

ABSTRACT
Genomic imprinting is an epigenetic phenomenon of differential allelic expression based on parental origin. To date, 263 imprinted genes have been identified among all investigated mammalian species. However, only 21 have been described in sheep, of which 11 are annotated in the current ovine genome. Here, we aim to i) use DNA/RNA high throughput sequencing to identify new monoallelically expressed and imprinted genes in day 135 ovine fetuses and ii) determine whether maternal diet (100%, 60%, or 140% of National Research Council Total Digestible Nutrients) influences expression of imprinted genes. We also reported strategies to solve technical challenges in the data analysis pipeline. We identified 80 monoallelically expressed, 13 new putative imprinted genes, and five known imprinted genes in sheep using the 263 genes stated above as a guide. Sanger sequencing confirmed allelic expression of seven genes, CASD1, COPG2, DIRAS3, INPP5F, PLAGL1, PPP1R9A, and SLC22A18. Among the 13 putative imprinted genes, five were localized in the known sheep imprinting domains of MEST on chromosome 4, DLK1/GTL2 on chromosome 18 and KCNQ1 on chromosome 21, and three were in a novel sheep imprinted cluster on chromosome 4, known in other species as PEG10/SGCE. The expression of DIRAS3, IGF2, PHLDA2, and SLC22A18 was altered by maternal diet, albeit without allelic expression reversal. Together, our results expanded the list of sheep imprinted genes to 34 and demonstrated that while the expression levels of four imprinted genes were changed by maternal diet, the allelic expression patterns were un-changed for all imprinted genes studied.
KEYWORDS: Genomic imprinting, allelic-specific gene expression, maternal nutrition, ovine
Introduction
Genomic imprinting refers to the epigenetic phenomenon in which certain genes are expressed in a parent-of-origin-specific manner and play critical roles in fetal growth as well as post-natal development and metabolism [1]. The imprinted alleles are silenced or reduced in expression compared to the non-imprinted and expressed alleles [2]. Imprinted genes tend to be located in clusters. Those in the same cluster are usually regulated by the same imprinting control region (ICR) [3]. Several mechanisms are involved in the control of allelic expression, including DNA allelic methylation, noncoding RNA and/or histone modifications [4]. Genomic imprinting is an evolutionary puzzle because monoallelic expression can expose deleterious recessive mutations, which are normally protected by diploidy [5]. However, imprinting may have a selective advantage because it has been maintained throughout mammalian evolution [6]. The identification of the full catalog of imprinted genes in different mammalian species will greatly facilitate the understanding of the evolutionary roles of genomic imprinting [7].
To date, 186 [8,9] and 112 [10] (http://www.geneimprint.com/site/genes-by-species) imprinted genes have been identified in mice and humans, respectively. However, only 49, 25, and 21 have been reported in cattle [11], pigs [12], and sheep [2,13], respectively. Although the general properties and regulations of imprinting are conserved across species [14], the identities of imprinted genes often are not. For example, only 51 imprinted genes are common between humans and mice. Therefore, it is imperative to identify imprinted genes in each specific species.
Next generation sequencing (NGS) technologies, including genome-wide DNA sequencing (DNA-seq) and transcriptome-wide RNA sequencing (RNA-seq), have been increasingly utilized for in-depth analysis and detection of novel imprinted genes in both humans and mice [15–17]. While high throughput, such studies require completion of genome sequencing and annotation, intensive bioinformatics, and careful independent validation (e.g., Sanger sequencing) to reduce false positives [7,11,18]. The recent completion of the sheep genome and improved annotation abilities provide a great opportunity to identify new imprinted genes in this understudied species.
Poor maternal nutrition, either over- or restricted-feeding during pregnancy [19], has been shown to cause abnormal DNA methylation and expression of a few imprinted genes, such as IGF2R and H19 in ovine fetuses [20]. NGS, however, has the power to simultaneously determine expression changes of all known imprinted genes, which has yet to be conducted in sheep. The objectives of this study were to identify new sheep imprinted genes and to investigate the impact of maternal diets on the expression of all ovine imprinted genes by fetal organs at days 135 of gestation, when the fetuses undergo rapid growth and ample fetal samples can be collected.
Results
High throughput identification of informative single nucleotide polymorphisms
Using single nucleotide polymorphisms (SNPs), the parental origin of an allele in the fetus can be assigned. Informative SNPs are those i) present in mRNAs (expressed), ii) homozygous in ram and heterozygous in fetuses, and iii) expressed at read counts of 20 or greater. They are essential in determining the origin of a parental allele of genes. However, mapping at SNP locations can introduce alignment bias towards the reference alleles because the reads of the alternative alleles may be treated as mismatches and discarded by the mapping tool [7]. To minimize such bias, we artificially built a pseudo-genome (named ‘alternative genome’) by flipping the reference/alternative alleles at all SNP sites from the dbSNP database (sheep 9940) of the sheep reference genome. DNA-seq reads of rams and RNA-seq reads of their respective fetuses were aligned to both the reference and alternative genomes for SNP calling (Figure 1). By comparing the homozygous SNPs in each ram to the heterozygous SNPs in his fetuses, we identified a total of 146,487 unique informative SNPs (represented by SNP1 in Figure S1). These informative SNPs were annotated to 15,298 genes, yielding on average of 9.6 informative SNPs per gene. The parental origins of these informative SNPs were determined using the rams’ genotypes as shown in Figure S1. To further reduce alignment bias, we used Samtools mpileup (version 1.4) [21] to calculate the allele-specific read counts for each informative SNP which were then averaged between the two genomes. Using this approach, we successfully reduced the mapping bias at informative SNP locations to <1% (Figure 2).
Figure 1.
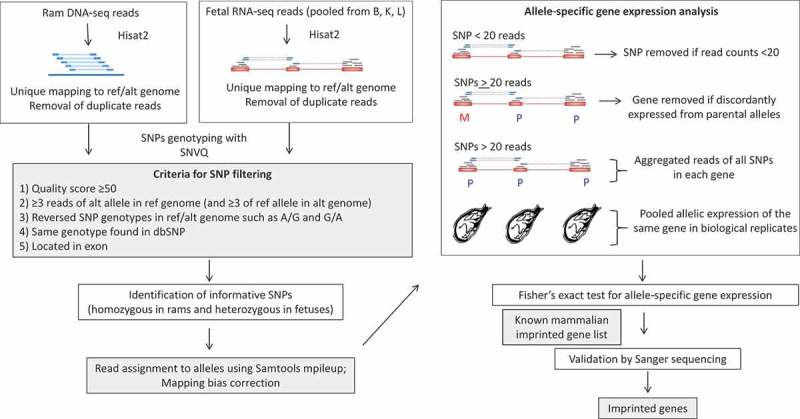
Data analysis pipeline used in this study. Left panels are the bioinformatics pipeline for SNP calling and informative SNPs identification. Right panels show the determination of monoallelically expressed genes in ovine fetuses and validation of putative imprinted genes. Details are presented in sections of Materials and Methods: SNP calling from DNA- and RNA-seq data, Identification of informative SNPs, and Differential allele-specific gene expression and statistical analysis. SNP: single nucleotide polymorphism; ref: reference genome; alt: alternative genome; B: Brain; K: Kidney; L: Lung; dup: duplication; M: maternally expressed; P: paternally expressed. Blue and red boxes: genomic DNA and exons; blue lines with blue boxes: RNA sequence reads; dash lines: mapped gaps in RNA-seq reads; Hisat2: Alignment software; SNVQ: SNP calling software; Samtools mpileup: software to assign read counts to alleles.
Figure 2.
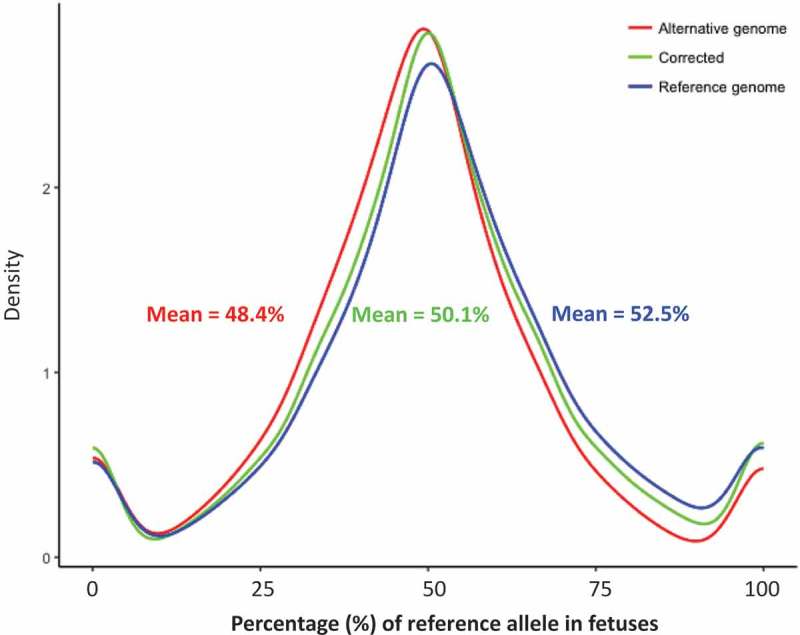
Correction of the RNA-seq alignment bias in the genome. Density plot of the percentage of reference allele’s read counts in the reference genome (blue), alternative genome (red), and after alignment bias correction (green).
Allele-specific gene expression
Fisher’s exact test was programmed to identify genes with allelic expression bias of ≥70%, which also had a read coverage ≥20 at each informative SNPs in at least one tissue type. We identified 4,537 such allelic-differentially expressed genes with a q-value <0.05. Eighty of these genes had significant allelically biased expression of the same parental alleles in all examined tissues (brain, kidney, and lung) and were thus classified as monoallelically expressed (Table S1). Among these, 19 and 61 preferentially expressed the paternal and maternal alleles, respectively. To decrease potential false positives, we conservatively used previously identified imprinted gene as a guide. By combining all imprinted genes in the human, mouse, cow, pig, and sheep we obtained a total of 263 unique imprinted genes, 119 of which have been annotated in the sheep genome (Table S2). Between these 119 and the 4,537 allelic differentially expressed genes, 18 were common and eight and ten were paternally and maternally expressed, respectively (Table 1). Five of the 18 were known to be imprinted in sheep, the other 13 were known to be imprinted in other species and were here designated as putatively imprinted (Table 1). Although there are 21 previously reported imprinted genes in sheep, only 11 of them are annotated in Oar_v4.0. Therefore, 5 out of 11 (45.6%) of imprinted genes were verified using just three tissues at one developmental stage. Each of the 18 genes were individually inspected using the Integrative Genomics Viewer (IGV) [22] to confirm their correct alignments. Read counts from parental alleles of informative SNPs within the genes in each tissue were summarized in Table S3. Nine of these – COPG2, GATM, GRB10, IGF2R, INPP5F, PEG3, PON3, PPPIR9A, and WARS – had more than three informative SNPs that showed significant differential allelic expression in multiple tissues and animals, firmly demonstrating their consistent parent-of-origin specific expression status and, therefore, mostly likely imprinted.
Table 1.
Summary of the confirmed/putative imprinted genes in sheep.
 |
‘-’: no informative SNPs in this animal at this gene. ‘ND’: expression of the informative SNP is Not Detectable (read counts lower than 20; not reliable for allelic expression determination).
Pink or blue: exclusively/predominately expressing the maternal or the paternal allele. Numbers: % of paternal allele expression [paternal allele reads/(paternal allele reads + maternal allele reads)]. Genes in bold: previously known sheep imprinted genes.
The rest of the 80 monoallelically expressed genes may include genes that are only imprinted in sheep. Using the list of known imprinted genes as a guide, however, does not allow us to make such determination. Yet, this conservative approach avoids any potential false positives while expanding ovine imprinting information.
Validation of the putative imprinted genes
To confirm the 13 putative imprinted genes identified above, we quantified their allelic expre-ssion using an independent method – Sanger sequencing. PCR products of seven genes: maternally expressed CASD1, COPG2, PPP1R9A, and SLC22A18 (Figure 3) and paternally expressed DIRAS3, INPP5F, and PLAGL1 (Figure 4) were successfully generated and their allelic expression patterns were verified. The other six genes could not be independently verified by this alternative approach because of the close proximity of informative SNPs to the edge of exons and difficulty in primer design.
Figure 3.
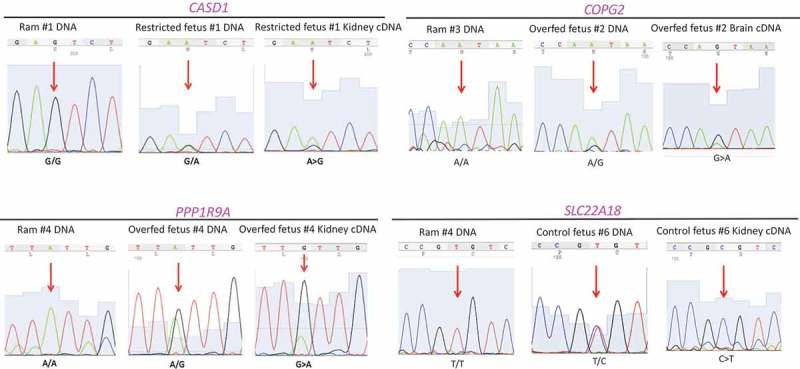
Validation of putative imprinted genes (maternally expressed) using ram and fetal DNA as well as fetal cDNA: CASD1, COPG2, PPP1R9A, and SLC22A18. Red arrows: locations of the informative SNPs. All SNPs were confirmed homozygous in rams and heterozygous in fetuses. Gene expression in cDNA of fetal tissues were allelically biased.
Figure 4.
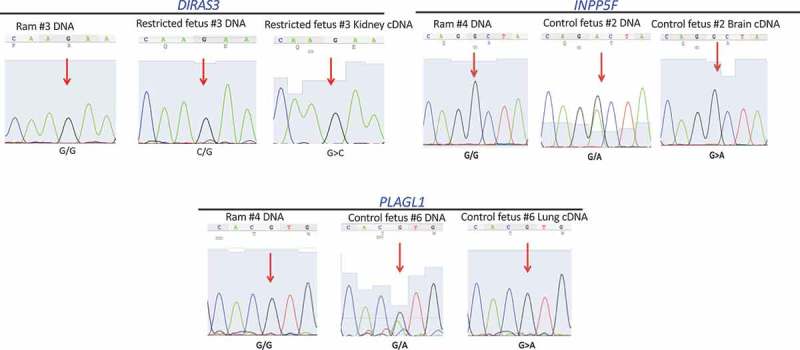
Validation of putative imprinted genes (paternally expressed) using ram and fetal DNA as well as fetal cDNA: DIRAS3, INPP5F, and PLAGL1. Red arrows: locations of the informative SNPs. All SNPs were confirmed homozygous in rams and heterozygous in fetuses. Allelic expression was determined using cDNA from fetal tissues.
A novel imprinted cluster in sheep
Most of imprinted genes were found to reside in clusters of approximately one megabase [1]; therefore, discovery of novel imprinted genes often uses the already established clusters as a guide [11]. We generated a genome visualization of the known ovine imprinted, monoallelically expressed, and putative imprinted genes identified in our analysis (Figure 5). The 80 monoallelically expressed genes were mostly distributed sporadically throughout the genome. Due to the limited information on imprinted clusters in sheep, we do not exclude the possibility that some of the 80 monoallelically expressed genes may be located in imprinted clusters yet to be identified.
Figure 5.
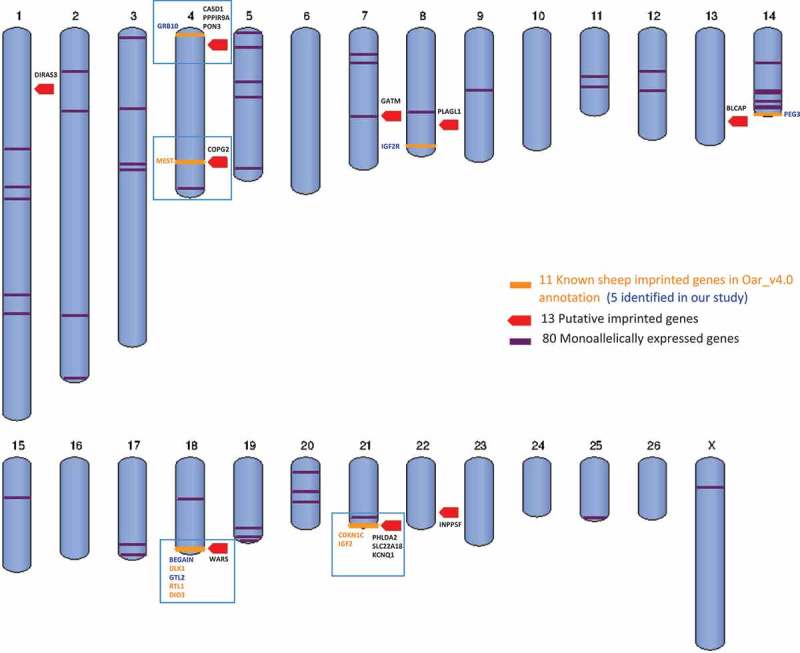
Visualization of the 11 known sheep imprinted genes in the ovine genome Oar_v4.0 annotation (orange), the five genes verified in our study (orange mark, blue text), the 13 putative imprinted genes identified here (red mark), the 80 monoallelically expressed genes (purple lines, some are overlapped), and the four imprinted clusters (blue boxes).
Among the 13 putative imprinted genes, maternally expressed genes CASD1, PPP1R9A and paternally expressed PON3 formed a novel sheep imprinted cluster located close to but not in the sheep known maternally expressed domain MEG1/GRB10 (Figure 6). This novel large imprinted cluster had been characterized as PEG10/SGCE in the mouse [23] and human [24], indicating it is conserved and likely important in development [25]. Moreover, COPG2 and WARS are located in the MEST domain and DLK1/GTL2 domain on chromosomes 4 and 18 (which also contains five other known imprin-ted genes; Figure 6), respectively. PHLDA2, SLC22A18, and KCNQ1 are located in KCNQ1 domain, next to IGF2/H19 domain on chromosome 21, known to be imprinted in sheep (Figure 6). The remaining five genes are located sporadically throughout the sheep genome and their associations with imprinted clusters, if any, are yet to be defined.
Figure 6.
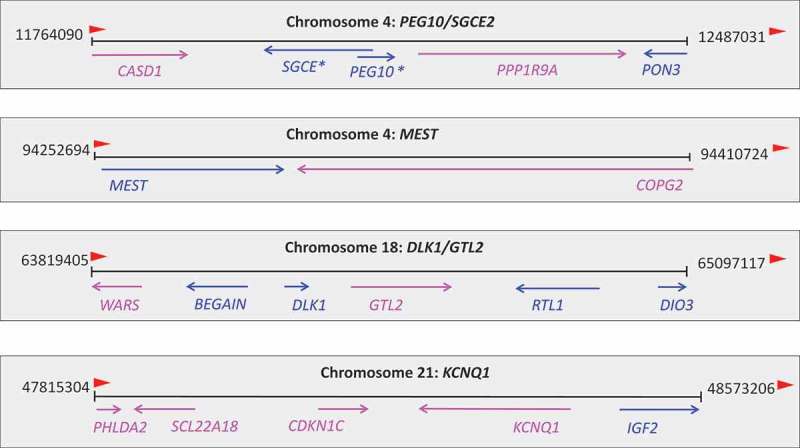
Genes and their parental expression patterns in the four imprinted clusters in sheep. Pink: maternally expressed; blue: paternally expressed. Arrow indicates the gene expression direction. *known imprinted in humans/mice.
Tissue-specific expression of imprinted genes in fetal organs
Imprinted genes have unique tissue- and developmental stage-specific expression patterns. Nearly all are established during fetal development [26,27]. Day 135 of gestation in sheep corresponds to the maximal fetal growth which allows ample tissue quantities [28]. To be conservative and avoid false positives, we did not intend to identify genes imprinted in some but not in other tissues in this study. We did, however, determine the tissue-specific expression levels of imprinted genes in control (Con) fetuses (Figure 7), overfed (Over) and restricted (Res) fetuses (Figure S2, Table S4.2).
Figure 7.
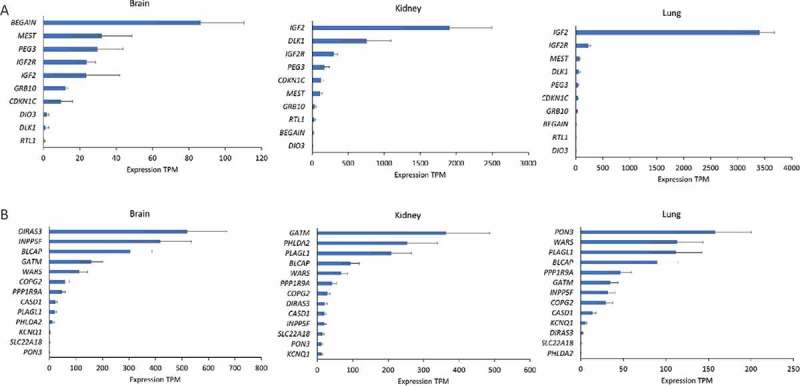
Expression levels (transcripts per million, TPM ± SD) of sheep imprinted genes in the brain, kidney, and lung tissue of day 135 ovine fetuses from ewes fed a control diet. (a) The 10 coding and previously known imprinted genes in the sheep genome. (b) The 13 putative imprinted genes identified in our study.
Expression levels [transcripts per million (TPM); Table S4] of sheep known (Figure 7(a)) and putative coding imprinted (Figure 7(b)) genes in the brain, kidney, and lung of fetuses from mothers of control diet exhibited tissue specificity. The fetal mitogen IGF2, for example, was expressed at the highest level in the lung and kidney among all imprinted genes. The genes DLK1 and GATM in the kidney, DIRAS3, INPP5F, and BLCAP in the brain were also among the highest expressed. While the tissue-specific expression of imprinted genes [29] have been reported previously in various species, they were mostly conducted using real time PCR which only gives relative values, while TPM from RNA-seq provides a close estimate to the absolute expression values after correcting for transcriptome size and gene length, allowing the visualization of expression differences among different genes across samples.
Effects of maternal nutrition on expression of imprinted genes in ovine fetuses
We compared the allelic expression of the 18 imprinted genes in fetal organs from the three maternal nutrition groups (Table 1). Although not all 18 had informative SNPs or expression values in all groups, no allelic expression reversal was observed in any fetal organ under any nutrition status. However, maternal nutrition did affect the levels of expression of the imprinted genes DIRAS3, IGF2, PHLDA2, and SLC22A18, in fetal organs (Table 2 and Figure S2). Specifically, the paternally expressed IGF2 gene, which promotes fetal growth [30], was downregulated in the brain of fetuses from mothers of restricted diet (Res) compared to controls (Con). The maternally expressed PHLDA2 was also downregulated in fetal brains but upregulated in fetal lungs of both Res and overfed (Over) groups compared to Con. This gene has been shown to be involved in placental growth [31] and its overexpression led to low birth weight in humans [32]. The paternally expressed DIRAS3 and maternally expressed SLC22A18 are inhibitors for cell proliferation and growth [33,34], and were both downregulated in lungs of the Res group and kidneys of the Over group compared to controls, respectively. Interestingly, three of the four affected genes – PHLDA2, SLC22A18, and IGF2 – are located near the imprinted cluster of KCNQ1 and IGF2/H19 on chromosome 21, indicating this domain is highly responsive to maternal diet changes.
Table 2.
Levels of differentially expressed imprinted genes in tissues of fetal sheep from mothers of different nutrition.
| Tissue | Genes | Control (TPM) | Overfed (TPM) | Log2 FC* | Restricted (TPM) | Log2 FC* |
|---|---|---|---|---|---|---|
| Brain | PHLDA2 | 19.36 | 0.44 | −1.71 | 0.47 | −6.32 |
| Brain | IGF2 | 23.61 | 38.3 | 0.69 | 5.22 | −2.07 |
| Kidney | SLC22A18 | 16.40 | 9.55 | −1.24 | 10.56 | −0.64 |
| Lung | PHLDA2 | 0.19 | 8.28 | 6.58 | 1.31 | 2.23 |
| Lung | DIRAS3 | 3.06 | 2.37 | −0.37 | 1.28 | −1.11 |
FC*: fold change over the expression levels in controls. Log2 FC: calculated by using bootstrapping and considered significant if greater than 1.
Discussion
Genomic imprinting in sheep is an underdeveloped area of research, despite the importance of the sheep in agriculture in many regions of the world and its frequent use as a model for human pregnancy and fetal development [35]. Our study is the first to employ NGS and bioinformatics to identify sheep imprinted genes and the effects of maternal diets on fetal imprinting. We identified 80 genes that consistently monoallelically expressed the same parental allele more than the other in all fetal tissues from all treatment groups. These 80 contain potential candidate imprinted genes in the sheep for future studies. Recent NGS studies identified more than 1,300 imprinted loci in mouse brain [36,37]; however, most were due to false positives [18]. To avoid such problems, we used the combined list of imprinted genes from all studied species to conservatively guide our data-mining. This approach, however, does not permit us to discover genes that are only imprinted in the ovine. Nonetheless, the list is the most comprehensive by combining information from five species and the conservative method generated five sheep known and 13 new putative imprinted genes, increasing the prior list of 21 by as much as 62%. Our results demonstrate the power of bioinformatics in genomic imprinting studies.
Imprinted domains
A unique feature of genomic imprinting is that imprinted genes tend to cluster as a result of long-range regulation by the imprinting control regions [1]. In sheep, the previously identified 21 imprinted genes are mostly clustered on chromosomes 18 and 21. From the 13 new putative imprinted genes, we identified a new sheep imprinted cluster on chromosome 4 (Figure 6), known as the PEG10/SGCE domain in humans [38], mice [39], and bovine [11]. Unfortunately, we did not have informative SNPs to study the expression of the two core genes, PEG10/SGCE, in this domain. Putative new imprinted genes CASD1, PPP1R9A, and PON3 were located in this large imprinting domain. The imprinted status of these three genes was supported by many informative SNPs from multiple tissues and animals in our data, strong evidence for their parental expression bias. All three are maternally expressed in mice [40] and located adjacent to the paternally expressed SGCE and PEG10 [23]. CASD1, like other maternally expressed genes in this domain, such as CALAR, is highly expressed in the brain and encodes for a glycosyl transferase [40]. However, in humans, CASD1 and PON3 are biallelically expressed [24], and PPP1R9A is imprinted in skeletal muscle but not in the brain [25]. The data in sheep are more similar to those in the mouse [40]. Although CASD1 was expressed in all three tissues studied (Figure 7), only the kidney had sufficient read counts at this informative SNP for the determination of maternal allele expression. This pattern of expression was also confirmed by Sanger sequencing. PPP1R9A is important for early development of extraembryonic tissues [25] and is similarly expressed in all tissues examined (Figure 7). PON3 belongs to an enzyme family associated with high-density lipoprotein that is believed to protect against the onset of atherogenesis [23]. Although it was nearly negligible in the brain and kidney, it was highly expressed in the lung in ovine fetuses (Figure 7). Such expression was also observed in human tissues [41].
COPG2 was located in the MEST cluster on chromosome 4 (Figure 6), which was previously known as the MEST/COPG2 imprinted domain in humans and mice [42]. The maternal allele of COPG2 gene is expressed in mice but the human COPG2 escapes genome imprinting although it is adjacent to the MEST gene [43]. In bovine, COPG2 was found to biallelically express in fetal tissues [44]. However, in our analysis of ovine fetuses, COPG2 showed preferential expression from the maternal allele in both brain and lung. Such lack of conservation of genomic imprinting in closely related species may lead to challenges and modification of the currently most plausible imprinting hypothesis,‘the parental conflict hypothesis’ [44], because not all imprinted genes fit in this model.
Our analysis placed the gene WARS near the DLK1/GTL2 imprinting domain on chromosome 18 (Figure 6), which also contains the widely-studied sheep Callipyge (CLPG) locus [45], expressed from the dominant paternal allele [46]. Six other imprinted genes have been identified in this region: paternally expressed DLK1, DAT, and RTL1 (also known as PEG11), and maternally expressed GTL2 (also known as MEG3), PEG11AS, and MEG8. Another paternally expressed gene BEGAIN, albeit located 138 kb proximally from the imprinted DLK1 gene, is not controlled by the ICR of the DLK1/GTL2 domain [47]. Paternally expressed WARS is located 150 kb downstream of BEGAIN in sheep and encodes a protein linking amino acids with nucleotide triplets in tRNA. It is believed to be one of the first proteins that appeared in evolution [provided by RefSeq, Jul 2008]. In the mouse, WARS is also paternally expressed [18]. However, it may not be controlled by the DLK1/GTL2 LRCE in the sheep due to its relative location to the gene BEGAIN (Figure 6).
Three other putative imprinted genes, SCL22A18, PHLDA2, and KCNQ1, are located on chromosome 21 (Figure 6) in the KCNQ1 domain, which contains several maternally expressed genes [48,49]. In our analysis, these three ovine genes also showed preferential expression from the maternal allele. The region is highly involved in fetal growth regulation [30] and was found to be affected by maternal diet in our study. Although members of this cluster are subjected to regulation by the same ICR, their expression levels varied dramatically. For example, PHLDA2 situates close to the KCNQ1 gene, yet they had the highest and lowest expression levels, respectively, in the kidney among all imprinted genes (Figure 7). This may suggest that allelic expression pattern and overall gene expression levels are regulated by different mechanisms.
Effects of maternal diets on expression of imprinted gene
Maternal stressors induce changes in expression of the fetal genome, which can permanently alter the offspring’s physiology, development, metabolism, and growth [50], as reported in mice and rats [51,52]. Understanding the effect of poor maternal nutrition in ovine fetal development is not only relevant to agriculture [53], but also to modeling for human pregnancy and fetal development. Restricted- and over-feeding of pregnant ewes were found to alter gene expression [28]. As seasonal breeders, ewes enter pregnancy in late fall or early winter, and usually have sufficient food in both quantity and quality. Nevertheless, during late gestation, when fetal growth is the most rapid, food becomes scarce, leading to nutrition restriction. Alternatively, the practice of flushing before and during the breeding season can result in overfeeding [50]. Such nutrient imbalance has been shown to severely impair fetal and placental development [50]. Interestingly, we found most of the disturbed gene expression were located in the KCNQ1 and IGF2/H19 clusters, consistent with the role of IGF2 as a major fetal growth regulator [54]. Diets of pregnant ewes containing different starch/fiber/protein portions have been shown to change the CpG methylation levels of specific imprinted genes such as IGF2R and H19 [20]. Our data also showed that despite the dramatic maternal diet changes, the allelic expression pattern was not affected, further suggesting that gene expression levels and imprinted patterns may be regulated through different epigenetic mechanisms.
Recommendations for future throughput imprinting studies
First, in order to avoid false positives raised by potential allelic drop-outs during amplification-based RNA-seq library preparation [55], we conservatively removed heterozygous SNPs between nucleotides that were not complementary even though parents were homozygous for the nucleotides (e.g., GG in fetus and TT in ram, SNP2 in Figure S1). This filtering reduced the numbers of informative SNP per gene. It is suggested that a non-amplification based hybrid capture NGS may circumvent this issue in library preparation [55]. Moreover, a number of informative SNPs were located too close to the edge of exons, making it difficult to design PCR primers. Consequently, validating them by Sanger sequencing proved difficult. Under this circumstance, new animals with different informative SNPs can be used. Allelic drop-outs may also affect PCR of Sanger sequencing. Digital Droplet PCR for absolute quantification of target informative SNPs may avoid this problem [56].
Second, a number of SNPs that were heterozygous in fetuses were also heterozygous in the parents (e.g., CA in fetus and CA in ram, SNP3 in Figure S1). These SNPs were not informative which reduced the number of informative SNPs and partially caused the lack of confirmation of the 6 known imprinted genes in the sheep genome. To alleviate this problem, more animals with different genetic background are required, as well as a better version of sheep genome annotation.
Third, most studies to discover imprinted genes employ reciprocal crosses between two closely related strains/breeds/species. While this design generates high frequencies of informative SNPs, parental allelic expression may be caused by species differences, not just imprinting. To overcome this problem, increasing the number of crosses from animals of the same species is the most relevant and preferred design.
Fourth, cis-expression quantitative trait loci (eQTL) confers monoallelic expression in all crosses. Even using known mammalian imprinted genes as a guide, we cannot rule out that the putative imprinted genes may contain eQTL. On the other hand, the list of 80 monoallelically expressed genes likely contain more imprinted genes in addition to eQTL because they consistently expressed the same parental allele among several different crosses. Nonetheless, reciprocal crosses are necessary to firmly distinguish these two types of monoallelic expression.
Lastly, in the original experimental design, we included the fetal cotyledons because many genes are only imprinted in the placenta. However, cotyledon samples are mostly contaminated with caruncles [57]. We also found similar cross contaminations in our samples and were not able to include them in our study. Therefore, other strategies such as microdissection or single-cell RNA-seq have to be used in order to reliably study placental imprinting.
Materials and methods
Tissue sample collection
All animal protocols [58,59] were reviewed and approved by the University of Connecticut Institutional Animal Care and Use Committee. Animal breeding, feeding, and sample collection were described in Pillai et al., 2017. Briefly, Western white-faced ewes (n = 12) were mated with Dorset rams (n = 4). Ewes were individually housed beginning 20 days after mating. Pregnancy was confirmed by ultrasound at day 28.5 ± 0.4 of gestation [58] if a ewe was not re-marked by a ram; day 0 represents the initial marking of the ewe by the ram. On day 30 of gestation, pregnant ewes were randomly assigned to control 100% (Con), restricted 60% (Res) or overfed 140% (Over) based on the National Research Council (NRC) total digestible nutrients (TDN) for ewes pregnant with twins. Ewes were euthanized at day 135 of gestation (n = 4 per diet), and 15 fetuses were used (Con: n = 7, including 3 sets of twins; Res: n = 4; Over: n = 4). Brain, kidney and lung samples were collected from all fetuses. Whole-blood samples were obtained from the four rams. Tissues were flash frozen in liquid nitrogen and were stored at −80°C until RNA extraction.
Whole-genome DNA- and RNA-sequencing
Genomic DNA of ram whole blood samples and fetal tissues were isolated using Qiagen DNeasy Blood & Tissue Kits (Qiagen, 69,504). The ram DNA was sent to Novogene (Novogene Co., Ltd.) for library preparation and sequencing. In brief, the DNA-seq library was prepared using the Illumina Truseq Nano DNA HT sample preparation kit (Illumina, FC-121–4003) with a 350 bp target insert size. Libraries were sequenced with 2 × 150 bp paired-end reads on HiSeq 2000 platform (Illumina). On average, 186.7 million raw read pairs were obtained for genotyping from each ram.
Total RNA was extracted from day 135 fetal brain, lung and kidney, using Trizol and RNAeasy kit (Qiagen, 74,104) with three quality controls: NanoDrop (Thermo Fisher Scientific), agarose gel electrophoresis and Qubit 2.0 (Thermo Fisher). Library preparation was carried out using TruSeq RNA library prep kit (Illumina, RS-122–2001, RS-122–2002), which selected mRNA using Oligo d(T) with magnetic beads and built 2 × 75 bp paired-end cDNA libraries. The libraries were quantified using real-time PCR. Agilent 2100 Bioanalyzer (Agilent) was used to assess the size distribution and to determine the RNA integrity number (RIN) in each sample (Table S5). All RNA samples for sequencing had the RIN value greater or equal to 7. Overall, we obtained 2,149 million raw sequencing reads that passed filtering from three sequencing runs of 45 fetal tissue samples. A total of 1,160, 576 and 413 million raw sequencing reads that passed filtering were obtained for sequencing runs 1, 2 and 3, respectively. An average of 23.8 million read pairs per sample was generated on a NextSeq 500 System (Illumina).
SNP calling from DNA- and RNA-seq data
We adapted the computational pipeline from the SNPiR [single nucleotide polymorphisms (SNPs) in RNA-seq data] [11,60] to solve several technical challenges in the identification of monoallelically expressed genes from RNA-seq data. Among those challenges are alignment bias of RNA-seq reads and filtering potential false positive SNPs. Heterozygosity can increase mapping bias because a read from the non-reference allele is considered a mismatch, resulting in a low mapping rate [7]. To minimize such alignment bias to the reference allele in the genome, we artificially built a pseudo-genome (named ‘alternative genome’) by flipping the reference/alternative alleles in all SNP sites based on known sheep dbSNP (sheep 9940). Raw genomic DNA-seq reads were trimmed by Trimmomatic (version 0.33) [61] to remove the universal sequencing adaptors of Illumina with a minimum Phred score of 20 and minimal length of 30 bp. We then mapped the filtered DNA-seq reads using Hisat2 aligner (version 2.0.5) [62] to both sheep reference genome Oar_v4.0 and the alternative genome. Only uniquely aligned reads were kept. The mapped reads and mapping rates of rams and fetuses in the two genomes were summarized in Table S6. The Picard Tool Mark Duplicates (2.12.0) [63] was used to remove the PCR duplicates. SNVQ (NGS Tools version 2.0.0) [64] was used to accurately detect the SNPs in the ram genome. To reduce potential false positive calls, the following parameters were used for SNP filtering (Figure 1): i) a minimum quality score of 50 at the SNP position, ii) a minimum of three reads aligned at the SNP using both the reference and alternative genomes, iii) reversed genotypes of called SNP when reference/alternative genomes were switched (e.g., A/G in reference genome; while G/A in alternative genome), iv) SNP present and consistent with that in the sheep dbSNP (sheep 9940) database, and v) SNP located in an exon.
Methods similar to the DNA-seq analysis for trimming, mapping and duplication removal were used for the RNA-seq data. The SNPs in fetuses were called at the individual fetus level, i.e., RNA-seq reads in the three tissue samples (brain, kidney and lung) of the same fetus were pooled to increase read coverage at each SNP site. The same SNP filtering criteria were applied as in the ram DNA-seq data.
Identification of informative SNPs
After genotyping SNPs of both the rams and their fetuses, we designated SNPs that were homozygous in the rams but heterozygous in their respective fetuses as informative SNPs (SNP1 in Figure S1). We then assigned the reads to the two parental alleles using Samtools mpileup (version 1.4) [21] and averaged the allele-specific read counts using the reference/alternative genomes.
Differential allele-specific gene expression and statistical analysis
When calculating the allele-specific gene expression (Figure 1), we only used informative SNPs that had total read counts of 20 or greater from each of the two parental alleles. This is because low read coverage may have a large variance in differential allelic expression estimation, potentially generating false positive differences [7]. We then identified the expressed parental allele of all SNPs in the same gene and removed genes which contained discordant parental allele expression (i.e., a mixture of maternally and paternally expressed informative SNPs in the same gene). Next, we aggregated the allele-specific reads for all informative SNPs in each gene to increase the sensitivity of imprinted gene prediction, as previously suggested [11], and pooled allelic expression of the same gene in biological replicates. The Fisher’s exact test was used to examine if an allele was expressed by more than 70% of total read counts from both alleles combined with a false discovery rate (FDR) ≤ 0.05.
Mammalian imprinted gene lists
The known mammalian (human, mouse, bovine, sheep, and pig) imprinted genes were obtained from three well-defined databases, including Imprinted Gene Database, (http://www.geneimprint.com/site/genes-by-species). Catalogue of Parent of Origin Effects (http://igc.otago.ac.nz/Search.html), and Mouse Book Database (http://www.mousebook.org/imprinting-gene-list). Additionally, we incorporated 18 novel imprinted genes identified recently in mice [8] and 23 in bovine [11] to create a more current and comprehensive list of imprinted genes (Table S2). This list was used to limit the number of imprinted genes found in the sheep.
Differential gene expression analysis across maternal diets
RNA-seq reads from 15 fetuses (Con: n = 7; Res: n = 4; Over: n = 4) were trimmed and aligned to Oar_v4.0 using Hisat2 version 2.0.5 aligner [62]. The percentages of mapped reads for all samples are summarized in Table S7 and the average multiple aligned rate is 90.3%. IsoEM version 1.1.5 [65] was used to quantify levels of gene expression to transcripts per kilobase million (TPM) using default parameters. TPM normalizes for gene length first and then for sequencing depth. This unit was preferred to RPKM because it normalizes transcriptome sizes. When comparing levels of gene expression across different samples, TPM allows more appropriate comparisons [66]. Differentially expressed genes (DEGs) between Con and Over or Con and Res were determined using IsoDE version 2 [67]. The test was preformed separately in brain, kidney, and lung. In each comparison, genes were deemed differentially expressed if they showed a P value <0.05 and confident log2 fold change (FC) >1. DEGs that are in the lists of sheep known/putative imprinted genes (Table 2), the 80 monoallelically expressed genes (Table S8.1), and the mammalian known imprinted genes (Table S8.2) were subsequently pulled from the total DEG list.
Sanger sequencing
The DNA of fetuses and their respective rams and the cDNA of the specific fetal tissue in which the gene was expressed monoallelically were all amplified by PCR. All primers used are in Table S9. The PCR products were sent to Eton Bioscience for Sanger sequencing.
Data access
The raw read FASTQ files for DNA/RNA-seq reads and informative SNP averaged read count files are available at Gene Expression Omnibus (GEO https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE111306.
Funding Statement
This work was supported by the USDA -ARS grant: 1265-31000-091-02S, the USDA Multi-state regional grant: W2171/3171, National Institute of Food and Agriculture, U.S. Department of Agriculture, under award number 2013–01919, Department of Education of Xinjiang Uygur Autonomous Region Scholarship: 2016-3-0036 and Studying Abroad program for Excellent Ph.D. Students of Guangxi Zhuang Autonomous Region: 2014-2.
Acknowledgments
The authors thank Zoetis for donating the controlled intravaginal drug release devices (CIDRs) used for estrus synchronization, Dr. Kanokwan Srirattana for help with PCR work, and the UConn Livestock staff, Dr. Thomas Hoagland, Victor Delaire and the animal science undergraduate students for the animal care during this experiment.
Disclosure statement
No potential conflict of interest was reported by the authors.
Ethics approval
All animal protocols were reviewed and approved by the University of Connecticut Institutional Animal Care and Use Committee.
Authors’ contributions
XCT, JD and ZJ designed the study; AJ, SP, MH, KM, SR, KG and SZ performed animal breeding, feeding and care; necropsy, and sample collection; KF, MZ, JD, EJ and HJ performed the experiment; JD, SS and IM analyzed the data; and JD and XCT wrote the original manuscript. AJ, MH, KF, KM, EJ and SR review & editing the manuscript. All authors read and approved the final manuscript.
Supplementary material
Supplemental data can be accessed here
References
- 1.Bartolomei MS, Ferguson-Smith AC.. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol. 2011;3(7):a002592. doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Doherty AM, MacHugh DE, Spillane C, et al. Genomic imprinting effects on complex traits in domesticated animal species. Front Genet. 2015;6:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koerner MV, Pauler FM, Huang R, et al. The function of non-coding RNAs in genomic imprinting. Dev Camb Engl. 2009;136:1771–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delaval K, Feil R. Epigenetic regulation of mammalian genomic imprinting. Curr Opin Genet Dev. 2004;14:188–195. [DOI] [PubMed] [Google Scholar]
- 5.Wilkins JF, Haig D. What good is genomic imprinting: the function of parent-specific gene expression. Nat Rev Genet. 2003;4:359–368. [DOI] [PubMed] [Google Scholar]
- 6.Tian X.(Cindy). Bovine Epigenetics and Epigenomics In: James E. Womack, editor. Bovine Genomics. John Wiley & Sons, Inc; 2012. p. 144–168. doi: 10.1002/9781118301739.ch11. [DOI] [Google Scholar]
- 7.Wang X, Clark AG. Using next-generation RNA sequencing to identify imprinted genes. Heredity. 2014;113:156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andergassen D, Dotter CP, Wenzel D, et al. Mapping the mouse Allelome reveals tissue-specific regulation of allelic expression. eLife. 2017;6:e25125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blake A, Pickford K, Greenaway S, et al. MouseBook: an integrated portal of mouse resources. Nucleic Acids Res. 2010;38:D593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morison IM, Paton CJ, Cleverley SD. The imprinted gene and parent-of-origin effect database. Nucleic Acids Res. 2001;29:275–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Z, Hagen DE, Wang J, et al. Global assessment of imprinted gene expression in the bovine conceptus by next generation sequencing. Epigenetics. 2016;11:501–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bischoff SR, Tsai S, Hardison N, et al. Characterization of conserved and nonconserved imprinted genes in swine. Biol Reprod. 2009;81:906–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei Y, Su J, Liu H, et al. MetaImprint: an information repository of mammalian imprinted genes. Dev Camb Engl. 2014;141:2516–2523. [DOI] [PubMed] [Google Scholar]
- 14.Hanna CW, Kelsey G. The specification of imprints in mammals. Heredity. 2014;113:176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barbaux S, Gascoin-Lachambre G, Buffat C, et al. A genome-wide approach reveals novel imprinted genes expressed in the human placenta. Epigenetics. 2012;7:1079–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luedi PP, Dietrich FS, Weidman JR, et al. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007;17:1723–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Soloway PD, Clark AG. A survey for novel imprinted genes in the mouse placenta by mRNA-seq. Genetics. 2011;189:109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeVeale B, Van Der Kooy D, Babak T. Critical evaluation of imprinted gene expression by RNA-Seq: a new perspective. PLoS Genet. 2012;8:e1002600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffman ML, Reed SA, Pillai SM, et al. Physiology and endocrinology symposium: the effects of poor maternal nutrition during gestation on offspring postnatal growth and metabolism. J Anim Sci 2017; 95:2222–2232. [DOI] [PubMed] [Google Scholar]
- 20.Lan X, Cretney EC, Kropp J, et al. Maternal diet during pregnancy induces gene expression and DNA methylation changes in fetal tissues in sheep. Front Genet. 2013;4:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinforma Oxf Engl. 2011;27:2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ono R, Shiura H, Aburatani H, et al. Identification of a large novel imprinted gene cluster on mouse proximal chromosome 6. Genome Res. 2003;13:1696–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monk D, Wagschal A, Arnaud P, et al. Comparative analysis of human chromosome 7q21 and mouse proximal chromosome 6 reveals a placental-specific imprinted gene, TFPI2/Tfpi2, which requires EHMT2 and EED for allelic-silencing. Genome Res. 2008;18:1270–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakabayashi K, Makino S, Minagawa S, et al. Genomic imprinting of PPP1R9A encoding neurabin I in skeletal muscle and extra-embryonic tissues. J Med Genet. 2004;41:601–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Babak T, DeVeale B, Tsang EK, et al. Genetic conflict reflected in tissue-specific maps of genomic imprinting in human and mouse. Nat Genet. 2015;47:544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thurston A, Taylor J, Gardner J, et al. Monoallelic expression of nine imprinted genes in the sheep embryo occurs after the blastocyst stage. Reprod Camb Engl. 2008;135:29–40. [DOI] [PubMed] [Google Scholar]
- 28.Peñagaricano F, Wang X, Rosa GJ, et al. Maternal nutrition induces gene expression changes in fetal muscle and adipose tissues in sheep. BMC Genomics. 2014;15:1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baran Y, Subramaniam M, Biton A, et al. The landscape of genomic imprinting across diverse adult human tissues. Genome Res. 2015;25:927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reik W, Constancia M, Dean W, et al. imprinting in development and disease. Int J Dev Biol. 2000;44:145–150. [PubMed] [Google Scholar]
- 31.Frost JM, Moore GE. The importance of imprinting in the human placenta. PLoS Genet. 2010;6:e1001015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis RM, Cleal JK, Ntani G, et al. Relationship between placental expression of the imprinted PHLDA2 gene, intrauterine skeletal growth and childhood bone mass. Bone. 2012;50:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang S, Chang IS, Lin W, et al. ARHI (DIRAS3), an imprinted tumour suppressor gene, binds to importins and blocks nuclear import of cargo proteins. Biosci Rep. 2009;30:159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang B, Liu T, Wu T, et al. microRNA-137 functions as a tumor suppressor in human non-small cell lung cancer by targeting SLC22A18. Int J Biol Macromol. 2015;74:111–118. [DOI] [PubMed] [Google Scholar]
- 35.Barry JS, Anthony RV. The pregnant sheep as a model for human pregnancy. Theriogenology. 2008;69:55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gregg C, Zhang J, Weissbourd B, et al. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science. 2010;329:643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gregg C, Zhang J, Butler JE, et al. Sex-specific parent-of-origin allelic expression in the mouse brain. Science. 2010;329:682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kainz B, Shehata M, Bilban M, et al. Overexpression of the paternally expressed gene 10 (PEG10) from the imprinted locus on chromosome 7q21 in high-risk B-cell chronic lymphocytic leukemia. Int J Cancer. 2007;121:1984–1993. [DOI] [PubMed] [Google Scholar]
- 39.Wiley CD, Matundan HH, Duselis AR, Isaacs AT, Vrana PB Patterns of hybrid loss of imprinting reveal tissue- and cluster-specific regulation. PloS One. 2008;3:e3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Babak T, Deveale B, Armour C, et al. Global survey of genomic imprinting by transcriptome sequencing. Curr Biol CB. 2008;18:1735–1741. [DOI] [PubMed] [Google Scholar]
- 41.Fagerberg L, Hallström BM, Oksvold P, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics MCP. 2014;13:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee YJ, Park CW, Hahn Y, et al. Mit1/Lb9 and Copg2, new members of mouse imprinted genes closely linked to Peg1/Mest(1). FEBS Lett. 2000;472:230–234. [DOI] [PubMed] [Google Scholar]
- 43.Yamasaki K, Hayashida S, Miura K, et al. The novel gene, gamma2-COP (COPG2), in the 7q32 imprinted domain escapes genomic imprinting. Genomics. 2000;68:330–335. [DOI] [PubMed] [Google Scholar]
- 44.The KH. COPG2, DCN, and SDHD genes are biallelically expressed in cattle. Mamm Genome Off J Int Mamm Genome Soc. 2005;16:545–552. [DOI] [PubMed] [Google Scholar]
- 45.Jiang C, Characterization YZ, Status I. Tissue distribution of porcine GTL2 gene. Agric Sci China. 2009;8:216–222. [Google Scholar]
- 46.Freking BA, Murphy SK, Wylie AA, et al. Identification of the single base change causing the callipyge muscle hypertrophy phenotype, the only known example of polar overdominance in mammals. Genome Res. 2002;12:1496–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smit MA, Tordoir X, Gyapay G, et al. BEGAIN: a novel imprinted gene that generates paternally expressed transcripts in a tissue- and promoter-specific manner in sheep. Mamm Genome Off J Int Mamm Genome Soc. 2005;16:801–814. [DOI] [PubMed] [Google Scholar]
- 48.Lewis A, Green K, Dawson C, et al. Epigenetic dynamics of the Kcnq1 imprinted domain in the early embryo. Dev Camb Engl. 2006;133:4203–4210. [DOI] [PubMed] [Google Scholar]
- 49.Umlauf D, Goto Y, Cao R, et al. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of polycomb group complexes. Nat Genet. 2004;36:1296–1300. [DOI] [PubMed] [Google Scholar]
- 50.Wu G, Bazer FW, Wallace JM, et al. Board-invited review: intrauterine growth retardation: implications for the animal sciences. J Anim Sci. 2006;84:2316–2337. [DOI] [PubMed] [Google Scholar]
- 51.Cooney CA, Dave AA, Wolff GL. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr. 2002;132:2393S–2400S. [DOI] [PubMed] [Google Scholar]
- 52.Waterland RA, Travisano M, Tahiliani KG, et al. Methyl donor supplementation prevents transgenerational amplification of obesity. Int J Obes. 2005. 2008;32:1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Begum G, Stevens A, Smith EB, et al. Epigenetic changes in fetal hypothalamic energy regulating pathways are associated with maternal undernutrition and twinning. FASEB J. 2012;26:1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Demetriou C, Abu-Amero S, Thomas AC, et al. Paternally expressed, imprinted insulin-like growth factor-2 in chorionic villi correlates significantly with birth weight. PLoS One. 2014;9:e85454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jennings LJ, Arcila ME, Corless C, et al. Guidelines for validation of next-generation sequencing–based oncology panels: a joint consensus recommendation of the association for molecular pathology and college of american pathologists. J Mol Diagn. 2017;19:341–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gutiérrez-Aguirre I, Rački N, Dreo T, et al. Droplet digital PCR for absolute quantification of pathogens. Methods Mol Biol Clifton NJ. 2015;1302:331–347. [DOI] [PubMed] [Google Scholar]
- 57.Bridger PS, Haupt S, Klisch K, et al. Validation of primary epitheloid cell cultures isolated from bovine placental caruncles and cotyledons. Theriogenology. 2007;68:592–603. [DOI] [PubMed] [Google Scholar]
- 58.Jones AK, Gately RE, McFadden KK, et al. Transabdominal ultrasound for detection of pregnancy, fetal and placental landmarks, and fetal age before day 45 of gestation in the sheep. Theriogenology. 2016;85:939–945.e1. [DOI] [PubMed] [Google Scholar]
- 59.Pillai SM, Jones AK, Hoffman ML, et al. Fetal and organ development at gestational days 45, 90, 135 and at birth of lambs exposed to under- or over-nutrition during gestation. Transl Anim Sci. 2017;1:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Piskol R, Ramaswami G, Li JB. Reliable identification of genomic variants from RNA-seq data. Am J Hum Genet. 2013;93:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma Oxf Engl. 2014;30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pertea M, Kim D, Pertea GM, et al. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11:1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Broad Institute. Picard tools - by broad institute [Internet]. Available from: http://broadinstitute.github.io/picard/
- 64.Duitama J, Srivastava PK, Măndoiu II. Towards accurate detection and genotyping of expressed variants from whole transcriptome sequencing data. BMC Genomics. 2012;13:S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nicolae M, Mangul S, Măndoiu II, et al. Estimation of alternative splicing isoform frequencies from RNA-Seq data. Algorithms Mol Biol AMB. 2011;6:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research. 2015;4:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Al Seesi S, Tiagueu YT, Zelikovsky A, et al. Bootstrap-based differential gene expression analysis for RNA-Seq data with and without replicates. BMC Genomics. 2014;15(Suppl 8):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.