

Abstract
Consanguineous populations of the Arabian Peninsula have been underrepresented in global efforts that catalogue human exome variability. We sequenced 291 whole exomes of unrelated, healthy native Arab individuals from Kuwait to a median coverage of 45X and characterised 170,508 single-nucleotide variants (SNVs), of which 21.7% were ‘personal’. Up to 12% of the SNVs were novel and 36% were population-specific. Half of the SNVs were rare and 54% were missense variants. The study complemented the Greater Middle East Variome by way of reporting many additional Arabian exome variants. The study corroborated Kuwaiti population genetic substructures previously derived using genome-wide genotype data and illustrated the genetic relatedness among Kuwaiti population subgroups, Middle Eastern, European and Ashkenazi Jewish populations. The study mapped 112 rare and frequent functional variants relating to pharmacogenomics and disorders (recessive and common) to the phenotypic characteristics of Arab population. Comparative allele frequency data and carrier distributions of known Arab mutations for 23 disorders seen among Arabs, of putative OMIM-listed causal mutations for 12 disorders observed among Arabs but not yet characterized for genetic basis in Arabs, and of 17 additional putative mutations for disorders characterized for genetic basis in Arab populations are presented for testing in future Arab studies.
Introduction
Characterising the patterns of genetic variation within and among human populations is crucial to understand human evolutionary history and the genetic basis of disorders1. Many global genome-wide genotyping and whole-genome sequencing studies (such as the Human Genome Diversity Project1,2, the 1000 Genomes Project (1KGP)3,4 and the UK10K project5) have been undertaken to catalogue genetic variation. Coding exonic regions, though estimated to encompass only approximately 1–2% of the genome, harbour the most functional variation and contain almost 85% of the known disease-causing pathogenic variants6,7; therefore, several global whole-exome sequencing studies have also been undertaken8–10. Such large-scale global projects have revealed that human populations harbour a large amount of rare variations which exhibit little homology between diverged populations3,9–17, Mendelian and rare genetic disorders are often associated with rare coding variants. Likewise, common markers associated with complex disorders too can vary in frequency across populations18. Considering that population-specific differences in allele frequencies are of clinical importance, it is fundamental to catalogue them in diverse ethnic populations19.
The Arabian Peninsula holds a strategic place in the early human migration routes out of Africa20–22. The Peninsula was instrumental in shaping the genetic map of current global populations because the first Eurasian populations were established here23. The ancestry of indigenous Arabs can largely be traced back to ancient lineages of the Arabian Peninsula23,24. The Arab population is heterogeneous but well-structured3,24–26. For example, the Kuwaiti population comprises three genetic subgroups, namely KWP (largely of West Asian ancestry representing Persians with European admixture), KWS (city-dwelling Saudi Arabian tribe ancestry) and KWB (tent-dwelling nomadic Bedouins characterised by the presence of 17% African ancestry)24. Further, the Qatari population also comprises similar subgroups with the third group displaying a much higher African ancestry25. The Greater Middle Eastern Variome study26 detected several ancient founder populations and continental & sub-regional admixture in the extended region of Greater Middle East (comprising the Gulf region, North Africa and Central Asia); the study further stated that the ancestral Arab population from Arabian Peninsula could be observed in nearly all of the GME regions possibly as a result of the Arab conquests in the seventh century.
Consanguinity in the Arab region has made the population vulnerable to a plague of recessive genetic disorders. An increased burden of runs of homozygosity has been observed in populations from Kuwait24 and the extended region of Greater Middle East26. An overwhelming proportion (63%) of the disorders documented in the Catalogue for Transmission Genetics in Arabs (CTGA)27 follows a recessive mode of inheritance28. Studying consanguineous populations lead to identifying causal mutations for Mendelian disorders29,30 and rare familial (monogenic) forms of common complex disorders31. These studies also paved the way to evaluate the role of consanguinity and environmental factors in complex lifestyle disorders, such as obesity and type 2 diabetes, cases of which are rapidly increasing in the Arabian Peninsula32,33. Thus, studying consanguineous populations is important to human medical genetics research26,34,35.
Despite consanguinity, diversity and admixture in its populations, the region is poorly represented in global genomic surveys. Even larger databases, such as the Exome Aggregation Consortium (ExAC)8 and the Genome Aggregation Database (gnomAD)8, are deficient in representing Middle Eastern populations. Although the Greater Middle East (GME) Variome project26 provides whole exome data of 1,111 individuals from six GME regions, the region of Arabian Peninsula is represented by only 214 samples and the sub-region of Kuwait by only 45 samples.
In our previous studies, we sequenced and analysed thirteen exomes from the KWS group36 and representative whole genomes from each of the three subgroups of the Kuwaiti population36–38. In this study, we extended the study by sequencing whole exomes of 291 native Kuwaiti Arab individuals representing the three population subgroups. We further analysed the data to infer the extent of exome variability in the Kuwaiti population and to delineate its impact on population substructures of Kuwait and medical genetics of the region.
Results
Exome variants discovered in the Kuwaiti population
The 291 exomes were sequenced to a median coverage of 45X, with an average of 80% of the target base pairs having at least 15X coverage. ‘Missingness’ rate (referring to the percent of samples where information was missing) of 1.8% was obtained leading to genotyping call rate of 98.2%. Totally, 173,849 (including 2,626 non-autosomal) variants were identified (Table 1 and Supplementary Table S1), 12.16% of which were novel. The call set included 170,508 single-nucleotide variants (SNVs) and 3,341 insertions and deletions (indels). 11.85% of the SNVs and 28% of the indels were novel. The observed aggregate transition/transversion (Ti:Tv) ratio of 3.22 was within the acceptable range for whole-exome sequencing variants39,40. A heterozygous to homozygous variant genotype ratio of 0.63 was obtained indicating that the population skews towards homozygosity with its inbreeding nature.
Table 1.
Statistics of variants observed in Kuwaiti exomes.
| All variants | Kuwaiti ‘population-specific’ variants@ | Kuwaiti ‘Population-specific’ variants seen in ≥2 individuals from the study cohort | ||||
|---|---|---|---|---|---|---|
| Number of variants | Average number of variants per individual | Number of variants | Average number of variants per individual | Number of variants | Average number of variants per individual | |
| Total variants (% novel) | 173849 (12.16) | 14767 | 57691 (36.63) | 365 | 9870 (34.73) | 189 |
| SNVs (% novel) | 170508 (11.85) | 14557 | 55644 (36.3) | 331 | 9429 (35.03) | 168 |
| Indels (% novel) | 3341 (28.07) | 210 | 2047 (45.72) | 34 | 441 (28.34) | 21 |
| Ti:Tv | 3.22 | 3.374 | 2.7 | 2.44 | 2.7 | 2.37 |
| ‘Personal’ SNVs# | 37044 | 129 | 37044 | 129 | 0 | 0 |
| Rare SNVs (excluding ‘personal’ variants) | 86446 | 908 | 17579 | 109 | 8408 | 74 |
| Low-frequency SNVs | 21277 | 1553 | 883 | 27 | 883 | 27 |
| Common SNVs | 25741 | 11969 | 138 | 67 | 138 | 67 |
| Missense SNVs$ | 91204 | 9187 | 33504 | 184 | 5559 | 87 |
| Synonymous SNVs$ | 70955 | 10385 | 19159 | 126 | 3377 | 70 |
| Stop gain SNVs$ | 1425 | 65 | 721 | 3 | 113 | 1 |
| Stop loss SNVs$ | 95 | 9 | 22 | 0 | 4 | 0 |
| LoF& SNVs$ | 1645 | 73 | 843 | 4.5 | 131 | 2 |
Ti:Tv, transition/transversion ratio.
@Variants from our cohort that were not seen in 1KGP were termed as ‘Kuwaiti population-specific’ variants.
#Personal SNVs are those that are observed only in a single exome from the study cohort and not seen in the data sets of 1KGP or GME. These are indeed “private mutations” and remain so until the mutations are observed in further exomes/genomes sequenced in future studies.
&Loss-of-function (LoF) variants represent the sum total of stop gain, stop loss, frameshift and splicing variants. LoF variants are expected to correlate with complete loss of function of the affected transcripts, including stop codon-introducing (nonsense) or splice site-disrupting single-nucleotide variants (SNVs), insertion/deletion (indel) variants predicted to disrupt a transcript’s reading frame or larger deletions removing either the first exon or >50% of the protein-coding sequence of the affected transcript.
$These were calculated using all the identified SNVs including the personal variants.
Validation of SNP calls
The validity of the SNP calls was confirmed by utilizing an in-house genome-wide genotype data set on 269 (of the sequenced 291) samples derived using the Illumina HumanOmniExpress BeadChip (Illumina Inc, USA). In an average, 13,175 variants could be compared per sample. The concordance rate of the SNP calls between the exome sequencing data and genome-wide genotype data was >99.7% (see Supplementary Table S2). The observed concordance rate in our study is on par with those reported in literature: Kenna et al.40 reported a genotype concordance rate of 98.9% on comparing the accuracy of genotypes inferred using Illumina high throughput sequencing platforms with genotypes ascertained using Illumina BeadChips. The disagreements in the SNP calls were seen more often with heterozygous SNPs than with homozygous SNPs. As is the practice41, we choose not to remove the inconsistent calls.
Principal component analysis of variants in the merged set of exome variants from Kuwait and global populations
The scatter plot of the first two principal components of the merged data set of exome variants from Kuwait, 1KGP global populations, and Qatar is presented in Fig. 1. The plot affirmed the heterogeneity of the Kuwaiti Arab population as comprising three substructures24 and inferred the regional affinity.
Figure 1.

Scatter plot of the first two principal components of the merged data set of exome variants from the three Kuwaiti substructures and from regional (Qatar) and 1KGP global populations.
Classifications of observed SNVs
50.7% of the identified SNVs were ‘rare’, 12.5% were ‘low-frequency’ and 15.1% were ‘common’. Up to 21.7% of the SNVs were ‘personal’ (found in only one Kuwaiti exome and not seen in the data sets of 1KGP Phase 3 and GME). Alternate allele was the major allele in 4.2% of the identified SNVs; 0.22% of the SNVs were fixed for the alternate allele, having a non-reference frequency of 100%. Among the identified SNVs, 53.5% were missense, 41.61% were synonymous and approximately 1% were loss-of-function (LoF). 55,644 of the identified SNVs were ‘population-specific’, 60% of which were missense; 9,429 of these 55,644 population-specific variants were polymorphic (seen in ≥2 exomes from the study cohort and not seen in 1KGP), most of which were ‘rare’ (8408 out of 9429); of the remaining 46,215 variants, 37,044 were ‘personal’ and 9171 were seen in one exome from the study cohort and were also seen in GME data set. On average, 14,557 SNVs and 210 indels were seen in every Kuwaiti individual. The average number of ‘personal variants’ per individual was 129. Population-specific missense variants per individual were more than synonymous changes (184 versus 126). The average number of LoF variants per Kuwaiti individual was 73, of which 4.5 were specific to the Kuwaiti population.
Homozygous LOF variants and “inactivated genes”
We had observed 1645 putative LoF SNVs (Table 1) in Kuwaiti exomes from 291 healthy individuals of Arab ethnicity. 186 of these 1645 LoF SNVs were homozygous and they were harbored in 179 genes (See Supplementary Table S3). Of the 186 homozygous LoF SNVs, 27 were with MAF <1% and another 9 were with MAF (≥1% and <2%). Sulem et al.42, by way of performing whole-genome sequencing of 2,636 Icelanders and chip-imputing a further 101,584 Icelanders, had identified a set of rare (MAF <2.0%) homozygous LoF variants in 1,171 genes. In a similar manner, the Exome Aggregation Consortium (ExAC) data set of 60,706 sequenced individuals identified 2,068 genes that were inactivated8; The GME26 consortium, by way of analyzing 354 exomes of healthy individuals, identified 301 genes with rare homozygous LoF variants of which 50 genes overlapped the Icelandic gene list and 94 overlapped the ExAC gene list of inactivated genes. Upon comparing the homozygous LoF variants from the Kuwaiti exomes with the above-mentioned data sets of inactivated genes, we found 23 genes (PNPLA1, ULBP3, OR8K3, RAD52, APOBEC1, PDIA2, WDR87, SIGLEC1, COL9A2, OTOF, SULTIC3, COQ2, MROH2B, FAM81B, UNC93A, DNAH11, PXDNL, OR4D10, SLC22A24, RNASE9, C17orf77, CARD14 and SLC5A4) in common with Icelandic data set, 3 genes (EML1, WWTR1 and PPFIA1) in common with ExAC data set and six genes (COL9A2, SLC5A9, FAM81B, GGT6, EFCAB13 and SLC5A4) in common with GME data set. Upon considering only those LoFs with <2% MAF in Kuwaiti exomes, the number of genes in common with Icelandic data got reduced to 8 - PNPLA1 (MAF_KWT of the LoF: 1.03), ULBP3 (0.52%), OR8K3 (1.5%), RAD52 (1.3%), APOBEC1 (0.34%), PDIA2 (1.3%), WDR87 (0.34%) and SIGLEC1 (0.34%)); upon considering only the rare (MAF <1%) homozygous LoF variants in Kuwaiti exomes, only one gene (EML1 (0.69%)) was seen in common with ExAC data set; and none with GME data set. GME work reported more genes as common with the Icelandic/ExAC data sets as they also considered indels along with SNVs to derive the list of LOFs while we considered only the SNVs.
Comparison with Greater Middle East (GME) Variome data
Results of comparing the variants observed in our study with those reported in GME populations26 are presented in Table 2. Up to 64% of the SNVs identified in our study were seen common with GME – the remaining 36% of variants not seen in GME are expected to enlarge the variome of the GME region. GME provided supporting evidence to designate up to 25% of Kuwaiti population-specific singleton mutations (seen in only one exome from the study cohort) as genuine SNVs. Up to as high as 58% of the population-specific polymorphic variants observed in Kuwaiti exomes were also seen in GME variome.
Table 2.
Comparing the Kuwaiti Arab whole-exome variants with Greater Middle East (GME) whole-exome Variome.
| SNV Category | Total observed in Kuwaiti Arabs | Present in GME variome | Present only in the “Arabian Peninsula” subregion of GME |
|---|---|---|---|
| All | 170508 | 109058 (64%) | 4351 |
| All:Personal | 37044 | 0 (0%) | 0 |
| Kuwaiti “Population-specific” | 55644 | 14660 (26.3%) | 2393 |
| Kuwaiti “Population-specific polymorphic” variants seen in ≥2 individuals from the study cohort | 9429 | 5474 (58%) | 793 |
Extent of variability in Kuwaiti exomes
In each of the categories of ‘all’, ‘known’, ‘novel’ and ‘Kuwaiti population-specific’ variants, the observed number of variants increased linearly with increasing number of sequenced exomes and did not reach a plateau (Fig. 2). A similar trend was observed when the three subgroups were examined individually (Supplementary Fig. S1). However, when the population-specific variants were divided into ‘personal’ and ‘population-specific polymorphic’ variants, population-specific variants shared by more than one individual reached a plateau.
Figure 2.
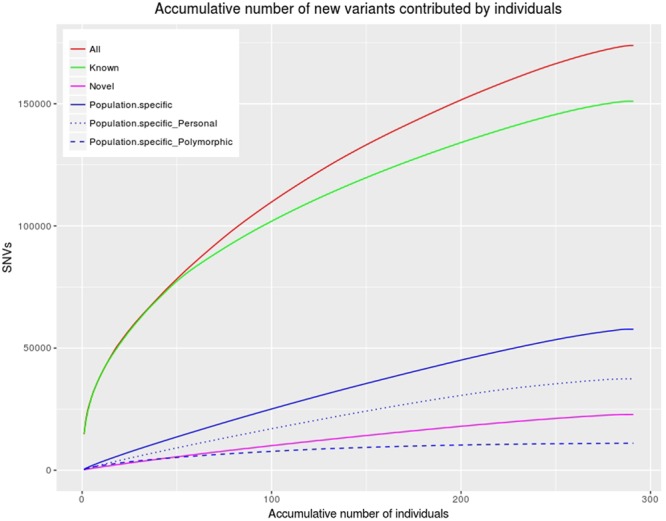
Distribution of total number of single-nucleotide variants (SNVs) upon step-wise addition of exomes. The red line represents the number of all variants found as the number of sequenced exomes increased. The green line represents the number of known variants among all variants found. The orange line represents the number of novel variants among all variants found. The blue line represents the number of population-specific variants among all variants found. Population-specific ‘personal’ variants observed in only one Kuwaiti exome and not seen in either 1KGP or GME are represented by the dotted line; population-specific ‘polymorphic’ variants observed in more than one Kuwaiti exome are represented by the dashed line.
Variants significantly differentiating the three population subgroups of Kuwait
Results of pFst likelihood ratio tests for allele frequency differences between the three subgroups based on 142,626 autosomal variants are presented in Supplementary Fig. S2. Three variants significantly distinguished KWP from KWB: rs2289043_A > G (UNC5C) (pFst = 3.28 × 10−6), mostly prevalent in admixed Americans (75%) and Europeans (71%); rs3739310_T > G (KIAA1456) (pFst = 4.40 × 10−5), frequently found in East Asians (78%) and Europeans (77%); and rs764374986_G > A (AKAP12) (pFst = 5.21 × 10−5), a rare variant occurring mostly in Africans (0.01%) from gnomAD data set (the variant is absent in 1kGP data set). Three variants significantly distinguished KWS from KWB:rs1150360_A > G (FAM76B) (pFst = 3.89 × 10−5), frequent in Africans (93%); rs138408584_G > C (PHRF1) (pFst = 6.97 × 10−5), rare in Europeans (~1%); and rs1043730_G > T (TRAF3IP2) (pFst = 9.57 × 10−5), present at 99% frequency in Africans and East Asians. Two variants significantly distinguished KWP from KWS: rs35840170_C > T (FBN3) (pFst = 7.63 × 10−5), present at ~20% frequency in East Asians and admixed Americans; and rs7956133_G > T (FAM216A) (pFst = 9.51 × 10−5) present at a frequency of 15% in Africans.
SAFD variants with significant allele frequency differences between the Kuwaiti and 1KGP global populations; and analysis of their population-wide occurrence
Examination of the SNVs seen in common between Kuwaiti exomes and 1KGP phase 3 exome data for significant allele frequency differences led to identifying 6,186 SAFD variants. Functional characterization of these variants is presented in Supplementary Table S4. Of these 6,186 SAFD variants, 2,960 were missense, 2,913 were synonymous, 20 were stop-gain and 26 were LoF. Extent of LoF variants among the SAFD SNVs was only 0.4% while it was 1.7% among the ‘all’ SNVs. Population-wide occurrence of the identified SAFD variants was investigated to determine the pairing occurrence of Kuwaiti population subgroups in the context of maximum allele frequency (Supplementary Fig. S3 and Fig. 3). (a) Analysis of the 5,140 SAFD variants, derived using gnomAD populations: The number of variants showing maximum allele frequency in KWS, KWP and KWB subgroups were 2885, 543 and 1712, respectively. In KWS, 38% of the 2885 variants showed maximum allele frequency in Ashkenazi Jews and 19% in Africans. In KWP, 35% had maximum allele frequency in Ashkenazi Jews and 33% in South Asians. In KWB, 61% variants showed maximum allele frequency in Africans. (b) Analysis of 6186 SAFD variants, derived using 1KGP populations: Coupling observed with South Asians and Africans was confirmed. KWB paired with Africans in 21% of 1,056 variants; KWP and KWS paired with South Asians in 31% of 1355 and 17% of 3775 variants, respectively. Furthermore, coupling with Europeans, which was not seen in the analysis using gnomAD populations, was observed in 53% of KWS variants, 52% of KWP variants and 41% of KWB variants.
Figure 3.

The occurrence pattern of pairing Kuwaiti populations with (a) the gnomAD or (b) the 1KGP global populations as populations with maximum allele frequency. X-axis: percentage of pairing occurrence of Kuwaiti populations and gnomAD (A) or 1KGP (B) global populations as populations with maximum allele frequency, Y-axis: Kuwaiti populations (KWT-All Kuwaitis; KWB-Bedouins; KWP-Persians; KWS-Saudi Arabian tribe). gnomAD global populations: AFR, Africans/African Americans; AMR, admixed Americans; ASJ, Ashkenazi Jewish; EAS, East Asians; FIN, Finnish; NFE, Non-Finnish Europeans; OTH, Other population not assigned; SAS, South Asians. 1KGP global populations: AFR, African; AMR, Admixed American; EAS, East Asian; EUR, European; SAS South Asian. (c) Considers only the variants with minor allele frequency (MAF) of >5% (n = 3887) and pairing with 1KGP global populations.
Validation of the genetic relatedness implied by analysis for population-wide occurrence of SAFD variants
In order to further explore the observed coupling in maximum allele frequency between Kuwaitis and other populations (including the Ashkenazi Jews), Kuwaiti exome data was merged with the data sets from Ashkenazi Jews43, Qatar44 and 1KGP phase 3. Upon applying quality control steps and LD-pruning the combined data set of coding-region variants, a total of 896 variants from 3,336 individuals was obtained. Genetic differentiation of Kuwaiti subpopulation groups in terms of regional and continental populations was assessed by way of calculating mean pairwise FST (Supplementary Fig. S4, Supplementary Table S5). Lowest degree of differentiation was observed between Kuwaiti subpopulation groups and Qataris (KWB FST = 0.0005, KWP FST = 0.0027, KWS FST = 0.0023) followed with Ashkenazi Jews (KWB FST = 0.0103, KWP FST = 0.0071, KWS FST = 0.0104) and Europeans (KWB FST = 0.0143, KWP FST = 0.0093, KWS FST = 0.0155). Scatter plots resulting from principal component analysis (PCA) of the merged data set are presented in Figs 4 and 5. Consistent with the FST analysis, the Kuwaiti population were seen dispersed over the Qataris, Ashkenazi Jewish and Europeans (Fig. 4). A clear dispersal of these populations was seen in the three-dimensional PCA plot (Fig. 5 and the interactive three-dimensional plot available at http://dgr.dasmaninstitute.org/exome_pca/).
Figure 4.

Two-dimensional principal component analysis (PCA) plots showing the dispersal of Kuwaitis over the Qataris, Ashkenazi Jewish and Europeans.
Figure 5.

Three-dimensional principal component analysis (PCA) plots showing the dispersal of Kuwaitis over the Qataris, Ashkenazi Jewish and Europeans. The interactive three-dimensional plot is available at http://dgr.dasmaninstitute.org/exome_pca/).
‘Rare and deleterious’ variants and their clinical significance
The analysis pipeline that examined the Kuwaiti exomes for deleterious variants which are rare in both 1KGP and ExAC data sets yielded a list of 46 variants (41 unique disorders of which 20 were reported in CAGS database) – comprising 35 pathogenic (for rare disorders), 1 drug response, 10 risk factors (1 corresponding to a rare but multifactorial disorder and the remaining 9 to complex and common disorders) (Table 3). Of these 46 variants, 43 variants remained rare in Kuwaiti exomes; and three variants reached an MAF value characterizing low-frequency variants (rs1800553/ABCA4/Risk-Factor:2.41%, rs61742245/VKORC1/Drug-response:1.04%, rs11909217/LIPI/Risk-Factor:1.72%). Pathogenic variants: The 35 pathogenic variants mapped to 32 genes and to 32 unique single-gene disorders; 28 of the 35 variants follow autosomal recessive (AR) and the remaining follow autosomal dominant (AD) mode of inheritance. In 16 instances of these 35 pathogenic variants, the disorders were observed in Arab population (as annotated in CAGS database27). Drug response variant: the VKORC1 variant was associated with warfarin resistance in AD mode. Risk factor variants: The 10 risk factor variants mapped to 9 genes and to susceptibility to 8 unique disorders. The inheritance patterns were seen to be mostly autosomal dominant (in three instances, can be AR along with AD). In instances of 4 of the 10 risk factor variants, the disorders were observed in Arab population as per CAGS database.
Table 3.
46 rare and deleterious variants (pathogenic and risk factors)@ seen in Kuwaiti Exomes.
| dbSNP; Ref/risk; gene | Inheritance mode (AD:autosomal dominant; AR:autosomal recessive; MF:multifactorial) | Disorder (PMIM); No of OMIM-listed genes for the disorder; annotation pertaining to whether the disorder is rare or common$ | RAFs: KWT/1KGP/GME; Ratios of RAFs: KWT-1KGP/KWT-GME | Carrier status in Kuwaiti exomes (rr/rR/RR) | Disorder seen in CAGS populations (YES/NO)? |
|---|---|---|---|---|---|
| A. PATHOGENIC and rare disorders CAUSAL VARIANTS (n = 34) | |||||
| rs137853054-G/A; PARK2 | AR | Parkinson disease, juvenile, type 2 (PMIM:600116)92; Single gene; Rare disorder. | 0.0034/0.0002/0.0025; 17.0000/1.3503 | 0/2/288 | Yes |
| rs34424986-G/A; PARK2 | AR. This and the above both participate in compound heterozygosity | Parkinson disease, juvenile, type 2. (PMIM:600116). Single gene; Rare disorder. | 0.0034/0.0004/-; 8.5000/- | 0/2/289 | Yes |
| rs121912615-A/C; SI | AR. In compound heterozygosity state | Sucrase-isomaltase deficiency (PMIM:222900). Single gene; Rare disorder. | 0.0052/0.0004/0.0025; 13.0000/2.0651 | 0/3/283 | No |
| rs200487396-G/A; COL12A1 | AD | Bethlem myopathy 2 (PMIM:616471); Single gene; Rare disorder. | 0.0018/0.0002/-; 9.0000/- | 0/1/279 | No |
| rs143137713-G/C; GYG1 | AR | Polyglucosan body myopathy 2 (PMIM:616199). Single gene; Rare disorder. | 0.0017/0.0004/-; 4.2500/- | 0/1/287 | No |
| rs148772854-C/T; RYR1 | AR. The variant participates in compound heterozygosity. | Minicore myopathy with external ophthalmoplegia (PMIM:255320). Single gene; Rare disorder. | 0.0017/0.006/0.0015; 0.2833/1.1243 | 0/1/290 | No |
| rs61757582-G/A; DHCR7 | AR | Smith-Lemli-Opitz syndrome (PMIM:270400). Single gene; Rare disorder. | 0.0018/0.0002/-; 9.0000/- | 0/1/284 | Yes |
| rs61754375-G/A; TYR | AR | Tyrosinase-negative oculocutaneous albinism, Type IA (PMIM:203100). Single gene; Rare disorder. | 0.0017/0.0002/-; 8.5000/- | 0/1/286 | Yes |
| rs61753185-G/A; TYR | AR | Tyrosinase-negative oculocutaneous albinism, Type IA (PMIM:203100). Single gene; Rare disorder. | 0.0017/0.0004/0.0005; 4.2500/3.3730 | 0/1/290 | Yes |
| rs104894313-C/T; TYR | AR | Oculocutaneous albinism type 1B (PMIM:606952). Single gene; Rare disorder. | 0.0017/0.002/0.0049; 0.8500/0.3476 | 0/1/285 | No |
| rs121434513-G/C; PNKD | AD | Paroxysmal Nonkinesigenic Dyskinesia 1 (PMIM: 118800). Single gene; Rare disorder. | 0.0017/0.0002/0.0005; 8.5000/3.3531 | 0/1/289 | Yes |
| rs121964924-A/G; DPYS | AR | Dihydropyrimidinase deficiency (PMIM:222748). Single gene; Rare disorder. | 0.0017/0.0002/0.0020; 8.5000/0.8424 | 0/1/289 | Yes |
| rs28940872-C/T; ACADS | AR. This variant is seen in compound heterozygosity. | Short-chain acyl-CoA dehydrogenase (SCAD) deficiency (PMIM:606885). Single gene; Rare disorder. | 0.0017/0.0002/0.0015; 8.5000/1.1251 | 0/1/289 | Yes |
| rs148211042-C/T; ABCB6 | AD | Pseudohyperkalemia, familial, 2, due to red cell leak (PMIM:609153). Single gene; Rare disorder. | 0.0017/0.0002/0.0010; 8.5000/1.6882 | 0/1/290 | No |
| rs58331765-C/T; ABCA4 | AR | Stargardt disease 1, Juvenile (PMIM:248200)93. Single gene; Rare disorder. | 0.0034/0.0028/0.0005; 1.2143/6.7460 | 0/2/288 | Yes |
| rs35152987-C/A; HBD | AR. | delta Thalassemia – it is usually associated with a single gene of HBD. It does not have clinical manifestation but is usually associated with beta thalassemia. Thalassemia is a rare disorder. | 0.0052/0.001/0.0086; 5.2000/0.6075 | 0/3/287 | No |
| rs114368325-G/A; CYP24A1 | AR. This variant is seen in compound heterozygosity. | Idiopathic hypercalcemia of infancy or hypercalcemia, infantile, 1; hcinf1 (PMIM:143880). Single gene; Rare disorder. | 0.0017/0.0004/0.0005; 4.2500/3.3730 | 0/1/290 | No |
| rs538881762-C/T; TENM4 | AD | Tremor, hereditary essential, 5 (ETM5). (PMIM:616736). Single gene. GARD lists ETM as NOT a rare disorder but lists the subtypes as rare. | 0.0018/0.0006/-; 3.0000/- | 0/1/276 | No |
| rs116100695-G/A; PKLR | AR | Pyruvate kinase deficiency of red cells (PMIM:266200). Single gene; Rare disorder. | 0.0034/0.0016/0.0096; 2.1250/0.3547 | 0/2/289 | Yes |
| rs6063-C/T; FGG | AR. Found in double heterozygosity | Fibrinogen Milano XII, digenic; Dysfibrinogenemia (PMIM:616004). Three genes. Rare disorder. | 0.0052/0.0028/0.0050; 1.8571/1.0328 | 0/3/286 | No |
| rs121908736-G/A; ADA | AR, SM. The variant can also occur in compound heterozygous state | Partial adenosine deaminase deficiency (PMIM:102700). Single gene; Rare disorder. | 0.0035/0.0018/0.0005; 1.9444/6.9444 | 0/2/285 | Yes |
| rs41295338-G/T; TGM1 | AR | AR congenital ichthyosis 1 (PMIM:242300). Single gene; Rare disorder. | 0.0034/0.0022/0.0060; 1.5455/0.5627 | 0/2/288 | Yes |
| rs28941785-C/T; CTH | AR. | Cystathioninuria (PMIM:219500). Single gene; Rare disorder. | 0.0035/0.0026/0.0065; 1.3462/0.5347 | 0/2/281 | Yes |
| rs77010315-C/A; SLC36A2 | AR, DR | Iminoglycinuria, digenic (PMIM:242600). Single gene; Rare disorder. | 0.0076/0.005/0.0025; 1.5200/3.0159 | 0/4/258 | No |
| rs56208331-G/A; GATA4 | AD | Tetralogy of fallot; TOF. (PMIM:187500). Single gene; Rare disorder. | 0.0034/0.0034/0.0025; 1.0000/1.3503 | 0/2/289 | Yes |
| rs121908970-C/T; MYO15A | AR. The variant was seen in hemizygosity state. | Deafness, with Smith-Magenis syndrome (PMIM: 600316). Single gene; Rare disorder. | 0.0018/0.001/0.0010; 1.8000/1.7875 | 0/1/274 | No |
| rs34324426-C/T; PEX6 | AR. The variant is in compound heterozygosity state. | Heimler syndrome 2 (PMIM:616617). Single gene; Rare disorder. | 0.0017/0.001/0.0015; 1.7000/1.1243 | 0/1/290 | No |
| rs104893836-T/C; GNRHR | AR. The variant is in compound heterozygosity state. | Hypogonadotropic hypogonadism 7 without anosmia. (PMIM:146110). Single gene; Rare disorder. | 0.0017/0.0012/0.0055; 1.4167/0.3069 | 0/1/290 | Yes |
| rs5907-G/A; SERPIND1 | AD | Thrombophilia due to Heparin cofactor II deficiency (PMIM:612356). Single gene. Inherited disorder | 0.0017/0.0012/-; 1.4167/- | 0/1/290 | No |
| rs113418909-A/T; SERPINA6 | AR,AD | Corticosteroid-binding globulin deficiency (PMIM:611489). Single gene; Rare disorder. | 0.0017/0.0014/0.0045; 1.2143/0.3751 | 0/1/290 | No |
| rs121909293-C/T; CTRC | AD | Pancreatitis, chronic, susceptibility to (PMIM:167800). 5 genes. Hereditary Pancreatitis, that leads to chronic form, as rare disorder. | 0.0017/0.0016/-; 1.0625/- | 0/1/290 | No |
| rs137941190-C/T; DCPS | AR. Found in compound heterozygous state. | Al-raqad syndrome (PMIM:616459). Single gene; Rare disorder. | 0.0017/0.002/0.0005; 0.8500/3.3730 | 0/1/288 | No |
| rs28940885-C/T; GALE | AR/AD heterogeneity | Galactose Epimerase Deficiency (PMIM:230350). Single gene; Rare disorder. | 0.0017/0.002/0.0025; 0.8500/0.6751 | 0/1/290 | Yes |
| rs73015965-A/G; PLG | AR. Participates in compound heterozygosity | Plasminogen deficiency, type I (PMIM:217090). Single gene; Rare disorder. | 0.0017/0.0022/0.0006; 0.7727/2.8286 | 0/1/289 | No |
| rs200879436-T/C; CEP152 | AR. The variant is seen in compound heterozygous state | Seckel syndrome 5 (PMIM:613823). Single gene; Rare disorder. | 0.0017/0.0034/0.0015; 0.5000/1.1251 | 0/1/288 | No |
| DRUG RESPONSE (n = 1) | |||||
| rs61742245-C/A; VKORC1 | AD | Warfarin resistance (PMIM:122700). Single gene; Polymorphisms in other genes, some of which have not been identified, have a smaller effect on warfarin metabolism. Rare disorder. Poor quality of anticoagulation with warfarin has been reported across Kuwait94 | 0.0104/0.0004/0.0137; 26.0000/0.7619 | 1/4/284 | No |
| C. Risk Factor and rare disorders | |||||
| rs139512218-G/T; SPRY4 | AD | Hypogonadotropic hypogonadism 17 with or without anosmia (PMIM: 615266). Single gene. CHH is a rare reproductive disorder but multifactorial (involving genes of FGF pathway). | 0.0035/0.0024/0.0010; 1.4583/3.4757 | 0/2/282 | No |
| D. RISK FACTORS (n = 9) – Common or multifactorial complex disorders | |||||
| rs11554495-C/A; KRT8 | AR | Cirrhosis, cryptogenic (PMIM:215600). Two genes. ClinVar annotation is pathogenic but we annotate it as risk factor as this disorder is probably common. GARD does not list this as rare disorder as it affects more than 600,000 people in USA | 0.0053/0.001/0.0027; 5.3000/1.9910 | 0/3/281 | No |
| rs1800553-C/T; ABCA4 | AD | Macular degeneration, age-related, 2 (AMD2), susceptibility to (PMIM:153800). Single gene; however, AMD is a complex trait. GARD lists this as NOT a rare disorder. ClinVar annotates this as pathogenic’; however, it is a susceptibility variant for a complex trait and hence it is risk factor. | 0.0241/0.0032/0.0211; 7.5313/1.1396 | 1/12/278 | No |
| rs11909217-C/T; LIPI | AD | Susceptibility to Hypertriglyceridemia, Familial. (PMIM: 145750). Two genes -LIPI and APOA5. Arab study reports a different variant 3’ UTR C > G from APOC395. This is an inherited common disorder. | 0.0172/0.006/0.0131; 2.8667/1.3138 | 0/10/280 | Yes |
| rs114817817-C/T; SRGAP1 | AD | Thyroid cancer, nonmedullary 2, susceptibility to (PMIM:188470). Three genes. Most common form of thyroid cancer. | 0.0017/0.0006/0.0070; 2.8333/0.2412 | 0/1/285 | Yes |
| rs72470545-G/A; HTRA2 | AD | Parkinson disease 13, Autosomal Dominant, susceptibility to (PMIM: 610297). Single gene. It is a complex neuro-degenerative disorder. | 0.0069/0.0036/0.0070; 1.9167/0.9789 | 0/4/287 | No |
| rs28932472-G/C; POMC | AR,AD,MF | Obesity, early-onset, susceptibility to (PMIM: # 601665). Several genes. Common disorder. Arab study reports another variant rs1042713 (Arg16Gly) in ADRB2 gene96 for this disorder; which is associated with susceptibility to nocturnal asthma in OMIM and our study. | 0.0017/0.0024/0.0005; 0.7083/3.3730 | 0/1/287 | Yes |
| rs34911341-C/T; GHRL | AR,AD,MF | Obesity, susceptibility to (PMIM: 601665). Several genes. Common disorder. | 0.0017/0.0026/0.0035; 0.6538/0.4823 | 0/1/290 | Yes |
| rs138292988-G/A; AP1S3 | AD | Psoriasis 15, pustular, susceptibility to (PMIM:616106). Single gene. Psoriasis is NOT a rare disorder; pustular psoriasis is a rare form Psoriasis. Susceptibility is the keyword. | 0.0017/0.0024/0.0015; 0.7083/1.1251 | 0/1/289 | No |
| rs116107386-A/C; AP1S3 | AD | Psoriasis 15, pustular, susceptibility to (PMIM:616106). Single gene. Psoriasis is NOT a rare disorder; pustular psoriasis is a rare form Psoriasis. | 0.0018/0.003/0.0050; 0.6000/0.3575 | 0/1/279 | No |
All the 46 variants were seen annotated in OMIM and ClinVar for clinical significance.
@Clinical significance of a variant was checked by way of using the evidences presented in OMIM and ClinVar (see Methods).
$Various resources that were examined to ascertain whether the disorder is rare or common: Catalogue of Transmission Genetics in Arabs (available at http://cags.org.ae/ctga/), Genetic and Rare Disease (GARD) Information Centre (available at https://rarediseases.info.nih.gov/diseases/), Genetics Home Reference (available at https://ghr.nlm.nih.gov/), Medscape (available at https://geneaware.clinical.bcm.edu/GeneAware/AboutGeneAware/DiseaseSearch.aspx) and literature.
Pathogenic variants and high MAF in Kuwaiti exomes
Five of the identified “rare & deleterious” variants that were annotated “pathogenic” for clinical significance in ClinVar were seen to possess risk allele frequencies of ≥1% in Kuwaiti exomes as opposed to <1% in 1KGP populations. ClinVar defines “pathogenic” variants as those that are interpreted for Mendelian disorders; or as those that have low penetrance. It is also possible that a variant in ClinVar can have an erroneous or conflicting classification. Cassa et al.45 examined 81,432 “pathogenic” variants from HGMD7 in a data set of whole-genome sequences of 1.092 individuals from 1KGP project and found that 4.62% of the tested variants to possess an MAF of ≥1% and 3.5% of the tested variants to possess an MAF of ≥5%; they concluded that many of these variants are probably erroneous findings or have lower penetrance than previously expected. It is also possible that such high frequency pathogenic variants are indeed of the type “increased susceptibility” and not of the type “causal”; it is also possible that the disorders with such high MAF “pathogenic” variants are not really “rare” but are either “common” or “more prevalent in the study population”; that it is also possible that the frequent variants have evidence to cause a disease when inherited in compound heterozygous state and have insufficient evidence to lead to a disease in homozygotes. The five “pathogenic” variants that were seen in Kuwaiti exomes with an MAF of ≥1% are as follows:
(a) Four variants retained as pathogenic for rare disorders: (i) rs79204362 (MAF_KWT:1.03% and MAF_1KGP: 0.42%) associated with Early onset of Glaucoma: ClinVar annotated this variant as Pathogenic based on evidence from literature studies and as of uncertain significance based on clinical testing. The disorder was supposed to be rare (1 in 10,000) in European-based populations and of higher frequency in Middle East 51–100 per 100,000 (i.e. 5 in 10,000); CTGA reported a high incidence rate of 1 in 2,500 in Saudi Arabian population. Thus, the MAF that is seen marginally higher at 1.03% was acceptable. (ii) rs61732874_C > A (MAF_KWT:1.55% and MAF_1KGP: 0.18%) associated with Familial Mediterranean fever (FMF): ClinVar annotated this as Pathogenic/likely-pathogenic based both on literature evidence and clinical testing. FMF is a rare disorder in European population; however, it is no longer a rare disorder in certain populations such as Japan (see Table 7). CAGS also listed the incidence as 51–100 per 100,000 in Arab population; CAGS further mentioned that estimates of the incidence of FMF in specific eastern Mediterranean populations ranged from 1 in 2000 to 1 in 100, depending on the population studied. Thus, the MAF that is seen at 1.55% was acceptable. (iii) rs61757294 (MAF_KWT:15.19% and MAF_1KGP:5.3%) associated with Corticosterone Methyloxidase Type II Deficiency, a rare genetic disorder: ClinVar annotated this variant as pathogenic based on evidence from literature publication and benign based on clinical testing records. The variant was found in patients of Iranian Jewish ancestry. This had DR mode of inheritance – both this variant and another one rs289316 need to be homozygous; thus, the observed high MAF was acceptable; in fact, the variant was a common variant in 1KGP as well. (iv) rs12021720 (MAF_KWT:13.47% and MAF_1KGP:10.9%) associated with Maple syrup urine disease, intermediate, type II (a rare genetic disorder): ClinVar annotated this variant as pathogenic based on literature evidence and as benign with clinical testing as source of annotation. Though the incidence rate world-wide is 1 in 185,000, CAGS reported an incidence rate of 2 in 10,000 in Bahrain - still the higher MAF is not justified. This mutation was seen in one of the three patients from the study46 and the patient was a compound heterozygote for a C to G transversion at nucleotide 309 in exon 4 [rs121965001] and a G to A transition at nucleotide 1165 in exon 9 [rs12021720], causing an Ile-to-Met substitution at amino acid 37 and a Gly-to-Ser substitution at amino acid 323, respectively. Thus, the high frequency of MAF at one of the two variants of the compound heterozygotes was acceptable for pathogenic variant; in fact, the variant was a common variant in 1KGP as well. (b) One variant retained as pathogenic for rare disorder but with a suggestion that they can be “likely benign”: rs61751507 (MAF_KWT:7.47% and MAF_1KGP:2.7%) associated with Carboxypeptidase N deficiency, which is possibly a complex disorder. ClinVar annotated this variant as pathogenic with evidence from literature publication and benign based on information clinical testing. The study47 found this pathogenic variant in just one patient and hence it may be considered as of insufficient evidence. Hence this variant can be considered as “Likely Benign”.
Table 7.
Arab disorders (annotated in CAGS) for which the OMIM-listed risk variants were seen in Kuwaiti exomes.
| SNPs from Kuwaiti exomes seen annotated in OMIM against the CAGS disorder; Ref > risk alleles | Disorder | ClinVar annotation for the disorder (CAGS OMIM Id) |
Incidence Rates | Inheritance | Carrier Frequencies (rr/rR/RR) |
RAF(KWT/1KGP/GME) | Ratios (KWT-1KGP /KWT-GME) |
|---|---|---|---|---|---|---|---|
| A. 7 Pathogenic variants for rare disorders | |||||||
| rs12021720 C > T p.Gly384Ser DBT |
Maple syrup urine disease, intermediate, type ii (PMIM:248600). It is a rare genetic disorder. OMIM lists single gene for the Type II MSUD | Pathogenic for the subtype. | AR | 5/67/218 | 0.1347/0.1082/0.12 The literature publication, cited in OMIM, reporting this variant as pathogenic, found it in compound heterozygosity with another variant [rs121965001] in the patient. Thus, the high MAF at one of the two variants of the compound heterozygotes is perhaps acceptable for pathogenic variant. |
0.97/0.98 | |
| rs79204362 C > T p.Arg368His CYP1B1 |
Glaucoma, early-onset, digenic (PMIM: 231300). OMIM lists single gene. This is a rare disorder but reaches high prevalence in Saudi Arabian populations | Pathogenic | 11–50 | AR | 0/6/284 | 0.0103/0.0042/0.0137 Incidence rate in Middle East is 51–100 per 100,000 (i.e. 5 in 10,000); CTGA reports as high as 1 in 2,500 in Saudi Arabian population. Accordingly, MAF is seen marginally higher at 1.03% |
2.47/0.76 |
| rs61732874 C > A p.Ala744Ser MEFV |
Familial Mediterranean fever (PMIM: 249100). OMIM lists single gene. However, this can be a common disorder in certain populations. | Pathogenic/Likely Pathogenic | 51–100 | AR | 0/9/278 | 0.0155/0.0018/0.0126 FMF is a rare disorder in Europe; however, it is no longer a rare disorder in populations such as Japan105. CAGS lists an incidence of 51–100 per 100,000 in Arab population; estimates of the incidence in specific eastern Mediterranean populations range from 1 in 2000 to 1 in 100, depending on the population studied. Accordingly, MAF is seen marginally higher at 1.55%. |
8.60/1.23 |
| rs121908530 G > A p.Gly156Arg AGXT |
Hyperoxaluria, primary, type i (PMIM: 259900). Rare disorder OMIM lists single gene. |
Pathogenic | 0/1/290 | 0.0017/./. | ./. | ||
| rs118204113 G > A p.Ala252Thr HMBS |
Porphyria, acute intermittent (PMIM:176000). Rare form of porphyria. OMIM lists single gene. |
Pathogenic | AD | 0/1/289 | 0.0017/./0.0005 | 0./3.41 | |
| rs587776954 A > G p.Met1Val; LoF C12ORF57 |
Temtamy syndrome (PMIM:218340). GARD lists this as a rare disorder. OMIM lists single gene. |
Pathogenic | AR | 0/2/288 | 0.0034/./0.0030 | 0./1.14 | |
| rs4149584 C > T p.Arg121Gln TNFRSF1A |
Periodic fever, familial, autosomal dominant (PMIM:142680). Rare disorder OMIM lists single gene. |
Pathogenic | AR | 0/4/287 | 0.0069/0.0069/0.0121 | 1.00/0.57 | |
| B.10 Risk Factor/association variants | |||||||
| B. 1. 1 Risk factors for supposedly rare disorders | |||||||
| rs61747728 C > T p.Arg229Gln NPHS2 |
Nephrotic syndrome, type 2, susceptibility to (PMIM: 600995). OMIM lists single gene. Rare disorder. | Risk factor Rare disorder; however, Zaki et al.106 concluded that the incidence of this disease seemed to be higher among Arab children (as seen in Kuwaiti hospitals over 5 years period) than in Western countries. |
11–50 | AR | 0/8/281 | 0.0138/0.0146/0.0121 | 0.94/1.14 |
| B. 2. 9 Risk factors/associations for common disorders | |||||||
| rs3135506 G > C p.Ser19Trp APOA5 |
Hypertriglyceridemia Familial, susceptibility to (PMIM: 145750). OMIM lists two genes. This is an inherited common disorder. | Risk factor for Common disorder | AD | 1/22/267 | 0.0412/0.0557/0.0706 | 0.74/0.58 | |
| rs2476601 G > A p.Arg620Trp PTPN22 |
Diabetes mellitus Type 1, insulin-dependent, susceptibility to (PMIM:222100). OMIM lists multiple genes. Complex disorder And associated with susceptibility to Systemic lupus erythematosus as well (PMIM:152700). |
Risk factor. Also risk factor for many other disorders such as SLE | AR | 0/5/280 | 0.0292/ 0.0274/0.0131 | 1.00/0.98 | |
| rs1799945 C > G p.His63Asp HFE |
Hemochromatosis, type 1 - microvascular complications of diabetes, susceptibility to, 7, included (PMIM:235200). OMIM lists two genes. Common disorder. | Risk factor | >100 | AR | 4/63/224 | 0.122/0.0731/0.1198 | 1.67/1.02 |
| rs17158558 C > T p.Arg982Cys RET |
Hirschsprung disease, susceptibility to, 1 (PMIM 142623). OMIM lists single gene. Complex disorder. |
Risk factor | AD | 0/15/273 | 0.0258/0.0220/0.0459 | 1.17/0.56 | |
| rs121918219 G > A p.Arg274Gln VANGL1 |
Neural tube defects, susceptibility to (PMIM: 182940). OMIM lists two genes. The average incidence of NTDs is 1/1000 births, with a marked geographic variation107. |
Risk factor. | >100 | AD | 0/1/289 | 0.0017/./0.0010 | ./1.71 |
| rs1800858 G > A p.Ala45Ala Synonymous RET |
Hirschsprung disease, susceptibility to, 1; HSCR1 (PMIM: 142623). CAGS reports an incidence rate of 1 per 5000 live births. OMIM lists single gene. |
Risk factor | AD | 25/128/138 | 0.3058 /0.2464/0.2593 | 1.24/1.1793 | |
| rs1801131 T > G p.Glu470Ala MTHFR |
Schizophrenia; sczd (PMIM: 181500) OMIM lists multiple genes. Complex trait. |
Risk factor | AD | 31/128/132 | 0.3265/0.3264/0.3353 | 1.00/0.97 | |
| rs2282440 G > A p.Thr329Ile SDC3 |
Obesity (PMIM: 601665). OMIM lists multiple genes. Complex trait. | Association | AD | 0/11/280 | 0.0189/0.0189/0.0282 | 1.00/0.67 | |
| rs77775126 C > T p.Thr373Ile RP1 |
Retinitis pigmentosa 1; RP1 (PMIM: 180100). OMIM lists single gene. It is a common disorder. The disorder is seen in CAGS with prevalence as approximately 1/3,000 to 1/5,000. | Risk Factor by way of inference – since we associate “pathogenic” only with Mendelian disorder. | AR | 0/11/280 | 0.0189/0.0189/0.0181 | 1.00/1.04 | |
| C. 6 Pathogenic variants that already appeared in the list of RARE & DELETERIOUS variants (see Table 3) | |||||||
| rs104893836 T > C p.Gln106Arg GNRHR |
Hypogonadotropic hypogonadism 7 without anosmia (PMIM:146110). OMIM lists single gene. MEDSCAPE lists Hypogonadotropic hypogonadism as rare genetic disorder. | Pathogenic | AR | 0/1/290 | 0.0017/0.0012/0.0055 | 1.43/0.31 | |
| rs58331765 C > T p.Val931Met ABCA4 |
Stargardt disease 1 (PMIM:248200). OMIM lists two genes. Rare disorder. |
Likely pathogenic/Pathogenic. | AR | 0/2/288 | 0.0034/0.0028/0.0005 | 1.23/6.82 | |
| rs61753185 G > A p.Arg77Gln TYR |
Tyrosinase-negative oculocutaneous albinism, Type IA (PMIM:203100). OMIM lists single gene. Rare disorder. |
Pathogenic for Tyrosinase-negative oculocutaneous albinism | AR | 0/1/290 | 0.0017/0.0004/0.0005 | 4.30/3.41 | |
| rs61754375 G > A p.Arg299His TYR |
Tyrosinase-negative oculocutaneous albinism, Type IA (PMIM: 203100). OMIM lists single gene. Rare disorder. |
Pathogenic for Tyrosinase-negative oculocutaneous albinism | AR | 0/1/286 | 0.0017/0.0002/. | 8.60/. | |
| rs34424986 G > A p.Arg275Trp PARK2 |
Parkinson disease 2, autosomal recessive juvenile; PARK2 (PMIM:600116). OMIM lists single gene. Rare disorders. | Pathogenic | 0/2/289 | 0.0034/0.0034/. | 1.00/. | ||
| rs56208331 G > A p.Asp426Asn GATA4 |
Tetralogy of fallot; (PMIM:187500). OMIM lists single gene. Rare disorder. |
Pathogenic | AD | 0/2/289 | 0.0034/0.0034/0.0025 | 1.00/1.36 | |
| D. 2 risk factor variants that already appeared in Tables 3 and 6 | |||||||
| rs121918530 A > G p.Asn261Ser MEF2A |
Coronary artery disease/myocardial infarction (PMIM: 608320). OMIM lists single gene. However, it is a complex disorder. | Risk factor by way of inference | AD | 0/6/284 | 0.0103/0.0004/0.0020 | 25.82/5.12 | |
| rs11558538 C > T p.Thr105Ile HNMT |
Asthma, susceptibility to (PMIM: 600807). Common disorder OMIM lists multiple genes. | Risk factor | AD | 7/55/212 | 0.1186/0.0595/0.0968 | 1.99/1.23 | |
Missense variants rare within global populations but common within Kuwaiti population
170 SNVs were identified as rare in global populations but common in Kuwaiti exomes; 85 of these were missense variants (Supplementary Table S6). The 85 variants were of two categories: (a) A set of 20 variants harboured in genes annotated for disorders in OMIM: However, these 20 variants were not of any pathogenic value as ClinVar annotated these variants as either ‘benign’ or ‘conflicting interpretation’. Not surprisingly, the REVEL scores in these instances (except in 2 instances – GLDC variant at around 0.8; and the DPYD variant at 0.4) were seen low at ≤0.3. (b) A set of 65 variants harboured in genes NOT annotated for any disorder in OMIM: Association with phenotypes was seen with only one of these 85 variants; the TTC38 variant rs117135869 (REVEL = 0.621; MAF_KWT: 5.0%; MAF_1KGP: 0.58%) has been recently identified as a novel metabolic quantitative trait loci (mQTLs) in a cohort from Middle Eastern population48; this variant was seen in 29 of the Kuwaiti exomes in the heterozygous form.
Missense variants mapping to drug-binding domains and were of pharmacogenomic relevance
We identified 21 missense SNVs that mapped to a set of 130 drug-binding domains reported in literature49 and were annotated in PharmGKB50 (Table 4). These 21 variants had impact on the efficacy of drugs used largely for treating common disorders (such as heart failure, hypertension, Chemotherapy, neoplasms, diabetes, nephrosclerosis, rheumatoid arthritis, asthma, pulmonary diseases, schizophrenia, tobacco use disorder, heroin dependence, sickle-cell anemia, and HIV). Furthermore, literature survey revealed that 7 of these 21 pharmacogenomic variants were associated with complex disorders in Arab studies (Table 5).
Table 4.
21 missense variants mapping to drug-binding domains and of pharmacogenomic relevance (efficacy, dosage toxicity).
| Chr:Position | dnSNP ID; variant change: amino acid change | Gene | KWT_RAF/1KGP_ RAF/GME_RAF; Ratio KWT-1KGP/KWT-GME RAFs | Ref_risk | Affecting Allele(s) for the drug response | Drug response to which is impacted | Disease/Condition |
|---|---|---|---|---|---|---|---|
| 4:3006043 | rs1024323; c.425C > T: Ala142Val | GRK4 | 0.3718/0.3732/0.3887; 0.9962/0.9565 | C_T | T | atenolol | Hypertension |
| CC | metoprolol | Hypertensive Nephrosclerosis | |||||
| CC | metoprolol | Hypertensive Nephrosclerosis | |||||
| 17:7579472 | rs1042522; c.215C > G: Pro72Arg | TP53 | 0.4425/0.5429/.; 0.8150/. | G_C | CG + GG | cisplatin; paclitaxel | Stomach neoplasms |
| 5:148206440 | rs1042713; c.46A > G: Arg16Gly | ADRB2 | 0.5513/0.5244/0.5645; 1.0513/0.98 | G_A | AA | terbutaline | Asthma |
| AA | salmeterol | Asthma | |||||
| A | Beta Blocking Agents | Heart Failure | |||||
| G | hydrochlorothiazide | Hypertension | |||||
| A | indacaterol | Chronic obstructive pulmonary disease | |||||
| 5:148206473 | rs1042714; c.79C > G: Gln27Glu | ADRB2 | 0.2246/ 0.2053/0.2412; 1.094/0.9312 | G_C | GG | terbutaline | Asthma |
| G | Beta Blocking Agents | Heart Failure | |||||
| C | indacaterol | Chronic obstructive pulmonary disease | |||||
| 15:75012985 | rs1048943; c.1384A > G: Ile462Val | CYP1A1 | 0.0653/0.1334/0.0619; 0.4895/1.0542 | T_C | C | warfarin | warfarin |
| 10:96798749 | rs10509681; c.1196A > G: Lys399Arg | CYP2C8 | 0.1198/0.0457/0.1047; 2.6197/1.1438 | T_C | C | rosiglitazone | Diabetes |
| C | repaglinide | Diabetes | |||||
| C | paclitaxel | Cancer(chemotherapy) | |||||
| 7:75615006 | rs1057868; c.1508C > T: Ala503Val | POR | 0.2609/0.2861/0.2808; 0.9120/0.9292 | C_T | CT + TT | tacrolimus | Kidney transplantation |
| 17:37879588 | rs1136201; c.1963A > G: Ile655Val | ERBB2 | 0.0975/0.1214/0.1255; 0.8032/0.7770 | A_G | G | trastuzumab | Breast cancer |
| 10:96827030 | rs11572080; c.416G > T: Arg139Lys | CYP2C8 | 0.1237/0.0457/0.1013; 2.7054/1.2211 | C_T | T | paclitaxel | Cancer(chemotherapy) |
| T | rosiglitazone | Diabetes | |||||
| T | repaglinide | Diabetes | |||||
| T | paclitaxel | Neoplasms | |||||
| 2:21263900 | rs1367117; c.293C > T: Thr98Ile | APOB | 0.1448/0.1693/0.1727; 0.8553/0.8386 | G_A | AG | Irbesartan | Hypertension |
| GG | Irbesartan | Hypertension | |||||
| 11:113270828 | rs1800497; c.2137G > A: Glu713Lys | ANKK1 | 0.1713/0.3257/0.1908; 0.5259/0.8975 | G_A | A | risperidone | Schizophrenia |
| A | lithium | Bipolar disorder | |||||
| A | antipsychotics | Schizophrenia | |||||
| A | Drugs used in nicotine dependence | Tobacco use disorder | |||||
| AA + AG | aripiprazole | Schizophrenia | |||||
| AA | methadone | Heroin dependence | |||||
| 5:148206885 | rs1800888; c.491C > T: Thr164Ile | ADRB2 | 0.0121/0.0040/0.0156; 3.0325/0.7759 | C_T | CT | terbutaline | Asthma |
| T | Beta Blocking Agents | Heart Failure | |||||
| 3:124456742 | rs1801019; c.638G > C: Gly213Ala | UMPS | 0.1632/0.1859/0.1767; 0.8780/0.9236 | G_C | C | fluorouracil | Colonic neoplasms |
| 4:3039150 | rs1801058; c.1457T > C: Val486Ala | GRK4 | 0.2293/0.3067/0.292; 0.7476/0.7079 | T_C | T | atenolol | Hypertension |
| CT | metoprolol | Hypertensive Nephrosclerosis | |||||
| CT | metoprolol | Hypertensive Nephrosclerosis | |||||
| 1:237048500 | rs1805087; c.2756A > G: Asp919Gly | MTR | 0.2603/0.2183/0.2021; 1.1929/1.2881 | A_G | A | folic acid; hydroxychloroquine; methotrexate; sulfasalazine | Rheumatoid Arthritis |
| 7:55229255 | rs2227983; c.1562G > A: Arg521Lys | EGFR | 0.2759/0.2921/0.2784; 0.9443/0.9907 | G_A | A | cetuximab | Colorectal neoplasms |
| 22:42525772 | rs28371706; c.320C > T: Thr107Ile | CYP2D6 | 0.0446/0.0591/.; 0.7553/. | G_A | A | codeine | Sickle Cell Anemia |
| AA + AG | nevirapine | HIV | |||||
| 4:2990499 | rs2960306; c.194G > T: Arg65Leu | GRK4 | 0.3711/0.3125/0.3620; 1.1875/1.0250 | G_T | T | atenolol | Hypertension |
| GT + TT | metoprolol | Hypertensive Nephrosclerosis | |||||
| TT | metoprolol | Hypertensive Nephrosclerosis | |||||
| 19:41522715 | rs3211371; c.1459C > T: Arg487Cys | CYP2B6 | 0.0423/0.0535/0.0624; 0.7896/0.6767 | C_T | TT | nevirapine | HIV |
| TT | methadone | Heroin dependence | |||||
| 19:41512841 | rs3745274; c.516G > T: Gln172His | CYP2B6 | 0.2759/0.3157/0.3026; 0.8738/0.9116 | G_T | TT | efavirenz | HIV |
| T | nevirapine | HIV | |||||
| TT | methadone | Heroin dependence | |||||
| GT + TT | nevirapine | HIV | |||||
| T | nevirapine | HIV | |||||
| GT | nevirapine | HIV | |||||
| TT | nevirapine | HIV | |||||
| TT | nevirapine | HIV | |||||
| GT | nevirapine | HIV | |||||
| TT | nevirapine | HIV | |||||
| 1:230845794 | rs699; c.803T > C: Met268Thr | AGT | 0.583/0.7051/0.5534; 0.8268/1.0535 | A_G | G | Antihypertensives | Hypertension |
Table 5.
Subset of 7 of the identified pharmacogenomic variants (from Table 4) that were also reported in Arab studies as relating to complex disorders.
| Pharmaco-genomic variant | Gene | KWT_RAF/1kGP_RAF/GME_RAF | Ratio KWT_1KGP/Ratio KWT_GME | Ref_risk allele | Drug to which response is reported | Disorder treated with the drug | Disorder with which Arab studies associate the variant |
|---|---|---|---|---|---|---|---|
| rs1805087 | MTR | 0.260/0.218/0.202 | 1.1929/1.2881 | A_G | Folic acid; hydroxy-chloroquine; methotrexate; sulfasalazine. | Rheumatoid arthritis | Autism – North Iran97. GG seems to be the risk factor. GG seems to be the risk factor. Complex trait. |
| rs1042522 | TP53 | 0.443/0.543/- | 0.8150/- | G_C | Cisplatin; paclitaxel | Stomach neoplasms | Susceptibility to Breast Cancer in Tunisia98. Complex trait. |
| rs1800497 | ANKK1 | 0.1713/0.326/0.191 | 0.5259/0.8975 | G_A | Risperidone | Schizophrenia | Risk of Schizophrenia in Egyptians99. CC and CT genotypes are the risk factors. Complex trait. |
| rs1801058 | GRK4 | 0.229/0.307/0.292 | 0.7476/0.7079 | T_C | Atenolol | Hypertension | Risk of myocardial infarction among hypertensive subjects in Jordan100. Complex trait. |
| rs699 | AGT | 0.583/0.705/0.553 | 0.8268/1.0535 | A_G | Anti-hypertensive | Hypertension | Reduced life-span through genetic susceptibilities to both Essential Hypertension and Myocardial Infarcation in UAE101. Complex trait. |
| rs1042713 | ADRB2 | 0.551/0.5244/0.565 | 1.0513/0.98 | A_G | Terbutaline | Asthma | Susceptibility to early onset obesity, insulin resistance etc. in Saudi Arabia102. the subjects who carry Gly16 homozygote genotype are in higher risk. Complex trait. |
| rs1042714 | ADRB2 | 0.225/0.205/0.241 | 1.0941/0.9312 | G_C | Terbutaline | Asthma | Associated with coronary artery disease/myocardial infarction in Saudi Arabia103,104. both the C/G and G/G genotypes are significantly associated with CAD. Complex trait. |
SAFD variants and their clinical relevance
For 230 of the 6,186 SAFD variants, ClinVar database provided annotation relating to clinical significance (Supplementary Fig. S5 and Supplementary Table S7). These 230 variants were from 186 unique genes, for 162 of which Inheritance patterns were known; 91 were AR and 63 were AD. 206 of these 230 variants were benign or likely benign. The disorders related to the genes harbouring the benign variants were often single-gene disorders and familial, hereditary and congenital. The 24 non-benign variants (Table 6) were from 21 unique genes associated with 20 unique disorders.
Table 6.
List of 24 SAFD variants annotated for clinical significance in ClinVar and OMIM.
| Chr: Position; dbSNP_ID_Ref_Alt; Gene | KUWAITI | 1KGP | gnomAD | Inheritance mode | Disease name; PMIM; number of OMIM listed genes; Is the disorder seen in Arab population?@ | |||
|---|---|---|---|---|---|---|---|---|
| KWT_MAF | Max_Pop (MAF) | 1KGP_MAF | Max_Pop (MAF) | gnomAD_MAF | Max_Pop (MAF) | |||
| A. 2 “Pathogenic” variants as per ClinVar annotation | ||||||||
| 8:143994266; rs61757294_ A_G c.1157T > C; p.Val386Ala CYP11B2 | 0.1519 This has DR mode of inheritance – both this and another variant rs289316 need to be homozygous; hence this higher MAF is alright though the variant is “pathogenic”. | KWS (0.1698) | 0.0531 | EUR (0.1093) | 0.0846 | ASJ (0.1163) | DR (both rs61757294 and rs28931609 need to be homozygous) | Corticosterone methyloxidase type 2 deficiency. PMIM: 610600; Single gene. Rare Genetic disorder. Yes, disorder seen in Arab country. |
| 10:101829514 rs61751507_ C_T c.533G>A; p.Gly178Asp CPN1 | 0.0747 (high MAF for “pathogenic” variant). ClinVar annotates this variant as pathogenic based on one literature publication which reports this variant in just one patient. Thus, it is possible that the significance is of insufficient evidence and that the variant may become “likely benign”. | KWS (0.0981) | 0.0266 | AMR (0.0634) | 0.0423 | AMR (0.0668) | AR | Carboxypeptidase N deficiency PMIM: 212070 Single gene. Familial Carboxypeptidase N deficiency is a rare disorder. |
| B. 4 Drug response variants as per ClinVar annotation | ||||||||
| 16:31105945 rs61742245_ C_A c.106G>T; p.Asp36Tyr VKORC1 | 0.0104 | KWB (0.0294) | 0.0004 | EUR (0.0010) | 0.0024 | ASJ (0.0384) | AD | Warfarin resistance PMIM: 122700 Multiple genes and rare disorder. Appears already in Table 3 as drug response. |
| 8:18257854 rs1801280_ T_C c.208G>A; p.Asp70Asn NAT2 | 0.3927 | KWP (0.3879) | 0.2927 | EUR (0.4493) | 0.3821 | FIN (0.4668) | AR Forms part of NAT2*5B haplotype | Slow acetylator due to N-acetyltransferase enzyme variant. Toxicity to the drugs of cisplatin or cyclophosphamide. PMIM: 243400 Yes, disorder seen in Arab country. |
| 10:96702047 rs1799853_ C_T c.430C>T; p.Arg144Cys CYP2C9 | 0.1181 | KWS (0.1262) | 0.0479 | EUR (0.1243) | 0.0926 | ASJ (0.1357) | AD | Warfarin sensitivity PMIM: 122700 Multiple genes. |
| 19:15990431 rs2108622_ C_T c.1297G>A; p.Val433Met CYP4F2 | 0.4102 | KWS (0.4541) | 0.2368 | SAS (0.4131) | 0.2735 | SAS (0.3978) | Na | Acenocoumarin response – Dosage. Warfarin sensitivity. PMIM: 122700 Multiple genes. |
| C. 14 Risk factor variants | ||||||||
| C. 1. 7 Risk factor variants as per ClinVar annotation for complex disorders | ||||||||
| 2:138759649 rs11558538_ C_T c.314C>T; p.Thr105Ile HNMT | 0.1259 | KWS (0.1495) | 0.0595 | SAS (0.1053) | 0.1008 | FIN (0.1601) | AD | Asthma, susceptibility to; PMIM: 600807 Multiple genes. Yes, disorder seen in Arab country. |
| 4:100268190 rs283413_ C_A# $ c.232G>T; p.Gly78Arg ADH1C | 0.0653 | KWP (0.1071) | 0.0072 | SAS (0.0174) | 0.0157 | ASJ (0.0633) | IC,Mu | Parkinson’s disease, susceptibility to; PMIM: 168600 Multiple genes. |
| 5:95751785 rs6232_ T_C c.661A>G; p.Asn221Asp PCSK1 | 0.0594 | KWB (0.0909) | 0.0210 | SAS (0.0501) | 0.0390 | SAS (0.0659) | ? | Obesity, susceptibility to, Body mass index quantitative trait locus 12 PMIM: 612362. OMIM lists single gene but in reality, BMI is associated with multiple genes. |
| 10:64415184 rs7076156_ G_A# & c.1130-972G>A; p.Ala62Pro ZNF365 | 0.3351 | KWS (0.4450) | 0.1288 | EUR (0.2734) | 0.2044 | ASJ (0.2795) | Na | Uric acid nephrolithiasis, susceptibility to; PMIM: 605990. OMIM lists single gene but this is a multifactorial disorder. |
| 14:104165753 rs861539_ G_A c.1849-1239G>A; p.Thr241Met XRCC3 | 0.3864 | KWS (0.4450) | 0.2169 | EUR (0.3936) | 0.2904 | ASJ (0.4015) | AD | Cutaneous malignant melanoma 6, susceptibility to PMIM: 613972. OMIM lists single gene but this is a multifactorial disorder. |
| 17:5485367 rs12150220_ A_T c.464T>A; p.Leu155His NLRP1 | 0.4377 | KWP (0.4643) | 0.1921 | EUR (0.4443) | 0.3674 | ASJ (0.4744) | AR | Vitiligo-associated multiple autoimmune disease susceptibility 1; PMIM: 606579. OMIM lists single gene but this is a multifactorial disorder. |
| 17:48437456 rs6504649_ C_G c.2402C>G; p.Thr801Arg XYLT2 | 0.4414 | KWB (0.4706) | 0.2510 | EUR (0.4006) | 0.3312 | ASJ (0.4536) | AR | Pseudoxanthoma elasticum, modifier of severity; PMIM: 264800. OMIM lists multiple genes: XYLT1, XYLT2, ABCC6. Yes, disorder seen in Arab country. |
| C. 2. 7 Risk factor variants as per our inference but annotated as “pathogenic” in ClinVar annotation | ||||||||
| 9:116153891 rs1800435_ C_G c.177G>C; p.Lys59Asn ALAD | 0.1241 | KWB (0.1912) | 0.0635 | SAS (0.1585) | 0.0830 | ASJ (0.2207) | AR | Aminolevulinate dehydratase, ALAD*1/ALAD*2 allele at this position associated with susceptibility to lead poisoning. PMIM: 612740. Lead poisoning is becoming a common disease. Single gene. |
| 10:54531242 rs5030737_ G_A c.154C>T; p.Arg52Cys MBL2 | 0.0790 | KWP (0.0902) | 0.0272 | EUR (0.0596) | 0.0558 | ASJ (0.1032) | AD | Mannose-binding lectin deficiency. PMIM: 614372 Single gene. Complex trait |
| 15:100230557 rs121918530_ A_G c.782A>G; p.Asn261Ser MEF2A | 0.0103 | KWB (0.0294) | 0.0004 | EUR (0.0020) | 0.0008 | NFE (0.0015) | AD | Coronary artery disease/myocardial infection PMIM: 608320 Single gene. Complex trait |
| 17:12899902 rs5030739_ C_T c.1621G>A; p.Ala541Thr ELAC2 | 0.0842 | KWS (0.1055) | 0.0232 | SAS (0.0501) | 0.0349 | ASJ (0.0510) | AR or AD? Has to be seen in compound heterozygous state with another variant rs4792311. | Susceptibility to Prostate cancer, hereditary, 2′ PMIM: 614731 Single gene. Complex trait |
| 17:12915009 rs4792311_ G_A c.650C>T; p.Ser217Leu ELAC2 | 0.3552 | KWP (0.3611) | 0.2145 | EUR (0.3151) | 0.2742 | ASJ (0.3699) | AR or AD? Has to be seen in compound heterozygous state with the previous variant of rs5030739. | Susceptibility to Prostate cancer, hereditary, 2′; PMIM: 614731 Single gene. Complex trait |
| 17:79767715 rs1801483_ G_A c.118G>A; p.Gly40Ser GCGR | 0.0378 | KWS (0.0505) | 0.0042 | EUR (0.0149) | 0.0075 | ASJ (0.0120) | AD | Diabetes mellitus type 2, non-insulin dependent; PMIM: 125853; Multiple genes. Multifactorial complex disorder. Yes, disorder seen in Arab country. |
| 18:55373793 rs34719006_ C_T c.208G>A; p.Asp70Asn ATP8B1 | 0.0258 | KWS (0.0413) | 0.0018 | AFR (0.0045) | 0.0031 | ASJ (0.0096) | AD | Cholestasis of pregnancy; PMIM: 147480 Single gene. However, it is the most common liver disease unique to pregnancy. Complex trait. |
| D. 2 variants associated with complex traits by GWAS studies followed by clinical testing as per ClinVar annotation | ||||||||
| 2:27730940 rs1260326_ C_T# c.1337T>C; p.Leu446Pro GCKR | 0.3945 | KWS (0.4352) | 0.2933 | EAS (0.4812) | 0.3667 | ASJ (0.5344) | Not available | Fasting plasma glucose level quantitative trait locus 5 PMIM: 613463 Single gene (but, FPG levels are associated with multiple genes). |
| 11:68846399 rs35264875_ A_T c.1450A>T; p.Met484Leu TPCN2 | 0.1832 | KWP (0.2260) | 0.0996 | SAS (0.2055) | 0.1593 | FIN (0.2980) | Not available | Skin/hair/eye pigmentation, variation in, SHEP10 PMIM: 612267; OMIM lists single gene (but, the variations in skin/hair/eye pigmentation variations are associated with multiple genes). |
| E. 2 Protective variants as per ClinVar annotation | ||||||||
| 4:100260789rs698_ T_C c.1048A > G; p.Ile350Val ADH1C This variant is in LD with the variant R271Q (corresponding to the variant listed in the next row rs1693482) that is responsible for the differences in enzymatic differences | 0.3137 | KWS (0.3515) | 0.2143 | EUR (0.4046) | 0.3470 | FIN (0.5169) | Ic, Mu | Alcohol dependence, protection against; PMIM: 103780. Multiple genes. |
| 4:100263965 rs1693482_ C_T c.815G>A; p.Arg272Gln ADH1C | 0.3296 | KWS (0.3830) | 0.2143 | EUR (0.4046) | 0.3462 | FIN (0.5167) | Ic, Mu | Alcohol dependence, protection against; PMIM: 103780. Multiple genes. |
@Disorders relating to the following variants rs61757294 (CYP11B2, Pathogenic, DR, Corticosterone methyloxidase type 2 deficiency). rs1801483 (GCGR, reannotated as risk factor, AD form T2D), rs11558538 (HNMT, Risk factor, AD, susceptibility to asthma), rs1801280 (NAT2, drug response, AR, Slow acetylator due to N-acetyltransferase enzyme variant) and rs6504649 (XYLT2, Risk Factor, AR, Pseudoxanthoma elasticum, modifier of severity) are seen annotated in CAGS as observed in many Arab countries.
Seven of these 24 non-benign SAFD variants were seen annotated in ClinVar as “Pathogenic”; however, either the associated disorder was common/complex or more prevalent in the study population or the patient carrying the variant was annotated in OMIM as susceptible to the disorder (which is usually a common disorder). Going by the practice that “pathogenic” variants are related to Mendelian disorders, we considered the variants associated with common disorders as risk factors. (i) rs1800435_G > C (MAF_KWT:12.41%; MAF_1KGP:6.4%) associated with “Aminolevulinate dehydratase, alad*1/alad*2 polymorphism susceptibility to lead poisoning ALAD porphyria”. ClinVar annotated this as pathogenic based on literature evidence and likely benign based on clinical testing. It increases the risk for lead poisoning. ALAD porphyria is a very rare genetic metabolic disease; however, quoting from the CDC report on lead poisoning – “There are approximately half a million U.S. children ages 1–5 with blood lead levels above 5 micrograms per deciliter (µg/dL), the reference level at which CDC recommends public health actions be initiated”, lead poisoning is no longer a rare disorder. Susceptibility is the keyword, and we reannotated this variant as risk factor. (ii) rs5030737 (MAF_KWT:7.90%; MAF_1KGP:2.8%) associated with Mannose-binding lectin deficiency, which is a complex trait. ClinVar annotated the variant as pathogenic based on literature reference; since we associate “pathogenic” to Mendelian disorders, we reannotated this variant as risk factor. (iii) rs121918530 (MAF_KWT:1.03%; MAF_1KGP:0.04%)associated with coronary artery disease/myocardial infarction, which is a complex multifactorial disorder. ClinVar annotated this variant as pathogenic based on literature evidence and likely benign based on clinical testing. since we associate “pathogenic” to Mendelian disorders, we reannotated this variant as risk factor. (iv) rs5030739 (MAF_KWT:8.42%; MAF_1KGP:2.32%) associated with “Prostate cancer hereditary 2, susceptibility to” (Complex trait). ClinVar annotated this variant as pathogenic based on literature evidence and benign based on clinical testing. The cited literature suggested increased risk of prostate cancer; ‘susceptibility to’ was the keyword. Thus, this variant was considered as risk factor. It was also the case that this variant has to appear in compound heterozygosity with the next listed variant of rs4792311. We reannotated this variant as risk factor. (v) rs4792311 (MAF_KWT:35.52%; MAF_1KGP:21.5%) associated with “Prostate cancer hereditary 2, susceptibility to” (Complex trait). ClinVar annotated this variant as pathogenic based on literature evidence and benign based on clinical testing. The cited literature suggested increased risk of prostate cancer; ‘susceptibility to’ is the keyword. Thus, this variant was considered as risk factor. It was also the case that this variant had to appear in compound heterozygosity with the previous listed variant of rs5030739. We reannotated this variant as risk factor. (vi) rs1801483 (MAF_KWT:3.78%; MAF_1KGP:0.42%) associated with Diabetes mellitus type 2, non-insulin dependent (a multifactorial complex disorder). ClinVar annotated the variant as pathogenic (based on literature evidence). Considering that we associate “pathogenic” only with Mendelian disorder, we reannotated the variant as risk factor. (vii) rs34719006 (MAF_KWT:2.58%; MAF_1KGP:0.18%) associated with Cholestasis of pregnancy, which is a most common liver disease unique to pregnancy. (Complex trait). ClinVar annotated this variant as pathogenic based on evidence from literature study and annotated as with conflicting evidence between likely benign (clinical testing), uncertain significance (Clinical testing). Considering that we associate “pathogenic” only with Mendelian, we reannotated this variant as risk factor.
This set of 24 SAFD variants with clinical significance was distributed onto (a) A set of 2 pathogenic variants (rs61757294 and rs61751507) with AR mode inheritance; the MAF of these two variants in Kuwaiti exomes were uncharacteristic of pathogenic variants (see above for more details); (b) A set of 4 drug response variants one of which was AR); (c) A set of 14 risk variants and 2 protective variants for complex traits (3 were AR); and (d) A set of two variants associated with phenotype traits through GWAS studies. Five of the disorders associated with the SAFD variants were seen annotated in CAGS as observed in Arab countries (see Table 6).
Assessing the Loss-of-Function SAFD Variants for clinical significance
We had identified 26 LoF SAFD variants (Supplementary Table S4); as many as 15 of these were stop-gain, seven were start-loss and the remaining four were splice site mutations. None of these 26 SAFD LoF variants was seen annotated for disorder in OMIM; however, the GWAS Catalog51 listed one of these variants namely rs2228015-C from CCR7 gene as associated with the complex phenotype trait of lymphocyte counts (at genome-wide significant p-value of 6E-09).
CAGS disorders for which the OMIM-listed causal variants were seen in Kuwaiti exomes
We further examined the CAGS database for disorders observed in Kuwait at any incidence rate and for disorders seen in any of the Arab countries at incidence rates of ≥11 per 100,000. CAGS database provided the Phenotype MIM number using which we retrieved the OMIM-reported causal variants and checked for their occurrences in Kuwaiti exomes. For 25 disorders, the OMIM-reported variants were seen in Kuwaiti exomes (Table 7); eight of these 25 disorders had already been seen in the analysis for functional variants. Except in one instance (rs1800858), all the variants were missense. 13 of these variants were “pathogenic” and the remaining 12 were “risk factor” variants. 18 of these disorders were observed in Kuwait and the remaining in other Arab countries.
Scrutinization of the identified variants against Arab mutations reported in Arab studies
Analyses performed so far in the study indicated that disorders relating to 20 instances of rare & deleterious variants, 16 of which were pathogenic variants for rare disorders and 4 were risk factor variants for complex disorders (see Table 3), 7 instances of pharmacogenomic variants that were associated with complex disorders in Arab studies (see Table 5), 5 instances of SAFD variants (see Table 6), and 17 additional instances from the analysis of CAGS disorders were seen in Arab population (see Table 7). During the analysis, we also found in Kuwaiti exomes two recessive mutations (namely rs1801133 & rs1801131 from MTHFR – see Table 8) associated with recessive early onset of susceptibility to Type 2 diabetes in Arab population. We set upon to identify which of these variants were also reported in Arab studies for the corresponding disorder. Upon performing literature survey and manual examination of the bibliography data presented in CAGS database, these variants could be classified onto the following categories (Table 8): (a) 16 Instances where the OMIM-listed variants identified in Kuwaiti exomes were also reported as Arab mutations in Arab studies. 9 of these were pathogenic variants for rare disorder; 1 was drug response; and 6 were risk factors for complex disorders. (b) 7 Instances where the identified pharmacogenomic variants in Kuwaiti exomes were also observed as associated with disorders in Arab studies. These were drug response variants to complex disorders. (c) 12 instances of disorders where the genetic basis at SNV level had not been reported in Arab studies. 7 of these were pathogenic variants for rare disorders and 5 were risk factors for complex disorders. (d) 10 Instances where the Arab studies reported variants different from the OMIM-listed variants observed in Kuwaiti exomes; however, the Arab reported variants were from the same gene. The Arab variants were generally seen in OMIM but not in our exomes. Eight of these variants were pathogenic for rare disorders and two were risk factors for complex disorders. (e) 7 Instances of variants (from 4 disorders) where the Arab studies reported variants from genes different from those of the OMIM-listed variants observed in Kuwaiti exomes. In general, the different gene and the different mutations were listed in OMIM for the disorder but were not seen in Kuwaiti exomes. All the 7 variants were risk factors for complex disorders.
Table 8.
Evaluation of the identified variants for observation as Arab mutations in Arab studies.
| dbSNP ID; Ref_risk alleles; gene | DISORDER; (PMIM); Number of OMIM-listed genes for the disorder; Reference for the Arab study. | Clin. Signific.; CAGS inciden. (per 100,000) | Inheritance mode of the disorder |
CARRIER FREQ (rr/rR/RR) |
RAF (KWT_RAF/1KGP_RAF/GME_RAF |
RATIO (KWT-1KGP/KWT-GME) |
|---|---|---|---|---|---|---|
| A. 16 Instances wherein the identified functional variants match with the variants reported in arab studies. | ||||||
| 4 Rare & deleterious variants – Pathogenic and rare disorder. | ||||||
| rs137853054 (G_A) PARK2 |
Parkinson disease, juvenile, type 2; (PMIM:600116). GARD lists this as rare disorder. OMIM lists single gene92; | Pathogenic and rare disorder | AR | 0/2/288 | 0.0034/0.0002/0.0025 | 17/1.3503 |
| rs61754375 (G_A) TYR |
Tyrosinase-negative oculocutaneous albinism, Type IA; (PMIM:203100) – OMIM lists single gene108; | Pathogenic and rare disorder | AR | 0/1/286 | 0.0017/0.0002/. | 8.5/. |
| rs121964924 (A_G) DPYS |
Dihydropyrimidinase deficiency; (PMIM:222748) – OMIM lists single gene109; | Pathogenic and rare disorder | AR | 0/1/289 | 0.0017/0.0002/0.0020 | 8.5/0.8424 |
| rs58331765 (C_T) ABCA4 |
Stargardt disease 1; (PMIM:248200) – OMIM lists two genes93; | Pathogenic and rare disorder | AR | 0/2/288 | 0.0034/0.0028/0.0005 | 1.2143/6.7460 |
| 2 Safd variants | ||||||
| rs61757294 (A_G) CYP11B2 |
Corticosterone methyloxidase type 2 deficiency. (PMIM: 610600)110. this cited study is on Iranian-Jewish origin patients. | Pathogenic and rare disorder | DR; this and rs28931609 to be homozygous. | ?? | 0.1519/0.0531/? For variants of DR mode of inheritance higher MAF is alright though it is a pathogenic variant. |
2.8606/? |
| rs1801280 (T_C) NAT2 | Slow acetylator due to N-acetyltransferase enzyme variant; (PMIM:243400) – OMIM lists single gene111; | Drug Response | AR | 41/134/100 | 0.3927/0.2927/0.4340 | 1.3416/0.9047 |
| 8 Cags arab disorders | ||||||
| rs79204362 (C_T) CYP1B1 |
Early onset of Glaucoma, digenic; (PMIM: 231300) – OMIM lists single gene112; This is a rare disorder but reaches high prevalence in Saudi Arabian populations. | Pathogenic and rare disorder; 11–50 | AR | 0/6/284 | 0.010/0.004/0.013 CTGA reports 1 in 2,500 among Saudi Arabians. MAF is only marginally higher at 1.03%. |
2.4587/0.7553 |
| rs61732874 (C_A) MEFV |
Familial Mediterranean Fever (FMF) Recessive; (PMIM: 249100) – OMIM lists single gene113; | Pathogenic; rare disorder 51–100 | AR | 0/9/278 | 0.015/0.001/0.012 FMF is a rare disorder in Europe; it is no longer rare in certain populations105. CAGS’ incidence is 51–100; varies 1/2000 to 1/100 in eastern Mediterranean populations. Thus, MAF of 1.55% is acceptable for pathogenic variant. |
8.6026/1.2281 |
| rs121908530 (G_A) AGXT |
Type 1 primary Hyperoxaluria (PMIM: 259900) – OMIM lists single gene114; | Pathogenic; rare disorder | ? | 0/1/290 | 0.001718/./. | ./. |
| rs587776954 (A_G) C12orf57 |
Temtamy syndrome (PMIM:218340) – OMIM lists single gene115; | Pathogenic; rare disorder | AR | 0/2/288 | 0.003436/./0.003021 | ./1.1374 |
| rs61747728 (C_T) NPHS2 |
Susceptibility to Nephrotic syndrome, type 2 (PMIM: 600995) – OMIM lists single gene116; | Risk factor for rare disorder. 11–50 | AR | 0/8/281 | 0.01375/0.0145767/0.012085 | 0.9433/1.1377 |
| rs1799945 (C_G) HFE |
Type 1 Hemochromatosis - microvascular complications of diabetes, susceptibility to, 7, included (PMIM:235200). OMIM lists two genes117–119; | Risk factor for common disorder; >100 | AR | 4/63/224 | 0.122/0.073/0.119 | 1.6693/1.0180 |
| rs1801131 (T_G) MTHFR |
Schizophrenia; sczd (PMIM:181500) – OMIM lists several genes120; | Risk Factor; complex disorder; >100 | AD | 31/128/132 | 0.3265/0.3264/0.3353 | 1.0001/0.9736 |
| rs77775126 (C_T) RP1 |
Retinitis pigmentosa 1 (PMIM: 180100) – OMIM lists single gene121,122. | Risk factor by inference for common disorder; 11–50 | AR | 0/11/280 | 0.0189/0.0189/0.0181 | 0.9999/1.0426 |
| 2 Variants that came up during the examination for arab study variants | ||||||
| rs1801133 (G_A) MTHFR |
Susceptibility to T2DM in Lebanese123,124. | Risk Factor; complex disorder | AR | 12/81/197 | 0.1810/0.2454/0.2557 | 0.7376/0.7077 |
| rs1801131 (T_G) MTHFR |
Protective effect: T2DM in Israel Jews and South Indians123,125. | Risk Factor; complex disorder | AD | 31/128/132 | 0.3265/0.2494/0.3353 | 1.3091/0.9737 |
| B. 7 Instances wherein the identified pharmacogenomic variants are implicated in arab disorders through arab studies. in all the instances, multiple genes are involved in the disorder. | ||||||
| rs1042713 (A_G) ADRB2 |
PharmGKB: drug response to asthma. OMIM: susceptibility to asthma, nocturnal PMIM:600807). Arab study: susceptibility to early onset obesity and insulin resistance96,102. | Drug response; complex disorder | AD | 82/126/55 | 0.5513/0.5244/0.5645 A->G (Arg16Gly) is causative; hence the frequencies of the risk allele G rather than the minor allele are listed. | 1.0513/0.98 |
| rs1042714 ADRB2 |
PharmGKB: drug response to asthma. OMIM: susceptibility to obesity and to childhood asthma. PMIM: 601665. Arab study: for Coronary artery disease/myocardial infarction103,104. | Drug response; complex disorder | AR | 19/90/176 | 0.2246/0.2053/0.2412 | 1.094/0.9312 |
| rs1805087 (A_G) MTR |
PharmGKB: drug response to Rheumatoid arthritis. OMIM: does not list. Arab study: for Autism – North Iran97. | Drug response; complex disorder | GG is the risk factor. | 23/105/162 | 0.2603/0.2183/0.2021 | 0.2603/0.2021 |
| rs1042522 (G_C) TP53 |
PharmGKB: drug response to stomach neoplasm. OMIM: smoking related accelerated decline in lung function PMIM:608852; Arab study: in susceptibility to Breast Cancer in Tunisia98. | Drug response; complex disorder | ?? | 57/140/90 | 0.4425/0.5429/. | 0.815/. |
| rs1800497 (G_A) ANKK1 |
PharmGKB: drug response to Schizophrenia. OMIM: Taq1A polymorphism associated with neuropsychiatric disorders. Arab study: risk factor for Schizophrenia (PMIM:181500) in Egyptians99. | Drug response; complex disorder | AD? | 7/85/197 | 0.1713/0.3257/0.1908 | 0.5259/0.8975 |
| rs1801058 (T_C) GRK4 |
PharmGKB: drug response to Hypertension. OMIM: does not list. Arab study: risk factor for myocardial infarction among hypertensive subjects in Jordan100. | Drug response; complex disorder | AD? | 16/101/173 | 0.2293/0.3067/0.292 | 0.7476/0.7079 |
| rs699 (A_G) AGT |
PharmGKB: drug response to Hypertension. OMIM: susceptibility to Essential hypertension (PMIM:145500). Arab study: Reduced life-span through genetic susceptibilities to Hypertension and Myocardial Infarction in UAE101. | Drug response; complex disorder | Mu? | 91/127/47 | 0.583/0.7051/0.5534 | 0.8268/1.0535 |
| C. 12 Instances wherein the genetic basis for the disorder has not been reported in arab studies. | ||||||
| 4 Rare & deleterious variants | ||||||
| rs28940872 (C_T) ACADS |
Scad deficiency (PMIM:606885). Single gene listed in OMIM. | Pathogenic; rare disorder; | AR | 0/1/289 | 0.0017/0.0002/0.0015 | 8.5/1.1251 |
| rs28941785 (C_T) CTH |
Cystathioninuria (PMIM:219500). Single gene listed in OMIM. | Pathogenic; rare disorder; | AR. | 0/2/281 | 0.0035/0.0026/0.0065 | 1.3461/0.5347 |
| rs28940885 (C_T) GALE |
Galactose Epimerase Deficiency (PMIM:230350). Single gene listed in OMIM. | Pathogenic; rare disorder; | AR | 0/1/290 | 0.0017/0.002/0.0025 | 0.85/0.6751 |
| rs114817817 (C_T) SRGAP1 |
Susceptibility to Thyroid cancer, nonmedullary 2, (PMIM:188470). OMIM lists three genes for this disorder. | Risk Factor; common disorder | AD; (AR,AD,MF) | 0/1/285 | 0.0017/0.0006/0.0070 | 2.8333/0.2415 |
| 1 Safd variants | ||||||
| rs6504649 (C_G) THR801ARG XYLT2 |
Pseudoxanthoma elasticum, modifier of severity. (PMIM:264800). OMIM lists three genes. | Risk Factor; common disorder | AR | 60/136/94 | 0.4414/0.251/0.3962 | 1.7586/1.1139 |
| 8 7 Cags arab variants | ||||||
| rs118204113 (G_A) HMBS |
Acute intermittent porphyria (PMIM:176000). Single gene listed in OMIM. | Pathogenic; rare disorder | AD | 0/1/289 | 0.001718/./0.000504 | ./3.4087 |
| rs4149584 (C_T) TNFRSF1A |
Familial periodic fever, autosomal dominant. (PMIM:142680). Single gene listed in OMIM. | Pathogenic; rare disorder; 51–100 | AR | 0/4/287 | 0.0069/0.0069/0.0121 | 1.0000/0.5687 |
| rs56208331 (G_A) GATA4 |
Tetralogy of fallot (PMIM:187500). Single gene listed in OMIM. | Pathogenic; rare disorder; 51–100 | AD | 0/2/289 | 0.0034/0.0034/0.0025 | 0.9998/1.3645 |
| rs104893836 (T_C) GNRHR |
Hypogonadotropic hypogonadism 7 without anosmia. (PMIM:146110). Single gene listed in OMIM. | Pathogenic; rare disorder | AR | 0/1/290 | 0.0017/0.0012/0.0055 | 1.4167/0.3069 |
| rs2476601 (A_G) PTPN22 |
Susceptibility to T1DM (PMIM:222100). Also associated with susceptibility to Systemic lupus erythematosus (PMIM:152700). OMIM lists multiple genes. | Risk Factor; common disorder | AR | 0/5/280 | 0.0292/ 0.0274/0.0131 | 1.00/0.98 |
| rs17158558 (C_T) RET |
Susceptibility to hirschsprung disease 1 (PMIM 142623). Single gene listed in OMIM. | Risk Factor; common disorder | AD | 0/15/273 | 0.02577/0.0219649/0.045867 | 1.1732/0.5618 |
| rs121918219 (G_A) VANGL1 |
Susceptibility to neural tube defects. (PMIM: 182940); OMIM lists 5 genes. | Risk Factor; common disorder; >100 | AD | 0/1/289 | 0.001718/./0.001007 | ./1.7060 |
| D. 10 Instances wherein different mutations from the same gene are seen in arab studies (generally seen in omim but not in our exomes). | ||||||
| 7 Rare & deleterious variants | ||||||
| rs121908736 (G_A) ADA | Partial adenosine deaminase deficiency (PMIM:102700). Arab studies: CAGS reports another mutation from the same gene p.Arg282 > Gln126. |
Pathogenic; rare disorder | AR, SM. Compound heterozygosity | 0/2/285 | 0.0035/0.0018/0.0005 | 1.9444/6.9444 |
| rs61757582 (G_A) DHCR7 |
Smith-Lemli-Opitz syndrome (PMIM:270400). Arab studies: N287K (861 C > A); R352Q (1055 G > A) (rs121909768; seen in OMIM); and R352L (1055 G > T)127. |
Pathogenic; rare disorder | AR | 0/1/284 | 0.0018/0.0002/. | 9/. |
| rs61753185$ (G_A) TYR |
Tyrosinase-negative oculocutaneous albinism, Type IA (PMIM:203100). Arab studies: c.817 G > C/p.W272C (rs62645902; not seen in OMIM)108,128. | Pathogenic; rare disorder | AR | 0/1/290 | 0.0017/0.0004/0.0005 | 4.25/3.3730 |
| rs116100695 (G_A) PKLR |
Pyruvate kinase deficiency of red cells (PMIM:266200). Arab studies: 1058 C > T; Thr353Met (rs74315362 seen in OMIM)129. | Pathogenic; rare disorder | AR | 0/2/289 | 0.0034/0.0016/0.0096 | 2.125/0.3547 |
| rs41295338 (G_T) TGM1 |
AR congenital ichthyosis 1 (PMIM:242300). Arab studies: Compound heterozygosity for missense mutations (R141H- rs121918718, R142H-rs121918719), (p.Tyr136Ter-rs1057517836), (p.Ser326Cysfs*8), and Leu362Arg130,131 | Pathogenic; rare disorder | AR | 0/2/288 | 0.0034/0.0022/0.0060 | 1.5454/0.5627 |
| rs121434513 (G_C) PNKD alias MR1 |
Paroxysmal Nonkinesigenic Dyskinesia 1 (PMIM: 118800). Arab studies: c.20 C > T; A7V rs121434512 – seen in OMIM132. | Pathogenic; rare disorder | AD | 0/1/289 | 0.0017/0.0002/0.0005 | 8.5/3.3530 |
| rs34424986 (G_A) PARK2 |
Parkinson disease, juvenile, type 2. (PMIM:600116). Arab studies: exon 4 deletion and a 2-base AG deletion in exon 2 (101–102) from the same gene associated with the disorder133. Not seen in OMIM. This table reports another variant rs137853054 seen in Arab study and in Kuwaiti exomes133–135. | Pathogenic; rare disorder 11–50 | AR | 0/2/289 | 0.0034/0.0004/. | 8.5/. |
| 3 Cags arab disorders | ||||||
| rs12021720 (T_C) DBT |
Maple syrup urine disease, intermediate, type II (PMIM:248600). Arab studies: c.1281 + 1 G > T in one patient from UAE. Not seen in OMIM136. | Pathogenic; rare disorder | AR | 5/67/218 | 0.1347/0.1082/0.12 The variant is in compound heterozygosity with [rs121965001]46. Hence high MAF is acceptable. |
0.97/0.98 |
| rs1800858 (G_A) RET |
Susceptibility to Hirschsprung disease 1; HSCR1 (PMIM: 142623). Arab studies: c.1852T > C Cys618Arg mutation rs76262710. Seen in OMIM (for neoplasia)137,138. | Risk Factor; common disorder; 11–50 | AD | 25/128/138 | 0.3058/0.2464/0.2593 | 1.24/1.1793 |
| rs121918530 (A_G) MEF2A |
Coronary artery disease/myocardial infarction (PMIM:600660). Arab studies: (rs1059759 G > C) in Saudi Arabian patients139. | Risk factor by inference for complex disorder. | AD | 0/6/284 | 0.01031/0.000399361/0.002014 | 25.8162/5.119 |
| E. 7 instances wherein the Arab studies report different mutation from different gene (but associated with the disorder in OMIM). These disorders involve multiple genes. In general, the different gene and the different mutations are seen in OMIM associated with the disorder. The Arab mutations are not seen in Kuwaiti exomes. | ||||||
| rs11909217 (C_T) LIPI RARE & DELETERIOUS |
Susceptibility to Hypertriglyceridemia, Familial (PMIM: 145750). OMIM lists LIPI and APOA5 for the disorder. Arab study: 3′ UTR C > G variant from APOC3 (not seen in OMIM or our exomes)95. This is an inherited common disorder. | Risk factor by inference; common disorder. | AD | 0/10/280 | 0.0172/0.006/0.01309 | 2.8667/1.3138 |
| rs3135506 (G_C) APOA5 CAGS VARIANT |
Risk factor; Common disorder | AD | 1/22/267 | 0.04124/0.0557109/0.070565 | 0.7402/0.5844 | |
|
rs28932472 (G_C)
POMC RARE & DELETERIOUS |
Obesity, early-onset, susceptibility to (PMIM: 601665). OMIM lists several genes. Arab study: rs1042713: Arginine 16 Glycine (Arg16Gly) polymorphism in ADRB2 gene; which is associated with susceptibility to nocturnal asthma in OMIM and our study. rs1042713 A_G: (AA:55, AG:126, GG:82) & (MAF_KWT, 1KGP, GME: 0.5513/0.5244/0.5645)102. | Risk Factor; common disorder |
AR,AD,MF | 0/1/287 | 0.0017/0.0024/0.0005 | 0.7083/3.3730 |
| rs34911341 (C_T) GHRL RARE & DELETERIOUS |
Risk Factor; common disorder | AR,AD,MF | 0/1/290 | 0.0017/0.0026/0.0035 | 0.6538/0.4823 | |
| rs2282440 (G_A) SDC3 CAGS variant |
Association variant; common disorder; >100 | AD | 0/11/280 | 0.0189/0.0189/0.0282 | 0.9999/0.6703 | |
| rs11558538 (C_T) HNMT SAFD variant |
Susceptibility to Asthma (PMIM: 600807). OMIM lists several genes. Arab studies: 786 T > C [rs2070744] from the 5’ flanking region of NOS3 (OMIM lists this variant but the variant is not seen in Kuwaiti exomes)140. |
Risk Factor for common disorder | AD | 7/55/212 | 0.1259/0.0595/0.0968 | 2.1159/1.3009 |
| rs1801483 (G_A) GCGR SAFD Variant |
T2DM (PMIM: 125853). Arab studies: Ala222Val (rs1801133 C > T AR/AD) mutation from MTHFR gene in Lebanese patients with T2DM and nephropathy123,124. The rs1801133 is seen in Kuwaiti exomes (MAF:KWT/1KGP/GME = 0.1810/0.2454/0.2557; AA/AG/GG: 12/81/197). Another MTHFR variant Glu429Ala (rs1801131 T_G AR) in homozygous form has protective effect in Israeli Jews123 and South Indians125. The rs1801131 variant is seen in Kuwaiti exomes (MAF: KWT/1KGP/GME = 0.3265/0.2494/0.3353; GG/GT/TT: 31/128/132). |
Risk factor by way of inference | AD | 0/22/269 | 0.0378/0.0042/0.0311 | 9/1.2148 |
Identified variants and the associated complex disorders
Examining our in-house genome-wide association study (GWAS) data (on 1351 native Kuwaiti Arab individuals genotyped on Illumina HumanOmniExpress BeadChip and 1900 native Kuwaiti Arab individuals genotyped on Illumina HumanCardio-Metabo BeadChip) for the presence of the variants identified through exome analysis revealed that 27 of the identified OMIM-listed causal variants present in Kuwaiti exomes were also seen in the GWAS data. Allele frequencies and carrier distributions as seen in exomes data set and GWAS data set are presented (Table 9). 13 of these 27 variants were associated with disorders observed in Arab studies. The 27 variants were pharmacogenomic (11), SAFD (9) and CAGS (5 + 2) variants for complex disorders. The allele frequencies among the data sets of Kuwait exome, Kuwait GWAS and GME were comparable with each other; and the carrier distributions were similar between the Kuwaiti exomes and GWAS data sets.
Table 9.
Comparison of allele frequencies & carrier distribution of the reported variants from Kuwaiti exome data set with a larger data set of our in-house genome-wide genotype data.
| dbSNP ID; Ref_risk; gene | Type of variant & whether the variant has been observed in Arab studies | Associated disorder | Risk allele frequency in Kuwaiti exomes/GME & Carrier distribution in Kuwaiti exomes | Risk allele frequency in Kuwaiti GWAS data set & Carrier distribution in GWAS data |
|---|---|---|---|---|
| rs699 (A_G) | Pharmacogenomic variant for hypertension; | PharmGKB listed this variant as drug response for Hypertension). (OMIM lists this variant as susceptibility to Essential hypertension (PMIM:145500). Arab study implicates this in Reduced life-span through genetic susceptibilities to both Essential Hypertension and Myocardial Infarction in UAE101. | 0.583/0.5534 | 0.5658 |
| AGT | Arab mutation | 91/127/47 | 439/653/260 | |
| rs1805087 (A_G) | Pharmacogenomic variant for Rheumatoid arthritis. | Arab study implicates this in Autism – North Iran97. | 0.2603/0.2021 | 0.2262 |
| MTR | Arab variant. | 23/105/162 | 77/456/819 | |
| rs1367117 (G_A) | Pharmacogenomic variant for hypertension | Hypertension | 0.1448/0.1727 | 0.1647 |
| APOB | 5/74/211 | 33/379/939 | ||
| rs2960306 (G_T) | Pharmacogenomic variant for hypertension. | Hypertension | 0.371/0.362 | 0.3607 |
| GRK4 | 42/129/166 | 182/612/559 | ||
| rs1024323 (C_T) | Pharmacogenomic variant for hypertension | Hypertension | 0.3718/0.3887 | 0.3544 |
| GRK4 | 33/137/103 | 172/608/562 | ||
| rs1801058 (T_C) | Pharmacogenomic variant for hypertension | Arab study implicates this as risk factor for myocardial infarction among hypertensive subjects in Jordan100. PharmGKB listed this variant as drug response for Hypertension). (OMIM does not list this variant). | 0.229/0.292 | 0.2627 91/529/733 |
| GRK4 | Arab mutation | 16/101/173 | . | |
| rs1042713 (A_G) | Pharmacogenomics variant for asthma. | Susceptibility to early onset obesity, insulin resistance etc.) in Saudi Arabia102. | 0.5513/0.5645 | 0.5814 |
| ADRB2 | Arab mutation | OMIM listed this variant for susceptibility to asthma, nocturnal PMIM:600807). | 82/126/55 | 376/533/196 |
| rs1800888 (C_T) | Pharmacogenomics variant | Drug response for asthma and heart failure | 0.01211/0.0156 | 0.014 |
| ADRB2 | 0/7/282 | 2/33/1316 | ||
| rs10509681 (T_C) | Pharmacogenomics variant for diabetes and cancer. | Drug response for Rosiglitazone and Repaglinide | 0.1197/1047 | 0.1223 |
| CYP2C8 | 3/63/222 | 32/266/1053 | ||
| rs1048943 (T_C) | Pharmacogenomics variant. | Warfarin sensitivity | 0.065/0.0619 | 0.042 |
| CYP1A1 | 3/32/256 | 4/101/1216 | ||
| rs1042522 (G_C) | Pharmacogenomics variant. | Drug response for stomach neoplasm. | 0.443/. | 0.4039 |
| TP53 | Arab variant | Susceptibility to Breast Cancer in Tunisia98 | 57/140/90 | 236/620/496 |
| rs861539 (G_A) | SAFD variant | Cutaneous malignant melanoma 6, susceptibility to | 0.3864/0.3836 | 0.3843 |
| XRCC3 | AD | PMIM: 613972 | 35/151/100 | 166/517/422 |
| rs35264875 (A_T) | SAFD variant | Skin/hair/eye pigmentation, variation in, SHEP10 | 0.1832/0.1677 | 0.1906 |
| TPCN2 | Association by GWAS | PMIM: 612267; | 12/72/178 | 59/397/895 |
| rs1260326 (C_T) | SAFD variant | FPG quantitative trait locus 5 | 0.3945/0.5388 | 0.469 |
| GCKR | PMIM: 613463 | 47/134/108 | 312/645/396 | |
| rs7076156 (G_A) | SAFD variant | Uric acid nephrolithiasis, susceptibility to; | 0.3351/0.28 | 0.3045 |
| ZNF365 | PMIM: 605990 | 33/129/129 | 130/562/659 | |
| rs5030737 (G_A) | SAFD variant | Mannose-binding protein deficiency. Complex trait | 0.0790/0.0535 | 0.0673 |
| MBL2 | AD | PMIM: 614372 | 2/39/231 | 6/167/1177 |
| rs6232 (T_C) | SAFD variant. | Obesity, susceptibility to, Body mass index quantitative trait locus 12 | 0.0594/0.0524 | 0.046 |
| PCSK1 | PMIM: 612362 | 1/32/253 | 2/119/1227 | |
| rs1801280 (T_C) | SAFD variant | Slow acetylator due to N-acetyltransferase enzyme variant. Toxicity to the drugs of cisplatin or cyclophosphamide. | 0.3927/0.434 | 0.433 |
| NAT2 | PMIM: 243400 | 41/134/100 | 182/483/313 | |
| AR | Arab mutation. | |||
| rs4792311 (G_A) | SAFD variant | Prostate cancer, hereditary, 2′; Complex trait |
0.3552/0.3092 | 0.3396 |
| ELAC2 | PMIM: 614731 | 30/146/114 | 167/583/602 | |
| AR or AD? | ||||
| Has to be in compound heterogeneity with rs5030739 | ||||
| rs2108622 (C_T) | SAFD variant – drug response. | Acenocoumarin response – Dosage. Warfarin sensitivity PMIM: 122700 | 0.4102/0.3983 | 0.4146 |
| CYP4F2 | 53/127/104 | 239/643/469 | ||
| rs1801131 (T_G) | CAGS; Arab mutation | Schizophrenia; sczd. (PMIM:181500). OMIM lists several genes. | 0.3265/0.3353 | 0.3527 |
| MTHFR | 31/128/132 | 146/659/544 | ||
| rs4149584 (C_T) | CAGS variant | Periodic fever, familial, autosomal dominant. (PMIM:142680). | 0.0069/0.0121 | 0.0113 |
| TNFRSF1A | Genetics basis for the disorder has not been reported in Arab studies. AR | 0/4/287 | 0/13/606 | |
| rs2282440 (G_A) SDC3 |
CAGS variant AD |
Obesity, early-onset, susceptibility to (PMIM: 601665). | 0.0189/0.0282 0/11/280 |
0.0147 0/11/361 |
| rs17158558 (C_T) | CAGS variant | Hirschsprung disease, susceptibility to, 1 (PMIM 142623). Complex disorder. Genetics basis is not reported in Arab studies. | 0.0260/0.0459 | 0.0325 |
| RET | AD | 0/15/273 | 2/84/1266 | |
| rs1799945 (C_G) | CAGS variant | Hemochromatosis, type 1 -microvascular complications of diabetes, susceptibility to, 7, included. (PMIM:235200). It is a common disorder. | 0.1219/0.1198 | 0.1156 |
| HFE | AR | 4/63/224 | 23/264/1061 | |
| rs1801133 (G_A) MTHFR |
AR Arab mutation |
Susceptibility to T2DM in Lebanese123,124 | 0.1810/0.2557 12/81/197 |
0.255 511/89/751 |
| rs1801131 (T_G) MTHFR |
AD Arab mutation |
Protective effect T2DM in Israel Jews123 and in South Indians. |
0.3265/0.3353 31/128/132 |
0.3527 147/660/546 |
Discussion
In this study, exomes from 291 healthy, unrelated native Kuwaiti Arabs were analysed to identify 170,508 SNVs and 3,341 indels. 12% of SNVs and 28% of indels were novel. One-third of the identified SNVs were population-specific, and 21.7% were ‘personal’ (observed in only one Kuwaiti exome and not seen in GME or 1KGP), consistent with the results of other studies on ethnic populations, including those from Qatar44, Spain17 and Denmark12. 53% of the identified SNVs were missense, and an average of 1.3% of the 14,557 SNVs that each person carried were predicted to affect protein function. Allele frequencies in 6,186 SAFD variants were significantly different from those observed in 1KGP populations.
Recent population genetic analyses have demonstrated that humans harbour an abundance of rare & deleterious variations, with >80% of all coding variants having a frequency of ≤1%10,14,52. In this study, a majority (51%) of the identified SNVs in Kuwaiti exomes were rare. Of the identified 55,644 population-specific SNVs, only 138 were ‘common’, and the rest were ‘rare’ or ‘low-frequency’. Up to 60% of the population-specific variants were missense changes, and 51% of LoF variants were population-specific (some of which were polymorphic). These observations support the notion that coding variants with allele frequency of <1% show increased population-specificity and are enriched for functional variants13. Human populations have experienced recent explosive growth, expanding by at least three orders of magnitude over the past 400 generations; such a rapid recent growth along with weak purifying selection has increased the load of rare variants, many of which are deleterious and relevant for understanding disease risks14,16.
On average, nearly 10.4% of Kuwaiti population-specific variants found in every Kuwaiti individual were homozygous; this extent of homozygosity, which is higher than that observed in other ethnic populations (such as the value of 7.05% in Spanish17), reflects the higher rate of consanguinity practised among the Kuwaiti Arab population. The GME study26 demonstrated an increased burden of runs of homozygosity in Greater Middle East populations; our previous works had shown that Kuwaiti population is heterogeneous (placed between populations that have large amount of ROH and the ones with low ROH) with the KWS subgroup as highly endogamous24. An average of 73 LoF variants (of which 4.5 were Kuwaiti-specific) were seen per individual. Observed disease-causing mutations failing to cause disease in at least a proportion of the individuals who carry them has been extensively discussed53. On an average, only 4.67% of Kuwaiti-specific LoF variants per individual were seen homozygous (as opposed to the expected 10.4%) and such a reduced homozygosity among LoF variants may explain the reduced penetrance.
Rare homozygous loss of function variants are supposed to exhibit strong signs of selective pressure. Of the genes harboring the identified 36 rare (MAF <2.0%) homozygous putative LoF variants observed in Kuwaiti exomes, only 8 were seen common with published list of inactivated genes from Icelanders42 and only 1 was common with the list from ExAC8. These findings suggest that the set of non-clinically relevant loss-of-function variants is far from being complete26 and consideration of ethnic populations with consanguinity as in the GME study and our study can augment the list of human knock-out events.
We previously catalogued36 exome variants from 15 native Kuwaiti individuals of KWS subgroup (city-dwelling Saudi Arabian tribe ancestry24) and postulated that further samples were needed to capture the full spectrum of exome variability. The present study indicated that our previous work captured only a portion (22%) of variability. The repertoire of ‘all’ SNVs and ‘population-specific’ variants increased with the number of samples sequenced and did not reach a plateau (Fig. 2). However, once population-specific variants were divided into personal and genuine polymorphic variants, the later reached a plateau. These data suggested that most of the Kuwaiti-specific polymorphisms within coding regions were restricted to approximately 10,000 positions (Fig. 2, dashed blue line).
Utility of whole-exome data in population structure analysis produces results congruent to those obtained using genome-wide genotype data54. In this study, principal component analysis of the merged data set of exome variants from Kuwait, 1KGP global populations, and Qatar confirmed the existence of three subgroups (Fig. 1) previously derived from genome-wide genotype data24 in Kuwaiti population. The KWB subgroup showed greater genetic affinity towards African populations, and the other two clearly demarcated subgroups (namely KWP and KWS) were between South Asian and European populations. Furthermore, the three substructures of the Qatari population25 lied akin to Kuwaitis. These results were supported by evidence from pFST likelihood ratio tests, which identified variants that differentiated the subgroups. The population-wide occurrence of Kuwaiti SAFD variants in the context of maximum allele frequency populations indicated pairing of Kuwaiti individuals mostly with Europeans and Ashkenazi Jewish populations from 1KGP phase 3 and gnomAD data sets. Such genetic relatedness among Middle Eastern, European and Ashkenazi Jewish populations was further confirmed through performing population genetic analyses (FST and PCA) by way of including genotype data from Ashkenazi Jews as well (Figs 4 and 5). Notably, in line with previous studies43, population genetic analysis presented in this study demonstrated the genetic relatedness among Middle Eastern, European and Ashkenazi Jewish populations.
The Kuwaiti exomes, presented in this study, included 46 clinically significant deleterious variants that are rare in global populations and in Kuwaiti exomes (except for three) (pathogenic: 35; drug response:1; risk factor: 10). 28 of the 36 pathogenic variants followed AR mode of inheritance and the 7 of the 10 risk factor variants followed AD mode of inheritance. Disorders associated with 20 of the 46 variants were seen in Arab populations. Three of the 46 variants reached an MAF value characterizing low-frequency variants in Kuwaiti exomes; two of these three were risk factor and one was drug response variant; and the allele frequencies were comparable with GME data set (see Table 3)-(rs61742245-VKORC1:1.04%,1.37%; rs1800553-ABCA4:2.41%,2.1%; rs11909217-LIPI:1.72%,1.31%). The three variants indicating high risk ratios in Kuwaiti exomes for disease pathogenesis and response to medication were: the VKORC1 variant was associated with warfarin resistance (AD) (heterozygous in four individuals and homozygous recessive in one individual), the ABCA4 variant was associated with susceptibility to age-related macular degeneration (AD) (heterozygous in 12 individuals and homozygous recessive in one individual), and the LIPI variant was associated with susceptibility to hypertriglyceridemia (AD) (heterozygous in 10 individuals). Mendelian and rare genetic disorders as well as monogenic forms of common complex diseases are often associated with rare coding variants. The rare coding variants can have remarkably different allelic frequencies in different ethnic populations compared with the 1KGP populations10,55. The data presented above reported rare variants associated with not only rare Mendelian disorders but also with complex disorders. This observation is in agreement with literature reports on many examples of rare and low-frequency variants associated with complex phenotype traits and common disorders (a review of some of the relevant studies are as listed in Table 1 in Schork et al.56). An interesting example of rare & deleterious “risk factor” variants associated with increasing risk for complex disorders was the GHRL variant (rs34911341-C/T; Arg51gln) which OMIM associated with susceptibility to the complex disorder of obesity (along with genes such as POMC, SDC3 and ADRB2); the variant was originally seen in 6.13% of 96 unrelated Swedish female subjects of morbid obesity (BMI 42.3 ± 3.4 Kg/m2)57. The variant had been seen in GME data set and in one individual from our study cohort; incidentally, the individual was morbidly obese female with a BMI of 44.3 kg/m2; Though our study cohort consisted 48 morbidly obese female individuals, only one of them carried this GHRL allele.
The study identified in Kuwaiti exomes a set of 21 missense SNVs (that were predominantly ‘common’ in both Kuwaiti exomes and 1KGP populations as well as in GME) mapping to drug-binding domains and were of pharmacogenomic relevance (relating to complex disorders, such as sickle-cell anaemia, hypertension, diabetes, asthma, cancer and chemical dependence). 7 of these 21 variants were also observed as Arab mutations associated with complex disorders in Arab populations (Table 5). Of the 21 pharmacogenomic SNVs, the CYP2C8*3 variants encoding two linked amino acid substitutions46 were particularly evident (Table 4) in Kuwaiti exomes; risk allele frequencies at these two variants were 12% in Kuwaiti exomes, 10.4% in GME and 4.6% in 1KGP; the risk alleles co-segregated in 33 individuals in our study cohort. CYP2C8 has emerged as a significant pharmacogene58,59 and is responsible for biotransformation of 5% of currently used drugs that undergo phase 1 hepatic metabolism60. The CYP2C8*3 variants regulate the dosage of the diabetes drugs rosiglitazone and repaglinide61,62. The minor alleles of the CYP2C8*3 variants were also associated with decreased metabolism of paclitaxel59. The ADRB2 variant (occurring with an MAF of 1.2% in Kuwaiti exomes, 1.6% in GME and 0.4% in 1KGP) regulates the efficacy of the asthma drug terbutaline and beta-blocking agents used to treat heart failure63. ADRB2 variant had also been correlated with the risk of type 2 diabetes, obesity and hypertension63; six individuals from our study cohort carried risk allele at this variant.
The identified 24 SAFD variants (all of which were missense variants – see Table 6) with clinical significance included (a) two pathogenic variants (with AR mode of inheritance) associated with the rare disorders of Corticosterone methyloxidase type 2 deficiency and Carboxypeptidase N deficiency); (b) four drug response’ variants associated with toxicity to the drugs of cisplatin or cyclophosphamide and with response to anti-coagulation drugs; (c) sixteen risk/protective’ variants associated with complex traits (ex. asthma, Parkinson’s disease, obesity, nephrolithiasis, melanoma 6, and alcohol dependency); and (d) two ‘Associated’ variants relating to traits of FPG levels and skin/hair/eye pigmentation. The gnomAD populations that showed highest MAFs at these 24 variants were Ashkenazi Jews (15 instances) and Europeans including Finnish (6 instances). The 1KGP populations that showed the highest MAFs were Europeans (14) and South Asians (7). As expected, a major number of these variants were ‘common’ (20 in Kuwaiti exomes and 14 in 1KGP data). The associated disorders are common in the region - Cholestasis of pregnancy (associated with the ATP8B1 variant), the most common AD disorder in pregnant women, has an incidence rate of 0.8%–1.46% in South Asian populations64; Hereditary prostate cancer (associating with the two ELAC2 variants) is one of the most prevalent cancers in Kuwait65; The corticosterone methyloxidase type 2 deficiency (associating with the CYP11B2 variant) is more common in people of Iranian Jewish ancestry66; and Coronary artery disease (associating with the MEF2A variant) has an incidence rate of approximately 6% in the Saudi Arabian population67.
In addition, two other SNVs from our analysis for functional variants were seen associated with quantitative traits in GWA studies – an SAFD LoF variant rs2228015/CCR7 associated with the complex hematological trait of lymphocyte count in European-ancestry people68 and a missense variant (rare in 1KGP but common in Kuwaiti exomes) rs117135869/TTC38 associated with a novel complex metabolic quantitative trait loci (mQTLs) in a cohort from Middle Eastern population48. Further search for presence in Kuwaiti exomes of OMIM-listed causal variants relating to CAGS disorders led to a list of additional 17 variants (see Table 7); 7 of these variants were “pathogenic” and the remaining 10 were “risk factor” variants. The analysis identified a total of 25 CAGS disorders for which the OMIM-listed causal variants were seen in Kuwaiti exomes; such a poor turnout of only 25 was probably due to the small size of the cohort.
Of the 112 variants of clinical significance discussed so far, as many as 44 were ‘common’ variants in Kuwaiti exomes. Very often these common variants were relating to complex disorders. In this study, we did not ourselves delineate the variants associated with complex disorders; we rather just examined whether and which of the functional variants identified in our study were annotated in OMIM, ClinVar, PharmGKB, and literature as associated with complex (or rare) disorders. A question arose as to whether the study cohort of 291 exomes had enough power. Of the 112 variants, 27 variants (comprising 1 rare, 3 low-frequency and 23 common variants) were also seen in our in-house GWAS data set of larger sample size; the set of 27 variants comprised 11 pharmacogenomic, 9 SAFD, 5 CAGS and the two MTHFR variants associated with susceptibility to T2DM (Table 9). The MAF at these variants were comparable among the Kuwaiti exomes, GWAS data, and the GME data set; the carrier distributions were also comparable with one another among the Kuwaiti exomes and GWAS data set.
Finally, disorders relating to 52 of the identified variants were observed in Arab population. Inheritance modes associated with these 52 variants were: 28 autosomal recessive, 15 autosomal dominant, and 9 ambiguous. 25 of these variants were relating to ‘rare’ and 27 were relating to complex disorders. This study (based on 291 exomes) provided data on 23 known Arab mutations for 23 disorders seen in Arab populations, data on 12 putative mutations for 12 disorders observed but not yet characterized for genetic basis in Arab population, and data on 17 additional putative mutations for disorders characterized for genetic basis in Arab populations. This data is useful for testing in future case-control studies.
Capturing the extent of genetic variation in Middle East region is poorly represented in global studies. However, the Greater Middle Eastern (GME) Variome Consortium26 has recently made a notable effort to address this concern by way of capturing genetic variations from exomes of 1,111 unrelated and supposedly healthy individuals from Northwest and Northeast Africa, Turkish peninsula, Syrian desert, Arabian Peninsula and Persia & Pakistan. The GME data set included 214 exomes from Arabian Peninsula (AP), of which 45 are from Kuwaiti population. Our study consisting of 291 Kuwaiti samples, sourced from the 3 Kuwaiti population subgroups, complements and augments the GME genetic variation data by way of presenting a higher number of exomes representing a single state of AP namely Kuwait. It is further the case that the GME study discovered and presented the variegated genetic architecture in GME populations; this is complemented by the population genetics results from our study from a relatively larger sample set of native Arabs living in a single state from the Peninsula. The GME study demonstrated the utility of the GME exome data set in discovering the genetic basis of Mendelian disorders in Greater Middle Eastern populations; our study provides data on Arab mutations for 23 disorders and points to 31 OMIM-listed variants relating to disorders seen in Arab populations for testing in future case-control studies.
A potential limitation of this study arises from the number of exomes sequenced. Though the number of population-specific variants seemed to saturate with 291 exomes, the total number of “all” identified variants did not saturate (Fig. 2); this indicates that we need to sequence furthermore samples to sufficiently represent the Arab population from Kuwait. It is further the case that variants associated with only a small set of disorders observed in the region were seen in the reported Kuwaiti exome data.
In conclusion, the presented assessment of 291 exomes of unrelated healthy individuals unveiled the prevalence of rare as well as common variants related to various Mendelian disorders and common complex diseases that are predominantly inherited as recessive. The inclusion of different genome data sets in our analyses highlighted similarities in allele frequencies among Arabs and Jews, and among nomadic Bedouins and Africans. Furthermore, our data corroborates the Kuwaiti population substructures previously determined by genome-wide genotype data; the results on population structures from Kuwait is generally in agreement with the variegated genetic architecture seen in Greater Middle Eastern populations26. The striking occurrence of pharmacogenomic variants relating to common complex disorders, underlines the importance and need for cataloguing genetic variants in similar Arab populations of the Middle East region. This study is a significant addition to regional data resources (such as GME26) and global resources (such as 1kGP3,4) on human exome variability; however, a wide range of similar studies in the region are warranted to support genomic discoveries in medical and population genetics at the regional and global levels26.
Methods
Ethics Statement
The protocols used in the study were approved by the International Scientific Advisory Board and the Ethical Review Committee at Dasman Diabetes Institute, Kuwait. Written informed consent was obtained from participants before collecting blood samples. Identities of the participants were protected from public exposure, and samples/data were processed anonymously. All methods were performed in accordance with the relevant guidelines and regulations.
Selection of subjects for whole-exome sequencing
To capture the extent of exome variation in the entire Kuwaiti population, 291 healthy, unrelated native Kuwaiti individuals from the study cohorts used in our earlier studies were selected24,36,69,70. At the time of recruitment, all participants in this study were healthy and deemed free of Mendelian or rare genetic disorders, cognition or physical disability, mental retardation or chronic disorders, such as cancer. Distribution of the selected participants in three subgroups of Kuwaiti population24 was as follows: 109 in KWS (Saudi Arabian tribe ancestry), 126 in KWP (Persian ancestry) and 34 in KWB (nomadic Bedouin ancestry).
Whole-exome sequencing
High-quality DNA samples were enriched for exomes using TruSeq Exome Enrichment kit and the Nextera Rapid Capture Exome kit (Illumina Inc. USA). The captured libraries were then clustered using TruSeq Paired Cluster Kit V3 (Illumina Inc. USA) and sequenced in HiSeq 2000 using Illumina’s Sequence by Synthesis technology as 100 paired-end reads.
Exome data analysis
The HugeSeq71 computational pipeline was used to automate the variant discovery process. Sequence reads were aligned to the reference human genome build hg19 using BWA72. Prior to variant calling, alignment files were processed using the Genome Analysis Toolkit (GATK)73. Post-alignment procedures included PCR duplicate removal, local realignment around known indels and base quality recalibration. Best practices for the GATK workflow were followed, and standard hard filtering parameters74,75 were used for variant discovery from the processed alignment files. Variant calling on each sample’s BAM file was performed using HaplotypeCaller followed by joint genotyping analysis of the resultant gVCFs to create raw SNV and indel VCFs. Variants called in the sequenced exomes were restricted to intervals covered by both TruSeq (163 samples) and Nextera (128 samples) Exome Enrichment kits. To improve the quality of the data set, the resulting variant call sets were filtered by setting sample variant thresholds at ≥10X depth, <180X depth and genotype quality of >20. Variants with allele balance of <30% were removed to filter out sites where the fraction of non-reference reads was too low. Hardy–Weinberg Equilibrium was assessed using an exact test, as defined by Wigginton et al.76, and excluded sites with p-values of <10−5. Lastly, all variants with a call rate of <90% were excluded. Thus, after the variant quality filtering steps, only the consensus of variants determined using both kits appeared in the final VCFs.
Classifying the variants
The Ensembl genome database build 75 was used as reference for gene annotation. SNP Variation Suite (SVS) v8.7.1 from Golden Helix Inc77 was used to derive functional classifications of the identified variants. The identified SNVs and indels were categorised as ‘known’ and ‘novel’ based on the content of the single-nucleotide polymorphism database of dbSNP14678. Variants already reported in dbSNP146 were annotated as ‘known’, and the others were annotated as ‘novel’. Variants observed in only a single exome from the study cohort and not seen in 1KGP or GME data sets were annotated as ‘personal’. Variants (excluding the ‘personal’) that were not observed in 1KGP phase 3 data were annotated as ‘population-specific’, and population-specific variants observed in more than one exome from the study cohort were annotated as ‘population-specific polymorphic’ variants. Variants leading to stop gain, stop loss, frameshift and damage in splice sites were annotated to cause LoF (loss of function). Variants were classified as ‘rare’ if MAF was <1% (personal variants were not considered as rare), as ‘low-frequency’ if MAF was 1–5% and as ‘common’ if MAF was ≥5%.
Principal component analysis of the merged set of exome variants from Kuwaiti and global populations
The 1KGP phase 3 exomes of 2,504 individuals from 26 populations, covering the four continents of Asia, Africa, America and Europe and 100 exomes of Qatari individuals44 were considered along with Kuwaiti exomes. An LD-pruned (LD threshold of 0.5) data set of 20,215 variants (having MAF of ≥5%) observed in all three Kuwaiti subgroups and the regional and global populations was created. Golden Helix SVS software v8.7.1 was used to perform principal component analysis with the merged data set.
pFST likelihood ratio tests: Comparison of allele frequency distribution among the Kuwaiti population subgroups
Reference alleles and alternate alleles were binned to set the standard for ‘Kuwaiti exome’. In order to detect alleles driving differentiation among the three Kuwaiti subpopulation groups of KWP, KWS and KWB, pFST likelihood ratio tests79 for allele frequency differences in autosomal variants (filtered for missingness rate and deviation from Hardy–Weinberg equilibrium) were performed.
Identification of SNVs with significant differences (SAFD variants) in allele frequencies between Kuwaiti and global populations
Autosomal SNVs observed in both Kuwaiti exomes and 1KGP phase 3 exomes4 were identified, and SNVs for which minor alleles were not observed in Kuwaiti exomes were excluded. SAFD variants that exhibited significant allele frequency differences were identified by performing one-sided binomial exact tests (allele frequencies in 1KGP global populations were considered as ‘expected’), followed by Bonferroni correction. A p-value threshold of 0.05 was used to assess the significance of allele frequency differences. ClinVar80 data resource was used to assess the clinical significance of the identified SAFD variants. In the context of population structure analyses, populations from gnomAD data set8 were also used to compare allele frequency distributions. The comprehensive scrutinization of population-wide occurrence was performed by considering the paired incidence of populations with maximum allele frequency.
Principal Component Analysis of the merged set of Kuwaiti exomes, Ashkenazi Jews, 1KGP phase 3 and Qatar and FST analysis
We combined Kuwaiti exomes with the data sets from Ashkenazi Jews43, 1KGP phase 34 and Qatar44. The combined data set of coding-region variants was cleaned and LD-pruned to obtain a total of about 896 variants and 3,336 individuals representing world populations. Principal component analysis (PCA) was performed using smartpca in the EIGENSOFT software package (v 6.1.4)81,82. Two-dimensional and three-dimensional scattered PCA plots were created using RStudio83 (v 1.1.423). Mean pairwise FST values and the matrix between populations were generated using PLINK84 (v 1.9). The FST heatmap was created using RStudio (v 1.1.423).
Examining OMIM and ClinVar annotations for inferring clinical significance of SNVs
OMIM and ClinVar should mention the Kuwaiti exome SNV, with literature evidence and citation reference, as an associated variant for a disorder; the dbSNP identifier of the SNV and the observed risk allele should be mentioned as such in the OMIM and ClinVar annotation80,85. The clinical significance for the variant should be mentioned consistently with the same term (such as ‘pathogenic’ or ‘risk factor’ or ‘protective’ or ‘drug response’) in all the records for the disorder; it should not be the case that few records list the significance as ‘pathogenic’ and few other records list as ‘benign’ or ‘conflicting interpretation’ for the disorder; ClinVar records listing “not specified” for the data item of ‘conditions’ were not considered. As is the practice86, in cases of ClinVar variants with conflicting annotation for clinical significance, evidence from a peer-reviewed publication and manually curation (OMIM) takes precedence over evidence from clinical testing submissions. ClinVar defines “Pathogenic” variants as those that are interpreted for Mendelian disorders; or as those that have low penetrance; “Drug response” variants as those that affect drug response, and not a disease; “Risk factor” variants as those that are interpreted not to cause a disorder but to increase the risk; “Association” variants as those that were identified in a GWAS study and further interpreted for their clinical significance; “Protective” variants that decrease the risk of a disorder, including infections; and “Susceptibility to” variants that increase the risk of a disorder. In those instances, wherein ClinVar annotated a variant as “pathogenic” but the associated disorder was “complex or common or more prevalent in the study population” or the patient carrying the variant was annotated in OMIM as susceptible to the disorder (which is often a common disorder), we reannotated the variant as “Risk factor by inference”.
Classifying disorders as ‘rare’ or ‘complex’
Various resources that were examined to ascertain whether the disorder is rare or common: Catalogue of Transmission Genetics in Arabs (available at http://cags.org.ae/ctga/), Genetic and Rare Disease (GARD) Information Centre (available at https://rarediseases.info.nih.gov/diseases/), Genetics Home Reference (available at https://ghr.nlm.nih.gov/), Medscape (available at https://geneaware.clinical.bcm.edu/GeneAware/AboutGeneAware/DiseaseSearch.aspx) and literature.
Examining the Kuwaiti exomes for rare, deleterious and pathogenic variants
‘Known’ missense and LoF SNVs having MAF of <1% in the 1KGP phase 3 data4 and ExAC database8 were catalogued. Of these, only the variants annotated as damaging by both SIFT87 and PolyPhen-288 tools were retained. The Kuwaiti exomes were examined for such variants. As an additional step, the resulting variants were filtered based on their Combined Annotation-Dependent Depletion score89 to prioritise functional, deleterious and pathogenic variants across many functional categories, effect sizes and genetic architectures. A scaled score of ≥20 was applied to retrieve only those variants that were predicted to be among the top 1% of deleterious variants in the human genome. The above set of variants were screened for clinical significance using the OMIM85 and ClinVar80 databases.
Variants found to be ‘rare’ within global populations but ‘common’ within Kuwaiti population
A data set of SNVs that are rare in 1KGP phase 3 populations but common within Kuwaiti exomes was created. For such missense variants, scores predicting their pathogenicity were calculating using the REVEL90 software.
Examining the pharmacogenomic relevance of Kuwaiti exome variants
Variants of pharmacogenomic relevance were delineated using the resources built upon the concept of druggable genome originally formulated by Hopkins and Groom49 and PharmGKB50. From the data set of variants derived for Kuwaiti exomes, missense SNVs (with MAF of >1%) that are not deleterious (i.e. SIFT and PolyPhen-2 scores were outside the deleteriousness range) were mapped to protein domains (using InterPro91) and checked for inclusion in the list of 130 domains reported by Hopkins and Groom. From the resulting set of variants mapping to drug-binding domains, only those for which pharmacogenomic annotation was available in PharmGKB database were retained.
Electronic supplementary material
Acknowledgements
The authors thank Maisa Mahmoud for help with recruiting participants. Members of the Tissue Bank Core Facility are acknowledged for sample processing and DNA extraction. The authors acknowledge the Kuwait Foundation for Advancement of the Sciences for funding this study (RA-2016-026; RA-2010-005).
Author Contributions
T.A.T. and O.A. performed the study design for this work and directed the study components. D.A. sequenced all the exomes; S.E.J. processed all the sequenced exomes. S.E.J. and M.E. performed all the data analysis and participated significantly in interpreting the results. P.H. and A.M.C. participated in data analysis; further, P.H. made significant contributions to the analysis relating to assignment of samples to population substructures. D.T. was involved in participant recruitment, collecting samples and managing phenotype information. S.D. was involved in supervising sample processing, DNA extraction and sample data management. T.A.T., S.E.J., M.E. and D.A. developed the manuscript. J.T. critically reviewed and approved the original manuscript. F.A. provided the required resources, critically reviewed and approved the manuscript.
Data Availability
The 291 individual VCF files were combined to include a genotype (ref/ref, ref/alt, /alt/alt) for each exome. The resulting VCF file containing genotypes for the final variant set of 173,849 SNVs and indels is available online at ftp://dgr.dasmaninstitute.org.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Sumi Elsa John and Dinu Antony contributed equally.
Osama Alsmadi and Thangavel Alphonse Thanaraj jointly supervised this work.
Contributor Information
Osama Alsmadi, Email: OA.12163@KHCC.JO.
Thangavel Alphonse Thanaraj, Email: alphonse.thangavel@dasmaninstitute.org.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-34815-8.
References
- 1.Cavalli-Sforza LL. The DNA revolution in population genetics. Trends Genet. 1998;14:60–65. doi: 10.1016/S0168-9525(97)01327-9. [DOI] [PubMed] [Google Scholar]
- 2.Cavalli-Sforza LL. The Human Genome Diversity Project: past, present and future. Nat Rev Genet. 2005;6:333–340. doi: 10.1038/nrg1596. [DOI] [PubMed] [Google Scholar]
- 3.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.1000 Genomes Project Consortium A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.UK10K Consortium The UK10K project identifies rare variants in health and disease. Nature. 2015;526:82–90. doi: 10.1038/nature14962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nature genetics. 2003;33(Suppl):228–237. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- 7.Stenson PD, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 8.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu W, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tennessen JA, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coventry A, et al. Deep resequencing reveals excess rare recent variants consistent with explosive population growth. Nature communications. 2010;1:131. doi: 10.1038/ncomms1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nature genetics. 2010;42:969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 13.Marth GT, et al. The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson MR, et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fakhro KA, et al. The Qatar genome: a population-specific tool for precision medicine in the Middle East. Hum Genome Var. 2016;3:16016. doi: 10.1038/hgv.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keinan A, Clark AG. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science. 2012;336:740–743. doi: 10.1126/science.1217283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dopazo J, et al. 267 Spanish Exomes Reveal Population-Specific Differences in Disease-Related Genetic Variation. Mol Biol Evol. 2016;33:1205–1218. doi: 10.1093/molbev/msw005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ioannidis JP, Ntzani EE, Trikalinos TA. ‘Racial’ differences in genetic effects for complex diseases. Nature genetics. 2004;36:1312–1318. doi: 10.1038/ng1474. [DOI] [PubMed] [Google Scholar]
- 19.Bustamante CD, Burchard EG, De la Vega FM. Genomics for the world. Nature. 2011;475:163–165. doi: 10.1038/475163a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cabrera, V., Abu-Amero, K., Larruga, J. & González, A. In The Evolution of Human Populations in Arabia Vertebrate Paleobiology and Paleoanthropology (eds Michael D. Petraglia & Jeffrey I. Rose) Ch. 6, 79–87 (Springer Netherlands, 2010).
- 21.Rose, J. & Petraglia, M. In The Evolution of Human Populations in Arabia Vertebrate Paleobiology and Paleoanthropology (eds Michael D. Petraglia & Jeffrey I. Rose) Ch. 1, 1–12 (Springer Netherlands, 2010).
- 22.Armitage SJ, et al. The southern route “out of Africa”: evidence for an early expansion of modern humans into Arabia. Science. 2011;331:453–456. doi: 10.1126/science.1199113. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez-Flores JL, et al. Indigenous Arabs are descendants of the earliest split from ancient Eurasian populations. Genome Res. 2016;26:151–162. doi: 10.1101/gr.191478.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alsmadi O, et al. Genetic substructure of Kuwaiti population reveals migration history. PloS one. 2013;8:e74913. doi: 10.1371/journal.pone.0074913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunter-Zinck H, et al. Population genetic structure of the people of Qatar. Am J Hum Genet. 2010;87:17–25. doi: 10.1016/j.ajhg.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scott EM, et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nature genetics. 2016;48:1071–1076. doi: 10.1038/ng.3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tadmouri GO, Al Ali MT, Al-Haj Ali S, Al Khaja N. CTGA: the database for genetic disorders in Arab populations. Nucleic acids research. 2006;34:D602–606. doi: 10.1093/nar/gkj015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tadmouri GO, et al. Consanguinity and reproductive health among Arabs. Reproductive Health. 2009;6:17–17. doi: 10.1186/1742-4755-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujikura K. Global Carrier Rates of Rare Inherited Disorders Using Population Exome Sequences. PloS one. 2016;11:e0155552. doi: 10.1371/journal.pone.0155552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarthy MI, MacArthur DG. Human disease genomics: from variants to biology. Genome Biol. 2017;18:20. doi: 10.1186/s13059-017-1160-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peltonen L, Perola M, Naukkarinen J, Palotie A. Lessons from studying monogenic disease for common disease. Human molecular genetics. 2006;15(Spec No 1):R67–74. doi: 10.1093/hmg/ddl060. [DOI] [PubMed] [Google Scholar]
- 32.Ng M, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384:766–781. doi: 10.1016/S0140-6736(14)60460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Channanath AM, Farran B, Behbehani K, Thanaraj TA. State of diabetes, hypertension, and comorbidity in Kuwait: showcasing the trends as seen in native versus expatriate populations. Diabetes care. 2013;36:e75. doi: 10.2337/dc12-2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bittles AH, Black ML. Evolution in health and medicine Sackler colloquium: Consanguinity, human evolution, and complex diseases. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(Suppl 1):1779–1786. doi: 10.1073/pnas.0906079106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erzurumluoglu AM, Shihab HA, Rodriguez S, Gaunt TR, Day IN. Importance of Genetic Studies in Consanguineous Populations for the Characterization of Novel Human Gene Functions. Ann Hum Genet. 2016;80:187–196. doi: 10.1111/ahg.12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alsmadi O, et al. Genome at juncture of early human migration: a systematic analysis of two whole genomes and thirteen exomes from Kuwaiti population subgroup of inferred Saudi Arabian tribe ancestry. PloS one. 2014;9:e99069. doi: 10.1371/journal.pone.0099069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.John SE, et al. Kuwaiti population subgroup of nomadic Bedouin ancestry-Whole genome sequence and analysis. Genom Data. 2015;3:116–127. doi: 10.1016/j.gdata.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thareja, G. et al. Comprehensive analysis of a personal genome of Persian ancestry from Kuwait (2014).
- 39.Freudenberg-Hua Y, et al. Single nucleotide variation analysis in 65 candidate genes for CNS disorders in a representative sample of the European population. Genome Res. 2003;13:2271–2276. doi: 10.1101/gr.1299703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kenna KP, et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. Journal of medical genetics. 2013;50:776–783. doi: 10.1136/jmedgenet-2013-101795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodriguez-Flores JL, et al. Exome sequencing of only seven Qataris identifies potentially deleterious variants in the Qatari population. PloS one. 2012;7:e47614. doi: 10.1371/journal.pone.0047614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sulem P, et al. Identification of a large set of rare complete human knockouts. Nature genetics. 2015;47:448–452. doi: 10.1038/ng.3243. [DOI] [PubMed] [Google Scholar]
- 43.Bray SM, et al. Signatures of founder effects, admixture, and selection in the Ashkenazi Jewish population. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16222–16227. doi: 10.1073/pnas.1004381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodriguez-Flores JL, et al. Exome sequencing identifies potential risk variants for Mendelian disorders at high prevalence in Qatar. Hum Mutat. 2014;35:105–116. doi: 10.1002/humu.22460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cassa CA, Tong MY, Jordan DM. Large numbers of genetic variants considered to be pathogenic are common in asymptomatic individuals. Hum Mutat. 2013;34:1216–1220. doi: 10.1002/humu.22375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsuruta M, et al. Molecular basis of intermittent maple syrup urine disease: novel mutations in the E2 gene of the branched-chain alpha-keto acid dehydrogenase complex. J Hum Genet. 1998;43:91–100. doi: 10.1007/s100380050047. [DOI] [PubMed] [Google Scholar]
- 47.Cao H, Hegele RA. DNA polymorphism and mutations in CPN1, including the genomic basis of carboxypeptidase N deficiency. J Hum Genet. 2003;48:20–22. doi: 10.1007/s100380300003. [DOI] [PubMed] [Google Scholar]
- 48.Yousri NA, et al. Whole-exome sequencing identifies common and rare variant metabolic QTLs in a Middle Eastern population. Nature communications. 2018;9:333. doi: 10.1038/s41467-017-01972-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 50.Thorn CF, Klein TE, Altman RB. PharmGKB: the Pharmacogenomics Knowledge Base. Methods Mol Biol. 2013;1015:311–320. doi: 10.1007/978-1-62703-435-7_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacArthur J, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog) Nucleic acids research. 2017;45:D896–D901. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cherepanova NS, Leslie E, Ferguson PJ, Bamshad MJ, Bassuk AG. Presence of epilepsy-associated variants in large exome databases. J Neurogenet. 2013;27:1–4. doi: 10.3109/01677063.2013.772176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet. 2013;132:1077–1130. doi: 10.1007/s00439-013-1331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Belkadi A, et al. Whole-exome sequencing to analyze population structure, parental inbreeding, and familial linkage. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:6713–6718. doi: 10.1073/pnas.1606460113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mathieson I, Reich D. Differences in the rare variant spectrum among human populations. PLoS Genet. 2017;13:e1006581. doi: 10.1371/journal.pgen.1006581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schork NJ, Murray SS, Frazer KA, Topol EJ. Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev. 2009;19:212–219. doi: 10.1016/j.gde.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ukkola O, et al. Mutations in the preproghrelin/ghrelin gene associated with obesity in humans. The Journal of clinical endocrinology and metabolism. 2001;86:3996–3999. doi: 10.1210/jcem.86.8.7914. [DOI] [PubMed] [Google Scholar]
- 58.Aquilante CL, Niemi M, Gong L, Altman RB, Klein TE. PharmGKB summary: very important pharmacogene information for cytochrome P450, family 2, subfamily C, polypeptide 8. Pharmacogenet Genomics. 2013;23:721–728. doi: 10.1097/FPC.0b013e3283653b27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dai D, et al. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics. 2001;11:597–607. doi: 10.1097/00008571-200110000-00006. [DOI] [PubMed] [Google Scholar]
- 60.Totah RA, Rettie AE. Cytochrome P450 2C8: substrates, inhibitors, pharmacogenetics, and clinical relevance. Clinical pharmacology and therapeutics. 2005;77:341–352. doi: 10.1016/j.clpt.2004.12.267. [DOI] [PubMed] [Google Scholar]
- 61.Bidstrup TB, Bjornsdottir I, Sidelmann UG, Thomsen MS, Hansen KT. CYP2C8 and CYP3A4 are the principal enzymes involved in the human in vitro biotransformation of the insulin secretagogue repaglinide. Br J Clin Pharmacol. 2003;56:305–314. doi: 10.1046/j.0306-5251.2003.01862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pedersen RS, Damkier P, Brosen K. The effects of human CYP2C8 genotype and fluvoxamine on the pharmacokinetics of rosiglitazone in healthy subjects. Br J Clin Pharmacol. 2006;62:682–689. doi: 10.1111/j.1365-2125.2006.02706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Litonjua AA, et al. Very important pharmacogene summary ADRB2. Pharmacogenet Genomics. 2010;20:64–69. doi: 10.1097/FPC.0b013e328333dae6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abedin P, Weaver JB, Egginton E. Intrahepatic cholestasis of pregnancy: prevalence and ethnic distribution. Ethn Health. 1999;4:35–37. doi: 10.1080/13557859998173. [DOI] [PubMed] [Google Scholar]
- 65.Salim EI, et al. Cancer epidemiology and control in the arab world - past, present and future. Asian Pac J Cancer Prev. 2009;10:3–16. [PubMed] [Google Scholar]
- 66.Rosler A, White PC. Mutations in human 11 beta-hydroxylase genes: 11 beta-hydroxylase deficiency in Jews of Morocco and corticosterone methyl-oxidase II deficiency in Jews of Iran. J Steroid Biochem Mol Biol. 1993;45:99–106. doi: 10.1016/0960-0760(93)90128-J. [DOI] [PubMed] [Google Scholar]
- 67.Al-Nozha MM, et al. Coronary artery disease in Saudi Arabia. Saudi Med J. 2004;25:1165–1171. [PubMed] [Google Scholar]
- 68.Astle WJ, et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell. 2016;167:1415–1429 e1419. doi: 10.1016/j.cell.2016.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hebbar P, et al. Genetic risk variants for metabolic traits in Arab populations. Sci Rep. 2017;7:40988. doi: 10.1038/srep40988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hebbar P, et al. The TCN2 variant ofrs9606756 [Ile23Val] acts as risk loci for obesity-related traits and mediates by interacting with Apo-A1. Obesity. 2017;25:1098–1108. doi: 10.1002/oby.21826. [DOI] [PubMed] [Google Scholar]
- 71.Lam HY, et al. Detecting and annotating genetic variations using the HugeSeq pipeline. Nat Biotechnol. 2012;30:226–229. doi: 10.1038/nbt.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.DePristo Mark A, Banks Eric, Poplin Ryan, Garimella Kiran V, Maguire Jared R, Hartl Christopher, Philippakis Anthony A, del Angel Guillermo, Rivas Manuel A, Hanna Matt, McKenna Aaron, Fennell Tim J, Kernytsky Andrew M, Sivachenko Andrey Y, Cibulskis Kristian, Gabriel Stacey B, Altshuler David, Daly Mark J. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics43, 11 10 11–33, 10.1002/0471250953.bi1110s43 (2013). [DOI] [PMC free article] [PubMed]
- 76.Wigginton JE, Cutler DJ, Abecasis GR. A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet. 2005;76:887–893. doi: 10.1086/429864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.SNP & Variation Suite (Version 8.7.1) [Software] (Golden Helix, Inc.).
- 78.Sherry ST, et al. dbSNP: the NCBI database of genetic variation. Nucleic acids research. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kronenberg Z, S. M. & Yandell, M. Association testing with GPAT++, https://github.com/zeeev/vcflib/wiki/Association-testing-with-GPAT (2014).
- 80.Landrum MJ, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic acids research. 2016;44:D862–868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nature genetics. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 83.RStudio: Integrated Development Environment for R v. 1.1.423 (RStudio, Inc., Boston, MA, 2016).
- 84.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick’s Online Mendelian Inheritance in Man (OMIM) Nucleic acids research. 2009;37:D793–796. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tarailo-Graovac M, Zhu JYA, Matthews A, van Karnebeek CDM, Wasserman WW. Assessment of the ExAC data set for the presence of individuals with pathogenic genotypes implicated in severe Mendelian pediatric disorders. Genet Med. 2017;19:1300–1308. doi: 10.1038/gim.2017.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 88.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ioannidis NM, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet. 2016;99:877–885. doi: 10.1016/j.ajhg.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hunter S, et al. InterPro: the integrative protein signature database. Nucleic acids research. 2009;37:D211–215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hattori N, et al. Point mutations (Thr240Arg and Gln311Stop) [correction of Thr240Arg and Ala311Stop] in the Parkin gene. Biochemical and biophysical research communications. 1998;249:754–758. doi: 10.1006/bbrc.1998.9134. [DOI] [PubMed] [Google Scholar]
- 93.Allikmets R, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nature genetics. 1997;15:236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 94.Zubaid M, et al. Quality of anticoagulation with warfarin across Kuwait. Hellenic J Cardiol. 2013;54:102–106. [PubMed] [Google Scholar]
- 95.Tas S. Strong association of a single nucleotide substitution in the 3’-untranslated region of the apolipoprotein-CIII gene with common hypertriglyceridemia in Arabs. Clin Chem. 1989;35:256–259. [PubMed] [Google Scholar]
- 96.Al-Balushi K, Zadjali F, Al-Sinani S, Al-Zadjali AM, Bayoumi R. Frequencies of the Arg16Gly, Gln27Glu and Thr164Ile Adrenoceptor beta2 Polymorphisms among Omanis. Sultan Qaboos Univ Med J. 2015;15:e486–490. doi: 10.18295/squmj.2015.15.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Haghiri R, Mashayekhi F, Bidabadi E, Salehi Z. Analysis of methionine synthase (rs1805087) gene polymorphism in autism patients in Northern Iran. Acta Neurobiol Exp (Wars) 2016;76:318–323. doi: 10.21307/ane-2017-030. [DOI] [PubMed] [Google Scholar]
- 98.Arfaoui A, et al. Role of p53 Codon72 SNP in breast cancer risk and anthracycline resistance. Anticancer Res. 2015;35:1763–1769. [PubMed] [Google Scholar]
- 99.Arab AH, Elhawary NA. Association between ANKK1 (rs1800497) and LTA (rs909253) Genetic Variants and Risk of Schizophrenia. BioMed Research International. 2015;2015:8. doi: 10.1155/2015/821827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Alsheyab F, Tiboura GR, Bataineh A, Banihani R. The link between salt sensitivity determined by polymorphism in G protein-coupled receptor kinase 4 (GRK4) gene, and myocardial infarction. 2013;25:107–112. [Google Scholar]
- 101.Frossard PM, et al. Associations of angiotensinogen gene mutations with hypertension and myocardial infarction in a gulf population. Clin Genet. 1998;54:285–293. doi: 10.1034/j.1399-0004.1998.5440405.x. [DOI] [PubMed] [Google Scholar]
- 102.Daghestani MH, et al. Arginine 16 Glycine Polymorphism in beta2-Adrenergic Receptor Gene is Associated with Obesity, Hyperlipidemia, Hyperleptinemia, and Insulin Resistance in Saudis. Int J Endocrinol. 2012;2012:945608. doi: 10.1155/2012/945608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Abu-Amero KK, Al-Boudari OM, Mohamed GH, Dzimiri N. The Glu27 genotypes of the beta2-adrenergic receptor are predictors for severe coronary artery disease. BMC Med Genet. 2006;7:31. doi: 10.1186/1471-2350-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Toraih E, Hussein MH, Badran DI. Beta2-Adrenergic Receptor Gene Polymorphisms in Egyptian Patients with Acute Myocardial Infarction. Advances in Molecular Biology. 2014;2014:11. doi: 10.1155/2014/471635. [DOI] [Google Scholar]
- 105.Migita K, et al. Familial Mediterranean fever is no longer a rare disease in Japan. Arthritis Res Ther. 2016;18:175. doi: 10.1186/s13075-016-1071-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zaki Mohammed, Helin Ingemar, Manandhar Darma S., Hunt Milroy C. J., Khalil Ahmed F. Primary nephrotic syndrome in Arab children in Kuwait. Pediatric Nephrology. 1989;3(2):218–220. doi: 10.1007/BF00852914. [DOI] [PubMed] [Google Scholar]
- 107.Salih MA, Murshid WR, Seidahmed MZ. Epidemiology, prenatal management, and prevention of neural tube defects. Saudi Med J. 2014;35(Suppl 1):S15–28. [PMC free article] [PubMed] [Google Scholar]
- 108.Zahed L, et al. Molecular basis of oculocutaneous albinism type 1 in Lebanese patients. J Hum Genet. 2005;50:317–319. doi: 10.1007/s10038-005-0257-5. [DOI] [PubMed] [Google Scholar]
- 109.Hamajima N, et al. Dihydropyrimidinase deficiency: structural organization, chromosomal localization, and mutation analysis of the human dihydropyrimidinase gene. Am J Hum Genet. 1998;63:717–726. doi: 10.1086/302022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pascoe L, Curnow KM, Slutsker L, Rosler A, White PC. Mutations in the human CYP11B2 (aldosterone synthase) gene causing corticosterone methyloxidase II deficiency. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:4996–5000. doi: 10.1073/pnas.89.11.4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Magalon H, et al. Population genetic diversity of the NAT2 gene supports a role of acetylation in human adaptation to farming in Central Asia. European journal of human genetics: EJHG. 2008;16:243–251. doi: 10.1038/sj.ejhg.5201963. [DOI] [PubMed] [Google Scholar]
- 112.Vincent AL, et al. Digenic inheritance of early-onset glaucoma: CYP1B1, a potential modifier gene. Am J Hum Genet. 2002;70:448–460. doi: 10.1086/338709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bernot A, et al. Non-founder mutations in the MEFV gene establish this gene as the cause of familial Mediterranean fever (FMF) Human molecular genetics. 1998;7:1317–1325. doi: 10.1093/hmg/7.8.1317. [DOI] [PubMed] [Google Scholar]
- 114.Rinat C, Wanders RJ, Drukker A, Halle D, Frishberg Y. Primary hyperoxaluria type I: a model for multiple mutations in a monogenic disease within a distinct ethnic group. J Am Soc Nephrol. 1999;10:2352–2358. doi: 10.1681/ASN.V10112352. [DOI] [PubMed] [Google Scholar]
- 115.Zahrani F, Aldahmesh MA, Alshammari MJ, Al-Hazzaa SA, Alkuraya FS. Mutations in c12orf57 cause a syndromic form of colobomatous microphthalmia. Am J Hum Genet. 2013;92:387–391. doi: 10.1016/j.ajhg.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ozaltin F, et al. Disruption of PTPRO causes childhood-onset nephrotic syndrome. Am J Hum Genet. 2011;89:139–147. doi: 10.1016/j.ajhg.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Roth M, et al. Absence of the hemochromatosis gene Cys282Tyr mutation in three ethnic groups from Algeria (Mzab), Ethiopia, and Senegal. Immunogenetics. 1997;46:222–225. doi: 10.1007/s002510050265. [DOI] [PubMed] [Google Scholar]
- 118.Settin A, El-Bendary M, Abo-Al-Kassem R, El Baz R. Molecular analysis of A1AT (S and Z) and HFE (C282Y and H63D) gene mutations in Egyptian cases with HCV liver cirrhosis. J Gastrointestin Liver Dis. 2006;15:131–135. [PubMed] [Google Scholar]
- 119.Sassi R, et al. Prevalence of C282Y and H63D mutations in the haemochromatosis (HFE) gene in Tunisian population. Ann Genet. 2004;47:325–330. doi: 10.1016/j.anngen.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 120.Lajin B, Alhaj Sakur A, Michati R, Alachkar A. Association between MTHFR C677T and A1298C, and MTRR A66G polymorphisms and susceptibility to schizophrenia in a Syrian study cohort. Asian J Psychiatr. 2012;5:144–149. doi: 10.1016/j.ajp.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 121.Khaliq S, et al. Novel association of RP1 gene mutations with autosomal recessive retinitis pigmentosa. Journal of medical genetics. 2005;42:436–438. doi: 10.1136/jmg.2004.024281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Al-Rashed M, et al. RP1 and retinitis pigmentosa: report of novel mutations and insight into mutational mechanism. Br J Ophthalmol. 2012;96:1018–1022. doi: 10.1136/bjophthalmol-2011-301134. [DOI] [PubMed] [Google Scholar]
- 123.Shpichinetsky V, et al. The association between two common mutations C677T and A1298C in human methylenetetrahydrofolate reductase gene and the risk for diabetic nephropathy in type II diabetic patients. J Nutr. 2000;130:2493–2497. doi: 10.1093/jn/130.10.2493. [DOI] [PubMed] [Google Scholar]
- 124.Nemr R, et al. Differential contribution of MTHFR C677T variant to the risk of diabetic nephropathy in Lebanese and Bahraini Arabs. Clin Chem Lab Med. 2010;48:1091–1094. doi: 10.1515/CCLM.2010.228. [DOI] [PubMed] [Google Scholar]
- 125.Ramanathan, G., Harichandana, B., Kannan, S., Elumalai, R. & Paul, S. Association between end-stage diabetic nephropathy and MTHFR (C677T and A1298C) gene polymorphisms. Nephrology (Carlton), 10.1111/nep.13208 (2017). [DOI] [PubMed]
- 126.Hellani A, Almassri N, Abu-Amero KK. A novel mutation in the ADA gene causing severe combined immunodeficiency in an Arab patient: a case report. J Med Case Rep. 2009;3:6799. doi: 10.1186/1752-1947-3-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Al-Owain M, et al. Smith-Lemli-Opitz syndrome among Arabs. Clin Genet. 2012;82:165–172. doi: 10.1111/j.1399-0004.2011.01742.x. [DOI] [PubMed] [Google Scholar]
- 128.Hattab FN, Amin WM. Papillon-Lefevre syndrome with albinism: a review of the literature and report of 2 brothers. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:709–716. doi: 10.1016/j.tripleo.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 129.Neubauer B, et al. Point mutations in the L-type pyruvate kinase gene of two children with hemolytic anemia caused by pyruvate kinase deficiency. Blood. 1991;77:1871–1875. [PubMed] [Google Scholar]
- 130.Wakil SM, et al. Novel mutations in TGM1 and ABCA12 cause autosomal recessive congenital ichthyosis in five Saudi families. Int J Dermatol. 2016;55:673–679. doi: 10.1111/ijd.13279. [DOI] [PubMed] [Google Scholar]
- 131.Bastaki F, et al. Summary of mutations underlying autosomal recessive congenital ichthyoses (ARCI) in Arabs with four novel mutations in ARCI-related genes from the United Arab Emirates. Int J Dermatol. 2017;56:514–523. doi: 10.1111/ijd.13568. [DOI] [PubMed] [Google Scholar]
- 132.Hempelmann A, Kumar S, Muralitharan S, Sander T. Myofibrillogenesis regulator 1 gene (MR-1) mutation in an Omani family with paroxysmal nonkinesigenic dyskinesia. Neuroscience letters. 2006;402:118–120. doi: 10.1016/j.neulet.2006.03.048. [DOI] [PubMed] [Google Scholar]
- 133.Gouider-Khouja N, et al. Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family. Clinical, genetic and pathological study. Parkinsonism Relat Disord. 2003;9:247–251. doi: 10.1016/S1353-8020(03)00016-6. [DOI] [PubMed] [Google Scholar]
- 134.Tassin J, et al. Chromosome 6-linked autosomal recessive early-onset Parkinsonism: linkage in European and Algerian families, extension of the clinical spectrum, and evidence of a small homozygous deletion in one family. The French Parkinson’s Disease Genetics Study Group, and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Am J Hum Genet. 1998;63:88–94. doi: 10.1086/301934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Myhre R, et al. Significance of the parkin and PINK1 gene in Jordanian families with incidences of young-onset and juvenile parkinsonism. BMC Neurol. 2008;8:47. doi: 10.1186/1471-2377-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Al-Shamsi A, Hertecant JL, Al-Hamad S, Souid AK, Al-Jasmi F. Mutation Spectrum and Birth Prevalence of Inborn Errors of Metabolism among Emiratis: A study from Tawam Hospital Metabolic Center, United Arab Emirates. Sultan Qaboos Univ Med J. 2014;14:e42–49. doi: 10.12816/0003335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Peretz, H. et al. Cys 618 Arg mutation in the RET proto-oncogene associated with familial medullary thyroid carcinoma and maternally transmitted Hirschsprung’s disease suggesting a role for imprinting. Hum Mutat10, 155–159, doi:10.1002/(SICI)1098-1004(1997)10:2<155::AID-HUMU7>3.0.CO;2-J (1997). [DOI] [PubMed]
- 138.Sztriha L, et al. Frameshift mutation of the zinc finger homeo box 1 B gene in syndromic corpus callosum agenesis (Mowat-Wilson syndrome) Neuropediatrics. 2003;34:322–325. doi: 10.1055/s-2003-44671. [DOI] [PubMed] [Google Scholar]
- 139.Elhawari S, et al. A study of the role of the Myocyte-specific Enhancer Factor-2A gene in coronary artery disease. Atherosclerosis. 2010;209:152–154. doi: 10.1016/j.atherosclerosis.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 140.Djidjik R, et al. Constitutive nitric oxide synthase gene polymorphisms and house dust mite respiratory allergy in an Algerian patient group. Tissue Antigens. 2008;71:160–164. doi: 10.1111/j.1399-0039.2007.00976.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 291 individual VCF files were combined to include a genotype (ref/ref, ref/alt, /alt/alt) for each exome. The resulting VCF file containing genotypes for the final variant set of 173,849 SNVs and indels is available online at ftp://dgr.dasmaninstitute.org.