

Abstract
All living things have evolved to sense changes in their environment in order to respond in adaptive ways. At the cellular level, these sensing systems generally involve receptor molecules at the cell surface, which detect changes outside the cell and relay those changes to the appropriate response elements downstream. With the advent of experimental technologies that can track signalling at the single-cell level, it has become clear that many signalling systems exhibit significant levels of ‘noise,’ manifesting as differential responses of otherwise identical cells to the same environment. This noise has a large impact on the capacity of cell signalling networks to transmit information from the environment. Application of information theory to experimental data has found that all systems studied to date encode less than 2.5 bits of information, with the majority transmitting significantly less than 1 bit. Given the growing interest in applying information theory to biological data, it is crucial to understand whether the low values observed to date represent some sort of intrinsic limit on information flow given the inherently stochastic nature of biochemical signalling events. In this work, we used a series of computational models to explore how much information a variety of common ‘signalling motifs’ can encode. We found that the majority of these motifs, which serve as the basic building blocks of cell signalling networks, can encode far more information (4–6 bits) than has ever been observed experimentally. In addition to providing a consistent framework for estimating information-theoretic quantities from experimental data, our findings suggest that the low levels of information flow observed so far in living system are not necessarily due to intrinsic limitations. Further experimental work will be needed to understand whether certain cell signalling systems actually can approach the intrinsic limits described here, and to understand the sources and purpose of the variation that reduces information flow in living cells.
Keywords: information theory, cell signalling, noise
1. Introduction
Signalling networks enable cells to sense information about their environment in order to adapt appropriately to changing conditions. Quantifying the reliability of communication has long been the domain of information theory [1]. Information theoretic concepts have been relevant to many areas of biology for quite some time (most notably neuroscience and bioinformatics) [2–8], but have not been applied to systems biology until relatively recently. It is becoming increasingly clear, however, that information theory is relevant for understanding information transmission via signal transduction networks and the corresponding cell-fate decisions that are made on the basis of environmental cues [9–13]. In the context of cell signalling, environmental information like the concentration of some nutrient or cytokine corresponds to the input to the channel, S, and the output can be quantified as some downstream molecular or phenotypic response, R [9]. The relevant quantity for measuring information transmission in signalling networks is the mutual information:
which is ultimately a non-parametric measure of the correlation between the signal, S, and corresponding downstream response, R. The mutual information is generally quantified in units of bits, which arises from employing a base 2 logarithm in the calculation [1,14]. Furthermore, estimation of the mutual information requires only that the signal variable and the response variable in question are measured; no underlying mechanistic knowledge of the signalling network is necessary. However, the mutual information depends on the input signal distribution (i.e. 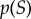 ) and thus does not necessarily reveal the underlying information transmission capabilities of the channel that are typically of primary interest. The relevant quantity for characterizing the limits of information transmission through some arbitrary signalling channel is the channel capacity, C, which is defined as the supremum of the mutual information over all possible probability distributions of the signal:
) and thus does not necessarily reveal the underlying information transmission capabilities of the channel that are typically of primary interest. The relevant quantity for characterizing the limits of information transmission through some arbitrary signalling channel is the channel capacity, C, which is defined as the supremum of the mutual information over all possible probability distributions of the signal:
This quantity is a property of the channel itself, and it is the upper limit on the amount of information that can be transmitted through a channel [1,14]. Practically speaking, any computational framework that aims to estimate C from experimental data cannot try all possible input distributions. Firstly, the data are always restricted to a finite set of input signals used in the experiment (call this finite set 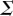 ). The supremum above is then estimated by ‘trying’ a finite set of possible probability distributions and choosing the one that maximizes the mutual information [9,10,15] (call this finite set of probability distributions
). The supremum above is then estimated by ‘trying’ a finite set of possible probability distributions and choosing the one that maximizes the mutual information [9,10,15] (call this finite set of probability distributions 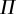 ). Technically speaking, calculations of the ‘channel capacity’ are thus just lower-bound estimates of C given these two finite sets (which we have previously defined as
). Technically speaking, calculations of the ‘channel capacity’ are thus just lower-bound estimates of C given these two finite sets (which we have previously defined as 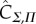 [15]). In this work, we follow the convention of calling these estimates C (e.g. [9,10]), but the limitations of estimating C from data should be kept in mind (see Methods).
[15]). In this work, we follow the convention of calling these estimates C (e.g. [9,10]), but the limitations of estimating C from data should be kept in mind (see Methods).
To date, there have been a number of studies examining the flow of information in intracellular signal transduction. Levchenko and co-workers measured the information transmitted in the form of nuclear-localized NF- B given some level of stimulation by TNF-
B given some level of stimulation by TNF-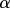 [9]. In this case, the response was measured at a particular point in time corresponding to the approximate peak in NF-
[9]. In this case, the response was measured at a particular point in time corresponding to the approximate peak in NF- B localization at 30 min following stimulation. Despite the relative importance of this signalling network in governing cellular decisions, they ultimately found that the amount of information that can be transmitted by this network is less than 1 bit, meaning that this particular signal–response pair is incapable of reliably making even binary decisions. Other studies have explored whether cells employ strategies to decrease the noise responsible for these seemingly low values (e.g. using dynamical trends as response to stimulus instead of a single point in time, or fold-change detection instead of concentration/copy number of chemical species) [10,16–19], or if noisy responses can be useful for responses at the level of cellular populations instead of individual cells [15]. While these investigations have contributed greatly to our understanding of information transmission in specific cases, a full understanding of the general properties of information transmission in signalling networks have not yet been realized. For instance, the majority of signalling networks studied so far have estimated channel capacities ranging between 0.5 and 2.5 bits of information at the single-cell level [9,10,15,17–19]. It is currently unclear if these values are indicative of all signalling networks or if 2.5 bits of information represents an intrinsic upper limit on intracellular information transmission [15].
B localization at 30 min following stimulation. Despite the relative importance of this signalling network in governing cellular decisions, they ultimately found that the amount of information that can be transmitted by this network is less than 1 bit, meaning that this particular signal–response pair is incapable of reliably making even binary decisions. Other studies have explored whether cells employ strategies to decrease the noise responsible for these seemingly low values (e.g. using dynamical trends as response to stimulus instead of a single point in time, or fold-change detection instead of concentration/copy number of chemical species) [10,16–19], or if noisy responses can be useful for responses at the level of cellular populations instead of individual cells [15]. While these investigations have contributed greatly to our understanding of information transmission in specific cases, a full understanding of the general properties of information transmission in signalling networks have not yet been realized. For instance, the majority of signalling networks studied so far have estimated channel capacities ranging between 0.5 and 2.5 bits of information at the single-cell level [9,10,15,17–19]. It is currently unclear if these values are indicative of all signalling networks or if 2.5 bits of information represents an intrinsic upper limit on intracellular information transmission [15].
In this work, we have begun to characterize the limits of information transmission in intracellular signal transduction in order to develop a theoretical understanding of cellular decision-making in the context of information theory. In particular, we start by focusing on atomistic signalling motifs (e.g. ligand–receptor interactions) and then progress to slightly larger networks that still achieve a dynamic steady state. In order to do this, we developed a framework for consistent comparison of information transmission in distinct systems. In the presence of only intrinsic biochemical noise, we find that motifs like the ligand–receptor interaction or post-translational modification cycles can encode more than 5 bits of information with reasonable bounds on protein copy numbers and parameters, which is far more than has ever been observed experimentally. As networks become more complicated (i.e. post-translational modification cycles coupled in, say, a kinase cascade), information content degrades as it is transmitted through the network. Nonetheless, cascades and other motifs can encode well over 3 bits of information, again much higher than has been observed experimentally. Our findings thus indicate that low levels of information transmission may be strongly influenced by ‘extrinsic noise’ in protein levels, as has been suggested previously [10,15]. By providing comparative estimates of the bounds of information transmission in these simple signalling motifs, our work also provides an intuitive framework for future experiments aimed at characterizing cellular decision-making processes that occur via larger, more complex networks (e.g. Wnt- or IGF-induced signalling). Finally, since this work examines specific signalling motifs, we also investigate whether or not certain biochemical trade-offs regulate or limit the flow of information through signalling networks in certain circumstances (e.g. saturation of enzymes in a covalent modification cycle tends to reduce information transmission). In addition to providing a platform for future work into quantifying information transmission in signalling networks, we expect that this work is fundamental to understanding why and how certain networks transmit specific levels of information.
2. Results
2.1. Framework
Prior approaches for data collection and estimation of information theoretic quantities have not generally considered a number of factors that can impact these estimates [9,10,12,13,15], such as the range or density of the sampled signal values. Since such factors can have a large impact on the estimation of the mutual information or channel capacity, any attempt to consistently compare different signalling networks must control for these effects. In order to systematically investigate the upper limits of information transmission in cells, we first developed a simple framework to control elements of the sampling of signal values that could impact estimation of information theoretic quantities.
In order to develop this framework, we considered a simple model of a signalling network defined only by a sigmoid function, commonly known as the ‘Hill function’ in biochemistry:
where R and S denote response and signal, respectively, n controls the ultrasensitivity of the response and K is the signal value resulting in half-maximal response. This model also includes a noise term,  , which is sampled from a Gaussian distribution with mean
, which is sampled from a Gaussian distribution with mean 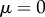 , and some chosen standard deviation,
, and some chosen standard deviation,  [15]. We chose this model to develop our framework for a number of reasons beyond its simplicity. For one, this model removes any assumptions about the underlying reaction network motif, allowing us to vary parameters like ultrasensitivity and the level of noise independently. Additionally, it turns out that many signalling motifs produce steady-state dose–response trends that are reminiscent of a sigmoid function, including phosphorylation cycles and kinase cascades [20,21], so this model captures many of the key features that will characterize the specific signalling motifs considered below. We proceeded to numerically sample signal and response pairs from this model; the resulting dose–response data can be seen in figure 1a for distinct levels of ultrasensitivity. Figure 1b shows the same data, but transformed to use indices for the signal in place of the raw values, where the minimum signal value for some arbitrary signal–response dataset is assigned an index of 0 and the maximum is assigned an index of
[15]. We chose this model to develop our framework for a number of reasons beyond its simplicity. For one, this model removes any assumptions about the underlying reaction network motif, allowing us to vary parameters like ultrasensitivity and the level of noise independently. Additionally, it turns out that many signalling motifs produce steady-state dose–response trends that are reminiscent of a sigmoid function, including phosphorylation cycles and kinase cascades [20,21], so this model captures many of the key features that will characterize the specific signalling motifs considered below. We proceeded to numerically sample signal and response pairs from this model; the resulting dose–response data can be seen in figure 1a for distinct levels of ultrasensitivity. Figure 1b shows the same data, but transformed to use indices for the signal in place of the raw values, where the minimum signal value for some arbitrary signal–response dataset is assigned an index of 0 and the maximum is assigned an index of 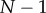 , where N is the number of unique signal values sampled. This transformation conserves the underlying correlation between the signal and response (and thus does not alter mutual information calculations), but facilitates visual comparison between distinct datasets. Using datasets generated from this model, we can examine how both the selection of distinct ranges of signal values and the number of sampled signal values impact estimates of the channel capacity. To calculate those estimates, we used a software package that we recently developed [15]. This package implements an approach to estimating the channel capacity broadly similar to that employed by Levchenko and co-workers [9]; further details may be found in the Methods, in the electronic supplementary material and in [15].
, where N is the number of unique signal values sampled. This transformation conserves the underlying correlation between the signal and response (and thus does not alter mutual information calculations), but facilitates visual comparison between distinct datasets. Using datasets generated from this model, we can examine how both the selection of distinct ranges of signal values and the number of sampled signal values impact estimates of the channel capacity. To calculate those estimates, we used a software package that we recently developed [15]. This package implements an approach to estimating the channel capacity broadly similar to that employed by Levchenko and co-workers [9]; further details may be found in the Methods, in the electronic supplementary material and in [15].
Figure 1.

Characterizing the information in a simple signalling model. Error bars in this and subsequent figures denote 95% confidence intervals in the channel capacity estimation procedure. (a) Transition zone dose–response data from the Hill function model sampled with 32 signal values for two different values of n but identical levels of noise (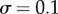 ). (b) The data from panel (a) but with the signal values mapped to indices for comparative analysis. Note that while the transition zone shrinks with increasing ultrasensitivity, normalization to the width of the transition zone reveals the similarity of the two datasets. (c) Channel capacity of the simple model for two levels of noise as a function of a shifting range of signal values. In both cases, the maximum information transmission occurs when
). (b) The data from panel (a) but with the signal values mapped to indices for comparative analysis. Note that while the transition zone shrinks with increasing ultrasensitivity, normalization to the width of the transition zone reveals the similarity of the two datasets. (c) Channel capacity of the simple model for two levels of noise as a function of a shifting range of signal values. In both cases, the maximum information transmission occurs when 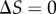 . (d) Channel capacity as a function of the density of signal values sampled in the transition zone. The minimum signal density for optimal information transmission depends on the noisiness of the channel. The entropy of the signal distribution is shown (black dotted line), denoting an upper bound to the channel capacity. (Online version in colour.)
. (d) Channel capacity as a function of the density of signal values sampled in the transition zone. The minimum signal density for optimal information transmission depends on the noisiness of the channel. The entropy of the signal distribution is shown (black dotted line), denoting an upper bound to the channel capacity. (Online version in colour.)
Since collection of data spanning all possible signal values is obviously experimentally (and computationally) infeasible for any signalling network, we first characterized how the estimated channel capacity would change as the range of sampled signal values changes. Intuitively, the majority of information should be within what we term the increasing regime or the transition zone of some arbitrary dose–response dataset [6]. In most sigmoidal dose–response curves, the range of signal values corresponding to this transition zone classically spans values bounded by 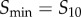 and
and 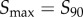 , which are the signal values resulting in 10% and 90% of the maximum response, respectively, after subtracting the baseline response [21,22]. This eliminates a large range of signal space in which the mean response is not changing significantly with signal (figure 1a). We examined whether the choice of these bounds (i.e. taking this range to be between
, which are the signal values resulting in 10% and 90% of the maximum response, respectively, after subtracting the baseline response [21,22]. This eliminates a large range of signal space in which the mean response is not changing significantly with signal (figure 1a). We examined whether the choice of these bounds (i.e. taking this range to be between 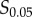 and
and 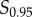 or
or 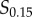 and
and 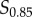 ) impacts the estimation of information transmission, and found that the effects are minimal (see the electronic supplementary material).
) impacts the estimation of information transmission, and found that the effects are minimal (see the electronic supplementary material).
In the above analysis, we assumed that the set of sampled signal values should be ‘centred’ within the transition zone. To examine how shifting the window of signal space alters the channel capacity, we first fixed the number of uniformly sampled signal values to be 32. We defined 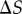 to be the shift between the centre of the transition zone (
to be the shift between the centre of the transition zone (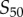 ) and the median sampled signal value. If
) and the median sampled signal value. If 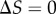 , we are sampling all 32 points within the
, we are sampling all 32 points within the 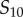 to
to 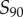 transition zone; if
transition zone; if 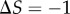 , then we have shifted the set of sampled points one unit to the left of this transition zone. As expected, shifting the range of sampled signal values away from the transition zone causes a reduction in information transmission for models exhibiting both low (
, then we have shifted the set of sampled points one unit to the left of this transition zone. As expected, shifting the range of sampled signal values away from the transition zone causes a reduction in information transmission for models exhibiting both low (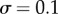 ) and high (
) and high ( ) levels of noise (figure 1c). This can be explained by the sigmoid shape of the dose–response curve: when the mean signal value is much less than or greater than K, mean responses from distinct signal values become more similar. When sampling outside of the transition zone, correlation, and thus information, between signal and response is lost.
) levels of noise (figure 1c). This can be explained by the sigmoid shape of the dose–response curve: when the mean signal value is much less than or greater than K, mean responses from distinct signal values become more similar. When sampling outside of the transition zone, correlation, and thus information, between signal and response is lost.
Next, we characterized how the number of signal values uniformly sampled within the transition zone changes the information transmission. Generally, the number of signal values chosen to experimentally characterize a dose–response relationship is essentially arbitrary [9,10,15], but this number can greatly impact estimation of the channel capacity. Since the mutual information (and by extension, the channel capacity) can be written as a difference of entropies:
(where 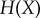 denotes the entropy of the random variable
denotes the entropy of the random variable 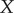 [14]), the entropy of the signal distribution,
[14]), the entropy of the signal distribution, 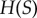 , is clearly upper bound on the mutual information. This limit can be reached with a uniform signal distribution when the signal values sampled produce pairwise disjoint response distributions, resulting in perfect information transmission between signal and response (figure 1d). As the density of sampled signals increases, however, overlap between the response distributions from neighbouring signal values prevents the mutual information from reaching the
, is clearly upper bound on the mutual information. This limit can be reached with a uniform signal distribution when the signal values sampled produce pairwise disjoint response distributions, resulting in perfect information transmission between signal and response (figure 1d). As the density of sampled signals increases, however, overlap between the response distributions from neighbouring signal values prevents the mutual information from reaching the 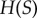 upper bound. There is thus some sufficiently dense sampling of signal values in the transition zone beyond which the mutual information does not increase. In the case of our simple model, sampling 32 signal values is sufficient for reliable channel capacity estimation for a model with relatively low noise (
upper bound. There is thus some sufficiently dense sampling of signal values in the transition zone beyond which the mutual information does not increase. In the case of our simple model, sampling 32 signal values is sufficient for reliable channel capacity estimation for a model with relatively low noise (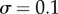 ) and sampling merely 8 is sufficient for a case with higher levels of nose (
) and sampling merely 8 is sufficient for a case with higher levels of nose (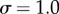 , figure 1d).
, figure 1d).
Using the above analysis as a guide, we can suggest an approach that allows for a reliable and systematic characterization of the limits on how much information can be transmitted through some arbitrary signalling network. For data collection, the first step is to estimate the signal value resulting in half-maximal response and sample numerous signal values around this value. From this dataset, a Hill function (or other appropriate function) can be parametrized through simple least-squares fitting techniques. This fit can then be employed as a guide to determine the signal values (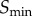 and
and 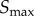 ) that correspond to the bounds of the transition region for the particular system under investigation. Given those bounds, the next step involves increasing the set of sampled signal values within the transition region that ‘saturates’ the information transmission as seen in figure 1d. While we employ this systematic approach to explore the information theoretic properties of computational models of signalling systems in this work, this general approach could easily be applied in an experimental setting, allowing for more straightforward comparison of information theoretic estimates from experimental data [9,10,15].
) that correspond to the bounds of the transition region for the particular system under investigation. Given those bounds, the next step involves increasing the set of sampled signal values within the transition region that ‘saturates’ the information transmission as seen in figure 1d. While we employ this systematic approach to explore the information theoretic properties of computational models of signalling systems in this work, this general approach could easily be applied in an experimental setting, allowing for more straightforward comparison of information theoretic estimates from experimental data [9,10,15].
2.2. Information in binary interactions
We first focused on the simplest motif present in signal transduction networks: a reversible binding interaction between two molecules. Perhaps the most quintessential example of this kind of interaction is between an extracellular ligand and a transmembrane receptor. Such interactions generally form the ‘first step’ of the cell signalling process that leads to the downstream cellular response [9,10,15,23,24]. While binary interactions can transmit information at other stages of signal transduction (i.e. through other types of allosteric interactions, or nucleation of the formation of a macromolecular complex [24]), we refer to this case as the ‘ligand–transmembrane receptor’ (LT) model (see figure 2a for a schematic of this motif). We proceeded to examine the information transfer between the extracellular ligand concentration/copy number (the signal) and the concentration/copy number of the ligand-bound form of the receptor at steady state (the response). The model itself is composed of only two reactions: association and dissociation of the ligand (L) and the transmembrane receptor (T). In the first case, we assume a fixed total number of receptor molecules (denoted 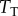 ) and we vary the input signal by changing the amount of total ligand available to bind the receptor (
) and we vary the input signal by changing the amount of total ligand available to bind the receptor (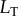 ). One benefit of this simple system is that we can analytically determine the bounds of the transition zone (in terms of total ligand concentration,
). One benefit of this simple system is that we can analytically determine the bounds of the transition zone (in terms of total ligand concentration, 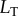 ) by solving the standard binding isotherm for distinct levels of bound receptor:
) by solving the standard binding isotherm for distinct levels of bound receptor:
 |
where 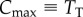 is the maximum possible number of ligand–receptor complexes and
is the maximum possible number of ligand–receptor complexes and 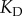 is the dissociation constant that characterizes the strength of the interaction.
is the dissociation constant that characterizes the strength of the interaction.
Figure 2.

(a) Schematic of the ‘ligand–transmembrane receptor’ (LT) motif. This represents a simple, reversible binary interaction between two molecules (in this case, the ‘L’ and ‘T’ molecules). While we consider this a model of a ligand binding a receptor, this could represent any reversible binding interaction in cell signalling. (b) The number of molecules in the system directly corresponds to the amount of noise in the response (which in this case is the fraction of bound receptor). (c) Similar to figure 1, we observe ‘saturation’ of information with sufficiently densely sampled signal values that depends on the level of noise in response. (d) The LT model that includes molecular turnover (i.e. synthesis and degradation of both L and T) exhibits the same trends as in (a) but the relative level of noise is higher. (e) A log-linear relationship exists between the number of molecules in the system (inversely proportional to the variability in response) and the channel capacity. (Online version in colour.)
We should note that this model is meant to represent a generic signalling motif, rather than a specific biological case in a real signalling network, and so we did not attempt to parametrize the model using a specific example or by fitting the model to a particular dataset. As with all stochastic reaction systems in biology, the amount of ‘intrinsic noise’ in the LT model depends critically on the total copy number of the proteins involved [25–28]. We thus estimated the channel capacity for a variety of values of 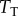 ranging from
ranging from 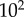 to
to 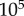 (figure 2). This corresponds to reasonable range of copy numbers for biological systems ranging from bacterial to yeast and mammalian cells [24,29–32]. Keeping the
(figure 2). This corresponds to reasonable range of copy numbers for biological systems ranging from bacterial to yeast and mammalian cells [24,29–32]. Keeping the 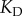 constant for all of the values of
constant for all of the values of 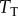 would mean that the level of saturation of the binding interaction would change dramatically as copy numbers of
would mean that the level of saturation of the binding interaction would change dramatically as copy numbers of 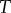 increased. In other words, if we had a
increased. In other words, if we had a 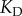 of, say,
of, say, 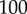 nM, then the binding interaction would be unsaturated for
nM, then the binding interaction would be unsaturated for 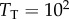 and very saturated for
and very saturated for 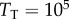 . To isolate the influence of copy numbers on information flow, we thus varied the
. To isolate the influence of copy numbers on information flow, we thus varied the 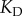 with
with 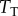 ; for simplicity, we changed
; for simplicity, we changed 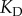 so that
so that 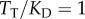 for all cases, which is a reasonable first-order approximation for binding affinities [31,33]. Assuming this interaction occurs in a yeast cell volume [24,31,34], this would correspond to a
for all cases, which is a reasonable first-order approximation for binding affinities [31,33]. Assuming this interaction occurs in a yeast cell volume [24,31,34], this would correspond to a 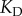 of approximately
of approximately 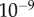 to
to 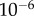 M, well within the range of biologically observed
M, well within the range of biologically observed 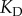 values. While the parameters we employ thus represent a reasonable scenario for a binary interaction in biology, we leave detailed exploration of the relationship between affinity and information flow in such a system to future work. Further details of the parameters we used may be found in the electronic supplementary material.
values. While the parameters we employ thus represent a reasonable scenario for a binary interaction in biology, we leave detailed exploration of the relationship between affinity and information flow in such a system to future work. Further details of the parameters we used may be found in the electronic supplementary material.
To facilitate construction and analysis of many models, we used the rule-based modelling language Kappa [31,35–39] to encode the model, and we simulated this system using the associated exact stochastic simulator [37,40]. These simulators use the standard ‘Doob–Gillespie’ approach to characterize the intrinsic fluctuations due to random chemical events in this kind of system [41]. A template rule-based model file for this (and all of our models) is provided as electronic supplementary material, and further details on the simulations may be found in the Methods and electronic supplementary material. We ran simulations for a variety of 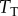 values, varying
values, varying 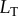 as the signal and using the concentration of the LT complex at steady state as the response. Since changing the labels of the signal values does not alter the correlation between signal and response (and thus has no impact on the mutual information), we mapped the signal values (i.e. the discrete set of
as the signal and using the concentration of the LT complex at steady state as the response. Since changing the labels of the signal values does not alter the correlation between signal and response (and thus has no impact on the mutual information), we mapped the signal values (i.e. the discrete set of 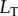 ’s used) to a set of indices both for simpler labelling of the signal values and visualization of the data. The resulting dose–response curves are shown in figure 2b.
’s used) to a set of indices both for simpler labelling of the signal values and visualization of the data. The resulting dose–response curves are shown in figure 2b.
Following the framework outlined above, we varied the number of signal values sampled in the transition zone and estimated the channel capacity for each dataset. As can be seen in figure 2b, lower copy numbers of the receptor result in higher levels of noise, which is a direct consequence of the increase in stochastic effects at lower copy numbers. As in our simple Hill function model of cell signalling, we found that there is a ‘saturating’ density of signal values beyond which there is no increase in the information transmitted, and that this density depends on the variability in response of the system (figure 2c). At these saturating densities we observe strikingly high levels of information transmission (about 6 bits) compared to values previously calculated from experimental datasets [9,15].
The above model assumed that both the total number of receptors and ligand molecules was fixed at a constant value. In living systems, receptor and ligand numbers are generally not fixed, but rather kept (relatively) constant through a combination of synthesis and degradation [15,24]. Since synthesis and degradation are themselves inherently stochastic events, fluctuations induced by molecular turnover could impact information transfer in this system [25,26]. To account for this, we added a simple set of ‘birth–death’ reactions to our models, explicitly including both synthesis and degradation of the ligand and the receptor. This led to a slightly more complex, but still analytically tractable binding curve from which we determined the signal bounds of the transition zone (see the electronic supplementary material).
The resulting dose–response datasets can be seen in figure 2d, and the signal density-dependent channel capacity trends can be seen in the electronic supplementary material. As anticipated, the additional variability introduced by molecular turnover reduced information transmission. For saturated signal density, the loss in information was approximately 0.5 bits regardless of the receptor copy number (figure 2e). In general, the trends observed for the model without synthesis and degradation were conserved in this model. As implied in figure 2c, there is clearly a scaling relationship between the channel capacity of the system and the number of receptors. Indeed, we found a significant log-linear relationship between the channel capacity (at saturating signal density) and receptor copy number in the range of receptor numbers we tested (figure 2e). In other words, regardless of the model in question, it takes an increase of one order of magnitude in receptor number to encode 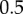 bits more information in the
bits more information in the 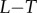 interaction. Increases in information transmission, in this case, are thus offset by considerable costs in terms of the energy required to synthesize and maintain a large population of receptor molecules at the cell surface.
interaction. Increases in information transmission, in this case, are thus offset by considerable costs in terms of the energy required to synthesize and maintain a large population of receptor molecules at the cell surface.
Energy cost aside, using these results to extrapolate to even larger numbers of receptors, we can estimate a hard upper bound on information transmission in cells. Since the ligand–receptor binding motif is the first step for virtually all signalling networks, downstream sensing of that ligand concentration is essentially limited by the amount of information encoded in this first step [9]. We estimate that for an interaction with over 1 million receptors (an extreme upper bound, being nearly an order of magnitude larger than what has been experimentally realized for human signalling networks [42]), the amount of information that can be transmitted is approximately 8 bits (or 7.5 bits with molecular turnover). While this is unlikely to be realized in vivo since ligand–receptor interactions are generally only a part of larger signalling networks, this gives us a point of reference for understanding the limitations of information transmission through even the simplest signalling systems.
2.3. Information in futile cycles
We next focused on the standard chemical modification motif for signalling: a covalent modification cycle (which we term the ‘GK loop’ due to Goldbeter & Koshland’s pioneering mathematical characterization of the cycle) [21]. This model’s kinetics have been thoroughly characterized mathematically for a number of operating regimes (e.g. saturated and unsaturated) and we can use this broad understanding to our advantage [11,43–45]. While the model encompasses any type of reversible post-translational modification of a protein substrate, our terminology will primarily reflect that of phosphorylation, where K, P and W denote kinase, phosphatase and substrate, respectively. A schematic of this model can be found in figure 3a. In this system, the input signal that governs the steady-sate level of substrate phosphorylation, and the value we use as the signal for our information theory calculations, is the ratio of maximum velocities of the enzymes [21,43–45]:
where 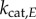 is the catalytic rate of a given enzyme
is the catalytic rate of a given enzyme 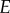 , and
, and 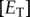 is the concentration of that enzyme. While this ratio could be varied by changing any of the variables above, changing the copy numbers of K and P would change both the signal itself and the relative influence of copy number on stochastic effects. We thus varied the value of the signal S by changing
is the concentration of that enzyme. While this ratio could be varied by changing any of the variables above, changing the copy numbers of K and P would change both the signal itself and the relative influence of copy number on stochastic effects. We thus varied the value of the signal S by changing 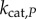 , which also allowed us to more finely sample signal space for highly ultrasensitive responses, and to more effectively characterize the transition zone (see the electronic supplementary material). For our information theoretic calculations, we take the response to be the concentration of unbound, phosphorylated substrate [21,43,44].
, which also allowed us to more finely sample signal space for highly ultrasensitive responses, and to more effectively characterize the transition zone (see the electronic supplementary material). For our information theoretic calculations, we take the response to be the concentration of unbound, phosphorylated substrate [21,43,44].
Figure 3.

(a) Schematic of a futile cycle, with a kinase ‘K’ acting to phosphorylate a substrate ‘W’ and a phosphatase ‘P’ catalysing the removal of the phosphoryl group. While a kinase/phosphatase pair are shown here, this motif represents any covalent modification cycle of this type. We term this the ‘Goldbeter–Koshland’ (GK) loop. (b) Dose–response datasets for GK loops. Here, the incoming signal represents the kinase activity relative to the phosphatase, and the response is the number of free, phosphorylated (active) substrate molecules. Three different levels of enzyme saturation are shown; black is least saturated, green is most saturated. Note that both ultrasensitivity and the relative level of noise increase with increasing saturation. (c) Channel capacity as a function of enzyme saturation. 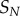 denotes number of signal values sampled. As expected from (b), increased saturation (i.e. the points to the left on the curve) results in lower information transmission due to higher variability in the response distribution. (d,e) Identical to (b,c) but the model includes synthesis and degradation. Note that the much higher levels of noise in the saturated case lead to even lower levels of information transfer. (Online version in colour.)
denotes number of signal values sampled. As expected from (b), increased saturation (i.e. the points to the left on the curve) results in lower information transmission due to higher variability in the response distribution. (d,e) Identical to (b,c) but the model includes synthesis and degradation. Note that the much higher levels of noise in the saturated case lead to even lower levels of information transfer. (Online version in colour.)
While this model is only slightly more complex than the LT model described above (with six total chemical species, rather than three), it can exhibit much more variety in its dose–response behaviour. In particular, as the substrate concentration is increased relative to the 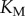 ’s of the enzymes, the steady-state response of phosphorylated substrate becomes extremely ultrasensitive [21]. Our measure of enzyme saturation is
’s of the enzymes, the steady-state response of phosphorylated substrate becomes extremely ultrasensitive [21]. Our measure of enzyme saturation is 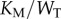 , where
, where 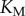 is the Michaelis constant (taken to be equivalent for both enzymes for simplicity) and
is the Michaelis constant (taken to be equivalent for both enzymes for simplicity) and 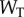 is total substrate concentration. Traditionally, altering the saturation of an enzyme is done by increasing the amount of total substrate relative to a fixed
is total substrate concentration. Traditionally, altering the saturation of an enzyme is done by increasing the amount of total substrate relative to a fixed 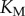 , but, as we saw in the previous section, changes in copy number correspond to changes in information transmission. We thus varied saturation by varying the association rate between the enzymes and the substrate (thus changing
, but, as we saw in the previous section, changes in copy number correspond to changes in information transmission. We thus varied saturation by varying the association rate between the enzymes and the substrate (thus changing 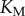 ). For every total copy number of substrate, we varied
). For every total copy number of substrate, we varied 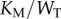 between
between 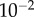 and
and 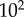 (i.e. saturated to not saturated). By changing
(i.e. saturated to not saturated). By changing 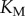 instead of
instead of 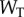 we can independently explore the effect of copy number and saturation on information flow in the GK loop. As with the LT model, we focus here on a biologically meaningful range of parameters, and not a specific biological system; further details on the parameters for this model may be found in the electronic supplementary material, and an example model file is available as additional electronic supplementary material. We applied the methodology described above to estimate the transition zone for any given value of
we can independently explore the effect of copy number and saturation on information flow in the GK loop. As with the LT model, we focus here on a biologically meaningful range of parameters, and not a specific biological system; further details on the parameters for this model may be found in the electronic supplementary material, and an example model file is available as additional electronic supplementary material. We applied the methodology described above to estimate the transition zone for any given value of 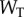 and
and 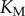 in this model: we first sampled the response distributions for 20 points in signal space (i.e. 20 different values of
in this model: we first sampled the response distributions for 20 points in signal space (i.e. 20 different values of 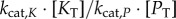 ), taking care to capture the entire transition zone in this range of points, and then fit the data to a Hill function to determine
), taking care to capture the entire transition zone in this range of points, and then fit the data to a Hill function to determine 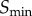 and
and 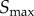 . Finally, as with the LT model, we varied the copy numbers of the components in the system, while keeping the ratio of components fixed:
. Finally, as with the LT model, we varied the copy numbers of the components in the system, while keeping the ratio of components fixed: 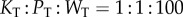 .
.
As we observed with the LT model, changing the copy numbers of the signalling components altered the variability of response to signal, and so we found a positive correlation between increased copy number and increased channel capacity. We also see, similar to the behaviour in our initial Hill function model, that increased ultrasensitivity (induced by enzyme saturation) tightens the bounds of the transition zone. If we again transform the discrete signal values to indices, we can visually compare how response distributions differ for a particular (relative) signal in the transition zone as seen in figure 3b. Dose–response trends also emerge, revealing that saturated enzymes produce increased noise in the response for signal values that produce a half-maximal response. This, in turn, reduces the amount of information present in the system, from nearly 5.5 bits in an unsaturated cycle to just above 3 bits (figure 3c).
In many realistic physiological contexts, total copy numbers are likely to be maintained by continued synthesis and degradation of the components of the system (substrate, kinase and phosphatase). Adding active synthesis and degradation to the model generally increases variability in the response (figure 3d) and decreases channel capacity (figure 3e), as in the case of the LT model. In the GK loop, however, the difference between unsaturated and saturated enzymes becomes exaggerated when molecular turnover is included in the model, from approximately 4 bits for the unsaturated case to less than 1 bit for the saturated case (figure 3e). This is due to the fact that the saturated response is ultrasensitive (figure 3b), so small fluctuations in kinase copy number that arise from synthesis and degradation can move the system between the low and high response regimes when the signal is near this transition. This generates massive fluctuations in the level of phosphorylated protein (i.e. large variability in the response distribution) and low channel capacities.
These findings reveal that, in certain parameter regimes, a GK loop can encode 4–5 bits of information, much higher than have been observed experimentally [9,15]. In different parameter regimes, however, the GK loop encodes such little information that binary decisions would be impossible to make reliably. In addition to controlling the shape of the dose–response curve, saturation can also control information flow, allowing even a motif as simple as the GK loop to transmit high levels of information when required (as in the case of, say, chemotaxis) while amplifying noise when necessary (i.e. to control the responses of cellular populations) [15].
2.4. Information in kinase cascades
While useful for gaining an understanding of basic information transmission properties, the atomistic signalling motifs described above are rarely present in isolation. On the contrary, eukaryotic organisms in general, and metazoans in particular, exhibit increasingly complex cellular signalling networks [46,47]. One of the most conserved motifs in these more complex networks is the kinase cascade, which is present in both simple eukaryotes, such as yeast [24,48], and complex multicellular organisms like humans [49]. To examine how more complex networks transmit information, we constructed a set of rule-based models that embody the core of a kinase cascade. We have two similar, but distinct model types: one that employs a scaffold protein and one that does not [50], termed the scaffold and solution models, respectively. Both involve kinases which are sequentially phosphorylated and phosphatases assigned to dephosphorylate a specific kinase (to prevent saturation due to substrate-sharing) [44] (figure 4a,b). We also varied the number of successive kinases in both the scaffold and solution models, and we refer to this number as the ‘depth’, 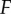 , of the cascade. All kinetic parameters in these models are broadly based on those found in the yeast MAPK kinase cascade. As a result of using these parameters, the kinases are unsaturated [24] in this model. Additional details on the model can be found in the electronic supplementary material, and an example rule-based model file is provided as additional electronic supplementary material.
, of the cascade. All kinetic parameters in these models are broadly based on those found in the yeast MAPK kinase cascade. As a result of using these parameters, the kinases are unsaturated [24] in this model. Additional details on the model can be found in the electronic supplementary material, and an example rule-based model file is provided as additional electronic supplementary material.
Figure 4.

(a) Solution cascade model where activation of 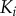 only depends on binding active
only depends on binding active 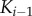 . (b) Scaffold-based cascade model where activation of kinase
. (b) Scaffold-based cascade model where activation of kinase 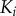 depends on having active
depends on having active 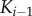 and
and 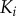 bound simultaneously to the scaffold, S. (c) Scaffold model dose–response trends generated using the VTZ approach (see text). The dotted lines denote the transition zone of the final kinase (which is used in the FTZ approach to generate dose–response data for all
bound simultaneously to the scaffold, S. (c) Scaffold model dose–response trends generated using the VTZ approach (see text). The dotted lines denote the transition zone of the final kinase (which is used in the FTZ approach to generate dose–response data for all 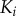 ). (d) As (c) but with the solution model. (Online version in colour.)
). (d) As (c) but with the solution model. (Online version in colour.)
We then generated dose–response datasets to examine the flow of information through these networks. In order to do this, we examined the information transmission between the signal and each kinase’s activity in the cascade: 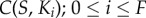 . With these data, we can begin to understand how changes in the structure of the signalling network (i.e. the presence or absence of a scaffold) and increases in network size (i.e. increasing
. With these data, we can begin to understand how changes in the structure of the signalling network (i.e. the presence or absence of a scaffold) and increases in network size (i.e. increasing 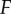 ) can influence information transmission. We considered cascades of depth
) can influence information transmission. We considered cascades of depth 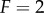 , 3 and 4. The response of various kinases in the cascade can occur in very different regimes of input signal, particularly in the solution model, where each successive kinase in the cascade responds to progressively smaller input signals (figure 4c) [44]. To deal with this effect, we used two approaches to calculate the channel capacity. In the first approach, we found the min and max signals (
, 3 and 4. The response of various kinases in the cascade can occur in very different regimes of input signal, particularly in the solution model, where each successive kinase in the cascade responds to progressively smaller input signals (figure 4c) [44]. To deal with this effect, we used two approaches to calculate the channel capacity. In the first approach, we found the min and max signals (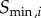 and
and 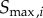 ) for each kinase independently. Since the transition zone in this case varies with the kinase in question, we call this the variable transition zone (VTZ) approach. The other approach involves finding the transition zone bounds for the final kinase,
) for each kinase independently. Since the transition zone in this case varies with the kinase in question, we call this the variable transition zone (VTZ) approach. The other approach involves finding the transition zone bounds for the final kinase, 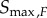 and
and 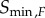 , and using the resulting range of signal values for calculating all the channel capacities for all the intermediate kinases in the cascade. We call this the fixed transition zone (FTZ) approach. This latter approach is of significant interest since the final kinase in these types of cascades is typically responsible directly or indirectly for initializing some sort of transcriptional programme that will govern behavioural changes in response to some stimulus [20,24].
, and using the resulting range of signal values for calculating all the channel capacities for all the intermediate kinases in the cascade. We call this the fixed transition zone (FTZ) approach. This latter approach is of significant interest since the final kinase in these types of cascades is typically responsible directly or indirectly for initializing some sort of transcriptional programme that will govern behavioural changes in response to some stimulus [20,24].
From these dose–response datasets, we noticed a few key differences between the two models. First, the copy number of active final kinase for any given signal value is substantially higher in the solution model than the scaffold model, reaching approximately 90% activation at the upper bound of the transition zone (see the electronic supplementary material). This is likely due to the increase in the number of reaction events required for activation to occur in the scaffold model; activation of a given kinase 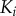 requires independent binding of both
requires independent binding of both
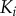 and
and 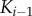 to the scaffold, whereas only one binding event is needed in the solution model. Second, as mentioned above, the response of each successive kinase to incoming signal becomes increasingly sensitive to signal (i.e. the responses are shifted to the left in figure 4c) in the solution model, and this does not occur in the scaffold model (figure 4d). This means that the sets of signals considered in the VTZ and FTZ approach are quite different for the solution model, but are essentially the same in the scaffold model. The fact that scaffold-based cascades can exhibit similar responses for both the first and last kinase in the cascade has been noted before, and has been referred to as dose–response alignment or DoRA [20].
to the scaffold, whereas only one binding event is needed in the solution model. Second, as mentioned above, the response of each successive kinase to incoming signal becomes increasingly sensitive to signal (i.e. the responses are shifted to the left in figure 4c) in the solution model, and this does not occur in the scaffold model (figure 4d). This means that the sets of signals considered in the VTZ and FTZ approach are quite different for the solution model, but are essentially the same in the scaffold model. The fact that scaffold-based cascades can exhibit similar responses for both the first and last kinase in the cascade has been noted before, and has been referred to as dose–response alignment or DoRA [20].
We also found significant differences between these models in terms of the information transfer between the input signal and the response of the various intermediate kinases for each intermediate in the cascade when 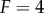 (figure 5). With one exception, the information transmission at each stage in the scaffold model is lower than the corresponding stage in the solution model for the VTZ approach. This is likely due to the lower magnitude of
(figure 5). With one exception, the information transmission at each stage in the scaffold model is lower than the corresponding stage in the solution model for the VTZ approach. This is likely due to the lower magnitude of 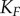 response in the scaffold model (see the electronic supplementary material); stochastic effects in this portion of response space would be much greater than that in the solution model (note the larger amount of noise in the dose–response curve in figure 4d versus 4c). We observe monotonically decreasing channel capacities as information progresses through the cascade in both models. Interestingly, information transmission appears to drop more quickly but levels off at deeper stages in the scaffold model, whereas the decrease is more uniform in the solution model.
response in the scaffold model (see the electronic supplementary material); stochastic effects in this portion of response space would be much greater than that in the solution model (note the larger amount of noise in the dose–response curve in figure 4d versus 4c). We observe monotonically decreasing channel capacities as information progresses through the cascade in both models. Interestingly, information transmission appears to drop more quickly but levels off at deeper stages in the scaffold model, whereas the decrease is more uniform in the solution model.
Figure 5.

(a,b) Channel capacity as a function of cascade depth for data VTZ (a) and FTZ (b) models. Distinctive behaviour arises with relatively minor differences between the models; the trends in the scaffold model are qualitatively identical for both VTZ and FTZ models, whereas the solution model exhibits strikingly different behaviour due to the increased sensitivity to signal of its downstream components. (Online version in colour.)
If we calculate channel capacities using the FTZ approach, where the range of signals considered in the calculation is taken based only on the response of the final kinase 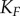 , we observe further differences between the models. Because the transition zones for the kinases in the cascade basically do not vary at all for the scaffold model (figure 4d), the FTZ approach gives the same result as the VTZ result for this case (figure 5). By contrast, the channel capacity between the signal and early kinases in the FTZ approach for the solution model is low, since the transition zone for the final kinase aligns poorly with the ideal transition zones for the upstream kinases (figure 4c). Most signal values sampled for the upstream kinases are significantly lower than the presumed half-maximal signal value of the
, we observe further differences between the models. Because the transition zones for the kinases in the cascade basically do not vary at all for the scaffold model (figure 4d), the FTZ approach gives the same result as the VTZ result for this case (figure 5). By contrast, the channel capacity between the signal and early kinases in the FTZ approach for the solution model is low, since the transition zone for the final kinase aligns poorly with the ideal transition zones for the upstream kinases (figure 4c). Most signal values sampled for the upstream kinases are significantly lower than the presumed half-maximal signal value of the 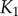 and
and 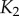 intermediates’ dose–response curve, and thus the channel capacity increases with the kinase’s position in the cascade (figure 5b). At first glance, this may appear to violate the data processing inequality, which states that information content cannot be increased during transmission through a channel. However, this is not the case in our model, since the signal and observed intermediates do not form a Markov chain [14].
intermediates’ dose–response curve, and thus the channel capacity increases with the kinase’s position in the cascade (figure 5b). At first glance, this may appear to violate the data processing inequality, which states that information content cannot be increased during transmission through a channel. However, this is not the case in our model, since the signal and observed intermediates do not form a Markov chain [14].
It is important to note that the difference between the FTZ and VTZ result for the solution model is more than just an artefact of calculating channel capacities via simulation. In a physiological context, a cell will be exposed to a particular signal distribution, and that distribution may change depending on the situation faced by an organism. In this model, we assume that only 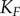 has a physiologically relevant activity, but in a highly complex signalling network with high amounts of crosstalk, like the networks found in human cells, each kinase might act on other substrates or have other functions [46,47]. In that case, each kinase in a solution cascade would encode information from highly distinct regions of the signal space, a property that could be leveraged physiologically.
has a physiologically relevant activity, but in a highly complex signalling network with high amounts of crosstalk, like the networks found in human cells, each kinase might act on other substrates or have other functions [46,47]. In that case, each kinase in a solution cascade would encode information from highly distinct regions of the signal space, a property that could be leveraged physiologically.
While these results are specific to the parameter sets we considered (e.g. unsaturated enzymes allow much greater information transmission in cascades, as observed in the GK loop model), our results from the scaffold and solution model indicate that distinct configurations of signalling cascades’ underlying interaction networks can impact information transmission. In general, these increasingly complex models of kinase cascades still exhibit a capacity for far more effective information transmission (greater than 4 bits) than have been observed in experimental data of cellular responses to signal.
2.5. Information in more realistic networks
While the above sections focused on several motifs common to cell signalling networks, we now focus on two cases in which these motifs, or related sets of signalling events, are composed into realistic information sensing and processing systems. The first case we consider is the two-component signalling (TCS) system from bacteria. These systems generally consist of two proteins: a sensor histidine kinase (HK) and a response regulator (RR) [29,51–53]. The HK is usually an integral membrane protein that senses an incoming signal, while the RR is a transcription factor that becomes active after phosphorylation by the HK. While TCS systems are similar to the GK loops considered above, the underlying biochemistry is quite different: in TCS, the HK autophosphorylates itself in response to an incoming signal, then transfers that phosphoryl group to the RR after binding it. TCS systems also lack independent phosphatase enzymes. Instead, the inactive (unphosphorylated) HK serves as the phosphatase and dephosphorylates the RR [29,51,53]. The HK thus acts as both kinase and phosphatase for its substrate, depending on its own activation state. A schematic of TCS signalling is shown in figure 6a.
Figure 6.

(a) Schematic of a two-component signalling system. Some input signal ‘I’ modulates the autposphorylation rate of the histidine kinase (HK). The phosphorylated HK can transfer the phosphoryl group to the response regulator (RR), thus activating it. The unphosphorylated HK acts as the phosphatase in this system. (b) Dose–response data for TCS motifs with different copy numbers of HK molecules. (c) Channel capacity values for various versions of the TCS model. As with the GK model, the amount of information transfer varies with both saturation and component copy number. The numbers boxed in red represent the models whose parameters are closest to experimental data from the EnvZ/OmpR TCS that is responsible for osmoregulation in bacteria such as E. coli. (d) Dose–response data for key observables in a rule-based model of the early events in EGFR signalling. (e) Channel capacities for the observables shown in (d). Note that while the dataset in (c) is composed of 32 signal values, the channel capacities reported here were calculated using 256 signal values to reach sufficiently dense signal value sampling in the transition zone. The need for high signal density is due to the large number of EGFR molecules in the system, a phenomenon we also observed in the LT model (figure 2). (Online version in colour.)
TCS represents an ideal model system for studying a realistic case of information transfer in biology. These relatively simple systems span the entirety of a cell signalling pathway, from detection of the signal to the activation of a transcription factor that will alter gene expression levels. There is also generally very little crosstalk between TCS pathways, simplifying the analysis [29,47,52]. Finally, several TCS pairs have been studied extensively using experimental techniques, making it possible to use experimentally determined rate parameters in our models [29,51,52,54]. To study this system, we adapted a previously implemented system of differential equations [29] to use rule-based stochastic simulations in order to introduce realistic levels of intrinsic noise into the system, and varied the existing kinetic parameters and protein copy numbers within ranges appropriate for bacterial systems [29,54]. Further information on parameters may be found in the electronic supplementary material, and the rule-based model itself is provided as additional electronic supplementary material.
In this model, we take the autophosphorylation rate of the HK to be the input signal and the amount of unbound, phosphorylated RR as the response, consistent with the biology of TCS signalling [29,51,52]. As expected, increasing protein copy numbers significantly increases channel capacities (figure 6b,c). Furthermore, increased saturation again induces a decrease in information, due mainly to a reduction in the fraction of RR that are phosphorylated as saturation increases [29]. Most notably, however, we can use the parameters that most closely reflect existing experimental data and estimate the channel capacity of the HK–RR pair from which the original model was derived: EnvZ and OmpR, respectively [51,54,55] (figure 6c, red box). Notably, these values are higher than nearly all experimentally characterized networks [9,15], showing that individual bacterial cells can, at least in principle, obtain relatively high quantities of information from extracellular stimuli.
We then turned our attention to a much more complex eukaryotic interaction network: receptor tyrosine kinase signalling cascades. One quintessential example is the epidermal growth factor (EGF) signalling network whose activity depends on ErbB2/HER transmembrane receptor dimerization. Owing to the extensive crosstalk present in human networks, a complete model of the EGF network does not exist [46,47]. Here, we focus on a previously published rule-based model that explored the early events of the EGF signalling networks using exact stochastic simulation of all relevant chemical species [30]. The kinetic parameters of this model were either directly measured or determined through comparison to experimental data by the original authors, and we did not alter those parameters for our simulations. We adapted that model for use with our information theory framework to characterize how information about EGF concentration (which we took to be the input signal) is transmitted through this network to the responses of several key downstream signalling species. In particular, we focused on ligand-induced EGF receptor (EGFR) dimerization, autophosphorylated EGFR (which is required for recruitment of effector proteins), and active Sos (the downstream-most component in this model) (figure 5d). This model contains nearly 200 000 EGFR molecules and we found that in order to accurately estimate the information transmission, we required a high signal density in the transition zone, sampling 256 distinct signal values from the transition zone in order to reach a ‘saturated’ channel capacity estimation. The model file we used for these simulations is provided as additional electronic supplementary material.
We observed high information transfer among the initial steps of the cascade: EGFR dimerization in response to EGF stimulation produced nearly 6.5 bits of information, close to the upper bound estimated from the LT model. From this point, the stochasticity of the interactions and lower copy number of other components, such as Sos, reduces the information transmission (figure 6d,e). However, information transmission through the entirety of the network was greater than 3 bits. This is quite high compared to experimentally determined values; however, it is important to note that Sos recruitment is by no means the final step of the cascade in vivo. Sos is then responsible for activating the MAPK pathway in metazoan signalling, and we have seen that information transfer can vary significantly, depending on the kinetics of the kinase in question (figure 3c,e). It is enough, however, to see that even with a model containing moderate signalling complexity (over 350 signalling species as opposed to 3 in the LT model or 6 in the GK model), reliable information transmission is possible.
3. Discussion
This work begins to address a fundamental question in the study of signal transduction and cellular decision-making: why do signalling networks transmit specific amounts of information? Here, we focused on characterizing the intrinsic limits of information transmission through signalling networks with the goal of providing context for information theoretic values estimated from experimental data. We found that models of simple signalling motifs, as well as larger, more realistic networks, are capable of transmitting substantially higher amounts of information than has been estimated experimentally; to date, the highest information transmission estimated for individual signalling networks in eukaryotic cells (that we are aware of) is less than 2.5 bits for measurements made at single time points [9,10,15,17–19].
There are a few possibilities as to why this might be the case. First, it is possible that the specific networks that have been examined experimentally using information theory do not require high levels of information transmission in order to achieve their intended function, and thus did not evolve to exist near the intrisic limits characterized here. This raises the possibility that other networks exist that transmit much more information than has been previously characterized. We would predict that networks with the highest information transmission will be for networks that control a continuous response variable (i.e. expression of a set of genes at a specific level, or direction of movement in chemotaxis) or those that control cell-fate decisions with high entropy (i.e. the differentiation of a pluripotent stem cell into a variety of cell fates) [15]. Second, the observed low levels of information in in vivo signal transduction could be a reflection of ‘extrinsic noise’ in a cell population [23,25,26]. The models we considered only included the intrinsic randomness of biochemical reaction events; however, other environmental factors can contribute to overall variability in response to signal, including noise in the signal distribution itself [15]. Finally, we showed in prior work that low information transmission (even values below 1 bit) can be useful when transmitting information to cellular populations is paramount. We found that there exists a fundamental trade-off between information transmission to single cells and cellular populations, and that there is some optimal level of noise to maximize population-level information transmission given certain conditions [15]. Regardless of the reason for low observed information transmission experimentally, we were able to examine how common signalling motifs, like post-translational modification cycles, could be potential mechanisms for the regulation of information transmission (figure 3).
In order to compare information transmission values from data obtained by simulation of multiple signalling motifs, we introduced a novel framework for the consistent application of information theoretic concepts to systems biology. While quantities such as the mutual information do not depend on the underlying structure of the reaction network between signal and response, they do depend on how the joint signal–response distribution is characterized (figure 1), meaning that dose–response data from distinct networks must be obtained using a standard methodology. The framework developed here can be applied both to simulated and experimental datasets and is crucial to make relevant comparisons of estimated values, such as those from different cell types or cell lines, organisms, or even networks with recombinant proteins (which could prove to be useful in the construction of de novo networks via synthetic biology). The results described above are an excellent example of the utility of this framework; we were able to characterize the upper limits of information transmission in networks of varying size and interconnectivity through stochastic simulation of rule-based models. The channel capacities for the initial atomistic signalling motif models that we examined (binary, physical interaction between macromolecules and chemical modification of macromolecules via enzymes) are generally integrated into more complex network architectures and can therefore provide perspective for analysis of larger networks, which are common in metazoan cells. For example, since examination of the covalent modification cycle revealed that enzyme saturation reduces information transmission, we restricted our analysis of the larger cascade models (which employ a variant of this motif) to those with unsaturated enzymes since we were primarily concerned with the upper limits of information transmission. In general, this framework provides a basis for understanding and comparing how various features (e.g. molecular copy numbers, kinetic parameters) influence the quantity of information transmitted through signalling networks, and it can serve as a standard for future application of information theory to quantification of information in signal transduction from both models and experiments.
We expect that this framework is just a starting point for broader application of information theory to systems biology. In particular, fully understanding how information flows through a network will likely require an explicit consideration of dynamics. While a useful starting point, quantities like the mutual information represent levels of information at particular points in time. In this work, we focused on networks whose components exhibited distinct steady-state responses; however the response of some signalling networks, such as those that exhibit perfect adaptation, are not well characterized by steady-state response [56]. It is clear that the framework presented here must be further developed, since cellular decision-making rarely waits until steady state is reached (at least on the molecular level) [20]. Previous examples of information theory applied to in vivo signalling networks circumvented this problem by defining a particular point in time to measure response, a logical solution when there exists some particular time at which an interesting event occurs, such as peak nuclear NF- B localization [9]. Another possibility is to define the response as a set of multiple time points from a dynamical response; however it is currently unclear if any molecular mechanisms would allow cells to both remember the previous activation state of a protein and then integrate that memory with the current activation level in order to actually extract that information [10,17]. More complex quantities, such as the transfer entropy [2,7], have been derived to examine how information flows through a system over time, eliminating the need for making arbitrary choices about when to take measurements. Adapting these quantities for use with high-resolution time course data will undoubtedly elucidate additional principles of information flow in signal transduction [10,17]. Extracellular signals and their corresponding cellular responses lie at the core of what cells have evolved to do: adapt to changing environmental conditions by altering their phenotype. Ultimately, we expect that development and application of systematic approaches such as the one presented here will form the basis for what becomes a rigorous theory of information transmission through signalling networks.
B localization [9]. Another possibility is to define the response as a set of multiple time points from a dynamical response; however it is currently unclear if any molecular mechanisms would allow cells to both remember the previous activation state of a protein and then integrate that memory with the current activation level in order to actually extract that information [10,17]. More complex quantities, such as the transfer entropy [2,7], have been derived to examine how information flows through a system over time, eliminating the need for making arbitrary choices about when to take measurements. Adapting these quantities for use with high-resolution time course data will undoubtedly elucidate additional principles of information flow in signal transduction [10,17]. Extracellular signals and their corresponding cellular responses lie at the core of what cells have evolved to do: adapt to changing environmental conditions by altering their phenotype. Ultimately, we expect that development and application of systematic approaches such as the one presented here will form the basis for what becomes a rigorous theory of information transmission through signalling networks.
4. Methods
4.1. Estimating the channel capacity
Our approach to calculating the mutual information and channel capacity is based loosely on the approach developed in [9]. Briefly, our algorithm uses resampling techniques to deal with the problem that calculations using finite datasets tend to overestimate the mutual information [3,8,57]. We first define a number of bins in signal and response space and construct a contingency table (i.e. a two-dimensional histogram) to estimate the joint distribution of signal and response (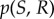 ). We then use this contingency table to calculate the mutual information. We perform this procedure for resamplings of the data with fewer and fewer total data points, and extrapolate the dependency between the size of the dataset and the mutual information to estimate what the mutual information would be if the dataset were infinite in size. We then iterate over the number of bins used for the calculation, since the number of bins has a large impact on the estimate of the mutual information. We control for artificial inflation of the estimate due to high numbers of bins by calculating the mutual information for a randomized dataset given some number of bins and checking to see if the information is not significantly different from 0. Finally, to estimate the channel capacity, we weight the signal distribution of the data using a large set of unimodal and bimodal probability distributions, and find the signal distribution that produces the maximal mutual information. Further details of this approach may be found in the electronic supplementary material, and an even more extensive description of the approach can be found in [15]. The software we developed is freely available as an open-source project on Github (https://github.com/ryants/EstCC).
). We then use this contingency table to calculate the mutual information. We perform this procedure for resamplings of the data with fewer and fewer total data points, and extrapolate the dependency between the size of the dataset and the mutual information to estimate what the mutual information would be if the dataset were infinite in size. We then iterate over the number of bins used for the calculation, since the number of bins has a large impact on the estimate of the mutual information. We control for artificial inflation of the estimate due to high numbers of bins by calculating the mutual information for a randomized dataset given some number of bins and checking to see if the information is not significantly different from 0. Finally, to estimate the channel capacity, we weight the signal distribution of the data using a large set of unimodal and bimodal probability distributions, and find the signal distribution that produces the maximal mutual information. Further details of this approach may be found in the electronic supplementary material, and an even more extensive description of the approach can be found in [15]. The software we developed is freely available as an open-source project on Github (https://github.com/ryants/EstCC).
4.2. Model definition and simulation
4.2.1. Rule-based modelling
We used both the Kappa [36] and BioNetGen [39] rule-based modelling languages for model construction. Generally speaking, rule-based frameworks allow one to encode human- and machine-readable interactions between molecular species in the cell in the form of rules, where a rule may specify numerous distinct reactions between chemical species [31,35–39]. Rule-based modelling differs from traditional ODE-based approaches by preventing the need to a priori enumerate all possible chemical species when defining the model. The models generated are also generally easier to share and modify, and allow for the application of specific simulation and analysis tools that leverage the syntactic nature of the rules themselves [35–39,58,59]. In addition, numerous peripheral software packages have been developed that use rule-based formalisms either as an underlying engine with convenient bindings for other languages (e.g. PySB [60]) or providing services complementary to the modelling itself (e.g. translation [61] or fitting [62,63]). These languages are embedded within software suites that contain specific methods for exact stochastic simulation of chemical kinetics based on Gillespie’s algorithm [37,40,41,64], allowing us to easily incorporate the intrinsic noise of event-based biochemical reactions into our information theoretic calculations.
We have provided template models, written in the Kappa language (v. 3), for all signalling motifs and systems considered here as electronic supplementary material.
4.2.2. Model parametrization
The scope of this work is to quantify the capacity for common network motifs to enable cells to respond to their environment, and to provide context for future work in specific signalling networks. Thus the models used in this work (with the exceptions of the two-component signalling and EGFR models) contain parameters that are not specific to some signalling system in a particular organism, but that are representative of classes of signalling networks (e.g. covalent modification cycles or MAPK-like kinase cascades). We use estimates derived from various other literature sources to parametrize the models, often rounding to the nearest order of magnitude. We thus did not parametrize the majority of our models with respect to specific biological datasets. We leave an in-depth analysis of the parameter sensitivity of information flow within these models to future work.
Information on the parameter values can be found throughout the main and electronic supplementary material text. Default values for the parameters can be found in the rule files themselves.
Supplementary Material
Supplementary Material
Acknowledgements
The authors thank J. Christian J. Ray for many helpful discussions.
Data accessibility
This article has no additional data.
Authors' contributions
R.S. and E.J.D. designed the research presented here, evaluated the results and wrote the manuscript. R.S. performed all simulations and analyses.
Competing interests
We declare we have no competing interests.
Funding
This work was supported in part by NSF grant no. MCB-1412262 to E.J.D.
References
- 1.Shannon CE. 1948. A mathematical theory of communication. Bell Syst. Tech. J. 27, 379–423. ( 10.1002/j.1538-7305.1948.tb01338.x) [DOI] [Google Scholar]
- 2.Wollstadt P, Martínez-Zarzuela M, Vicente R, Díaz-Pernas FJ, Wibral M. 2014. Efficient transfer entropy analysis of non-stationary neural time series. PLoS ONE 9, e102833 ( 10.1371/journal.pone.0102833) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strong S, Koberle R, de Ruyter van Steveninck R, Bialek W. 1998. Entropy and information in neural spike trains. Phys. Rev. Lett. 80, 197–200. ( 10.1103/PhysRevLett.80.197) [DOI] [Google Scholar]
- 4.Brenner N, Bialek W, de Ruyter van Steveninck R. 2000. Adaptive rescaling maximizes information transmission. Neuron 26, 695–702. ( 10.1016/S0896-6273(00)81205-2) [DOI] [PubMed] [Google Scholar]
- 5.Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Favera R, Califano A. 2006. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinf. 7, S7 ( 10.1186/1471-2105-7-s1-s7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laughlin S. 1981. A simple coding procedure enhances a neuron’s information capacity. Zeitschrift für Naturforschung Section C: Biosciences 36, 910–912. ( 10.1515/znc-1981-9-1040) [DOI] [PubMed] [Google Scholar]
- 7.Schreiber T. 2000. Measuring information transfer. Phys. Rev. Lett. 85, 461–464. ( 10.1103/PhysRevLett.85.461) [DOI] [PubMed] [Google Scholar]
- 8.Steuer R, Kurths J, Daub CO, Weise J, Selbig J. 2002. The mutual information: detecting and evaluating dependencies between variables. Bioinformatics 18, S231–S240. ( 10.1093/bioinformatics/18.suppl_2.S231) [DOI] [PubMed] [Google Scholar]
- 9.Cheong R, Rhee A, Wang CJ, Nemenman I, Levchenko A. 2011. Information transduction capacity of noisy biochemical signaling networks. Science 334, 354–358. ( 10.1126/science.1204553) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selimkhanov J, Taylor B, Yao J, Pilko A, Albeck J, Hoffmann A, Tsimring L, Wollman R. 2014. Accurate information transmission through dynamic biochemical signaling networks. Science 346, 1370–1373. ( 10.1126/science.1254933) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine J, Kueh HY, Mirny L. 2007. Intrinsic fluctuations, robustness, and tunability in signaling cycles. Biophys. J. 92, 4473–4481. ( 10.1529/biophysj.106.088856) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mehta P, Goyal S, Long T, Bassler BL, Wingreen NS. 2009. Information processing and signal integration in bacterial quorum sensing. Mol. Syst. Biol. 5, 325 ( 10.1038/msb.2009.79) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyons SM, Prasad A. 2012. Cross-talk and information transfer in mammalian and bacterial signaling. PLoS ONE 7, e34488 ( 10.1371/journal.pone.0034488) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cover TM, Thomas JA. 1991. Elements of information theory. New York, NY: Wiley-Interscience. [Google Scholar]
- 15.Suderman R, Bachman JA, Smith A, Sorger PK, Deeds EJ. 2017. Fundamental trade-offs between information flow in single cells and cellular populations. Proc. Natl Acad. Sci. USA 114, 5755–5760. ( 10.1073/10.1073/pnas.1615660114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee REC, Walker SR, Savery K, Frank DA, Gaudet S. 2014. Fold change of nuclear NF-κB determines TNF-induced transcription in single cells. Mol. Cell. 53, 867–879. ( 10.1016/j.molcel.2014.01.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Potter GD, Byrd TA, Mugler A, Sun B. 2017. Dynamic sampling and information encoding in biochemical networks. Biophys. J. 112, 795–804. ( 10.1016/j.bpj.2016.12.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Q, Gupta S, Schipper DL, Kowalczyk GJ, Mancini AE, Faeder JR, Lee REC. 2017. NF-κB dynamics discriminate between TNF doses in single cells. Cell Syst. 5, 638–645. ( 10.1016/j.cels.2017.10.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keshelava A, Solis GP, Hersch M, Koval A, Kryuchkov M, Bergmann S, Katanaev VL. 2018. High capacity in G protein-coupled receptor signaling. Nat. Commun. 9, 1256 ( 10.1038/s41467-018-02868-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu RC. et al. 2008. Negative feedback that improves information transmission in yeast signalling. Nature 456, 755–761. ( 10.1038/nature07513) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldbeter A, Koshland DE. 1981. An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl Acad. Sci. USA 78, 6840–6844. ( 10.1073/pnas.78.11.6840) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ventura AC. et al. 2014. Utilization of extracellular information before ligand-receptor binding reaches equilibrium expands and shifts the input dynamic range. Proc. Natl Acad. Sci. USA 111, E3860–E3869. ( 10.1073/pnas.1322761111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. 2009. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459, 428–432. ( 10.1038/nature08012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suderman R, Deeds EJ. 2013. Machines vs. ensembles: effective MAPK signaling through heterogeneous sets of protein complexes. PLoS Comput. Biol. 9, e1003278 ( 10.1371/journal.pcbi.1003278) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elowitz MB. 2002. Stochastic gene expression in a single cell. Science 297, 1183–1186. ( 10.1126/science.1070919) [DOI] [PubMed] [Google Scholar]
- 26.Swain PS, Elowitz MB, Siggia ED. 2002. Intrinsic and extrinsic contributions to stochasticity in gene expression. Proc. Natl Acad. Sci. USA 99, 12795–12800. ( 10.1073/pnas.162041399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paulsson J. 2004. Summing up the noise in gene networks. Nature 427, 415–418. ( 10.1038/nature02257) [DOI] [PubMed] [Google Scholar]
- 28.Hilfinger A, Paulsson J. 2011. Separating intrinsic from extrinsic fluctuations in dynamic biological systems. Proc. Natl Acad. Sci. USA 108, 12167–12172. ( 10.1073/pnas.1018832108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rowland MA, Deeds EJ. 2014. Crosstalk and the evolution of specificity in two-component signaling. Proc. Natl Acad. Sci. USA 111, 5550–5555. ( 10.1073/pnas.1317178111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blinov ML, Faeder JR, Goldstein B, Hlavacek WS. 2006. A network model of early events in epidermal growth factor receptor signaling that accounts for combinatorial complexity. BioSystems 83, 136–151. ( 10.1016/j.biosystems.2005.06.014) [DOI] [PubMed] [Google Scholar]
- 31.Deeds EJ, Krivine J, Feret J, Danos V, Fontana W. 2012. Combinatorial complexity and compositional drift in protein interaction networks. PLoS ONE 7, e32032 ( 10.1371/journal.pone.0032032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghaemmaghami S, Huh W, Bower K, Howson R, Belle A, Dephoure N, O’Shea EK, Weissman JS. 2003. Global analysis of protein expression in yeast. Nature 425, 737–741. ( 10.1038/nature02046) [DOI] [PubMed] [Google Scholar]
- 33.Maslov S, Ispolatov I. 2007. Propagation of large concentration changes in reversible protein-binding networks. Proc. Natl Acad. Sci. USA 104, 13655–13660. ( 10.1073/pnas.0702905104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jorgensen P, Nishikawa JL, Breitkreutz BJ, Tyers M. 2002. Systematic identification of pathways that couple cell growth and division in yeast. Science 297, 395–400. ( 10.1126/science.1070850) [DOI] [PubMed] [Google Scholar]
- 35.Hlavacek WS, Faeder JR, Blinov ML, Posner RG, Hucka M, Fontana W. 2006. Rules for modeling signal-transduction systems. Sci. Signal. 2006, re6 ( 10.1126/stke.3442006re6) [DOI] [PubMed] [Google Scholar]
- 36.Danos V, Feret J, Fontana W, Harmer R, Krivine J. 2007. Rule-based modelling of cellular signalling. In Concurrency theory: CONCUR 2007 (eds L Caires, VT Vasconcelos). Lecture Notes in Computer Science, vol. 4703, pp. 17–41. Berlin, Germany: Springer; (doi:10.1007/978-3-540-74407-8_3) [Google Scholar]
- 37.Danos V, Feret J, Fontana W, Krivine J. 2007. Scalable simulation of cellular signaling networks. In Programming languages and systems (ed. Z Shao). Lecture Notes in Computer Science, vol. 4807, pp. 139–157. Berlin, Germany: Springer; (doi:10.1007/978-3-540-76637-7_10) [Google Scholar]
- 38.Feret J, Danos V, Krivine J, Harmer R, Fontana W. 2009. Internal coarse-graining of molecular systems. Proc. Natl Acad. Sci. USA 106, 6453–6458. ( 10.1073/pnas.0809908106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chylek LA, Harris LA, Tung CS, Faeder JR, Lopez CF, Hlavacek WS. 2013. Rule-based modeling: a computational approach for studying biomolecular site dynamics in cell signaling systems. Wiley Interdiscip. Rev.: Syst. Biol. Med. 6, 13–36. ( 10.1002/wsbm.1245) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sneddon MW, Faeder JR, Emonet T. 2011. Efficient modeling, simulation and coarse-graining of biological complexity with NFsim. Nat. Methods 8, 177–183. ( 10.1038/nmeth.1546) [DOI] [PubMed] [Google Scholar]
- 41.Gillespie DT. 1977. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 81, 2340–2361. ( 10.1021/j100540a008) [DOI] [Google Scholar]
- 42.Lauffenburger DA, Linderman JJ. 1996. Receptors: models for binding, trafficking, and signalling. Int. J. Biochem. Cell Biol. 28, 1418 ( 10.1016/s1357-2725(97)89771-3) [DOI] [Google Scholar]
- 43.Rowland MA, Harrison B, Deeds EJ. 2015. Phosphatase specificity and pathway insulation in signaling networks. Biophys. J. 108, 986–996. ( 10.1016/j.bpj.2014.12.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rowland MA, Fontana W, Deeds EJ. 2012. Crosstalk and competition in signaling networks. Biophys. J. 103, 2389–2398. ( 10.1016/j.bpj.2012.10.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomez-Uribe C, Verghese GC, Mirny LA. 2007. Operating regimes of signaling cycles: statics, dynamics, and noise filtering. PLoS Comput. Biol. 3, e246 ( 10.1371/journal.pcbi.0030246) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kirouac DC, Saez-Rodriguez J, Swantek J, Burke JM, Lauffenburger DA, Sorger PK. 2012. Creating and analyzing pathway and protein interaction compendia for modelling signal transduction networks. BMC Syst. Biol. 6, 29 ( 10.1186/1752-0509-6-29) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rowland MA, Greenbaum JM, Deeds EJ. 2017. Crosstalk and the evolvability of intracellular communication. Nat. Commun. 8, 16009 ( 10.1038/ncomms16009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen RE, Thorner J. 2007. Function and regulation in MAPK signaling pathways: lessons learned from the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 1773, 1311–1340. ( 10.1016/j.bbamcr.2007.05.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kiel C, Serrano L. 2009. Cell type-specific importance of ras-c-raf complex association rate constants for MAPK signaling. Sci. Signal. 2, ra38 ( 10.1126/scisignal.2000397) [DOI] [PubMed] [Google Scholar]
- 50.Good MC, Zalatan JG, Lim WA. 2011. Scaffold proteins: hubs for controlling the flow of cellular information. Science 332, 680–686. ( 10.1126/science.1198701) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Batchelor E, Goulian M. 2003. Robustness and the cycle of phosphorylation and dephosphorylation in a two-component regulatory system. Proc. Natl Acad. Sci. USA 100, 691–696. ( 10.1073/pnas.0234782100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Skerker JM, Prasol MS, Perchuk BS, Biondi EG, Laub MT. 2005. Two-component signal transduction pathways regulating growth and cell cycle progression in a bacterium: a system-level analysis. PLoS Biol. 3, e334 ( 10.1371/journal.pbio.0030334) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ray JCJ, Igoshin OA. 2010. Adaptable functionality of transcriptional feedback in bacterial two-component systems. PLoS Comput. Biol. 6, e1000676 ( 10.1371/journal.pcbi.1000676) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cai SJ, Inouye M. 2002. EnvZ-OmpR interaction and osmoregulation in Escherichia coli. J. Biol. Chem. 277, 24155–24161. ( 10.1074/jbc.m110715200) [DOI] [PubMed] [Google Scholar]
- 55.Qin L, Yoshida T, Inouye M. 2001. The critical role of DNA in the equilibrium between OmpR and phosphorylated OmpR mediated by EnvZ in Escherichia coli. Proc. Natl Acad. Sci. USA 98, 908–913. ( 10.1073/pnas.031383098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muzzey D, Gómez-Uribe CA, Mettetal JT, van Oudenaarden A. 2009. A systems-level analysis of perfect adaptation in yeast osmoregulation. Cell 138, 160–171. ( 10.1016/j.cell.2009.04.047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Treves A, Panzeri S. 1995. The upward bias in measures of information derived from limited data samples. Neural Comput. 7, 399–407. ( 10.1162/neco.1995.7.2.399) [DOI] [Google Scholar]
- 58.Danos V, Feret J, Fontana W, Krivine J. 2008. Abstract interpretation of cellular signalling networks In Verification, model checking, and abstract interpretation (eds F Logozzo, DA Peled, LD Zuck). Lecture Notes in Computer Science, vol. 4905, pp. 83–97. Berlin, Germany: Springer; ( 10.1007/978-3-540-78163-9_1) [DOI] [Google Scholar]
- 59.Harmer R, Danos V, Feret J, Krivine J, Fontana W. 2010. Intrinsic information carriers in combinatorial dynamical systems. Chaos 20, 037108 ( 10.1063/1.3491100) [DOI] [PubMed] [Google Scholar]
- 60.Lopez CF, Muhlich JL, Bachman JA, Sorger PK. 2013. Programming biological models in Python using PySB. Mol. Syst. Biol. 9, 646 ( 10.1038/msb.2013.1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suderman R, Hlavacek WS. 2017. TRuML: a translator for rule-based modeling languages. In Proc. 8th ACM Int. Conf. on Bioinformatics, Computational Biology, and Health Informatics, Boston, MA, USA, 20–23 August 2017, pp. 372–377. New York, NY: ACM; ( 10.1145/3107411.3107471) [DOI] [Google Scholar]
- 62.Shockley EM, Vrugt JA, Lopez CF. 2018. PyDREAM: high-dimensional parameter inference for biological models in python. Bioinformatics 34, 695–697. ( 10.1093/bioinformatics/btx626) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thomas BR, Chylek LA, Colvin J, Sirimulla S, Clayton AHA, Hlavacek WS, Posner RG. 2016. BioNetFit: a fitting tool compatible with BioNetGen, NFsim and distributed computing environments. Bioinformatics 32, 798–800. ( 10.1093/bioinformatics/btv655) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suderman R, Mitra ED, Lin YT, Erickson KE, Feng S, Hlavacek WS. In press Generalizing Gillespie’s direct method to enable network-free simulations. Bull. Math. Biol. ( 10.1007/s11538-018-0418-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This article has no additional data.