reporter in a variety of mammalian cell lines. (A) The indicated cell lines were treated with Ac42AzGlc (200 μM) or DMSO vehicle for 6 h. Total cell lysates were then collected and subjected to CuAAC with alkyne-rhodamine, and labeled proteins were visualized using in-gel fluorescence scanning.
Figure 4.
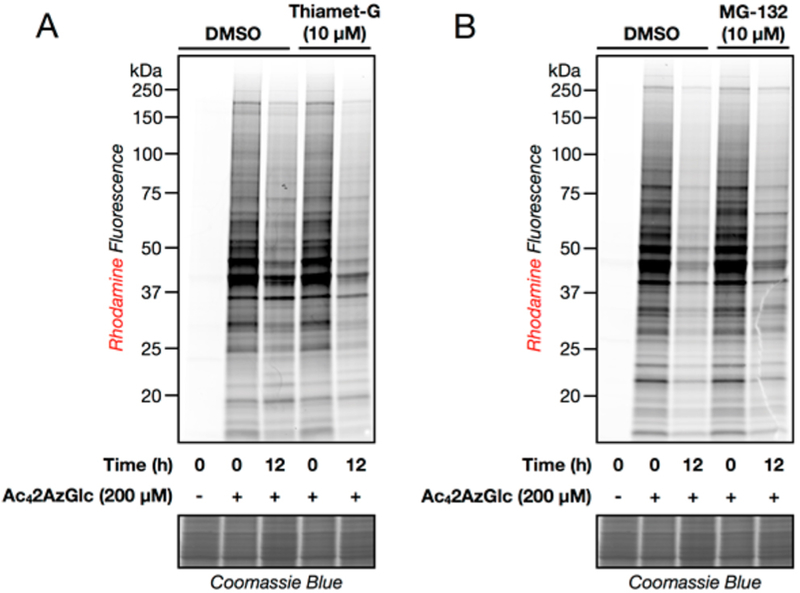
Characterization of the dynamics of 2AzGlc labeling. (A) Protein labeling by 2AzGlc is not enzymatically removed by O-GlcNAcase (OGA). HeLa cells were treated with 200 μM Ac42AzGlc for 6 h. At this time, the medium was exchanged for fresh medium containing 200 μM Ac4GlcNAc and either the OGA inhibitor Thiamet-G (10 μM) or DMSO. Cells were collected at indicated times, subjected to CuAAC with alkyne-rhodamine, and analyzed by in-gel fluorescence scanning. (B) Labeling by 2AzGlc is not notably stabilized by proteasome inhibition. HeLa cells were treated with 200 μM Ac42AzGlc for 6 h. At this time, the medium was exchanged for fresh medium containing 200 μM Ac4GlcNAc and either the proteasome inhibitor MG132 (10 μM) or DMSO. Cells were collected at the indicated times, subjected to CuAAC with alkyne-rhodamine, and analyzed by in-gel fluorescence scanning.
Finally, to explore the potential mechanism underlying the toxicity of Ac42AzGlc treatment, we looked to the elegant experiments performed by the Yarema lab who were exploring the off-target effects of monosaccharides modified by short-chain fatty acids.22 Although the exact biological mechanism behind the toxicity of certain acetylated monosaccharides are not entirely known, the authors demonstrated that the toxicity of these molecules could be eliminated by revealing the 6-hydroxyl functionality, which increases the flux of the carbohydrate into cellular biosynthetic pathways. To test whether this observation could explain the toxicity of Ac42AzGlc, we synthesized the 6-OH analog (Ac32AzGlc, Figure 5A). First, we examined whether aqueous conditions would result in migration of the 4-O-acetate to the 6-hydroxyl group, as can be observed under certain reaction conditions. To accomplish this, we dissolved Ac32AzGlc in a 5:1 mixture of D2O and deuterated DMSO and incubated the mixture for 16 h at RT. After this length of time, the solution was diluted to 1:1 D2O and deuterated DMSO and analyzed by proton NMR. A comparison of this spectrum to a freshly prepared sample in the same solvent composition showed essentially no differences (Supporting Information), demonstrating that any acetate migration is minimal and below the detection limit of NMR. Next, treatment with either NIH3T3 or H1299 cells with Ac32AzGlc resulted in robust labeling of proteins, as visualized by in-gel fluorescence (Figure 5B). However, unlike Ac42AzGlc, the 6-OH analog showed no cellular toxicity at all of the concentrations tested (Figure 5C). These results support an inherent toxicity mechanism that is largely unrelated to protein labeling by 2AzGlc as an O-GlcNAc reporter.
Figure 5.
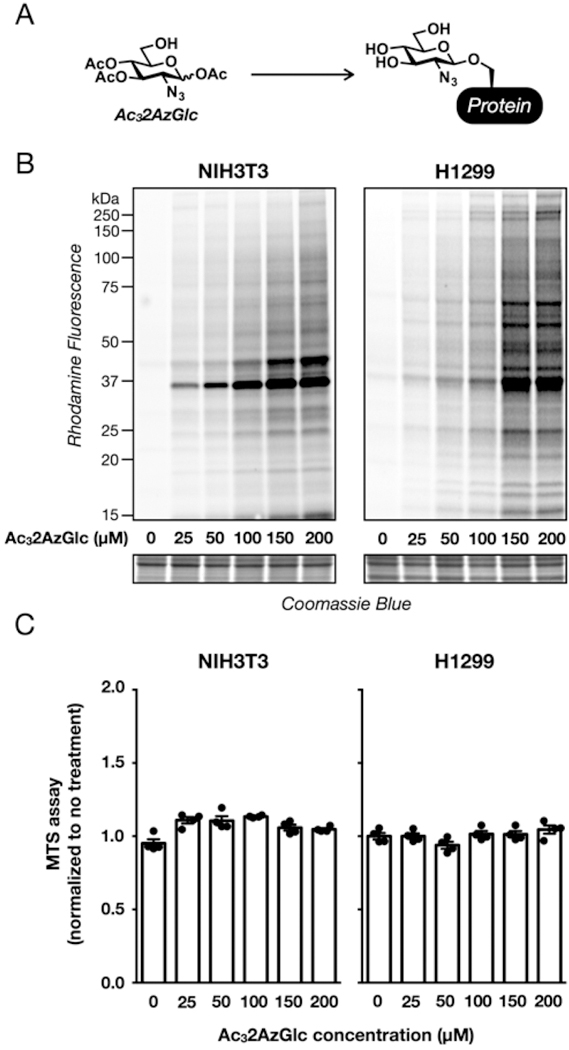
Acetylation pattern and not labeling by 2AzGlc responsible for cellular toxicity. (A) Proteins can also be labeled by Ac32AzGlc, which bears a free hydroxyl at the 6-OH position. (B) Ac32AzGlc-treatment results in labeling of a wide range of proteins. NIH3T3 or H1299 cells were treated with the indicated concentrations of Ac32AzGlc for 16 h, followed by CuAAC with alkyne-rhodamine and visualization of labeled proteins by in-gel fluorescence scanning. (C) No concentrations of Ac32AzGlc are toxic to mammalian cells. NIH3T3 or H1299 cells were treated with the indicated concen-trations of Ac32AzGlc for 16 h, and their viability was then measured using an MTS assay. Quantitated data are the average of four separate biological experiments, and error bars represent ± SEM from the mean of biological replicates (n = 4).
Conclusions.
The development of new MCRs for glycans has continued to grow the chemical toolbox for the investigation of glycobiology. In particular, these reporters have been key for the visualization and identification of glycoproteins. We have been focused on understanding the selectivity of different MCRs, including some that would not be predicted to label proteins based on the previously established biosynthetic and salvage pathways. Continuing that theme here, we performed the cellular characterization of 2AzGlc as a chemical reporter of glycosylation. Using bioorthogonal chemistry, we show that 2AzGlc labels a range of proteins in total cell lysate (Figures 1B and 3) but does not label cell-surface glycans at a detectable level (Figure 2A), suggesting that it might be selective for O-GlcNAc modifications. This selectivity was further supported by an unbiased proteomics experiment that used 2AzGlc to enrich a total of 361 proteins, 73.4% of which had been previously identified in other O-GlcNAcylation proteomics experiments and only one exclusively extracellular or lumenal glycoprotein (Figure 2C and Table S1). The ability for 2AzGlc to be transferred by OGT from the corresponding UDP-2AzGlc sugar donor in cells is somewhat surprising, given the ternary crystal structures of OGT in complex with its peptide and donor-sugar substrates, as well as the structure with the corresponding products.27 These structures show that the 2-acetamide group of GlcNAc undergoes significant movement during the enzymatic reaction and that the amide nitrogen contributes a hydrogen-bond to the phosphate of the UDP leaving group. Additionally, the same authors show that in vitro OGT will accept UDP-GalNAc but not UDP-Glc or UDP-2-keto-Glc.27 However, another structural and biochemical study found that OGT accepts UDP-GlcNAcF3 (2-N-trifluoroacetamide), which they interpreted as evidence that the electronics of the 2-acetamide are not key for catalysis.28 Given these data, the 2-azido group might either be functionally benign or could participate in weak hydrogen bonds, given its unique electronics.29 We also confirmed a statement from the Bertozzi lab that 2AzGlc is toxic to mammalian cells (Figure 1B), although they did not show any supporting data.23 Here, we demonstrate that this toxicity can be eliminated by deprotection of the 6-position to reveal the hydroxyl group, which the Yarema lab has shown increased flux of other monosaccharide analogs into the corresponding biosynthetic pathways. Given the mechanism of OGA, we also explored the reversibility of the 2AzGlc modifications. We used a pulse-chase experiment to demon-strate that 2AzGlc is not removed from proteins by OGA (Figure 4A). As stated above, this is not necessarily surprising, given the requirement for anchimeric assistance in the enzymatic mechanism.21 Interestingly, although a loss of 2AzGlc labeling is not OGA-dependent, the rate of loss is similar to other MCRs that are removed by OGA, such as 6AzGlcNAc,8 suggesting to us that the 2AzGlc-labeled proteins might be degraded. However, a pulse-chase experiment with the proteasomal inhibitor MG-132 only showed a modest stabilization of the fluorescent signal (Figure 4B), indicating that another pathway exists to remove these modifications. Further experiments will be needed to directly test this hypothesis. Our proteomics data show that the vast majority of identified proteins have been previously characterized as being potentially O-GlcNAcylated by a variety of chemical and biological techniques (Table S1, proteins highlighted in blue). This indicates that the specificity for protein substrates between 2AzGlc and other MCRs is similar. However, we cannot rule out that different proteins are more readily modified by OGT with a reporter that lacks the 2-acetamido functionality versus an MCR like 6AzGlcNAc. Therefore, we suggest that 6AzGlcNAc be used for studies aimed at the biology of O-GlcNAcylation and that all potentially modified proteins be confirmed by either antibodies or the chemoenzymatic modification strategy developed by the Hsieh-Wilson lab.30 In summary, 2AzGlc is the second MCR that has been characterized to be selective for O-GlcNAcylation. It also further highlights the promiscuity of OGT, which now includes 2AzGlc, as well as several N-acetyl-glucosamine monosacchar-ides5−9,31 and could have interesting implications for the modification of intracellular proteins by natural, non-N-acetyl-glucosamine monosaccharides that can be biosynthetically transformed to the corresponding UDP-sugar donors.
EXPERIMENTAL PROCEDURES
General.
All reagents used for chemical synthesis were purchased from Sigma-Aldrich, Alfa Aesar, or EMD Millipore unless otherwise specified and used without further purification. All anhydrous reactions were performed under an argon or nitrogen atmosphere. Analytical thin-layer chromatography (TLC) was conducted on EMD Silica Gel 60 F254 plates with detection by ceric ammonium molybdate (CAM), anisaldehyde, or UV. For flash chromatography, 60 Å silica gel (EMD) was utilized. 1H spectra were obtained at 400, 500, or 600 MHz on Mercury 400 or VNMRS-500 or −600 Varian spectrometers. Chemical shifts are recorded in ppm (δ) relative to solvent. 13C spectra were obtained at 100, 125, or 150 MHz on the same instruments.
Cell Lines.
NIH3T3 cells stably expressing either HA-tagged O-GlcNAc Transferase (OGT) or an empty vector were generated using Amphopack293 retroviral packaging cell lines according to manufacturer procedure (Clontech).
Synthesis of Chemical Reporters.
Known compounds Thiamet-G, Ac4GalNAz, Ac4GlcNAz, Ac42AzGlucose, and azido-rhodamine were synthesized according to literature procedures.8,26,32,33
Synthesis of 1,3,4,6-Tetra-O-acetyl-2-azido-2-deoxyglucose (Ac42AzGlc, 2).
2-Azido-2-Deoxyglucose (1).
Commercially available glucosamine hydrochloride was subjected to azide formation using a diazo transfer reaction according to a literature procedure.34,1H NMR (500 MHz, deuterium oxide) of β-anomer: δ 4.71 (d, J = 8.1 Hz, 1H), 3.92−3.82 (m, 1H), 3.81−3.70 (m, 1H), 3.54−3.43 (m, 3H), 3.28 (dd, J = 9.6, 8.2 Hz, 1H). The product was used in the subsequent reaction without further characterization.
1,3,4,6-Tetra-O-acetyl-2-azido-2-deoxyglucose (Ac42AzGlc, 2).
Compound 1 was resuspended in pyridine and acetic anhydride. A catalytic amount of dimethylaminopyridine was added and the reaction stirred at RT for 16 h. Upon completion, the reaction was concentrated, and the crude was resuspended in dichloromethane. The organic layer was washed with equivolume 1 M HCl (1×), saturated sodium bicarbonate (×2), water (×2), and brine (1×). The washed organic layer was then concentrated and subjected to column chromatography to yield the pure product. NMR characterization of this known compound was consistent with previous reports.23,1H NMR (500 MHz, CDCl3) of β-anomer: δ 5.55 (d, J = 8.5 Hz, 1H), 5.15−5.00 (m, 2H), 4.34−4.25 (m, 1H), 4.11−4.05 (m, 1H), 3.84−3.77 (m, 1H), 3.71−3.62 (m, 1H), 2.19 (s, 3H), 2.09 (s, 3H), 2.07 (d, 3H), 2.02 (s, 3H).
Synthesis of 1,3,4-Tri-O-acetyl-2-azido-2-deoxyglucose (Ac32AzGlc, 5).
6-tert-Butyldimethylsilyl-2-azido-2-deoxyglucose (6TBS2AzGlc, 3).
Compound 1 (668 mg, 3.26 mmol) was dissolved in dry DMF (8 mL) under nitrogen at RT. Imidazole (667 mg, 9.8 mmol) and tert-butyldimethylsilyl chloride (613 mg, 4.07 mmol) were added and the reaction stirred for 16 h. The reaction was diluted in ethyl acetate and washed with sodium bicarbonate (2×), water (2×), and brine (1×). The organic layer was dried with sodium sulfate, filtered, and concentrated by a vacuum. The compound was purified by column chromatography (25% ethyl acetate/hexanes) to afford the product as a light brown syrup (327 mg, 31%). 1H NMR (400 MHz, CDCl3) of α-,β-anomer mixture: 5.18 (d, J = 3.4 Hz, 1H), 4.55 (d, J = 8.0 Hz, 1H), 4.01 (dd, J = 14.0, 6.0 Hz, 1H), 3.95 (t, J = 9.5 Hz, 1H) 3.86−3.65 (m, 6H), 3.53−3.46 (m, 2H), 3.37 (t, J = 9.5 Hz 1H), 3.31−3.25 (m, 1H), 3.20−3.14 (m, 2H), 0.81 (s, 18H), 0.00 (s, 12H). 13C NMR (125 MHz, CDCl3): δ (ppm) 95.89, 91.59, 76.91, 74.47, 72.45, 71.18, 70.08, 66.33, 63.9, 62.99, 25.77, −5.80. ESI-MS calculated for C12H26N3O5Si [M + H]: 320.1636. Found: 319.5972.
1,3,4-Tri-O-acetyl-6-tert-butyldimethylsilyl-2-azido-2-deoxyglucose (Ac36TBS2AzGlc, 4).
Compound 3 (150 mg, 0.47 mmol) was dissolved in pyridine (2 mL) and cooled to 0 °C. Acetic anhydride (200 uL, 2.11 mmol) was added, and the reaction warmed to RT over 16 h. The reaction was diluted in DCM and washed with sodium bicarbonate (2×), water (2×), and brine (1×). The organic layer was dried with sodium sulfate, filtered, and concentrated by a vacuum before purification by column chromatography (10% ethyl acetate:-hexanes) to afford the product as a light brown syrup (170 mg, 81%). 1H NMR (400 MHz, CDCl3) of α-, β-anomer mixture: δ (ppm) 6.28 (d, J = 3.6 Hz, 1H), 5.52 (d, J = 8.6 Hz, 1H), 5.46−5.40 (m, 1H), 5.14 (dd, J = 10.2, 9.4 Hz, 1H), 5.08−5.04 (m, 2H), 3.91−3.85 (m, 1H), 3.76−3.70 (m, 1H), 3.69−3.58 (m, 6H), 2.16 (d, J = 1.8 Hz, 6H), 2.09 (d, J = 4.7 Hz, 6H), 2.03−1.99 (m, 6H), 0.86 (s, 18H), 0.04−0.03, (m, 12H). 13C NMR (125 MHz, CDCl3): δ (ppm) 169.7, 169.14, 168.38, 92.23, 89.83, 74.95, 73.01, 72.17, 71.08, 68.01, 62.34, 61.53, 60.19, 25.51, 20.52, −5.72. ESI-MS calculated for C18H31N3O8SiNa [M + Na]+: 468.1773. Found: 468.1766.
1,3,4-Tri-O-acetyl-2-azido-2-deoxyglucose (Ac32AzGlc, 5).
Compound 4 (170 mg, 0.38 mmol) was dissolved in THF (2 mL) and cooled to 0 °C. Glacial acetic acid (445 uL) was added followed by the addition of 1.0 M TBAF in THF (7.8 mL), and the reaction was allowed to warm to RT over 16 h. The solution was diluted with ethyl acetate; washed with sodium bicarb (2×), water (2×), and brine (1×); and dried with sodium sulfate. The resulting mixture was filtered, concentrated by a vacuum, and purified by column chromatography (40% ethyl acetate/hexanes) to afford product as a yellow syrup (107 mg, 85%). 1H NMR (400 MHz, CDCl3) of α-,β-anomer mixture: δ (ppm) 6.27 (d, J = 3.7 Hz, 1H), 5.54 (d, J = 8.5 Hz, 1H), 5.32 (dd, J = 10.5, 9.3 Hz, 1H), 4.95−4.90 (m, 1H), 4.56−4.47 (m, 2H), 4.21 (dd, J = 12.5, 1.7 Hz, 1H), 4.21 (dd, 12.5, 2.3 Hz, 1H), 3.92−3.87 (m, 1H), 3.65−3.48 (m, 6H), 2.20−2.18 (m, 12H), 2.12 (s, 6H). 13C NMR (125 MHz, CDCl3): δ (ppm) 171.28, 170.95, 168.34, 92.35, 89.91, 74.86, 72.73, 72.03, 68.27, 62.16, 59.92, 20.53. ESI-MS calculated for C12H18N3O8 [M + H]+: 332.1088. Found: 332.1085.
Cell Culture.
NIH3T3 and MEF cells were cultured in high glucose DMEM media (HyClone, ThermoScientific) enriched with 10% fetal calf serum (FCS, HyClone, ThermoScientific). MDA-MB-231, MDA-MB-468, HeLa, and MCF-7 were cultured in high glucose DMEM (HyClone, ThermoScientific) enriched with 10% fetal bovine serum (FBS HyClone, ThermoScientific). H1299 and HCT15 cells were cultured in RPMI 1640 medium (HyClone, ThermoScientific) enriched with 10% FBS. A549 cells were cultured in F-12K medium (HyClone, ThermoScientific) enriched with 10% FBS. MCF10A and SH-SY5Y were cultured in a 1:1 mixture of F-12 and DMEM media (HyClone, ThermoScientific) enriched with 10% FBS. All cell lines were maintained in a humidified incubator at 37 °C and 5.0% CO2.
Metabolic Labeling.
To cells at 80−85% confluency, high- or low-glucose media containing Ac4GlcNAz, Ac4GalNAz, Ac42AzGlc (1000 stock in DMSO), or DMSO vehicle was added as indicated. For chase experiments, low-glucose media was replaced with high-glucose media supplemented with 200 μM Ac4GlcNAc (Sigma) with or without 20 μM Thiamet-G.
Preparation of NP-40-Soluble Lysates.
The cells were collected by trypsinization and pelleted by centrifugation for 4 min at 2000g, followed by washing 2× with PBS (1 mL). Cell pellets were then resuspended in 100 μL of 1% NP-40 lysis buffer [1% NP-40, 150 mM NaCl, 50 mM triethanolamine (TEA) pH 7.4] with Complete, Mini, EDTA-free Protease Inhibitor Cocktail Tablets (Roche) for 20 min and then centrifuged for 10 min at 10 000g at 4 °C. The supernatant (soluble cell lysate) was collected, and the protein concentration was determined by BCA assay (Pierce, ThermoScientific).
Cu(I)-Catalyzed [3 + 2] Azide−Alkyne Cycloaddition (CuAAC).
Cell lysate (200 μg) was diluted with 1% NP-40 lysis buffer to obtain a desired concentration of 1 μg μL−1. Newly made click chemistry cocktail (12 μL) was added to each sample [alkynyl-rhodamine tag (100 μM, 10 mM stock solution in DMSO); tris(2-carboxyethyl)phosphine hydrochloride (TCEP; 1 mM, 50 mM freshly prepared stock solution in water); tris[(1-benzyl-1-H-1,2,3-triazol-4-yl)methyl]amine (TBTA; 100 μM, 10 mM stock solution in DMSO); CuSO4·5H2O (1 mM, 50 mM freshly prepared stock solution in water)] for a total reaction volume of 200 μL. The reaction was gently vortexed and allowed to sit at RT for 1 h. Upon completion, 1 mL of ice cold methanol was added to the reaction, and it was placed at −20 °C for 2 h to precipitate proteins. The reactions were then centrifuged at 10 000g for 10 min at 4 °C. The supernatant was removed, the pellet was allowed to air-dry for 15 min, and then 50 μL of 4% SDS buffer (4% SDS, 150 mM NaCl, 50 mM TEA pH 7.4) was added to each sample. The mixture was sonicated in a bath sonicator to ensure complete dissolution, and 50 μL of 2× SDS-free loading buffer (20% glycerol, 0.2% bromophenol blue, 1.4% β-mercaptoethanol, pH 6.8) was then added. The samples were boiled for 5 min at 97 °C, and 40 μg of protein was then loaded per lane for SDS-PAGE separation (any Kd, criterion Gel, Bio-Rad).
In-Gel Fluorescence Scanning.
Following SDS-PAGE separation, gels were scanned on a Typhoon 9400 Variable Mode Imager (GE Healthcare) using a 532 nm for excitation and 30 nm bandpass filter centered at 610 nm for detection.
MTS Assay.
NIH-3T3 and H1299 cells (1 × 104 cells) were plated per well in a 96-well, white bottom dish 24 h before treatment with 0− 200μM Ac4GlcNAc or Ac32AzGlucose for 16 h under low glucose media conditions (1 g/L). CellTiter 96 aqueous non-radioactive cell proliferation assay (Promega, Madison, WI) was used according to the provided protocol. Absorbance at 490 nm was read using a BioTek Synergy H4Multi-Mode Microplate reader.
Flow Cytometry of Cell-Surface Labeling with DBCO-Biotin.
H1299 cells grown in six-well plates at 80−85% confluency were treated with 200 μM Ac4GlcNAc, Ac4GlcNAz, Ac4GalNAz, or Ac32AzGlc in triplicate for 16 h, at which time the medium was removed and cells were gently washed with PBS before being detached from the plate with 1 mM EDTA in PBS. Cells were collected by centrifugation (5 min, 300g at 4 °C) and were washed three times with PBS (5 min, 300g at 4 °C). Cells were then resuspended in 200 μL of PBS containing DBCO-biotin (Click Chemistry Tools, 60 μM) for 1 h, after which time they were washed three times with PBS (5 min, 300g at 4 °C) before being resuspended in ice-cold PBS containing fluorescein isothiocynate (FITC) conjugated avidin (Sigma, 5 μg/mL, min at 4 °C). Cells were then washed three times in PBS (5 min, 300g at 4 °C) before being resuspended in 400 μL of PBS for flow-cytometry analysis. A total of 10 000 cells [dead cells were excluded by treatment with propidium iodide (2.5 μg mL−1 in water, 30 min)] were analyzed on a BD SORP LSRII Flow Cytometer using the 488 nm argon laser.
Biotin Enrichment and On-Bead Trypsinolysis.
NIH3T3 or H1299 cell pellets labeled with Ac36AzGlcNAc, Ac32AzGlc, or Ac4GlcNAc for 16 h were resuspended in 200 μL of H2O, 60 μL of PMSF in H2O (250 mM), and 500 μL of 0.05% SDS buffer (0.05% SDS, 10 mM TEA pH 7.4, 150 mM NaCl) with Complete Mini protease inhibitor cocktail (Roche Biosciences). To this was added 8 μL of benzonase (Sigma), and the cells were incubated on ice for 30 min. Then, 4% SDS buffer (2000 μL) was added, and the cells were briefly sonicated in a bath sonicator followed by centrifugation (20 000g for 10 min at 15 °C). Soluble protein concentration was normalized by BCA assay (Pierce, ThermoScientific) to 1 mg mL−1, and 10 mg of total protein was subjected to the appropriate amount of click chemistry cocktail containing alkyne-PEG3-biotin (5 mM, Click Chemistry Tools) for 1 h, after which time 10 volumes of ice-cold MeOH were added. Precipitation proceeded for 2 h at −20 °C. Precipitated proteins were centrifuged at 5200g for 30 min at 0 °C and washed three times with 40 mL of ice-cold MeOH, with resuspension of the pellet each time. The pellet was then air-dried for 1 h. To capture the biotinylated proteins by streptavidin beads, the air-dried protein pellet was resuspended in 2 mL of resuspension buffer (6 M urea, 2 M thiourea, 10 mM HEPES at pH 8.0) by bath sonication. To cap cysteine residues, 100 μL of freshly made TCEP (200 mM stock solution, Thermo) was then added and the mixture incubated for 30 min, followed by 40 μL of freshly prepared iodoacetamide (1 M stock solution, Sigma) and incubation for a further 30 min in the dark. Steptavadin beads (250 μL of a 50% slurry per sample, Thermo) were washed two times with 1 mL of PBS and one time with 1 mL resuspension buffer and resuspended in resuspension buffer (200 μL). Each sample was combined with streptavadin beads and incubated on a rotator for 2 h. These mixtures were then transferred to Mini Bio-Spin columns (Bio-Rad) and placed on a vacuum manifold. Captured proteins were then washed with agitation 5× with resuspension buffer (10 mL), 5× with PBS (10 mL), 5× with 1% SDS in PBS (10 mL), 30× with PBS (1 mL per wash, vacuum applied between each wash), and 5× 2 M urea in PBS (1 mL per wash, vacuum applied between each wash). Beads were then resuspended in 2 M urea in PBS (1 mL), transferred to screw-top tubes, and pelleted by centrifugation (2000g for 2 min). At this time, 800 μL of the supernatant was removed, leaving a volume of 200 μL. To this bead mixture was added 2 μL of CaCl2 (200 mM stock, 1 mM final concentration) and 2 μL of 1 mg mL−1 sequence grade trypsin (Promega) and incubated at 37 °C for 18 h. The resulting mixtures of tryptic peptides and beads were transferred to Mini Bio-Spin columns (Bio-Rad), and the eluent was collected by centrifugation (1000g for 2 min). Any remaining peptides were eluted by the addition of 100 μL of 2 M urea in PBS followed by centrifugation as immediately above. The tryptic peptides were then applied to C18 spin columns (Pierce) according to manufacturer’s instructions, eluted with 70% acetonitrile in H2O, and concentrated to dryness on a speedvac.
LC-MS/MS Proteomic Analysis.
Peptides were desalted on a trap column following separation on a 12 cm/75um reversed phase C18 column (Nikkyo Technos Co., Ltd. Japan). A 3 h gradient increasing from 10% B to 45% B in 3 h (A, 0.1% formic acid; B, acetonitrile/0.1% formic acid) was delivered at 150 nL min− 1. The liquid chromatography setup (Dionex, Boston, MA, USA) was connected to an Orbitrap XL (Thermo, San Jose, CA, USA) operated in top-5-mode. Acquired tandem MS spectra (CID) were extracted using ProteomeDiscoverer v. 1.3 (Thermo, Bremen, Germany) and queried against the human Uniprot protein database using MASCOT 2.3.02 (Matrixscience, London, UK). Peptides fulfilling a Percolator calculated 1% false discovery rate threshold were reported. All LC-MS/MS analyses were carried out at the Proteomics Resource Center at The Rockefeller University, New York, NY, USA. Excel files containing identified proteins will be made available upon request.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank H. Molina (Rockefeller University) for assistance with the proteomic analysis. B.W.Z and K.N.C. were fellows of the National Science Foundation Graduate Research Fellowship Program (DGE-0937362). This research was supported by the National Science Foundation (CHE-1506503 to M.R.P.), Susan G. Komen for the Cure (CCR14299333 to M.R.P.), and the American Cancer Society (RSG-14-225-01-CCG to M.R.P.), and in part by the National Cancer Institute of the U.S. National Institutes of Health (CCSG P30CA014089). Flow cytometry was performed in the USC Flow Cytometry Core Facility. LC-MS/MS proteomic analysis was performed at the Rockefeller University Proteomics Resource Center.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.6b00877.
Supporting figures and proteomic data tables (PDF)
REFERENCES
- (1).Grammel M, and Hang HC (2013) chemical reporters for biological discovery. Nat. Chem. Biol 9, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Chuh KN, and Pratt MR (2015) Chemical methods for the proteome-wide identification of posttranslationally modified proteins. Curr. Opin. Chem. Biol 24, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chuh KN, Batt AR, and Pratt MR (2016) Chemical Methods for Encoding and Decoding of Posttranslational Modifications. Cell Chemical Biology 23, 86–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Patterson DM, Nazarova LA, and Prescher JA (2014) Finding the right (bioorthogonal) chemistry. ACS Chem. Biol 9, 592–605. [DOI] [PubMed] [Google Scholar]
- (5).Boyce M, Carrico IS, Ganguli AS, Yu S-H, Hangauer MJ, Hubbard SC, Kohler JJ, and Bertozzi CR (2011) Metabolic cross-talk allows labeling of O-linked {beta}-N-acetylglucosamine-modified proteins via the N-acetylgalactosamine salvage pathway. Proc. Natl. Acad. Sci. U. S. A 108, 3141–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zaro BW, Yang Y-Y, Hang HC, and Pratt MR (2011) Chemical reporters for fluorescent detection and identification of O-GlcNAc-modified proteins reveal glycosylation of the ubiquitin ligase NEDD4−1. Proc. Natl. Acad. Sci. U. S. A 108, 8146–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bateman LA, Zaro BW, Chuh KN, and Pratt MR (2013) N-Propargyloxycarbamate monosaccharides as metabolic chemical reporters of carbohydrate salvage pathways and protein glycosylation. Chem. Commun 49, 4328–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chuh KN, Zaro BW, Piller F, Piller V, and Pratt MR (2014) Changes in metabolic chemical reporter structure yield a selective probe of O-GlcNAc modification. J. Am. Chem. Soc 136, 12283–12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Vocadlo D, Hang H, Kim E, Hanover J, and Bertozzi C (2003) A chemical approach for identifying O-GlcNAc-modified proteins in cells. Proc. Natl. Acad. Sci. U. S. A 100, 9116–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zaro BW, Chuh KN, and Pratt MR (2014) Chemical Reporter for Visualizing Metabolic Cross-Talk between Carbohydrate Metabolism and Protein Modification. ACS Chem. Biol 9, 1991–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Torres CR, and Hart GW (1984) Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem 259, 3308–3317. [PubMed] [Google Scholar]
- (12).Holt GD, and Hart GW (1986) The subcellular distribution of terminal N-acetylglucosamine moieties. Localization of a novel protein-saccharide linkage, O-linked GlcNAc. J. Biol. Chem 261, 8049–8057. [PubMed] [Google Scholar]
- (13).Zachara NE, and Hart GW (2002) The emerging significance of O-GlcNAc in cellular regulation. Chem. Rev 102, 431–438. [DOI] [PubMed] [Google Scholar]
- (14).Vocadlo DJ (2012) O-GlcNAc processing enzymes: catalytic mechanisms, substrate specificity, and enzyme regulation. Curr. Opin. Chem. Biol 16, 488–497. [DOI] [PubMed] [Google Scholar]
- (15).Shafi R, Iyer SP, Ellies LG, O’Donnell N, Marek KW, Chui D, Hart GW, and Marth JD (2000) The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc. Natl. Acad. Sci. U. S. A 97, 5735–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).O’Donnell N, Zachara NE, Hart GW, and Marth JD (2004) Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol. Cell. Biol 24, 1680–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sinclair DAR, Syrzycka M, Macauley MS, Rastgardani T, Komljenovic I, Vocadlo DJ, Brock HW, and Honda BM (2009) Drosophila O-GlcNAc transferase (OGT) is encoded by the Polycomb group (PcG) gene, super sex combs (sxc). Proc. Natl. Acad. Sci. U. S. A 106, 13427–13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Yang YR, Song M, Lee H, Jeon Y, Choi E-J, Jang H-J, Moon HY, Byun H-Y, Kim E-K, Kim DH, Lee MN, Koh A, Ghim J, Choi JH, Lee-Kwon W, Kim KT, Ryu SH, and Suh P-G (2012) O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell 11, 439–448. [DOI] [PubMed] [Google Scholar]
- (19).Ma Z, and Vosseller K (2013) O-GlcNAc in cancer biology. Amino Acids 45, 719–733. [DOI] [PubMed] [Google Scholar]
- (20).Yuzwa SA, and Vocadlo DJ (2014) O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer’s disease and beyond. Chem. Soc. Rev 43, 6839–6858. [DOI] [PubMed] [Google Scholar]
- (21).Macauley MS, Whitworth GE, Debowski AW, Chin D, and Vocadlo DJ (2005) O-GlcNAcase uses substrate-assisted catalysis: kinetic analysis and development of highly selective mechanism-inspired inhibitors. J. Biol. Chem 280, 25313–25322. [DOI] [PubMed] [Google Scholar]
- (22).Aich U, Campbell CT, Elmouelhi N, Weier CA, Sampathkumar SG, Choi SS, and Yarema KJ (2008) Regioisomeric SCFA Attachment to Hexosamines Separates Metabolic Flux from Cytotoxicity and MUC1 Suppression. ACS Chem. Biol 3, 230–240. [DOI] [PubMed] [Google Scholar]
- (23).Saxon E, Luchansky SJ, Hang HC, Yu C, Lee SC, and Bertozzi CR (2002) Investigating cellular metabolism of synthetic azidosugars with the Staudinger ligation. J. Am. Chem. Soc 124, 14893–14902. [DOI] [PubMed] [Google Scholar]
- (24).Hang HC, Yu C, Kato DL, and Bertozzi CR (2003) A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc. Natl. Acad. Sci. U. S. A 100, 14846–14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kerns RJ, and Wei P (2012) Synthetic Methods To Incorporate α-Linked 2-Amino-2-Deoxy-D-Glucopyranoside and 2-Amino-2-Deoxy-D-Galactopyranoside Residues into Glycoconjugate Structures, in Glycobiology and Drug Design, pp 235–263, American Chemical Society, Washington, DC. [Google Scholar]
- (26).Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ, and Vocadlo DJ (2008) A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol 4, 483–490. [DOI] [PubMed] [Google Scholar]
- (27).Lazarus MB, Jiang J, Gloster TM, Zandberg WF, Whitworth GE, Vocadlo DJ, and Walker S (2012) Structural snapshots of the reaction coordinate for O-GlcNAc transferase. Nat. Chem. Biol 8, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Schimpl M, Zheng X, Borodkin VS, Blair DE, Ferenbach AT, Schuttelkopf AW, Navratilova I, Aristotelous T, Albarbarawi O, Robinson DA, Macnaughtan MA, and van Aalten DMF (2012) O-GlcNAc transferase invokes nucleotide sugar pyrophosphate participation in catalysis. Nat. Chem. Biol 8, 969–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tchertanov L (1999) Structural metrics relationships in covalently bonded organic azides. Acta Crystallogr., Sect. B: Struct. Sci 55, 807–809. [DOI] [PubMed] [Google Scholar]
- (30).Clark PM, Dweck JF, Mason DE, Hart CR, Buck SB, Peters EC, Agnew BJ, and Hsieh-Wilson LC (2008) Direct In-Gel Fluorescence Detection and Cellular Imaging of O-GlcNAc-Modified Proteins. J. Am. Chem. Soc 130, 11576–11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Li J, Wang J, Wen L, Zhu H, Li S, Huang K, Jiang K, Li X, Ma C, Qu J, Parameswaran A, Song J, Zhao W, and Wang PG (2016) An OGA-Resistant Probe Allows Specific Visualization and Accurate Identification of O-GlcNAc-Modified Proteins in Cells. ACS Chem. Biol 11, 3002–3006. [DOI] [PubMed] [Google Scholar]
- (32).Saxon E, and Bertozzi C (2000) Cell surface engineering by a modified Staudinger reaction. Science 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- (33).Charron G, Zhang MM, Yount JS, Wilson J, Raghavan AS, Shamir E, and Hang HC (2009) Robust Fluorescent Detection of Protein Fatty-Acylation with Chemical Reporters. J. Am. Chem. Soc 131, 4967–4975. [DOI] [PubMed] [Google Scholar]
- (34).Goddard-Borger ED, and Stick RV (2007) An Efficient, Inexpensive, and Shelf-Stable Diazotransfer Reagent: Imidazole-1-sulfonyl Azide Hydrochloride. Org. Lett 9, 3797–3800. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.