

ABSTRACT
Horvath’s epigenetic clock consists of 353 CpGs whose methylation levels can accurately predict the age of individuals. Using bioinformatics analysis, we investigated the conformation, energy characteristics and presence of tentative splice sites of the sequences surrounding the epigenetic clock CpGs, in relation to the median methylation changes in different ages, the presence of CpG islands and their position in genes. Common characteristics in the 100 nt sequences surrounding the epigenetic clock CpGs are G-quadruplexes and/or tentative splice site motifs. Median methylation increases significantly in sequences which adopt less stable structures during transcription. Methylation is higher when CpGs overlap with G-quadruplexes than when they precede them. Median methylation in epigenetic clock CpGs is higher in sequences expressed as single products rather than in multiple products and those containing single donors and multiple acceptors. Age-related methylation variation is significant in sequences without G-quadruplexes, particularly those producing low stability nascent RNA and those with splice sites. CpGs in sequences close to transcription start sites and those which are possibly never expressed (hypothetical proteins) undergo similar extent of age-related median methylation decrease and increase. Preservation of methylation is observed in CpG islands without G-quadruplexes, contrary to CpGs far from CpG islands (open sea). Sequences containing G-quadruplexes and RNA pseudoknots, determining the recognition by H3K27 histone methyltransferase, are hypomethylated. The presented structural DNA and co-transcriptional RNA analysis of epigenetic clock sequences, foreshadows the association of age-related methylation changes with the principle biological processes of DNA and histone methylation, splicing and chromatin silencing.
KEYWORDS: DNA methylation, aging, epigenetic clock, G-quadruplex, alternative splicing, co-transcriptionally formed RNA structures, hypothetical proteins, tentative splice sites
Introduction
It has been recently shown that DNA methylation and its topography are generally modified by aging [1,2] and aging-related diseases [3–5]. However, the characteristics of the CpGs that undergo aging-dependent methylation changes and the mechanisms involved in this process are poorly described [6]. Furthermore, while DNA methylation regulates splicing of about 22% of alternative exons [7], the mechanism by which this occurs remains obscure [7–12]. Finally, there is evidence, albeit very limited, that DNA methylation is associated to DNA conformational characteristics [13,14].
Horvath [15] identified 353 CpGs, the methylation level of which can be used to predict the chronological age in humans. Some of the critical CpGs, in which methylation deregulation has been associated with aging, belong to important genes but, for approximately 20% of the epigenetic clock sequences, their protein products are not experimentally verified, and their functions remain unknown (hypothetical proteins). These aging-related ‘CpG markers’ might reflect critical methylation loci and processes related to the structural characteristics of the sequences in which age-related methylation deterioration occurs [10,11]. Identification of the common sequence characteristics of these CpGs could be useful for understanding the processes involved in DNA methylation, particularly during aging, as well as the way conformation, energy, and splice site selection, all independently related to methylation, are combined in the actual set of epigenetically modified sequence ensemble. This knowledge could contribute to our understanding of aging-related deregulations.
DNA can assume a variety of structures deviating from the canonical B-DNA helix. G-rich sequences can be assembled into so called G-quadruplexes (G4s), which consist of stacked planar quartets of guanines that are stabilized by Hoogsteen hydrogen bonds. These structures have been identified as sites of epigenetic, transcriptional, and translational regulation. G4s have been implicated in the deregulation of DNA methyltransferases (DNMTases) IIIA and IIIB activity [10,11,16–21], alternative expression [22], and splicing [23]. RNA can exist in an even greater variety of folded structures. For example, RNA pseudoknots are structures in which single stranded regions or loops connect two helical segments [24]. Pseudoknots are critical for RNA splicing [12,25,26] and the recognition of RNA editing factors [27]. They are considered responsible for changes in expression profiles [28–30]. Although there is an established correlation between methylation, CpG frequency, chromatin states, and gene expression, there is no such correlation between conformational features of DNA and RNA and the methylation sensitive areas during aging [31].
We presently studied the structural characteristics of the sequences neighboring the epigenetic clock CpGs (G4s and pseudoknots) and the thermodynamic stability of the conformations these sequences assume during co-transcriptional helical unwinding. In addition, we investigated the presence of tentative splice sites, their correlation, and multiplicity in the sequences neighboring the epigenetic clock CpGs. We report on the association of these characteristics with methylation and age-related methylation changes and we discuss the implications on the DNA methylation mechanisms and its shortcomings during aging and how they could contribute to the design of more targeted epigenetic clock tools for studying aging.
Results
Figure 1 summarizes the analysis followed in order to identify the association of age-related methylation extent and deviations in epigenetic clock CpGs with the presence of characteristic conformation and sequence elements, which have been associated either with epigenetic modifications or splicing.
Figure 1.

Sequence of tasks for evaluating the association of age-related methylation deviation in epigenetic clock CpGs with the presence G-quadruplexes, co-transcriptionally formed RNA characteristics, and the presence of tentative splice sites close to the epigenetic clock CpGs.
G4s, co-transcriptional folding of RNA, and methylation in the epigenetic clock sequence ensemble
G4 frequency in sequences surrounding epigenetic clock CpGs
The epigenetic clock sequence ensemble among young and old individuals was analyzed with respect to the frequency of G4s and the co-transcriptional RNA conformation, including the presence of pseudoknots, which are considered critical for the regulation of genome expression (Table S1). In order to evaluate the impact of G4s we first analyzed their frequency and compared it to the previously reported genomic G4 frequency [32] as well as their enrichment around the epigenetic clock CpGs. In addition, we determined the optimal size of the sequence to be investigated for the remaining of the study. In the epigenetic clock sequences outside transcription start sites (TSS), the G4 frequency (0.546 for the epigenetic sequence ensemble) exceeds the average frequency reported for the human genome close to transcribed regions (0.4) [32]. The G4 frequency in the epigenetic clock sequence ensemble is higher adjacent to TSS (0.6 for CpGs in the TSS200nt upstream region and 0.636 for the CpGs in the TSS1500nt upstream region). In addition, the frequency increases in sequences that extend 40, 50, 60 or 100 nt on each side of epigenetic clock CpGs (Figure 2(B)). Unless otherwise stated, we used sequences ± 50 nt centered at the epigenetic clock CpGs throughout this study to detect the presence of conformational motifs and other expression-related sequence motifs.
Figure 2.
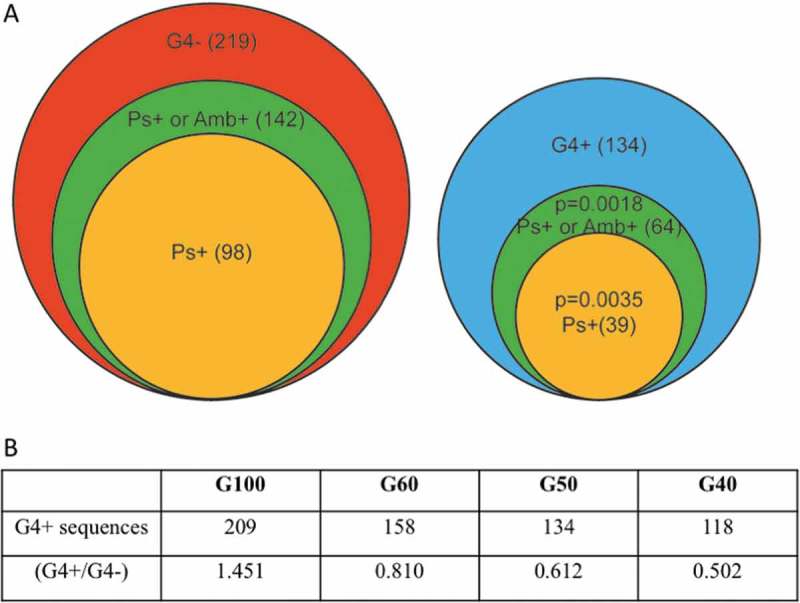
A Number of sequences forming pseudoknots (Ps+), or ambivalent sequences (Amb+, sequences predicted to assume both pseudoknot and non-pseudoknot conformations of < 1kcal/mol difference between the most stable non-pseudoknot conformation and the next less stable pseudoknot conformation), in sequences predicted to form G4s (G4+, right) or without G4s (G4-, left) among the epigenetic clock sequence ensemble (± 50 nt from clock CpGs, G50). B Number of sequences containing G-quadruplexes in different sequence sizes and the corresponding ratio of G4+/G4- sequences. P values obtained by chi-squared test.
G4 presence and DNA methylation among young individuals
G4s are very frequently detected among the epigenetic clock sequence ensemble (Figure 2(A)). Although median CpG methylation among young individuals is higher in sequences lacking G4s (Figure 3(A)), this association is not significant. G4s were also frequently detected in the strand complementary to that of the epigenetic clock CpGs (results not shown). In these sequences, however, the G4 presence does not vary with methylation and was therefore not included in the following analysis.
Figure 3.
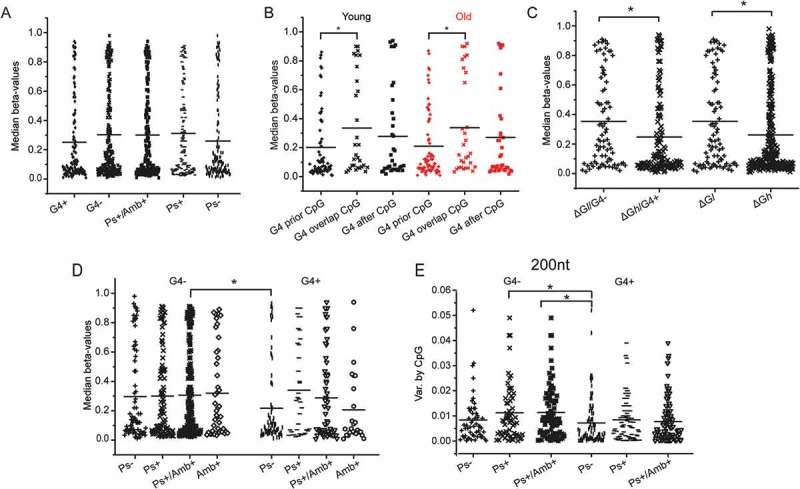
Dot plots indicating the median CpG methylation and methylation variance by CpG in: A. Young individuals with respect to the propensity of the surrounding sequence (CpG ± 50 nt) to form G4s (G4+) and pseudoknots (Ps), or ambivalent sequences (Amb+), B. Young (black) and old (red) individuals with respect to the relative the positions of CpG and G4s in the surrounding sequence (CpG ± 50 nt), C. Young individuals with respect to the relative folding stability and propensity of the surrounding sequence (± 50 nt) to form G4s (ΔGl < 16.35 kcal/mol, ΔGh > 16.35 kcal/mol), D. Young individuals with respect to the combined presence of G4 and pseudoknots in the surrounding sequence (CpG ± 50 nt). E. Variance of methylation by CpG relative to the sequence propensity to form G4s, identified in different sequence lengths (± 50 nt and ± 100 nt). Statistically significant differences by Student’s t-test are shown by *.
It should be noted that, the G4 frequency is not directly proportional to the CpG frequency, which is known to have an impact on DNA methylation. In CpG-dense sequences that have a higher than average CpG content (> 16.8 CpGs per sequence), the average G4 frequency is 0.533/sequence, while in CpG-poor sequences that have a lower than average CpG content (5.6 CpGs/sequence), the corresponding G4 frequency is 0.265. Thus, the presence of G4s was studied as an independent parameter for methylation.
A detailed analysis of the relative position of G4s with respect to the epigenetic clock CpGs revealed that their distance affects methylation in both age categories (Figure 3(B)). Sequences where the G4 precedes the epigenetic clock CpG exhibit significantly lower median methylation both among younger (P = 0.027) and older individuals (P = 0.032). This is not observed in any other case.
Profiling RNA folding during transcription
The propensity of the investigated sequences to fold into conformations that might affect the rate of transcription was then evaluated by identifying sequences that can co-transcriptionally form complex structures, including pseudoknots. Pseudoknots are associated with the formation of long-range interactions, which probably contribute to the identification of specific sequence motifs suitable for RNA processing, such as those associated with splicing. Two computational tools for identifying pseudoknots were used, Hotknots and Kinefold. Although the number of epigenetic clock sequences positive for pseudoknots (Ps) was significantly higher based on Kinefold analysis (results obtained by manual submission, Table S2) compared to those obtained by Hotknots, the latter was more useful for independently analyzing the impact of splice sites on methylation and was therefore used in the remaining analysis, unless otherwise noted. Based on the analysis by Hotknots, 137 of the 353 sequences are predicted to form pseudoknots (Figure 2(A)). Sequences predicted to contain Ps exhibit higher overall methylation, though the observed methylation differences were not significant (Figure 3(A)). Some sequences were predicted to form both pseudoknot and non-pseudoknot conformations of very similar stability (< 1 kcal/mol difference between the most stable non-pseudoknot conformation and the next less stable pseudoknot conformation, Table S1). These sequences will hereon be referred to as ‘ambivalent’ sequences (Amb), since the presence of methylation could potentially change the equilibrium between their pseudoknot and non-pseudoknot conformations [33]. The ambivalent pseudoknots are frequent among the epigenetic clock sequence ensemble (Figure 2(A)) and were further studied together with pseudoknots, unless otherwise noted. The G4-/Ps+ or Amb+ sequences are the most prevalent category of sequences identified (142/353, P = 0.018, Figure 2(A)).
The thermodynamic stability of the co-transcriptionally folded RNA product given by Hotknots was also recorded (Table S1) and its impact on methylation in younger individuals was analyzed. In this case, the epigenetic clock sequences were categorized into two balanced groups of low and high energy (energy threshold: ΔG = -16.35 kcal/mol). The free energy of the predicted single-stranded conformations assumed by epigenetic clock sequences was found to be related to the extent of methylation. Methylation was higher when G4s were not formed and the stability of the co-transcriptionally formed structures was lower (P = 0.011, Figure 3(C)). Thus, the combination of the absence of G4s (G4-) and low ΔG (ΔGl) might be an additional marker for higher methylation.
‘Accessibility’ of epigenetic clock CpGs and methylation changes during aging
G4 is not an independent determining factor for methylation changes during aging. G4 positive sequences are more frequently methylated among older individuals relative to G4 negative sequences, but the association is not significant (P = 0.108). However, the methylation increase among G4 positive sequences during aging is smaller relative to the observed decrease, but this association is also not significant (P = 0.206).
On the contrary, median methylation is higher in the absence of G4s, when pseudoknots can be efficiently formed co-transcriptionally by the nascent RNA and becomes significantly lower in the presence of G4s and absence of pseudoknots (P = 0.037, Figure 3(D)). Thus, significantly higher methylation is observed in the epigenetic clock CpGs, which i) are present in non-compressed sequences (G4 represents a form of tentative helical compression) and, ii) fold, co-transcriptionally, into RNA structures of low stability, or form long-range interactions. The combination of these elements (G4-/Ps+ or Amb+) could be considered as a structure ‘accessible’ or ‘open’ to methylation, revealing temporarily exposed single-stranded DNA sequences, which do not re-anneal fast due to the low RNA folding stability. These differences become more significant when 200 nt sequences instead of the 100 nt sequences are examined (Figure 3(E), P = 0.005).
The association of the G4 presence and the RNA folding characteristics, both significantly related to DNA methylation, with DNA methylation decrease or increase as a result of aging, was then evaluated. According to our analysis (Table 1) there is a significant tendency for methylation decrease among all epigenetic clock CpGs (P = 0.007), particularly among G4- sequences (P = 0.01). All G4- sequences show similar age-related differences. However, sequences that are ‘closed’ to methylation (G4+/Ps-) undergo either methylation increase or decrease to the same extent (P = 0.709). The extent of methylation decrease is related to the presence of Ps (or Amb) or the stability of the co-transcriptionally formed RNA. Sequences that can form G4 and produce RNA with pseudoknots (G4+/Ps+ or Amb+) undergo the strongest hypomethylation (7.1 median methylation decrease and 2.4 median methylation increase). Methylation decrease and increase are equally frequent among sequences without splice sites (P = 0.456, Table 2). Significant variation of methylation is observed in sequences with low CpG frequency (outside CpG islands). The methylation decrease is more frequent among sequences which produce co-transcriptionally comparably less stable RNA structures (mean ΔG = 19.259 for sequences which get less methylated vs. mean ΔG = 21.008 for more methylated sequences, P = 0.009). Sequences recognized as ‘true’ enhancers according to ENCODE project, were not significantly associated with G4 (P = 0.746) and either positive or negative methylation changes during aging.
Table 1.
Median methylation (beta values) differences between young and old subjects relative to the presence of G4, RNA co-transcriptional structures and stability and the presence of tentative splice sites.
| Median methylation decrease x102 (sequences) | Median methylation increase x102 (sequences) | p* | |
|---|---|---|---|
| All sequences | 4.66 (148) | 2.86 (177) | 0.007 |
| G4- | 4.2 (99) | 2.7(102) | 0.01 |
| G4-/ΔGh | 3.93(60) | 2.7 (67) | 0.13 |
| G4-/ΔGl | 4.73(38) | 2.69(35) | 0.017 |
| G4-/Ps+ or Amb+ | 4.31 (70) | 2.96 (62) | 0.075 |
| G4-/Ps-/Amb- | 4.07(29) | 2.3(40) | 0.069 |
| G4+ | 5.5(49) | 3.09(75) | 0.099 |
| G4+/Ps-/Amb- | 3.14 (20) | 3.56 (43) | 0.709 |
| G4+/Ps+ or Amb+ | 7.1(29) | 2.4(32) | 0.085 |
| CpG< median average | 5.45 (92) | 3.32 (69) | 0.003 |
| CpG> median average | 4.92 (21) | 3.71 (75) | 0.21 |
*Student’s t-test
ΔGh > 16.35 kcal/mol; ΔGl < 16.35 kcal/mol
Table 2.
Median methylation changes as a function of aging (young – old individuals) in epigenetic clock CpGs relative to CpG islands, transcription start and tentative splice sites. Correlation to the presence of G4, tentative splice sites and characteristics of corresponding nascent RNA products.
| Median methylation decrease by CpG x102 (Sequences) | Median methylation increase by CpG x102 (Sequences) | p-value* | G4 (Frequency) |
Total sequences | Median ΔG | Ps+ or Amb+ (Frequency) | Total Splice sites (Frequency per sequence) |
|
|---|---|---|---|---|---|---|---|---|
| CpG island | 2.034 (34) |
2.965 (91) |
0.195 | - | 135 | - | - | 189 (1.400) |
| CpG island G4+ | 3.122 (17) |
3.147 (58) |
0.982 | - | 75 | 25.53 | 28 (0.373) |
101 (1.347) |
| CpG island G4- | 0.947 (17) |
2.890 (40) |
0.041 | - | 60 | 20.69 | 33 (0.55) |
88 (1.467) |
| Open Sea | 6.256 (31) |
2.644 (25) |
0.005 | 16 (0.246) |
65 | 17.90 | 37 (0.569) |
95 (1.462) |
| TSS1500 | 4.168 (41) |
2.968 (41) |
0.191 | - | 89 | - | - | 133 (1.494) |
| TSS1500 G4+ | 2.64 (10) |
3.845 (17) |
0.434 | - | 28 | 22.99 | 17 (0.643) |
37 (1.321) |
| TSS1500 G4- | 4.661 (31) |
2.346 (24) |
0.048 | - | 61 | 17.50 | 44 (0.685) |
96 (1.574) |
| TSS200 only | 3.553 (15) |
2.963 (19) |
0.665 | 14 (0.333) |
42 | 20.12 | 29 (0.69) |
32 (0.762) |
| Including 5’ UTRs | 3.946 (41) |
2.251 (45) |
0.046 | 41 (0.461) |
89 | 20.83 | 47 (0.528) |
136 (1.528) |
| Sequences without tentative splice sites | 3.819 (42) |
2.952 (36) |
0.456 | 25 (0.291) |
86 | 17.58 | 52 (0.604) |
0 |
| Sequences with tentative splice sites | 4.314 (106) |
2.872 (141) |
0.008 | 109 (0.405) |
269 | 20.94 | 154 (0.572) |
510 (1.896) |
| Hypothetical proteins | 2.359 (22) |
2.718 (27) |
0.70 | 21 (0.396) |
53 | 21.00 | 28 (0.528) |
55 (1.038) |
*Student’s t-test
Based on the analysis by Kinefold, the structures of the more stable nascent RNA sequences formed during transcription (Figure 4) exhibit common characteristics with regard to their free energy and gene location (low energy Figure 4(A-E), high energy, Figure 4(F-J), Table S3), the presence of G4s and pseudoknots in proximity with epigenetic clock CpGs. The second category of sequences which assume more complex and stable structures show very small methylation decrease upon aging, in contrast to sequences A, C, and D, in which the CpGs are located in sequences which assume unstable RNA structures. A common characteristic, in most cases, appears to be the presence of a stable secondary structure next to the epigenetic clock CpG.
Figure 4.
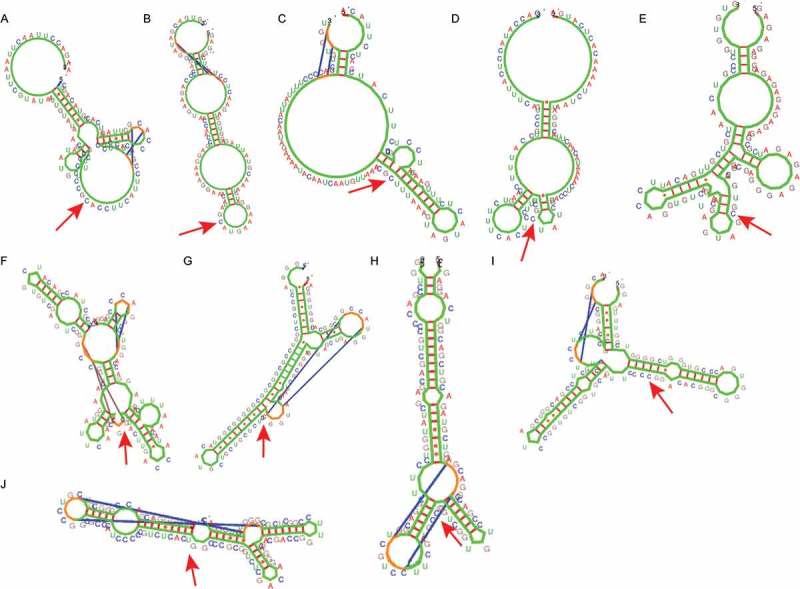
Characteristic Kinefold structures of ± 50 nt next to the methylation site. Structures (A-E). ΔGl, open-sea sequences, G4-; A, C, D: methylation decrease by aging; B, E: methylation increase by aging. Structures (F-J). G4+, Ps- (according to Hotknots), ΔGh (> 18.14), methylation decrease by aging. The arrows indicate the epigenetic clock CpGs.
Alternative expression, presence and multiplicity of tentative splice sites and methylation
Tentative splice sites can be frequently detected in a variety of sequences. However, only few of them are selected. Recent evidence shows that methylation participates in the splice site selection process [34]. In addition, aging is related to reduced transcriptional fidelity and a high frequency of missplicing [35] and alternative gene expression. We presently evaluated the association of the epigenetic clock sequence ensemble with splicing related-parameters, by determining the following:
Frequency and methylation of epigenetic clock CpGs expressed as alternative or single products
For this purpose, CpGs were classified according to their participation in a single RNA product or in multiple RNA products (Table S1). Using this criterion, it is observed that almost half of the epigenetic clock CpGs are observed in multiple RNA products and are less methylated relative to those observed as single products (P = 0.006, Figure 5(A). Sequences producing co-transcriptionally a single RNA product of less stable conformation (Single/ΔGl) are more methylated compared to those producing multiple products of higher stability (Multiple/ΔGh, P = 0.006, Figure 5(A)). Significant differences in methylation are observed in sequences, which are open to methylation and produce single products (Single Open vs. Single Closed, P = 0.039, Figure 5(A)).
Figure 5.
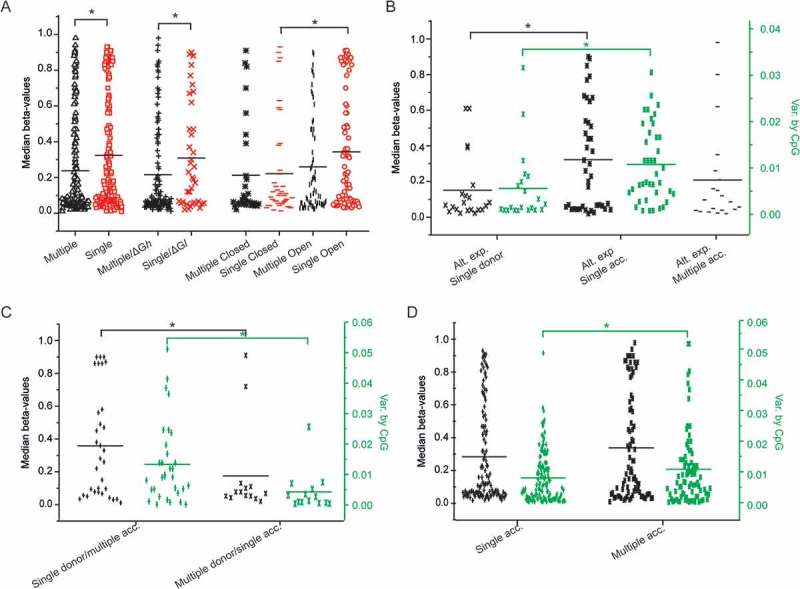
Dot plots of median methylation and methylation variance by CpG among young individuals relative to the sequence expression, the energy of the transcript, its conformational characteristics and the presence of tentative splice sites. A. Dot plots of CpG methylation in sequences predicted to be alternatively (black) or singly (red) expressed in relation to their propensity to form G4s and Pseudoknots (G4-/Ps+: open conformation, G4+/Ps-: closed conformation) and the relative folding stability (ΔGl, ΔGh) of the corresponding transcript. B. Dot plots of median CpG methylation (black) and methylation variance by CpG (green) in sequences containing single tentative donors (no acceptors) and single or multiple acceptors (no donors), C. Dot plots of median CpG methylation (black) and methylation variance by CpG (green) in sequences containing different combinations of tentative splice sites. D. Dot plots of median CpG methylation (black) and methylation variance by CpG (green) in sequences containing single acceptors and multiple acceptors regardless of the presence of donors. Statistically significant differences by Student’s t-test are shown by *.
Presence of tentative strong splice sites
According to the analysis performed by ESEfinder [36,37] (Table S1), the epigenetic clock sequence ensemble contains a large number of tentative strong donor (5’ splice sites) and acceptor sites (3’ splice sites). Strong associations of median methylation and methylation variance with the presence of tentative donor and acceptor sites are observed in several cases, including:
i) the presence of a single donor vs. single acceptor in sequences for which multiple products have been isolated (P = 0.018, Figure 5(B)), ii) the presence of single tentative donor and multiple acceptors vs. multiple donors and a single acceptor in the same sequence (P = 0.019, Figure 5(C)) and, iii) sequences with single acceptors, irrespective of the presence of donor sites vs. those containing multiple acceptors (P = 0.046, Figure 5(D)). Thus, methylation is probably associated with the availability of the donor and the selection of the acceptor by the splicing apparatus (spliceosome). Furthermore, it also poses a limit in the use of the splice sites, since sequences expressed as single or unique products are significantly more methylated than those that are found in different variants (or products).
Methylation relative to the spatial organization of genes, the presence of CpG islands, G4s, RNA conformation, and stability of tentative splice sites
The frequency and distribution of the parameters examined previously (G4s, splice sites, co-transcriptional RNA stability, and pseudoknot formation) in association with methylation varies depending on the site where the epigenetic clock CpG is located with respect to the whole gene organization and the presence of CpG islands. In this part of the study, we provide a comprehensive analysis of these elements using two specific sequence categories as examples: open sea sequences, removed from CpG islands (Figure 6(A,B)), and TSS1500 (Figure 7(A,B)), which frequently contain CpG islands. Finally, all data regarding parameters, which were previously examined, were evaluated with respect to the methylation changes they undergo from the young to the old individuals (methylation decrease, i.e., negative methylation changes during aging or methylation increase, i.e., positive methylation changes during aging – Table 2).
Figure 6.
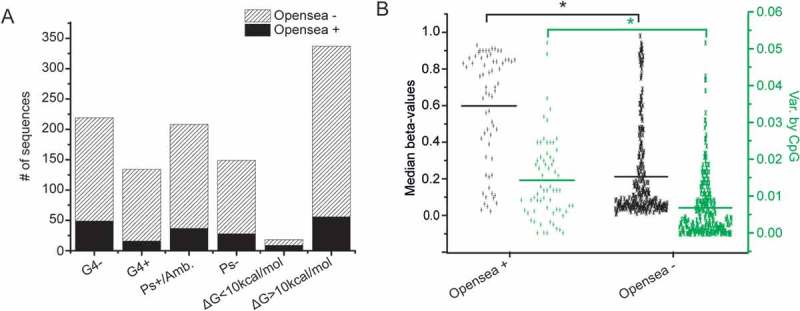
Open sea sequences. A. Frequency of G4, pseudoknots and relative low folding stability of the corresponding transcripts in open sea and non-open sea sequences (P values obtained by chi-squared test). B. Dot plots of median methylation and methylation variance by CpG in open sea and non-open sea sequences among young individuals. Statistically significant differences by Student’s t-test are shown by *.
Figure 7.
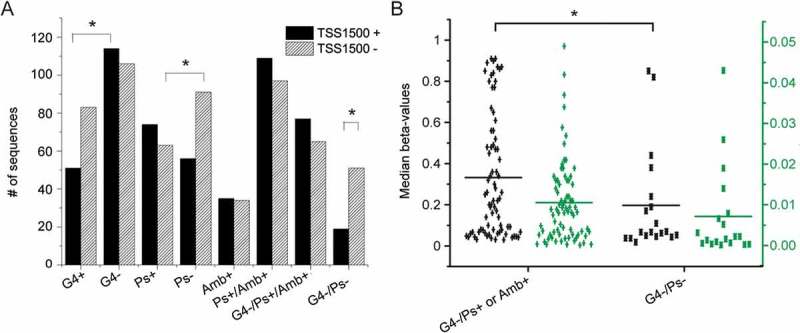
Characteristics of the 1500 nt regions upstream transcription start sites (TSS1500). A. Frequency of G4, Pseudoknots, ambivalent motifs and their combinations (P values obtained by chi-squared test) in sequences predicted (black) or not (striped) to be loci of transcription initiation. B. Dot plots of median methylation (black) and methylation variance by CpG (green) among young individuals in TSS sequences with ‘open’ or ‘closed’ conformation. Statistically significant differences by Student’s t-test are shown by *.
Exceptionally high methylation among young individuals is observed in sequences described as ‘open sea’, i.e., regions removed from CpG islands and without enrichment in CpG content. The frequency of G4s in these sequences is significantly lower compared to all other sequences and not associated with methylation. These sequences form co-transcriptionally RNA structures of low stability, but the pseudoknot frequency in the RNA products is similar to the remaining sequences (Figure 6(A) and Table 2). Half of the lowest energy RNA conformations (ΔG < 10 kcal/mol), which can be formed co-transcriptionally by the epigenetic clock sequences, belong to the open sea category (Figure 6(A)). The formation of low stability, single stranded conformations by open sea sequences, in addition to the lower frequency of G4s compared to all other sequences (P = 0.0017, Figure 6(A)) could probably account for the exceptionally high median methylation in these sequences (P < 0.0001, Figure 6(B)) and the very significant methylation fluctuations that develop upon aging (median methylation decrease 6.256 x 10-2 vs. median methylation increase 2.644 x 10-2, Table 2).
In TSS1500 sequences the G4 frequency is lower and the pseudoknot frequency predicted by Hotknots is higher compared to the remaining sequences (P = 0.012 and P = 0.009, respectively, Figure 7(A)). These differences in the G4+ and Ps- frequencies are also related to very significant differences in methylation (P < 0.001, Figure 7(B)). Similar findings are observed in gene body sequences (Figure S1).
Age-related methylation deviations, CpG gene position, and proximity to CpG islands
In the following section, we evaluated the median methylation changes that are observed in individual CpGs during aging and their association with the parameters that were previously analyzed (G4 presence, pseudoknots and nascent RNA stability, presence of splice sites, relative position in genes, and relation to CpG islands) (Table 2). It is evident from Table 2 that in CpG islands methylation differences (decrease vs. increase) are observed only when some of the parameters previously analyzed are considered. Thus, in CpG islands, significant variation of methylation among the epigenetic clock CpGs during aging is only related to the absence of G4 (very small methylation decrease in G4- CpG islands, 0.947 x 10-2, versus 2.890 x 10-2 increase, P = 0.041). Open sea sequences, which are characterized by low G4 frequency, produce nascent RNA of very low stability and have more potential splice sites, relative to other gene regions (e.g., TSS200), exhibit a pronounced methylation decrease (6.256 x 10-2). These facts indicate that the presence of CpG islands is not the sole determinant for methylation changes. It should be noted that median methylation decrease by aging is not directly related to the absolute methylation levels at younger age (results not shown).
The distance from TSS is also related to the median methylation changes upon aging. In TSS200, aging leads to moderate methylation decrease in epigenetic clock CpGs (3.553 x 10-2 median decrease), contrary to the significant differences observed in sequences that extend further from TSS (TSS1500). The presence of G4 is related to significant differences in TSS1500 methylation changes during aging (median methylation decrease in TSS1500/G4+: 2.64 x 10-2 vs. TSS1500/G4-: 4.661 x 10-2).
Median methylation increase exhibits small and insignificant variation as a function of aging, regardless of the CpG location (median methylation increase 3.845 x 10−2 to 2.251 x 10-2). As expected, sequences that are occasionally, but not exclusively, observed in 5’ UTRs and contain very frequent tentative splice sites (similarly to open sea sequences), and G4 frequency comparable to TSS1500, exhibit median methylation increase upon aging (Table 2) similar to the two previous sequence categories (2.346 x 10-2 for TSS1500, 2.963 x 10-2 for TSS200, and 2.251 x 10-2 for 5’UTRs).
Sequences coding for hypothetical proteins, which are probably never expressed as stable products, are very frequent among the epigenetic clock sequence ensemble (approximately 20%). These sequences undergo age-associated median methylation changes of similar extent (2.359 x 10-2 median decrease and 2.718 x 10-2 increase, respectively); these values are also similar to the median methylation increase for all CpGs, particularly those lacking G4s.
Discussion
The epigenetic clock CpGs are subject to characteristic changes of methylation during the aging process. Analysis of the sequences neighboring these CpGs reveals the frequent presence of elements that have been shown to affect DNA methylation or have an impact on transcription. According to our data, CpG methylation varies widely depending on the CpG distance from CpG islands and presence of nearby transcription start sites, as well as the presence of G4s in the surrounding sequences and the conformation and stability of the co-transcriptionally produced RNA. Although changes of methylation in enhancer regions are critical in cellular adaptation [38] and a variety of pathological conditions, the epigenetic clock sequences identified by Horvath contain a limited number of such regions and their presence is not correlated with significant variation of methylation during aging. Additional important parameters are the formation of low stability nascent RNA transcripts, frequently folding into pseudoknots and their localization with respect to splice factor binding sites. The relative position of these motifs affects the level of methylation of the epigenetic clock CpGs in young individuals with respect to aging. The methylation changes in these sequences which are frequently alternatively expressed, probably contribute to transcription infidelity and are responsible for their role as markers of aging according to Horvath [15]. Notably, these elements are neither related to methylation increase nor decrease in all cases.
G-quadruplexes are characteristic recognition motifs for proteins involved in aging, e.g., the telomere binding protein TRF2 [39], and in development, e.g., the polycomb repressive complex 2 histone methyltransferase (PRC2) of H3K27 histone [40]. PRC2, which binds to thousands of transcripts in vivo, also shows high affinity for RNA containing G4s or loops and bulges [40]. Our results show that CpGs in epigenetic clock sequences which possess G4 in addition to the above characteristics and could thus be characterized as potential PRC2 RNA target sites, undergo a methylation decrease during aging. Teschendorff et al [41] previously reported that PRC2 sequences are mostly hypermethylated by aging. However, only 12% of the hypermethylated CpGs reported by Teschendorff et al [41]are included among the epigenetic clock CpGs. Based on our results, it appears that CpGs proximal to G4s undergo an age-related methylation decrease when associated with certain RNA characteristics. G4 proximity alone does not however appear to be an independent age determinant.
Similarly to somatic mutations responsible for certain diseases that undergo positive selection among patients, the methylation changes in epigenetic clock CpGs probably reflect mechanisms that contribute to the deviation of DNA methylation as a result of aging. This is more evident from a recent study published when our study was conducted [42]. The methylation changes in the numerous CpGs that belong to DNA sequences coding for hypothetical proteins could be considered as markers of the functional deterioration of at least some of the enzymes responsible for the maintenance of methylation during aging.
Few studies have been reported on the activity of DNMT I and DNMT IIIA and IIIB. According to Valinluck and Sowers [43], methylation infidelity arises from cytosine modification by halogenating and oxidizing factors that have a dual effect on DNMT I activity, leading to methylation increase or decrease in the first and second case, respectively. This process is probably responsible for the observed similar levels of median methylation decrease and increase in sequences coding for hypothetical proteins, as well as the basal methylation deviation in all other sequences. The impact of the oxidative processes is also reflected by inclusion of enzymes involved in the oxidation/reduction processes in the epigenetic clock sequence ensemble [NADPH oxidase 4, NADH ubiquinone (two sequences), dehydrogenase/reductase (SDR family) member 10, cytochrome P450].
On the other hand, the lack of methylation fidelity in association with the presence of G4s, which is most evident in methylation decrease of CpGs in open sea sequences, is probably related to perturbation of DNA methyltransferases (DNMTases) IIIA and IIIB activities in these sites. In vitro studies have recently reported that DNA methylation is influenced by the presence of G4s. This is related to the negative effect of G4s on the specificity of DNMTases IIIA and IIIB within specific sequence length limits and leads to strand-specific non-CpG methylation [16,17]. In addition, the presence of G4s probably has a negative effect on DNA accessibility [16], through G4-binding ligands [44] or G4-specific proteins [23], which participate in epigenetic reprogramming [22]. Such proteins, specific for RNA G4s have been shown to block the RNA polymerase activity. A similar mechanism could be involved in the regulation of DNA polymerase activity.
Finally, the rate of transcription and co-transcriptional splicing most probably influences the efficiency of methylation maintenance and de novo methylation. The efficiency of the above-mentioned enzymes to add methyl groups to DNA is affected by several parameters, such as the nascent RNA structure and stability, the presence and multiplicity of splice sites, the restricted methylation decrease in G4- CpG islands, and the relative position of the epigenetic clock CpGs with respect to G4s. For example, G4s, which are formed when the DNA strands are separated (e.g., during replication or transcription), could modify splicing by ‘stalling’ the RNA polymerase and, possibly, the DNA methylation process. In short, the combination of all these elements and the presence of specific sequence motifs that can be recognized by other DNA-binding factors [45] and RNA-binding proteins involved in expression and splicing are probably responsible for the critical methylation changes during aging.
The strand specificity of these phenomena, i.e., the observed lack of impact on methylation of the G4 presence in the antisense strand (C-rich strand) is in accordance with previous reports, according to which methyltransferase activity and methylation are strand specific [46]. Conformational ambivalence, a term presently introduced to describe structures that exhibit pseudoknot and non-pseudoknot structures of very similar free energy, is often significant according to the present analysis. Conformational ambivalence is important in most cases, in association with 5’ and 3’ splice sites, possibly involved in the selection process. However, conformational ambivalence is not correlated with methylation in TSS.
In conclusion, most of the elements discussed above are potentially responsible for the compromised fidelity of DNA methyltransferases in the presence of extensive methyl-cytosine destruction (oxidation, or halogenation) associated with the activity of DNMTI. Methylation infidelity is also probably enhanced by ‘perturbations’, such as specific DNA conformation elements (G4s) and by RNA polymerase ‘stalling’ as a result of complex nascent RNA structures and the presence of tentative splice sites [17]. The above complex scenario for the association of aging, methylation, and splicing selection, which emerges from studying the epigenetic clock sequence ensemble, provides useful grounds for further research on principle epigenetic mechanisms, their association with aging and various pathological conditions. Identification of the critical elements responsible for the loss of epigenetic surveillance is important for understanding the impact of CpG mutations, polymorphisms, and changes in their surrounding sequences and for designing appropriate diagnostic tools. A better understanding of the mechanisms underlying these processes will also help us identify the environmental factors that play a regulatory role in formulating our epigenome [11,47,48] and their effects on epigenetic heredity [49]. Moreover, it improves our understanding of the splicing and alternative splicing processes, which are still poorly understood [50–52]. Finally, regardless of the epigenetic mechanisms affecting aging, the epigenetic clock sequence ensemble provides valuable evidence for the design of high-throughput technologies targeting each of these processes.
Materials and methods
Data concerning the probe identifiers of the epigenetic clock CpGs, their coordinates and products, the median methylation among young (< 35 years old) and old (> 55 years old) individuals, the methylation variance by CpG, and their relation to CpG islands were acquired from the publicly available supplementary material of the Horvath’s publication [15].
The epigenetic clock CpGs were analyzed with respect to different positional and context-based features in regions of variable lengths centered at the cytosine of the clock CpGs. First, the 27K array probe identifiers of these CpG loci were analyzed with respect to their position in gene regions, such as gene body, promoter, UTR, etc. Then, we extracted DNA sequences of different sizes adjacent to the clock CpGs. To examine the incidence of potential splice sites that may be affected by the presence of the clock CpGs in their consensus motifs, we built a Perl wrapper of ESEfinder [36,37], a Web tool that enables the detection of potential binding sites of SR proteins, as well as 5’ and 3’ splice sites. The results of the batch submissions were further processed to extract the frequency and strength of the identified 5’ and 3’ splice sites. Next, we analyzed the propensity of the 200, 120, 100, and 80 nt sequences surrounding the epigenetic clock CpGs to form pseudoknot and G4 structures. Hotknots [53,54] was applied to identify pseudoknot structures in the 100 nt region adjacent to clock CpGs, using Perl-based batch submissions to the Hotknots Web server. Folding predictions and their corresponding energy were also simulated using the local version of Kinefold [55]. Both tools predict the presence of co-transcriptionally formed pseudoknots in RNA, while Kinefold can also predict pseudoknots in DNA. Finally, to detect G4 structures we used the QGRS algorithm [56] over various sequence lengths centered at the clock CpGs. The outcome of the above tools was parsed and further processed using Perl scripts in order to integrate different levels of information and identify potential significant statistical associations. Differences in methylation levels and methylation variation upon aging were examined using a Student’s t-test. Dot plots indicating median were generated using Origin® 9.1.
Funding Statement
This work is not part of a funded project.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data can be accessed here
References
- 1.Ashapkin VV, Kutueva LI, Vanyushin BF.. Aging as an epigenetic phenomenon. Curr Genomics. 2017;18:385–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sutherland H, Macartney-Coxson D, Griffiths L, et al. Methylome ‐ wide association study of whole blood DNA in the Norfolk Island isolate identifies robust loci associated with age. Aging (Albany NY). 2017;9:753–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kato N, Loh M, Takeuchi F, et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat Genet. 2015;47:1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rönn T, Volkov P, Gillberg L, et al. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum Mol Genet. 2015;24:3792–3813. [DOI] [PubMed] [Google Scholar]
- 5.Johnson KC, Houseman EA, King JE, et al. Normal breast tissue DNA methylation differences at regulatory elements are associated with the cancer risk factor age. Breast Cancer Res. 2017;19:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun D, Yi SV. Impacts of chromatin states and long-range genomic segments on aging and DNA methylation. PLoS One. 2015;10:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lev Maor G, Yearim A, Ast G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015;31:274–280. [DOI] [PubMed] [Google Scholar]
- 8.Shukla S, Kavak E, Gregory M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deschênes M, Chabot B. The emerging role of alternative splicing in senescence and aging. Aging Cell. 2017;16:918–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peffers MJ, Goljanek-Whysall K, Collins J, et al. Decoding the regulatory landscape of ageing in musculoskeletal engineered tissues using genome-wide DNA methylation and RNASeq. PLoS One. 2016;11:1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cole JJ, Robertson NA, Rather MI, et al. Diverse interventions that extend mouse lifespan suppress shared age-associated epigenetic changes at critical gene regulatory regions. Genome Biol. 2017;18:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong JJL, Gao D, Nguyen TV, et al. Intron retention is regulated by altered MeCP2-mediated splicing factor recruitment. Nat Commun. 2017;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halder R, Halder K, Sharma P, et al. Guanine quadruplex DNA structure restricts methylation of CpG dinucleotides genome-wide. Mol Biosyst. 2010;6:2439. [DOI] [PubMed] [Google Scholar]
- 14.Gupta S, Pathak RU, Kanungo MS. DNA methylation induced changes in chromatin conformation of the promoter of the vitellogenin II gene of Japanese quail during aging. Gene. 2006;377:159–168. [DOI] [PubMed] [Google Scholar]
- 15.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cammas A, Millevoi S. RNA G-quadruplexes: emerging mechanisms in disease. Nucleic Acids Res. 2017;45:1584–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JH, Park SJ, Nakai K. Differential landscape of non-CpG methylation in embryonic stem cells and neurons caused by DNMT3s. Sci Rep. 2017;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith SS, Baker DJ, Jardines LA. A G4-DNA/B-DNA junction at codon 12 of c-Ha-ras is actively and asymmetrically methylated by DNA (cytosine-5) methyltransferase. Biochem Biophys Res Commun. 1989;160:1397–1402. [DOI] [PubMed] [Google Scholar]
- 19.Cree SL, Fredericks R, Miller A, et al. DNA G-quadruplexes show strong interaction with DNA methyltransferases in vitro. FEBS Lett. 2016;590:2870–2883. [DOI] [PubMed] [Google Scholar]
- 20.François M, Leifert WR, Tellam R, et al. Folate deficiency and DNA-methyltransferase inhibition modulate G-quadruplex frequency. Mutagenesis. 2016;31:409–416. [DOI] [PubMed] [Google Scholar]
- 21.Xu B, Zhao C, Chen Y, et al. Methyl substitution regulates the enantioselectivity of supramolecular complex binding to human telomeric G-Quadruplex DNA. Chem Eur J. 2014;20:16467–16472. [DOI] [PubMed] [Google Scholar]
- 22.Guilbaud G, Murat P, Recolin B, et al. Local epigenetic reprogramming induced by G-quadruplex ligands. Nat Chem. 2017;9:1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weldon C, Dacanay JG, Gokhale V, et al. Specific G-quadruplex ligands modulate the alternative splicing of Bcl-X. Nucleic Acids Res. 2017;46:886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Staple DW, Butcher SE. Pseudoknots: RNA structures with diverse functions. PLoS Biol. 2005;3:956–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moss WN, Dela-Moss LI, Kierzek E, et al. The 3′ splice site of influenza A segment 7 mRNA can exist in two conformations: a pseudoknot and a hairpin. PLoS One. 2012;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rieder LE, Staber CJ, Hoopengardner B, et al. Tertiary structural elements determine the extent and specificity of messenger RNA editing. Nat Commun. 2013;4:1–11. [DOI] [PubMed] [Google Scholar]
- 27.Holly AC, Pilling LC, Hernandez D, et al. Splicing factor 3B1 hypomethylation is associated with altered SF3B1 transcript expression in older humans. Mech Ageing Dev. 2014;135:50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tholstrup J, Oddershede LB, Sørensen MA. mRNA pseudoknot structures can act as ribosomal roadblocks. Nucleic Acids Res. 2012;40:303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bindewald E, Afonin KA, Viard M, et al. Multistrand structure prediction of nucleic acid assemblies and design of RNA switches. Nano Lett. 2016;16:1726–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reiling C, Khutsishvili I, Huang K, et al. Loop contributions to the folding thermodynamics of DNA straight hairpin loops and pseudoknots. J Phys Chem B. 2015;119:1939–1946. [DOI] [PubMed] [Google Scholar]
- 31.Day K, Waite LL, Thalacker-Mercer A, et al. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013;14:R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y, Du Z, Li N. Extensive selection for the enrichment of G4 DNA motifs in transcriptional regulatory regions of warm blooded animals. FEBS Lett. 2007;581:1951–1956. [DOI] [PubMed] [Google Scholar]
- 33.Shimada Y, Mohn F, Bühler M. The RNA-induced transcriptional silencing complex targets chromatin exclusively via interacting with nascent transcripts. Genes Dev. 2016;30:2571–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kucharski R, Maleszka J, Maleszka R, et al. A possible role of DNA methylation in functional divergence of a fast evolving duplicate gene encoding odorant binding protein 11 in the honeybee. Proc Biol Sci. 2016;283:718–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Wang Z, Ma T, et al. Alternative splicing in aging and age-related diseases. Transl Med Aging. 2017;1:32–40. [Google Scholar]
- 36.Cartegni L, Wang J, Zhu Z, et al. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith PJ, Zhang C, Wang J, et al. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet. 2006;15:2490–2508. [DOI] [PubMed] [Google Scholar]
- 38.Magnusson M, Larsson P, Xuchun E, et al. Rapid and specific hypomethylation of enhancers in endothelial cells during adaptation to cell culturing. Epigenetics. 2016;11:614–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Purohit G, Mukherjee AK, Sharma S, et al. Extratelomeric binding of the telomere binding protein TRF2 at the PCGF3 promoter is G-Quadruplex Motif-dependent. Biochemistry. 2018;57:2317–2324. [DOI] [PubMed] [Google Scholar]
- 40.Wang X, Kj G, Ar G, et al. Targeting of Polycomb repressive complex 2 to RNA by short repeats of consecutive Guanines. Mol Cell. 2017;65:1056–1067.e5. [DOI] [PubMed] [Google Scholar]
- 41.Teschendorff AE, Menon U, Gentry-Maharaj A, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19:371–384. [DOI] [PubMed] [Google Scholar]
- 43.Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–950. [DOI] [PubMed] [Google Scholar]
- 44.Hong S, Wang D, Horton JR, et al. Methyl-dependent and spatial-specific DNA recognition by the orthologous transcription factors human AP-1 and Epstein-Barr virus Zta. Nucleic Acids Res. 2017;45:2503–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rao S, Chiu TP, Kribelbauer JF, et al. Systematic prediction of DNA shape changes due to CpG methylation explains epigenetic effects on protein-DNA binding. Epigenetics and Chromatin. 2018;11:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zamiri B, Mirceta M, Bomsztyk K, et al. Quadruplex formation by both G-rich and C-rich DNA strands of the C9orf72 (GGGGCC)8•(GGCCCC)8 repeat: effect of CpG methylation. Nucleic Acids Res. 2015;43:10055–10064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dugué P-A, Bassett JK, Joo JE, et al. DNA methylation-based biological aging and cancer risk and survival: pooled analysis of seven prospective studies. Int J Cancer. 2018;142:1611–1619. [DOI] [PubMed] [Google Scholar]
- 48.Dugué P-A, Bassett JK, Joo JE, et al. Association of DNA methylation-based biological age with health risk factors and overall and cause-specific mortality. Am J Epidemiol. 2018;187:529–538. [DOI] [PubMed] [Google Scholar]
- 49.Guerrero-Bosagna C, Weeks S, Skinner MK. Identification of genomic features in environmentally induced epigenetic transgenerational inherited sperm epimutations. PLoS One. 2014;9:e100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malousi A, Kouidou S. DNA hy permethylation of alternatively spliced and repeat sequences in humans. Mol Genet Genomics. 2012;287:631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anastasiadou C, Malousi A, Maglaveras N, et al. Human epigenome data reveal increased CpG methylation in alternatively spliced sites and putative exonic splicing enhancers. DNA Cell Biol. 2011;30:267–275. [DOI] [PubMed] [Google Scholar]
- 52.Karambataki M, Malousi A, Maglaveras N, et al. Synonymous polymorphisms at splicing regulatory sites are associated with CpGs in neurodegenerative disease-related genes. NeuroMolecular Med. 2010;12:260–269. [DOI] [PubMed] [Google Scholar]
- 53.Ren J, Rastegari B, Condon A, et al. HotKnots : heuristic prediction of RNA secondary structures including pseudoknots. RNA. 2005;11:1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andronescu MS, Pop C, Condon AE. Improved free energy parameters for RNA pseudoknotted secondary structure prediction. RNA. 2010;16:26–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xayaphoummine A, Bucher T, Isambert H. Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots. Nucleic Acids Res. 2005;33:605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kikin O, D’Antonio L, Bagga PS. QGRS mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006;34:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.