

Abstract
Background
Glucocorticoid (GC) therapy is frequently used to treat rheumatoid arthritis due to potent anti-inflammatory actions of GCs. Direct actions of GCs on immune cells were suggested to suppress inflammation.
Objectives
Define the role of the glucocorticoid receptor (GR) in stromal cells for suppression of inflammatory arthritis.
Methods
Bone marrow chimeric mice lacking the GR in the hematopoietic or stromal compartment, respectively, and mice with impaired GR dimerisation (GRdim) were analysed for their response to dexamethasone (DEX, 1 mg/kg) treatment in serum transfer-induced arthritis (STIA). Joint swelling, cell infiltration (histology), cytokines, cell composition (flow cytometry) and gene expression were analysed and RNASeq of wild type and GRdim primary murine fibroblast-like synoviocytes (FLS) was performed.
Results
GR deficiency in immune cells did not impair GC-mediated suppression of STIA. In contrast, mice with GR-deficient or GR dimerisation-impaired stromal cells were resistant to GC treatment, despite efficient suppression of cytokines. Intriguingly, in mice with impaired GR function in the stromal compartment, GCs failed to stimulate non-classical, non-activated macrophages (Ly6Cneg, MHCIIneg) and associated anti-inflammatory markers CD163, CD36, AnxA1, MerTK and Axl. Mice with GR deficiency in FLS were partially resistant to GC-induced suppression of STIA. Accordingly, RNASeq analysis of DEX-treated GRdim FLS revealed a distinct gene signature indicating enhanced activity and a failure to reduce macrophage inflammatory protein (Mip)-1α and Mip-1β.
Conclusion
We report a novel anti-inflammatory mechanism of GC action that involves GR dimerisation-dependent gene regulation in non-immune stromal cells, presumably FLS. FLS control non-classical, anti-inflammatory polarisation of macrophages that contributes to suppression of inflammation in arthritis.
Keywords: arthritis, inflammation, corticosteroids, fibroblasts
Introduction
Rheumatoid arthritis (RA) is characterised by severe infiltration of various immune cells into the joints and a tight interplay of these immune cells with non-immune stromal cells, like fibroblast-like synoviocytes (FLS).1 2 Pro-inflammatory cytokines secreted by immune cells result in the activation, hyperproliferation and cytokine secretion of FLS. RA is mainly medicated with glucocorticoids (GC)3 4 due to their strong immune suppressive effects, however, long-term usage is compromised by severe side effects such as osteoporosis, insulin resistance and GC resistance.5 6 A better understanding of the cell type-specific actions of GCs will provide rationales for specialised treatment strategies to overcome these negative side effects.
GCs act via the glucocorticoid receptor (GR), a nuclear receptor that operates as ligand-induced transcription factor acting as a homodimer or monomer. The dimeric GR directly interacts with GC-response elements on the DNA, mainly inducing gene expression, a mechanism called transactivation. GR dimers are strong inducers of metabolic genes and were considered to be mainly responsible for side effects.7 The monomeric GR binds either directly to DNA8 9 or indirectly via transcription factors (eg, NF-κB and AP-1) leading in the latter case to transrepression of gene activity. Previously, transrepression in immune cells was considered to mediate the anti-inflammatory effects of GCs.7 This view, however, was challenged by recent studies with mice harbouring an impaired GR dimerisation (GRdim),10 which fail to suppress inflammation in response to GCs in lethal inflammation11 12 acute lung injury13 and antigen-induced arthritis (AIA).14
Non-immune cells, in particular FLS, were reported to exhibit a strong decrease of inflammatory mediators on GC treatment in vitro.15 16 Whether direct GC actions on these stromal cells contribute to the suppression of inflammation in vivo is still unknown. Thus, we addressed this question in a T cell independent model of arthritis, the serum transfer-induced arthritis (STIA) model.
Herein, we show that direct actions of GCs on immune cells are not sufficient to reduce inflammation in STIA, whereas intact GR dimerisation in stromal cells such as FLS is required to increase proportion of non-classical Ly6Cneg, MHCIIneg macrophages, and to suppress STIA.
Methods
Methods are available in the online supplementary methods.
annrheumdis-2017-212762supp010.docx (29.5KB, docx)
Statistics
Results are presented as mean±SEM and statistical analysis was performed with two-way analysis of variance (ANOVA), followed by Tukey’s post hoc test (multiple comparisons for qRT-PCR, flow cytometry and area under the curve), with one-way ANOVA (ankle swelling) and a two-tailed Student’s t-test (cytokine analysis, efferocytosis measurements and cartilage and bone destruction).
Results
Functional GR in stromal cells is crucial to mediate the anti-inflammatory effects of GC treatment
First, we tested whether GCs act directly on immune cells to suppress inflammation by using the STIA model. To eliminate radiosensitive immune cells, we irradiated wild-type (wt) recipient mice and reconstituted them with fetal liver cells of GR-deficient donor mice (GRdel→wt) and as control with cells of wt donor mice (wt →wt) (figure 1A). GRdel→wt mice were deficient for the GR in the haematopoietic compartment (online supplementary figure 1A). The absence of GR in radiosensitive immune cells did not lead to differences in the onset or progression of STIA in wt→wt and GRdel→wt mice (figure 1B–D). Surprisingly, daily GC treatment starting at day 4 efficiently reduced ankle thickness (figure 1B) and clinical score (figure 1C,D), as well as infiltrating immune cells (figure 1E) in wt→wt and GRdel→wt mice. Thus, deletion of the GR in radiosensitive immune cells had no influence on GC therapy of STIA. Next, we generated mice with GR deletion in the radioresistant compartment by using irradiated tamoxifen-inducible GRflox; Rosa-26-Cre ERT (‘GRnull’) mice as recipients and reconstituted them with wt bone marrow (wt→GRnull and wt→wt, figure 1F) (online supplementary figure 1B). GR-deficient mice with a GR competent haematopoietic system (wt→GRnull) had a strongly attenuated response to dexamethasone (DEX) treatment compared with DEX-treated wt→wt controls assessed by ankle thickness (figure 1G), clinical score (figure 1H,I) and cellular infiltration (figure 1J). Cartilage and bone injury was strongly reduced by DEX in wt→wt mice, and to a lesser extent in wt→GRnull mice (online supplementary figure 2). In conclusion, GR expression in stromal cells is crucial for anti-inflammatory action of GCs.
Figure 1.

GR deficiency in stromal cells abrogates anti-inflammatory effects of glucocorticoid (GC) treatment in serum transfer-induced arthritis (STIA). (A) Scheme of fetal liver cell transfer into irradiated wt recipient mice. Mice were either reconstituted with wt donor fetal liver cells (wt→wt) or with GR-deficient donor fetal liver cells (GRdel→wt). (B) Ankle thickness and (C) clinical score of mice treated with PBS (black and red) or DEX (grey and blue). (D) AUC of the clinical score of mice treated with PBS (black and red) or DEX (grey and blue). (E) H&E staining of paraffin sections of hind paws of wt→wt and GRdel→wt mice (scale bar=200 µm); asterisks show the area of inflammation. Group size n=5 mice per group. (F) Scheme of bone marrow transplantation of irradiated wt and GR-deficient recipients with wt donor bone marrow cells (wt→wt and wt→GRnull). (G) Ankle thickness and (H) clinical score of mice treated with PBS (black and red) or DEX (grey and blue). (I) AUC of the clinical score of mice treated with PBS (black and red) or DEX (grey and blue). (J) H&E staining of paraffin sections of hind paws of wt→wt and wt→GRnull mice (scale bar=200 µm); asterisks show area of inflammation. Group size n=5 mice per group. AUC, area under the curve; DEX, dexamethasone; GR, glucocorticoid receptor; ns, not significant; PBS, phosphate buffered saline; wt, wild type.
annrheumdis-2017-212762supp001.pdf (10MB, pdf)
annrheumdis-2017-212762supp002.jpg (855.5KB, jpg)
{kind=link}
The anti-inflammatory actions of GC treatment require intact GR dimerisation in stromal cells
To test the impact of intact GR dimerisation on exogenous GC exposure in STIA, we analysed mice with an impaired GR dimerisation (GRdim). In contrast to wt mice, GRdim mice were resistant to DEX treatment in STIA reflected by sustained inflammation (figure 2A–D). Cartilage and bone injury was less reduced in DEX-treated GRdim mice as compared with wt mice (online supplementary figure 3).
Figure 2.

Intact GR dimerisation in stromal cells is crucial for proper anti-inflammatory glucocorticoid (GC) actions in serum transfer-induced arthritis (STIA). (A) Ankle thickness and (B) clinical score of wt mice and mice with an impaired GR dimerisation (GRdim) treated with PBS (black and red) or DEX (grey and blue). (C) AUC for the clinical score of wt and GRdim mice treated with PBS (black and red) or DEX (grey and blue). (D) H&E staining of paraffin sections of hind paws of wt and GRdim mice (scale bar=200 µm); asterisks show area of inflammation. Group size n=5–7 mice per group. (E) Scheme of bone marrow transplantation of irradiated wt and GRdim recipient mice that were reconstituted with wt bone marrow cells (wt→wt and wt→GRdim). (F) Ankle thickness and (G) clinical score of mice treated with PBS (black and red) or DEX (grey and blue). (H) AUC of the clinical score of mice treated with PBS (black and red) or DEX (grey and blue). (I) H&E staining of paraffin sections of hind paws of wt→wt and wt→GRdim mice (scale bar=200 µm); asterisks show area of inflammation. Group size n=4–6 mice per group. AUC, area under the curve; DEX, dexamethasone; GR, glucocorticoid receptor; ns, not significant; PBS, phosphate buffered saline; wt, wild type.
annrheumdis-2017-212762supp003.jpg (769.7KB, jpg)
{kind=link}
To address whether the necessity of GR dimerisation for suppression of inflammation is restricted to stromal cells, we analysed lethal irradiated GRdim mice that received wt bone marrow (figure 2E, online supplementary figure 1C). In contrast to control mice (wt→wt), GRdim mice with a wt haematopoietic compartment (wt→GRdim) did not respond to DEX treatment in STIA (figure 2F–I). Thus, GR dimerisation in stromal cells is essential to suppress inflammation in arthritis.
Loss of GR dimerisation in stromal cells has no effect on cytokine suppression
Since suppression of cytokines is a hallmark of the anti-inflammatory activities of GCs, we determined serum cytokine levels after 72 hours of DEX treatment in wt→wt and wt→GRdim chimeric mice. Despite the fact that wt→GRdim mice were resistant to GC therapy (figure 3A, online supplementary figure 1D), we observed a reduction of interleukin (IL)-1β, tumour necrosis factor (TNF)α and interferon-γ in both wt→wt and wt→GRdim mice (figure 3B). Thus, GC-mediated suppression of cytokines is insufficient to fully suppress STIA.
Figure 3.

Impaired GR dimerisation in stromal cells abrogates suppression of serum transfer-induced arthritis (STIA) but does not affect cytokine suppression by glucocorticoids (GC). (A) Ankle thickness and clinical score of irradiated wt and GRdim recipient mice reconstituted with wt bone marrow (wt→wt and wt→GRdim) treated for 72 hours with PBS (black and red) or DEX (grey and blue). (B) Serum cytokine levels of IL-1β, TNFα and IFNγ are suppressed in both wt→wt and wt→GRdim mice after DEX treatment. *p<0.05, group size n=5–6 mice per group. DEX, dexamethasone; GR, glucocorticoid receptor; IFN, interferon; IL, interleukin; PBS, phosphate buffered saline; TNF, tumour necrosis factor; wt, wild type.
GR dimers in stromal cells induce non-classical, non-activated macrophages
Flow cytometry analysis of joints of wt→wt and wt→GRdim mice after DEX treatment (figure 4 and online supplementary figure 5) revealed a significant reduction of the fraction of infiltrating leucocytes (CD45pos) and myeloid cells (CD11bpos) in DEX-treated wt→wt mice, but not in wt→GRdim mice (figure 4A). Neutrophils (CD45pos, CD11bpos, Ly6Gpos) and T cells (CD45pos, CD3pos) were not changed in wt→wt or wt→GRdim mice (figure 4A). In addition, the fraction of classical activated macrophages (Ly6Cpos, F4/80pos and F4/80pos, MHCIIpos) of CD11bpos cells (figure 4B) was also unchanged. Strikingly, we detected a strong and significant induced fraction of non-classical (CD11bpos, Ly6Cneg, F4/80pos) and non-activated macrophages (CD11bpos, F4/80pos, MHCIIneg) (figure 4B). This was supported by an increased ratio of non-activated macrophages to activated macrophages (MHCIIneg/MHCIIpos) in DEX-treated wt→wt mice, but not in wt→GRdim mice (figure 4C).
Figure 4.

Glucocorticoids (GC) reduce the percentage of CD45pos and CD11bpos cells and increase the fraction of non-classical, non-activated macrophages after 72 hours ofDEX treatment during serum transfer-induced arthritis (STIA) by a GR dimer-dependent mechanism in stromal cells. (A) Haematopoietic cells (CD45pos) and myeloid cells (CD11bpos) presented as percentage of single cells, and neutrophils (Ly6Gpos) and T cells (CD3pos) presented as percentage of CD45pos cells in the ankles of irradiated wt and GRdim mice reconstituted with wt bone marrow (wt→wt (black) and wt→GRdim (white)) treated with PBS or DEX for 72 hours. (B) Classical macrophages (Ly6Cpos, F4/80pos), non-classical macrophages (Ly6Cneg, F4/80pos), activated macrophages (MHCIIpos, F4/80pos) and non-activated macrophages (MHCIIneg, F4/80pos) in wt→wt (black) and wt→GRdim (white) mice after PBS or DEX treatment for 72 hours presented as percentage of CD11bpos cells and (C) the ratio of non-activated macrophages versus activated macrophages. *p<0.05; **p<0.01; ***p<0.001 by two-way analysis of variance (ANOVA) followed Tukey’s multiple comparison correction, group size n=5–6 mice per group. DEX, dexamethasone; GR, glucocorticoid receptor; Mø, macrophages; PBS, phosphate buffered saline.
annrheumdis-2017-212762supp005.pdf (1.7MB, pdf)
Accordingly, anti-inflammatory macrophage marker gene expression (CD163, CD36, AnxA1, Axl and MerTK) was induced in DEX-treated wt→wt mice but not in wt→GRdim mice (figure 5A,B). Of note, complete GRdim mice treated with DEX showed a comparable lack of upregulation of CD163, CD36 and Axl mRNA expression (online supplementary figure 6). Thus, in STIA DEX treatment increases the fraction of non-classical, non-activated anti-inflammatory macrophages dependent on GR dimerisation in stromal cells.
Figure 5.

The GR dimer in stromal cells mediates the induction of genes of anti-inflammatory macrophages after DEX treatment in serum transfer-induced arthritis (STIA). (A) Quantitative RT-PCR of isolated ankle mRNA extracts showing the regulation of CD163, CD36, AnxA1, Axl and MerTK after 72 hours of DEX treatment in wt→wt and wt→GRdim mice. Group size n=5–6 mice per group. (B) Quantitative RT-PCR of isolated ankle mRNA extracts showing the regulation of CD163, CD36, AnxA1, Axl and MerTK after 10 days of DEX treatment in wt→wt and wt→GRdim mice. Group size n=4–6 mice per group. *P<0.05; **P<0.01; ***P<0.001 by two-way analysis of variance (ANOVA) followed Tukey’s multiple comparison correction. DEX, dexamethasone; GR, glucocorticoid receptor; PBS, phosphate buffered saline.
annrheumdis-2017-212762supp006.pdf (523.7KB, pdf)
GR deficiency in FLS delays GC-induced suppression of STIA
Next, we addressed to which extent the GR in FLS mediates the GC-induced suppression of inflammation in STIA. We therefore crossed GRflox mice to Col1a2CreERT mice17 to generate tamoxifen-inducible GRflox; Col1a2-CreERTtg/+ mice (GRCol1a2-CreERT). We observed a strong reduction of GR mRNA expression in FLS isolated from the joints (online supplementary figure 7A) and reduced GR protein expression in cadherin-11 positive cells (online supplementary figure 7B). Subsequently, we subjected these mice to STIA and started DEX treatment after STIA induction when an ankle thickness of 0.93±0.12 and 0.89±0.11 mm was reached in mutant and control mice. Strikingly, DEX treatment of GRCol1a2-CreERT mice resulted in a delayed and reduced reduction of the ankle thickness and clinical score compared with GRflox littermates (figure 6A,B). Histological analysis revealed ongoing infiltration of immune cells in the joints of DEX-treated GRCol1a2-CreERT mice, where inflammatory infiltration was resolved in DEX-treated GRflox mice (figure 6C). Thus, deletion of the GR in FLS reduces the anti-inflammatory responsiveness towards DEX treatment in STIA.
Figure 6.

GR deficiency in FLS results in delayed suppression of STIA after DEX treatment and impaired GR dimerisation in FLS leads to a distinct gene expression pattern after DEX treatment. (A) Ankle thickness and clinical score of tamoxifen-inducible GRCol1a2-CreERT and GRflox control mice treated with PBS (black and red) or DEX (grey and blue). (B) AUC of the clinical score of tamoxifen-inducible GRCol1a2-CreERT and GRflox control mice treated with PBS (black and red) or DEX (grey and blue). (C) H&E staining of paraffin sections of hind paws of GRCol1a2-CreERT and GRflox control mice. Original magnification: 5× (scale bar=200 µm), asterisks show the area of inflammation. Group size n=5–7 mice. *P<0.05 by two-way analysis of variance (ANOVA) followed Tukey’s multiple comparison correction. (D) Differentially expressed genes of wt and GRdim FLS treated with interleukin (IL)-1β and DEX by RNASeq analysis. n=3. (E) Cytokine multiplex analysis of supernatants of wt and GRdim FLS treated with control medium, DEX, IL-1β or in combination with DEX and IL-1β. n=4. *p<0.05 and **p<0.01 by two-way ANOVA followed Tukey’s multiple comparison correction. (F) Scheme of glucocorticoid (GC)-induced suppression of STIA. AUC, area under the curve; DEX, dexamethasone; FLS, fibroblast-like synoviocytes; GR, glucocorticoid receptor; ns, not significant; PBS, phosphate buffered saline; STIA, serum transfer-induced arthritis; TNF, tumour necrosis factor; wt, wild type.
annrheumdis-2017-212762supp007.jpg (3.7MB, jpg)
{kind=link}
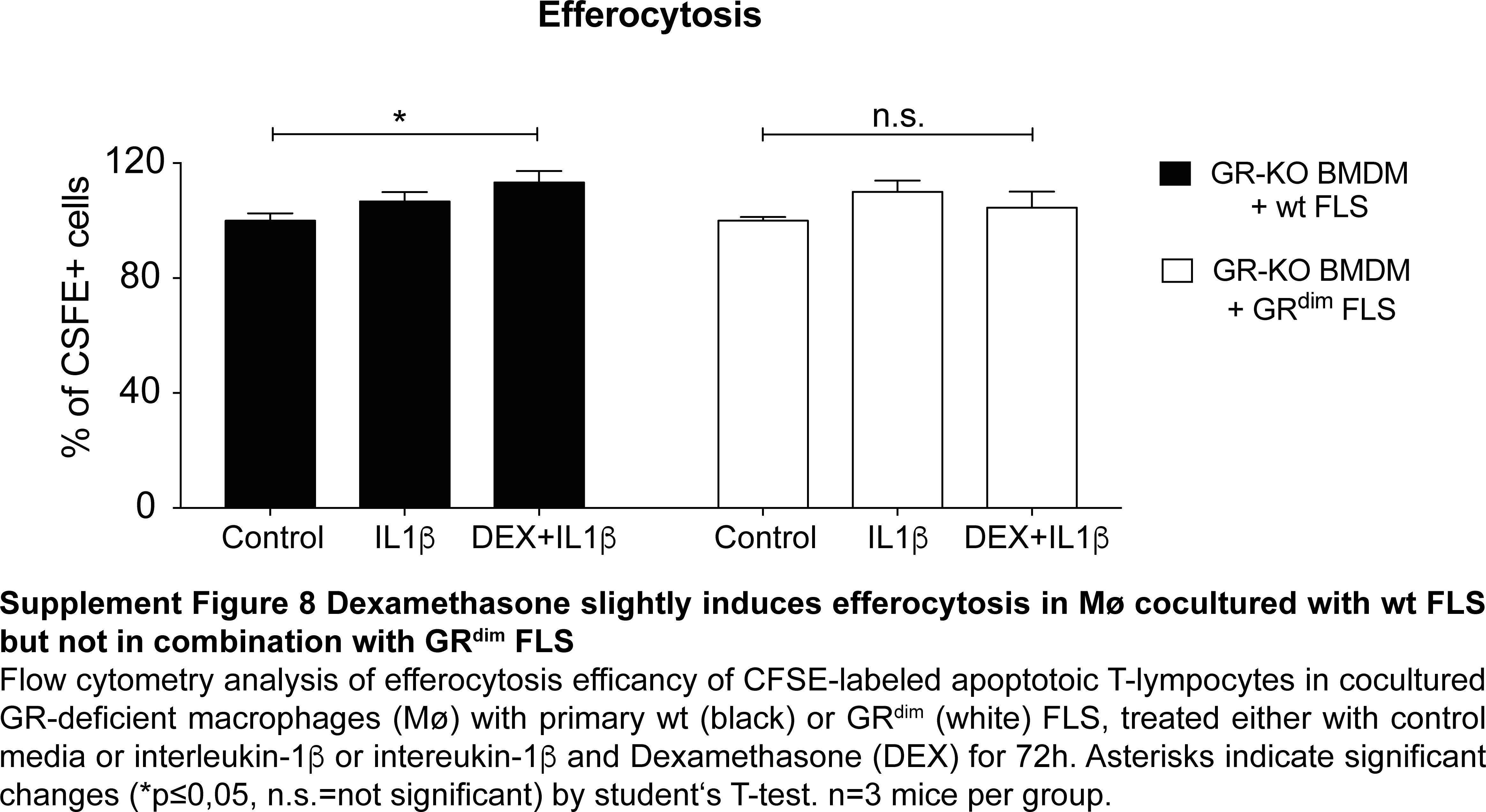
As FLS sustain inflammation in arthritis in tight interplay with macrophages, we subsequently analysed to which extent impaired GR dimerisation in FLS changes the effects of GCs on macrophage function. We therefore cultured wt and GRdim FLS with GR-deficient macrophages treated with IL1β and DEX and determined their capacity of efferocytosis of labelled apoptotic T lymphocytes. DEX-treated cocultures of wt FLS and macrophages showed a small, but significant increase of efferocytosis that was absent in cultures of macrophages with GRdim FLS (online supplementary figure 8). Next we analysed the transcriptome of wt and GRdim FLS treated with IL-1β and DEX by RNASeq. We found 346 genes upregulated and 212 genes downregulated in GRdim FLS compared with wt FLS. Among these genes, classically known GR-target genes such as Gilz, Metallothionein-1 and 2 and Fkbp5 were strongly induced in wt FLS treated with IL-1β and DEX, whereas in GRdim FLS these genes were significantly less induced (online supplementary figure 9A). Enriched KEGG pathways included cell cycle, cell adhesion, extracellular matrix (ECM)-receptor interaction and others, indicating a higher activity of GRdim FLS exposed to DEX (online supplementary figure 9C). Interestingly, we found macrophage-associated chemokines "macrophage inflammatory protein -1α and -β" (Mip-1α and Mip-1β) strongly downregulated in wt but not in GRdim FLS (online supplementary figure 9B). These results were confirmed by a multiplex analysis of wt and GRdim FLS supernatants (figure 6E). However, classical inflammatory cytokines, like IL-6 and TNFα, were sufficiently suppressed by DEX in both wt and GRdim FLS (figure 6E).
annrheumdis-2017-212762supp008.jpg (893.7KB, jpg)
{kind=link}
annrheumdis-2017-212762supp009.pdf (550.1KB, pdf)
Taken together FLS with compromised GR dimerisation are less efficient to induce macrophage efferocytosis, have a distinct gene signature including enriched KEGG pathway for cell cycle and cell adhesion, and are impaired in DEX-mediated chemokine regulation.
Discussion
Here we show for the first time that stromal cells, presumably FLS, are essential for suppression of inflammation by GCs in an arthritis mouse model that closely resembles the pathology of the human disease. In STIA, GR deficiency in immune cells is dispensable for anti-inflammatory effects of GCs. These findings challenge the general assumption that the GR mediates anti-inflammatory actions mainly by direct actions on immune cells. We could further show that functional GR dimerisation in stromal cells is crucial to mediate the anti-inflammatory effects, emphasising the importance of GR dimerisation for mediating GC actions in multiple models of arthritis such as AIA, G6PI arthritis14 and STIA. The spontaneous resolution of STIA in the absence of DEX, however, is not affected by an impaired GR dimerisation (data not shown).
Intriguingly, repression of inflammatory cytokines by GCs in a therapeutic setting was not affected by impaired GR dimerisation in stromal cells, despite an ongoing joint inflammation. In addition, we could show that DEX treatment from day 0 onwards does not prevent initiation of STIA, but strongly impairs progression of disease comparable to therapeutic conditions (online supplementary figure 4). Thus, although cytokines like IL-1β and TNFα are shown to be important for the induction of STIA18 their decline alone is not enough to inhibit progression of inflammation. This finding is important considering the increasing focus on cytokine inhibitors as treatment for patients with arthritis. Recombinant IL1 receptor antagonist treatment in patients with RA shows only weak effects.19 Also, TNFα inhibitors failed to exert effective disease remission and 20%–40% of patients are not responding at all to the therapy.20 However, cytokine blockade, achieved with different functional antibody treatments, can result in positive outcomes,20 and cytokine suppression has shown to affect bone and cartilage destruction in arthritis.21 Here we observed a strong attenuation of DEX-mediated reduction in bone and cartilage damage, when GR in stromal cells was compromised, indicating that reduction of cytokines alone is not sufficient to reduce STIA-induced damage.
annrheumdis-2017-212762supp004.pdf (549.7KB, pdf)
Since the immune cells in the bone marrow chimeric wt→GRdim mice are still expressing the functional GR we hypothesise that the suppression of these cytokines is mediated by a direct effect of DEX on immune cells. This is supported by findings that mice with a macrophage-specific GR deletion or a complete impaired GR dimerisation are not able to suppress lipopolysaccharide-induced cytokine expression after DEX treatment in vitro.22 23 Of note, GCs did not alter the percentage of classical activated macrophages (F4/80pos, Ly6Cpos and F4/80pos, MHCIIpos), but reduced the fraction of CD45pos and CD11bpos cells. Thus, they might be the targets of cytokine suppression. However, we could show that GCs increased the relative abundance of non-classical, non-activated macrophages (F4/80pos, Ly6Cneg and F4/80pos, MHCIIneg).
These anti-inflammatory acting macrophages were previously shown to be essential for the resolution of STIA and collagen-induced arthritis.24 25 In line with the induction of the percentage of anti-inflammatory macrophages we detected only in wt mice an increased expression of several anti-inflammatory markers known to be associated with increased phagocytosis and efferocytosis activity (CD163, CD36, AnxA1, Axl and MerTK). For instance, AnxA1-deficient mice have decreased levels of phagocyting cells.26 CD36 silencing attenuates efferocytosis27 and loss of MerTK and Axl results in a lack of apoptotic cell clearance, induction of autoimmunity28 and aggravated STIA.29 Accordingly, cocultures of FLS with macrophages failed to increase efferocytosis in the presence of GRdim FLS. Taken together, the failure of wt→GRdim mice to increase these factors upon DEX treatment suggests an impaired clearance of apoptotic cells after DEX treatment, resulting in an ongoing inflammation. This emphasises the possibility to improve therapeutic strategies by inducing the generation of non-classical, non-inflammatory macrophages.
We suggest that the induction of therapeutically important anti-inflammatory macrophages can be indirectly stimulated by actions of stromal cells, in particular FLS. So far, FLS were mainly characterised as drivers of chronic inflammation in RA due to their hyperproliferation and secretion of multiple inflammatory mediators such as cytokines, matrix metalloproteinases and other factors.30 It is also well known that FLS contribute substantially to the TNF response of macrophages31 and support monocyte survival and integrity in the lining structure.32 We also could show a slightly increased efferocytosis efficiency of Mø cocultured with wt FLS compared with those that were cocultured with GRdim FLS, when treated with IL-1β and DEX. GRdim FLS show a specific gene signature induced by DEX exposure. Cell cycle-associated factors and their regulators, but also cell adhesion molecules are in general higher expressed, suggesting a higher proliferative and migratory activity of FLS. Whether this is the reason for failing to trigger anti-inflammatory macrophages in vivo in GRdim mice requires further investigation. In addition, GRdim FLS are resistant to DEX-mediated suppression of macrophage-recruiting cytokines Mip-1α and Mip-1β. Interestingly, Mip-1α-deficient mice are protected from collagen induced arthritis (CIA), which was independent of serum TNFα levels that were equal in wt and Mip-1α-deficient mice.33 This is supported by anti-Mip-1α antibody treatment of CIA mice resulting in the attenuation of CIA.34 Taken together, these data imply an important crosstalk between FLS and macrophages in GC-induced suppression of STIA.
So far, GC actions on FLS were considered as anti-inflammatory due to the attenuation of expression of inflammatory mediators15 16 mediated by the monomeric GR. Challenging this view, we provide here the concept that FLS can mediate anti-inflammatory effects evoked by GCs and their dimerised receptor by increasing anti-inflammatory macrophage populations (figure 6F).
The action of GR in FLS and stromal cells is essential; the action of GR in macrophages themselves does not suffice to reduce inflammation. Accordingly, liposomal-packed prednisolone only reduced inflammatory gene expression but did not induce non-classical, anti-inflammatory macrophages.35 Supporting our conclusion, prednisolone-carrying liposomes targeting synovial lining cells strongly increase therapeutic benefits in collagen-induced arthritis36 and adjuvant arthritis in rats.37
In summary, our data revealed a major role of stromal cells such as FLS in GC-mediated suppression of arthritis to induce anti-inflammatory acting macrophages. Exploiting the GC-induced anti-inflammatory crosstalk between stromal cells and the immune system will provide novel rationales for therapies with higher efficiency and may avoid side effects of steroid therapy.
Acknowledgments
We are grateful to the staff of the animal facilities of the University of Ulm, our animal welfare officer Dr Sibylle Ott and in particular to Birgit Widmann. We thank Sandra Förtsch of the Institute for Molecular Psychosomatics for providing IHC reagents.
Footnotes
UB and JPT contributed equally.
Handling editor: Josef S Smolen
Contributors: MK performed experiments, processed the data and wrote the manuscript. SC performed experiments and processed the data. SV and GC performed experiments. LF analysed the data. WB provided materials. GK provided materials and revised the manuscript. UB designed the study, performed experiments, processed the data and wrote the manuscript. JPT designed the study and wrote the manuscript. UB and JPT contributed equally to the study and share correspondence.
Funding: This study was funded by the Boehringer Ingelheim Foundation (to JPT), Deutsche Forschungsgemeinschaft (DFG, Priority Program Immunobone 1468, Tu 220/6-1, 6-2, Collaborative Research Centre 1149, C02/INST 40/492-1, Trilateral Consortium Tu 220/12-1 to JPT).
Competing interests: None declared.
Patient consent: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: All data are in the manuscript and supplementary figures, except the raw data of the RNASeq study. These data will be made available upon publication.
Presented at: Parts of this manuscript were orally presented at the 2016 ACR/ARHP Annual Meeting and as such the abstract was published in an online supplementary of Arthritis & Rheumatology: Koenen M, Baschant U, Culemann S, Kockmann T, Kaltenbach HM, Vettorazzi S, Nanni P, Roschitzki B, Auf dem Keller U, Tuckermann JP. Glucocorticoid receptor dimerization in stromal cells modulates macrophage polarization during serum transfer-induced arthritis [abstract]. Arthritis Rheumatol. 2016;68 (suppl 10).
References
- 1. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med 2011;365:2205–19. 10.1056/NEJMra1004965 [DOI] [PubMed] [Google Scholar]
- 2. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev 2010;233:233–55. 10.1111/j.0105-2896.2009.00859.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hench PS. The reversibility of certain rheumatic and non-rheumatic conditions by the use of cortisone or of the pituitary adrenocorticotropic hormone, 1950. [Google Scholar]
- 4. van Everdingen AA, Jacobs JW, Siewertsz Van Reesema DR, et al. . Low-dose prednisone therapy for patients with early active rheumatoid arthritis: clinical efficacy, disease-modifying properties, and side effects: a randomized, double-blind, placebo-controlled clinical trial. Ann Intern Med 2002;136:1–12. 10.7326/0003-4819-136-1-200201010-00006 [DOI] [PubMed] [Google Scholar]
- 5. Hartmann K, Koenen M, Schauer S, et al. . Molecular actions of glucocorticoids in cartilage and bone during health, disease, and steroid therapy. Physiol Rev 2016;96:409–47. 10.1152/physrev.00011.2015 [DOI] [PubMed] [Google Scholar]
- 6. Ogawa A, Johnson JH, Ohneda M, et al. . Roles of insulin resistance and beta-cell dysfunction in dexamethasone-induced diabetes. J Clin Invest 1992;90:497–504. 10.1172/JCI115886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schäcke H, Berger M, Rehwinkel H, et al. . Selective glucocorticoid receptor agonists (SEGRAs): novel ligands with an improved therapeutic index. Mol Cell Endocrinol 2007;275:109–17. 10.1016/j.mce.2007.05.014 [DOI] [PubMed] [Google Scholar]
- 8. Lim HW, Uhlenhaut NH, Rauch A, et al. . Genomic redistribution of GR monomers and dimers mediates transcriptional response to exogenous glucocorticoid in vivo. Genome Res 2015;25:836–44. 10.1101/gr.188581.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schiller BJ, Chodankar R, Watson LC, et al. . Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biol 2014;15:418 10.1186/s13059-014-0418-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reichardt HM, Kaestner KH, Tuckermann J, et al. . DNA binding of the glucocorticoid receptor is not essential for survival. Cell 1998;93:531–41. 10.1016/S0092-8674(00)81183-6 [DOI] [PubMed] [Google Scholar]
- 11. Vandevyver S, Dejager L, Van Bogaert T, et al. . Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J Clin Invest 2012;122:2130–40. 10.1172/JCI60006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kleiman A, Hübner S, Rodriguez Parkitna JM, et al. . Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. Faseb J 2012;26:722–9. 10.1096/fj.11-192112 [DOI] [PubMed] [Google Scholar]
- 13. Vettorazzi S, Bode C, Dejager L, et al. . Glucocorticoids limit acute lung inflammation in concert with inflammatory stimuli by induction of SphK1. Nat Commun 2015;6:7796 10.1038/ncomms8796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baschant U, Frappart L, Rauchhaus U, et al. . Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in T cells. Proc Natl Acad Sci U S A 2011;108:19317–22. 10.1073/pnas.1105857108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hardy RS, Hülso C, Liu Y, et al. . Characterisation of fibroblast-like synoviocytes from a murine model of joint inflammation. Arthritis Res Ther 2013;15:R24 10.1186/ar4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gossye V, Elewaut D, Bougarne N, et al. . Differential mechanism of NF-kappaB inhibition by two glucocorticoid receptor modulators in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 2009;60:3241–50. 10.1002/art.24963 [DOI] [PubMed] [Google Scholar]
- 17. Singh K, Maity P, Krug L, et al. . Superoxide anion radicals induce IGF-1 resistance through concomitant activation of PTP1B and PTEN. EMBO Mol Med 2015;7:59–77. doi:10.15252/emmm.201404082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ji H, Pettit A, Ohmura K, et al. . Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. J Exp Med 2002;196:77–85. 10.1084/jem.20020439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burger D, Dayer JM, Palmer G, et al. . Is IL-1 a good therapeutic target in the treatment of arthritis? Best Pract Res Clin Rheumatol 2006;20:879–96. 10.1016/j.berh.2006.06.004 [DOI] [PubMed] [Google Scholar]
- 20. Mikuls TR, Weaver AL. Lessons learned in the use of tumor necrosis factor-alpha inhibitors in the treatment of rheumatoid arthritis. Curr Rheumatol Rep 2003;5:270–7. 10.1007/s11926-003-0005-9 [DOI] [PubMed] [Google Scholar]
- 21. Jung SM, Kim KW, Yang CW, et al. . Cytokine-mediated bone destruction in rheumatoid arthritis. J Immunol Res 2014;2014:1–15. 10.1155/2014/263625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tuckermann JP, Kleiman A, Moriggl R, et al. . Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J Clin Invest 2007;117:1381–90. 10.1172/JCI28034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bhattacharyya S, Brown DE, Brewer JA, et al. . Macrophage glucocorticoid receptors regulate Toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood 2007;109:4313–9. 10.1182/blood-2006-10-048215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park SY, Lee SW, Lee SY, et al. . SIRT1/adenosine monophosphate-activated protein kinase α signaling enhances macrophage polarization to an anti-inflammatory phenotype in rheumatoid arthritis. Front Immunol 2017;8 10.3389/fimmu.2017.01135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Misharin AV, Cuda CM, Saber R, et al. . Nonclassical Ly6C(-) monocytes drive the development of inflammatory arthritis in mice. Cell Rep 2014;9:591–604. 10.1016/j.celrep.2014.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hannon R, Croxtall JD, Getting SJ, et al. . Aberrant inflammation and resistance to glucocorticoids in annexin 1 −/− mouse. Faseb J 2003;17:253–5. 10.1096/fj.02-0239fje [DOI] [PubMed] [Google Scholar]
- 27. Kim W, Lee HN, Jang JH, et al. . 15-Deoxy-Δ12,14-prostaglandin J2 exerts proresolving effects through nuclear factor E2-related factor 2-induced expression of CD36 and heme oxygenase-1. Antioxid Redox Signal 2017;27:1412–31. 10.1089/ars.2016.6754 [DOI] [PubMed] [Google Scholar]
- 28. Seitz HM, Camenisch TD, Lemke G, et al. . Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J Immunol 2007;178:5635–42. 10.4049/jimmunol.178.9.5635 [DOI] [PubMed] [Google Scholar]
- 29. Waterborg CEJ, Través PG, Beermann S, et al. . 03.05 The tam receptors axl and mer play a protective role in a temporal and spatial manner in inflammatory arthritis. Ann Rheum Dis 2017;76:A31. [Google Scholar]
- 30. Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol 2013;9:24–33. 10.1038/nrrheum.2012.190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Donlin LT, Jayatilleke A, Giannopoulou EG, et al. . Modulation of TNF-induced macrophage polarization by synovial fibroblasts. J Immunol 2014;193:2373–83. 10.4049/jimmunol.1400486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kiener HP, Watts GF, Cui Y, et al. . Synovial fibroblasts self-direct multicellular lining architecture and synthetic function in three-dimensional organ culture. Arthritis Rheum 2010;62:742–52. 10.1002/art.27285 [DOI] [PubMed] [Google Scholar]
- 33. Chintalacharuvu SR, Wang JX, Giaconia JM, et al. . An essential role for CCL3 in the development of collagen antibody-induced arthritis. Immunol Lett 2005;100:202–4. 10.1016/j.imlet.2005.03.012 [DOI] [PubMed] [Google Scholar]
- 34. Kasama T, Strieter RM, Lukacs NW, et al. . Interleukin-10 expression and chemokine regulation during the evolution of murine type II collagen-induced arthritis. J Clin Invest 1995;95:2868–76. 10.1172/JCI117993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hofkens W, Schelbergen R, Storm G, et al. . Liposomal targeting of prednisolone phosphate to synovial lining macrophages during experimental arthritis inhibits M1 activation but does not favor M2 differentiation. PLoS One 2013;8:e54016 10.1371/journal.pone.0054016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Metselaar JM, van den Berg WB, Holthuysen AE, et al. . Liposomal targeting of glucocorticoids to synovial lining cells strongly increases therapeutic benefit in collagen type II arthritis. Ann Rheum Dis 2004;63:348–53. 10.1136/ard.2003.009944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vanniasinghe AS, Manolios N, Schibeci S, et al. . Targeting fibroblast-like synovial cells at sites of inflammation with peptide targeted liposomes results in inhibition of experimental arthritis. Clin Immunol 2014;151:43–54. 10.1016/j.clim.2014.01.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
annrheumdis-2017-212762supp010.docx (29.5KB, docx)
annrheumdis-2017-212762supp001.pdf (10MB, pdf)
annrheumdis-2017-212762supp002.jpg (855.5KB, jpg)
annrheumdis-2017-212762supp003.jpg (769.7KB, jpg)
annrheumdis-2017-212762supp005.pdf (1.7MB, pdf)
annrheumdis-2017-212762supp006.pdf (523.7KB, pdf)
annrheumdis-2017-212762supp007.jpg (3.7MB, jpg)
annrheumdis-2017-212762supp008.jpg (893.7KB, jpg)
annrheumdis-2017-212762supp009.pdf (550.1KB, pdf)
annrheumdis-2017-212762supp004.pdf (549.7KB, pdf)