

Abstract
Maintenance of tissue integrity in skeletal muscle requires the immunomodulatory and regenerative functions of muscle-resident regulatory T cells (Tregs). Chronic skeletal muscle infections, such as with Toxoplasma gondii disrupt normal immuno-regulatory networks and lead to pathogenic changes in Treg function. Specifically, Tregs during chronic T. gondii infection reinforce an inflammatory macrophage bias that exacerbates injury in skeletal muscle. In this study, we investigated whether the aberrations in skeletal muscle Treg function during chronic infection could be overcome by treatment with Treg-related factors associated with enhanced muscle regeneration during sterile injury. We show treatment of chronically infected mice with the Treg promoting therapies, interleukin-2 complexed with anti-IL-2 antibody or interleukin-33 (IL-33), did not restore macrophage dynamics or muscle function, respectively, in vivo. However supplementation of known Treg-derived factors, interleukin-10 (IL-10) and amphiregulin (Areg) improved muscle function and skewed macrophages toward a restorative phenotype in the presence of chronic infection. These shifts in macrophage phenotype are coupled with enhanced physiologic parameters of regeneration. Together, these data suggest that while Treg-mediated immuno-regulation is compromised during chronic skeletal muscle infection, supplementation of canonical Treg-derived factors such as IL-10 and Areg can restore immunologic balance and enhance muscle repair.
Keywords: Tregs, Macrophages, Muscle Injury, Muscle Infection, IL10, Amphiregulin
INTRODUCTION
Skeletal muscle disease and pathology can result in a wide range of functional consequences for patients from focal muscle weakness to severely debilitating complications (1–4). Infection represents a major source of acquired myopathies in large part due to the plethora of pathogens capable of causing secondary myopathy, including bacteria, viruses, parasites, and fungi (1). Treatment of underlying bacterial or fungal infections can often lead to acute recovery (1). However, for many viruses and parasites, poor druggability and effective immune evasion results in chronic infection and long-term myopathy (1, 5–7). Furthermore, while some pathogens can directly cause skeletal muscle damage, previous studies, in other sites of infection, have shown that infection can lead to immunologic scarring and reprogramming that potentiates aberrant immune-mediated pathology (8). For these reasons, a greater understanding of the interplay between infection, skeletal muscle, and tissue-specific immunity is necessary to treat myopathies refractory to antimicrobial therapies.
A full complement of skeletal muscle immunity is necessary to maintain tissue health and integrity over a lifetime. At homeostasis, intramuscular leukocytes are present up to 500–2,000 cells/mm2 histologically in adult murine muscles of which the majority are quiescent myeloid cells and the minority are CD4+ T cells, CD8+ T cells, regulatory T cells (Tregs), eosinophils, and neutrophils (9). In response to injury, these cells rapidly activate to promote regeneration. Notably, macrophages are critical to regulating distinct steps in the regenerative process (10–12). Inflammatory macrophages facilitate an early phase of repair characterized by IFNγ-mediated inflammation, clearance of dead and necrotic debris and early activation of muscle progenitor cells (satellite cells) (13). The subsequent emergence of restorative macrophages is important for immuno-regulation, extracellular matrix deposition, and driving differentiation and growth during myogenesis (13). Recently, Tregs have emerged as a key player in skeletal muscle regeneration (14–17). Specifically, Tregs accumulate with the same kinetics as restorative macrophages (16). Ablation of Tregs reduces the transition of inflammatory macrophages to restorative macrophages during repair, leading to prolonged damage (16). Additionally, diminished Treg responsiveness was characterized in age-related muscle pathology (17). This pathology was due to a diminishment of ST2 expressing Tregs and corrected by IL-33 supplementation as increased IL-33:ST2 signaling promoted Treg accumulation, a regenerative transcriptional program, and tissue regeneration (17). Furthermore, skeletal muscle Tregs produce both the immunosuppressive cytokine, IL-10, and the epidermal growth factor-like ligand, amphiregulin (Areg), following sterile injury (16). Both IL-10 and Areg have been independently shown to be critical for driving efficacious repair (10, 16).
Chronic infections of the skeletal muscle have previously been shown to severely alter the tissue immune landscape and compromise immune-regulatory networks (7, 18–20). Toxoplasma gondii is an obligate intracellular parasite capable of establishing a chronic infection in the CNS and striated muscle tissues (21). A robust interferon-gamma (IFNγ)-mediated immune response is necessary to control infection (21). Recently, we showed chronic skeletal muscle infection with Toxoplasma gondii drastically alters skeletal muscle immunity and leads to long-term muscle damage and myositis (18). Tregs decrease in frequency and also highly express the Th1-lineage specific transcription factor Tbet in infected skeletal muscle (18). Furthermore, in this setting, Tregs pathogenically promote long-term muscle damage and accumulation of inflammatory macrophages in the skeletal muscle (18).
In this present study, we addressed whether pathogenic Treg function can be compensated for by therapeutic treatment of Treg-promoting or Treg-derived factors. To accomplish this, we administered Treg-related factors previously shown to enhance muscle regeneration, IL-2 complexed with anti-IL-2 antibody, IL-33, IL-10, and Areg. Interestingly, IL-2 treatment preferentially expanded a Tbet expressing Treg population and was associated with a injurious increase in inflammatory macrophages, in support of our previous findings (18). Mice treated with IL-33 did not exhibit changes in the constitution of macrophage subsets in the muscle, while IL-10 and Areg treated mice exhibited an increase in restorative macrophages. However, only Areg treatment led to a reduction in Tbet expression in Tregs. Further, in Areg-treated mice, these changes were coupled with significantly improved physiological parameters of skeletal muscle health. Our findings implicate a role for IL-10 and Areg as potential therapeutic targets in chronically inflamed settings of skeletal muscle.
MATERIALS AND METHODS
Mice.
8–10 week old female C57BL/6 mice were obtained from Taconic Farms (Germantown, NY). All procedures involving mice were reviewed and approved by the Institutional Animal Care and Use Committee at the University at Buffalo.
Oral infection with Toxoplasma gondii.
Tissue cysts for oral infection were prepared from the brain homogenates of mice chronically infected (≥30 days post-infection) with RFP-expressing ME49 cysts (graciously provided by Michael Grigg). Cyst counts were enumerated using a fluorescence microscope. 8–10 week old female mice were orally gavaged with five ME49 cysts.
Muscle Injections.
For IL-33 treatment, anesthetized mice were injected intramuscular (i.m.) with 300ng/tibialis anterior (TA) (3μl) of recombinant murine IL-33 (Biolegend) followed by 2μg/mouse intraperitoneal (i.p.) (200μl) every other day thereafter until time of analysis. For IL-10 treatment, anesthetized mice were injected i.m. with 500ng/TA (3μl) of recombinant murine IL-10 (Peprotech) and 1μg/mouse i.p. (200μl) every other day thereafter until time of analysis. For amphiregulin treatment, anesthetized mice were injected i.m. with 1μg/TA (3μl) of recombinant murine amphiregulin (R&D Systems) and 7μg/mouse i.p. (200μl) every other day thereafter until time of analysis. Control mice were injected with PBS.
IL-2 Complex treatment.
Recombinant murine IL-2 (1.5μg, Peprotech) was incubated with anti-IL-2 (15μg, JES6–1A12, eBioscience) for 5 minutes at room temperature. IL-2 complex (IL-2c) was administered i.p. for 5 consecutive days. Control mice were injected with PBS.
Isolation of tissue lymphocytes from organ tissues.
Skeletal muscle was harvested from PBS-perfused mice and minced in digestion media (RPMI, 1% penicillin-streptomycin, 1mM sodium pyruvate, 0.1% β-mercaptoethaol, 25mM HEPES, 150μg/ml DNase I [Sigma-Aldrich], 1mg/ml Collagenase II [Invitrogen], and 1U/ml Dispase [Invitrogen]). Tissues were digested at 37˚C for 55 minutes. Mononuclear cells were enriched by passing digested tissue through a 70μΜ filter, and performing a percoll gradient purification (37.5% Percoll [GE healthcare]/62.5% Hank’s buffered saline solution [HBSS]). Single-cell suspensions were resuspended in 10% media (RPMI with 10% FBS, 1% penicillin-streptomycin, 1mM sodium pyruvate, 0.1% β-mercaptoethaol, 25mM HEPES).
Spleens were harvested and passed through a 70μM filter. Red blood cells were lysed in ACK lysing buffer (Lonza) and resuspended in 10% media.
Extracellular and Intracellular flow cytometric analysis of tissue lymphocytes.
Single-cell suspensions were stained with Live/Dead Fixable Aqua dead cell stain (Life Technologies) and an extracellular antibody cocktail in HBSS. Cells were subsequently fixed and permeabilized (Intracellular Fixation and Permeabilization Buffer Set, eBioscience). Afterwards, samples were stained with eBioscience Permeabilization Buffer containing intracellular antibody stains. Final resuspension was performed in FACS Buffer (PBS, 1% bovine serum albumin [Sigma-Aldrich], 2mM EDTA [Life Technologies]) for acquisition. For stains containing biotinylated antibodies, streptavidin staining was performed separately immediately following primary antibody staining. Absolute numbers were calculated using CountBright absolute counting beads (Life Technologies).
Antibodies.
Antibodies used were: anti-TCRβ-APC-Cy7 (BD Pharmigen, clone H57–597), anti-CD44-BV605 (BD Horizon, clone IM7), anti-CD4-PE-Cy7 (BD Pharmigen, clone RM4–5), anti-CD8α-PE (BD Pharmigen, clone H35–17.2), anti-CD8β-PerCP-Cy5.5 (BioLegend, clone YTS156.7.7), anti-T1/ST2-biotin (MD Bioproducts, clone DJ8), Streptavidin-PE (ebioscience), anti-Foxp3-FITC (eBioscience, clone FJK-16s), anti-Tbet-ef660 (eBioscience, clone eBio4B10), anti-Ki67-AF700 (BD Pharmigen, clone B56), anti-Ly6C-PerCp-Cy5.5 (eBioscience, clone HK1.4), anti-CD11b-BV605 (BD Horizon, clone M1/70), anti-Ly6G-PECF94 (BD Horizon, clone 1A8), anti-ICOS-PE-Cy5 (eBiosicence, clone 7E.17G9), anti-CD25-PerCP-Cy5.5 (BD Pharmigen, clone, PC61), anti-IFNγ-BV650 (BD Horizon, clone XMG1.2), anti-CD45-V450 (BD Horizon, clone 30-F11), anti-Nos2-AF488 (eBioscience, clone CXNFT), anti-CD68-PE-Cy7 (eBioscience, clone FA-11), and anti-CD206-APC (BioLegend, clone C068C2). Flow cytometry data was acquired using a BD LSRFortessa Cell Analyzer and analyzed using FlowJo version 10.4.2 (Tree Star, Ashland, OR).
Muscle functional strength testing.
Mice were placed on a 1 × 1 cm wire screen. The screen was slowly inverted and held above a padded container. Muscle strength was scored based on time-elapsed between full inversion and falling with a maximum of 60 seconds.
Quantification of parasite burden.
Tissues were isolated and preserved in RNAlater (Qiagen) for RNA isolation with Trizol (Life Technologies) or DNA extraction with the Qiagen DNeasy Blood and Tissue kit. Total parasite burden was quantified by qPCR-amplification of a T. gondii specific gene, B1 from genomic DNA isolated from tissues (Forward: 5’-TCCCCTCTGCTGGCGAAAAGT-3’, Reverse: 5’-AGCGTTCGTGGTCAACTATCGATTG - 3’). Ct values were compared to a standard curve constructed from B1 amplification of known T. gondii genomic DNA concentrations. Tachyzoite and bradyzoite parasite burden was quantified by qRT-PCR of Sag1 (Forward:5’- ATCGCCTGAGAAGCATCACTG-3’, Reverse: 5’-CGAAAATGGAAACGTGACTGG-3’), Eno1 (Forward: 5’-GGTATTGATATGCTTATGGTGGAG-3’, Reverse: 5’-GCGATGTATTTGTATAGTGGTAGG-3’), and Bag1 (Forward: 5’-GGGATGTACCAAGCATCCTG-3’, Reverse: AGGGTAGTACGCCAGAGCAA-3’), respectively. TgActin (Forward: 5’-ATGTATGTCGCTATCCAGGCCGTT - 3’, Reverse: 5’ - TGATCTTCATGGTGGAAGGAGCCA −3’) was used as a housekeeping gene.
qRT-PCR of muscle differentiation factors.
Tissues from experimental mice were isolated and preserved in RNAlater (Qiagen) for RNA isolation by Trizol extraction. Isolated RNA was converted to cDNA (iScript, BD Biosciences) assayed for muscle differentiation factors Pax7 (5’-GACTCGGCTTCCTCCATCTC-3’, Reverse: 5’-AGTAGGCTTGTCCCGTTTCC-3’), Pax3 (Forward: 5’-ACTACCCAGACAATTTACACCAGG-3’, Reverse: 5’-AATGAGATGGTTGAAAGCCATCAG-3’), Igf1 (Forward: 5’-GTGTGGACGAGGGGCTTTTACTTC-3’, Reverse: 5’-GCTTCAGTGGGGCACAGTACATCTC-3’), Myf5 (Forward: 5’-GAACAGGTGGAGAACTATTA-3’, Reverse: 5’-GCACATGCATTTGATACATCAG-3’), Myod (Forward: 5’-GAGCGCATCTCCACAGACAG-3’, Reverse: 5’-AAATCGCATTGGGGTTTGAG-3’), and Myog (Forward: 5’-CCAGTACATTGAGCGCCTAC-3’, Reverse: 5’-ACCGAACTCCAGTGCATTGC-3’) by real-time PCR (iTaq Universal SYBR Green supermix, BD Biosciences). Ct values were normalized the housekeeping gene Gapdh (Forward: 5’-CCCACTCTTCCACCTTCGATG-3’, Reverse: 5’-GTCCACCACCCTGTTGCTGTAG-3’).
Muscle histopathology.
Skeletal muscle TA muscles were perfusion fixed in 4% paraformaldehyde (Sigma-Aldrich) and then isolated. Fixed tissue samples were cryopreserved in 30% sucrose, frozen in OCT (Sakura) and cryosectioned at 20μm. Sections were stained with Wheat Germ Agglutinin (WGA) AF488 (1:100, Thermo Scientific) and DAPI (Sigma). Images were acquired with a Zeiss AxioImager.Z1 microscope. Image analysis was performed on FIJI. Cross-sectional area was quantified by segmentation analysis and subsequent measurement. Percent damaged area was determined by binarization of WGA stained area as a percentage of total muscle area. For binarization analysis, only the tibialis anterior tissue was analyzed (peripheral tissue/connective tissue debris was excluded from analysis). Centralized nuclei were enumerated and normalized to total muscle area.
Statistics.
All statistics were generated using Graphpad Prism v6.0c.
RESULTS
IL-2c treatment fails to expand a population of suppressive Tregs and results in increased inflammation
Previously, we demonstrated that Tregs during chronic T. gondii infection promote tissue pathology and an accumulation of inflammatory macrophages within skeletal muscle (18). However the factors that contribute to the acquisition of pathogenic functions during chronic infection remains unclear. Interestingly, expansion of Tregs through the administration of recombinant IL-2 complexed to an anti-IL-2 antibody (clone JES6–1A12) (IL-2c) alleviates immune-mediated pathology during acute T. gondii infection and in murine models of muscular dystrophy and experimental myasthenia gravis (14, 22–24). We therefore asked if treatment with IL-2c could reconstitute a population of suppressive Tregs to reduce inflammatory macrophages and enhance restorative macrophages during chronic infection. As expected, administration of IL-2c in chronically infected mice significantly increased both the frequency and absolute number of Tregs in the skeletal muscle (Figure 1A, B). This is associated with increased proliferation of Tregs, as indicated by the proliferation marker Ki67 (Figure 1B). A distinguishing trait of the majority (~60%) of Tregs during chronic infection is the expression of the Th1-lineage specific transcription factor, Tbet (18). Recent molecular studies demonstrated IL-2c preferentially expand Tregs due to the ability of JES6–1A12 to block IL-2Rβ while enhancing IL-2Rα (CD25) signaling, which is highly expressed on Tregs (25). During chronic infection, the majority of CD25+ Tregs co-express Tbet (Figure 1C). Further, double positive Tbet+CD25+ Tregs express the highest levels of ICOS which has also been shown to enhance IL-2 signaling (Figure 1C) (26, 27). As such, we find treatment led to a preferential expansion of Tbet+ Tregs versus Tbet- Tregs, increasing from 60% to 75% (Figure 1D). Given that Foxp3-CD4+ conventional T cells (Tconv) also express CD25 upon activation, we assessed the level of off-target effects of treatment on this subset. Unsurprisingly, we also observed an expansion of the total number of Tconv with IL-2c (Supplemental Figure 1A) however the proportion of IFNγ directly ex vivo remained similar (Supplemental Figure 1B). The treatment resulted in a reduced frequency of Tconv to Tregs due to the preference for Treg expansion (Figure 1A, Supplemental Figure 1A).
Figure 1. IL-2c treatment preferentially expands injurious Tbet+Tregs and inflammatory macrophages in skeletal muscle during chronic T. gondii infection.
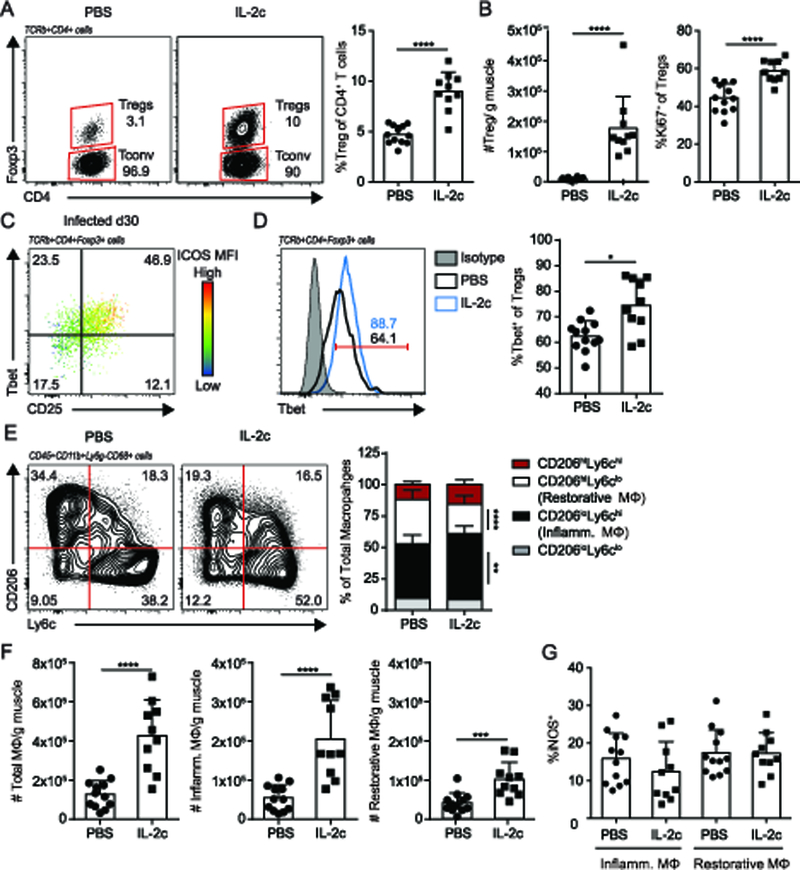
T. gondii infected mice were administered IL-2-anti-IL-2 complex (IL-2c) treatment 24 days post-infection for 5 consecutive days. Mice were used for analysis one-day following the final treatment (30 dpi) (A) Representative flow plots (left) and graphical summary (right) of skeletal muscle Treg (TCRβ+CD4+Foxp3+) expansion versus Tconv (TCRβ+CD4+Foxp3-) (B) Absolute number of skeletal muscle Tregs (left) and Ki67 expression in Tregs (C) Representative flow plot of Tbet, CD25, and ICOS (color scale, MFI) expression in skeletal muscle Tregs 30 days post-infection (dpi) with T. gondii (ME49). (D) Representative flow plot overlay (left) and graphical summary (right) of Tbet expression in skeletal muscle Tregs from PBS (black line) and IL2-treated (blue line) mice versus. Tbet-antibody isotype control is displayed in gray. (E) Representative flow plot (left) and graphical summary (right) of skeletal muscle macrophage (CD45+CD11b+Ly6g-CD68+) subsets by CD206 and Ly6c expression (F) Absolute number of total macrophages (MΦ), inflammatory (inflamm.) MΦ (CD206loLy6chi), and restorative MΦ (CD206hiLy6clo) (G) Quantification of iNOS expression by flow cytometric analysis in inflammatory and restorative MΦ. Results are cumulative of n ≥ 2 independent experiments of n ≥ 4/group/experiment; error bars represent SD. *p < 0.5, **p < 0.01, ***p < 0.001, ****p < 0.0001, Mann-Whitney U test (A, B, D, G), Student t test (F), Kruskal-Wallis H test (E).
We next assessed the effect of IL-2c-mediated preferential expansion of Tbet+ Tregs on macrophage phenotype in the skeletal muscle. IL-2c treatment is accompanied by an increase in the proportion of inflammatory (CD206loLy6chi) macrophages and a concomitant decrease in restorative (CD206hiLy6clo) macrophages (Figure 1E). While the absolute number of both inflammatory and restorative macrophages increased, the extent to which inflammatory macrophages increased surpasses that of restorative macrophages (Figure 1F). Thus, the shift in macrophage bias toward inflammatory macrophages is largely driven by increases in the absolute number of inflammatory macrophages (Figure 1E, F). A functional readout for the inflammatory phenotype of macrophages is the production of inducible nitric oxide synthase (iNOS), which catalyzes the production of nitric oxide radicals. Following IL-2c treatment, the proportion of iNOS expressing macrophages remained similar between treatment groups (Figure 1G). Together, these findings show that IL-2c treatment does not resolve skeletal muscle inflammation. Further, these results suggest that IL-2c treatment, which is successful during acute T. gondii infection and other models, does not enhance Treg function to sufficiently overcome the inflammation found in infected skeletal muscle.
IL-33 supplementation increases restorative macrophages but does not improve skeletal muscle function
Previously, IL-33:ST2 signaling in Tregs has been shown to reinforce a regenerative transcriptional program in muscle, heighten Treg-mediated immunosuppression, and enhance adaptation to inflammatory environments (17, 28, 29). In aged skeletal muscle with weak regenerative capacity and an absence of ST2 expressing Tregs, IL-33 supplementation promoted tissue repair associated with increases in ST2+ Treg accumulation and functional signatures (17). Thus, we tested whether reinforcing regenerative Treg function during chronic infection indirectly leads to restoration of muscle function and restorative macrophage bias by an intramuscular injection of IL-33 followed by subsequent intraperitoneal injections to increase the tissue specificity of treatment, as in ((16), Figure 2A). Unlike in aged muscle, IL-33 administration during chronic infection did not lead to a significant increase in Treg accumulation either by absolute number or frequency (Figure 2B). Treatment also did not increase Treg proliferation (Figure 2B). Similar to Tregs from uninfected CTX-injured muscle, Tregs in infected muscle expressed comparable levels of ST2, the receptor for IL-33, indicating Tregs were capable of IL-33-signaling in this setting (Figure 2C). In support of this, IL-33 treatment showed a trend in increasing ST2 expression in Tregs in chronically infected muscle (Figure 2C). Further, a greater frequency of Tregs expressed RORγt following IL-33 administration during chronic infection (Figure 2D). Previously, RORγt+ Tregs have been found in association with the reparative phase of tissue repair following toxin-induced muscle injury (30). These changes did not coincide with alterations to local Tconv or CD8+ T cells immunity in the skeletal muscle (Figure 2E). However, administration of IL-33 did lead to a systemic increase in Tconv and CD8+ T cells as indicated by splenic numbers (Supplemental Figure 2).
Figure 2. IL-33 treatment improves restorative macrophages but not function.
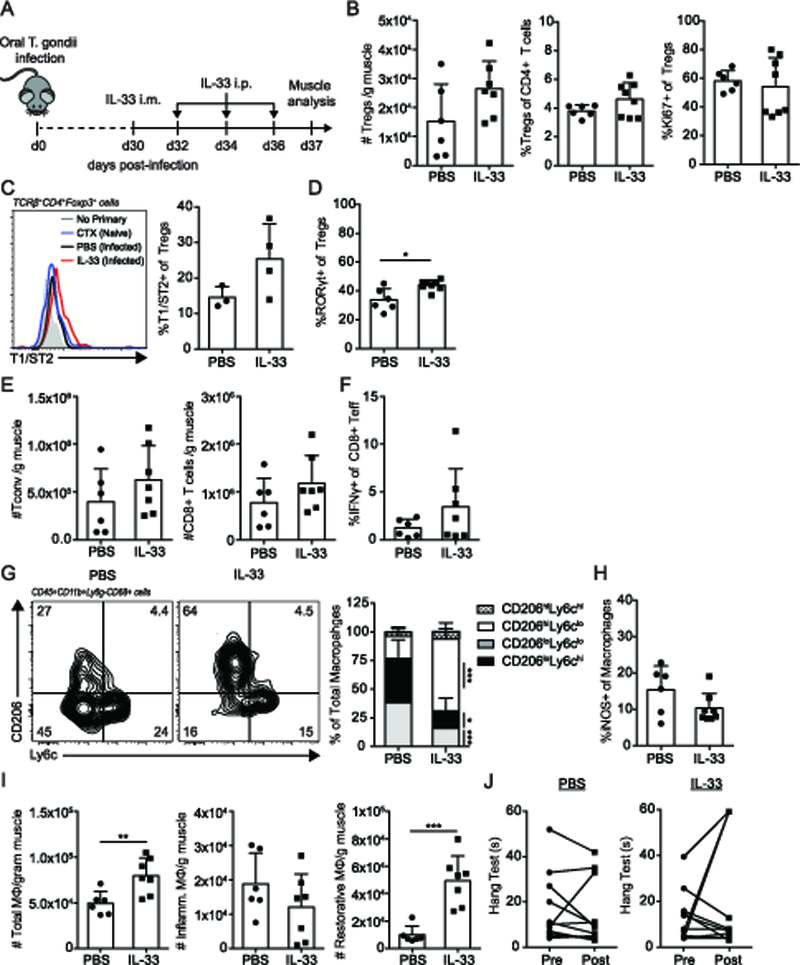
Mice were infected with 5 ME49 T. gondii cysts. 30-days post infection, IL-33 (or PBS control) treatment was administered by intramuscular administration Followed by subsequent intraperitoneal boosters every other day for three treatments. Muscle was analyzed 7 days post-treatment. (A) IL-33 treatment regimen during chronic infection. (B) Graphical summaries of absolute number of Tregs, frequency of Tregs of total CD4+ T cells, and Ki67 expression in Tregs by flow cytometric analysis (C) Representative flow plot of T1/ST2 expression on Tregs from no primary (T1/ST2-biotin) staining control, uninfected CTX-injured, PBS-treated chronically infected, and IL-33-treated chronically infected muscle samples (left) and graphical summary (right), results are representative of single experiment of 3–4 mice/group (D) Graphical summary of RORγt expression in skeletal muscle Tregs by flow cytometric analysis (E) Absolute numbers of Tconv and CD8+ T cells in treated skeletal muscle (F) IFNγ expression in CD44+CD8+ T cells (G) Representative flow plots (left) and graphical summary (right) of MΦ subsets and (H) iNOS expression in total MΦ by flow cytometric analysis. (I) Absolute number of total, inflammatory, and restorative MΦ per gram muscle (J) Inverted screen test of PBS- and IL-33-treated mice pre and post treatment (max: 60s). Results are cumulative of n = 2 independent experiments of n ≥ 3–4/group/experiment (B, E-J); error bars represent SD. *p < 0.5, **p < 0.01, ***p < 0.001, Mann-Whitney U test (B-D, F, H), Student t test (B, E, I, J), Kruskal-Wallis H test (G).
We next addressed whether enhancing Treg regulatory and reparative functions via IL-33:ST2 signaling during chronic infection lead to a reduction in inflammatory macrophages and tissue damage. Interestingly, the distribution of inflammatory and restorative macrophages shifted dramatically in favor of restorative macrophages in both frequency and absolute number (Figure 2G, I). iNOS expression from total macrophages remained constant (Figure 2H). However, despite the acquisition of a more regenerative phenotype in both Tregs and macrophages, this was insufficient to drive significant improvement in skeletal muscle function assessed through an inverted screen test (Figure 2J). Together, this data suggests IL-33-mediated changes to skeletal muscle immunity alone does not improve skeletal muscle fitness during chronic infection.
IL-10 treatment improves inflammation and muscle function in the skeletal muscle
We next asked whether supplementation of Treg-derived factors decrease underlying inflammation and improve muscle function. The cytokine IL-10 is critical to facilitate efficacious skeletal muscle repair (10). Among its many immunosuppressive functions, IL-10 is able to directly alternatively activate macrophages, which potentiates repair (31–33). Skeletal muscle Tregs were previously shown to produce more IL-10 during injury than splenic Tregs, however, the extent to which Treg-derived IL-10 contributes to wound repair is unknown (16). To determine whether supplementation of IL-10 alleviates skeletal muscle damage during chronic infection, we used the therapeutic approach in (16) to increase the tissue specificity of treatment by first administering IL-10 directly by intramuscular injection followed by subsequent intraperitoneal injections (Figure 3A). Given the immunosuppressive effects of IL-10, we began by assessing whether treatment affected local and systemic T cell compartments. IL-10 administration resulted in a reduction in splenic Tconv but not Treg and CD8+ T cell numbers (Supplemental Figure 3A). In contrast, within the skeletal muscle, Treg, Tconv, and CD8+ T cell numbers remained unaltered by treatment (Figure 3B). Phenotypically, IL-10 supplementation did not result in a reduction in Tbet+ Tregs and Tconv or IFNγ+ Tconv in spleen and muscle (Figure 3C, Supplemental Figure 3B, C). However, the proportion of macrophages skewed toward restorative macrophages (Figure 3D). This significant shift in macrophage bias is accounted for by the cumulative effect of non-significant changes in the absolute numbers of both inflammatory and restorative macrophages (Figure 3E). iNOS expression in total macrophages is not altered due to IL-10 treatment (Figure 3F). As active immune surveillance is crucial to limit T. gondii reactivation, we assessed whether a reduced inflammatory response due to IL-10 treatment resulted in parasitic reactivation. Importantly, the anti-inflammatory effects of IL-10 treatment did not lead to increases in skeletal muscle parasite burden during the 7 days of treatment (Figure 3G). Next, we ascertained whether the immunologic shift towards restorative macrophages resulted in improved tissue function. Interestingly, IL-10 treatment improved muscle function on hang test (Figure 3H). Together, these results suggest that IL-10 treatment can improve both inflammation and skeletal muscle function during chronic infection without having a detrimental impact on parasite burden.
Figure 3. IL-10 treatment increases restorative macrophage bias and function in chronically infected skeletal muscle.
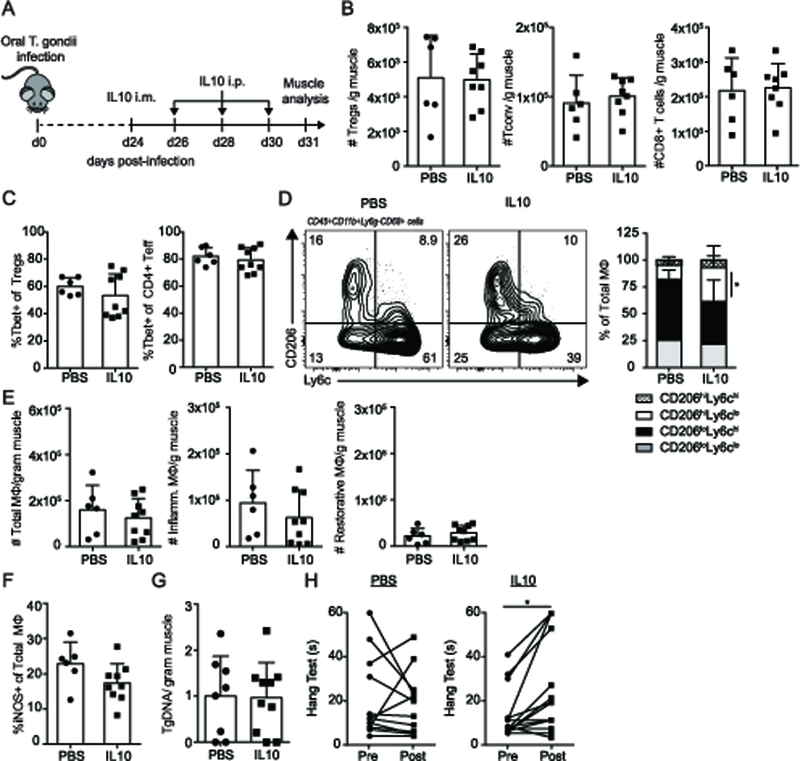
Mice were infected with 5 ME49 T. gondii cysts. 24-days post infection, IL-10 (or PBS control) treatment was administered by intramuscular administration followed by subsequent intraperitoneal boosters every other day for three treatments. Muscle was analyzed 7 days post-treatment. (A) IL-10 treatment regimen during chronic infection. (B) Graphical summaries of absolute number of skeletal muscle Treg and Tconv (C) Graphical summary of Tbet expression in skeletal muscle Tregs and Tconv by flow cytometric analysis (D) Representative flow plots (left) and graphical summary (right) of skeletal muscle MΦ subsets (E) Absolute number of total, inflammatory, and restorative MΦ per gram muscle (F) iNOS expression in total MΦ by flow cytometric analysis (G) Skeletal muscle parasite burden assessed by qPCR quantification of T. gondii specific B1 gene (H) Inverted screen test of PBS- and IL-10-treated mice pre and post treatment (max: 60s). Results are cumulative of n = 3 independent experiments of n = 2–5/group/experiment; error bars represent SD. *p < 0.5, **p < 0.01, Mann-Whitney U test (C, F), Student t test (B, E, G), Kruskal-Wallis H test (D), ANCOVA (H).
Amphiregulin alleviates immunologic and physiologic parameters of skeletal muscle fitness
Given that IL-10 improved muscle function and mitigated inflammation, we asked whether this was restricted to IL-10 or if other Treg-derived factors could also impact muscle health during chronic infection. Previously, skeletal muscle Tregs have been shown to express high amounts of amphiregulin (Areg) in response to sterile injury (16). Further, Areg plays an important role in skeletal muscle regeneration, as its absence hampers wound repair (16). Functionally, Areg can directly stimulate satellite cells as well as enhance the suppressive capacity of Tregs (16, 34, 35). We asked whether treatment with Areg to chronically infected mice improved measures of function and inflammation. As with the IL-10 treatment, we used the therapeutic approach in (16) to increase the tissue specificity of treatment by first administering Areg directly by intramuscular injection followed by subsequent intraperitoneal injections (Figure 4A). We first sought to determine whether Areg treatment led to changes in skeletal muscle immunity that facilitates tissue repair. Interestingly, while the frequency and number of Tregs remained the same in muscle, muscle that had been treated with Areg exhibit decreased Treg Tbet expression that is not associated a concurrent decrease in Tbet expression in conventional CD4+ T cells (Figure 4 B-D). This change is associated with a shift in the distribution of macrophage subsets in favor of restorative CD206hiLy6clo macrophages (Figure 4E). This is due to an increased absolute number of restorative macrophages as the number of pro-inflammatory macrophages remained unchanged (Figure 4F). Administration of Areg in chronically infected mice improved hang test scores compared to PBS treated controls (Figure 4G). Importantly, these changes in immunity did not affect total parasite burden, which remained unchanged (Figure 4H). Analysis of T. gondii stage-specific transcripts suggest Areg treatment promotes an environment that skews toward parasite encystation (bradyzoites- eno1, bag1) rather than reactivation (tachyzoites- sag1) (Figure 4I). These results show that Areg treatment can foster immunological changes adherent to a reparative program that leads to improved function without compromising control of parasitic infection.
Figure 4. Therapeutic administration of amphiregulin increases restorative MΦ and reduces Tbet expression in Tregs during infection.
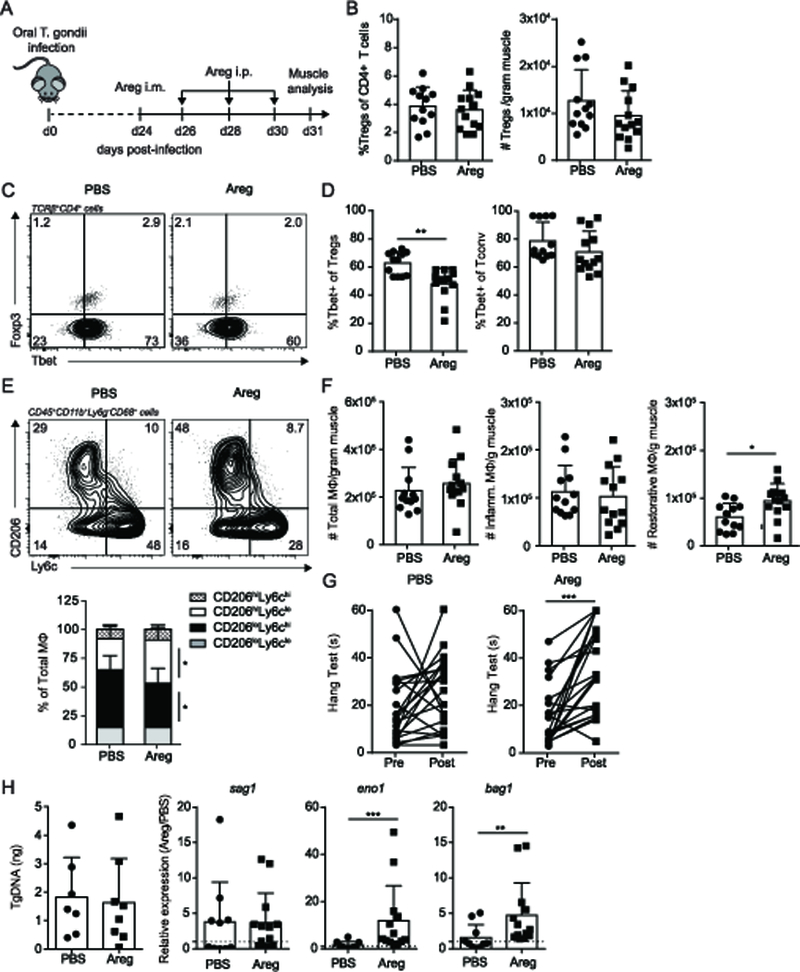
Mice were infected with 5 ME49 T. gondii cysts. 24-days post infection, Areg (or PBS control) treatment was administered by intramuscular administration followed by subsequent intraperitoneal boosters every other day for three treatments. Muscle was analyzed 7 days post-treatment. (A) Areg treatment regimen during chronic infection. (B) Graphical summaries of frequency (left) and absolute number (right) of skeletal muscle Treg (C) Representative flow plot and (D) graphical summary of Tbet expression in skeletal muscle Tregs and Tconv by flow cytometric analysis (E) Representative flow plots (top) and graphical summary (bottom) of skeletal muscle MΦ subsets (F) Absolute number of total, inflammatory, and restorative MΦ per gram muscle (G) Inverted screen test of PBS- and Areg-treated mice pre and post treatment (max: 60s). (H) Skeletal muscle total parasite burden assessed by qPCR quantification of T. gondii specific B1 gene (left) and stage specific transcripts sag1 (tachyzoite), eno1 (bradyzoite), and bag1 (bradyzoite) (right). For sag1, eno1, and bag1, statistics were performed on log-transformed values. Results are cumulative of n = 3 independent experiments of n ≥ 4 group/experiment; error bars represent SD. *p < 0.5, **p < 0.01, Mann-Whitney U test (B, D), Student t test (B, F, H), Kruskal-Wallis H test (E), ANCOVA (G).
Due to the ability of Areg to directly affect the myogenicity of muscle progenitors (satellite cells), we next asked whether the improvements we observed in function and inflammation were associated with quantitative differences in physiological parameters of myogenesis following Areg treatment. We first examined the expression of myogenic regulatory factors that direct the molecular progression of myogenesis. Areg treatment led to increased expression of pax7, suggestive of an increase in satellite cells (Figure 5A). Further, Areg treatment induced the expression of positive regulators of myotube activation and differentiation, myf5 and myog respectively (Figure 5A). Pax3, a paralogue of pax7 is decreased with Areg treatment. However, Pax3+ stem cells represent a minor fraction of the overall myogenic precursor pool. To examine morphologic features, we compared the number of regenerating fibers, fiber size distribution, and percent total damaged area between PBS- and Areg-treated mice. Muscle fiber size analysis showed the distribution of the muscle fibers skews significantly toward larger fibers with Areg treatment (Figure 5C). These changes are reflective of an improved progression of regeneration as fibers begin to differentiate and hypertrophy. They are also in agreement with the overexpression of myf5, myod, and myog observed transcriptionally (Figure 5A). Finally, Areg-treated muscles exhibited a decreased total area of damage/inflammation than their PBS-treated counterparts (Figure 5B, D). Collectively, these data suggest that Areg treatment results in a meaningful improvement in both physiologic and immunologic indices of muscle function during chronic infection.
Figure 5. Therapeutic administration of amphiregulin myogenesis during infection.
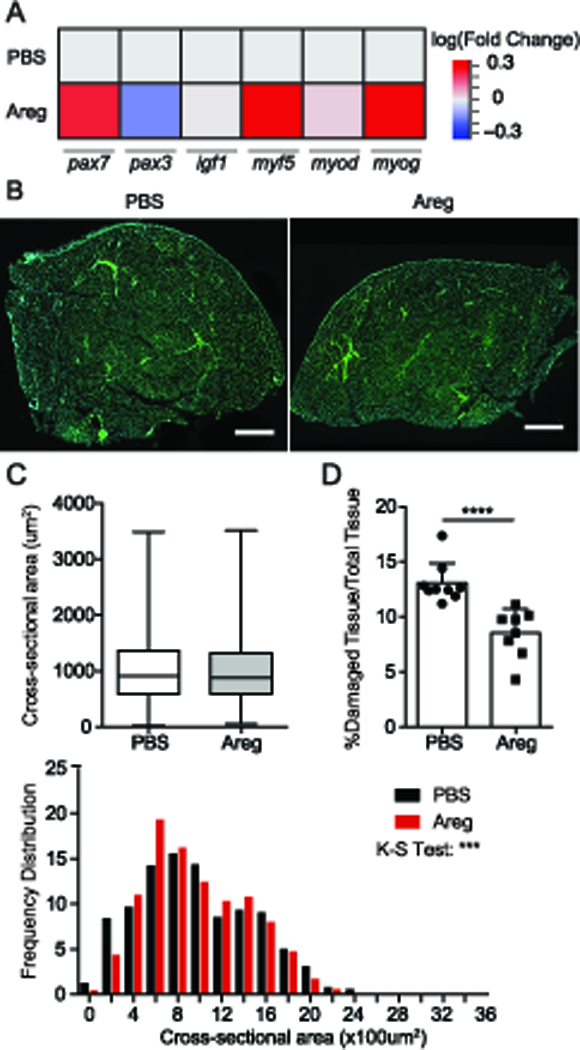
(A) Whole muscle mRNA expression of muscle-repair associated genes, pax7, pax3, igf1, myf5, myod, and myog normalized to gapdh relative to PBS-treated skeletal muscle. (B) WGA (green) and DAPI (blue) staining of skeletal muscle sections. Scale Bars: 500um (C) Average (top) and frequency distribution (bottom) of myofiber cross-sectional area. Error bars represent maximum and minimum (D) Percent damaged area relative to total tissue area determined by percent occupied space of binarized WGA staining. Only the tibialis anterior tissue was analyzed. Peripheral tissue/connective tissue debris were excluded from analysis (asterisk). Results are representative of three experiments of n ≥ 4/group/experiment; error bars represent SD. *p < 0.5, **p < 0.01, ***p < 0.001, ****p < 0.0001, Student t test (C), Mann-Whitney U test (D), Kolmogorov-Smirnov test (C).
DISCUSSION
Skeletal muscle immunity is important for maintaining tissue health both at homeostasis and in response to injury and infection. Chronic infections pose a major challenge for many tissues as it leads to long-term alterations of the local immune landscape, which can have detrimental implications for tissue fitness and overall function (7, 18–20). Previously, we showed chronic T. gondii infection leads to long-term skeletal muscle damage associated with disruption of both Treg and macrophage compartments (18). Specifically, Tregs promote a pathogenic accumulation of inflammatory macrophage accumulation within skeletal muscle (18). This is in sharp contrast to the previously reported role of Tregs to promote restorative macrophages during sterile injury and in the mdx mouse model of muscular dystrophy (14, 16). In this study, we demonstrate that therapeutic administration of Treg repair-associated factors, IL-10 and Areg, can overcome the tissue damage and macrophage bias reinforced by pathogenic Tregs during chronic skeletal muscle infection while maintaining disease latency. Our results imply an important role for these factors in supporting the muscle repair program in chronically infected and inflamed settings where Treg function may be aberrant.
Given that Tregs promote skeletal muscle pathology during chronic infection, we asked if we could enhance tissue fitness by supplementing Treg-enhancing (IL-2 and IL-33) or Treg-derived (IL-10 and Areg) agents known to participate in tissue repair. Preferential expansion of Tregs via administration of IL-2c (JES6–1A12) has previously been shown to be efficacious in delaying onset or reducing disease severity in diabetes, allergy, and experimental autoimmune encephalitis (36–38). In skeletal muscle, IL-2c treatment alleviates damage in muscular dystrophy (mdx) and experimental myasthenia mouse models (14, 22). IL-2c-induced Treg expansion in these settings suppresses immune-mediated pathology (14, 22, 37–39). Previously, restoration of Tregs by IL-2c treatment showed enhanced susceptibility to acute T. gondii, Listeria monocytogenes, and vaccinia virus infections due to diminished Th1-mediated host resistance (23, 24). In contrast, our results showed a failure to expand a population of suppressive Tregs. Differences in the type, strength, and duration of inflammation within the skeletal muscle are likely the major factors that underlie the antithetical roles of Tregs observed between sterile injury-repair, acute infection, and chronic infection. The ability of Tregs to adapt to local inflammatory environments is well documented (40, 41). In the setting of Th1 inflammation, such as that established by T. gondii infection, Tregs can express the Th1-lineage specific marker Tbet, which enables enhanced trafficking and survival (24, 42–44). Interestingly, the ability of Tregs to adapt to local inflammation is essential to control overwhelming Th1 inflammation during acute T. gondii infection (43). However, the consequences of Tbet expression in Tregs during T. gondii infection is unknown. We show that IL-2c treatment not only increases the overall frequency of skeletal muscle Tregs during chronic infection, but preferentially expands the Tbet+ subset within these Tregs. The expansion of Tbet+ Tregs is associated with the enhanced predominance of inflammatory macrophages in skeletal muscle and greater tissue pathology. These findings build upon our previous findings that skeletal muscle Tregs are injurious during chronic infection (18).
It is also possible that elevated numbers of IFNγ producing cells due to IL-2c treatment could lead to the increased polarization toward an inflammatory macrophage phenotype observed in the muscle. Under instances of normal Treg function, these effects could be compensated for by the preferentially expanded Treg compartment (~11-fold expansion of Tregs versus ~9-fold expansion of Tconv) with IL-2c treatment. However, the inability of the expanded Tregs to prevent further increases in Tconv would further reinforce the notion that Treg function is defective within this inflamed environment. Our findings suggest long-term acquisition of previously adaptive inflammatory traits in Tregs may lead to detrimental outcomes within the tissue during chronic inflammation.
The alarmin, IL-33, plays a critical role in tissue protection at many sites including the intestine, lung, CNS, and adipose tissue in response to injury (45). Notably, a large proportion of non-lymphoid organ Tregs are enriched for ST2 expression, the receptor for IL-33 (45). IL33:ST2 signaling has been shown to enhance proliferation of ST2+Treg (17, 28, 46, 47). Further, supplementation of IL-33 improves the tissue-repair response in aged muscle, in part though promoting accumulation of Tregs at sites of injury and promoting a reparative transcriptional program within Tregs (17). We show that IL-33 treatment to chronically infected muscle did not result in Treg accumulation, however, did increase RORγt expression with a trend in increased ST2 expression. Increases in the expression of either of these factors has independently been associated with tissue repair (17, 30). Further, macrophages shifted towards a more restorative phenotype following IL-33 administration. Despite these changes, function remained unaltered. This is in contrast to IL-10 and Areg treatments which lead to improvements in both restorative immunity and tissue function. Notably, whereas IL-10 treatment lead to a reduction in systemic T cell numbers, IL-33 lead to an increase. It is possible that the lack of improvement assessed by functional testing could be secondary to increased immunopathology. Additionally, our data suggests the tissue specific response to IL-33 during T. gondii infection may be distinct and requires further investigation. Together, our data suggests IL-33 mediated effects alone are inadequate to improve the overall fitness of muscle during chronic infection.
In the muscle, a major mode by which Tregs may affect macrophage polarization during regeneration is through the production of IL-10 (10, 16). IL-10 null mice have increased Th1 inflammation and transition poorly to restorative macrophages in disuse injury (10). Previous reports show Tregs from naïve regenerating muscle produce IL-10, and thus potentially represent an important source of IL-10 during repair (16). We previously showed that skeletal muscle Tregs from chronic T. gondii infection also produce IL-10, likely in response to increased inflammation at these sites (18). However, given the ongoing damage and excessive inflammation, these results imply Treg-derived IL-10 may not be sufficient to confer protective effects of IL-10 in infected skeletal muscle. Instead, either increased production of IL-10 by Tregs or production by other sources may be necessary. In support of this, our findings show IL-10 treatment improves both muscle function and increases restorative macrophages. Interestingly, while IL-10 treatment did not change Tconv numbers in skeletal muscle, it lead to a systemic decrease in Tconv while not significantly altering CD8+ T cells. The simultaneous availability of both CD4+ and CD8+ T cells is key to restricting parasite reactivation during chronic infection due to their combined ability to produce IFNγ (51). While, long-term decreases in CD4+ T cells is correlated with parasite reactivation in HIV patients, CD4+ or CD8+ T cell depletion alone does not result in parasite reactivation, increased brain pathology or mortality in mouse models (51). In agreement with this, the parasite burden in the skeletal muscle remains unaltered by IL-10 treatment despite systemic reduction of Tconv. Thus, at the dose of IL-10 given to elicit therapeutic relief of the skeletal muscle, inhibition of systemic and local T-cell immunity are not sufficient to lead to reactivation of infection.
Amphiregulin directly participates in skeletal muscle wound repair mainly through promoting satellite cell myogenicity (16). However, in other systems Areg signaling enhances the suppressive capacity of Tregs as well as resolves injury and inflammation during infection (16, 34, 35). Consistent with its ability to act on satellite cells, we show that therapeutic administration of Areg leads to an enhanced myogenic molecular profile as evidenced by increased whole-tissue expression of the satellite cell specific marker Pax7, as well as markers of myotube activation and differentiation, Myf5 and Myog, respectively. These molecular changes are reflected in augmented morphological indicators of tissue fitness. The efficacy of regeneration is largely driven by skeletal muscle immunity. Accordingly, improved physiologic parameters correlate with an increased number of restorative macrophages. Lending further support to the notion that long-term Tbet expression in Tregs is deleterious, we show tissue improvements following Areg treatment are associated with a decrease in Tbet+ Tregs, but not Tbet+ Tconv. Collectively, Areg treatment alleviates infection-induced damage and inflammation in the skeletal muscle during chronic T. gondii infection. Previously, insulin-like growth factor 1 was shown to enhance epidermal growth factor receptor signaling in the presence of epidermal growth factor family members such as Areg (52). Infected mice responding to injury have decreased transcript levels of insulin-like growth factor 1 on the whole tissue level compared to uninfected injured muscle (data not shown). We previously showed that skeletal muscle Tregs during chronic infection produce more Areg than their naïve counterparts (18). However, it is possible that the therapeutic benefit of Areg supplementation acts in part by overcoming a defect in Areg sensitivity that underlies the inability to fully resolve new and ongoing damage during chronic infection. Additionally, Areg production is not limited to Tregs, but can been produced by multiple sources, among which include epithelial cells, mast cells, type 2 innate lymphoid cells, eosinophils, and activated CD4+ T cells (53). The contribution of Areg from these sources has yet to be characterized in skeletal muscle.
Chronic inflammation is a hallmark feature of many myopathies, not limited to infectious myositis, with the potential to dysregulate homeostatic immune-mediated processes. Treg function and fitness is critical to maintaining immunologic balance, however these functions may be corrupted in the cases severe or long-lasting inflammation (8, 18, 24, 54–56). Thus, therapeutic modulation or mimicry of Treg function during chronic injuries and infections are attractive targets to improve skeletal muscle health. Our current findings demonstrate chronic infection-induced disruptions to immune-regulatory networks governing skeletal muscle tissue integrity can be therapeutically overcome by supplementation of Treg repair-associated products, IL-10 and Areg but not Treg-modulating, IL-2c or IL-33. In fact, treatment with IL-2c during chronic infection leads to increased disrepair, in opposition to other injury models. Notably, IL-10 and Areg treatment does not impact the latency of chronic infection in our model. Together with our previous findings, we show the efficacy of individual Treg-targeted therapies is highly context-dependent. However, leveraging the effects of downstream products such as IL-10 and Areg may serve as therapeutic avenues during chronic skeletal muscle infection, potentially in conjunction with antimicrobial therapy when necessary. Continued insight into how chronic infection and inflammation reshapes local immunity and interacts with the tissue niche will embolden efforts toward the development of future efficacious therapies.
Supplementary Material
AKNOWLEDGEMENTS
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institute of Health under Award Number R21AI128284. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We thank the Confocal Microscope and Flow Cytometry Core Facility at the Jacobs School of Medicine for their technical assistance; and Dr. John Grainger and Dr. Joanne Konkel for helpful discussion and the critical reading of this manuscript.
This work was supported by the NIH Institutes of Health Grant AI128284 (E.A.W.)
Abbreviations used in this article:
- Areg
amphiregulin
- IL-2c
IL-2 complex
- iNOS
inducible NO synthase
- TA
tibialis anterior
- Tconv
conventional T cell
- Tregs
regulatory T cells
- WGA
wheat germ agglutinin
REFERENCES
- 1.Crum-Cianflone NF 2008. Bacterial, Fungal, Parasitic, and Viral Myositis. Clin. Microbiol. Rev. 21: 473–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drescher C, Konishi M, Ebner N, and Springer J 2016. Loss of muscle mass: Current developments in cachexia and sarcopenia focused on biomarkers and treatment. Int. J. Cardiol. 202: 766–72. [DOI] [PubMed] [Google Scholar]
- 3.Dalakas MC, and Hohlfeld R 2003. Polymyositis and dermatomyositis. Lancet 362: 971–82. [DOI] [PubMed] [Google Scholar]
- 4.Rayavarapu S, Coley W, Kinder TB, and Nagaraju K 2013. Idiopathic inflammatory myopathies: pathogenic mechanisms of muscle weakness. Skelet. Muscle 3: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Beshbishi SN, Ahmed NN, Mostafa SH, and El-Ganainy GA 2012. Parasitic infections and myositis. Parasitol. Res. 110: 1–18. [DOI] [PubMed] [Google Scholar]
- 6.Bruschi F, and Chiumiento L 2011. Trichinella inflammatory myopathy: host or parasite strategy? Parasit. Vectors 4: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crum-Cianflone NF 2010. Nonbacterial myositis. Curr. Infect. Dis. Rep. 12: 374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fonseca DM da TW Hand S-J Han MY Gerner AG Zaretsky AL Byrd OJ Harrison AM Ortiz M Quinones G Trinchieri JM Brenchley IE Brodsky RN Germain G Randolph J, and Belkaid Y 2015. Microbiota-Dependent Sequelae of Acute Infection Compromise Tissue-Specific Immunity. Cell 163: 354–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tidball JG 2017. Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol. 17: 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng B, Wehling-Henricks M, Villalta SA, Wang Y, and Tidball JG 2012. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol. 189: 3669–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Novak ML, and Koh TJ 2013. Phenotypic transitions of macrophages orchestrate tissue repair. Am. J. Pathol. 183: 1352–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villalta SA, Nguyen HX, Deng B, Gotoh T, and Tidball JG 2008. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 18: 482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rigamonti E, Zordan P, Sciorati C, Rovere-Querini P, and Brunelli S 2014. Macrophage plasticity in skeletal muscle repair. Biomed Res. Int. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villalta SA, Rosenthal W, Martinez L, Kaur A, Sparwasser T, Tidball JG, Margeta M, Spencer MJ, and Bluestone JA 2014. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci. Transl. Med. 6: 258ra142–258ra142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castiglioni A, Corna G, Rigamonti E, Basso V, Vezzoli M, Monno A, Almada AE, Mondino A, Wagers AJ, Manfredi AA, and Rovere-Querini P 2015. FOXP3+ T Cells Recruited to Sites of Sterile Skeletal Muscle Injury Regulate the Fate of Satellite Cells and Guide Effective Tissue Regeneration. PLoS One 10: e0128094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, and Mathis D 2013. A special population of regulatory T cells potentiates muscle repair. Cell 155: 1282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ, Benoist C, and Mathis D 2016. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity 44: 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin RM, Blair SJ, Warunek J, Reid R, Blader IJ, Wohlfert EA, Jin RM, Blair SJ, Warunek J, Heffner RR, Blader IJ, and Wohlfert EA 2017. Regulatory T Cells Promote Myositis and Muscle Damage in Toxoplasma gondii Infection. J. Immunol. 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarleton RL, and Tarleton RL 1990. Depletion of CD8+ T cells increases susceptibility and reverses vaccine-induced immunity in mice infected with Trypanosoma cruzi. J. Immunol. 144: 717–24. [PubMed] [Google Scholar]
- 20.Fabre MV, Beiting DP, Bliss SK, and Appleton JA 2009. Immunity to Trichinella spiralis muscle infection. Vet. Parasitol. 159: 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wohlfert EA, Blader IJ, and Wilson EH 2017. Brains and Brawn: Toxoplasma Infections of the Central Nervous System and Skeletal Muscle. Trends Parasitol. 33: 519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu R, Zhou Q, La Cava A, Campagnolo DI, Van Kaer L, and Shi F-D 2010. Expansion of regulatory T cells via IL-2/anti-IL-2 mAb complexes suppresses experimental myasthenia. Eur. J. Immunol. 40: 1577–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benson A, Murray S, Divakar P, Burnaevskiy N, Pifer R, Forman J, and Yarovinsky F 2012. Microbial infection-induced expansion of effector T cells overcomes the suppressive effects of regulatory T cells via an IL-2 deprivation mechanism. J. Immunol. 188: 800–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oldenhove G, Bouladoux N, a Wohlfert E, a Hall J, Chou D, Dos Santos L, O’Brien S, Blank R, Lamb E, Natarajan S, Kastenmayer R, Hunter C, Grigg ME, and Belkaid Y 2009. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 31: 772–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, Votavova P, Pepper M, Kovar M, and Garcia KC 2015. Antibodies to Interleukin-2 Elicit Selective T Cell Subset Potentiation through Distinct Conformational Mechanisms. Immunity 42: 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, Sanders D, Lacey C, Wang Y, Vence L, Hwu P, and Radvanyi L 2014. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J. Clin. Invest. 124: 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kornete M, Sgouroudis E, and Piccirillo CA 2012. ICOS-dependent homeostasis and function of Foxp3+ regulatory T cells in islets of nonobese diabetic mice. J. Immunol. 188: 1064–74. [DOI] [PubMed] [Google Scholar]
- 28.Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, Pott J, Griseri T, Bollrath J, Hegazy AN, Harrison OJ, Owens BMJ, Löhning M, Belkaid Y, Fallon PG, and Powrie F 2014. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 513: 564–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, Treuting PM, and Rudensky AY 2015. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 162: 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, Burzyn D, Ortiz-Lopez A, Lobera M, Yang J, Ghosh S, Earl A, Snapper SB, Jupp R, Kasper D, Mathis D, and Benoist C 2015. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science 349: 993–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sica A, and Mantovani A 2012. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122: 787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege J-L, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, and Wynn TA 2014. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 41: 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Couper KN, Blount DG, and Riley EM 2008. The Journal of Immunology. J. Immunol. 152: 4368–4374. [DOI] [PubMed] [Google Scholar]
- 34.Zaiss DMW, van Loosdregt J, Gorlani A, Bekker CPJ, Gröne A, Sibilia M, van Bergen en Henegouwen PMP, Roovers RC, Coffer PJ, and Sijts. AJAM 2013. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity 38: 275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang S, Zhang Y, Wang Y, Ye P, Li J, Li H, Ding Q, and Xia J 2016. Amphiregulin Confers Regulatory T Cell Suppressive Function and Tumor Invasion via the EGFR/GSK-3β/Foxp3 Axis. J. Biol. Chem. 291: 21085–21095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, and Bluestone JA 2008. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 28: 687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, Grey ST, and Sprent J 2009. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J. Exp. Med. 206: 751–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson MS, Pesce JT, Ramalingam TR, Thompson RW, Cheever A, and Wynn TA 2008. Suppression of murine allergic airway disease by IL-2:anti-IL-2 monoclonal antibody-induced regulatory T cells. J. Immunol. 181: 6942–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, Nakayama M, Rosenthal W, and Bluestone JA 2009. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 10: 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakaguchi S, a a Vignali D, Rudensky AY, Niec RE, and Waldmann H 2013. The plasticity and stability of regulatory T cells. Nat. Rev. Immunol. 13: 461–7. [DOI] [PubMed] [Google Scholar]
- 41.Hori S 2014. Lineage stability and phenotypic plasticity of Foxp3+ regulatory T cells. Immunol. Rev. 259: 159–72. [DOI] [PubMed] [Google Scholar]
- 42.Koch M Tucker-Heard a, G., Perdue NR, Killebrew JR, Urdahl KB, and Campbell DJ 2009. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 10: 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG, Pritchard GH, Silver JS, Bouladoux N, Stumhofer JS, Harris TH, Grainger J, Wojno EDT, Wagage S, Roos DS, Scott P, a Turka L, Cherry S, Reiner SL, Cua D, Belkaid Y, Elloso MM, and a Hunter C 2012. The cytokines interleukin 27 and interferon-γ promote distinct Treg cell populations required to limit infection-induced pathology. Immunity 37: 511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benson A, Murray S, Divakar P, Burnaevskiy N, Pifer R, Forman J, and Yarovinsky F 2012. Microbial infection-induced expansion of effector T cells overcomes the suppressive effects of regulatory T cells via an IL-2 deprivation mechanism. J. Immunol. 188: 800–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Molofsky AB, Savage AK, and Locksley RM 2015. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 42: 1005–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, and Turnquist HR 2014. IL-33 is an unconventional Alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J. Immunol. 193: 4010–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turnquist HR, Zhao Z, Rosborough BR, Liu Q, Castellaneta A, Isse K, Wang Z, Lang M, Stolz DB, Zheng XX, Demetris AJ, Liew FY, Wood KJ, and Thomson AW 2011. IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J. Immunol. 187: 4598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baumann C, V Bonilla W, Fröhlich A, Helmstetter C, Peine M, Hegazy AN, Pinschewer DD, and Löhning M 2015. T-bet- and STAT4-dependent IL-33 receptor expression directly promotes antiviral Th1 cell responses. Proc. Natl. Acad. Sci. U. S. A. 112: 4056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, Guo L, Yagi R, Yamane H, Punkosdy G, Feigenbaum L, Zhao K, and Paul WE 2012. The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity 37: 660–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanhere A, Hertweck A, Bhatia U, Gökmen MR, Perucha E, Jackson I, Lord GM, and Jenner RG 2012. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat. Commun. 3: 1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gazzinelli R, Xu Y, Hieny S, Cheever A, and Sher A 1992. Simultaneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii. J. Immunol. 149: 175–80. [PubMed] [Google Scholar]
- 52.Worster DT, Schmelzle T, Solimini NL, Lightcap ES, Millard B, Mills GB, Brugge JS, and Albeck JG 2012. Akt and ERK control the proliferative response of mammary epithelial cells to the growth factors IGF-1 and EGF through the cell cycle inhibitor p57Kip2. Sci. Signal. 5: ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zaiss DMW, Gause WC, Osborne LC, and Artis D 2015. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 42: 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, Tanaka S, Bluestone JA, and Takayanagi H 2014. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 20: 62–68. [DOI] [PubMed] [Google Scholar]
- 55.Blatner NR, Mulcahy MF, Dennis KL, Scholtens D, Bentrem DJ, Phillips JD, Ham S, Sandall BP, Khan MW, Mahvi DM, Halverson AL, Stryker SJ, Boller A-M, Singal A, Sneed RK, Sarraj B, Ansari MJ, Oft M, Iwakura Y, Zhou L, Bonertz A, Beckhove P, Gounari F, and Khazaie K 2012. Expression of RORγt marks a pathogenic regulatory T cell subset in human colon cancer. Sci. Transl. Med. 4: 164ra159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, Fehling HJ, and Bluestone JA 2013. Self-antigen-Driven Activation Induces Instability of Regulatory T Cells during an Inflammatory Autoimmune Response. Immunity 39: 949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.